Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(3):869-881. doi:10.7150/ijbs.56152 This issue Cite
Research Paper
MLKL inhibits intestinal tumorigenesis by suppressing STAT3 signaling pathway
Qun Zhao1,3 ![]() , Xinran Cheng1, Jian Guo1, Yun Bi1, Li Kuang2, Jianhua Ren2, Jing Zhong1, Longrui Pan1, Xudong Zhang1, Yang Guo1, Yongqiang Liu4, Shu Jin5, Yan Tan1, Xianjun Yu1
, Xinran Cheng1, Jian Guo1, Yun Bi1, Li Kuang2, Jianhua Ren2, Jing Zhong1, Longrui Pan1, Xudong Zhang1, Yang Guo1, Yongqiang Liu4, Shu Jin5, Yan Tan1, Xianjun Yu1 ![]()
1. Laboratory of Inflammation and Molecular Pharmacology, Hubei Key Laboratory of Embryonic Stem Cell Research, School of Basic Medical Sciences & Biomedical Research Institute, Hubei University of Medicine, Shiyan 442000, China.
2. Department of Oncology, Affiliated Dongfeng Hospital, Hubei University of Medicine, Shiyan 442000, China.
3. State Key Laboratory of Molecular Oncology, National Cancer Center, National Clinical Research Center for Cancer, Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100021, China.
4. Institute of Clinical Pharmacology, Guangzhou University of Chinese Medicine, Guangzhou 510405, China.
5. Department of Gastroenterology, Taihe Hospital, Hubei University of Medicine, Shiyan 442000, China.
Received 2020-11-19; Accepted 2021-1-15; Published 2021-2-17
Abstract

Mixed lineage kinase domain-like protein (MLKL) plays an important role in necroptosis, but the role and mechanism of MLKL in intestinal tumorigenesis remain unclear. Here, we found that hematopoietic- and nonhematopoietic-derived MLKL affected intestinal inflammation, but nonhematopoietic-derived MLKL primarily inhibited intestinal tumorigenesis. Loss of MLKL enhanced intestinal regeneration and the susceptibility to intestinal tumorigenesis in Apcmin/+ mice by hyperactivating the Janus kinase 2 (JAK2)/ signal transducer and activator of transcription 3 (STAT3) axis. Furthermore, MLKL deficiency increased interleukin-6 (IL-6) production in dendritic cells. Administration of anti-IL-6R antibody therapy reduced intestinal tumorigenesis in Apcmin/+Mlkl-/- mice. Notably, low MLKL expression in human colorectal tumors, which enhanced STAT3 activation, was associated with decreased overall survival. Together, our results reveal that MLKL exhibits a suppressive effect during intestinal tumorigenesis by suppressing the IL-6/JAK2/STAT3 signals.
Keywords: Intestinal tumorigenesis, MLKL, IL-6/STAT3, Anti-IL-6R antibody therapy.
Introduction
Colorectal cancer is the third most common cancer and is associated with high mortality worldwide [1]. Various risk factors is associated with the initiation and progression of colorectal cancer, including genetic mutations, inflammation, gut microbiota and epigenetic modification [2]. Although approximately 90% of patients can be cured at the early stage, colorectal cancer is usually detected at an advanced stage [3-4]. Therefore, identify promising targets for predicting colorectal cancer risk and disease progression is needed.
Necroptosis is caspase-independent necrosis that requires the receptor-interacting protein kinase 1 (RIPK1) protein and RIPK3. RIPK1 and RIPK3 interacts via the RIP homotypic interaction motifs (RHIMs) and facilitates the formation of amyloid signaling complex RIPK1-RIPK3 necrosome during necroptosis. MLKL, an important downstream mediator, is activated by RIPK3 and then oligomers to disrupt the membrane integrity, leading to necroptotic cell death [5-7]. Recent studies have shown that multiple key mediators of necroptosis are involved in the progression of tumors [8-9]. Ripk3-deficient mice are susceptible to intestinal tumorigenesis [10-11]. Notably, reduced MLKL expression in tumors is significantly correlated with poor overall survival [12]. Recently, we reported that loss of MLKL accelerated colitis-associated tumorigenesis [13]. However, the precise mechanisms and signaling pathway by which MLKL participates in intestinal tumorigenesis remain unclear. Notably, colitis-associated tumorigenesis represents only 1% of colorectal cancer, and sporadic intestinal tumors are more prevalent [14]. Thus, it is necessary to elucidate the role and the underlying mechanisms of MLKL in sporadic intestinal tumorigenesis.
Signal transducer and activator of transcription 3 (STAT3) as a transcription factor that regulates a large number of physiological and pathological cellular processes [15-18]. Hyperactivation of STAT3 plays an important role in numerous malignant tumors [19-20]. Accumulating evidences have reported that persistent activation of STAT3 promotes colorectal cancer progression [18, 20]. Interleukin-6 (IL-6), a prominent member of the IL-6 family of cytokines, forms a complex with signaling receptors [21]. IL-6 binds to IL-6Rα and forms IL-6-IL-6Rα complexes, subsequently interacts with the membrane-associated gp130 subunit, leading to the phosphorylation of Janus kinase 2 (JAK2) [22]. JAK2 activation promotes the phosphorylation, nuclear translocation, and transcriptional activation of STAT3 [23].
In the present study, we revealed that loss of MLKL increased the susceptibility to intestinal regeneration and tumorigenesis through hyperactivation of IL-6/JAK2/STAT3 signaling. Notably, anti-IL-6R antibody therapy effectively suppressed intestinal tumorigenesis and STAT3 activation in Apcmin/+Mlkl-/- mice. We further found that reduced MLKL expression was correlated with poor prognosis and STAT3 hyperactivation in colorectal tumors. Thus, our findings emphasized that loss of MLKL in intestinal tumors exhibited aggressive disease.
Materials and methods
Animal experiments
Mlkl-/- mice were generated and genotyped as previously described [13]. Mlkl-/- mice were crossed with Apcmin/+ mice to generate Apcmin/+Mlkl+/- and Mlkl+/- mice. Then, Apcmin/+Mlkl+/- mice were crossed with Mlkl+/- mice to generate Apcmin/+ and Apcmin/+Mlkl-/- mice. All the mice were on a C57BL/6 background and maintained in a specific pathogen-free (SPF) facility. Animal experiments were performed according from Institutional Animal Care and Use Committee of the Hubei University of Medicine (approval no. 2019-066).
AOM/DSS induced colorectal carcinogenesis
8-week-old mice were injected intraperitoneally with azoxymethane (AOM, 10 mg/kg). After 5 days, mice were treated 2.5% (m/v) dextran sodium sulfate (DSS) in normal drinking water for 7 days, and then administrated normal water for 14 days. The DSS-normal water treatment cycle was repeated three times. At the third cycle on day 80, the mice were euthanized and intestinal tumors were collected for further analysis.
Bone marrow transplant
To address the function hematopoietic or/and nonhematopoietic cells to intestinal tumorigenesis, chimeric mice were generated as previously described [24-25]. In brief, 6-week-old recipient WT mice or Mlkl-/- mice were lethally irradiated (9 Gy) to remove bone marrow cells, WT or Mlkl-/- recipient mice were injected with 1×107 bone marrow cells via the tail vein from WT mice or Mlkl-/- donor mice. WT→WT, WT→Mlkl-/-, Mlkl-/-→WT and Mlkl-/-→Mlkl-/- chimeric mice were fed water supplemented with 2 g/L neomycin sulfate for two weeks. Chimeric mice were subjected to establish AOM/DSS-induced colorectal tumor models after eight weeks of bone marrow reconstitution.
Determination of clinical scores
The scoring of stool consistency and occult blood was performed as previously described [26]. The disease activity index (DAI) was determined based on the combined score.
Tumor load
Tumor load was determined according to the published protocol [27]. Tumor load was calculated as follows: count tumor numbers, measure tumor size, and calculate tumor load per mouse.
Whole-body irradiation
Whole-body irradiation (WBI) was performed according to published protocol [28]. Eight-week-old mice (n=5 per groups) were subjected to 10-Gy WBI. At 3 and 5 days, the intestinal was subjected to H&E staining, and the regenerating villi length was analyzed in 10 fields per mouse. The intestinal tissues were collected for further analysis.
RNA-sequencing
Intestinal tissues were obtained and isolated RNA from 6-week-old Apcmin/+ and Apcmin/+Mlkl-/- mice. The libraries of mRNA were conducted in Illumina HiSeq X-ten system (Shanghai Bohao Biotechnology). The raw reads were filtered and mapped to the genome by Seqtk and TopHat, respectively [29]. Fragments gene were reassembled using HTSeq [30-31], and significant differentially expressed genes (DEGs) were clarified based on False Discovery Rate (FDR) (Q< 0.05) and fold-change >2 [32].
Quantitative reverse-transcriptase PCR (qRT-PCR)
Intestinal tissues were collected and homogenized in TRIzol reagent (Life Technologies) using a Mini-Rotor (Thermo Scientific). The transcript levels of genes were measured by qRT-PCR analysis.
Western blotting
Isolated intestines were homogenized using a Mini-Rotor (Thermo Scientific). Tissues were lysed and protein concentrations were quantified. The proteins were separated by SDS-PAGE gels and were detected using chemiluminescent substrate (Thermo Scientific). Antibodies for western blotting used included: rabbit anti-MLKL (#ab184718, Abcam, 1:1000), rabbit anti-phosphor-STAT3 (Tyr 705) (#9145, Cell Signaling Technology, CST, 1:1000), mouse anti-STAT3 (#9139, CST, 1:1000), rabbit anti-PCNA (#13110, CST, 1:1000), rabbit anti-Cleaved-Caspase-3 (#9661, CST, 1:1000), rabbit anti-Cyclin D1 (#2978, CST, 1:1000), rabbit anti-C-myc (#9402, CST, 1:1000), and rabbit anti-GAPDH (#GB11002, Wuhan Service Bio Technology Co., Ltd., 1:5000).
Intestinal tissue for the analysis of cytokine production
The intestinal tissues were homogenized and lysed. The lysates were centrifuged and the protein concentration was measured by ELISA (eBioscience) [33].
Isolation of intestine epithelia cells (IECs) and lamina propria lymphocytes (LPLs)
Isolation of intestinal epithelial cells and lamina propria lymphocytes was performed as previously described [34]. Intestines were cleared and then cut into pieces. The IECs were collected following shaking after incubation in cold PBS with 3 mM EDTA/1.5 mM DTT. The remaining tissues were further digested by collagenase type XI (Sigma) and DNase I (Sigma) at 37°C for 1 h, and the LPLs were isolated from the supernatants by Percoll. For IL-6 stimulation, intestinal tissues were digested by collagenase type I (Sigma) and Dispase II (Sigma) for 2 h. Cells were plated into 6-well plates coated with type I collagen (Invitrogen) overnight after washing at least three times.
Anti-IL-6R antibody treatment
4-week-old Apcmin/+ and Apcmin/+Mlkl-/- mice were intraperitoneal injected with 4 mg/kg tocilizumab (#A2012, Selleckchem; Houston, TX, USA) weekly. After 10 weeks of treatment, mice were euthanized, and the intestinal tumors were collected for further analysis.
Clinical specimens
Tissue microarrays data on 98 colorectal cancer samples and 79 adjacent samples were obtained from (Shanghai Outdo Biotech Co., Ltd). An anti-MLKL antibody (#ab184718, Abcam; 1:100) was used to detect the expression of MLKL in the immunohistochemical assays. The samples were scanned to obtain high-resolution digital images. The expression of MLKL was scored and quantified by pathologists. The quantified multiplicative index was determined as follows: 0 to 3: the average staining intensity; 0 to 4: extent of staining in the scores. The low MLKL staining indexes ranged from 0 to 4, and the higher MLKL staining indexes ranged from 4 to 12. Above experiments was approved by the ethics committee of Hubei University of Medicine (Hubei, China) (approval no. 2019-TH-027).
Statistical analysis
Data are expressed as the mean ± standard errors of the mean (SEM). Statistical significance was conduced using 2-tailed Student's t test, one-way ANOVA, log-rank test or Pearson's correlation coefficients test. P-values < 0.05 were considered statistically significant.
Results
MLKL in hematopoietic and nonhematopoietic cellular compartments mediates protection against intestinal tumorigenesis
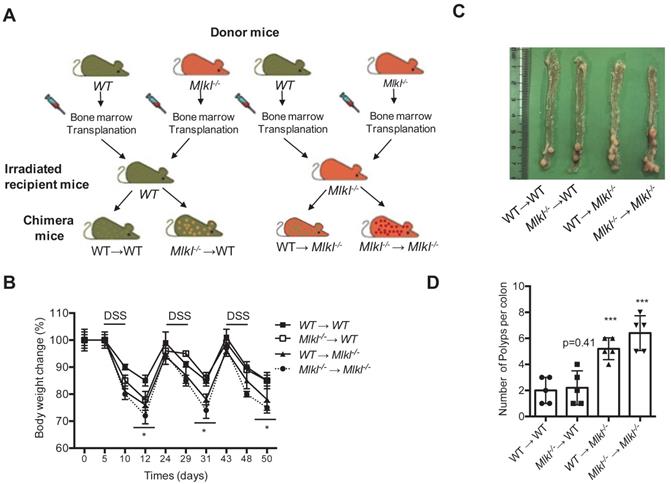
Dysplastic epithelial cells and myeloid cells are involved in intestinal tumorigenesis [35]. MLKL is widely expressed in various cell types and organs as shown by others studies and our previous findings [6, 36-37]. Thus, the function of MLKL in in hematopoietic and/or nonhematopoietic cellular compartments might be associated with intestinal tumorigenesis. To gather further evidence, we established four groups of chimeric mice (WT→WT, Mlkl-/- →WT, WT→Mlkl-/-, Mlkl-/-→Mlkl-/-) and were subjected to AOM/DSS-induced colorectal carcinogenesis (Figure 1A). During the first cycle of DSS treatment, the WT→Mlkl-/- and Mlkl-/- →WT chimeric mice showed similarly weight loss relative to Mlkl-/-→Mlkl-/- mice (Figure 1B), suggesting that MLKL in both hematopoietic and nonhematopoietic cellular compartments was necessary during this phase. During the second cycle of DSS, we found that the WT→Mlkl-/- mice, but not the Mlkl-/- →WT mice, showed more weight loss, suggesting that the effect of MLKL was mainly dependent on its expression in the nonhematopoietic cellular compartment during the later stage of intestinal tumorigenesis. In addition, clinical scores also confirmed these phenomenon (Figure S1 and Table S1). Correspondingly, the Mlkl-/-→Mlkl-/- mice exhibited more intestinal polyps than the WT→WT mice (Figure 1C, 1D). Moreover, the WT→Mlkl-/- mice had significantly more intestinal polyps than in WT→WT and Mlkl-/-→WT mice (Figure 1C, 1D). However, the number of intestinal polyps in the Mlkl-/-→WT mice was not significantly different from the WT→WT mice (Figure 1C, 1D). These results show an early role of MLKL in hematopoietic and nonhematopoietic cellular compartments and indicate that MLKL in nonhematopoietic cellular compartments is critical for the inhibition of intestinal polyp formation.
MLKL in hematopoietic and nonhematopoietic cellular compartments mediates protection against intestinal tumorigenesis. (A) Schematic overview of four chimeric mice: WT → WT, WT → Mlkl-/-, Mlkl-/-→ WT, and Mlkl-/- → Mlkl-/-. (B) Weight change in the chimeric mice of the AOM/DSS model. (C) Representative images of the intestinal tumors from chimeric mice on the day 80 of the AOM/DSS model. (D) Quantification of the polyp numbers in chimeric mice in the AOM/DSS model. * p < 0.05 and *** p < 0.001 versus WT → WT groups.
MLKL deficiency increases intestinal tumor burden in the Apcmin/+ model
APC mutations are frequently found in intestinal tumors patients [38]. Apcmin/+ mice develop spontaneous intestinal polyps and become a good animal model for investigating intestinal tumorigenesis [39]. To unveil the function of MLKL in intestinal tumorigenesis, we analyzed tumor progression in Apcmin/+Mlkl-/- mice and Apcmin/+ mice. The survival time was dramatically decreased in the Apcmin/+Mlkl-/- mice, and the median survival of these mice was only 127 days relative to the median survival of the Apcmin/+ mice, which was 185 days (Figure 2A). Macroscopic dissections showed increased lesions in the Apcmin/+Mlkl-/- mice compared to the Apcmin/+ mice at 12 weeks of age (Figure 2B). However, there were no overt defects in Mlkl-/- intestines compared with those of the WT mice during the 12-months of observations (Figure S2A). Moreover, no significant difference in proliferation and self-renewal was found in WT and Mlkl-/- intestines (Figure S2B), suggesting that MLKL was only functional under disease conditions. In addition, the Apcmin/+Mlkl-/- mice had markedly increased tumor numbers (Figure 2C) and tumor loads (Figure 2D). Notably, the tumor size distribution showed that tumors sizes were larger in the Apcmin/+Mlkl-/- mice compared to the Apcmin/+ mice (Figure 2E). H&E staining also indicated that larger polyps were found in the intestines of the Apcmin/+Mlkl-/- mice than in Apcmin/+ littermate controls (Figure 2F). The Apcmin/+ mice develop anemia and thymus atrophy, which is associated with severe intestinal tumorigenesis [24]. We found that anemia and thymus atrophy were significantly exacerbated in Apcmin/+Mlkl-/- mice compared to Apcmin/+ littermate controls, suggesting that loss of MLKL increased intestinal tumor development (Figure 2G, 2H). These results indicate that MLKL is dispensable for intestinal homeostasis under physiological condition and plays a protective role against intestinal tumorigenesis.
MLKL deficiency increases intestinal tumor burden in the Apcmin/+ model. (A) Survival of Apcmin/+ and Apcmin/+Mlkl-/- mice. (B) Polyps in representative intestines from Apcmin/+ and Apcmin/+Mlkl-/- mice. (C and D) Quantification of polyp formation (C) and tumor load (D) in 16-week-old Apcmin/+ and Apcmin/+Mlkl-/- intestines. (E) Distribution of tumor size in 16-week-old Apcmin/+ and Apcmin/+Mlkl-/- mice. (F) Images of the H&E-stained intestines from Apcmin/+ and Apcmin/+Mlkl-/- mice. (G and H) Hematocrit (G) and thymus weight (H) of 16-week-old WT, Mlkl-/-, Apcmin/+ and Apcmin/+Mlkl-/- mice. (I) Ki-67 staining of the representative intestines from age-matched WT, Mlkl-/-, Apcmin/+ and Apcmin/+Mlkl-/- mice. (J-K) Whole intestines and colonic crypts were isolated from 16-week-old Apcmin/+ and Apcmin/+Mlkl-/- mice. PCNA (J) and cleaved caspase-3 (K) were analyzed by western blotting. Scale bars: 50 μm. * p < 0.05, ** p < 0.01, *** p < 0.001 versus Apcmin/+ mice.
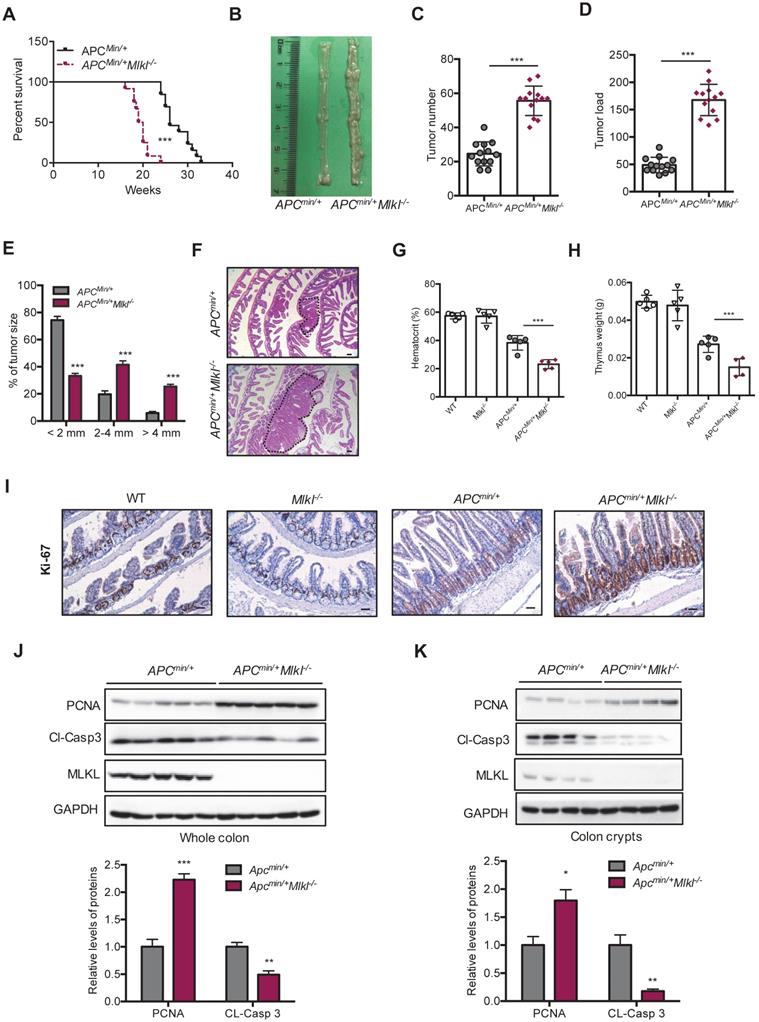
To investigate the mechanism by which MLKL deficiency promoted intestinal tumorigenesis in Apcmin/+Mlkl-/- mice, we examined cell proliferation by staining proliferation marker Ki-67. The results revealed that the level of Ki-67 in the Apcmin/+Mlkl-/- colonic crypts was significantly higher than in the Apcmin/+ mice, while no significant difference was found in WT and Mlkl-/- mice (Figure 2I). The observation of increased proliferation in the Apcmin/+Mlkl-/- mice was further supported by western blotting. Compared with the Apcmin/+ mice, the Apcmin/+Mlkl-/- mice exhibited increased levels of PCNA and decreased levels of cleaved caspase-3 (Figure 2J). Similar results were observed in colonic crypts from the Apcmin/+Mlkl-/- intestinal tumors (Figure 2K). These results suggest that loss of MLKL accelerates intestinal tumorigenesis in Apcmin/+ mice by promoting proliferation and preventing apoptosis.
Loss of MLKL enhances the activation of STAT3
To elucidate the mechanisms underlying loss of MLKL promoted intestinal tumorigenesis, microassay analysis was performed on the intestines of 6-week-old Apcmin/+ and Apcmin/+Mlkl-/- mice. Because the Apcmin/+Mlkl-/- mice showed similar histology compared to the Apcmin/+ mice at this time point, these gene alterations might be related to earlier events in intestinal tumorigenesis. A total of 5178 genes were upregulated and 1611 genes were downregulated in the Apcmin/+Mlkl-/- intestines compared to Apcmin/+ intestines (Figure S3A, S3B). Ingenuity Pathway Analysis (IPA) showed that significantly impacted disease processes, including cancer, intestinal injury, abnormalities and intestinal disease (Figure 3A). Moreover, cell death, survival and tumor morphology were enriched in the Apcmin/+Mlkl-/- mice (Figure 3A). To elucidate the MLKL-mediated signaling pathway, the upregulated genes were further functionally enriched by gene set enrichment analysis (GSEA) [40]. The results indicated that the STAT3 signaling pathway-related gene set was enriched in Apcmin/+Mlkl-/- mice intestines (Figure 3B). Further analysis of the genes related to STAT3 signaling indicated that the loss of MLKL significantly increased the levels of STAT3 target genes (Figure 3C). Moreover, phosphorylation of STAT3 (pSTAT3) was enhanced in the intestines of the 6-week-old Apcmin/+Mlkl-/- mice compared to those of the Apcmin/+ mice, but no remarkable increase in expression of pSTAT3 was observed in either the intestines of the WT or Mlkl-/- mice (Figure 3D). The expression of STAT3 target genes, Cyclin D1 and C-myc, was upregulated in the intestines of the Apcmin/+Mlkl-/- mice compared to Apcmin/+ mice (Figure 3D).
Loss of MLKL enhances the activation of STAT3. (A) Ingenuity Pathway Analysis (IPA) of the disease processes and cellular functions in 6-week-old Apcmin/+ and Apcmin/+Mlkl-/- mice. (B) GSEA demonstrated enrichment of STAT3 target genes. (C) Heat map showing a summary of the expression of STAT3 pathway target genes in 6-week-old Apcmin/+ and Apcmin/+Mlkl-/- mice. (D) Proteins isolated from 6-week-old Apcmin/+ and Apcmin/+Mlkl-/- mouse intestines were analyzed by western blotting to assess STAT3 activation. (E) Images of H&E-stained intestinal tissues in 3 and 5 days after WBI. (F) Villus length in WT and Mlkl-/- intestines were measured after WBI. (G) Proteins isolated from WT and Mlkl-/- mice during regeneration were analyzed by western blotting to assess STAT3 activation. (H) Gene expression of intestinal tissues during regeneration (days 0 and 3) in WT and Mlkl-/- mice. (I-J) Whole intestine tissues and colonic crypts from 16-week-old Apcmin/+ and Apcmin/+Mlkl-/- mice were lysed and measured the levels of Cyclin D1 and C-myc. Scale bars: 50 μm. * p < 0.05, ** p < 0.01, *** p < 0.001 versus Apcmin/+ mice.
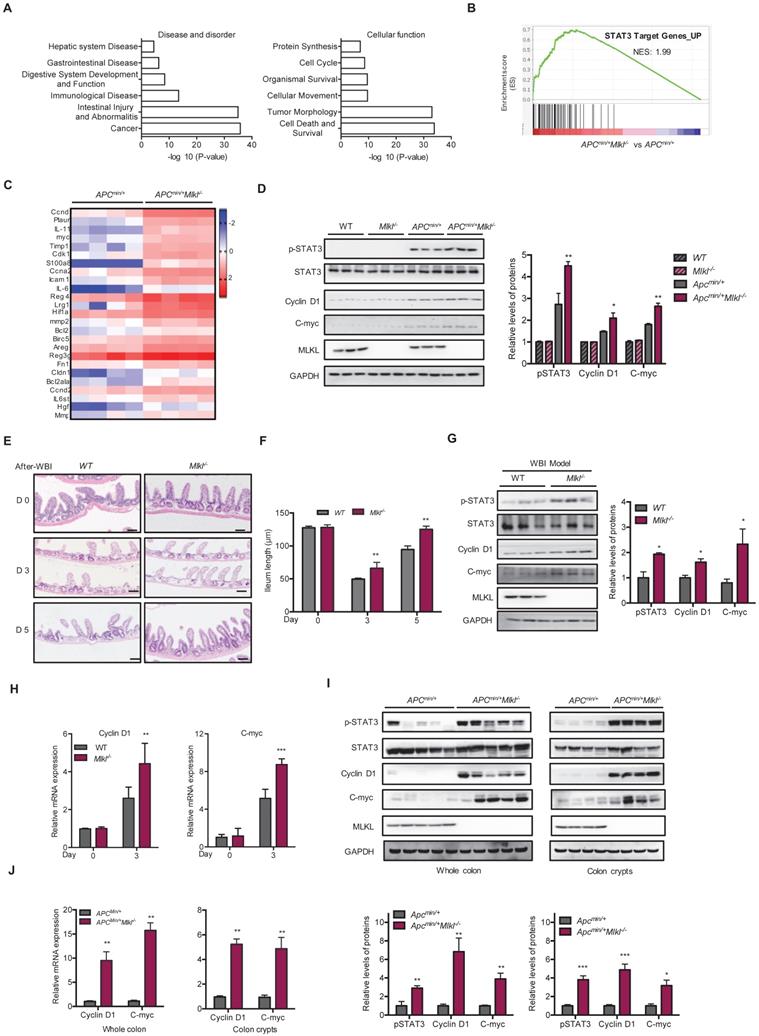
Enhancement of STAT3 activation promotes intestinal regeneration during tissue injury and damage [41]. We established whole-body irradiation (WBI)-induced intestinal injury and regeneration models to confirm the function of MLKL. Compared to that in the WT mice, the length of the villi in the Mlkl-/- mice was quickly recovered on days 3 and 5 after irradiation (Figure 3E, 3F), suggesting that MLKL deficiency accelerated intestinal regeneration process. There were higher protein levels of pSTAT3 in the Mlkl-/- intestines in the regeneration models (Figure 3G). The expression of Cyclin D1 and C-myc was upregulated in the intestines of the Mlkl-/- mice compared to those of the WT mice in the WBI-induced intestinal regeneration models (Figure 3G, 3H). In addition, the levels of the progenitor cell-associated genes Cd44 and Sox9 was also significantly increased (Figure S3C). To determine whether Apcmin/+Mlkl-/- mice exhibited higher regenerative potential during intestinal regeneration, we also performed WBI-induced regeneration experiments in Apcmin/+ and Apcmin/+Mlkl-/- mice. By day 3 after irradiation, the protein levels of pSTAT3 were enhanced in the intestines of Apcmin/+Mlkl-/- mice compared to the Apcmin/+ mice in the WBI-induced regeneration models (Figure S3D). Consistent with these findings, intestines of Apcmin/+Mlkl-/- mice exhibited higher levels of Cyclin D1 and C-myc compared with the intestines of Apcmin/+ mice (Figure S3D).
To further confirm the role of MLKL on STAT3 activation during intestinal tumorigenesis, we examined STAT3 activation in 16-week-old Apcmin/+Mlkl-/- and Apcmin/+ intestinal tumors. Western blotting revealed that the protein level of pSTAT3 were markedly increased in the intestinal tumors of the Apcmin/+Mlkl-/- mice compared with those of the Apcmin/+ mice (Figure 3I). In addition, higher levels of pSTAT3 were also observed in the Apcmin/+Mlkl-/- intestinal crypts (Figure 3I). Notably, protein expression of the STAT3 target genes Cyclin D1 and C-myc was increased in the intestines and crypts of the Apcmin/+Mlkl-/- mice (Figure 3I). Consistently, qRT-PCR results indicated that the mRNA levels of the Cyclin D1 and C-myc were significantly upregulated in the Apcmin/+Mlkl-/- mice compared to the Apcmin/+ mice (Figure 3J). Similar to the Apcmin/+ mouse model, the expression levels of pSTAT3, Cyclin D1 and C-myc were greatly elevated in the Mlkl-/-→Mlkl-/- and WT→Mlkl-/- intestinal polyps compared with the Mlkl-/-→WT and WT→WT intestinal polyps at day 80 of the AOM/DSS-induced colitis-associated tumorigenesis (Figure S3E). Collectively, our results indicate that loss of MLKL promotes STAT3 activation in intestinal regeneration and tumorigenesis.
MLKL deficiency exacerbates IL-6/STAT3 activation by promoting JAK2 phosphorylation
STAT3 activation is known to be required for IL-6 production, and IL-6/STAT3 signaling exerts a tumor-promoting function in intestinal tumorigenesis [18, 20]. We found higher levels of IL-6 in the Apcmin/+Mlkl-/- intestinal tumors than in those of the Apcmin/+ mice (Figure 4A). ELISA analysis further found higher levels of IL-6 protein in the Apcmin/+Mlkl-/- intestinal tumors (Figure 4B). To explore the cellular source of this increased IL-6, we separated laminar propria lymphocytes (LPLs) and intestinal epithelial cells (IECs) from the intestinal tumors of Apcmin/+Mlkl-/- and Apcmin/+ mice. The results showed that IL-6 was specifically upregulated in LPLs, but not in IECs (Figure 4C). Previous studies have shown that intestinal dendritic cells are responsible for IL-6 production [20, 42]. We thus isolated certain types of cells from LPLs in Apcmin/+Mlkl-/- and Apcmin/+ intestinal tumors. qRT-PCR results indicated that dendritic cells were responsible for IL-6 production (Figure 4D).
MLKL deficiency exacerbates IL-6/STAT3 activation by promoting JAK2 phosphorylation. (A) Quantitative analysis of IL-6 mRNA expression in Apcmin/+ and Apcmin/+Mlkl-/- intestinal tumors. (B) The protein levels of IL-6 in Apcmin/+ and Apcmin/+Mlkl-/- intestinal tumors as determined by ELISA. (C) Quantitative mRNA expression of IL-6 in isolated laminar propria lymphocytes (LPLs) and intestinal epithelial cells (IECs) of intestinal tumors from Apcmin/+ and Apcmin/+Mlkl-/- mice. (D) Macrophages, dendritic cells, and T cells isolated by flow cytometric cell sorting were analyzed to assess IL-6 mRNA expression by qRT-PCR. (E) LPS-stimulated WT and Mlkl-/- BMDCs were analyzed for quantitative mRNA expression of IL-6 by qRT-PCR (LPS, 100 ng/ml). (F) Mlkl-/- BMDCs were overexpressed with MLKL for 48 h, cells then were stimulated with LPS and analyzed the expression of IL-6 mRNA. (G) WT and Mlkl-/- BMDCs were pre-treated 10 μM ERK inhibitor U0126 for 2 h, and then cells then were stimulated with LPS and analyzed the expression of IL-6 mRNA by qRT-PCR. (H-I) Primary WT and Mlkl-/- IECs isolated from 3-week-old mice were stimulated with 20 ng/mL IL-6. Cell lysates were analyzed for pSTAT3, Cyclin D1 and C-myc. (J) WT and Mlkl-/- IECs were stimulated with 20 ng/mL IL-6. Cell lysates were analyzed for pJAK2 (Y1007/Y1008). (K) The effect of MLKL on the interaction of JAK2 with STAT3. WT and Mlkl-/- IECs were stimulated with 20 ng/mL IL-6. Cell lysates were subjected to immunoprecipitation and western blotting with indicated antibody. * p < 0.05, ** p < 0.01, *** p < 0.001.
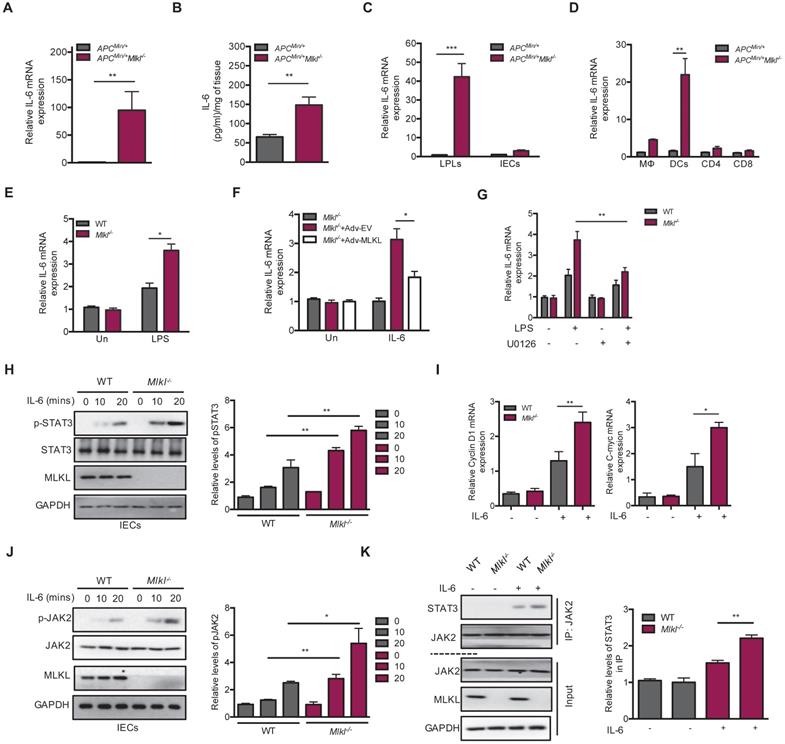
Our previous study revealed that MLKL deficiency promoted inflammatory responses via activating of ERK signaling in dendritic cells by [13]. We thus explored whether dendritic cells produced IL-6 in vitro and found that the level of IL-6 was higher in LPS-stimulated Mlkl-/- bone marrow-derived dendritic cells (BMDCs) (Figure 4E). In contrast, MLKL overexpression partially decreased LPS-induced IL-6 expression in the Mlkl-/- BMDCs (Figure 4F). To address the role of MEK/ERK activation in IL-6 production in Mlkl-/- BMDCs, the MEK/ERK inhibitor U0126 was used. U0126 blocked the LPS-induced IL-6 expression in the Mlkl-/- BMDCs (Figure 4G). Together, these data suggest that IL-6 is overproduced in MLKL-deficient dendritic cells through the activation of ERK signaling.
To dissect IL-6-mediated hyperactivation of STAT3 in Mlkl-/- epithelial cells, we isolated primary WT and Mlkl-/- IECs and stimulated them with extracellular IL-6. The results showed that the levels of p-STAT3 were higher in the Mlkl-/- IECs than in the WT IECs following treatment with IL-6 (Figure 4H). Similarly, higher expression levels of the Cyclin D1 and C-myc were observed in Mlkl-/- IECs (Figure 4I). Moreover, the IL-6-induced increase in pSTAT3 in the context of knocked down MLKL expression was also found in colorectal carcinoma HT-29 cells (Figure S4). Accumulating evidence suggests that phosphorylation and activation of JAK2 play a critical role in IL-6/STAT3 activation [23]. To explore whether MLKL interfered with the activation of JAK2, we assessed the levels of JAK2 phosphorylation in the Mlkl-/- and WT IECs following treatment with IL-6. As shown in Figure 4J, MLKL deficiency increased IL-6-induced JAK2 phosphorylation. Of note, co-immunoprecipitation experiments revealed that the interaction JAK2 and STAT3 was increased in Mlkl-/- IECs compared to WT IECs (Figure 4K). These data together indicate that MLKL deficiency promotes intestinal tumorigenesis through activation of the IL-6/JAK2/STAT3 axis.
Blocking IL-6 signaling inhibits intestinal tumorigenesis in Apcmin/+Mlkl-/- mice
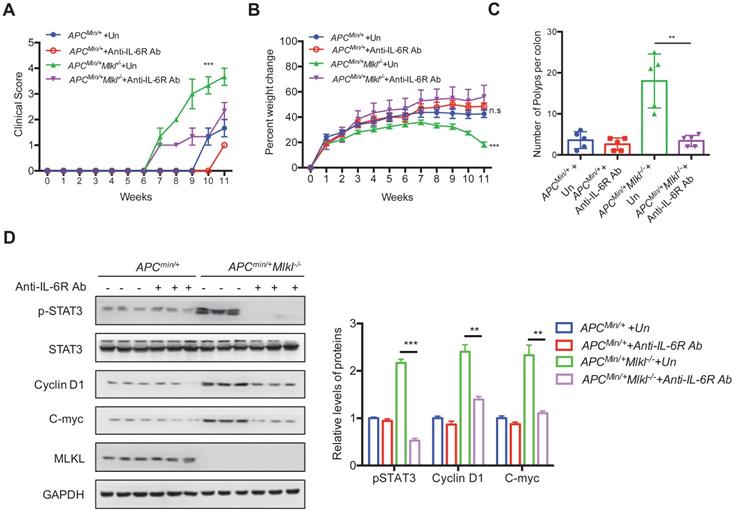
The above observations suggest that loss of MLKL leads to hyperactivation of IL-6 signaling. Next, we sought to explore whether the IL-6/STAT3 axis contributed to the increase in intestinal tumors in Apcmin/+Mlkl-/- mice. To this end, we used IL-6 receptor antibodies (anti-IL-6R) to block IL-6 signaling. Apcmin/+ and Apcmin/+Mlkl-/- mice were intraperitoneally injected with anti-IL-6R weekly for 10 weeks. The anti-IL6R-treated Apcmin/+Mlkl-/- mice exhibited a lower clinical score and significant weight gain compared to the untreated (UT) Apcmin/+Mlkl-/- mice (Figure 5A, 5B). However, the anti-IL-6R-treated Apcmin/+ mice exhibited mild changes in clinical score and body weight compared to the UT Apcmin/+ mice (Figure 5A, 5B). After IL-6R antibody treatment for 10 weeks, the anti-IL6R-treated Apcmin/+Mlkl-/- mice exhibited fewer intestinal polyps than the UT Apcmin/+Mlkl-/- group (Figure 5C and Figure S5A). Consistent with the decreased intestinal polyps in the anti-IL-6R-treated Apcmin/+Mlkl-/- mice, the anemia and thymus atrophy in the Apcmin/+Mlkl-/- mice were considerably alleviated after anti-IL-6R treatment (Figure S5B, S5C). Western blotting showed that the expression levels of pSTAT3, Cyclin D1 and C-myc was significantly suppressed in the anti-IL-6R-treated Apcmin/+Mlkl-/- mice compared to the UT Apcmin/+Mlkl-/- mice (Figure 5D). Thus, these results demonstrate that IL-6/STAT3 hyperactivation in Apcmin/+Mlkl-/- mice plays a causative role in increased intestinal polyps formation.
Blocking IL-6 signaling inhibits intestinal tumorigenesis in Apcmin/+Mlkl-/- mice. (A and B) 4-week-old Apcmin/+ and Apcmin/+Mlkl-/- mice were administrated with anti-IL6R antibody weekly for 10 weeks. Clinical score and the percent weight change were monitored. (C) Quantification of polyp formation in PBS- or anti-IL6R-treated mice as indicated. (D) Intestinal tissues were harvested from PBS-treated Apcmin/+Mlkl-/- and anti-IL6R-treated Apcmin/+Mlkl-/- mice and then lysed. The levels of pSTAT3, Cyclin D1 and C-myc were determined by western blotting. ** p < 0.01 and *** p < 0.001.
The levels of MLKL has prognostic implications in colorectal cancer
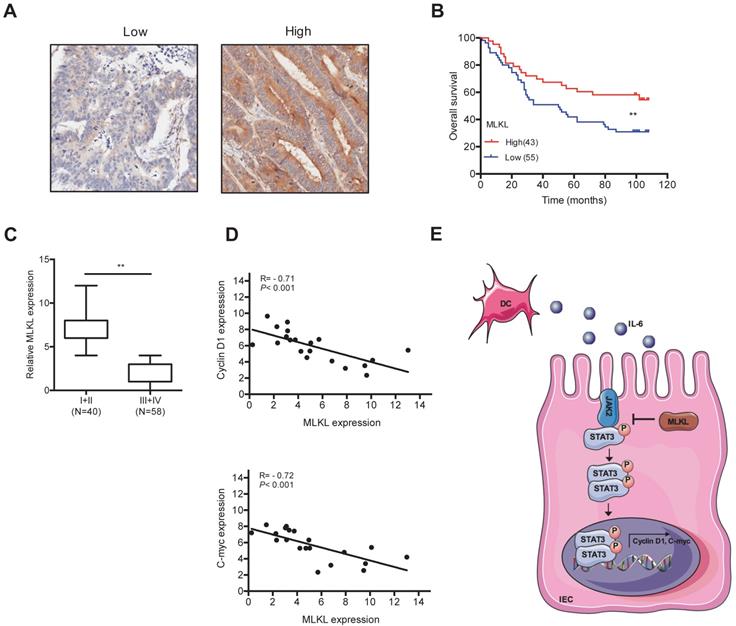
To examine the potential role of MLKL in human colorectal tumors, we analyzed MLKL expression colorectal tumor samples. Notably, Kaplan-Meier survival analysis indicated that low MLKL expression exhibited shorter overall survival than patients with high MLKL expression (Figure 6A, 6B). Moreover, the expression of MLKL was inversely linked to the stages of disease progression in colorectal tumors (Figure 6C). Given our findings that loss of MLKL increased activation of STAT3 in spontaneous intestinal tumorigenesis, we further examined the potential correlation between MLKL expression and STAT3 activation in human colorectal tumor samples. The levels of MLKL mRNA are negatively correlated with Cyclin D1 and C-myc expression (Figure 6D). These clinical findings indicate that low MLKL expression level may be prognostic for the malignant progression of colorectal cancer.
The levels of MLKL has prognostic implications in colorectal cancer. (A-B) Expression of MLKL in colorectal cancer samples detected by IHC. Overall survival of patients based on MLKL expression levels conducted by Kaplan-Meier plot. (C) Boxed plot of MLKL expression assessed at different clinical stages. (D) Pearson correlation analysis of MLKL and Cyclin D1 (P < 0.001; R = - 0.71) and MLKL and C-myc (P < 0.001; R = - 0.72) in human colorectal cancer. (E) Model of MLKL negatively impacts IL-6/STAT3 signaling in intestinal tumorigenesis. ** p < 0.01.
Discussion
In this study, we showed that loss of MLKL activated the STAT3 signaling pathway to accelerate intestinal regeneration and tumorigenesis. Mechanismly, we found that loss of MLKL increased the phosphorylation of JAK2 and promoted the association of JAK2 with STAT3, leading to hyperactivation of STAT3. Furthermore, IL-6 was upregulated in MLKL-deficient dendritic cells via increased ERK activation. In summary, MLKL exhibits a suppressive effect in the progression of intestinal tumors by regulating the IL-6/JAK2/STAT3 axis (Figure 6E).
MLKL is a major necroptosis regulatory protein that is widely expressed in tissues and organs [6, 36-37]. Increasing evidences suggest that MLKL is involved in pathophysiological conditions, including liver injury, insulin resistance and T2D, cholestasis, and hepatitis [43-46]. Experimental studies demonstrate that MLKL protects mice from against mucosal infection and promotes mucosal repair [13, 47]. Several recent reports have indicated that the prognostic value of MLKL protein expression in multiple malignant tumors [8-9]. We previously reported that the loss of MLKL led to increased susceptibility to colitis-associated tumorigenesis [13]. Herein, we demonstrated that loss of MLKL promoted intestinal tumor progression and increased tumor burden. Our results highlighted that the loss of MLKL activated the STAT3 signaling pathway. Moreover, in the absence of MLKL, high levels of phosphorylated STAT3 were found in intestinal regeneration and tumorigenesis. Sustained STAT3 activation contributed to tumor progression, aberrant JAK2/STAT3 has been detected in a variety of tumor types, suggesting that STAT3 exhibits promise as a drug target for cancer therapeutics [48]. We found that loss of MLKL strengthened the association of JAK2 with STAT3, thereby promoting STAT3 phosphorylation and intestinal tumorigenesis. Our study provides new insights into the role of MLKL serves as a negative regulator in intestinal tumorigenesis through suppressing STAT3 activation.
Several recent reports indicated that the lower levels of MLKL were correlated with poor overall survival in malignant tumors [12, 49-52]. Notably, we observed that reduced MLKL expression in patients with colorectal tumors was correlated with high histological grade and decreased overall survival. Furthermore, we saw that there was a negative relationship between MLKL and the STAT3 target genes expression in colorectal tumors. In agreement, we also found that the size of tumors in the Apcmin/+Mlkl-/- mice were larger than those in the Apcmin/+ mice. These data suggested that MLKL plays a tumor-suppressive role during intestinal tumorigenesis.
IL-6 forms a complex with the IL-6 receptor, leading to persistent activation of STAT3 [53]. The levels of IL-6 are dramatically increased in serum and tumor samples of humans and mice with colorectal cancer, and high IL-6 levels in colorectal cancers are found to correlate with poor prognosis [54-55]. IL-6 promotes the survival of colon carcinoma cells in vitro; however blocking IL-6 signal prevents tumor growth [56-57]. Mlkl-/- mice exhibited massive inflammatory responses and exhibited high IL-6 expression during colitis and colitis-associated tumorigenesis [13]. Herein, we found that IL-6 was elevated in the Apcmin/+Mlkl-/- intestine tumors. Further experiments demonstrated that immune cells, especially dendritic cells, were the main IL-6-producing cells in the Apcmin/+Mlkl-/- intestines. Interestingly, the activation of ERK pathway promoted IL-6 expression in Mlkl-/- dendritic cells, indicating that MLKL deficiency induces IL-6 production in dendritic cells by activating ERK. However, we cannot exclude the possibility that MLKL is involved in other cell types. The contribution of the cell-specific function of MLKL to the suppression of intestinal tumors needs to be further investigated using MLKL conditional knockout mice in future studies.
In summary, our data reveal that MLKL exhibit a suppressive effect during intestinal tumorigenesis via suppressing IL-6/STAT3 signaling pathway. Our results also highlight that interfering with IL-6/STAT3 axis could be a better therapeutic option for intestinal tumor patients with low MLKL expression.
Abbreviations
AOM: Azoxymethane; BMDCs: Bone marrow-derived dendritic cells; DAI: Disease activity index; DEGs: Differentially expressed genes; DSS: Dextran sodium sulfate; FDR: False Discovery Rate; IECs: Intestinal epithelial cells; IL-6: Interleukin-6; IL-6R: Interleukin-6 receptor; JAK2: Janus kinase 2; LPLs: Laminar propria lymphocytes; MLKL: Mixed lineage kinase domain-like protein; PCNA: Proliferating cell nuclear antigen; qRT-PCR: Quantitative reverse-transcriptase PCR; RHIMs: RIP homotypic interaction motifs; RIPK1: Receptor-interacting protein kinase 1; STAT3: Signal transducer and activator of transcription 3; UT: Untreated; WBI: Whole-body irradiation.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81902852, 81502548), the Natural Science Foundation of Hubei Provincial Department of Education (D20202101, D20182101), the Foundation of Health Commission of Hubei Province (WJ2019M053), the China Postdoctoral Science Foundation (2020M670220), the Biomedical Research Foundation, Hubei University of Medicine (HBMUPI201809), Faculty Development Grants from Hubei University of Medicine (2018QDJZR06), Open Project of Hubei Key Laboratory of Embryonic Stem Cell Research of Hubei University of Medicine (2020ESOF008), and the Student's Platform for Innovation and Entrepreneurship Training Program (202010929001).
Authorship contributions
Q. Z., and X. Y. conceived the study and designed the experiments. Q. Z., X. C., J. G., Y. B., L. K., J. R., J. Z., X. Z., and X. Y. performed experiments. Q. Z., L. P., Y. G., Y. L., S. J., Y. T., and X. Y. analyzed data. Q. Z., and X. Y. wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC. et al. Colorectal cancer statistics, 2020. CA Cancer J Clin. 2020;70:145-64
2. Islami F, Goding Sauer A, Miller KD, Siegel RL, Fedewa SA, Jacobs EJ. et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J Clin. 2018;68:31-54
3. Woolf SH. The best screening test for colorectal cancer-a personal choice. N Engl J Med. 2000;343:1641-3
4. Walsh JM, Terdiman JP. Colorectal cancer screening: scientific review. JAMA. 2003;289:1288-96
5. Sun L, Wang H, Wang Z, He S, Chen S, Liao D. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213-27
6. Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y. et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013;23:994-1006
7. Zhao J, Jitkaew S, Cai ZY, Choksi S, Li QN, Luo J. et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. P. Natl Acad. Sci. USA. 2012;109:5322-7
8. Najafov A, Chen H, Yuan J. Necroptosis and Cancer. Trends Cancer. 2017;3:294-301
9. Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan K. et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer. 2019;18:100
10. Yan G, Zhao H, Zhang Q, Zhou Y, Wu L, Lei J. et al. A RIPK3-PGE2 circuit mediates myeloid-derived suppressor cell-potentiated colorectal carcinogenesis. Cancer Res. 2018;78:5586-99
11. Bozec D, Iuga AC, Roda G, Dahan S, Yeretssian G. Critical function of the necroptosis adaptor RIPK3 in protecting from intestinal tumorigenesis. Oncotarget. 2016;7:46384-400
12. Li X, Guo J, Ding AP, Qi WW, Zhang PH, Lv J. et al. Association of mixed lineage kinase domain-like protein expression with prognosis in patients with colon cancer. Technol. Cancer Res. Treat. 2017;16:428-34
13. Zhao Q, Yu X, Li M, Liu Y, Han Y, Zhang X. et al. MLKL attenuates colon inflammation and colitis-tumorigenesis via suppression of inflammatory responses. Cancer Lett. 2019;459:100-11
14. Yashiro M. Ulcerative colitis-associated colorectal cancer. World J. Gastroenterol. 2014;20:16389-97
15. Jenkins BJ, Roberts AW, Greenhill CJ, Najdovska M, Lundgren-May T, Robb L. et al. Pathologic consequences of STAT3 hyperactivation by IL-6 and IL-11 during hematopoiesis and lymphopoiesis. Blood. 2007;109:2380-8
16. Miklossy G, Hilliard TS, Turkson J. Therapeutic modulators of STAT signalling for human diseases. Nat. Rev. Drug Discov. 2013;12:611-29
17. Gao B, Wang H, Lafdil F, Feng D. STAT proteins - key regulators of anti-viral responses, inflammation, and tumorigenesis in the liver. J. Hepatol. 2012;57:430-41
18. Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T. et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91-102
19. Corvinus FM, Orth C, Moriggl R, Tsareva SA, Wagner S, Pfitzner EB. et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia. 2005;7:545-55
20. Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S. et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer cell. 2009;15:103-13
21. Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015;16:448-57
22. Kishimoto T. Interleukin-6: from basic science to medicine-40 years in immunology. Annu. Rev. Immunol. 2005;23:1-21
23. Boulanger MJ, Chow DC, Brevnova EE, Garcia KC. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science. 2003;300:2101-4
24. Song XY, Gao HC, Lin YY, Yao YK, Zhu S, Wang JJ. et al. Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity. 2014;40:140-52
25. Ma X, Meng Z, Jin L, Xiao Z, Wang X, Tsark WM. et al. CAMK2 gamma in intestinal epithelial cells modulates colitis-associated colorectal carcinogenesis via enhancing STAT3 activation. Oncogene. 2017;36:4060-71
26. Zhao Q, Bi Y, Zhong J, Ren ZT, Liu YX, Jia JJ. et al. Pristimerin suppresses colorectal cancer through inhibiting inflammatory responses and Wnt/beta-catenin signaling. Toxicol. Appl. Pharm. 2020 386
27. Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat. Protoc. 2007;2:1998-2004
28. Yuan HR, Li N, Fu D, Ren J, Hui JY, Peng JJ. et al. Histone methyltransferase SETD2 modulates alternative splicing to inhibit intestinal tumorigenesis. J. Clin. Invest. 2017;127:3381-97
29. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105-11
30. Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166-9
31. Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25
32. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139-40
33. Zhao Q, Yu X, Zhang H, Liu Y, Zhang X, Wu X. et al. RIPK3 mediates necroptosis during embryonic development and postnatal inflammation in Fadd-deficient mice. Cell Rep. 2017;19:798-808
34. Song XY, Zhu S, Shi PQ, Liu Y, Shi YF, Levin SD. et al. IL-17RE is the functional receptor for IL-17C and mediates mucosal immunity to infection with intestinal pathogens. Nat. Immunol. 2011;12:1151-8
35. Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer. 2008;8:618-31
36. Alvarez-Diaz S, Dillon CP, Lalaoui N, Tanzer MC, Rodriguez DA, Lin A. et al. The pseudokinase MLKL and the kinase RIPK3 have distinct roles in autoimmune disease caused by loss of death-receptor-induced apoptosis. Immunity. 2016;45:513-26
37. Zhang X, Fan C, Zhang H, Zhao Q, Liu Y, Xu C. et al. MLKL and FADD are critical for suppressing progressive lymphoproliferative disease and activating the NLRP3 inflammasome. Cell Rep. 2016;16:3247-59
38. Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat. Rev. Cancer. 2001;1:55-67
39. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159-70
40. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545-50
41. Neufert C, Pickert G, Zh.eng Y, Wittkopf N, Warntjen M, Nikolaev A. et al. Activation of epithelial STAT3 regulates intestinal homeostasis. Cell Cycle. 2010;9:652-5
42. Dann SM, Spehlmann ME, Hammond DC, Iimura M, Hase K, Choi LJ. et al. IL-6-dependent mucosal protection prevents establishment of a microbial niche for attaching/effacing lesion-forming enteric bacterial pathogens. J. Immunol. 2008;180:6816-26
43. Wu X, Poulsen KL, Sanz-Garcia C, Huang E, McMullen MR, Roychowdhury S. et al. MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcohol-associated fatty liver and steatohepatitis. J. Hepatol. 2020;73:616-27
44. Xu H, Du X, Liu G, Huang S, Du W, Zou S. et al. The pseudokinase MLKL regulates hepatic insulin sensitivity independently of inflammation. Mol. Metab. 2019;23:14-23
45. Afonso MB, Rodrigues PM, Simao AL, Ofengeim D, Carvalho T, Amaral JD. et al. Activation of necroptosis in human and experimental cholestasis. Cell Death Dis. 2016;7:e2390
46. Gunther C, He GW, Kremer AE, Murphy JM, Petrie EJ, Amann K. et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J. Clin. Invest. 2016;126:4346-60
47. Yu SX, Chen W, Liu ZZ, Zhou FH, Yan SQ, Hu GQ. et al. Non-hematopoietic MLKL protects against Salmonella mucosal infection by enhancing inflammasome activation. Front. Immunol. 2018;9:119
48. Carpenter RL, Lo HW. STAT3 target genes relevant to human cancers. Cancers (Basel). 2014;6:897-925
49. He L, Peng K, Liu YZ, Xiong J, Zhu FF. Low expression of mixed lineage kinase domain-like protein is associated with poor prognosis in ovarian cancer patients. Oncotargets Ther. 2013;6:1539-43
50. Colbert LE, Fisher SB, Hardy CW, Hall WA, Saka B, Shelton JW. et al. Pronecrotic mixed lineage kinase domain-like protein expression is a prognostic biomarker in patients with early-stage resected pancreatic adenocarcinoma. Cancer. 2013;119:3148-55
51. Ruan JY, Mei L, Zhu Q, Shi G, Wang H. Mixed lineage kinase domain-like protein is a prognostic biomarker for cervical squamous cell cancer. Int. J. Clin. Exp. Patho. 2015;8:15035-8
52. Ertao Z, Jianhui C, Kang W, Zhijun Y, Hui W, Chuangqi C. et al. Prognostic value of mixed lineage kinase domain-like protein expression in the survival of patients with gastric caner. Tumour Biol. 2016;37:13679-85
53. Bauer S, Kerr BJ, Patterson PH. The neuropoietic cytokine family in development, plasticity, disease and injury. Nat. Rev. Neurosci. 2007;8:221-32
54. Heikkila K, Ebrahim S, Lawlor DA. Systematic review of the association between circulating interleukin-6 (IL-6) and cancer. Eur. J. Cancer. 2008;44:937-45
55. Li J, Huang L, Zhao H, Yan Y, Lu J. The role of interleukins in colorectal cancer. Int. J. Biol. Sci. 2020;16:2323-39
56. Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A. et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21:491-501
57. Becker C, Fantini MC, Wirtz S, Nikolaev A, Lehr HA, Galle PR. et al. IL-6 signaling promotes tumor growth in colorectal cancer. Cell Cycle. 2005;4:217-20
Author contact
![]() Corresponding authors: Qun Zhao, Laboratory of Inflammation and Molecular Pharmacology, Hubei Key Laboratory of Embryonic Stem Cell Research, School of Basic Medical Sciences & Biomedical Research Institute, Hubei University of Medicine, Shiyan 442000, China, E-mail: zhaoqunedu.cn; Xianjun Yu, Laboratory of Inflammation and Molecular Pharmacology, Hubei Key Laboratory of Embryonic Stem Cell Research, School of Basic Medical Sciences & Biomedical Research Institute, Hubei University of Medicine, Shiyan 442000, China, E-mail: xjyuedu.cn.
Corresponding authors: Qun Zhao, Laboratory of Inflammation and Molecular Pharmacology, Hubei Key Laboratory of Embryonic Stem Cell Research, School of Basic Medical Sciences & Biomedical Research Institute, Hubei University of Medicine, Shiyan 442000, China, E-mail: zhaoqunedu.cn; Xianjun Yu, Laboratory of Inflammation and Molecular Pharmacology, Hubei Key Laboratory of Embryonic Stem Cell Research, School of Basic Medical Sciences & Biomedical Research Institute, Hubei University of Medicine, Shiyan 442000, China, E-mail: xjyuedu.cn.