Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(15):2853-2867. doi:10.7150/ijbs.44365 This issue Cite
Research Paper
miR-550-1 functions as a tumor suppressor in acute myeloid leukemia via the hippo signaling pathway
Chao Hu1,2,3*, Mengxia Yu4*, Chenying Li1,2*, Yungui Wang1,2,3*, Xia Li1, Bryan Ulrich3, Rui Su2,5, Lei Dong2,5, Hengyou Weng2,5, Huilin Huang2,5, Xi Jiang2,3,5 ![]() , Jianjun Chen2,3,5
, Jianjun Chen2,3,5 ![]() , Jie Jin1
, Jie Jin1 ![]()
1. Department of Hematology, the First Affiliated Hospital, College of Medicine, Zhejiang University, 79 Qingchun Road, Hangzhou, 310003, P.R. China.
2. Department of Cancer Biology, University of Cincinnati, Cincinnati, OH 45219, USA.
3. Section of Hematology/Oncology, Department of Medicine, University of Chicago, Chicago, Illinois 60637, USA.
4. Department of Hematology, Affiliated Hangzhou First People's Hospital, Zhejiang University School of Medicine, 216 Huansha Road, Hangzhou, 310006, P.R. China.
5. Department of Systems Biology & the Gehr Family Center for Leukemia Research, Beckman Research Institute of City of Hope, Monrovia, CA 91016, USA.
*These authors contributed equally to this work.
Received 2020-1-29; Accepted 2020-7-23; Published 2020-9-1
Abstract

MicroRNAs (miRNAs) and N6-methyladenosine (m6A) are known to serve as key regulators of acute myeloid leukemia (AML). Our previous microarray analysis indicated miR-550-1 was significantly downregulated in AML. The specific biological roles of miR-550-1 and its indirect interactions and regulation of m6A in AML, however, remain poorly understood. At the present study, we found that miR-550-1 was significantly down-regulated in primary AML samples from human patients, likely owing to hypermethylation of the associated CpG islands. When miR-550-1 expression was induced, it impaired AML cell proliferation both in vitro and in vivo, thus suppressing tumor development. When ectopically expressed, miR-550-1 drove the G0/1 cell cycle phase arrest, differentiation, and apoptotic death of affected cells. We confirmed mechanistically that WW-domain containing transcription regulator-1 (WWTR1) gene was a downstream target of miR-550-1. Moreover, we also identified Wilms tumor 1-associated protein (WTAP), a vital component of the m6A methyltransferase complex, as a target of miR-550-1. These data indicated that miR-550-1 might mediate a decrease in m6A levels via targeting WTAP, which led to a further reduction in WWTR1 stability. Using gain- and loss-of-function approaches, we were able to determine that miR-550-1 disrupted the proliferation and tumorigenesis of AML cells at least in part via the direct targeting of WWTR1. Taken together, our results provide direct evidence that miR-550-1 acts as a tumor suppressor in the context of AML pathogenesis, suggesting that efforts to bolster miR-550-1 expression in AML patients may thus be a viable clinical strategy to improve patient outcomes.
Keywords: acute myeloid leukemia, miR-550-1, WWTR1, apoptosis, proliferation
Introduction
Acute myeloid leukemia (AML) is a form of cancer that arises when hematopoietic stem cells (HSCs) undergo oncogenic mutations. In the United States, 19,940 new AML cases are expected to be diagnosed, and 11,180 AML-associated deaths are expected to occur in 2020 (https://www.seer.cancer.gov/). While there have been countless efforts to develop novel therapeutic strategies suited to the treatment of AML, the majority of patients still suffer from poor outcomes, with recent reports estimating a 5-year survival rate of 40% among AML patients[1, 2]. As such, there is a clear need to better understand the molecular basis for AML in order to expedite the development of more efficacious therapeutic interventions.
MicroRNAs (miRNAs) are small 20-24 nucleotide non-coding RNA molecules that exhibit endogenous biological functionality via targeting specific downstream mRNAs [3]. These miRNAs mediate their activities through their interactions with the RNA-induced silencing complex (RISC), pairing with compatible bases in the 3' untranslated region (UTR) of target mRNAs. These interactions can result in either a suppression of mRNA translation, or a reduction in mRNA stability that lead to mRNA degradation, thus resulting in a marked reduction in target gene expression at the protein level [3, 4]. There is now countless evidence that specific miRNAs serve essential regulatory roles in both the context of normal physiology and disease pathogenesis, including leukemogenesis [5-8]. Importantly, improved understanding of these miRNAs has led to their utilization for the treatment of certain disorders [9]. In hepatitis C mouse models, knockdown of miR-122 led to reduced liver damage and viral load via owing to its ability to regulate several targets, such as mannan binding lectin serine protease 1 (MASP1) and prolyl 4-hydroxylase subunit α1 (P4HA1) [10]. Using locked nucleic acids (LNAs)-modified antimiR-122, preclinical studies were performed in an effort to treat hepatitis C infection. The results indicated this LNAs could bring about a significant reduced liver injure and reduction in infection burden [10]. In non-small cell lung cancer (NSCLC), Wiggins JF et al. [11] chemically synthesized miR-34a and a lipid-based transport vehicle, and found that this combination effectively blocked cell proliferation by targeting cyclin-dependent kinase 4 (CDK4) in vitro and in vivo. In our previous research, we have identified a set of miRNAs with specific regulatory roles in the context of the proliferation, differentiation, and apoptosis of AML cells. These miRNAs include miR-9, miR-22, miR-26a, miR-150, miR-495, miR-181, miR-126, miR-196b and the miR-17-92 cluster [12-21]. In light of our research into miR-150, we ultimately developed a novel FLT3 ligand-binding (amidoamine)-miR-150 nanoparticle (G7-FLT3L-miR-150 nanoparticle) [22], which specifically delivered miR-150 to FLT3-overexpressing AML cells by employing FLT3L as a guiding molecule. Through inhibiting the activation of PIM, AKT, ERK, and STAT5, this nanoparticle displayed a strong anti-leukemic effect in vitro and in vivo. Even so, however, the specific role of these miRNAs in AML is not completely understood.
WW-domain containing transcription regulator-1 (WWTR1), also known as the transcriptional co-activator with PDZ-binding motif (TAZ), was first detected based on its status as a 14-3-3 interacting protein [23]. WWTR1 and the paralogous Yes-associated protein (YAP) serve as central downstream regulatory factors in the Hippo signaling pathway, which modulates a wide range of cellular processes pertaining to cellular energy status, hypoxia, osmotic stress, tissue organ size, regeneration, homeostasis, and tumorigenesis [24, 25]. Indeed, elevated WWRT1 mRNA and protein expression have been found to be associated with the development of gastric, colorectal, breast, and lung cancers [26-30]. Consistent with this, elevated WWTR1 protein level has been determined to be a risk factor for the development of both glioblastoma multiforme and colorectal cancer [31, 32]. In one study, Justice et al. [33] also determined that there was an association in gastric cancer between increased WWTR1 expression and tumor TNM stage as well as incidence of lymph node metastasis. Wang et al.[27] further found that WWTR1 expression in NSCLC led to its regulation of Cyclin A and C transforming growth factor (CTGF), which in turn led to a disruption of apoptosis in neoplastic cells. Recent work has revealed that large tumor suppressor (LATS)-mediated YAP/WWTR1 phosphorylation was the key regulatory event controlling the activity of these proteins in cells [34]. Consistent with this, Jimenez-Velasco et al. [35] found LATS1 and LATS2 to be downregulated in leukemia as a consequence of their hypermethylation, and reduced LATS2 expression has been found to be associated with worse outcomes among leukemia patients. This suggests the possibility that a reduction in LATS1/2 activity may underlie the alterations in YAP/WWTR1 stabilization and activation in the context of leukemia. However, clarity is still needed regarding the mechanisms governing increased WWTR1 activity in AML.
In the present study, we for the first time provided evidence that miR-550-1 was significantly downregulated in AML. Moreover, when overexpressed, miR-550-1 was able to impair AML cell proliferation and oncogenesis both in vitro and in vivo owing to its ability to regulate the Hippo signaling pathway. We further found WWTR1 to be a direct miR-550-1 target, thereby at least partially explaining its role in regulating AML progression.
Materials and Methods
Cell line and patients' samples
We cultured MV4-11 and Kasumi-1 cells in RPMI-1640 (Invitrogen, Carlsbad, USA) containing 10% heat-inactivated FBS (Gibco, Grand Island, USA), 1% penicillin-streptomycin (Sigma-Aldrich, St Louis, USA) and 1% HEPES (Sigma-Aldrich). Murine progenitor cells were cultured in RPMI 1640 containing 10 ng/ml interleukin 3 (IL-3) (Peprotech, Rocky Hill, USA), 10 ng/ml IL-6 (Peprotech), 100 ng/ml stem cell factor (SCF) (Peprotech), 55 nM 2-mercaptoethanol (BME) (Sigma-Aldrich), 1% HEPES, 10% FBS, and 1% PS. The AML patients' samples were acquired from the First Affiliated Hospital of Zhejiang University and the University of Chicago Hospital with informed consent. The study was approved by the institutional review board of two hospital's ethics committee.
Measurement of cell viability
A MTT assay (Promega, Madison, USA) was used to measure viability based on provided directions. Briefly, MV4-11 and Kasumi-1 cells were plated into 96-well plates (10000 cells/100 μL), with dye solution added to wells at the indicated time points. After 4-hour 37°C incubation, stop buffers were added and cell absorbance was assessed the following day at 570 nm.
Flow cytometry
A BD LSRII Flow Cytometer was used in all analyses, and FlowJo v10 was used for data analysis. For measurements of apoptosis, 0.5×106 cells were stained with an Annexin V-APC Apoptosis Detection Kit (BD Biosciences, San Diego, USA) based on provided directions.
For cell cycle analyses, 0.5×106 cells were fixed overnight at 4°C in 75% ethanol, washed thrice in PBS, and stained using propidium iodide for 20 minutes.
For immunophenotyping analyses, BM, PB, and spleen cells (0.5×106) were collected, washed thrice using PBS, and stained at 4°C with antibodies specific for CD11b, CD117, CD45.1, CD45.2, and Gr-1 (BD Biosciences) for 20 minutes. After two additional washed, cells were then fixed in a fixation buffer prior to analysis.
Western blotting
A total of 5×106 cells were washed in cold PBS prior to lyse with RIPA buffer (Pierce, Rockford, USA) supplemented with PMSF, EDTA, and protease inhibitors. Samples underwent 30 min of centrifugation at 12000×g, after which supernatants were isolated and loaded in equal protein amounts (30-50 ug) onto gels for SDS-PAGE analysis. After separation, proteins were transferred onto PVDF membranes which were then blocked with 5% skim milk in TBST for 1 h, followed by probing overnight at 4°C with anti-β-ACTIN (#3700), anti-WWTR1 (#83669), anti-PARP (#9532), anti-AKT (#4685), anti-p-AKT (#4060), anti-CDK6 (#13331), anti-Rb (#9309), anti-p-Rb (#8516), anti-E2F1 (#3742), anti-CCND1 (#2978), anti-BCL-2 (#15071), anti-p27 (#3686) (Cell Signal Technology, Beverly, USA), or anti-c-myc (Abcam, Cambridge, USA, ab32072). A peroxidase-conjugated secondary antibody was then applied to blot for 1 h, followed for four washed with TBST, after which chemi-luminescence (ECL, Pierce) was used to detect protein bands.
Quantitative RT-PCR
A miRNeasy kit (Qiagen, Frederick, USA) was used for extracting total RNA from 1×106 cells based on provided directions. cDNA was then synthesized from 1 µg of this RNA via M-MLV reverse transcriptase (Invitrogen, Carlsbad, CA, USA). A 7900HT real-time PCR system (Applied Biosystems, Foster City, USA) was employed for qPCR analyses, with SYBR Green used to setup triplicate reactions assessing relative mRNA expression. For miRNA expression, TaqMan qPCR was conducted according to provided directions (Applied Biosystems). The 2-ΔΔCt method was used to calculate miRNA and mRNA relative expression, which was normalized to endogenous levels of U6 and GAPDH, respectively.
Plasmid and virus production
The pri-miR-550-1 sequence was amplified by PCR from healthy human BM mononuclear cells (MNCs). Primers with mutated sequences (Table 1) were then used to generate the indicated mutant miR-550-1 template, and these wild type (WT) and mutant miR-550-1 sequences were thereafter cloned into the MSCV-PIG vector (MSCV-Puromycin-IRES-GFP vector) (Cold Spring Harbour Laboratory, USA) in order to overexpress these two miRNA isoforms. These sequences were inserted between the XhoI (CTCGAG) and EcoRI (GAATTC) sites in this vector. For WWTR1-CDS vectors, the WT sequence was amplified from healthy human BM MNCs prior to insertion into the pCDH vector (SBI, Mountain View, USA). The MSCVneo-MLL-AF9 plasmid was kindly provided by Dr. Scott Armstrong.
Oligonucleotide sequences
| Name | Sequence (5'->3') |
|---|---|
| miR-550-1 precursor F | CTGGTGCAGTGCCTGAGGGAGTAAG |
| miR-550-1 precursor R | CTTACTCCCTCAGGCACTGCACCAG |
| mutant miR-550-1 F | CTGGTGCAGCATTCAAGGGAGTAAG |
| mutant miR-550-1 R | CTTACTCCCTTGAATGCTGCACCAG |
| WWTR1-3'UTR F | GGGCACTAGTATTCGACCTGATTTACAGTTTC |
| WWTR1-3'UTR R | TATTACGCGTTGAGATCAGGAGTTTGAGAAC |
| WWTR1-qPCR F | GATCCTGCCGGAGTCTTTCTT |
| WWTR1-qPCR R | CACGTCGTAGGACTGCTGG |
| WWTR1-CDS-F | TTCTATCTAGAATGAATCCGGCCTCGG |
| WWTR1-CDS-R | TTTTGCGGCCGCTTACAGCCAGGTTAGAA |
F: forward primer; R: reverse primer.
One day prior to transfection, 5 ×105 HEK293T cells were plated into 60-mm dishes. Retroviruses were then produced via transfecting cells with vector DNA and a packaging vector (PCL-Eco or PCL-Ampho) with the Effectene Transfection Kit (Qiagen). The WWTR1 overexpressing lentivirus was generated via co-transfection of the WWTR1-pCDH plasmid and packaging lentivirus vectors (pRSV-Rev, pMDLg/pRRE and pMD2.G). At 48 and 72 h post-transfection, cellular supernatants were harvested and filtered through a 0.45 μm cellulose acetate filter prior to storage.
Target gene analysis
miRNA target gene predictions were made through the use of the PITA (http://genie.weizmann.ac.il/pubs/mir07/), miRBase Targets (http://microrna.sanger.ac.uk), TargetScan (http://www.targetscan.org), and miRanda (http://www.microrna.org) miRNA-target gene prediction databases.
Dual luciferase reporter and mutagenesis assay
We conducted dual luciferase reporter and mutagenesis assays based on a modified version of a previously reported protocol [16]. The WWTR1 3'-UTR sequences containing putative miR-550-1-binding sites were synthesized via PCR utilizing the following primers: forward 5'-GGGCACTAGTATTCGACCTGATTTACAGTTTC-3' and reverse 5'- TATTACGCGTTGAGATCAGGAGTTTGAGAAC-3'. The resultant fragment underwent insertion into the pMIR-REPORT vector (Ambion, Austin, USA). In addition, a mutated version of this 3'-UTR fragment was generated using primers bearing the mutant sequence. A total of 6,000 HEK293T cells were plated per well of a 24-well plate in triplicate, and following overnight culture these cells were co-transfected with pMIR-REPORT-WWTR1 or mutant pMIR-REPORT-WWTR1 vectors and MSCV-PIG-miR-550-1, mutant MSCV-PIG-miR-550-1, or MSCV-PIG empty vectors (20ng each). The β-galactosidase vector (1ng) (Ambion) was additionally transfected into all experimental cells, and after a 48 h incubation all cells were lysed. Relative luciferase activity was then measured via a Dual-Light Combined Reporter Gene Assay System (Applied Biosystems).
Colony forming/replating assay
Colony formation assays were performed in accordance to a modified version of a previously reported protocol [16]. Donor murine BM cells were isolated from 6-week old B6.SJL (CD45.1) after injecting 5-fluorouracil (5-FU; 150mg/kg) for 5 days, and hematopoietic progenitor cells were isolated via a Mouse Lineage Cell Depletion Kit (Miltenyi Biotec Inc., Auburn, USA). These progenitor cells then underwent infection with the indicated viruses using a spinoculation protocol with the assistance of polybrene [36-38]. Following transduction, cells were incubated overnight at 37oC in fresh media, and this was then repeated the following day. Thereafter, 2 × 104 cells were plated in methylcellulose medium (Stem Cell Technologies Inc, Vancouver, Canada) containing 10 ng/ml IL-3, IL-6, granulocyte-macrophage colony-stimulating factor (GM-CSF), 30 ng/ml SCF, and 2 μg/ml puromycin and/or 1 mg/ml G418, as appropriate. After incubating for 7 days, colony formation in each of the experimental groups was assessed, and these colonies were then replated.
Primary and secondary bone marrow transplantations (BMT)
For BMT assays, donor mice were B6.SJL (CD45.1+), and recipient mice were C57BL/6 (CD45.2+). All animal experiments were approved by the local Institutional Animal Care and Use Committee (IACUC).
In the primary BMT assays, BM cells from healthy donor mice (B6.SJL) were transduced with the indicated retrovirus combinations (MSCV-PIG + MSCVneo-MLL-AF9, MSCV-PIG-miR-550-1 + MSCVneo-MLL-AF9, and mutant MSCV-PIG-miR-550-1 + MSCVneo-MLL-AF9). The resultant donor cells were then mixed with helper cells (BM cells from a healthy C57BL/6 mouse) at a ratio of 3 × 105 to 1 × 106 per recipient mouse. These cells were then injected into the tail vein of an 8-week old lethally irradiated (960 rads) recipient mouse. In secondary BMT assays, these lethally irradiated recipient mice were injected using leukemic BM cells that had been isolated from the initial primary recipient mice, and no helper cells were added.
Once recipient mice exhibited signs of systemic illness, peripheral blood (PB) samples were collected via tail bleeding in order to establish whole blood counts. Engraftment was evaluated via flow cytometry based on CD45.1 expression in PB samples. Moribund mice were euthanized, and liver, thymus, and spleen weight was determined. BM cells were collected from euthanized animals and prepared for cytospin slides, which then underwent Wright-Giemsa staining.
m6A dot blot assay
We isolated total RNA with the miRNeasy kit and quantified RNA levels. Next, RNA samples were spotted using a Bio-Dot Apparatus (Bio-Rad, Hercules, USA) on Amersham Hybond-N+ membranes (GE Healthcare, Chicago, USA), and a UV cross-linker was then used to cross-link them to this membrane. Membranes then underwent two washes using Milli-Q, followed by treatment for 10 min using 0.02% methylene blue (Sigma-Aldrich). Membranes were then rinsed until the dye was washed away from background regions, and dots of methylene blue were then imaged. Next, 5% nonfat dry milk was used for membrane blocking for 1 h, after which an antibody against m6A (1:2000 dilution, Synaptic Systems, Goettingen, Germany, #202003) was used to probe blots at 4°C overnight. Membranes were then washed thrice in TBST and probed at room temperature using HRP-conjugated goat anti-rabbit IgG for 1 h, prior to visualization with an ECL system.
Statistical analysis
SPSS v16 (IBM, Armonk, USA) was used to compare all experimental results via Student's t-tests or two-way ANOVAs. Data are given as means ± standard deviations (SDs) from at least three repeat experiments. The Kaplan-Meier approach was used to assess overall survival. P<0.05 was the threshold of statistical significance.
Results
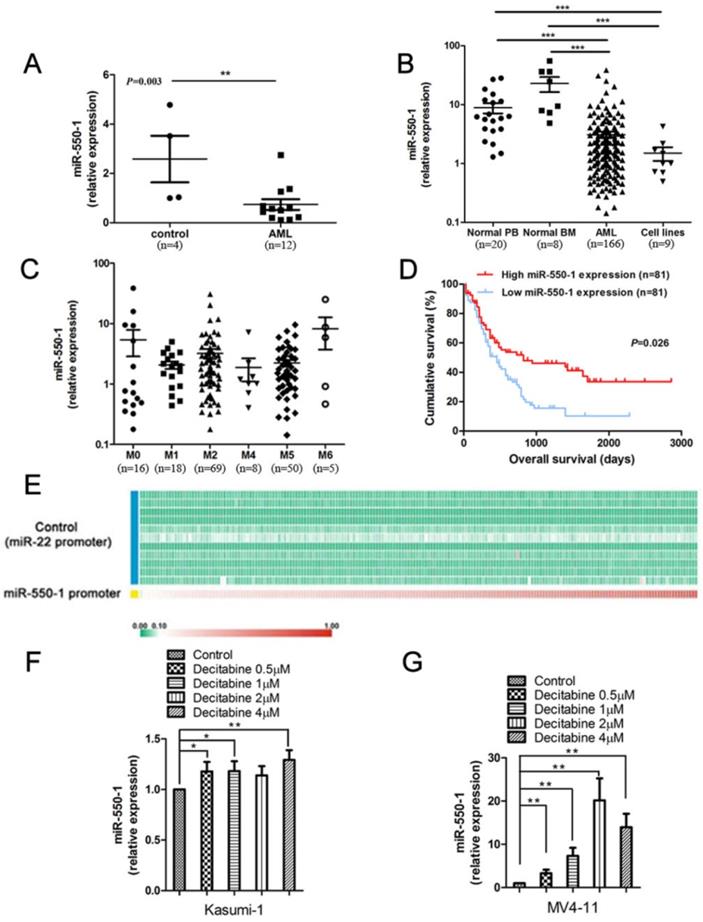
miR-550-1 is down-regulation in AML
We have previously detected a set of specific miRNAs that were down-regulated in AML samples with t(8; 21), inv(16), t(15; 17), or mixed lineage leukemia (MLL) rearrangements, relative to normal controls (NC) [21]. Of these previously identified miRNAs, miR-550-1 was among the most significantly down-regulated in the AML cohort. We therefore first confirmed that miR-550-1 was significantly down-regulated in AML patients using bone marrow (BM) mononuclear cell (MNC) samples from 12 patients with primary AML (from University of Chicago Hospitals), revealing a marked decrease in the expression of this miRNA as compared to NC samples (n=4) (P=0.003) (Fig. 1A). We then extended these findings to a larger 166 patient primary AML sample cohort (from the First Affiliated Hospital of Zhejiang University) (Table 2 and Table S1), using qPCR to confirm that miR-550-1 was significantly down-regulated in AML patients relative to NC samples, which included 8 normal BM samples and 20 normal peripheral blood (PB) samples (P<0.001) (Fig. 1B). Similarly, miR-550-1 expression was markedly decreased in 9 leukemic cell lines (including MV4-11, Kasumi-1, MOLM-13, MONOMAC-6, OCI-AML3, NB4, THP-1, HL-60, OCI-AML2) relative to controls (P<0.001) (Fig. 1B). In addition, the expression level of miR-550-1 was significantly increased at complete remission (CR) compared to initial diagnosis (P=0.010) (Figure S1). We did not find any significant differences with respect to miR-550-1 expression as a function of the French-American-British (FAB) subtype (Fig. 1C). Given its low expression in AML patients, we next sought to determine whether the level of miR-550-1 expression correlated with patient clinical outcomes. Using a 162 patient TCGA AML dataset, we divided patients based on whether they expressed high or low levels of miR-550-1, as determined based on the median expression level of this miRNA. In so doing we found that patients with lower miR-550-1 expression exhibited poorer overall survival (OS) than did those with higher expression (P=0.026; Fig. 1D). Together, these findings validated our previous results with respect to patterns of miR-550-1 in AML, and strongly suggested the possibility that this miRNA may serve as a tumor suppressive role in the context of leukemogenesis.
Human AML samples exhibit significantly decreased miR-550-1 expression and altered miR-550-1 DNA methylation. (A) Taqman qPCR was used to assess miR-550-1 expression in MNC samples from 12 AML patients and 4 normal controls (NC). (B) Taqman qPCR was used to assess miR-550-1 expression in MNC samples from 166 AML patients, as well as in 8 normal bone marrow (BM) samples, 20 samples of normal peripheral blood (PB), as well as 9 leukemic cell lines. (C) Relative miR-550-1 expression was assessed in various AML subtypes. (D) Reduced miR-550-1 expression levels were found to correlate with poorer overall survival in the TCGA dataset; (E) The pri-miR-550-1 promoter CpG islanded exhibited hypermethylation, unlike those of pri-miR-22 in 194 AML patient samples from the TCGA_194S dataset; (F, G) We treated the Kasumi-1 and MV4-11 cells using a range of DNA methylation inhibitor concentrations (0, 1, 2, or 4 µmol decitabine), revealing decitabine treatment to increased miR-550-1 expression relative to controls. Error bar indicates SD of triplicate experiments *, P<0.05; **, P<0.01; ***, P<0.001.
High and low miR-550-1 expressing AML patient characteristics
| miR-550-1 | P value | ||
|---|---|---|---|
| Low expression | High expression | ||
| Number | 83 | 83 | |
| Age | 0.740 | ||
| <60 | 55 | 57 | |
| ≥60 | 28 | 26 | |
| Gender | 0.431 | ||
| Male | 46 | 51 | |
| Female | 37 | 32 | |
| 2016 WHO classification | 0.732 | ||
| AML with recurrent genetic abnormalities | |||
| AML with mutated NPM1 | 23 | 25 | |
| AML with biallelic mutations of CEBPA | 10 | 10 | |
| AML not otherwise specified | |||
| AML with minimal differentiation | 8 | 6 | |
| AML without maturation | 4 | 3 | |
| AML with maturation | 18 | 19 | |
| Acute myelomonocytic leukemia | 5 | 1 | |
| Acute monoblastic/monocytic leukemia | 15 | 18 | |
| Pure erythroid leukemia | 0 | 1 | |
| Hb median (range) (g/L) | 81.0 (51.0-136.0) | 88.4 (50.0-135.0) | 0.041 |
| PLT median (range) (×109/L) | 46.5 (20.4-110.1) | 51.2 (22.1-109.2) | 0.580 |
| WBC median (range) (×109/L) | 12.5 (0.7-236.9) | 12.4 (0.2-222.5) | 0.936 |
| BM blasts median (range) % | 72.5 (28.4-90.5) | 70.2 (25.2-82.3) | 0.449 |
| Genes mutations, n (%) | |||
| NPM1 | 23 (27.7) | 25 (30.1) | 0.732 |
| CEBPADM | 10 (12.4) | 10 (12.4) | 1.000 |
| FLT3-ITD | 23 (27.7) | 18 (21.7) | 0.368 |
| DNMT3a | 12 (14.5) | 7 (8.4) | 0.223 |
| IDH1 | 5 (6.0) | 12 (14.5) | 0.073 |
| IDH2 | 14 (16.9) | 6 (7.2) | 0.056 |
WHO: World Health Organization; Hb: hemoglobin; PLT: platelet; WBC: white blood cell; BM: bone marrow; DM: double-allele mutation.
Patients were separated into groups based on their miR-550-1 expression level relative to the median value, with 83 each in the high and low expression groups.
Promoter methylation is known to be a key regulator of many miRNAs in the context of AML, including miR-126 and miR-375 [13, 39]. Whereas we have previously found the miR-22 promoter to be hypomethylated [15], when we assessed this TCGA dataset we found that the pri-miR-550-1 promoter region was hypermethylated (Fig. 1E). Consistent with these findings, when the MV4-11 and Kasumi-1 AML cell lines were treated using the hypomethylating agent, decitabine, miR-550-1 expression rose significantly (Fig. 1F, G). In addition, the methylation degree of miR-550-1 CpG islands was apparently inhibited with decitabine (Figure S2). These findings are therefore consistent with a model wherein miR-550-1 promoter hypermethylation in AML cells may lead to its reduced expression.
miR-550-1 suppresses tumor development in vitro
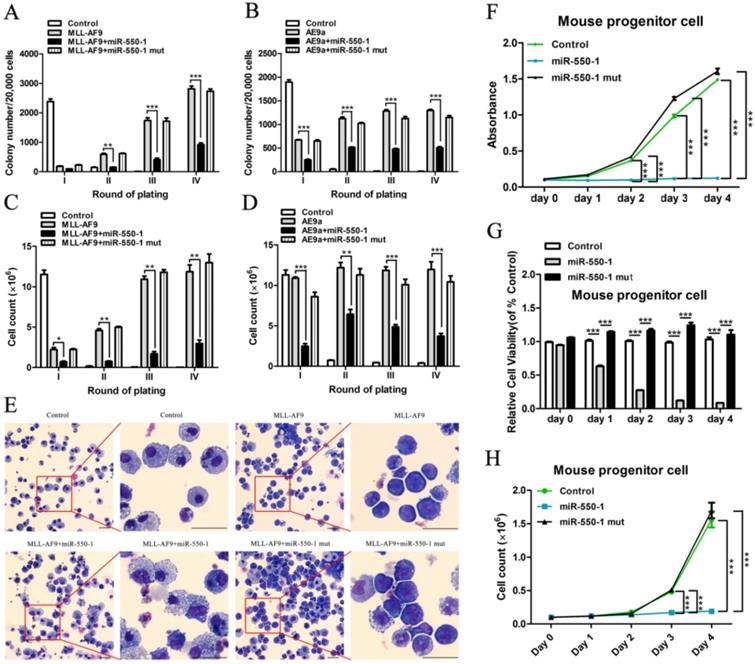
To explore the biological role of miR-550-1 in AML cells, we first conducted colony formation assays (CFA). To this end, MSCVneo-MLL-AF9 + MSCV-PIG (as control) [40], MSCVneo-MLL-AF9 + MSCV-PIG-miR-550-1 (i.e., MLL-AF9 + miR-550-1), MSCVneo-MLL-AF9 + MSCV-PIG-miR-550-1 mutant (i.e., MLL-AF9 + miR-550-1 mut), MSCVneo-AE9a (AML1-ETO9a fusion gene; a truncated AML1-ETO fusion gene causing rapid AML onset in mice [41]) + MSCV-PIG (as control), MSCVneo-AE9a + MSCV-PIG-miR-550-1 (i.e., AE9a + miR-550-1) or MSCVneo-AE9a + MSCV-PIG-miR-550-1 mutant (i.e., AE9a + miR-550-1 mut) were separately co-transduced into normal murine BM progenitor cells prior to replating on methylcellulose medium. Following a 7 day incubation period, the media was then replaced and equivalent culture conditions were maintained for each group. We observed a marked reduction in colony formation capabilities for BM progenitor cells induced with the MLL-AF9 or AE9a fusion proteins following induction of miR-550-1 expression (Fig. 2A-D). Indeed, such ectopic miR-550-1 expression led to a clear reduction in the viability and proliferation of MLL-AF9 fusion protein-induced murine BM progenitor cells (Fig. 2E-H). When the 6-base miR-550-1 seed sequence was mutated, this led to a complete ablation of the miR-550-1-dependent suppression of the proliferation of these AML-like cells (Fig. 2A-H), suggesting that miR-550-1 exerts these observed effects via a canonical base-pairing mechanism regulating target gene expression. Notably, when miR-550-1 was overexpressed, this led to a marked induction of cell differentiation (Fig. 2E).
miR-550-1 suppresses tumor cell growth in vitro. (A-D) MLL-AF9 or AE9a -induced colony formation following replating was dramatically reduced upon over-expression of wild-type but not mutant miR-550-1. (E) Forced ectopic expression of wild-type miR-550-1, but not of mutant miR-550-1, led to a significant enhancement of cell differentiation as determined via cytospin analyses. (F-H) Ectopic forced expression of wild-type but not mutant miR-550-1 led to a reduction in the viability and proliferation of murine progenitor cells. Error bar indicates SD of triplicate experiments. *, P<0.05; **, P<0.01; ***, P<0.001. Scale bar = 10 µm.
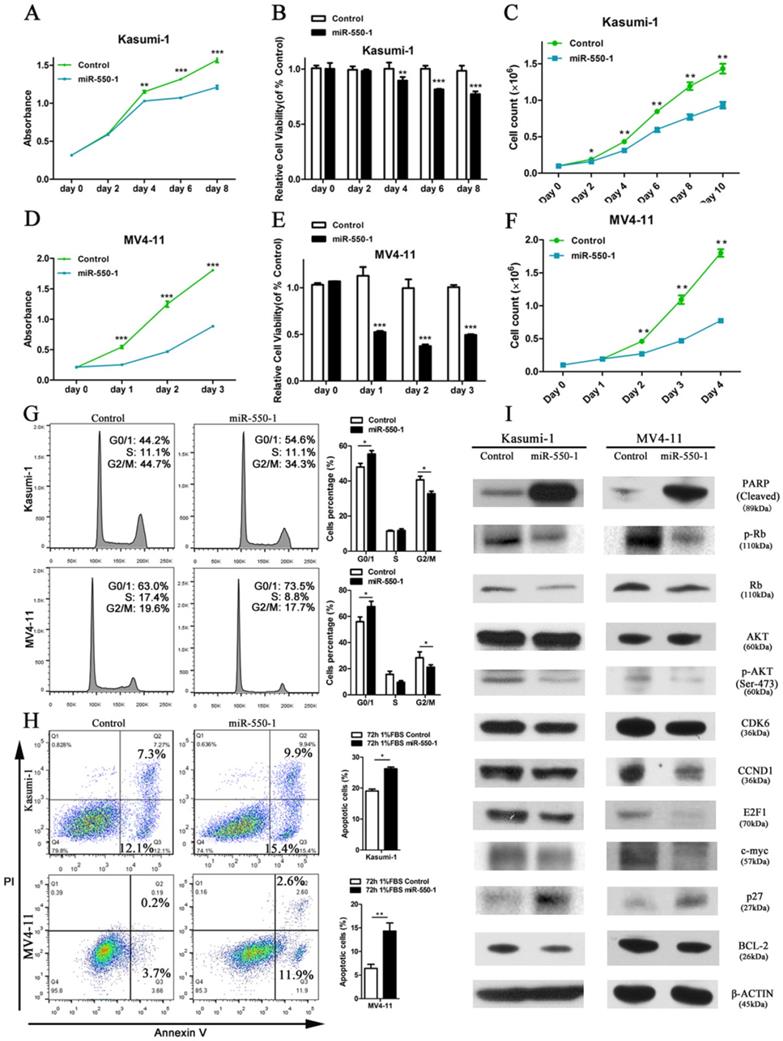
In order to assess the role of miR-550-1 in the context of AML biology, we next used the MV4-11 and Kasumi-1 human AML cell lines to conduct gain-of-function experiments. We found that forced ectopic miR-550-1 expression led to a clear reduction in the viability and proliferation for both of these cell lines (Fig. 3A-F). Furthermore, miR-550-1 overexpression led to a marked increase in the apoptotic cell death and G0/1 phase arrest (Fig. 3 G and H). Consistent with these results [42, 43], western blotting confirmed that the levels of the G0/1-S checkpoint regulatory molecules CCND1 and CDK2 were reduced upon ectopic miR-550-1 expression, as well as the levels of the cell cycle regulatory proteins p-Rb, E2F1, and P27. We also observed clear decreases in the levels of the proliferation-associated proteins p-AKT and c-myc, as well as the anti-apoptotic BCL-2 protein upon ectopic miR-550-1 expression, whereas levels of the pro-apoptotic PARP protein were markedly increased in these same cells (Fig. 3I). Together these results strongly suggest that miR-550-1 significantly reduces rates of AML cell proliferation and leukemic cell transformation proliferation in vitro.
Ectopic miR-550-1 expression reduced the viability and proliferation of Kasumi-1 and MV4-11 cells. (A, B) Using MTT assays revealed that ectopic miR-550-1 expression led to a marked reduction in the viability of Kasumi-1 cells beginning on day 4. (C) The inhibitory effects of ectopic miR-550-1 expression on Kasumi-1 cells were assessed. (D, E) Using MTT assays revealed that ectopic miR-550-1 expression led to a marked reduction in the viability of MV4-11 cells beginning on day 1. (F) The inhibitory effects of ectopic miR-550-1 expression on MV4-11 cells were assessed. (G) Ectopic miR-550-1 expression led to the induction of G0/1 phase arrest in both Kasumi-1 and MV4-11 cells. (H) miR-550-1 overexpression led to the apoptotic death of both Kasumi-1 and MV4-11 cells; (I) Western blotting assessment of proteins related to apoptosis, proliferation, and the cell cycle in Kasumi-1 and MV4-11 cells with or without elevated miR-550-1 expression. Error bar indicates SD of triplicate experiments. *, P<0.05; **, P<0.01; ***, P<0.001.
miR-550-1 inhibits AML leukemogenesis in vivo
We next explored whether miR-550-1 was able to suppress the development of leukemia in vivo as it did in vitro by using a primary BMT assay system. Briefly, we co-transduced murine BM progenitor cells (B6.SJL, CD45.1) with the MSCVneo-MLL-AF9 + MSCV-PIG (as control), MSCVneo-MLL-AF9 + MSCV-PIG-miR-550-1 (i.e., MLL-AF9 + miR-550-1), or MSCVneo-MLL-AF9 + MSCV-PIG- miR-550-1 mutant (i.e., MLL-AF9 + miR-550-1 mut) vectors. Transduced cells were then injected into the tail veins of recipient mice (C57BL/6, CD45.2). By flow cytometry, we found all mice in the MLL-AF9+miR-550-1 group displayed an apparent decline in the proportion of c-Kit+ blast cells in the BM, spleen (SP), and PB compared to control or MLL-AF9+miR-550-1 mutant group (Fig. 4A and B). Moreover, we then assessed engraftment, revealing that forced ectopic miR-550-1 expression was associated with a clear reduction in engraftment rates and tumor burdens in the BM, PB, liver, and spleen (Fig. 4C-G). Importantly, mice in the MLL-AF9 + miR-550-1 group exhibited a significantly longer latency for AML development as compared with either the control group or the MLL-AF9 + miR-550-1 mutant group (both P<0.05; Fig.4H). Together, these findings strongly suggest that inducing the expression of miR-550-1 in these cells leads to a significant reduction in the induction of primary leukemogenesis by the MLL-AF9 fusion gene.
miR-550-1 is able to disrupt MLL-fusion-mediated leukemogenesis in vivo. (A, B) Overexpression of wild-type but not mutant miR-550-1 reduced the proportion of c-Kit+ blast cells in the BM, SP and PB. (C, D) Induced expression of wild-type, but not mutant miR-550-1, led to a clear reduction of the engraftment frequency in the BM, SP and PB samples. Red line: isotype control, blue line: CD45.1. (E-G) miR-550-1 overexpression led to a clear reduction in PB, liver, and splenic tumor burden. (H) Animals in the MLL-AF9+miR-550-1 group had a markedly slower rate of leukemia development than did those in the primary BMT MLL-AF9 only group (P=0.035). (I) Leukemic development was markedly reduced for the secondary BMT MLL-AF9+miR-550-1 group relative to the MLL-AF9 only group (P=0.010); (J and K) Overexpression of wild-type but not mutant miR-550-1 led to a marked reduction in the colony formation capabilities of secondary BMT leukemic cells. Error bar indicates SD of triplicate experiments. *, P<0.05; **, P<0.01; ***, P<0.001.
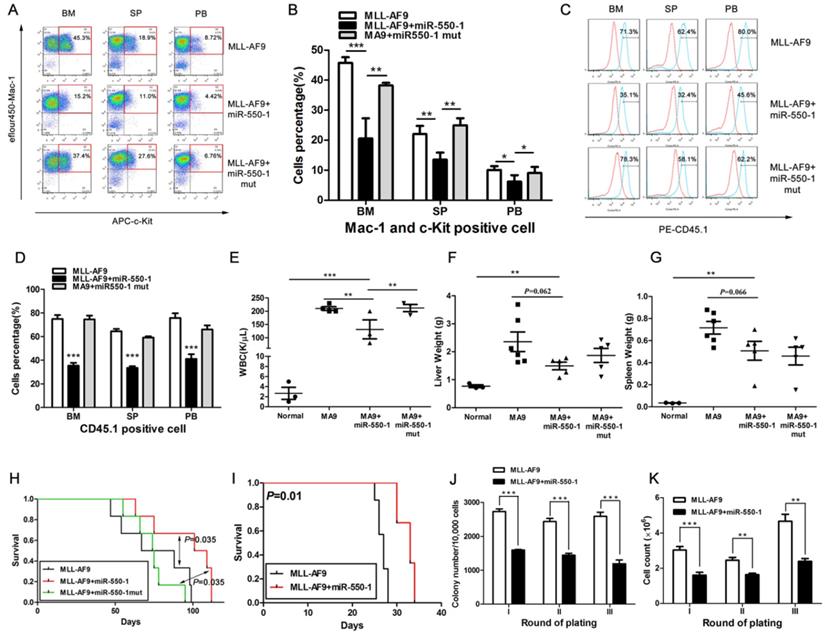
We additionally utilized secondary BMT assays as a means of assessing the importance of miR-550-1 in the maintenance of AML following its development. We found that mice in the miR-550-1 + MLL-AF9 group exhibited slower AML development than did those in the MLL-AF9 only group (median overall survival: 33 vs. 27 days, P=0.010; Fig. 4I), suggesting that suppressing miR-550-1 did contribute to the sustained maintenance of AML driven by the MLL-AF9 fusion gene. Secondary CFA further provided confirmation that miR-550-1 overexpression was linked to delayed leukemogenesis (Fig. 4J and K). Our results together therefore provide clear evidence for the role of miR-550-1 as a tumor suppressor in the context of AML development and maintenance.
Identification of potential miR-550-1 target genes
miRNAs are able to mediate their biological activities through the suppression of specific target genes. Given that our results suggested that miR-550-1 played a central role in suppressing the development of leukemia through regulating the cell cycle and inducing apoptosis, we next sought to identify potential miR-550-1 target genes linked to the promotion of apoptosis and G0/1 phase arrest. We therefore employed the TargetScan, PITA, miRanda, and miRBase programs in order to predict potential targets, identifying a total of 4,941 putative target genes identified by a minimum of 1 of these programs. Of the genes identified via this approach, WWTR1 was of potential interest as it scored highly among the predicted genes. WWTR1 has previously been found to serve as a key transcriptional co-activator in the Hippo signaling pathway, and there were multiple reports indicating that it could positively regulate both the cell cycle (specifically the G1/S transition) and mitochondrially-induced apoptosis in a range of cancer types [26, 28, 44]. We therefore first sought to assess WWTR1 mRNA expression in AML patient samples, revealing it to be significantly upregulation in these patients' samples relative to NC samples (P<0.05; Fig. 5A). Kaplan-Meier survival curves also confirmed that higher WWRT1 mRNA levels were correlated to poorer overall survival outcomes in these patients (P=0.030; Fig. 5B). This suggests that WWTR1 may serve as an oncogenic function in AML, and may also be a miR-550-1 target gene.
WWTR1 is a direct miR-550-1 target. (A) qPCR-mediated assessment of WWTR1 expression in AML and NC samples. (B) Lower WWTR1 expression was associated with improved overall survival in AML patients relative to higher expression (P=0.030). (C) There was a significant negative correlation between miR-550-1 and WWTR1 expression (r=-0.257, P=0.014). (D, E) Western blotting and qPCR confirmed the ability of miR-550-1 to inhibit endogenous WWTR1 expression. (F) Western blotting suggested that WTAP may be a miR-550-1 target. (G) miR-550-1 was able to significantly suppress luciferase activity for a vector bearing the WTAP 3'-UTR without affecting a control vector. (H) A m6A dot blot analysis in MV4-11 cells that were or were not overexpressing miR-550-1. (I) miR-550-1 target sites in the WWTR1 3'-UTR were mutated. (J) miR-550-1 led to a significant suppression of luciferase activity for a vector containing the WWTR1 3'-UTR without affecting a control vector. Error bar indicates SD of triplicate experiments. *, P<0.05; **, P<0.01.
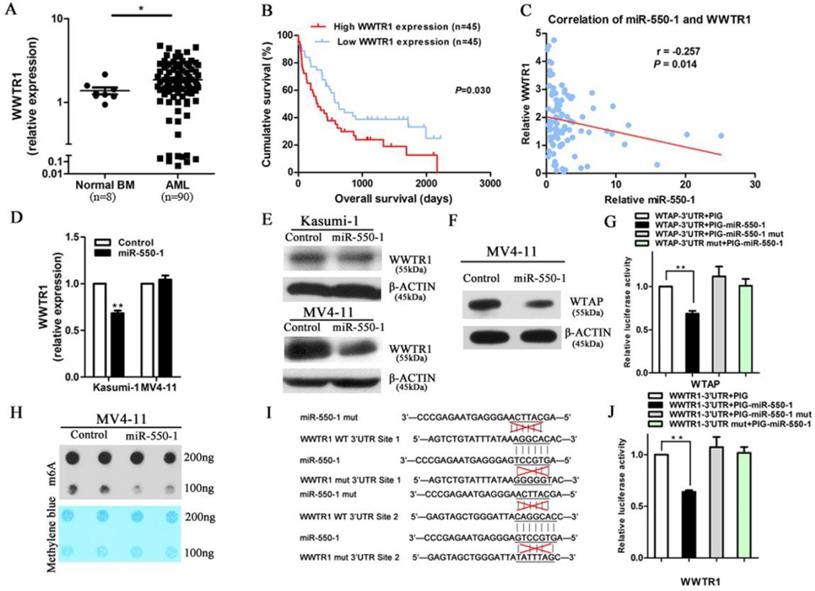
Using the TCGA and CALGB datasets, we performed in silico analyses revealing a negative correlation between miR-550-1 and WWTR1 expression (r=-0.173, P=0.021; r=-0.291, P=0.007, respectively). We additionally examined the expression of miR-550-1 and WWTR1 in 90 AML samples in our cohort, again revealing a significantly negative correlation between these two factors (r=-0.257, P=0.014; Fig. 5C). We also confirmed that the expression of WWTR1 was reduced at both the mRNA and protein level upon miR-550-1 overexpression in AML cells (Fig. 5D and E). Curiously, in the MV4-11 cells, the WWTR1 was downregulated only at the protein but not at the mRNA level upon miR-550-1 overexpression, suggesting that miR-550-1 might primarily affect the WWTR1 mRNA stability. Previous work suggested that Wilms tumor 1-associated protein (WTAP) targeting could lead to alterations in mRNA stability as a function of m6A modification [45, 46]. Our results also suggested that WTAP was a direct miR-550-1 target gene (Fig. 5F and G). Induced ectopic expression of miR-550-1 also led to a reduction in global mRNA m6A levels (Fig. 5H), potentially contributing at least in part to the reduced WWTR1 stability observed in our results.
To further confirm that there was a direct interaction between miR-550-1 and WWTR1, we next utilized a luciferase reporter assay system, generating luciferase reporter constructs bearing either a WT or mutated form of the WWTR1 3'-UTR in the pMIR-REPORT vector (Fig. 5I). When utilized in cells, we found that miR-550-1 was able to significantly decrease the luciferase activity associated with promoters bearing the WT WWTR1 3'-UTR (P=0.002), whereas they had no effect on those bearing the mutated WWTR1 3'-UTR (Fig. 5J). These results thus provide strong evidence supporting the fact that WWTR1 is a miR-550-1 target.
WWTR1 is a key miR-550-1 target in AML
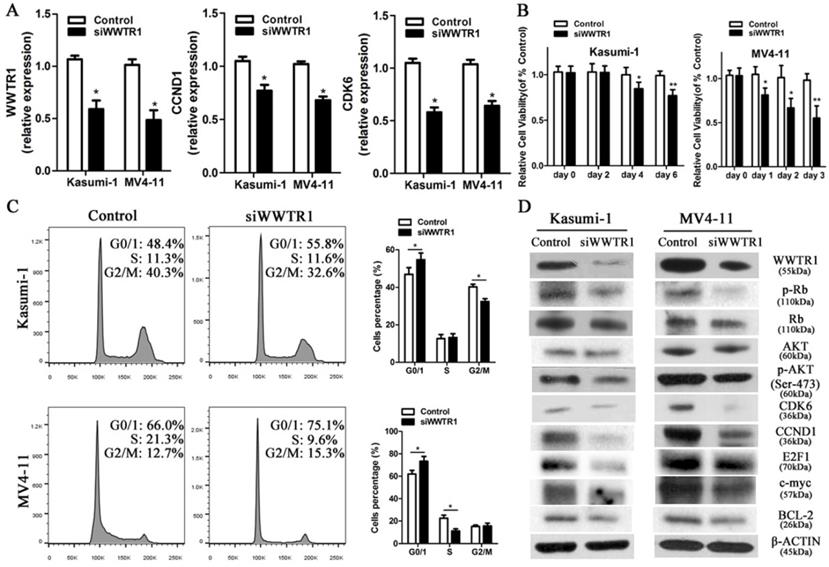
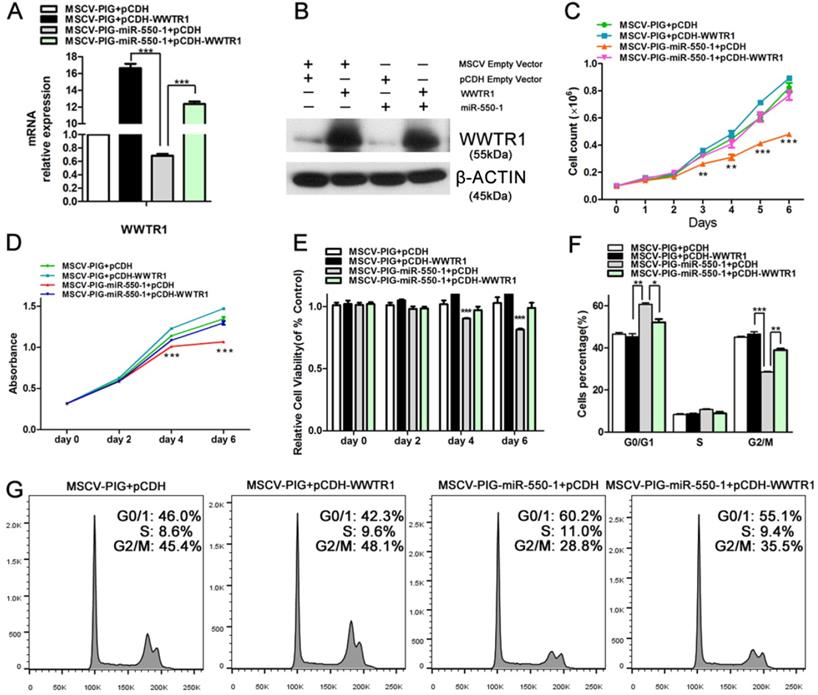
As WWTR1 is known to have important roles in the context of cancer [26, 47, 48], we next performed loss- and gain-of-function experiments in order to ascertain as to whether WWTR1 contributed to the anti-leukemic activity of miR-550-1. At present, the role of WWTR1 in AML has not been specifically assessed. We therefore employed a small interfering RNA construct to silence WWTR1 expression at the mRNA and protein level (Fig. 6A and D), and this led to a marked decrease in the proliferation of Kasumi-1 and MV4-11 cells, which instead underwent G0/1 arrest (Fig. 6B and C). We found that WWTR1 knockdown was linked to decreased protein levels of CCND1, CDK6, p-Rb, E2F1, BCL-2, p-AKT, and c-myc (Fig. 6D), phenocopying the anti-leukemic activity of miR-550-1. When WWTR1 was instead overexpressed in Kasumi-1 cells, this led to an increase in cell viability and proliferation, largely reversing the inhibitory effects of miR-550-1 (Fig. 7A-E). When WWTR1 and miR-550-1 were co-expressed, this led to a reversal in miR-550-1-mediated suppression of cell proliferation and G0/1-phase arrest in Kasumi-1 cells (Fig. 7F and G). Together, these findings thus suggest that miR-550-1-mediated suppression of WWTR1 expression at least partially governs the anti-leukemic activity of this miRNA.
Silencing WWTR1 partly recapitulates miR-550-1-associated phenotypes. (A) qPCR-mediated assessment of WWTR1, CCND1, and CDK6 expression in Kasumi-1 and MV4-11 cells following siWWTR1 transfection. (B) MTT-mediated assessment revealing that WWTR1 silencing led to a reduction in Kasumi-1 and MV4-11 cell viability. (C) Silencing of WWTR1 resulted in G0/1 phase arrest in both Kasumi-1 and MV4-11 cells. (D) Western blotting assessment of proteins related to apoptosis, proliferation, and the cell cycle in Kasumi-1 and MV4-11 cells with or without decreased WWTR1 expression. Error bar indicates SD of triplicate experiments. *, P<0.05; **, P<0.01.
Inducing WWTR1 expression can partially rescue miR-550-1-associated phenotypes. (A, B) Elevated ectopic WWTR1 expression was confirmed in miR-550-1-transfected or control Kasumi-1 cells by qPCR and Western blotting. (C) miR-550-1-mediated inhibition of cell proliferation was partially rescued via overexpressing WWTR1 in Kasumi-1 cells beginning on day 3. (D, E) MTT-mediated assessments revealed that elevated ectopic WWTR1 expression partly overcame miR-550-1-dependent reductions in cell viability beginning on day 4. (F, G) Elevated ectopic WWTR1 expression led to a partial rescue of miR-550-1-dependent G0/1 phase arrest. Error bar indicates SD of triplicate experiments. *, P<0.05; **, P<0.01; ***, P<0.001.
Discussion
Herein, we produce evidence that miR-550-1 plays a role in suppressing the development of AML and the proliferation of AML cells, instead promoting their apoptotic death. In humans, miR-550-1 is encoded in the 7p14.3 locus, which is a non-coding section of the ZNRF2 gene. Landgraf et al. first identified this miRNA via small RNA library sequencing in 2007, but it has not previously been studied in depth in the context of cancer [49]. To our knowledge, no previous reports have shown that miR-550-1 plays a vital role in leukemogenesis. We determined that miR-550-1 expression was decreased in two independent primary AML patient cohorts, and elevated miR-550-1 expression was associated with higher hemoglobin (Hb) levels (Table 2), potentially contributing to improved patient prognosis. However, at present there is no strong evidence regarding a link between miR-550-1 and Hb, and as such further research is required. Consistent with our findings regarding the methylation of the miR-550-1 region, we found that decitabine was able to partially reverse miR-550-1 downregulation, suggesting methylation of this region in AML patients contributed to miR-550-1 dysregulation.
In order to explore the specific mechanisms governing the link between miR-550-1 and reduced leukemia severity, we first conducted an in silico target analysis approach. We then, using luciferase reporter and mutagenesis assays, confirmed that WWTR1, which is an oncogene in solid tumors [28, 30, 31], was one key miR-550-1 target. Noguchi et al. [28] recently found that elevated WWTR1 expression was associated with poorer overall survival in those with NSCLC, and that it was confirmed to be an independent inferior element in NSCLC patients (HR=1.34, 95%CI 1.01-1.76, P=0.040). Similarly, breast cancer patients with elevated WWTR1 expression also exhibit poorer outcomes [30]. Furthermore, there is a positive correlation between WWTR1 expression and poorer prognosis, increased tumor invasion, and metastasis in those with gastric cardia adenocarcinoma [50]. Consistent with these past results, we observed significant upregulation of WWTR1 mRNA expression in AML, and this expression was negatively correlated with that of miR-550-1. YAP and TAZ have been reported to be transcriptional coactivators capable of recognizing cognate cis-regulatory elements via interactions with additional transcription factors, such as TEA domain family members (TEAD) [51, 52]. Li et al. [26] and Noguchi et al. [28] found WWTR1 to be capable of regulating the cell-cycle and apoptosis via regulating CCND1, CCND3, c-myc, BCL-2, and DKK1 expression, with WWTR1 knockdown resulting in a G1/S transition block in the cell cycle. YAP and TAZ have also been found to upregulate BCL-2-family member transcription, thereby suppressing the mitochondrial pathway of apoptosis [44]. Consistent with this, we found that both miR-550-1 overexpression and WWTR1 knockdown reduced CCND1, CDK6, p-Rb, E2F1, BCL-2, and p-AKT levels, and increased p27, and PARP levels. When ectopic WWTR1 lacking the 3'-UTR region was overexpressed, this significantly rescued the miR-550-1-induced G0/1-phase arrest observed in vitro. To date, the function of WWTR1 in AML was not previously determined, but our study thus highlights for the first time that WWTR1 is a key mediator related to the anti-leukemic activity of miR-550-1.
m6A is the most common methylation event modifying mRNA molecules in mammals, regulating a range of processes such as heat shock, differentiation, DNA damage responses, tissue development, and miRNA processing [53-55]. There is clear evidence that a disruption in m6A is linked to the pathogenesis of AML [56, 57]. WTAP is a m6A methyltransferase complex component, regulating m6A methyltransferase activity. Recent work indicates that WTAP is able to improve CDK2 stability via binding to the 3'UTR region, thereby enhancing cell proliferation in renal carcinoma [45]. Bansal et al. [58] found that WTAP knockdown reduced the proliferation of AML cells, suggesting it serves as an oncogenic function. Our work highlights, for the first time, that miR-550-1 mediates its anti-leukemic activity at least in part via decreasing WWTR1 stability by targeting WTAP.
Although we found that impairing WWTR1 mRNA stability, rather than promoting its degradation, was the primarily regulatory role of miR-550-1 in MV4-11 cells, further clarification is needed to determine why this effect was not identical in both cell lines. Whether miR-550-1/YAP/WWTR1 interact in a manner so as to form a negative feedback loop in AML remains unknown. Interestingly, a report by Chaulk et al. [59] suggested that nuclear YAP and WWTR1 together induced DICER complex activity, thus suggesting that these proteins might promote the maturation of certain pre-miRNAs into their mature forms in certain contexts. Liu et al. [60] also found circRNA-000425 to be a YAP/WWTR1 target using a circRNA microarray analysis, and revealed that YAP/WWTR1 were able to promote the oncogenic activity of miR-17 and miR-106b via inhibiting circRNA-000425 transcription. Additional experiments, however, are needed to determine whether YAP and WWTR1 regulate miR-550-1 expression. Our findings indicate that miR-550-1 is able to inhibit cell proliferation and promote apoptosis of AML cells via, at least in part, vying the direct targeting of the WWTR1 3'-UTR. Ultimately our findings both identify a novel tumor-suppressor miRNA, and also characterize previously unknown regulatory pathways governing WWTR1 expression in AML.
In summary, our study reveals the following: (1) elevated miR-550-1 expression is a favorable prognostic indicator in AML, and in AML patients it is at least partially dysregulated due to the hypermethylation promoter; (2) miR-550-1 is able to promote apoptosis and inhibit proliferation via regulation of the WTAP/WWTR1/BCL-2 and WTAP/WWTR1/CDK6/Rb/E2F1 pathways in AML; (3) m6A modifications are important for regulating the ability of miR-550-1 to target WWTR1. Our results thus demonstrate that miR-550-1 is a latent factor which suppresses AML, and as such enhancing expression of this miRNA may be a valuable therapeutic strategy in those with AML.
Abbreviations
AML: acute myeloid leukemia; miRNAs: microRNAs; HSC: hematopoietic stem cells; WWTR1: WW-domain containing transcription regulator-1; TAZ: transcriptional co-activator with PDZ-binding motif; YAP: yes-associated protein; CTGF: C transforming growth factor; LATS: large tumor suppressor; qRT-PCR: quantitative RT-PCR; BM: bone marrow; MNC: mononuclear cells; CFA: colony forming/replating assays; ALL: acute lymphoblastic leukemia.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was supported in part by grants from the National Natural Science Foundation of China (No. 81700137) and Zhejiang Provincial Natural Science Foundation of China (LQ19H080004, LY16H080004). This study was also supported in part by the National Institutes of Health (NIH) R01 grant (CA182528).
Authors' Contributions
Conception and design: Xi Jiang, Jie Jin, Jianjun Chen; Development of methodology: Xi Jiang, Chao Hu, Mengxia Yu, Yungui Wang, Chenying Li; Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): Chao Hu, Xia Li, Bryan Ulrich, Rui Su, Hengyou Weng, Huilin Huang; Analysis and interpretation of data: Chao Hu, Xi Jiang, Lei Dong; Writing, review, and/or revision of the manuscript: Chao Hu, Xi Jiang, Chenying Li; Administrative, technical, or material support: Xi Jiang, Jianjun Chen; Study supervision: Jie Jin, Xi Jiang, Jianjun Chen.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jiang X, Hu C, Ferchen K, Nie J, Cui X, Chen CH. et al. Targeted inhibition of STAT/TET1 axis as a therapeutic strategy for acute myeloid leukemia. Nat Commun. 2017;8:2099
2. Marcucci G, Mrozek K, Bloomfield CD. Molecular heterogeneity and prognostic biomarkers in adults with acute myeloid leukemia and normal cytogenetics. Curr Opin Hematol. 2005;12:68-75
3. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-97
4. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215-33
5. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522-31
6. Chen J, Odenike O, Rowley JD. Leukaemogenesis: more than mutant genes. Nat Rev Cancer. 2010;10:23-36
7. Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835-40
8. Xiao C, Rajewsky K. MicroRNA control in the immune system: basic principles. Cell. 2009;136:26-36
9. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203-22
10. Elmen J, Lindow M, Silahtaroglu A, Bak M, Christensen M, Lind-Thomsen A. et al. Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. 2008;36:1153-62
11. Wiggins JF, Ruffino L, Kelnar K, Omotola M, Patrawala L, Brown D. et al. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 2010;70:5923-30
12. Li Z, Huang H, Li Y, Jiang X, Chen P, Arnovitz S. et al. Up-regulation of a HOXA-PBX3 homeobox-gene signature following down-regulation of miR-181 is associated with adverse prognosis in patients with cytogenetically abnormal AML. Blood. 2012;119:2314-24
13. Li Z, Lu J, Sun M, Mi S, Zhang H, Luo RT. et al. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc Natl Acad Sci U S A. 2008;105:15535-40
14. Mi S, Lu J, Sun M, Li Z, Zhang H, Neilly MB. et al. MicroRNA expression signatures accurately discriminate acute lymphoblastic leukemia from acute myeloid leukemia. Proc Natl Acad Sci U S A. 2007;104:19971-6
15. Jiang X, Hu C, Arnovitz S, Bugno J, Yu M, Zuo Z. et al. miR-22 has a potent anti-tumour role with therapeutic potential in acute myeloid leukaemia. Nat Commun. 2016;7:11452
16. Jiang X, Huang H, Li Z, He C, Li Y, Chen P. et al. MiR-495 is a tumor-suppressor microRNA down-regulated in MLL-rearranged leukemia. Proc Natl Acad Sci U S A. 2012;109:19397-402
17. Li Z, Chen P, Su R, Li Y, Hu C, Wang Y. et al. Overexpression and knockout of miR-126 both promote leukemogenesis. Blood. 2015;126:2005-15
18. Mi S, Li Z, Chen P, He C, Cao D, Elkahloun A. et al. Aberrant overexpression and function of the miR-17-92 cluster in MLL-rearranged acute leukemia. Proc Natl Acad Sci U S A. 2010;107:3710-5
19. Li Z, Huang H, Chen P, He M, Li Y, Arnovitz S. et al. miR-196b directly targets both HOXA9/MEIS1 oncogenes and FAS tumour suppressor in MLL-rearranged leukaemia. Nat Commun. 2012;3:688
20. Huang H, Jiang X, Wang J, Li Y, Song CX, Chen P. et al. Identification of MLL-fusion/MYC dash, verticalmiR-26 dash, verticalTET1 signaling circuit in MLL-rearranged leukemia. Cancer Lett. 2016;372:157-65
21. Jiang X, Huang H, Li Z, Li Y, Wang X, Gurbuxani S. et al. Blockade of miR-150 maturation by MLL-fusion/MYC/LIN-28 is required for MLL-associated leukemia. Cancer Cell. 2012;22:524-35
22. Jiang X, Bugno J, Hu C, Yang Y, Herold T, Qi J. et al. Eradication of Acute Myeloid Leukemia with FLT3 Ligand-Targeted miR-150 Nanoparticles. Cancer Res. 2016;76:4470-80
23. Kanai F, Marignani PA, Sarbassova D, Yagi R, Hall RA, Donowitz M. et al. TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 2000;19:6778-91
24. Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol. 2011;13:877-83
25. Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 2015;15:73-9
26. Li Y, Yang FL, Zhu CF, Tang LM. Effect and mechanism of RNAi targeting WWTR1 on biological activity of gastric cancer cells SGC7901. Mol Med Rep. 2018;17:2853-60
27. Wang L, Chen Z, Wang Y, Chang D, Su L, Guo Y. et al. TR1 promotes cell proliferation and inhibits apoptosis through cyclin A and CTGF regulation in non-small cell lung cancer. Tumour Biol. 2014;35:463-8
28. Noguchi S, Saito A, Horie M, Mikami Y, Suzuki HI, Morishita Y. et al. An integrative analysis of the tumorigenic role of TAZ in human non-small cell lung cancer. Clin Cancer Res. 2014;20:4660-72
29. Bartucci M, Dattilo R, Moriconi C, Pagliuca A, Mottolese M, Federici G. et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene. 2015;34:681-90
30. Diaz-Martin J, Lopez-Garcia MA, Romero-Perez L, Atienza-Amores MR, Pecero ML, Castilla MA. et al. Nuclear TAZ expression associates with the triple-negative phenotype in breast cancer. Endocr Relat Cancer. 2015;22:443-54
31. Tian T, Li A, Lu H, Luo R, Zhang M, Li Z. TAZ promotes temozolomide resistance by upregulating MCL-1 in human glioma cells. Biochem Biophys Res Commun. 2015;463:638-43
32. Wang L, Shi S, Guo Z, Zhang X, Han S, Yang A. et al. Overexpression of YAP and TAZ is an independent predictor of prognosis in colorectal cancer and related to the proliferation and metastasis of colon cancer cells. PLoS One. 2013;8:e65539
33. Justice RW, Zilian O, Woods DF, Noll M, Bryant PJ. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995;9:534-46
34. Visser S, Yang X. LATS tumor suppressor: a new governor of cellular homeostasis. Cell Cycle. 2010;9:3892-903
35. Jimenez-Velasco A, Roman-Gomez J, Agirre X, Barrios M, Navarro G, Vazquez I. et al. Downregulation of the large tumor suppressor 2 (LATS2/KPM) gene is associated with poor prognosis in acute lymphoblastic leukemia. Leukemia. 2005;19:2347-50
36. Lavau C, Du C, Thirman M, Zeleznik-Le N. Chromatin-related properties of CBP fused to MLL generate a myelodysplastic-like syndrome that evolves into myeloid leukemia. EMBO J. 2000;19:4655-64
37. Lavau C, Luo RT, Du C, Thirman MJ. Retrovirus-mediated gene transfer of MLL-ELL transforms primary myeloid progenitors and causes acute myeloid leukemias in mice. Proc Natl Acad Sci U S A. 2000;97:10984-9
38. Lavau C, Szilvassy SJ, Slany R, Cleary ML. Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX-ENL. EMBO J. 1997;16:4226-37
39. Bi L, Zhou B, Li H, He L, Wang C, Wang Z. et al. A novel miR-375-HOXB3-CDCA3/DNMT3B regulatory circuitry contributes to leukemogenesis in acute myeloid leukemia. BMC Cancer. 2018;18:182
40. He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S. et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828-33
41. Yan M, Kanbe E, Peterson LF, Boyapati A, Miao Y, Wang Y. et al. A previously unidentified alternatively spliced isoform of t(8;21) transcript promotes leukemogenesis. Nat Med. 2006;12:945-9
42. Hu C, Yu M, Ren Y, Li K, Maggio DM, Mei C. et al. PP2A inhibition from LB100 therapy enhances daunorubicin cytotoxicity in secondary acute myeloid leukemia via miR-181b-1 upregulation. Sci Rep. 2017;7:2894
43. Wang L, You LS, Ni WM, Ma QL, Tong Y, Mao LP. et al. beta-Catenin and AKT are promising targets for combination therapy in acute myeloid leukemia. Leuk Res. 2013;37:1329-40
44. Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ. et al. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012;151:1457-73
45. Tang J, Wang F, Cheng G, Si S, Sun X, Han J. et al. Wilms' tumor 1-associating protein promotes renal cell carcinoma proliferation by regulating CDK2 mRNA stability. J Exp Clin Cancer Res. 2018;37:40
46. Horiuchi K, Umetani M, Minami T, Okayama H, Takada S, Yamamoto M. et al. Wilms' tumor 1-associating protein regulates G2/M transition through stabilization of cyclin A2 mRNA. Proc Natl Acad Sci U S A. 2006;103:17278-83
47. Choi W, Kim J, Park J, Lee DH, Hwang D, Kim JH. et al. YAP/TAZ Initiates Gastric Tumorigenesis via Upregulation of MYC. Cancer Res. 2018;78:3306-20
48. Ji J, Xu R, Zhang X, Han M, Xu Y, Wei Y. et al. Actin like-6A promotes glioma progression through stabilization of transcriptional regulators YAP/TAZ. Cell Death Dis. 2018;9:517
49. Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A. et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401-14
50. Wei J, Wang LC, Zhu J, Sun AQ, Yu GZ, Chen M. et al. The Hippo signaling effector WWTR1 is a metastatic biomarker of gastric cardia adenocarcinoma. Cancer Cell International. 2019 19
51. Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014;94:1287-312
52. Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B. et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol. 2015;17:1218-27
53. Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014;16:191-8
54. Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian SB. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591-4
55. Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res. 2018;28:507-17
56. Li Z, Weng H, Su R, Weng X, Zuo Z, Li C. et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell. 2017;31:127-41
57. Ianniello Z, Fatica A. N6-Methyladenosine Role in Acute Myeloid Leukaemia. Int J Mol Sci. 2018 19
58. Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Penalva LO. et al. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia. 2014;28:1171-4
59. Chaulk SG, Lattanzi VJ, Hiemer SE, Fahlman RP, Varelas X. The Hippo pathway effectors TAZ/YAP regulate dicer expression and microRNA biogenesis through Let-7. J Biol Chem. 2014;289:1886-91
60. Liu Z, Huang S, Cao Y, Yao Y, Li J, Chen J. et al. YAP1 inhibits circRNA-000425 expression and thus promotes oncogenic activities of miR-17 and miR-106. Biochem Biophys Res Commun. 2018;503:2370-5
Author contact
![]() Corresponding authors: Jie Jin, Department of Hematology, The First Affiliated Hospital of Zhejiang University, Hangzhou, Zhejiang 310003, China. E-mail: jiej0503edu.cn; Jianjun Chen, Department of Systems Biology & the Gehr Family Center for Leukemia Research, Beckman Research Institute of City of Hope, Monrovia, CA 91016, USA. E-mail: jianchenorg; or Xi Jiang, Department of Systems Biology & the Gehr Family Center for Leukemia Research, Beckman Research Institute of City of Hope, Monrovia, CA 91016, USA. E-mail: xjiangorg.
Corresponding authors: Jie Jin, Department of Hematology, The First Affiliated Hospital of Zhejiang University, Hangzhou, Zhejiang 310003, China. E-mail: jiej0503edu.cn; Jianjun Chen, Department of Systems Biology & the Gehr Family Center for Leukemia Research, Beckman Research Institute of City of Hope, Monrovia, CA 91016, USA. E-mail: jianchenorg; or Xi Jiang, Department of Systems Biology & the Gehr Family Center for Leukemia Research, Beckman Research Institute of City of Hope, Monrovia, CA 91016, USA. E-mail: xjiangorg.