Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Structure and Function of SRs
Role of SRs Family in Cancer
Role of SRs in Antitumor...
Conclusions and Perspectives
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(13):2442-2453. doi:10.7150/ijbs.46751 This issue Cite
Review
Serine/arginine-rich splicing factors: the bridge linking alternative splicing and cancer
Xiang Zheng2*, Qiu Peng4*, Lujuan Wang4*, Xuemei Zhang2, Lili Huang1, Jia Wang3 ![]() , Zailong Qin1
, Zailong Qin1 ![]()
1. Laboratory of Genetics and Metabolism, Maternal and Child Health Hospital of Guangxi Zhuang Autonomous Region; Guangxi Birth Defects Research and Prevention Institute, Nanning, Guangxi, 530003, China.
2. Department of Pathology, Affiliated Hospital of Guilin Medical University, Guilin, Guangxi, 541001, China.
3. Department of Immunology, Changzhi Medical College, Changzhi, Shanxi, 046000 China.
4. Cancer Research Institute, School of Basic Medical Science, Central South University, Changsha, Hunan, 410008, China.
*These authors contributed equally to this study.
Received 2020-4-16; Accepted 2020-6-22; Published 2020-7-6
Abstract

The serine/arginine-rich splicing factors (SRs) belong to the serine arginine-rich protein family, which plays an extremely important role in the splicing process of precursor RNA. The SRs recognize the splicing elements on precursor RNA, then recruit and assemble spliceosome to promote or inhibit the occurrence of splicing events. In tumors, aberrant expression of SRs causes abnormal splicing of RNA, contributing to proliferation, migration and apoptosis resistance of tumor cells. Here, we reviewed the vital role of SRs in various tumors and discussed the promise of analyzing mRNA alternative splicing events in tumor. Further, we highlight the challenges and discussed the perspectives for the identification of new potential targets for cancer therapy via SRs family members.
Keywords: serine and arginine-rich factors (SRs), SRSF, cancer, alternative splicing, splicing regulation, N6-methyladenosine (m6A), arginine methylation
Introduction
Alternative pre-mRNA splicing (AS) is an important gene regulation process that greatly enriches the repertoire of the transcriptome, contributing to transcriptomic and proteomic diversity [1]. AS is emerging as a pivotal step that tightly regulated in different tissues, differentiation stages and important cellular pathways in higher eukaryotes [2-4]. Growing studies showed that, AS process, which allows production of multiple mRNA variants from each gene and represents an evolutionary advantage for higher eukaryotes, and the alterations of AS contributing to various diseases including growth hormone deficiency, Parkinson's disease and cancer [5]. AS is a sophisticated and ubiquitous nuclear process, which is a natural source of cancer-causing errors in gene expression [6]. Increased high-throughput sequencing of the genome and transcriptome of cancers has unveiled a variety of means by which pre-mRNA splicing is altered in cancer [2]. As AS products are usually consistent with active roles in cancer, the alternative splicing process itself is a potential target for gene therapy.
Pre-mRNA splicing relies on the binding of spliceosome and some RNA-binding proteins (RBPs) to the pre-mRNA cis-acting element, and involves a series of RNA-RNA, RNA-protein, and protein-protein interactions [7-9]. However, there are also splicing regulatory elements, especially in the exons, including enhancers or silencers located within introns and exons region of gene (exonic or intronic splicing enhancers (ESE or ISE) or silencers (ISS or ESS)), regulating splicing by combining with the corresponding trans-acting factors [10]. As the key recognizers of these elements, serine and arginine-rich splicing factors (SRs) are concentrated in spliceosome and play vital roles in regulation of alternative splicing [11]. Extensive evidence has proved that SRs are involved in nearly every step of spliceosome assembly [12]. In addition, SRs have also been demonstrated to be associated with genomic stability, mRNA export, mRNA stability and translation [13, 14].
SRs, the important splicing regulators, belong to the serine arginine-rich protein family and canonically consisted of 12 members (SRSF1-12) [15]. The first SRs identified were SRSF1 (previously called SF2/ASF) and SRSF2 (previously called SC35) [16]. Most SRs are located exclusively in the nucleus, but some SRs (such as SRSF1, SRSF3 and SRSF7) can shuttle between nucleus and cytoplasm [17]. Because SRs play a pivotal role in transcription and translation, dysregulation of SRs greatly disrupts both DNA stability and normal protein expression pattern, then leads to abnormal biological function. Recently, it has been widely studied that SRs act as oncoproteins in various tumor types, such as lung, colon, breast, pancreas and leukemia cancer [18-21]. In this review, we summarized the function and mechanism of SRs in RNA splicing and their roles in tumor progression. We also discussed the role of SRs as potential therapeutic targets for cancer therapy.
Structure and Function of SRs
Structure of SRs
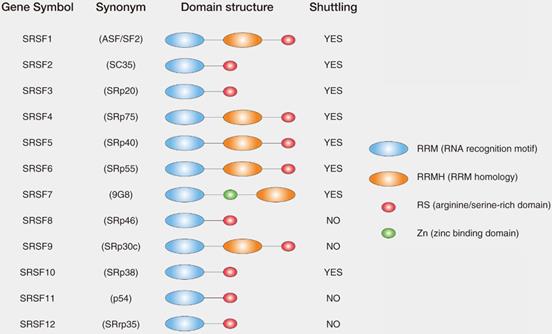
SRs are serine/arginine-rich splicing factors which are very conserved in plants and animals [22, 23]. The SRs were first discovered in the early 1990s as the essential splicing factors that influenced alternative splicing in Hela cells [24, 25]. SRs consist of 12 members in mammalian (SRSF1-12), which have the very similar domains. SRs are characterized by the presence of an N-terminal 1 or 2 RNA recognition domains (RRM domain) and a C-terminal domain enriched with the Arginine (R) and Serine (S) amino acid sequences (RS domain) [26] (Fig. 1). In general, RRM domains recognize RNA and determine the binding of SRs to RNA, while RS domains mediate diverse protein-protein and protein-RNA interactions regulated by phosphorylation [16].
List and domain structures of SRs. The domain structure of core SRs consist of RNA recognition motif (RRM), RRM homology (RRMH), arginine/serine-rich domain (RS), zinc binding domain (Zn). Synonym for SRs are also indicated in the parenthesis. Among 12 core SRs, SRSF1, SRSF2, SRSF3, SRSF4, SRSF5, SRSF6, SRSF7 and SRSF10 were reported to shuttle between nucleus and cytoplasm (shuttling SRs), whereas the other SRs have not been shown to have shuttling activity.
Function of SRs
Function of SRs in alternative pre-mRNA splicing
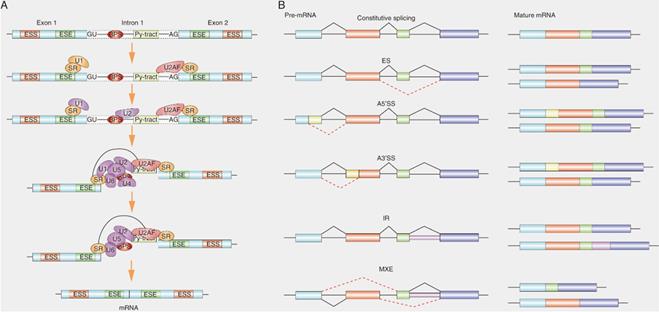
SRs commonly designate a family of RNA binding proteins that share common RNA binding motifs [15]. The unique RS domain is an important structure that distinguishes SRs from other RNA-binding proteins. As the ubiquitous splicing factors, SRs play an important role in both constitutive and alternative splicing functions. In the splicing reaction, the SRs bind to exonic splicing enhancers (ESE), recruit small nuclear ribonucleoprotein (snRNP) and its cofactors to the splicing sites [27], Next, SRs facilitate the U2 snRNA binding to the branch point region of pre-mRNA to form complex A, and then they also bridge the communication between these initial splice site recognition events in the pre-spliceosome and the mature spliceosome to form a bridge complex across the splicing sites by binding to U2AF and U1 70K, thereby promoting the recruitment of U4/U5/U6 snRNP to form complex B [28] (Fig. 2). In addition, the locations of SRs in pre-mRNA sites affect the splicing outcome. For example, exon-combined SRs act as enhancers, but intron-combined SRs may as suppressors [29]. Therefore, the location of SRs-pre-mRNA interactions affect the selection of splicing sites and the assembly of spliceosome [30].
Role of SRs in alternative splicing. (A) Graphical representation of the stepwise assembly of spliceosomal complexes. SRs bound to ESE (exonic splicing enhancer) elements recruit U2AF35 to an upstream 3' ss (splicing site) and U1 to the downstream 5' ss. SRs and U2AF2 bind, respectively, to the branch point site (BPS) and the polypyrimidine tract (Py-tract).Next, SRs recruit U2 to the BPS of pre-mRNA and then SRs form a bridge complex across two splicing sites by binding to U2AF at 3' ss and U1 at 5' binding site. Recruitment of the preassembled U4/U6/U5 tri-snRNP. Finally, major conformational changes, including release of snRNP, lead to spliceosome activation and formation of the mature mRNA. (B) Schematic depicting constitutive splicing and five examples of alternative splicing patterns are exons skipping (ES), alternative 5' splice sites (A5' SS), alternative 3' splice sites (A3'SS), intron retention (IR) and mutually exclusive exons (MXE). Shown on the right are the mature mRNA transcripts derived from each alternative splicing event.
Function of SRs in transcription
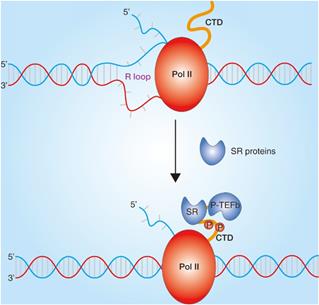
A large number of studies have found that SRs play a pivotal role in almost all processes of RNA metabolism. In addition to regulating RNA splicing, SRs also plays an important role in regulating transcription (Fig. 3) [31]. SRs are concentrated in nuclear speckles which are small subnuclear membraneless organelles or structures, also called the splicing factor compartments and are from there recruited to nascent transcripts transcribed by RNA polymerase II (Pol II) [32]. The interactions of SR-related proteins with the C-terminal domain of Pol II have been reported [33], and the SRs were also identified in the Pol II complex, which elucidates that SRs may contribute to transcriptional elongation [34]. Further studies show that SRSF2 could dynamically bridge P-TEFb to Pol II and thereby further catalyzes Pol II phosphorylation in C-terminal domain to promote transcriptional elongation in genes [35]. In addition, the co-transcriptional complex of SRs have been demonstrated to prevent the formation of R loops because of hybridization of the synthesized pre-mRNA to the complementary strand of the DNA template and thus to contribute to genome stability [14].
Role of SRs in transcription. Transcriptional regulation in chromatin. SRs bound to nascent RNA and impede the formation of R-loop and dynamically bridge P-TEFb to Pol II. Transcription elongation are regulated by Pol II phosphorylation in C-terminal repeat domain (CTD) and SRs interactions.
Function of SRs in the splicing of transcripts containing N6-methyladenosine (m6A) modifications
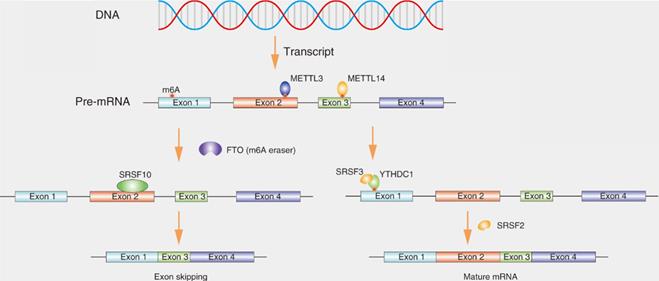
N6-methyladenosine (m6A) is the most prevalent internal RNA modification, especially within eukaryotic messenger RNAs (mRNAs), affecting various aspects of biological processes including RNA splicing (Fig. 4) [36]. m6A modifications are catalyzed by RNA methyltransferases, such as METTL3, METTL14 and WTAP (writers). The modifications are removed by the demethylases fat mass and obesity associated protein (FTO) and ALKB homolog 5 (ALKBH5) (erasers). The modifications are recognized by m6A-binding proteins, such as YTH domain-containing proteins and IGF2BPs (readers) [37]. Disruption of the dynamic of m6A can affect the levels of many RNAs, because the methylation-related molecules are located in nuclear speckles and interact with splicing factors which may be involved in modulating splicing [38].Recent studies indicate that the downregulation of ALKBH5 can affect the distribution of splicing factors, such as SRSF1, SRSF2 and SRPK1 [39]. FTO could mediate splicing of RUNX1 through recruiting SRSF2 [40]. Xiao et al. reported that YTHDC1 promotes exon inclusion in targeted mRNAs through recruiting pre-mRNA splicing factor SRSF3 while blocking SRSF10 bound to the region near the m6A sites, ultimately resulting in exon skipping owing to the failure of m6A modification [41]. Feng Z et al. also showed that METTL3 knockdown facilitates the formation of MyD88 isoform through regulating splicing [42].
SRs affects the splicing of transcripts containing N6-methyladenosine (m6A) modifications. m6A methylation of RNA is catalyzed by the methyltransferase METTL3 and METTL14. YTHDC1 could effectively binds to m6A site and recruit SRSF3 to pre-mRNA, leading to exon inclusion. If fat mass and obesity-associated (FTO) demethylates the m6A site, as a result, SRSF10 instead of SRSF2 binds to pre-mRNA, leading to exon skipping.
Role of SRs Family in Cancer
Almost all tumors have complex pathogenesis with the participation of multiple factors. The above-mentioned studies provide enough evidence to support the correlation between SRs and cancers. Because SRs are the master regulator of pre-mRNA splicing, dysregulation of SRs greatly damage DNA stability and normal expression pattern of protein, and then result in aberrant biological function, which indicates a critical role for SRs in cancer. For example, Li et al found that depletion of SRSF1 in vivo could induce DNA double-strand break and gross DNA recombination [14]. Similarly, Xiao et al also showed that loss of SRSF2 in mouse embryo fibroblasts induced double strand break and G2/M cell cycle arrest via regulating at least in part the p53 hyperphosphorylation and hyperacetylation [43]. Indeed, a series of experiments have demonstrated that SRs are aberrantly expressed in human cancers, participating in tumorigenesis and cancer metastasis (Fig. 5).
Role of SRs family in cancer. The diagram shows selected examples of genes with cancer-related alternatively spliced isoforms that due to the activity of specific SRs. Red arrowheads represent promotion and green lines represent inhibition.
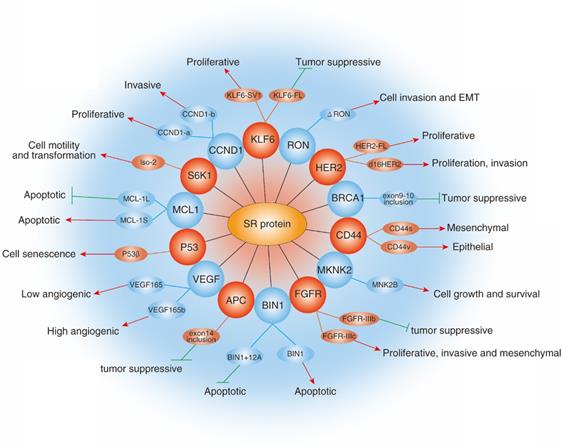
Abnormal expression of SRs and cancer
Growing evidence establish the relationship between the dysregulated expression of SRs and tumorigenesis which mainly owing to changing in the alternative splicing patterns of key genes. It has been reported that copy number of SRs are dramatically increased in various cancers, which are related to the high expression of SRs in cancers [18]. For example, SRSF6 has been found to be massively amplified in lung, colon and breast cancers [44]. Using both discovery and validation patient cohorts, Jiang L et al. reported that SRSF1 DNA copy number gain and mRNA over-expression were strongly associated with poor survival, and they further demonstrated that SRSF1 is important for tumorigenesis of SCLC (small cell lung cancer) and may play a key role in DNA repair and chemo-sensitivity both in vitro and in vivo. Moreover, SRSF1 promotes SCLC growth and survival by regulating PI3K/AKT and MEK/ERK signaling pathways, which are well-known as oncogenic signaling pathways [21]. Increased expression of SRs usually correlate with cancer progression, as in malignant ovarian tissue, the expression of SRSF1, SRSF2 and SRSF3 were significantly upregulated relative to normal tissue [45]. Park WC et al. found that the expression of SRSF7 was higher in gastric cancer than normal gastric tissue [46]. Song et al also observed that the level of SRSF7 was upregulated in NSCLC tissues, meanwhile, the expression level of SRSF7 was negatively correlated with miR-374b-5p and positively correlated with MALAT1. Overexpression of SRSF7 could reverse the effects of MALAT1 knockdown on proliferation, apoptosis, and invasion in NSCLC cells [47]. In addition, SRSF1 was found to be upregulated in colon, kidney, lung, liver, pancreas and breast tumour [18, 20, 48, 49]. Kim et al reported that the expression level of SRSF5 was markedly up-regulated in lung cancer samples compared with normal lung tissue samples and SRSF5 showed the higher detection accuracy (89%) than carcinoembryonic antigen(74%)(CEA, a commonly marker of non-small cells lung cancer (NSCLC) [50]. Yang et al also discovered that SRSF5 is overexpressed in oral squamous cell carcinoma tissues and cells. The expression of SRSF5 is controlled by an autoregulation mechanism [51]. Fu et al demonstrated that SRSF9 is frequently overexpressed in many types of tumors through immunohistochemistry staining, such as glioblastoma, colon adenocarcinoma, squamous cell lung carcinoma and malignant melanoma [52]. Other SRs, such as SRSF3, SRSF5 and SRSF6 also presented a higher expression level in colorectal cancer than normal tissue [46].
Abnormal expression of SRs often causes changes of cancer-related processes, such as epithelial-mesenchymal transition, metastasis and angiogenesis. Kong et al identified that SRSF6 promoted the EMT and metastasis in colorectal cancer, and found that the knockdown of SRSF6 significantly increased the expression of E-cadherin as well as reduced the expression of Snail, Vimentin, Fibronectin and MMP9 in SW620 cells [53]. In addition, Wan et al also found that SRSF6 could promote invasion and metastasis in colorectal cancer by inhibiting ZO-1 exon23 inclusion, which known to play an inhibitory role in colorectal cancer progression [54]. Recently, Zhou et al reported that SRSF1 is increased in glioma tissues and cell lines. Moreover, Further studies revealed that SRSF1-mediated splicing of the MYO1B increased the potential of proliferation, and metastasis in glioma cells by regulating the PDK1/AKT and PAK/LIMK pathway [55]. Angiogenesis is modulated by the balance of pro-angiogenic VEGF165 and anti-angiogenic VEGF165b splicing isoforms. Manetti et al demonstrated that SRSF6 could involve in distal splice-site selection as it bound around the distal splice-site and was up-regulated by TGFb, resulting in increased the expression of VEGF165b and inhibition of angiogenesis [56].
Dysregulated SRs' expression in cancer
| SRs | Cancer Type | Target Gene | Effects on Cancer Progression | References |
|---|---|---|---|---|
| SRSF1 | Breast, Lung, and Colon cancer | Cyclin D1, CASP9, BIN1, Mnk2 and RON1 | Cell proliferation, invasion, migration and apoptosis | [49, 63, 133-135] |
| SRSF2 | Lung and renal cancer | Caspase-8, BCL-x, MCL-1, Survivin and BIM | Apoptosis | [136, 137] |
| SRSF3 | Breast cancer | MCL-1 | Apoptosis | [66] |
| SRSF4 | Acute Myeloid Leukemia | Caspase-8 | Apoptosis | [19] |
| SRSF5 | Breast cancer | MCL-1 | Apoptosis | [66] |
| SRSF6 | Melanoma | BIM | Apoptosis | [138] |
| SRSF7 | Colon and lung cancer | Fas | Apoptosis | [47] |
| SRSF9 | Colon cancer | b-catenin | Proliferation | [52] |
| SRSF10 | Colon cancer | BCLAF1 | Apoptosis | [73] |
Splicing function of SRs and cancer
Most genes undergo alternative splicing, and the isoforms produced by alternative splicing are closely related to the progression and metastasis of cancers [57], such as pro-oncogenic and antiapoptotic splicing isoforms of MDM2, BCL(X), and VEGF genes [58]. In tumor cells, SRs cause abnormal splicing of many tumor-related genes, which affect cell proliferation, apoptosis and migration, and thereby promotes tumorigenesis and cancer development [59, 60]. Karni R et al. found that upregulated SRSF1 protein increased the splice isoform of BIN1 (bridging integrator-1). The resulting BIN1 isoforms cannot bind to the oncoprotein MYC, causing the BIN1 protein to lack tumor-suppressor activity [18]. A series of studies found that dysregulated expression of SRSF1 protein can also alter alternative splicing of many other tumor-related genes. For example, in prostate cancer, SRSF1 promotes the increase of oncogenic cyclin D1b splice isoform [61]. In lung cancer, SRSF1 promotes the expression of the anti-apoptotic caspase 9b splice isoform [62]. In breast cancer, SRSF1 regulates the alternative splicing of the pro-apoptotic gene BIM, which increases the expression of splice variant lacking the BH3 domain, thereby inhibiting apoptosis [63]. In addition, the alternative splicing regulated by SRSF1 in tumor cells also includes the proto-oncogenes MYBL2 (B-myb), MYB (C-myb) [64], the tumor suppressor gene BRCA1 [65], and the apoptosis-related gene Mcl1 [66], Vascular endothelial growth factor VEGF [67], cell migration-related gene MST1R (Ron) [49], etc. In addition, Kurokawa K et al. found that SRSF3 can regulate apoptosis by affecting alternative splicing of HIPK2 (homeodomain-interacting protein kinase-2) and produced a HIPK2 Δe8 isoform in colon cancer cells [68]. Liu et al found that compared with control, the expression of SRSF4 was obviously decreased in Acute Myeloid Leukemia (AML) patients. Moreover, an important correlation was observed between SRSF4 and the caspase-8 splicing isoforms in AML patients [19]. Shi J et al. reported that SRSF2 involved in the regulation of alternative splicing of the tumor suppressor gene KLF6 (Krüppel-like factor 6) and promoted the production of splice isoform containing KLF6 exon 1a. The isoform of KLF6 which cannot inhibit tumorigenesis due to the absence of the DNA binding domain [69]. SRSF6 can regulate the alternative splicing of tumor-related genes, such as the insulin receptor INSR, the kinase Mnk2 (MKNK2) and the tumor suppressor gene DLG-1, thereby mediating tumorigenesis [70-72]. Zhou X et al. showed that SRSF10 was up-regulated in colon cancer samples, and inhibition of SRSF10 could significantly inhibit the proliferation and tumorigenesis in colon cancer cells by regulating the expression of Bcl-2-associated transcription factor 1 (BCLAF1) isoform containing BCLAF1 exon5a [73]. Recently, Liu et al showed that SRSF10, was upregulated by human papillomavirus oncoproteins E6 and E7 through E2F1 pathway, and promoted the tumorigenesis of cervix. Further investigation that SRSF10 could promote the expression of CD47 to inhibit macrophage phagocytosis by promoting nuclear factor-κB activation by regulating the splicing of interleukin-1 receptor accessory protein exon 13.
Recent genome-wide analyses of cancer have revealed globally abnormal splicing profiles in mature mRNA [74, 75]. The molecular mechanisms of aberrant splicing in cancers may be mutations in SRs, such as SRSF2 [76]. SRSF2 mutation is found in 40% -50% of patients in chronic myelomonocytic leukaemia [77]. SRSF2 missense mutation are highly concentrate on at residue P95 or to a lesser extent at the surrounding residues (G93 to P107) [78]. SRSF2 canonically binds to C- and G-rich exonic splicing enhancers (ESEs) motifs to promote splicing [79]. The mutant SRSF2 preferentially recognizes C-rich CCNG ESE motif versus G-rich GGNG ESE motif and results in alternatively splicing of many mRNAs [80]. For example, Papaemmanuil et al reported that SRSF2 mutations altered the splicing of EZH2 mRNA that induced an in-frame stop codon in myeloid neoplasms. The abnormal EZH2 mRNA induces nonsense‑mediated decay (NMD) and leads to the downregulation of EZH2 protein and histone H3 lysine 27 trimethylation levels [81].
Translation function of SRs and cancer
We mentioned above that the nuclear shuttle SR proteins can directly regulate mRNA translation [82]. Sanford JR et al. reported that SRSF1 could stimulate translation both in vitro using a HeLa cell-free translation system and in xenopus oocytes system [83]. As an adaptor protein, SRSF1 recruits mTORC1 or PP2A to its bound mRNA, and increases the phosphorylation level of 4E-BP1. Phosphorylated 4E-BP1 no longer binds to the initiation factor eIF4E, which promotes mRNA translation initiation [84]. Karni R et al. have found that overexpression of SRSF1 could activate the mTORC1 signaling pathway, resulting in increased phosphorylation of its downstream target proteins S6K and 4E-BP1, and that the tumorigenesis of SRSF1 can be inhibited by the mTOR-specific inhibitor rapamycin [85]. Moreover, SRSF1 can activate the Ras-MAPK signaling pathway and increase cell transformation by increasing B-Raf expression [86]. Maslon MM et al. also performed a high-throughput deep sequencing analysis of polysomal fractions in cells overexpressing SRSF1 and identified approximately 1,500 mRNAs that were translational targets of SRSF1. These include mRNAs encoding proteins involved in cell cycle regulation, such as spindle, kinetochore and M phase proteins, which are essential for accurate chromosome segregation [87]. These findings suggest that the regulation of translational functions of the SRSF1 protein promotes, to a certain extent, SRSF1 protein-mediated cell transformation and tumorigenesis. Other shuttling SR proteins like SRSF7 and SRSF3 also have the function of regulating translation [88, 89]. For example, SRSF3 was reported to involve in the regulation of IRES (internal ribosomal entry site)-mediated translation initiation. Further investigation found that SRSF3 interacted with the RNA binding protein PCBP2 that bound to IRES sequences in the genomic RNAs of viruses and was necessary for viral translation. [88].
Post-translational modification of SRs and cancer
At least five types of posttranscriptional modifications are known in SRs, including methylation, acetylation, small ubiquitin-related modifier (SUMO), ubiquitination and phosphorylation [22]. There has been considerable interest in modulating splicing by modifying post-translational modifications (PTM) required for spliceosome function, and known that PTMs of a variety of splicing factors regulate spliceosome formation and splicing catalysis [90]. Arginine methylation has been detected on many RNA binding proteins, such as hnRNPs and SRs [91]. Arginine methylation in SRs was first reported on Npl3p in budding yeast [92]. In addition, Sinha R et al. reported that three methylated arginine residues were identified between the two RRMs in SRSF1, which appeared to play an important role in promoting nuclear import of the SRs. Further studies showed that arginine control the subcellular localization and the positive charge state. Mutations or block methylation and remove the positive charge lead to the cytoplasmic accumulation of SRSF1 [93]. Botti et al also showed that arginine methylation in SRSF5 is important for its shuttling capacity and mRNA export [94]. Ubiquitin proteasome pathway-mediated protein degradation is an important mechanism by which the cell regulates intracellular protein levels and functions. Vaishali et al reported that ubiquitination can regulate the expression of SRSF1 in normal and T cells [95]. In addition, Osman et al showed that SRSF5 protein is decreased drastically during cell differentiation by proteasome-mediated proteolysis [96]. Chen et al also showed that mutually exclusive ubiquitylation and acetylation of SRSF5 control tumor growth by regulating the alternative splicing of CCAR1 to generate the CCAR1S isoform, which promote tumor growth by promoting glucose consumption and acetyl-CoA production [97]. In addition, reversible phosphorylation in the splicing reaction was necessary. Phosphorylation of SRs modulates the shuttling of SRs to modulate splicing. Phosphorylation of SRs is necessary for spliceosome assembly, but dephosphorylation of SRs allows splicing catalysis to occur and initiates nuclear export of SRs [28, 98, 99]. Phosphorylation of the RS repeat domain could release the stable α-helical structure to form the "arginine claw" structures which is recognized by a SRs specific transportin (TRN-SR), thereby trafficking the SRs to nucleus where it involves in splicing [100]. Serine-arginine protein kinase (SRPK) can phosphorylate Arg-Ser dipeptide repeats of SRs and promote nuclear import [101]. Abnormal expression of SRPK1 damages the biological function of SRs, leading to aberrant splicing of pre-mRNA, which can contribute to the progression of cancers [102]. Matos et al reported that SRPK1 could regulate the alternative splicing of Rac1b that enhanced cell cycle progression via NF-kB signaling [103]. SRPK1 contributes to the splicing of Rac1b by inducing the phosphorylation of SRSF1 to further promote its entry into the nucleus [104]. Small ubiquitin-related modifier (SUMO) is a reversible and transient posttranslational protein modifier and involved in diverse biological processes, such as subcellular partitioning, transcription regulation, DNA damage repair, stress response and chromatin remodeling. Federico et al showed that the SRSF1 was a modulator of the sumoylation pathway. SRSF1 can enhance sumoylation of specific substrates by interacting with Ubc9. SRSF1 also can regulate SUMO E3 ligase protein inhibitor of activated STAT-1(PIAS1)-induced overall protein sumoylation by interacting with the PIAS1 [105]. In recent years, proteomic analysis has also shown extensive lysine acetylation in SRs, which may be an important nonhistone substrates recognized by histone acetyltransferases (HATs) in cells [106]. Edmond V et al. found that the acetyltransferase Tip60 acetylated SRSF2 on its lysine 52 residue inside the RNA recognition motif, thereby promoted its proteasomal degradation, while the deacetylase HDAC6 countered this acetylation and acted as a positive regulator of SRSF2 protein level [107].
Other functions of SRs in cancer
SRs regulate RNA metabolism at multiple levels. In addition to participating in the regulation of RNA splicing and translation efficiency, SRSF1 protein also plays a role in the nuclear export and stability of RNA. These functions may be involved in SRSF1-mediated tumorigenesis. For example, SRSF1 is highly expressed in non-small cell lung cancer. Down-regulating SRSF1 in non-small cell lung cancer cell lines can promote the occurrence of apoptosis, which is related to the down-regulation of anti-apoptotic protein survivin. Mechanism shows that SRSF1 specifically binds to survivin mRNA and regulates its expression at multiple levels. It not only relies on the mTOR signaling pathway to promote translation of survivin mRNA, but also stabilizes its mRNA level. The analysis of clinical tumor samples further confirms that there is a positive correlation between SRSF1 and survivin in non-small cell lung cancer. These results provide new data on the mTORC1- and survivin-dependent mechanisms of SRSF1-related carcinogenic potential, and suggest that the expression of SRSF1 and survivin is involved in NSCLC progression [108]. SRSF1 is also up-regulated in breast cancer. Overexpression of SRSF1 promotes cell transformation of breast epithelial cells both in vitro SRSF1-overexpressing MCF-10A cells and in vivo SRSF1-overexpressing COMMA-1D transplantation models, and promotes proliferation and inhibits apoptosis in three-dimensional culture cells, forming more acinar than normal cells [63]. Overexpression of SRSF1 in mammary epithelial cells not only regulates the alternative splicing of BIM and BIN1 to inhibit apoptosis, but also cooperates with the oncogene MYC to promote cell transformation [63]. Subsequent studies have further confirmed that MYC regulates its downstream splicing activity and tumorigenesis by promoting SRSF1 transcriptional activation [63].
Role of SRs in Antitumor Therapies
Because SRs play an important role in RNA metabolism, during the past decades, increasing strategies have been made to target the spliceosome for antitumor therapeutic approaches [109, 110]. As described above, it has been extensively reported that widespread changes in RNA splicing in cancer samples compared with normal samples, such as known pro-oncogenic and antiapoptotic isoforms of genes, including TP53, BCL (X), and VEGF [58, 111, 112]. Guyot et al. reported that anti-angiogenic forms of VEGF isoform VEGF165b can weaken the therapeutic effects of the anti-VEGF antibodies (bevacizumab/Avastin) in different tumor cell lines. [113]. These findings showed that regulating pre-RNA splicing of specific gene isoforms may serve as a new therapeutic strategy for cancer. Recently, a series of studies unveiled diverse compounds that targeted the spliceosome. Among the first compounds known to alter splicing are natural products (spliceostatin A, pladienolide B, and herboxidiene) that bind the SF3B complex [114, 115], and their derivatives including E7107 and H3B-8800 [116], which physically bind to the SF3B complex and inhibit pre-mRNA splicing at an early step in spliceosome assembly and thereby block splicing. In addition, Lee SC et al. showed that mutated SRSF2 are more sensitive to E7107 in comparison to cells bearing wild-type SRSF2 in leukemia, suggesting that cancers induced by spliceosome mutated factors was more sensitive to these drugs [117].
Chemoresistance and radioresistance are the major cause of cancer treatment failure. Splicing dysregulation plays key roles in tumorigenesis and the involvement of SRs in resistance of cancer to chemotherapy and radiotherapy remains elusive [118]. Recently, The expression level of SRSF1 was found increased in irradiation treated lung cancer cells, whereas interference of SRSF1 sensitized lung cancer cells to irradiation by regulating various cancer-related splicing, such as the splicing of PTEN-like mitochondrial phosphatase (PTPMT1) [118]. Maude et al reported that SRSF4, but not SRSF6 played an important role in anti-cancer drug cisplatin affected splicing, and knockdown of SRSF4 expression inhibited lots of the splicing alterations and cell death induced by cisplatin [119].
Alternative splicing regulation is also a key target for anti-tumor treatment. SRs are phosphorylated by SRPK1 and SRPK2 (SRs kinases), DNA topoisomerase I and U4/U6 small ribonucleoprotein PRP4 [120]. Through regulation of SRs phosphorylation, small molecule inhibitors that target kinases involved in alternative splicing are promising candidates for drugs [121]. Pilch B et al. found that NB-506, an inhibitor of topoisomerase I, could inhibit the phosphorylation of SRSF1 and modulate gene expression by controlling splicing [122]. In addition, Wu F et al. reported that antibody targeting SRPK1, a key regulator of SRs phosphorylation, significantly inhibited the growth, migration and invasion of non-small cell lung cancer [123]. PRP4 belongs to the serine/threonine protein kinase family, plays an important role in cell mitosis and splicing. PRP4 kinase has been identified as a potential therapeutic target in multiple tumors. For example, Duan et al found that inhibition of PRP4 activity by shRNAs could reverse paclitaxel resistance 5-10-fold in human ovarian cancer [124]. Jouhua et al also showed that inhibition of PRP4 could enhance paclitaxel activity in breast cancer cells [125].
In another example, symmetric arginine dimethylation (SDMA), catalyzed by PRMT5, is required for spliceosome assembly [126, 127]. Some studies shown that compounds inhibition of PRMT5 results in splicing inhibition and anticancer effects in a number of cancer types [128, 129]. Recent studies positioned RNA methylation as a major pathway in modulating the development of tumor due to its function in post-transcriptional regulation including alternative splicing. It is of great significance to identify chemical inhibitors targeting m6A modification-related enzymes, including METTL3, METTL14, FTO and ALKBH5, etc [130].
Conclusions and Perspectives
Alternative splicing is a vital step in gene expression and generates transcriptome diversity in eukaryotic cells. As the key regulators of cellular biology, SRs are extensively participated in regulating from RNA stability to RNA splicing, and even RNA modification(m6A), export and translation [14, 84, 131]. Moreover, the expression of most SRs in various tumor types is up-regulated, and therefore SRs promotes tumorigenesis as proto-oncogenes. SRs mediate tumorigenesis mainly by regulating alternative splicing of tumor-related genes, providing a large number of targets for tumor prevention and treatment. Since most of the current research is focused on a single SR protein, this also leads to some interesting problems that have not been well explained. For example, do SRs regulated targets have something in common? are these targets regulated by many SR proteins or specifically by one? is the regulatory mechanism similar for all these targets? At present, it is known that antisense oligonucleotides or small molecule drugs can be used to regulate some alternative splicing processes that mediate tumorigenesis [132]. These may be one of the measures to achieve tumor treatment by targeting SRs in tumorigenesis. However, due to relatively few animal models system for knock-out/knock-in SRs, the regulation of SRs is still incomplete, and more research is needed in this area to further determine the functions and mechanisms of different SRs in tumorigenesis, which will provide more abundant research ideas and treatment strategies for tumor treatment. In addition, with the broad application of bioinformatics and high-throughput sequencing technology in research, increasing numbers mutations of SRs have been found in tumors. What specific role do these mutations play in the progression of tumors? And whether repairing these mutations can be used as a potential treatment strategy is still unknown. Of course, I believe that with the advancement of research methods, more and more mysteries about SRs will be unveiled.
Abbreviations
SRs/SRSF: serine/arginine-rich splicing factors; AS: Alternative splicing; m6A: N6-methyladenosine; RBP: RNA-binding protein; ESE: exonic splicing enhancer; ISE: intronic splicing enhancer; ESS: exonic splicing silencer; ISS: intronic silencer; RRM: RNA recognition; hnRNP: heterogeneous nuclear ribonucleoparticle protein; snRNP: small nuclear ribonucleoprotein; P-TEFb: The positive transcription elongation factor b; METTL3: Methyltransferase-Like Protein 3; METTL14: Methyltransferase-Like Protein 14; WTAP: Wilms Tumor 1 Associated Protein; FTO: Fat Mass and Obesity-Associated Protein; ALKBH5: Alpha-Ketoglutarate-Dependent Dioxygenase AlkB Homolog 5; YTHDF: YTH Domain-Containing Family Protein 1; IGF2BP: Insulin Like Growth Factor 2 MRNA Binding Protein; RUNX1: RUNX Family Transcription Factor 1; YTHDC1: YTH Domain-Containing Protein 1; SCLC: small cell lung cancer; BIN1: Bridging Integrator 1; MYC: MYC proto-oncogene; BIM: Bcl-2-Like Protein 11; MYBL2: MYB Proto-Oncogene Like 2; MYB: MYB Proto-Oncogene; BRCA1: Breast Cancer Type 1 Susceptibility Protein; Mcl1: Myeloid Cell Leukemia Sequence 1; VEGF: Vascular Endothelial Growth Factor; MST1R: Macrophage Stimulating 1 Receptor; HIPK2: Homeodomain Interacting Protein Kinase 2; KLF6: Kruppel Like Factor 6; INSR: Insulin Receptor; Mnk2: MAPK Interacting Serine/Threonine Kinase 2; DLG-1: Discs Large MAGUK Scaffold Protein 1; MTORC1: Mechanistic Target of Rapamycin Kinase Complex 1; PP2A: Protein Phosphatase 2 Phosphatase Activator; S6K: Ribosomal Protein S6 Kinase B1; 4E-BP1: Eukaryotic Translation Initiation Factor 4E Binding Protein 1; MAPK: Mitogen-Activated Protein Kinase; B-Raf: B-Raf Proto-Oncogene Serine/Threonine Kinase; PTM: post-translational modifications; HATs: histone acetyltransferases; HDAC6: Histone Deacetylase 6; SF3B: Splicing Factor 3b Subunit; SRPK1: Serine/Arginine-Rich Protein-Specific Kinase 1; SRPK2: Serine/Arginine-Rich Protein-Specific Kinase 2; PRP4: Pre-MRNA Processing Factor 4; SDMA: symmetric arginine demethylation; RMT5: Protein Arginine Methyltransferase 5.
Acknowledgements
This work was supported in part by grants from the National Natural Science Foundation of China (81702919, 81802478), Guangxi Natural Science Foundation (2017GXNSFBA198071, 2018GXNSFBA050042), Guangxi Science and Technology Program Project (AD19245005), the Natural Science Foundation of Hunan Province (No.2020JJ5480) and the Applied Basic Research Programs of Science and Technology Commission Foundation of Shanxi Province (201901D211472).
Authors' contributions
XZ, QP, LW, XZ and LH collected the related paper and drafted the manuscript. JW and ZQ revised and finalized the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Adamia S, Pilarski PM, Bar-Natan M, Stone RM, Griffin JD. Alternative splicing in chronic myeloid leukemia (CML): a novel therapeutic target? Curr Cancer Drug Targets. 2013;13:735-48
2. Kalsotra A, Cooper TA. Functional consequences of developmentally regulated alternative splicing. Nat Rev Genet. 2011;12:715-29
3. Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB. et al. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc Natl Acad Sci U S A. 2008;105:20333-8
4. Yap K, Lim ZQ, Khandelia P, Friedman B, Makeyev EV. Coordinated regulation of neuronal mRNA steady-state levels through developmentally controlled intron retention. Genes Dev. 2012;26:1209-23
5. Sette C. Alternative splicing programs in prostate cancer. Int J Cell Biol. 2013;2013:458727
6. Venables JP. Aberrant and alternative splicing in cancer. Cancer Res. 2004;64:7647-54
7. Yan C, Hang J, Wan R, Huang M, Wong CC, Shi Y. Structure of a yeast spliceosome at 3.6-angstrom resolution. Science. 2015;349:1182-91
8. Wan R, Yan C, Bai R, Wang L, Huang M, Wong CC. et al. The 3.8 A structure of the U4/U6.U5 tri-snRNP: Insights into spliceosome assembly and catalysis. Science. 2016;351:466-75
9. Zhang X, Yan C, Hang J, Finci LI, Lei J, Shi Y. An Atomic Structure of the Human Spliceosome. Cell. 2017;169:918-29 e14
10. Singh RK, Cooper TA. Pre-mRNA splicing in disease and therapeutics. Trends Mol Med. 2012;18:472-82
11. Jeong S. SR Proteins: Binders, Regulators, and Connectors of RNA. Mol Cells. 2017;40:1-9
12. Savisaar R, Hurst LD. Purifying Selection on Exonic Splice Enhancers in Intronless Genes. Mol Biol Evol. 2016;33:1396-418
13. Huang Y, Gattoni R, Stevenin J, Steitz JA. SR splicing factors serve as adapter proteins for TAP-dependent mRNA export. Mol Cell. 2003;11:837-43
14. Li X, Manley JL. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell. 2005;122:365-78
15. Busch A, Hertel KJ. Evolution of SR protein and hnRNP splicing regulatory factors. Wiley Interdiscip Rev RNA. 2012;3:1-12
16. Manley JL, Krainer AR. A rational nomenclature for serine/arginine-rich protein splicing factors (SR proteins). Genes Dev. 2010;24:1073-4
17. Zahler AM, Neugebauer KM, Stolk JA, Roth MB. Human SR proteins and isolation of a cDNA encoding SRp75. Mol Cell Biol. 1993;13:4023-8
18. Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol. 2007;14:185-93
19. Liu J, Huang B, Xiao Y, Xiong HM, Li J, Feng DQ. et al. Aberrant expression of splicing factors in newly diagnosed acute myeloid leukemia. Onkologie. 2012;35:335-40
20. Anczukow O, Akerman M, Clery A, Wu J, Shen C, Shirole NH. et al. SRSF1-Regulated Alternative Splicing in Breast Cancer. Mol Cell. 2015;60:105-17
21. Jiang L, Huang J, Higgs BW, Hu Z, Xiao Z, Yao X. et al. Genomic Landscape Survey Identifies SRSF1 as a Key Oncodriver in Small Cell Lung Cancer. PLoS Genet. 2016;12:e1005895
22. Zhou Z, Fu XD. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma. 2013;122:191-207
23. Howard JM, Sanford JR. The RNAissance family: SR proteins as multifaceted regulators of gene expression. Wiley Interdiscip Rev RNA. 2015;6:93-110
24. Ge H, Zuo P, Manley JL. Primary structure of the human splicing factor ASF reveals similarities with Drosophila regulators. Cell. 1991;66:373-82
25. Krainer AR, Mayeda A, Kozak D, Binns G. Functional expression of cloned human splicing factor SF2: homology to RNA-binding proteins, U1 70K, and Drosophila splicing regulators. Cell. 1991;66:383-94
26. Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J. 2009;417:15-27
27. Sahebi M, Hanafi MM, van Wijnen AJ, Azizi P, Abiri R, Ashkani S. et al. Towards understanding pre-mRNA splicing mechanisms and the role of SR proteins. Gene. 2016;587:107-19
28. Cho S, Hoang A, Sinha R, Zhong XY, Fu XD, Krainer AR. et al. Interaction between the RNA binding domains of Ser-Arg splicing factor 1 and U1-70K snRNP protein determines early spliceosome assembly. Proc Natl Acad Sci U S A. 2011;108:8233-8
29. Shen M, Mattox W. Activation and repression functions of an SR splicing regulator depend on exonic versus intronic-binding position. Nucleic acids research. 2012;40:428-37
30. Erkelenz S, Mueller WF, Evans MS, Busch A, Schoneweis K, Hertel KJ. et al. Position-dependent splicing activation and repression by SR and hnRNP proteins rely on common mechanisms. Rna. 2013;19:96-102
31. Twyffels L, Gueydan C, Kruys V. Shuttling SR proteins: more than splicing factors. FEBS J. 2011;278:3246-55
32. Misteli T, Caceres JF, Spector DL. The dynamics of a pre-mRNA splicing factor in living cells. Nature. 1997;387:523-7
33. Yuryev A, Patturajan M, Litingtung Y, Joshi RV, Gentile C, Gebara M. et al. The C-terminal domain of the largest subunit of RNA polymerase II interacts with a novel set of serine/arginine-rich proteins. Proc Natl Acad Sci U S A. 1996;93:6975-80
34. Das R, Yu J, Zhang Z, Gygi MP, Krainer AR, Gygi SP. et al. SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol Cell. 2007;26:867-81
35. Lin S, Coutinho-Mansfield G, Wang D, Pandit S, Fu XD. The splicing factor SC35 has an active role in transcriptional elongation. Nat Struct Mol Biol. 2008;15:819-26
36. Dai D, Wang H, Zhu L, Jin H, Wang X. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018;9:124
37. Zhao W, Qi X, Liu L, Liu Z, Ma S, Wu J. Epigenetic Regulation of m(6)A Modifications in Human Cancer. Mol Ther Nucleic Acids. 2019;19:405-12
38. Yang Y, Sun B-F, Xiao W, Yang X, Sun H-Y, Zhao Y-L. et al. Dynamic m 6 A modification and its emerging regulatory role in mRNA splicing. Science Bulletin. 2015;60:21-32
39. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ. et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18-29
40. Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W. et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24:1403-19
41. Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF. et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell. 2016;61:507-19
42. Feng Z, Li Q, Meng R, Yi B, Xu Q. METTL3 regulates alternative splicing of MyD88 upon the lipopolysaccharide-induced inflammatory response in human dental pulp cells. J Cell Mol Med. 2018;22:2558-68
43. Xiao R, Sun Y, Ding JH, Lin S, Rose DW, Rosenfeld MG. et al. Splicing regulator SC35 is essential for genomic stability and cell proliferation during mammalian organogenesis. Mol Cell Biol. 2007;27:5393-402
44. Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol. 2005;6:635-45
45. Fischer DC, Noack K, Runnebaum IB, Watermann DO, Kieback DG, Stamm S. et al. Expression of splicing factors in human ovarian cancer. Oncol Rep. 2004;11:1085-90
46. Park WC, Kim HR, Kang DB, Ryu JS, Choi KH, Lee GO. et al. Comparative expression patterns and diagnostic efficacies of SR splicing factors and HNRNPA1 in gastric and colorectal cancer. BMC Cancer. 2016;16:358
47. Fu Y, Wang Y. SRSF7 knockdown promotes apoptosis of colon and lung cancer cells. Oncology letters. 2018;15:5545-52
48. de Miguel FJ, Sharma RD, Pajares MJ, Montuenga LM, Rubio A, Pio R. Identification of alternative splicing events regulated by the oncogenic factor SRSF1 in lung cancer. Cancer Res. 2014;74:1105-15
49. Ghigna C, Giordano S, Shen H, Benvenuto F, Castiglioni F, Comoglio PM. et al. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol Cell. 2005;20:881-90
50. Kim HR, Lee GO, Choi KH, Kim DK, Ryu JS, Hwang KE. et al. SRSF5: a novel marker for small-cell lung cancer and pleural metastatic cancer. Lung cancer. 2016;99:57-65
51. Yang S, Jia R, Bian Z. SRSF5 functions as a novel oncogenic splicing factor and is upregulated by oncogene SRSF3 in oral squamous cell carcinoma. Biochimica et biophysica acta Molecular cell research. 2018;1865:1161-72
52. Fu Y, Huang B, Shi Z, Han J, Wang Y, Huangfu J. et al. SRSF1 and SRSF9 RNA binding proteins promote Wnt signalling-mediated tumorigenesis by enhancing beta-catenin biosynthesis. EMBO molecular medicine. 2013;5:737-50
53. Kong J, Sun W, Li C, Wan L, Wang S, Wu Y. et al. Long non-coding RNA LINC01133 inhibits epithelial-mesenchymal transition and metastasis in colorectal cancer by interacting with SRSF6. Cancer letters. 2016;380:476-84
54. Wan L, Yu W, Shen E, Sun W, Liu Y, Kong J. et al. SRSF6-regulated alternative splicing that promotes tumour progression offers a therapy target for colorectal cancer. Gut. 2019;68:118-29
55. Zhou X, Wang R, Li X, Yu L, Hua D, Sun C. et al. Splicing factor SRSF1 promotes gliomagenesis via oncogenic splice-switching of MYO1B. The Journal of clinical investigation. 2019;129:676-93
56. Manetti M, Guiducci S, Romano E, Ceccarelli C, Bellando-Randone S, Conforti ML. et al. Overexpression of VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, leads to insufficient angiogenesis in patients with systemic sclerosis. Circulation research. 2011;109:e14-26
57. Oltean S, Bates DO. Hallmarks of alternative splicing in cancer. Oncogene. 2014;33:5311-8
58. Zhang J, Manley JL. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov. 2013;3:1228-37
59. David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 2010;24:2343-64
60. Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet. 2016;17:19-32
61. Paronetto MP, Cappellari M, Busa R, Pedrotti S, Vitali R, Comstock C. et al. Alternative splicing of the cyclin D1 proto-oncogene is regulated by the RNA-binding protein Sam68. Cancer Res. 2010;70:229-39
62. Olsson M, Zhivotovsky B. Caspases and cancer. Cell Death Differ. 2011;18:1441-9
63. Anczukow O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R. et al. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol. 2012;19:220-8
64. Kumar A, Baker SJ, Lee CM, Reddy EP. Molecular mechanisms associated with the regulation of apoptosis by the two alternatively spliced products of c-Myb. Mol Cell Biol. 2003;23:6631-45
65. Mixon M, Kittrell F, Medina D. Expression of Brca1 and splice variant Brca1delta11 RNA levels in mouse mammary gland during normal development and tumorigenesis. Oncogene. 2000;19:5237-43
66. Gautrey HL, Tyson-Capper AJ. Regulation of Mcl-1 by SRSF1 and SRSF5 in cancer cells. PLoS One. 2012;7:e51497
67. Amin EM, Oltean S, Hua J, Gammons MV, Hamdollah-Zadeh M, Welsh GI. et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell. 2011;20:768-80
68. Kurokawa K, Akaike Y, Masuda K, Kuwano Y, Nishida K, Yamagishi N. et al. Downregulation of serine/arginine-rich splicing factor 3 induces G1 cell cycle arrest and apoptosis in colon cancer cells. Oncogene. 2014;33:1407-17
69. Shi J, Hu Z, Pabon K, Scotto KW. Caffeine regulates alternative splicing in a subset of cancer-associated genes: a role for SC35. Mol Cell Biol. 2008;28:883-95
70. Scheper GC, Morrice NA, Kleijn M, Proud CG. The mitogen-activated protein kinase signal-integrating kinase Mnk2 is a eukaryotic initiation factor 4E kinase with high levels of basal activity in mammalian cells. Mol Cell Biol. 2001;21:743-54
71. Cohen-Eliav M, Golan-Gerstl R, Siegfried Z, Andersen CL, Thorsen K, Orntoft TF. et al. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J Pathol. 2013;229:630-9
72. Wang H, Lekbaby B, Fares N, Augustin J, Attout T, Schnuriger A. et al. Alteration of splicing factors' expression during liver disease progression: impact on hepatocellular carcinoma outcome. Hepatol Int. 2019;13:454-67
73. Zhou X, Li X, Cheng Y, Wu W, Xie Z, Xi Q. et al. BCLAF1 and its splicing regulator SRSF10 regulate the tumorigenic potential of colon cancer cells. Nat Commun. 2014;5:4581
74. Dvinge H, Bradley RK. Widespread intron retention diversifies most cancer transcriptomes. Genome medicine. 2015;7:45
75. Danan-Gotthold M, Golan-Gerstl R, Eisenberg E, Meir K, Karni R, Levanon EY. Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic acids research. 2015;43:5130-44
76. Dvinge H, Kim E, Abdel-Wahab O, Bradley RK. RNA splicing factors as oncoproteins and tumour suppressors. Nature reviews Cancer. 2016;16:413-30
77. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R. et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64-9
78. Hahn CN, Venugopal P, Scott HS, Hiwase DK. Splice factor mutations and alternative splicing as drivers of hematopoietic malignancy. Immunological reviews. 2015;263:257-78
79. Daubner GM, Clery A, Jayne S, Stevenin J, Allain FH. A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. The EMBO journal. 2012;31:162-74
80. Zhang J, Lieu YK, Ali AM, Penson A, Reggio KS, Rabadan R. et al. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci U S A. 2015;112:E4726-34
81. Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P. et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616-27 quiz 99
82. Sato H, Hosoda N, Maquat LE. Efficiency of the pioneer round of translation affects the cellular site of nonsense-mediated mRNA decay. Mol Cell. 2008;29:255-62
83. Sanford JR, Gray NK, Beckmann K, Caceres JF. A novel role for shuttling SR proteins in mRNA translation. Genes Dev. 2004;18:755-68
84. Michlewski G, Sanford JR, Caceres JF. The splicing factor SF2/ASF regulates translation initiation by enhancing phosphorylation of 4E-BP1. Mol Cell. 2008;30:179-89
85. Karni R, Hippo Y, Lowe SW, Krainer AR. The splicing-factor oncoprotein SF2/ASF activates mTORC1. Proc Natl Acad Sci U S A. 2008;105:15323-7
86. Shimoni-Sebag A, Lebenthal-Loinger I, Zender L, Karni R. RRM1 domain of the splicing oncoprotein SRSF1 is required for MEK1-MAPK-ERK activation and cellular transformation. Carcinogenesis. 2013;34:2498-504
87. Maslon MM, Heras SR, Bellora N, Eyras E, Caceres JF. The translational landscape of the splicing factor SRSF1 and its role in mitosis. Elife. 2014: e02028.
88. Bedard KM, Daijogo S, Semler BL. A nucleo-cytoplasmic SR protein functions in viral IRES-mediated translation initiation. EMBO J. 2007;26:459-67
89. Swartz JE, Bor YC, Misawa Y, Rekosh D, Hammarskjold ML. The shuttling SR protein 9G8 plays a role in translation of unspliced mRNA containing a constitutive transport element. J Biol Chem. 2007;282:19844-53
90. Obeng EA, Stewart C, Abdel-Wahab O. Altered RNA Processing in Cancer Pathogenesis and Therapy. Cancer Discov. 2019;9:1493-510
91. Schlossberg D, Brooks JB, Shulman J. Possibility of diagnosing meningitis by gas chromatography: cryptococcal meningitis. J Clin Microbiol. 1976;3:239-45
92. Siebel CW, Guthrie C. The essential yeast RNA binding protein Np13p is methylated. Proc Natl Acad Sci U S A. 1996;93:13641-6
93. Sinha R, Allemand E, Zhang Z, Karni R, Myers MP, Krainer AR. Arginine methylation controls the subcellular localization and functions of the oncoprotein splicing factor SF2/ASF. Mol Cell Biol. 2010;30:2762-74
94. Botti V, McNicoll F, Steiner MC. et al. Cellular differentiation state modulates the mRNA export activity of SR proteins. J Cell Biol. 2017Jul3;216(7):1993-2009 doi: 10.1083/jcb.201610051. Epub 2017 Jun 7
95. Moulton VR, Gillooly AR, Tsokos GC. Ubiquitination regulates expression of the serine/arginine-rich splicing factor 1 (SRSF1) in normal and systemic lupus erythematosus (SLE) T cells. J Biol Chem. 2014;289:4126-34
96. Breig O, Baklouti F. Proteasome-mediated proteolysis of SRSF5 splicing factor intriguingly co-occurs with SRSF5 mRNA upregulation during late erythroid differentiation. PLoS One. 2013;8:e59137
97. Chen Y, Huang Q, Liu W, Zhu Q, Cui CP, Xu L. et al. Mutually exclusive acetylation and ubiquitylation of the splicing factor SRSF5 control tumor growth. Nat Commun. 2018;9:2464
98. Tacke R, Chen Y, Manley JL. Sequence-specific RNA binding by an SR protein requires RS domain phosphorylation: creation of an SRp40-specific splicing enhancer. Proc Natl Acad Sci U S A. 1997;94:1148-53
99. Xiao SH, Manley JL. Phosphorylation-dephosphorylation differentially affects activities of splicing factor ASF/SF2. EMBO J. 1998;17:6359-67
100. Hamelberg D, Shen T, McCammon JA. A proposed signaling motif for nuclear import in mRNA processing via the formation of arginine claw. Proc Natl Acad Sci U S A. 2007;104:14947-51
101. Velazquez-Dones A, Hagopian JC, Ma CT, Zhong XY, Zhou H, Ghosh G. et al. Mass spectrometric and kinetic analysis of ASF/SF2 phosphorylation by SRPK1 and Clk/Sty. J Biol Chem. 2005;280:41761-8
102. Wu Q, Chang Y, Zhang L, Zhang Y, Tian T, Feng G. et al. SRPK1 Dissimilarly Impacts on the Growth, Metastasis, Chemosensitivity and Angiogenesis of Glioma in Normoxic and Hypoxic Conditions. Journal of Cancer. 2013;4:727-35
103. Matos P, Goncalves V, Jordan P. Targeting the serrated pathway of colorectal cancer with mutation in BRAF. Biochimica et biophysica acta. 2016;1866:51-63
104. Goncalves V, Henriques AF, Pereira JF, Neves Costa A, Moyer MP, Moita LF. et al. Phosphorylation of SRSF1 by SRPK1 regulates alternative splicing of tumor-related Rac1b in colorectal cells. Rna. 2014;20:474-82
105. Pelisch F, Gerez J, Druker J, Schor IE, Munoz MJ, Risso G. et al. The serine/arginine-rich protein SF2/ASF regulates protein sumoylation. Proc Natl Acad Sci U S A. 2010;107:16119-24
106. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC. et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834-40
107. Edmond V, Moysan E, Khochbin S, Matthias P, Brambilla C, Brambilla E. et al. Acetylation and phosphorylation of SRSF2 control cell fate decision in response to cisplatin. EMBO J. 2011;30:510-23
108. Ezponda T, Pajares MJ, Agorreta J, Echeveste JI, Lopez-Picazo JM, Torre W. et al. The oncoprotein SF2/ASF promotes non-small cell lung cancer survival by enhancing survivin expression. Clin Cancer Res. 2010;16:4113-25
109. Bonnal S, Vigevani L, Valcarcel J. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov. 2012;11:847-59
110. Effenberger KA, Urabe VK, Jurica MS. Modulating splicing with small molecular inhibitors of the spliceosome. Wiley Interdiscip Rev RNA. 2017 8
111. Surget S, Khoury MP, Bourdon JC. Uncovering the role of p53 splice variants in human malignancy: a clinical perspective. Onco Targets Ther. 2013;7:57-68
112. Paronetto MP, Passacantilli I, Sette C. Alternative splicing and cell survival: from tissue homeostasis to disease. Cell Death Differ. 2016;23:1919-29
113. Guyot M, Hilmi C, Ambrosetti D, Merlano M, Lo Nigro C, Durivault J. et al. Targeting the pro-angiogenic forms of VEGF or inhibiting their expression as anti-cancer strategies. Oncotarget. 2017;8:9174-88
114. Lee SC, Abdel-Wahab O. Therapeutic targeting of splicing in cancer. Nat Med. 2016;22:976-86
115. Agrawal AA, Yu L, Smith PG, Buonamici S. Targeting splicing abnormalities in cancer. Curr Opin Genet Dev. 2018;48:67-74
116. Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M. et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med. 2018;24:497-504
117. Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR. et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med. 2016;22:672-8
118. Sheng J, Zhao Q, Zhao J, Zhang W, Sun Y, Qin P. et al. SRSF1 modulates PTPMT1 alternative splicing to regulate lung cancer cell radioresistance. EBioMedicine. 2018;38:113-26
119. Gabriel M, Delforge Y, Deward A, Habraken Y, Hennuy B, Piette J. et al. Role of the splicing factor SRSF4 in cisplatin-induced modifications of pre-mRNA splicing and apoptosis. BMC Cancer. 2015;15:227
120. Giannakouros T, Nikolakaki E, Mylonis I, Georgatsou E. Serine-arginine protein kinases: a small protein kinase family with a large cellular presence. FEBS J. 2011;278:570-86
121. Fukuhara T, Hosoya T, Shimizu S, Sumi K, Oshiro T, Yoshinaka Y. et al. Utilization of host SR protein kinases and RNA-splicing machinery during viral replication. Proc Natl Acad Sci U S A. 2006;103:11329-33
122. Pilch B, Allemand E, Facompre M, Bailly C, Riou JF, Soret J. et al. Specific inhibition of serine- and arginine-rich splicing factors phosphorylation, spliceosome assembly, and splicing by the antitumor drug NB-506. Cancer Res. 2001;61:6876-84
123. Wu F, Li J, Du X, Zhang W, Lei P, Zhang Q. Chimeric antibody targeting SRPK-1 in the treatment of non-small cell lung cancer by inhibiting growth, migration and invasion. Mol Med Rep. 2017;16:2121-7
124. Duan Z, Weinstein EJ, Ji D, Ames RY, Choy E, Mankin H. et al. Lentiviral short hairpin RNA screen of genes associated with multidrug resistance identifies PRP-4 as a new regulator of chemoresistance in human ovarian cancer. Molecular cancer therapeutics. 2008;7:2377-85
125. Bauer JA, Ye F, Marshall CB, Lehmann BD, Pendleton CS, Shyr Y. et al. RNA interference (RNAi) screening approach identifies agents that enhance paclitaxel activity in breast cancer cells. Breast cancer research: BCR. 2010;12:R41
126. Brahms H, Meheus L, de Brabandere V, Fischer U, Luhrmann R. Symmetrical dimethylation of arginine residues in spliceosomal Sm protein B/B' and the Sm-like protein LSm4, and their interaction with the SMN protein. RNA. 2001;7:1531-42
127. Meister G, Eggert C, Buhler D, Brahms H, Kambach C, Fischer U. Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Curr Biol. 2001;11:1990-4
128. Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS. et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature. 2015;525:384-8
129. Banasavadi-Siddegowda YK, Welker AM, An M, Yang X, Zhou W, Shi G. et al. PRMT5 as a druggable target for glioblastoma therapy. Neuro Oncol. 2018;20:753-63
130. Vu LP, Cheng Y, Kharas MG. The Biology of m(6)A RNA Methylation in Normal and Malignant Hematopoiesis. Cancer Discov. 2019;9:25-33
131. Park SK, Jeong S. SRSF3 represses the expression of PDCD4 protein by coordinated regulation of alternative splicing, export and translation. Biochem Biophys Res Commun. 2016;470:431-8
132. Shilo A, Siegfried Z, Karni R. The role of splicing factors in deregulation of alternative splicing during oncogenesis and tumor progression. Mol Cell Oncol. 2015;2:e970955
133. Maimon A, Mogilevsky M, Shilo A, Golan-Gerstl R, Obiedat A, Ben-Hur V. et al. Mnk2 alternative splicing modulates the p38-MAPK pathway and impacts Ras-induced transformation. Cell reports. 2014;7:501-13
134. Shultz JC, Goehe RW, Murudkar CS, Wijesinghe DS, Mayton EK, Massiello A. et al. SRSF1 regulates the alternative splicing of caspase 9 via a novel intronic splicing enhancer affecting the chemotherapeutic sensitivity of non-small cell lung cancer cells. Molecular cancer research: MCR. 2011;9:889-900
135. Olshavsky NA, Comstock CE, Schiewer MJ, Augello MA, Hyslop T, Sette C. et al. Identification of ASF/SF2 as a critical, allele-specific effector of the cyclin D1b oncogene. Cancer Res. 2010;70:3975-84
136. Merdzhanova G, Edmond V, De Seranno S, Van den Broeck A, Corcos L, Brambilla C. et al. E2F1 controls alternative splicing pattern of genes involved in apoptosis through upregulation of the splicing factor SC35. Cell Death Differ. 2008;15:1815-23
137. Kedzierska H, Poplawski P, Hoser G, Rybicka B, Rodzik K, Sokol E. et al. Decreased Expression of SRSF2 Splicing Factor Inhibits Apoptotic Pathways in Renal Cancer. International journal of molecular sciences. 2016 17
138. Jiang CC, Lai F, Tay KH, Croft A, Rizos H, Becker TM. et al. Apoptosis of human melanoma cells induced by inhibition of B-RAFV600E involves preferential splicing of bimS. Cell Death Dis. 2010;1:e69
Author contact
![]() Corresponding authors: Zailong Qin, E-mail: qinzailongedu.cn, from Laboratory of Genetics and Metabolism, Maternal and Child Health Hospital of Guangxi Zhuang Autonomous Region, Guangxi Birth Defects Research and Prevention Institute, Nanning, Guangxi, 530003, China or Jia Wang, E-mail: jiaicqedu.cn, from Department of Immunology, Changzhi Medical College, Changzhi, Shanxi, 046000 China.
Corresponding authors: Zailong Qin, E-mail: qinzailongedu.cn, from Laboratory of Genetics and Metabolism, Maternal and Child Health Hospital of Guangxi Zhuang Autonomous Region, Guangxi Birth Defects Research and Prevention Institute, Nanning, Guangxi, 530003, China or Jia Wang, E-mail: jiaicqedu.cn, from Department of Immunology, Changzhi Medical College, Changzhi, Shanxi, 046000 China.