Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(2):623-634. doi:10.7150/ijbs.55020 This issue Cite
Research Paper
MEF2A-mediated lncRNA HCP5 Inhibits Gastric Cancer Progression via MiR-106b-5p/p21 Axis
Weiwei Chen1*, Kundong Zhang1*, Yuhan Yang1, Zengya Guo1, Xiaofeng Wang1, Buwei Teng2, Qian Zhao3 ![]() , Chen Huang1
, Chen Huang1 ![]() , Zhengjun Qiu1
, Zhengjun Qiu1 ![]()
1. Department of General Surgery, Shanghai General Hospital, Shanghai Jiaotong University School of Medicine, 100 Haining Road, Shanghai, 201600, China.
2. Lianyungang Clinical College of Nanjing Medical University/The First People's Hospital of Lianyungang, 6 Zhenhua East Road, Haizhou District, City of Lianyungang, Jiangsu Province, 222061, China.
3. Department of Pathophysiology, Key Laboratory of Cell Differentiation and Apoptosis of National Ministry of Education, Shanghai Jiao Tong University School of Medicine, Shanghai, 200025, China.
* These authors contributed equally to this work.
Received 2020-10-26; Accepted 2020-12-31; Published 2021-1-16
Abstract

Background: Long non-coding RNAs (lncRNAs) are deemed to be relevant to the tumorigenesis and development of a variety of tumors, containing gastric cancer (GC). The purpose of our investigations is to explore the character of HCP5 in GC.
Methods: HCP5 expression was detected by quantitative real-time polymerase chain reaction (qRT-PCR) in 62 matched GC tissues and corresponding para-carcinoma tissues. In vitro and in vivo functional assays were subjected to verify the biological effects of HCP5 after alteration of HCP5. Chromatin immunoprecipitation assay (CHIP) assays were conducted to confirm that myocyte enhancer factor 2A (MEF2A) could bind to HCP5 promoter regions and thereby induce HCP5 expression. Analysis of the latent binding of miR-106b-5p to HCP5 and p21 was made by bioinformatics prediction and luciferase reporter assays.
Results: Significant downregulation of HCP5 was detected in GC tissues. Negative correlation was determined between HCP5 expression level and tumor size and overall survival in GC patients. HCP5 depletion had a facilitating impact on proliferation, migration and invasion of GC cells. Consistently, overexpression of HCP5 came into an opposite effect. Moreover, we demonstrated that MEF2A could combine with the promoter region of HCP5 and thereby induce HCP5 transcription. Luciferase reporter assays revealed that HCP5 could compete with miR-106b-5p as a competing endogenous RNA (ceRNA) and upregulated p21 expression in GC.
Conclusions: MEF2A-mediated HCP5 could exert an anti-tumor effect among the development of GC via miR-106b-5p/p21 axis, which provides a novel target for GC therapy.
Keywords: HCP5, MEF2A, Competing endogenous RNA, miR-106b-5p/p21 axis, Gastric Cancer.
Introduction
Gastric cancer (GC) is known as the fifth most commonly diagnosed type of malignancy and the third primary cause of cancer-associated death globally, particularly in Eastern Asian [1, 2]. On account of lacking apparent symptoms and effective screening tools, a large proportion of GC patients are diagnosed at progressive stages or with distant metastasis [3]. For GC patients at early stages, the best treatment is surgical resection; for patients at advanced stage, the most important treatment is chemotherapy [4, 5]. Despite great improvement has been made in diagnosis and therapeutic strategies of GC so far, the prognosis for advanced-stage patients remained largely unsatisfactory owing to the current status that little is known about the concrete mechanism of gastric tumorigenesis and progression [6, 7]. Therefore, there is an urgent need to get a comprehensive understanding of GC and to find effective targets of clinical therapeutics to improve diagnosis and prognosis for GC.
Human transcriptome, approximately 98% of which, is composed of non-coding RNAs (ncRNAs) [8-10]. Long non-coding RNAs (lncRNAs) are deemed as a newly-discovered kind of ncRNAs whose transcripts are larger than 200 nucleotides, the capacity for protein-coding of which is limited or none [11, 12]. In recent years, research has showed that lncRNAs could function as pivotal regulators in diverse tumor process, such as proliferation, metastasis, apoptosis and cell differentiation [13-15]. Moreover, the progression in a variety of tumors has been discovered to be associated with aberrant expression of lncRNAs, including gastric tumorigenesis [16-19]. Notably, lncRNAs could exert vital effects on regulating gene expression in various manners, including histone modification, chromatin modification, splicing modulation, transcriptional and post-transcriptional processing [20-22]. Nevertheless, the regulatory molecular mechanisms of lncRNAs in GC are still not fully excavated and require further investigation.
In our research, we expounded a GC-related lncRNA HCP5, which was markedly downregulated in GC tissues. Moreover, alteration of HCP5 expression could regulate the characteristics of GC cells such as migration, invasion and proliferation in vitro and tumor growth in vivo. Herein, we aimed at illustrating the latent regulatory mechanisms involved in HCP5 in GC progression. Further research found that HCP5 downregulation in GC was mediated by transcription factor MEF2A. Next, HCP5 was discovered to compete with miR-106b-5p and thereby regulate p21 expression. Taken all together, our results elucidated the clinical relevance of HCP5 in GC, providing novel and deeper comprehension into the regulatory role of HCP5 acted as in GC tumorigenesis and progression.
Materials and methods
Tissue specimens
62 paired GC tissues and normal tissues were obtained from GC patients who received gastrectomy at Shanghai General Hospital. For RNA extraction, all specimens were stored at -80℃ after surgical excision. The study was approved by the Ethics Committee of Shanghai General Hospital and written informed consent was obtained before specimen collection.
Cell culture
Human GC cell lines (MGC-803, HGC-27, SGC-7901, MKN-28, MKN-45) and human normal gastric epithelial mucosa cell line (GES-1), were all purchased from the Culture Collection of Chinese Academy of Sciences (Shanghai, China). All above GC cells and GES-1 cells were maintained in RPMI-1640 medium (Basalmedia, China), and DMEM medium (Gibco, USA) was used to culture HEK-293T cells. Cells were cultured at 37℃ in 5% CO2 incubator supplemented with 10% fetal bovine serum (Sigma, China).
RNA extraction and quantitative RT-PCR assays
We extracted total RNA from cultured cell lines and tissues using TRIzol Reagent (Takara, Japan). For mRNA and lncRNA detection, reverse transcriptions were conducted by FastKing gDNA Dispelling RT SuperMix (TIANGEN, China). For reverse transcriptions of miRNA, miDETECT A Track miRNA qPCR Kit (Ruibo, China) was used. Then qRT-PCR assay was operated on the QuantStudio 5 real-time PCR system (Applied Biosystems, USA) with SYBR green reaction mix (Applied Biosystems, USA). For all of the qRT-PCRs, U6 and β-actin were deemed as an internal control. All process was repeated in triplicate and results were analyzed using the 2-ΔΔCT method. The primers used in this research were listed in Additional file 1: Table S2.
Cell counting kit-8 (CCK8) assay
96-well plates were seeded with 2000 cells after 48h transfection. Then, by adding CCK-8 solution (Dojindo Crop, Japan) at appropriate time, cell viability was evaluated by a microplate reader (BioTek Instruments, USA) after incubating at 37 °C for 2 h.
Colony formation assay
6-well plates were seeded with five hundred cells after 48h transfection, culturing at 37°C in 5% CO2 incubator. Next, PBS was used to wash cultured cells in triplicate and cells were fixed for 20 min in methanol. Crystal violet was used to stain cells for another 20 min.
Wound healing assays
A 200uL pipette tip was used for creating an artificial scratch when GC cells were seeded. After culturing in serum-free medium for 0h, 24h and 48h, typical images were captured.
Transwell assay
Upper chambers were plated with GC cells supplied with medium of no serum. For invasion assays, the lower chamber (Corning-Costar; pore size; 8um, USA) was covered with matrigel mix (Sigma, USA). After incubation for 24h or 48h, cells were fixed and stained. For visualization, images of cultured cells were collected and counted in random different five fields.
Xenografts in mice
Experiments on animals were approved by the Animal Care Committee of Shanghai General Hospital. BALB/c nude male mice at 4-week-old were used and SGC-7901 cells stably overexpressing HCP5 were injected subcutaneously into the back flank (2 x 106, 200ul). Tumor sizes were measured every 5 days using a caliper as soon as the tumors were measurable. The formula (length x width2)/2 was used to calculate the volume. Finally, the tumor weight was detected at the time that mice were sacrificed.
Cell transfection
Oligonucleotides used in our study were purchased (Genepharma, China). Sequences of oligonucleotides are shown in Additional file 1: Table S3. We transfected these Oligonucleotides by Lipofectamine 2000 (Invitrogen, USA). Transfection was carried out when cell density reaches a confluence of 60%-70%.
Chromatin Immunoprecipitation assay (ChIP)
In brief, 1% formaldehyde was used to cross-link GC cells for 10 min. Afterwards, glycine was used to quench cells. Sonication was used to lyse cells and cells were immunoprecipitated with MEF2A antibody (sc-17785X; Santa Cruz). Normal IgG antibody (sc-2025; Santa Cruz) was served as the negative treatment. Subsequently, we analyzed the precipitated chromatin DNA by qRT-PCR. The sequences of primers for CHIP are showed in Additional file 1: Table S2.
Western blot
RIPA (Beyotime, China) was used to isolate total proteins from MGC-803 and SGC-7901 cells. Extracted proteins were separated using 10% gel and then transferred onto a polyvinylidene fluoride (PVDF) membrane. Then, 5% skim milk was used to block the membrane. Next, the membrane was incubated with the primary antibody at 4°C overnight. The antibodies we used are showed in Additional file 1: Table S4. The secondary antibody was purchased from CST (1:4000, Cell Signaling Technology, USA).
Luciferase reporter assay
The complementary DNA (cDNA) of HCP5 was loaded into psiCHECK2 vector (Promega) (HCP5-wild). Mutations are made in the potential miR-106b-5p binding sites by Fast Mutagenesis kit V2 (Vazyme China) (HCP5-mut). The luciferase activity was measured via Dual-Luciferase Reporter Assay System (Promega, USA).
Bioinformatic analyses
The downstream miRNA targets of HCP5 were predicted using starBase, NPInter, RNAInter and miRcode databases. Moreover, RNAInter, starBase, TargetScan and miRDB were used to discover the targets of miR-106b-5p.
Statistical analysis
SPSS 22.0 and GraphPad Prism 8.2 were used for analysis of statistics. Data were showed as mean ± SD. Statistical analysis between two groups was conducted by Student's t-test. Pearson correlation coefficients were used to evaluate correlations of groups. As for our analyses, P<0.05 between groups is deemed to be statistically significant.
Results
HCP5 is significantly decreased in human gastric cancer tissues
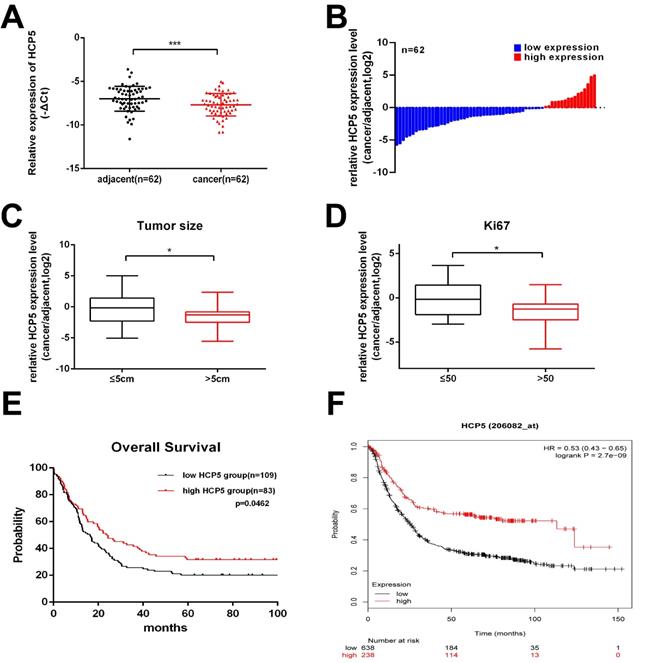
Firstly, to explore whether HCP5 is dysregulated in GC progress, HCP5 expression was detected in 62 paired GC tissues and corresponding para-carcinoma normal tissues by qRT-PCR. We observed that, compared to adjacent normal tissues, HCP5 in tumor tissues was markedly downregulated (Fig. 1A, B). Next, clinical relevance was analyzed between HCP5 expression level in gastric cancer and patients' clinical features. 62 GC patients were divided into two groups according to their tumor size and Ki67 index. As we can see, HCP5 expression was prominently reduced in patients with a larger tumor size and higher Ki67 index (Ki67 >50) (Fig. 1C, D). Moreover, further analysis revealed that HCP5 expression was notably positively correlated to better tumor differentiation and no statistically significant differences among other clinical parameters were found to be correlated with HCP5 in our study (Additional file 1: Table S1). Further, we analyzed datasets obtained from the R2 Platform database, from which negative correlation was found between HCP5 expression and overall survival in GC patients (Fig. 1E). Consistently, Kaplan-Meier plotter datasets manifested that HCP5 may serve as a promising prognostic biomarker as well (Fig. 1F). Collectively, these results manifested that HCP5 was markedly downregulated in GC and may be relevant to GC progression.
HCP5 is downregulated in GC. A HCP5 was detected in 62 paired GC tissues and adjacent normal tissues by qRT-PCR. B Fold changes (log2) of HCP5 were ranked from low to high. C, D HCP5 expression in GC based on tumor size (C) and ki67 index (D). E, F R2 OS (E) and Kaplan-Meier (F) curves based on HCP5 expression. Data are showed as mean ± SD. *p < 0.05, ***p < 0.001.
Altering HCP5 expression impacts the proliferation, migration and invasion of GC cells in vitro
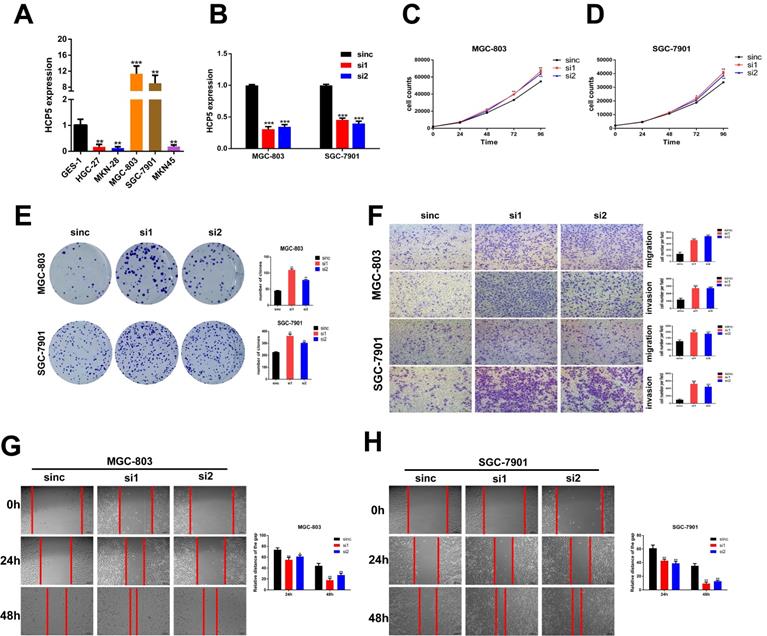
To make a deeper exploration to the biological features of HCP5 in GC malignant progression, we detected the HCP5 expression among normal gastric epithelium cell (GES-1) and GC cell lines by qRT-PCR (Fig. 2A). Since HCP5 expression in GC was notably decreased and negatively correlated with GC prognosis, we wondered whether loss-of-HCP5 in GC cell lines could exert an inhibitory effect on GC cells. The MGC-803 and SGC-7901 cells were selected for further research as they have relative high expression level of HCP5. Next, two independent siRNAs were transfected into the two selected cell lines and knockdown efficiency was detected by qRT-PCR (Fig. 2B). CCK-8 proliferation assays manifested that downregulation of HCP5 could notably facilitate GC cell proliferation (Fig. 2C, D). Besides, colony formation experiments suggested that silencing of HCP5 significantly increased the number of GC cell colonies (Fig. 2E). Furthermore, Transwell assays were conducted to assess whether HCP5 knockdown influenced cell migration and invasive abilities. Our research manifested that cell migration and invasion numbers were dramatically increased in both GC cells transfected with siHCP5 than in siNC group (Fig. 2F). Moreover, wound healing assays were performed to show that silencing of HCP5 enhanced GC cell migration ability (Fig. 2G, H).
Knockdown of HCP5 promotes GC cell proliferation, migration, and invasion. A HCP5 expression in 5 GC cell lines and GES-1 were analyzed by qRT-PCR. B Knockdown efficiency of silencing HCP5 in GC cells. C-E GC cell proliferation after HCP5 knockdown was detected using CCK8 assay (C, D) and colony formation assay (E). F-H Typical images of transwell assays (F) and wound healing assays (G, H) after HCP5 knockdown (100x, scale bar=100μm). Data are showed as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001.
In addition, we constructed pcDNA3.1 vector containing full-length lncRNA HCP5 to upregulate HCP5 expression by transfecting it into two selected GC cells (Fig. S1A). Our results manifested that overexpression of HCP5 could restrain the proliferation ability of GC cells by performing CCK8 assays and colony formation assays (Fig. S1B-D). Moreover, transwell assays showed that capabilities of GC cell migration and invasion were markedly restrained by HCP5 upregulation (Fig. S1E). Besides, wound healing assays manifested that the alteration of migration ability induced by overexpressing HCP5 was consistent with transwell migration assays (Fig. S1F, G). Our results clearly indicated that HCP5 downregulation facilitated GC cell proliferation, migration and invasion abilities and HCP5 overexpression exerted inverse effects. Therefore, HCP5 could act as a tumor suppressor in GC progression.
HCP5 overexpression inhibits gastric tumor growth in vivo
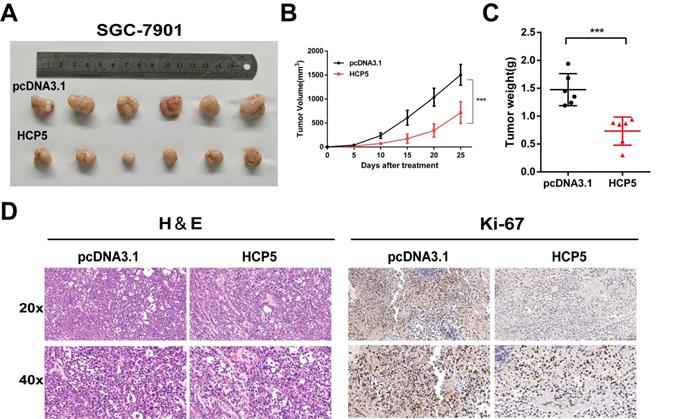
To gain insights into whether HCP5 could act as a critical role in vivo on tumor growth, we subcutaneously inoculated SGC-7901 cells stably overexpressing HCP5 into the back flank of male nude mice. Congruous with results in vitro, we found that the volumes of the xenograft tumors with HCP5 overexpression were markedly decreased (Fig. 3A). Moreover, tumor growth was effectively suppressed by HCP5 overexpression indicated by tumor growth curve (Fig. 3B). Next, tumor weight of HCP5 overexpression group was found dramatically reduced (Fig. 3C). For further study, hematoxylin-eosin (HE) staining and immunohistochemistry (IHC) for Ki67 were conducted, and it revealed that HCP5 overexpression led to a prominent decrease in Ki67 (Fig. 3D). In all, our results demonstrated that HCP5 overexpression significantly restrained GC tumor growth in vivo.
HCP5 overexpression inhibits gastric tumor growth in vivo. A Image of Xenograft tumors with stable HCP5 overexpression after harvested. B, C Tumor volume(B) and weight (C) were measured. D Represent images of IHC staining for H&E and Ki-67 of xenograft tumor tissue samples. Scare Bar=50um. Data are showed as the mean ± SD. ***p < 0.001.
MEF2A transcriptionally modulate HCP5 expression in GC cells
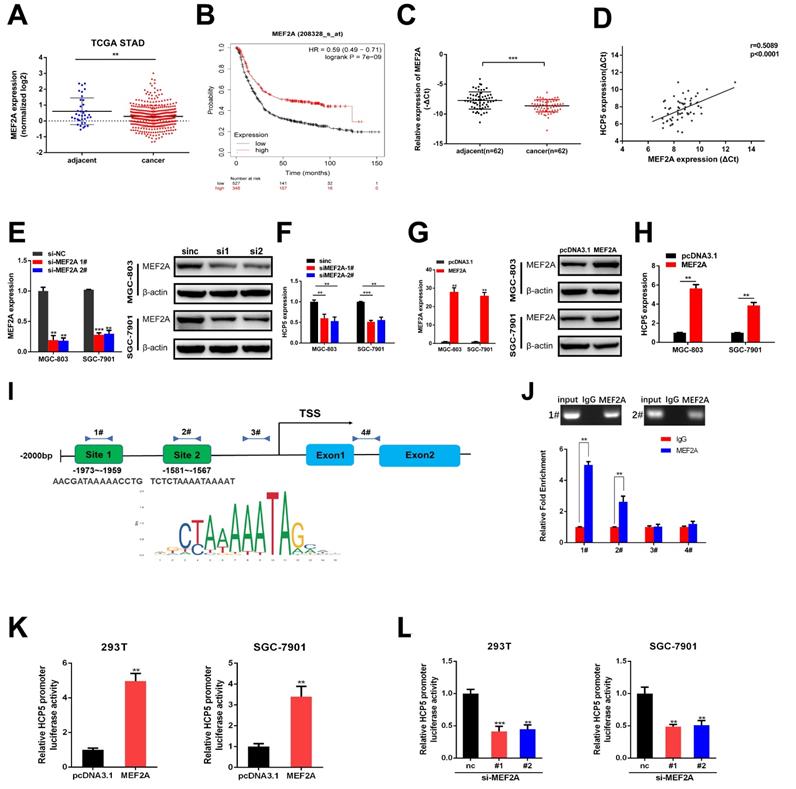
In order to discover the regulatory mechanism under HCP5 downregulation in GC, we used JASPAR database to predict latent transcription factors (TFs) that might interact with HCP5 promoter. MEF2A achieved the highest score of binding to the promoter of HCP5 among all the TFs that were predicted. By analyzing datasets from TCGA STAD, MEF2A was notably downregulated in GC tissues in comparison with adjacent normal tissues (Fig. 4A). Moreover, higher MEF2A expression is relevant to a better prognosis of GC patients based on Kaplan-Meier plotter database (Fig. 4B). Furthermore, we detected MEF2A expression in our own 62 paired GC specimens and results showed that MEF2A was dramatically downregulated and positively relevant to HCP5 in GC tissues (Fig. 4C, D). To gain insights into the correlation between MEF2A and HCP5, we silenced MEF2A in GC cells and found that the expression of HCP5 was decreased (Fig. 4E, F). Conversely, overexpression of MEF2A caused an increase in HCP5 expression (Fig. 4G, H). Based on the results predicted by JASPAR, there were two latent combining sites predicted on HCP5 promoters according to the binding motif of MEF2A (Fig. 4I). Importantly, results from ChIP assays revealed that MEF2A could directly combine with the promoter of HCP5 at site 1 and site 2 (Fig. 4J). To further verify that HCP5 could serve as a transcriptional target of MEF2A, we cloned HCP5 promoter into pGL4.27 vector, and luciferase activity was measured after co-transfecting luciferase reporter and pcDNA3.1-MEF2A which overexpressed MEF2A and siRNAs against MEF2A into SGC-7901 cells and HEK-293T cells. Results showed that MEF2A overexpression markedly increased luciferase activity while MEF2A knockdown markedly reduced luciferase activity (Fig. 4K, L). In all, our data revealed that HCP5 downregulation is modulated by MEF2A in GC.
HCP5 is modulated by MEF2A. A MEF2A expression from TCGA database. B OS curves based on MEF2A expression from Kaplan-Meier Plotter. C MEF2A expression in 62 paired GC tissues and corresponding adjacent tissues. D Analysis of correlation based on expression level between MEF2A and HCP5 in GC tissues. E-H HCP5 expression was detected by qRT-PCR after MEF2A alteration. I Predicted MEF2A-binding sites in HCP5 promoters. J ChIP assays were conducted to verify the combination between MEF2A and HCP5 promoters relative to normal IgG in MGC-803 cells. K, L Luciferase activity driven by HCP5-reporter vector was altered in HEK293T and SGC-7901 cells after MEF2A overexpression or knockdown. Data are showed as the mean ± SD. **p < 0.01, ***p < 0.001.
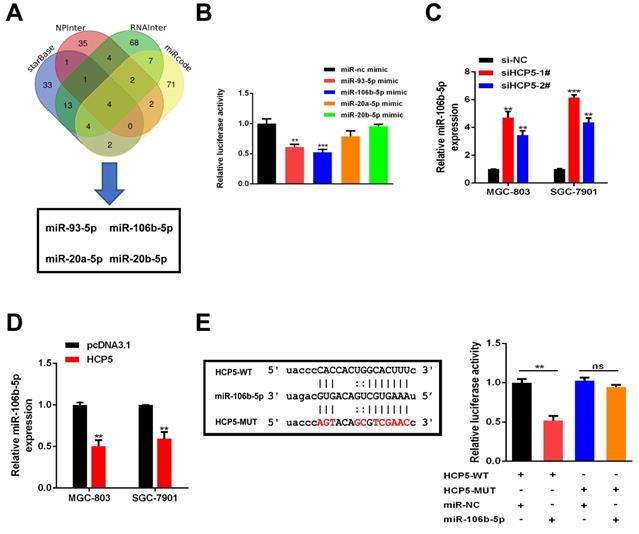
HCP5 functions as a ceRNA and sponges miR-106b-5p in gastric cancer cells
Plenty of lncRNAs have been found to function as a ceRNA to compete with microRNAs and modulate the expression of the target mRNAs. To further investigate how HCP5 exerted its effects, Bioinformatics prediction was conducted by starBase, NPInter, RNAInter and miRcode databases. Results showed that four miRNAs (miR-93-5p, miR-106b-5p, miR-20a-5p and miR-20b-5p) may serve as biological targets of HCP5 (Fig. 5A). TCGA databases revealed that miR-20a-5p, miR-106b-5p, and miR-93-5p were downregulated in GC, while miR-20b-5p made no difference (Fig. S2). Next, dual luciferase reporter assays were conducted to verify these predictions. Briefly, a luciferase plasmid psiCHECK2 comprising full-length HCP5 sequence, along with specific miRNA mimics, was co-transfected into HEK-293T cells. Results showed that HCP5-driven luciferase activity was merely restrained by miR-93-5p and miR-106b-5p mimics. Moreover, miR-106b-5p owned a stronger suppression effect of luciferase activity than miR-93-5p (Fig. 5B). Hence, we selected miR-106b-5p as a primary candidate for further research. HCP5 knockdown resulted in upregulating miR-106b-5p, whereas HCP5 overexpression resulted in the opposite effect (Fig. 5C, D). Besides, luciferase activity of HCP5-WT was remarkably decreased after transfecting miR-106b-5p mimics, whereas it was unable to change the activity of HCP5-MUT reporter vector, suggesting that miR-106b-5p might directly interact with HCP5 (Fig. 5E). Collectively, these results demonstrated that HCP5 physically interacts with miR-106b-5p and may serve as a ceRNA.
HCP5 sponges miR-106b-5p in GC cells. A Predictions of 4 potential targeted miRNAs of HCP5 from the four datasets (StarBase, NPInter, RNAInter, miRcode). B In HEK-293T cells, several miRNA mimics were co-transfected with psiCHECK2-HCP5 vectors. C, D MiRNA expression in GC cells after HCP5 knockdown(C) or overexpression(D). E HCP5-WT and HCP5-MUT were co-transfected with miR-106b-5p mimics or miR-NC mimics into HEK293T cell. Data are showed as the mean ± SD. **p < 0.01, ***p < 0.001.
HCP5 suppresses GC cell proliferation, migration and invasion of gastric via mediating miR-106b-5p
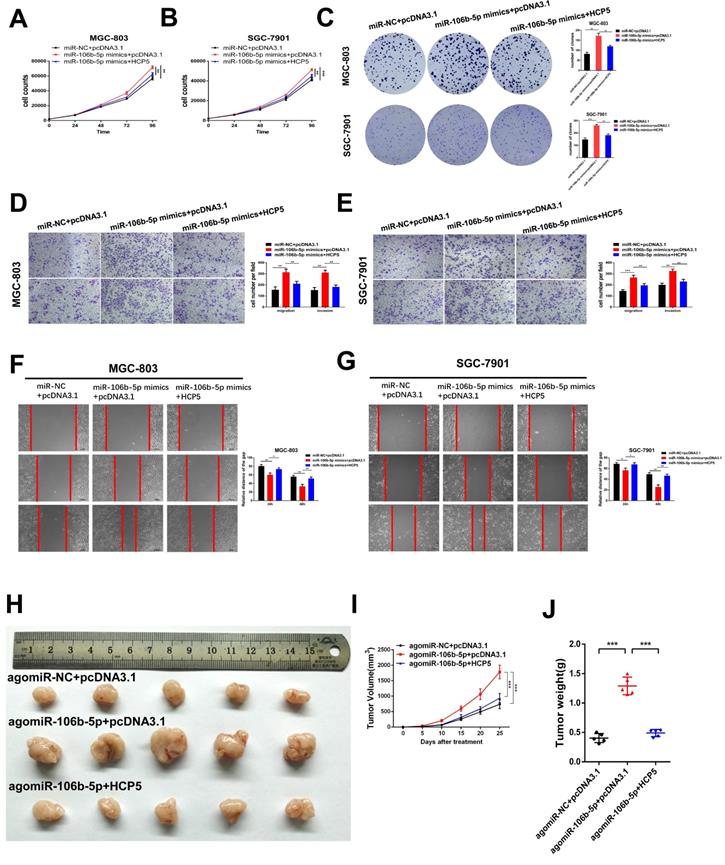
To deeply explore the biological reciprocities between HCP5 and miR-106b-5p in GC, rescue experiments were performed. MiR-106b-5p mimics and HCP5 overexpression plasmid were co-transfected into GC cells. As results showed, we found that the cell growth promotion and the increase in the number of cell colonies induced by upregulating miR-106b-5p was relieved by HCP5 overexpression (Fig. 6A-C). Moreover, by performing transwell assays and wound healing assay, miR-106b-5p was found to markedly enhance the abilities of GC cell migration and invasion. However, these effects were abrogated by overexpressing HCP5(Fig. 6D-G). To make a further understanding on the impact of miR-106b-5p in vivo and whether HCP5 could reverse these effects, miR-106b-5p agomir and pcDNA3.1-HCP5 overexpression plasmid were co-transfected into SGC-7901 cells. Subsequently, we inoculated subcutaneously the treated SGC-7901 cells and results revealed that miR-106b-5p overexpression dramatically facilitated xenograft tumor growth, whereas HCP5 overexpression partly interdicted the effects of enhanced tumorigenicity induced by miR-106b-5p (Fig. 6H-J). In all, our data indicated that the anti-tumor effect of HCP5 was partially mediated by negative regulation of miR-106b-5p.
HCP5 suppresses cell proliferation, migration and invasion of GC cells via mediating miR-106b-5p. A-C HCP5 rescued the promotion effect on proliferation induced by miR-106b-5p via performing CCK-8 experiments (A, B) and colony formation assays (C). D, E Transwell assays were used to show that HCP5 could rescue the potentiation induced by overexpressing miR-106b-5p. (Scare Bar=100um). F, G Rescue effects of HCP5 upregulation on the enhancement of migration resulted from miR-106b-5p in GC cells detected by wound healing assays (100x, Scare Bar=100um). H-J Image of xenograft tumors in different groups treated with agomiR-106b-5p, pcDNA3.1-HCP5, HCP5 and negative control groups. (H). Volume(I) and weight(J) of xenograft tumors. Data are showed as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001.
P21, a target gene of miR-106b-5p, is indirectly regulated by HCP5
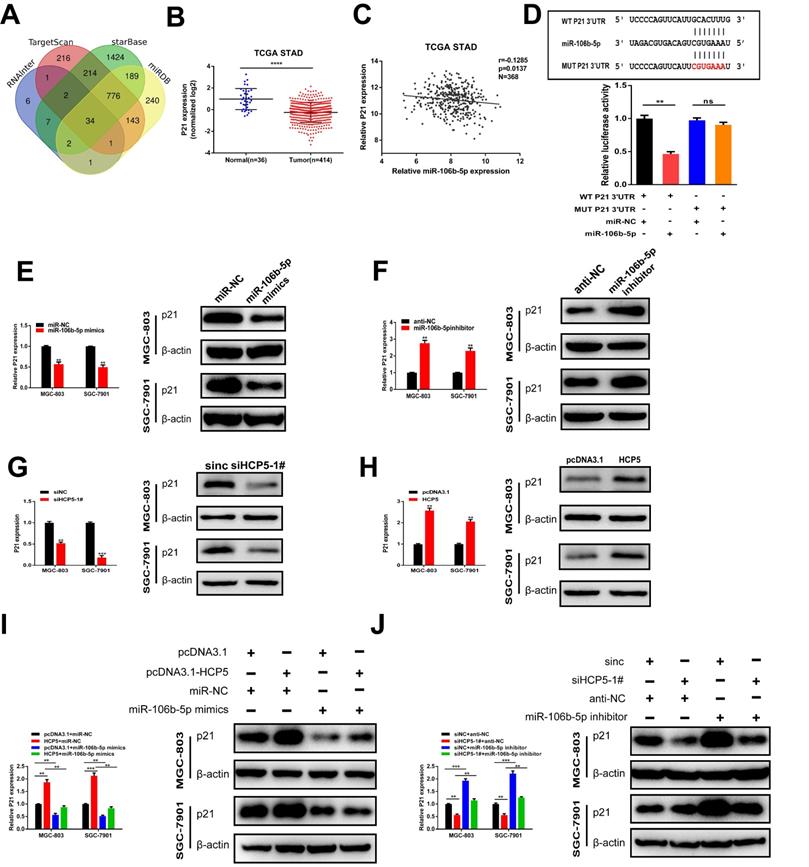
To further explore the regulatory network among HCP5, miR-106b-5p, and its specific downstream targets in GC, we used RNAInter, TargetScan, starBase, miRDB databases to predict potential targets of miR-106b-5p. As a result, 34 genes were predicted to be direct targets of miR-106b-5p, including p21 (Fig. 7A). In view of the results of previous research, p21 is regulated by miR-106b-5p negatively [23, 24]. TCGA database showed that p21 was downregulated in GC and negatively relevant to miR-106b-5p in GC tissues (Fig. 7B, C). Thus, we select p21 for our further research. Furthermore, 3'UTR-WT and 3'UTR-MUT sequences of p21 were loaded into psiCHECK2 and then mimics of miR-106b-5p were co-transfected into HEK-293T cells. Results revealed that significantly decreased luciferase activity was discovered in p21-wild rather than in p21-mut (Fig. 7D). Subsequently, to verify whether p21 was mediated by miR-106b-5p, miR-106b-5p mimics and inhibitors were transfected into GC cells, our data showed that alterations in mRNA and protein levels were seen on p21 induced by miR-106b-5p upregulation or downregulation (Fig. 7E, F). Since HCP5 can compete with miR-106b-5p, we wondered whether HCP5 could regulate p21 expression through targeting miR-106b-5p. We discovered that downregulation of HCP5 markedly decrease p21 expression on mRNA and protein levels in GC cells (Fig. 7G). Consistently, overexpression of HCP5 exerted the opposite effects (Fig. 7H). For the rescue experiments, we found that miR-106b-5p mimics could partially abrogate the increase effect caused by HCP5 overexpression on p21 mRNA and protein expression (Fig. 7I). Besides, miR-106b-5p downregulation counteracted the corresponding decrease in p21 expression caused by downregulation of HCP5 in MGC-803 and SGC-7901 cells (Fig. 7J). In conclusion, our results manifested that HCP5/miR-106b-5p axis could post-transcriptionally modulate p21 expression.
P21 is indirectly regulated by HCP5 by targeting miR-106b-5p. A Prediction of binding sites between MEF2A and HCP5 promoter via Targetscan, StarBase, RNAInter and miRDB. B P21 expression TCGA STAD database. C Analysis of relevance between miR-106b-5p and p21 from TCGA database. D MiR-106b-5p mimics was co-transfected with p21-3'UTR-WT and p21-3'UTR-MUT plasmids in HEK-293T cells. E, F Western blot and qRT-PCR were conducted to detect p21 expression in MGC-803 and SGC-7901 cells after miR-106b-5p alteration. G, H Western blot and qRT-PCR were conducted to detect p21 expression in GC cells after HCP5 alteration. I P21 expression in GC cells after different treatment. J P21 expression on mRNA and protein levels in GC cells after transfecting scrambled, si-HCP5-1#, miR-106b-5p inhibitor or si-HCP5-1#+miR-106b-5p inhibitor. Data are showed as the mean ± SD. **p < 0.01, ***p < 0.001.
Discussion
Emerging research has reported that lncRNAs could take part in the tumorigenesis and progression of tumor, containing GC [17, 19]. In our research, HCP5 was deemed as a critical GC-related lncRNA, in comparison with para-carcinoma normal tissues, which is markedly downregulated in GC tissues. Moreover, HCP5 expression is negatively associated with overall survival in GC patients, indicating that HCP5 is a latent prognostic factor for gastric cancer. Our results illuminated that HCP5 could exert an anti-tumor effect in GC by suppressing abilities of GC cell invasion, migration and proliferation in vitro and restrain growth of tumor in vivo.
HCP5, positioning between MICA and MICB genes, is located at the centromeric end of the HLA-B gene [25]. Previous research has considered HCP5 to be an oncogene in cervical cancer [26], glioma [27] and follicular thyroid carcinoma [28], while HCP5 downregulation was also found in malignancy like nasopharyngeal carcinoma [29], lung adenocarcinoma [30] and ovarian cancer [31]. As previous research shows, miR-199a-5p, a gene could also act as an oncogene in several cancer types whereas become a tumor suppressor in others [32]. The contradictory phenomenon probably owing to diverse genetic characteristics or pathological pathogenesis of different tumors. However, the biological effects and specific regulatory pathways of the role of HCP5 in gastric cancer remain unclear.
Recently, increasing research has reported that transcription of lncRNAs could be regulated by transcription factors as well [33, 34]. Herein, we put forward a possible mechanism responsible for HCP5 downregulation in GC. We firstly identified that transcription factor MEF2A could specifically bind to HCP5 promoter region via the combining site located -1973nt~ -1959nt and-1581nt~-1567nt and elucidated the mechanism by which MEF2A upregulated the expression of HCP5.
Increasing number of reports provide support for an extensive regulatory network inside ceRNAs, suggesting that ncRNAs which shared miRNA binding sites could regulate target RNA by competing for posttranscriptional control [35, 36]. We discovered that miR-106b-5p could directly interact with HCP5 through predictions of bioinformatics analysis and luciferase reporter assays. MiR-106b-5p was reported to participate in numerous cellular processes by targeting different mRNAs. For instance, miR-106b-5p exerts a promotion effect on hepatocellular carcinoma (HCC) metastasis by interacting with PTEN [37] and facilitates Glioma cell proliferation of by targeting Retinoblastoma-like protein 2 (RBL2) [38]. Moreover, upregulation of miR-106b-5p was detected among GC cells upon HCP5 knockdown, whereas HCP5 overexpression led to miR-106b-5p decrease, consistently with our hypothesis. In our research, miR-106b-5p was confirmed to act as a promoter of facilitating GC cell malignant biological properties. Rescue experiments further demonstrated that HCP5 could in part intercept the inhibitory impact caused by HCP5 on GC cells. Our results verified that HCP5 could interact with miR-106b-5p and partly block out the effects of miR-106b-5p and thereby inhibits GC progression.
Generally, lncRNAs exert its functions as a ceRNA mostly depend on the miRNA target. Hence, significantly important parts involved in the ceRNA regulatory network are downstream target mRNAs [36, 39]. Based on such considerations, bioinformatics databases were used to predict miR-106b-5p targets, and p21 was chosen as our research focus followed by validation using a luciferase reporter assay.
P21 is reported to take part in the inhibition of cyclin-dependent kinase activity. Moreover, downregulation of p21 is found among diverse variety of cancers, in which p21 could exert critical effects on regulation of tumor progression [40, 41]. It could suppress cell proliferation by blocking the activity of cyclin-dependent kinase (CDK) associated with G1/S transition [42, 43]. Hence, we hypothesized that HCP5 could compete with miR-106b-5p to upregulate p21 expression.
In conclusion, our results identified HCP5, a markedly downregulated lncRNA, was relevant to tumor size and Ki67 index of GC. Moreover, higher HCP5 expression is relevant to better overall survival, indicating that HCP5 could act as a GC prognostic factor. From functional assays, we discovered that alteration of lncRNA HCP5 could exert strong effects on proliferation, migration and invasion of GC cells. Furthermore, MEF2A was found to combine with HCP5 promoter regions and thereby regulate HCP5 expression in GC. Our research illuminated that, HCP5, an anti-tumor lncRNA, directly targeting miR-106b-5p, in turn, suppresses GC progression through upregulation of p21. Therefore, HCP5/miR-106b-5p/p21 axis may serve as a promising therapeutic target for clinically application in GC treatment.
Abbreviations
lncRNA: long non-coding RNA; GC: gastric cancer; qRT-PCR: real-time quantitative polymerase chain reaction; miRNA: microRNA; MEF2A: myocyte enhancer factor 2A; ceRNA: competing endogenous RNA; CHIP: chromatin immunoprecipitation assay; GES-1: gastric epithelial mucosa cell line; CCK-8: cell counting kit-8 proliferation assay; PVDF: polyvinylidene fluoride; cDNA: complementary DNA; HE staining: hematoxylin-eosin staining; IHC: immunohistochemical analysis; TF: transcription factor; HCC: hepatocellular carcinoma; RBL2: retinoblastoma-like protein 2; CDK: cyclin-dependent kinase; 3'UTR: 3′-untranslated region.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81974372) received by Z.J.Q., the National Natural Science Foundation of China (81772526) received by C.H., the National Natural Science Foundation of China (82002494) and Shanghai Sailing Program (20YF1440100) received by X.W.
Authors' contributions
Q.Z., C.H., Z.J.Q. and W.W.C. conceived and designed the research, analyzed the data, and revised the manuscript. W.W.C wrote the manuscript. W.W.C and K.D.Z conducted most of the experiments. Y.H.Y., Z.Y.G., X.F.W and B.W.T. performed part of the experiments. All authors reviewed and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians. 2018;68:394-424
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA: a cancer journal for clinicians. 2016;66:115-32
3. Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet (London, England). 2016;388:2654-64
4. El Abiad R, Gerke H. Gastric cancer: endoscopic diagnosis and staging. Surgical oncology clinics of North America. 2012;21:1-19
5. Wagner AD, Syn NL, Moehler M, Grothe W, Yong WP, Tai BC. et al. Chemotherapy for advanced gastric cancer. The Cochrane database of systematic reviews. 2017;8:Cd004064
6. Van Cutsem E, Ducreux M. Colorectal and gastric cancer in 2015: The development of new agents and molecular classifications. Nature reviews Clinical oncology. 2016;13:69-70
7. Shen L, Shan YS, Hu HM, Price TJ, Sirohi B, Yeh KH. et al. Management of gastric cancer in Asia: resource-stratified guidelines. The Lancet Oncology. 2013;14:e535-47
8. Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F. et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57-74
9. Amaral PP, Dinger ME, Mercer TR, Mattick JS. The eukaryotic genome as an RNA machine. Science (New York, NY). 2008;319:1787-9
10. Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D. et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223-7
11. Tsai MC, Spitale RC, Chang HY. Long intergenic noncoding RNAs: new links in cancer progression. Cancer Res. 2011;71:3-7
12. Nagano T, Fraser P. No-nonsense functions for long noncoding RNAs. Cell. 2011;145:178-81
13. Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26-46
14. Kung JT, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics. 2013;193:651-69
15. Amaral PP, Mattick JS. Noncoding RNA in development. Mammalian genome: official journal of the International Mammalian Genome Society. 2008;19:454-92
16. De Troyer L, Zhao P, Pastor T, Baietti MF, Barra J, Vendramin R. et al. Stress-induced lncRNA LASTR fosters cancer cell fitness by regulating the activity of the U4/U6 recycling factor SART3. Nucleic acids research. 2020;48:2502-17
17. Tang J, Yan T, Bao Y, Shen C, Yu C, Zhu X. et al. LncRNA GLCC1 promotes colorectal carcinogenesis and glucose metabolism by stabilizing c-Myc. Nature communications. 2019;10:3499
18. Yao RW, Wang Y, Chen LL. Cellular functions of long noncoding RNAs. Nat Cell Biol. 2019;21:542-51
19. Zhuo W, Liu Y, Li S, Guo D, Sun Q, Jin J. et al. Long Noncoding RNA GMAN, Up-regulated in Gastric Cancer Tissues, Is Associated With Metastasis in Patients and Promotes Translation of Ephrin A1 by Competitively Binding GMAN-AS. Gastroenterology. 2019;156:676-91.e11
20. Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nature reviews Genetics. 2009;10:155-9
21. Sun TT, He J, Liang Q, Ren LL, Yan TT, Yu TC. et al. LncRNA GClnc1 Promotes Gastric Carcinogenesis and May Act as a Modular Scaffold of WDR5 and KAT2A Complexes to Specify the Histone Modification Pattern. Cancer discovery. 2016;6:784-801
22. Zeng F, Wang Q, Wang S, Liang S, Huang W, Guo Y. et al. Linc00173 promotes chemoresistance and progression of small cell lung cancer by sponging miR-218 to regulate Etk expression. Oncogene. 2020;39:293-307
23. Petrocca F, Visone R, Onelli MR, Shah MH, Nicoloso MS, de Martino I. et al. E2F1-regulated microRNAs impair TGFbeta-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer cell. 2008;13:272-86
24. Dong X, Hu X, Chen J, Hu D, Chen LF. BRD4 regulates cellular senescence in gastric cancer cells via E2F/miR-106b/p21 axis. Cell death & disease. 2018;9:203
25. Kulski JK. Long Noncoding RNA HCP5, a Hybrid HLA Class I Endogenous Retroviral Gene: Structure, Expression, and Disease Associations. Cells. 2019 8
26. Yu Y, Shen HM, Fang DM, Meng QJ, Xin YH. LncRNA HCP5 promotes the development of cervical cancer by regulating MACC1 via suppression of microRNA-15a. European review for medical and pharmacological sciences. 2018;22:4812-9
27. Teng H, Wang P, Xue Y, Liu X, Ma J, Cai H. et al. Role of HCP5-miR-139-RUNX1 Feedback Loop in Regulating Malignant Behavior of Glioma Cells. Molecular therapy: the journal of the American Society of Gene Therapy. 2016;24:1806-22
28. Liang L, Xu J, Wang M, Xu G, Zhang N, Wang G. et al. LncRNA HCP5 promotes follicular thyroid carcinoma progression via miRNAs sponge. Cell death & disease. 2018;9:372
29. Low JS, Chin YM, Mushiroda T, Kubo M, Govindasamy GK, Pua KC. et al. A Genome Wide Study of Copy Number Variation Associated with Nasopharyngeal Carcinoma in Malaysian Chinese Identifies CNVs at 11q14.3 and 6p21.3 as Candidate Loci. PloS one. 2016;11:e0145774
30. Zhu TG, Xiao X, Wei Q, Yue M, Zhang LX. Revealing potential long non-coding RNA biomarkers in lung adenocarcinoma using long non-coding RNA-mediated competitive endogenous RNA network. Brazilian journal of medical and biological research = Revista brasileira de pesquisas medicas e biologicas. 2017;50:e6297
31. Liu N, Zhang R, Zhao X, Su J, Bian X, Ni J. et al. A potential diagnostic marker for ovarian cancer: Involvement of the histone acetyltransferase, human males absent on the first. Oncology letters. 2013;6:393-400
32. He XJ, Ma YY, Yu S, Jiang XT, Lu YD, Tao L. et al. Up-regulated miR-199a-5p in gastric cancer functions as an oncogene and targets klotho. BMC cancer. 2014;14:218
33. Sang B, Zhang YY, Guo ST, Kong LF, Cheng Q, Liu GZ. et al. Dual functions for OVAAL in initiation of RAF/MEK/ERK prosurvival signals and evasion of p27-mediated cellular senescence. Proc Natl Acad Sci U S A. 2018;115:E11661-e70
34. Yu T, Zhao Y, Hu Z, Li J, Chu D, Zhang J. et al. MetaLnc9 Facilitates Lung Cancer Metastasis via a PGK1-Activated AKT/mTOR Pathway. Cancer Res. 2017;77:5782-94
35. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353-8
36. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344-52
37. Shi DM, Bian XY, Qin CD, Wu WZ. miR-106b-5p promotes stem cell-like properties of hepatocellular carcinoma cells by targeting PTEN via PI3K/Akt pathway. OncoTargets and therapy. 2018;11:571-85
38. Liu F, Gong J, Huang W, Wang Z, Wang M, Yang J. et al. MicroRNA-106b-5p boosts glioma tumorigensis by targeting multiple tumor suppressor genes. Oncogene. 2014;33:4813-22
39. Karreth FA, Pandolfi PP. ceRNA cross-talk in cancer: when ce-bling rivalries go awry. Cancer discovery. 2013;3:1113-21
40. El-Deiry WS. p21(WAF1) Mediates Cell-Cycle Inhibition, Relevant to Cancer Suppression and Therapy. Cancer Res. 2016;76:5189-91
41. Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 2005;65:3980-5
42. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes & development. 1999;13:1501-12
43. Thaler S, Hähnel PS, Schad A, Dammann R, Schuler M. RASSF1A mediates p21Cip1/Waf1-dependent cell cycle arrest and senescence through modulation of the Raf-MEK-ERK pathway and inhibition of Akt. Cancer Res. 2009;69:1748-57
Author contact
![]() Corresponding authors: Zhengjun Qiu, MD, Department of General Surgery, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, 100 Haining Road, Shanghai 200080, People's Republic of China. Email: zhengjun.qiucn; Chen Huang, PhD, Department of General Surgery, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, 100 Haining Road, Shanghai 200080, People's Republic of China. Email: richard-hccom; Qian Zhao, PhD, Department of Pathophysiology Key Laboratory of Cell Differentiation and Apoptosis and National Ministry of Education, Shanghai Jiao Tong University School of Medicine, 280 South Chongqing Road, Shanghai 200025, People's Republic of China. Email: qzhaoedu.cn
Corresponding authors: Zhengjun Qiu, MD, Department of General Surgery, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, 100 Haining Road, Shanghai 200080, People's Republic of China. Email: zhengjun.qiucn; Chen Huang, PhD, Department of General Surgery, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, 100 Haining Road, Shanghai 200080, People's Republic of China. Email: richard-hccom; Qian Zhao, PhD, Department of Pathophysiology Key Laboratory of Cell Differentiation and Apoptosis and National Ministry of Education, Shanghai Jiao Tong University School of Medicine, 280 South Chongqing Road, Shanghai 200025, People's Republic of China. Email: qzhaoedu.cn