Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(4):1088-1100. doi:10.7150/ijbs.56800 This issue Cite
Research Paper
Genistein-3′-sodium sulfonate Attenuates Neuroinflammation in Stroke Rats by Down-Regulating Microglial M1 Polarization through α7nAChR-NF-κB Signaling Pathway
Chaoming Liu1,4, Song Liu1, Lijiao Xiong1,3, Limei Zhang1,2, Xiao Li1,2, Xingling Cao1,2, Jinhua Xue1,2, Liangdong Li1,3, Cheng Huang1,2 ![]() , Zhihua Huang1,2
, Zhihua Huang1,2 ![]()
1. Key Laboratory of Prevention and treatment of cardiovascular and cerebrovascular diseases of Ministry of Education, Gannan Medical University, Ganzhou 341000, China.
2. Department of Physiology, School of Basic Medical Sciences, Gannan Medical University, Ganzhou 341000, China.
3. First Affiliated Hospital of Gannan Medical University, Ganzhou 341000, China.
4. Department of Pathobiology, JiangXi College of Traditional Chinese Medicine, Fuzhou, 344000, China.
Received 2020-12-5; Accepted 2021-2-22; Published 2021-3-8
Abstract

Microglial M1 depolarization mediated prolonged inflammation contributing to brain injury in ischemic stroke. Our previous study revealed that Genistein-3′-sodium sulfonate (GSS) exerted neuroprotective effects in ischemic stroke. This study aimed to explore whether GSS protected against brain injury in ischemic stroke by regulating microglial M1 depolarization and its underlying mechanisms. We established transient middle cerebral artery occlusion and reperfusion (tMCAO) model in rats and used lipopolysaccharide (LPS)-stimulated BV2 microglial cells as in vitro model. Our results showed that GSS treatment significantly reduced the brain infarcted volume and improved the neurological function in tMCAO rats. Meanwhile, GSS treatment also dramatically reduced microglia M1 depolarization and IL-1β level, reversed α7nAChR expression, and inhibited the activation of NF-κB signaling in the ischemic penumbra brain regions. These effects of GSS were further verified in LPS-induced M1 depolarization of BV2 cells. Furthermore, pretreatment of α7nAChR inhibitor (α-BTX) significantly restrained the neuroprotective effect of GSS treatment in tMCAO rats. α-BTX also blunted the regulating effects of GSS on neuroinflammation, M1 depolarization and NF-κB signaling activation. This study demonstrates that GSS protects against brain injury in ischemic stroke by reducing microglia M1 depolarization to suppress neuroinflammation in peri-infarcted brain regions through upregulating α7nAChR and thereby inhibition of NF-κB signaling. Our findings uncover a potential molecular mechanism for GSS treatment in ischemic stroke.
Keywords: Microglia polarization, Neuroinflammation, Ischemic stroke, α7nAChR-NF-κB signaling, Genistein-3′-sodium sulfonate.
Introduction
Stroke is the third leading cause of death among human diseases, and over 80% of stroke results from brain ischemia [1, 2]. Currently, therapeutic strategies for ischemic stroke are limited: administration of tissue plasminogen activator (tPA) or mechanical thrombectomy to gain brain reperfusion. However, the therapeutic window of these approaches is exceptionally narrow: generally less than 8 h [1, 3-5]. Thus, seeking novel drugs or other therapeutic strategies are still the concerns of neuroscientists.
Ischemic stroke activates inflammatory cascades in the acute phase, resulting in increased production of pro-inflammatory cytokines and chemokines, aggregation and adhesion of inflammatory cells, impairing of the blood-brain barrier (BBB), and subsequently exacerbating brain damage [6, 7]. Post-stroke neuroinflammation is considered as a favorable target for the treatment of ischemic stroke [1, 3, 7, 8]. Microglia, one of the essential participants in neuroinflammation, is activated in a few minutes of ischemic stroke hitting [9, 10]. Activated microglial cells polarize to M2 phenotype in the early stage of ischemic stroke and subsequently switch to M1 phenotype [11]. The roles of M2 and M1 polarization of microglia in neuroinflammation are antagonistic: M2 phenotype microglial cells exert anti-inflammatory and neuroprotective effects by secreting transforming growth factor β (TGF-β) and interleukin-10 (IL-10), while M1 polarization produces pro-inflammatory cytokines such as interleukin-1β (IL-1 β), IL-6 and tumor necrosis factor α (TNF-α), which contribute to the disruption of BBB and the deterioration of brain injury [3, 12, 13]. Thus, reducing microglial M1 depolarization is a critical strategy to limit brain's inflammatory damage in ischemic stroke [14-18].
Cholinergic anti-inflammatory pathway (CAIP) is mediated by the stimulation of vagal nerve, which releases acetylcholine (ACh) to modulate cerebral inflammation mainly through α7 nicotinic acetylcholine receptor (α7nAChR) [14, 19, 20]. This receptor is expressed in the membrane of microglia, neurons, and endothelial cells in the brain. Activation of α7nAChR reduces microglial activation, inhibits the production of pro-inflammatory cytokines, and attenuates brain injury and functional deficits [6, 21, 22]. Moreover, it was reported that activation of α7nAChR inhibited nuclear factor kappa B (NF-κB) pathway including blocking the phosphorylation of IκB and thereby restraining the nucleus translocation of p65-NF- κB, a subunit of NF-κB [23, 24]. In ischemic stroke mice, α7nAChR agonist reduced the brain injury through decreasing the phosphorylation of p65-NF-κB protein in microglia [25-27], demonstrating that α7nAChR-mediated inhibition of NF-κB signaling is a crucial neuroprotective mechanism against ischemic brain injury.
Estrogen displays potent neuroprotective effects, whereas long-term estrogen treatment increases the incidence risk of breast cancer [28-30]. Therefore, phytoestrogens are seeing as an alternative to estrogen. Genistein is widely studied as a phytoestrogen which protects against ischemic stroke-induced brain injury. However, the bioavailability of genistein is limited because of its poor water and lipid solubility [31]. Genistein-3'-sodium sulfonate (GSS) is a sulfonated product of genistein, which has a better water solubility than genistein. Our previous research indicated that GSS treatment significantly alleviated cerebral edema and brain infarction in transient middle cerebral artery occlusion (tMCAO) rats [32]. Besides, we also found that Genistein has a therapeutic effect in tMCAO rat because it down-regulates inflammatory cytokines levels such as IL-1β, TNF-α and IL-6, demonstrating that GSS precursor has significant anti-inflammation effects [33]. 17β-Estradiol and progesterone promote microglial depolarization from M1 to M2 phenotype in ischemic stroke [34, 35]. Therefore, we hypothesized that GSS could inhibit microglial M1 depolarization through regulating α7nAChR-mediated inhibition of NF-κB pathway and thus suppress the neuroinflammation and protect against the brain injury in ischemic stroke. In this study, we investigated the effects of GSS on microglial M1 polarization and its underlying molecular mechanisms using tMCAO rats and lipopolysaccharide (LPS)-treated microglial cells.
Materials and methods
Animal and Drugs
Animal experiments were performed strictly following the guidelines of the Animal Care and Use Committee of Gannan Medical University. Sprague-Dawley (SD) rats (Male, 250 g) were purchased from Hunan SJA Laboratory Anima Company (Hunan, China) and were housed at 50-60% relative humidity and a temperature of 22-25℃. The rats drank sterile running water and took SPF grade chow freely. Before performing experiments, all rats were acclimatized to the laboratory environment for at least one week.
GSS was purchased from Shanghai Skychemical Co., Ltd (Shanghai, China), and the chemical structure of it was showed in our preview study [32]. It was dissolved in normal saline (NS) to 1 mg/mL concentration before use. Specific α7nAChR-antagonist α-bungarotoxin (α-BTX, #ab120542) was obtained from Abcam (Cambridge, United Kingdom). It was dissolved in 0.1 M phosphate buffer solution (PBS).
Reagents
Tris, sodium dodecyl sulfate (SDS), 30% acrylamide and protein lysis buffer (#R0010) were obtained from Solarbio Science & Technology Company (Beijing, China). TRzol was purchased from Ambion company (Texas, United States). Anti-CD11b (#ab1211, #ab133357), anti-IL-1β (#ab9787), anti-CD40 (#ab13545), anti-CD68 (#ab125212), and anti-α7nAChR (#ab10096) were purchased from Abcam (Cambridge, United Kingdom). Anti-IKK (#2682s), anti-phospho-IKK(#2697s), anti-P65-NF-κB (#242s), anti-phospho-P65--NF-κB (#3033s), anti-IκB (#4814s) and anti-phospho-IκB (#9246s) were purchased from Cell Signaling Technology (Massachusetts, United States). Anti-β-tubulin (#MA511732), AlexaFluor488 mouse secondary antibody (#A11029), AlexaFluor488 rabbit secondary antibody (#A11034), enhanced chemiluminescence reagent (ECL) Western blotting detection reagents (#32106), Reverse Transcription Master Mix kit (Invitrogen, #4374966), SYBR® Select Master Mix (Life Technologies, #4472908), 0.25% trypsin (Gibco, #25200-056), DMEM medium (Gibco, #31800-014), fetal bovine serum (FBS) (Gibco, #10099-141) and penicillin streptomycin combination (Gibco, #15140-122) were purchased from Thermo Fisher Scientific (Massachusetts, United States). 2-, 3-, 5-triphenyltetrazolium chloride (TTC, #T8877-25G) was purchased from Sigma (United States).
Transient middle cerebral artery occlusion and reperfusion (tMCAO) rat model and drug treatments
Rat tMCAO model was established, as described previously [32, 36]. Briefly, rats were anesthetized with 1% pentobarbital sodium (50 mg/kg, i.p.), and brain ischemia was induced by inserting a 5-cm-long nylon filament (diameter, 0.24-0.28 mm) into the middle cerebral artery for 2 h. Then, the filament was removed to allow reperfusion of the brain for 24 h. Sham rats were performed a comparable surgery as tMCAO rats but without the occlusion of the middle cerebral artery. GSS (1.0 mg/kg) was administrated via sublingual vein injection 10 min after ischemia. Since α-BTX is not able to pass through the blood-brain barrier, we injected it into the lateral ventricle at a dose of 0.5 µg/kg body weight 30 min before tMCAO surgery. Rats without α-BTX and GSS treatments were parallelly given equivalent vehicle (saline). The schematic diagram of animal treatments is shown in Fig S1A and B in supplementary materials. The doses of GSS and α-BTX were chosen based on previous study and our preliminary experiments [32].
Lateral ventricular injection
Rats were anesthetized with 1% pentobarbital sodium (50 mg/kg, i.p.) and fixed on a stereotactic apparatus (ZH-1-Lanxing, RWD, China). When bregma was exposed, a burr hole was prepared on the skull in the right hemisphere (1.6 mm lateral and 0.9 mm posterior to bregma) using a power drill (68605, ϕ 1.4 mm, RWD, China). Then an injector (5-μL micro-syringe, Hamilton, USA) connected with micro-pump (KDS310, Harvard Apparatus, USA) was placed its needle into the right lateral ventricle at 3.5 mm depth of sub-dural through the micro-hole. Placement of needle was performed using a stereotactic apparatus. The vehicle or the α-BTX was then administered into the lateral brain ventricle at a speed of 0.25 μL/min. The needle stayed in place for 5 min after injection to prevent backflow.
Neurological test
All rats were received Garcia assessment at 24 h after cerebral ischemia reperfusion (I/R) to evaluate neurobiological function. The evaluation includes six tests scoring on a scale from 3 to 18 [37]: spontaneous activity, symmetry in four limb movements, forepaw outstretching, climbing, body proprioception, and response to vibrissae touch. Repeat each test 3 times and take the average as an evaluation score.
TTC staining
After the neurological assessment, rats were anesthetized with 1% pentobarbital sodium (50 mg/kg, i.p.) and sacrificed. Rat brain was collected and performed 2,3,5-Triphenyltetrazolium chloride (TTC) staining. In brief, rat brain was consecutively sliced into six coronal sections (2 mm thickness) and then stained with 0.5% TTC solution for 30 min at 37℃ in a water bath shaker in the dark. During the staining, the slices were turned over every 5 min. After the staining, the slices were washed with PBS and fixed in 4% paraformaldehyde for 6 h. The infarcted area and brain area in TTC-stained brain slices were measured using ImageJ 1.37a. The corrected percentage of infarct volume was calculated as the formula: infarct volume (%) = (the area of contralateral hemisphere - the area of non-infarcted ipsilateral hemisphere) /(2 * the area of contralateral hemisphere) *100% [37].
Cell culture and treatments
BV2 microglial cells were purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). BV2 cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin in a cell incubator with 5.0% CO2 at 37°C. Cells were seeded in culture dishes at a density of 1×106 cells/mL and treated with LPS (1.0 μg/mL) for 24 h to induce M1 polarization. A schematic diagram for in vitro experiments is shown in figure S1 C and D in supplementary materials. These cell experiments were performed at least three independent times with at least three biological replicates.
To determine the effects of GSS on M1 polarization, cells were randomly allocated into four groups: control group (treated with vehicle), LPS group (treated with LPS), LPS+GSS group (treated with LPS plus GSS treatment, 10 μM), and GSS group (treated with GSS alone). Cells were treated with GSS for 24 h.
In order to determine whether α7nAChR-mediated NF-κB signaling was involved in the effects of GSS on microglial M1 polarization, cells were randomly divided into six groups: control group (treated with vehicle), LPS group (treated with LPS), LPS+GSS group (treated with LPS plus GSS treatment, 10 μM), GSS group (treated with GSS alone), α-BTX+LPS+GSS group (pretreated with α-BTX followed by LPS and GSS treatment), and α-BTX+LPS group (pretreated with α-BTX followed by LPS incubation). Cells were pretreated α-BTX (10 nM) 30 min before given LPS incubation.
Western blot
Ischemic penumbra brain tissues were collected for western blotting. Brain tissues were added protein lysis buffer and homogenized on ice for 1 h. BV2 cells were also lysed with protein lysis buffer on ice for 1 h. Then these samples were centrifuged at 12,000 g for 15 min to obtain the supernatants. The protein concentration in samples was measured by BCA kit according to the manufacturer's instructions. Equivalent total protein (30 μg) in each sample was separated in SDS/PAGE gel by electrophoresis and transferred to PVDF membrane. After blocking with 5% non-fat milk, the membrane was incubated with following primary antibodies overnight at 4℃: CD11b (1:1000), IL-1β (1: 500), CD40 (1:500), CD68 (1:500), α7nAChR (1:500), IKK (1:1000), phospho-IKK (1:500), P65-NF-κB (1:1000), phospho-p65-NF-κB (1:500), IκB (1:1000), phospho-IκB (1:500) and β-tubulin (1:2000). Then the membrane was incubated with respective secondary antibodies (1:5000) at room temperature for 1 h and then incubated with an enhanced chemiluminescence reagent for 1 min. Finally, protein band images were captured, and the gray-scale of each band was analyzed using Chemiluminescence Imaging System (Amersham™ Imager 600, United States). All experiments were run in triplicate and repeated a minimum of three times independently with consistent results.
Quantitative PCR (qPCR)
Total RNA was isolated from tissues or cells using TRIzol reagent according to the manufacturer's instructions. 4 μg total RNA was used to synthesize the first strand cDNA using Reverse Transcription Master Mix kit (SYBR® Select Master Mix). Then PCR reaction was run in triplicate and performed as following reaction system: cDNA 2 μL, 1x SYBRÒ 10 μL, 10 μM primer 2 μL, adding ddH2O to a total volume of 20 μL. Parameters for PCR reaction were as bellows: pre-denatured at 95℃ for 20 min follow by 40 cycles (desaturated at 95 ℃ for 10 s, annealed at 61 ℃ for 20 s, and extended at 72 ℃ for 25 s). PCR results were normalized to GAPDH and expressed as folds of sham or control group. The sequence of primers used in this study was shown in Table 1.
The sequence of primers.

ELISA assay
The concentration of IL-1β in the culture supernatant was determined using enzyme-linked immunosorbent assay (ELISA) kit (R&D systems, Minneapolis, MN) according to the manufacturer's instructions.
Immunofluorescence
Rats were anesthetized and fixed the whole animal with 4% paraformaldehyde through transcranial perfusion. The brain was then taken out, immersed in 4% paraformaldehyde, dehydrated with 10% sucrose, and cut into brain slices (30 μm thickness) by freezing microtome. Dried brain slices were hydrated with PBS at room temperature (RT) for 20 min, and then the slices were incubated in 0.3 % Triton X-100/PBS solution at RT for 10-15 min. After washed with PBS for 5 min×3 times, the slices were blocked with 3% BSA at RT for 30 min and incubated with anti-CD11b antibody (1:200) or α7nAChR (1:200) at 4℃ overnight. Then the slices were washed with PBS and incubated with AlexaFluor488 mouse secondary antibody (1:2000). Finally, the slices were stained with DAPI and sealed with 50% glycerin. The immunofluorescent images were captured under a fluorescence microscope (Carl Zeiss Lsm880, Germany).
Immunofluorescence for BV2 cells was performed as bellows: cells were fixed with cold methanol at -20℃ for 5 min, washed with PBS for 5 min×3 times. The subsequent staining procedure was the same as above brain slice immunofluorescence, except for incubating with anti-IL-1β (1:100) or anti-P65-NF-κB (1:100), and then incubated with AlexaFluor488 rabbit secondary antibody (1:2000).
Statistical analysis
Data were presented with mean ± standard deviation (SD). Statistical analysis was performed using SPSS 20.0 software. One-way ANOVA with Newman-Keuls test was used to compare the differences between the means of more than two groups. The value of P<0.05 was considered statistically significant.
Results
GSS treatment reduced brain injury and neurological deficit in tMCAO rats
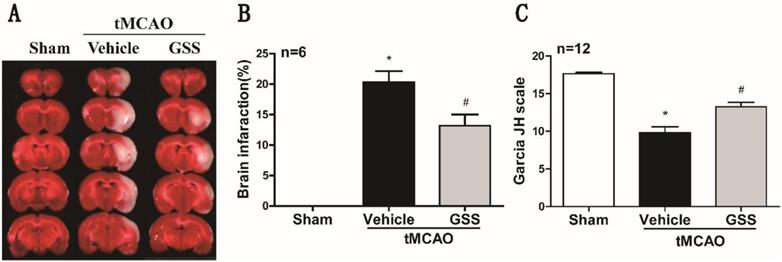
TTC staining and neurological deficit evaluation results showed that vehicle-administrated tMCAO rats (tMCAO group) developed significant brain infarction and neurological deficit at 24 h after I/R compared with sham rats, which were significantly inhibited by GSS treatment (Figure 1).
GSS treatment reduced brain infarct volume and improved neurologic function in tMCAO rats. Transient MCAO rats were treated with 1.0 mg/kg GSS or equivalent vehicle as indicated, then brain infarcted volume and neurological function scores were evaluated. (A) The representative images of TTC staining for infarcted brain were shown. (B) Analysis results of brain infarcted volume. (C) Analysis results of neurological function scores. Comparisons between groups were performed using one-way ANOVA followed by Newman-Keuls test. *P<0.05 versus sham group, #P<0.05 versus tMCAO group.
GSS treatment inhibited microglial M1 polarization in the brain peri-infarct area in tMCAO rats
Next, we used CD11b as a cell surface marker to determine microglial activation. As shown in Fig 2A, the morphology of microglia in sham group was ramified with a small cell body, which was in a resting phenotype, whereas most microglial cells were activated in the peri-infarct area of brain of tMCAO rats, characterized by a rod-shape morphology with a larger cell body, a fusion of cell nucleus, and thick or less number of cellular processes, which were significantly inhibited by GSS treatment. In the meantime, Real-time qPCR and western blot results showed that mRNA and protein expression of M1 microglial markers (CD11b, CD40 and CD68) significantly increased in tMCAO group. Whereas, these changes were significantly attenuated by GSS treatment in tMCAO rats (Figure 2B, D-I). Besides, GSS treatment also significantly inhibited the increase of IL-1β protein expression in the peri-infarct brain region induced by tMCAO insult (Figure 2B and C).
GSS treatment inhibited microglial M1 polarization and α7nAChR-NF-κB signaling activation in tMCAO rats. Microglial M1 depolarization and α7nAChR-NF-κB signaling activation in the ischemic penumbra region was determined 24 h I/R. (A) Microglia morphology was determined by immunofluorescent staining of CD11b (Scale bar = 20 μm, n=6). (B-F) Western results of IL-1β, CD11b, CD40 and CD68. (G-I) CD11b, CD40 and CD68 mRNA expression determined by qPCR. (J) Representative images of immunofluorescent staining of α7nAChR (Scale bar = 20 μm, n=6). (K-O) Western blot results of α7nAChR and NF-κB signaling proteins. (P-R), the qPCR results of IKK, IκB and P65-NF-κB mRNA expression. Comparisons between groups were carried out using one-way ANOVA followed by Newman-Keuls test. *P<0.05 versus sham group, #P<0.05 versus tMCAO group.
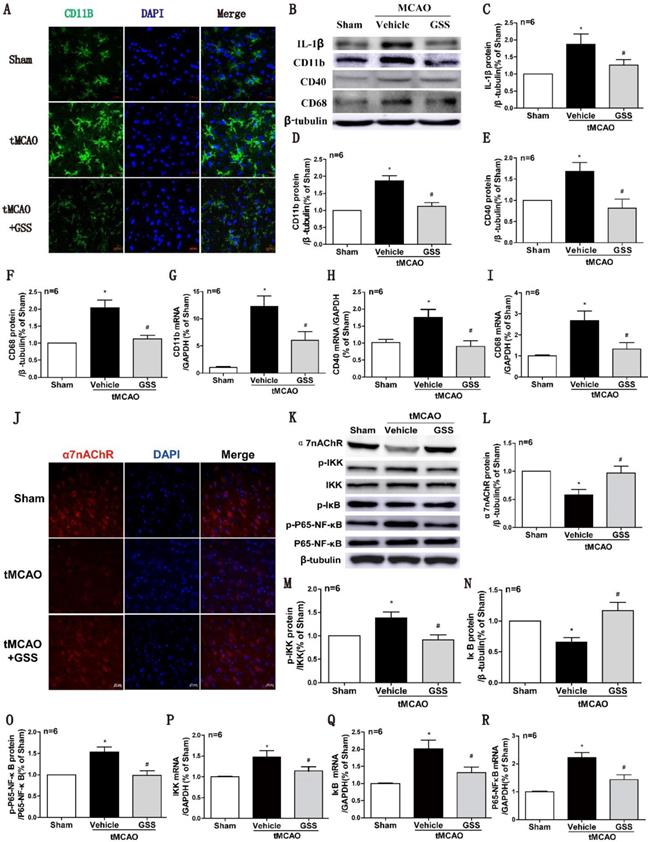
GSS treatment restored α7nAChR protein expression while suppressed NF-κB signaling in tMCAO rats
NF-κB signaling pathway is regulated by α7nAChR, attributing to M1 polarization of microglia [39, 40]. Real-time qPCR results showed that mRNA expression of IKK, IκB and P65-NF-κB in the ischemic penumbra significantly increased in tMCAO group compared with sham group. GSS treatment significantly inhibited tMCAO-induced increases of phosphorylated IKK and phosphorylated p65 protein, but restored IκB and α7nAChR protein expression in the ischemic penumbra region (Figure 3J-O). GSS treatment significantly reduced mRNA expressions of these genes in tMCAO rats (Figure 2P-R).
α-BTX pretreatment reversed the therapeutic effects of GSS treatment on brain injury and neurologic function in tMCAO rats. α-BTX (0.5 μg/kg) was administrated by stereotaxic injection 30 min before the tMCAO surgery. Brain infarct size was measured by TTC staining 24 h after I/R. (A) and (B) Representative images and analysis result of TTC staining for the evaluation of brain infarcted volume were shown, respectively. (C) Analysis result of neurological function scores. Comparisons between groups were carried out using one-way ANOVA followed by Newman-Keuls test. *P<0.05.
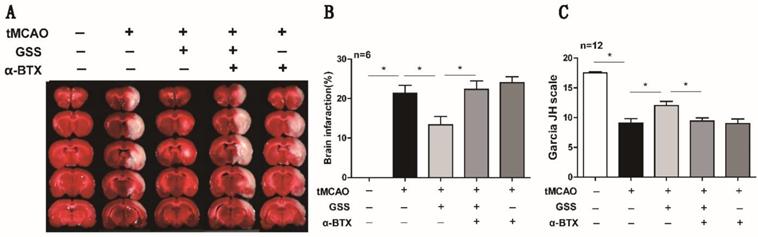
α7nAChR inhibitor blocked the protective effects of GSS against brain injury after tMCAO
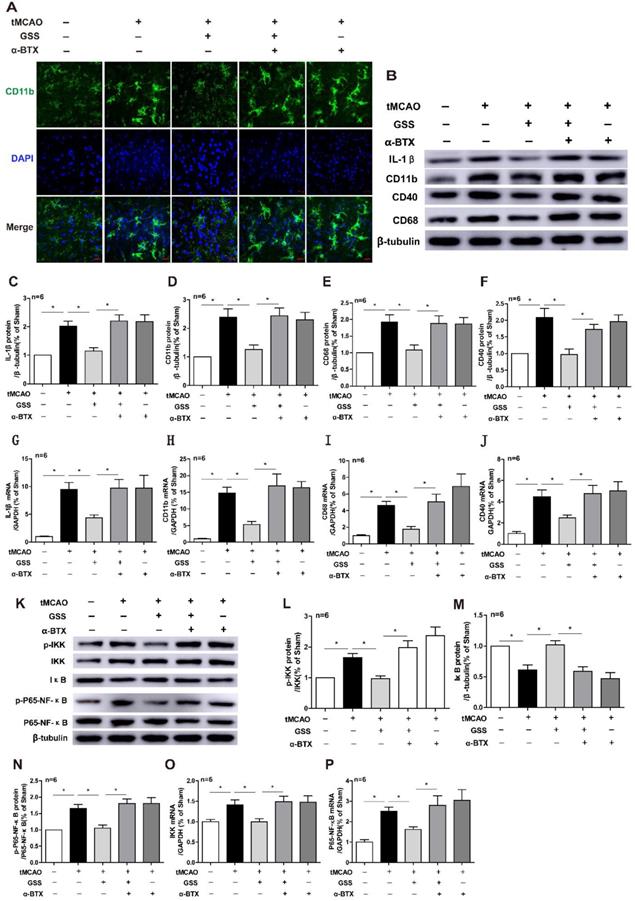
Next, we applied a specific α7nAChR inhibitor, α-BTX, to illustrate whether GSS inhibited microglial M1 polarization and brain injury through activating α7nAChR signaling. We found that α-BTX pretreatment alone did not change the infarction area and neurological deficit in tMCAO rats but significantly suppressed the therapeutic effects of GSS on brain injury and neurological function (Figure 3). Pretreatment with α-BTX also reversed the effects of GSS on the morphological change of microglia (Figure 4A) as well as mRNA and protein expression of M1 microglial markers (CD11b, CD40 and CD68) and IL-1β in the ischemic penumbra of tMCAO rats (Figure 4B-J). As shown in Fig 6A and B, we observed that α-BTX pretreatment significantly blocked the inhibitive effects of GSS on NF-κB signaling to exhibit as increasing mRNA expressions of IKK and P65-NF-κB and phosphorylation of IKK and p65 while reducing IκB protein expression to a comparable level as tMCAO group (Figure 4K-P).
α-BTX pretreatment inhibited the changes of microglia M1 polarization and α7nAChR-NF-κB signaling caused by GSS treatment in tMCAO rats. α-BTX (0.5 μg/kg) was administrated by stereotaxic injection 30 min before the tMCAO surgery. Microglia M1 depolarization and expression of α7nAChR-NF-κB signaling was measured 24 h after I/R. (A) Representative images of immunofluorescent staining of CD11b (Scale bar = 20 μm, n=6). (B-F) Western blot results of IL-1β, CD11b, CD68 and CD40. (G-J) qPCR results of CD11b, IL-1β, CD68 and CD40. (K-N) Western blot results of protein expression of NF-κB pathway. (O) and (P) qPCR results of IKK and P65-NF-κB. Comparisons between groups were carried out using one-way ANOVA followed by Newman-Keuls test. *P<0.05.
GSS treatment inhibited LPS-induced M1 polarization of microglial cells
We further used LPS-stimulated BV2 microglial cells to reaffirm the observed effects of GSS on M1 microglial polarization in vivo. ELISA results showed that LPS (1 μg/mL) stimulating for 24 h significantly upregulated the concentration of IL-1β in the cell culture supernatant compared with vehicle-treated cells. In contrast, the increase of IL-1β protein in the supernatant was significantly reduced by GSS (10 μM) treatment (Figure 5B). We also applied cell immunofluorescent staining and qPCR to validate the finding of ELISA. Consistently, the upregulation of IL-1β protein (Figure 5A) and mRNA expression (Figure 5F) in BV2 induced by LPS were significantly inhibited by GSS treatment. Besides, LPS stimulus significantly increased CD40 and CD68 mRNA expression in BV2 cells, which was significantly inhibited by GSS treatment (Figure 5G and H). Similarly, GSS treatment also significantly suppressed the increase of CD40 and CD68 protein expression induced by LPS (Figure 5C-E). GSS treatment alone did not significantly affect the expression of M1 polarization makers (IL-1β, CD40 and CD68) compared with that of control (Figure 5A-H).
GSS inhibited LPS-induced M1 polarization and α7nAChR-NF-κB signaling activation of BV2 microglial cells. M1 polarization and the mRNA or protein expression of α7nAChR-NF-κB signaling of LPS-stimulated BV2 cells was determined 24 h after GSS treatment. (A) Immunofluorescence results of IL-1β (Scale bar = 500 μm, n=6). (B) ELISA result of IL-1β. (C-E) Western blot results of CD40 and CD68. (F-H) qPCR results of IL-1 β, CD40 and CD68. (I) Immunofluorescence results of P65-NF-κB (Scale bar = 10 μm, n=6). (J-N) Western blot results of α7nAChR and NF-κB signaling proteins. Comparisons between groups were carried out using one-way ANOVA followed by Newman-Keuls test. *P<0.05 versus control group, #P<0.05 versus LPS group.
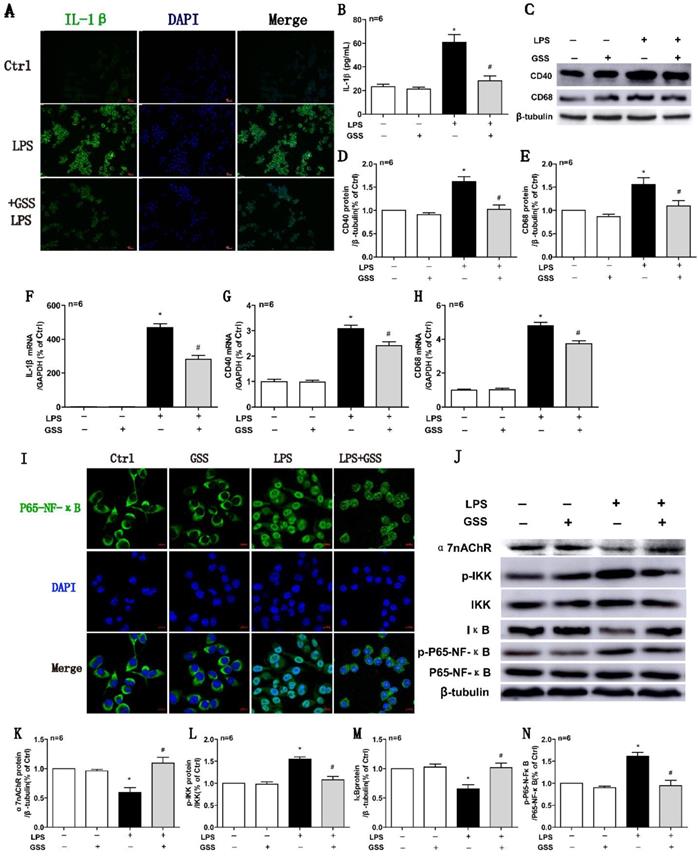
GSS treatment restored α7nAChR expression while blocked NF-κB signaling in LPS-stimulating BV2 cells
We determined the intracellular localization of P65-NF-κB using immunofluorescent staining. As shown in figure 5I, P65-NF-κB dominantly located in the cell nucleus after LPS stimulation, while it distributed mainly in the cytoplasm after GSS treatment in LPS-stimulated BV2 cells. Meanwhile, α7nAChR and IκB protein expression were significantly decreased, while expressions of p-IKK and p-P65-NF-κB were significantly increased in LPS-treated BV2 cells compared with vehicle-treated cells, which were significantly blocked by GSS treatment (Figure 5J-N). GSS treatment alone did not significantly affect α7nAChR expression and NF-κB signaling compared with that of control (Figure 5I-N).
α7nAChR blocker antagonized the protective effects of GSS against microglial M1 polarization and NF-κB signaling
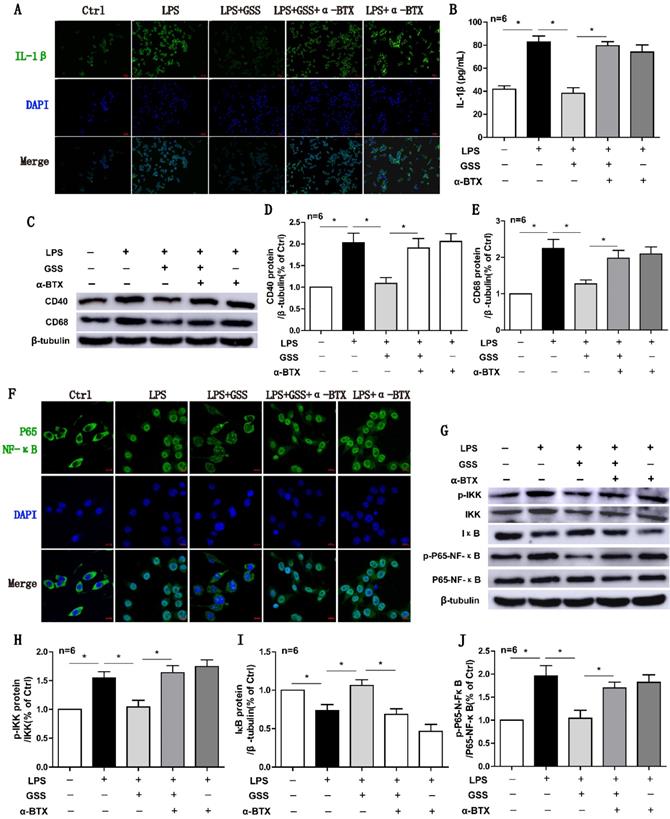
Finally, we used α-BTX (an α7nAChR blocker) to determine whether the anti-M1 polarization effects of GSS was attributed to α7nAChR-mediated signaling. Both immunofluorescent staining and ELISA results showed that α-BTX pretreatment significantly reversed the effects of GSS on IL-1β protein expression in LPS-stimulated BV2 cells (Figure 6A and B). At the same time, α-BTX pretreatment restored the protein expressions of CD40 and CD68, which were significantly decreased by GSS treatment in LPS-stimulated BV2 cells (Figure 6C-E). GSS treatment significantly inhibited the nucleus translocation of P65-NF-κB in LPS-stimulated BV2 cells. Whereas, these effects of GSS were significantly attenuated by α-BTX pretreatment (Figure 6F). Western blot results also showed α-BTX pretreatment insignificantly blunted the inhibitive effects of GSS on NF-κB signaling. As shown in figure 6G-J, GSS treatment significantly reduced the levels of p-IKK and p-P65-NF-κB but increased IκB protein expression compared with LPS-stimulated group. In contrast, α-BTX pretreatment dramatically blocked these effects of GSS (Figure 6A-J).
α7nAChR inhibitor reversed the effects of GSS on M1 polarization and NF-κB signaling in LPS-stimulated BV2 cells. Cells were pretreated α-BTX (500 μM) 30 min before giving LPS insult and GSS treatment. M1 depolarization and NF-κB signaling activation was determined 24 h after GSS treatment. (A) Immunofluorescence results of IL-1β in BV2 cells (Scale bar = 500 μm, n=6). (B) ELISA results of IL-1β. (C-E) Western blot results of CD40 and CD68. (F) Immunofluorescence results of P65-NF-κB (Scale bar = 10 μm, n=6). (G-J) Western blot results of NF-κB signaling proteins.Comparisons between groups were carried out using one-way ANOVA followed by Newman-Keuls test. *P<0.05.
Discussion
In the present study, we found that GSS treatment significantly reduced brain infarcted volume and improved neurological function by inhibiting microglial M1 depolarization-mediated inflammation in tMCAO rats, with an underlying mechanism through upregulating α7nAChR expression and thereby blocking NF-κB signaling.
Our previous study showed that GSS treatment reduced infarcted brain area and improved neurological recovery in tMCAO rats and the mechanism underlying the therapeutic effects was through protecting against neuronal apoptosis [32]. In that study, we found that GSS showed a dose-dependent effect, of which 1 mg/kg body weight had the best effect. Therefore, we adopted this dose in the present study. Besides, we also noticed that GSS treatment reduced MMP-3 and MMP-9 mRNA and protein expression [41], provoking us to investigate the anti-inflammation effects and its underlying molecular mechanism of GSS in ischemic stroke. Our results showed that GSS treatment reversed IL-1β expression in brain peri-ischemic regions and alleviated brain injury and neurological deficits in tMCAO rats. These results demonstrate that the GSS inhibits inflammatory responses in ischemic penumbra brain regions during ischemic stroke.
Neuroinflammation occurs rapidly after the onset of ischemic stroke and lasts for the whole disease process [42]. It was reported that prolonged inflammation exacerbates brain injury and postpones brain function recovery [43]. Microglia is one of the resident immune cells in the brain. Their activated states account for the dual effects of neuroinflammation in ischemic stroke: Microglial M2 phenotype is dominant in the early stages of ischemic stroke; it gradually switches to M1 phenotype [1]. M2-depolarized microglia exert anti-inflammation effects by producing IL-10 and TGF-β. In contrast, M1-depolarized microglia is proinflammatory. Thereby, inhibiting microglia depolarization to M1 phenotype is considered as a potential strategy for the treatment of ischemic stroke [3]. In this study, we used the microglial marker CD11b to determine the activation of microglia in the ischemic brain [44]. Our results showed that microglia in ischemic penumbra regions was significantly activated, showing a rod-shaped morphology with a large cell body [44]. In comparison, GSS treatment significantly inhibited the activation of microglia in ischemic penumbra regions where most microglia were ramified shape as showing in sham rats [45, 46]. In this study, we used CD40 and CD68 further verified microglial M1 depolarization [47-49]. Our results showed that CD40 and CD68 expression significantly increased in the petri-ischemic regions in tMCAO rats, while GSS treatment dramatically blocked the expression of these markers. Besides, we validated our findings through in vitro experiments: GSS treatment significantly blocked LPS-induced M1 depolarization and IL-1β expression in BV2 microglial cells. The M1 depolarized microglia are characterized by a set of markers (CD16, CD32, CD40, CD68 and CD86) and secrete proinflammatory cytokines such as IL-1β and TNF-α [50, 51]. Hence, our results demonstrate that GSS treatment suppresses microglial M1 depolarization in tMCAO rats. Our previous study showed that GSS increased IL-10 expression in peri-infarcted regions in ischemic stroke rats (unpublished data), implying that GSS might promote microglia depolarization to M2 phenotype, which requires future research.
NF-κB, as a critical transcription factor, contributes to neuroinflammation through regulating microglia activation. In canonical NF-κB activation pathways, phosphorylated IKK protein promotes the degradation of IκB protein, thereby resulting in the release and subsequent nucleus translocation of P65-NF-κB protein, which regulates a variety of target genes expression [52]. This study showed that GSS treatment significantly increased IκB protein expression but reduced the phosphorylation levels of IKK and P65-NF-κB protein in both tMCAO rats and LPS-stimulated BV2 cells. Moreover, GSS treatment also remarkedly inhibited the nucleus translocation of P65-NF-κB induced by LPS in BV2 cells. Our results suggest that GSS inhibits microglial M1 depolarization through blocking NF-κB signaling activation.
α7nAChR is a crucial receptor mediating cholinergic anti-inflammatory pathway. The stimulation of α7nAChR inhibits the activation of NF-κB signaling in microglia, reduced M1 microglia in the peri-infarct brain regions, and reduced brain injury in permanent MCAO (pMCAO) mice [2, 25]. In this study, we found that α7nAChR protein expression significantly decreased in the ischemic penumbra regions in tMCAO rats, while it was restored by GSS treatment. Besides, GSS treatment also significantly inhibited the decrease of α7nAChR expression induced by LPS in BV2 cells. These results indicate that GSS upregulates microglial α7nAChR function in ischemic stroke. After pretreatment with α7nAChR inhibitor, α-BTX, we found that GSS lost the protection against brain infarction and neurological dysfunction and the inhibitive effects on microglial M1 depolarization and NF-κB signaling in both in vivo and in vitro experiments. These findings demonstrate that α7nAChR-mediated inhibition of NF-κB signaling is a vital molecular mechanism underlying the regulation of microglial M1 depolarization by GSS.
Conclusions
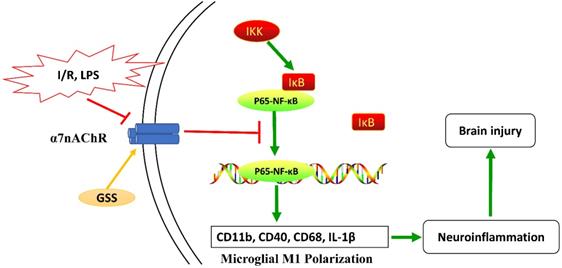
Taken together, as shown in Figure 7, our results from in vivo and in vitro experiments showed that GSS treatment upregulated α7nAChR and thereby inhibited the activation of NF-κB and microglial M1 depolarization, contributing to the neuroprotective effects in tMCAO rats. This study uncovers a novel molecular mechanism underlying the anti-ischemic stroke effects of GSS, demonstrating that GSS protected against brain ischemic injury by suppressing neuroinflammation via α7nAChR-mediated inhibition of NF-κB signaling.
Schematic diagram of the molecular mechanisms underly GSS against brain injury in tMCAO rats. GSS treatment upregulates α7nAChR and thereby inhibits NF-κB signaling, subsequently suppresses M1 depolarization-mediated neuroinflammation in brain penumbra regions, protecting against brain injuries in tMCAO rats.
Abbreviations
ACh: acetylcholine; α7nAChR: α7 nicotinic acetylcholine receptor; α-BTX: α-bungarotoxin; ANOVA: analysis of variance; BBB: blood-brain barrier; CAIP: Cholinergic anti-inflammatory pathway; ELISA: enzyme-linked immunosorbent assay; GSS: Genistein-3′-sodium sulfonate; IL-1β: interleukin-1β; IL-6: interleukin-6; IL-10: interleukin-10; LPS: lipopolysaccharide; NF-κB: nuclear factor kappa B; SD: Sprague-Dawley; TGF-β: transforming growth factor β; tMCAO: transient middle cerebral artery occlusion; TNF-α: tumor necrosis factor α; tPA: tissue plasminogen activator; TTC: 2,3,5-Triphenyltetrazolium chloride.
Supplementary Material
Supplementary figure S1.
Acknowledgements
This work was supported by the grants from the National Natural Science Foundation of China (No. 31360250, 81760654 and 31760290), Jiangxi Natural Science Foundation (No. 20122BAB205037), Bureau of Education (No. GJJ160968, GJJ150970), the Open Project of Key Laboratory of Prevention and treatment of cardiovascular and cerebrovascular diseases, Ministry of Education (No. XN201923), Ganzhou Science and Technology Guiding Project (No. GZ2015ZSF302) and Innovation Team Foundation of Gannan Medical University (No. TD201705).
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics approval and consent to participate
Animal experimental protocols were approved by the Gannan Medical University Animal Care and Use Committee. All animal experiments were performed following the Guidelines for the Care and Use of Laboratory Animals of China Medical University (No. 201745).
Authors' contributions
Zhihua Huang and Cheng Huang participated in the design of this study, and they both performed the statistical analysis. Chaoming Liu and Song Liu carried out the study and collected important background information. Song Liu works in Xiamen Haicang Biological Science and Technology Development after graduating from GMU. Lijiao Xiong drafted the manuscript. Limei Zhang, Xiao Li, Xingling Cao, Jinhua Xue and Liangdong Li provided assistances for data acquisition, data analysis and statistical analysis. All authors have read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jayaraj RL, Azimullah S, Beiram R. et al. Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation. 2019;16(1):142
2. Kim J, Thayabaranathan T, Donnan GA. et al. Global Stroke Statistics 2019. Int J Stroke. 2020;15(8):819-38
3. Dabrowska S, Andrzejewska A, Lukomska B. et al. Neuroinflammation as a target for treatment of stroke using mesenchymal stem cells and extracellular vesicles. J Neuroinflammation. 2019;16(1):178
4. Sahota P, Savitz SI. Investigational therapies for ischemic stroke: neuroprotection and neurorecovery. Neurotherapeutics. 2011;8(3):434-51
5. Barthels D, Das H. Current advances in ischemic stroke research and therapies. Biochim Biophys Acta Mol Basis Dis. 2020;1866(4):165260
6. Cai PY, Bodhit A, Derequito R. et al. Vagus nerve stimulation in ischemic stroke: old wine in a new bottle. Front Neurol. 2014;5:107
7. Xing C, Arai K, Lo EH. et al. Pathophysiologic cascades in ischemic stroke. Int J Stroke. 2012;7(5):378-85
8. Lambertsen KL, Finsen B, Clausen BH. Post-stroke inflammation-target or tool for therapy? Acta Neuropathol. 2019;137(5):693-714
9. Ma Y, Wang J, Wang Y. et al. The biphasic function of microglia in ischemic stroke. Prog Neurobiol. 2017;157:247-72
10. Kawabori M, Yenari MA. The role of the microglia in acute CNS injury. Metab Brain Dis. 2015;30(2):381-92
11. Hu X, Li P, Guo Y. et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43(11):3063-70
12. Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387-94
13. Zhao SC, Ma LS, Chu ZH. et al. Regulation of microglial activation in stroke. Acta Pharmacol Sin. 2017;38(4):445-58
14. Han B, Li X, Hao J. The cholinergic anti-inflammatory pathway: An innovative treatment strategy for neurological diseases. Neurosci Biobehav R. 2017;77:358-68
15. Al MA, Chauhan A, Qi S. et al. Microglial IRF5-IRF4 regulatory axis regulates neuroinflammation after cerebral ischemia and impacts stroke outcomes. Proc Natl Acad Sci. 2020;117(3):1742-52
16. Kumar A, Barrett JP, Alvarez-Croda DM. et al. NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain Behav Immun. 2016;58:291-309
17. Liu Z, Ran Y, Huang S. et al. Curcumin protects against ischemic stroke by titrating microglia/macrophage polarization. Front Aging Neurosci. 2017;9:233
18. Zhang H, Lu M, Zhang X. et al. Isosteviol sodium protects against ischemic stroke by modulating microglia/macrophage polarization via disruption of gas5/mir-146a-5p sponge. Sci Rep. 2019;9(1):12221
19. Wang H, Yu M, Ochani M. et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421(6921):384-88
20. Pohanka M. Alpha7 nicotinic acetylcholine receptor is a target in pharmacology and toxicology. Int J Mol Sci. 2012;13(2):2219-38
21. Shytle RD, Mori T, Townsend K. et al. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem. 2004;89(2):337-43
22. Egea J, Buendia I, Parada E. et al. Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem Pharmacol. 2015;97(4):463-72
23. Zoheir N, Lappin DF, Nile CJ. Acetylcholine and the alpha 7 nicotinic receptor: a potential therapeutic target for the treatment of periodontal disease? Inflamm Res. 2012;61(9):915-26
24. Wang H, Liao H, Ochani M. et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med. 2004;10(11):1216-21
25. Han Z, Shen F, He Y. et al. Activation of α-7 nicotinic acetylcholine receptor reduces ischemic stroke injury through reduction of pro-inflammatory macrophages and oxidative stress. Plos One. 2014;9(8):e105711
26. Han Z, Li L, Wang L. et al. Alpha-7 nicotinic acetylcholine receptor agonist treatment reduces neuroinflammation, oxidative stress, and brain injury in mice with ischemic stroke and bone fracture. J Neurochem. 2014;131(4):498-508
27. Xue X, Qu XJ, Yang Y. et al. Baicalin attenuates focal cerebral ischemic reperfusion injury through inhibition of nuclear factor κB p65 activation. Biochem Biophys Res Commun. 2010;403(3-4):398-404
28. COLDITZ Graham A. Hormones and breast cancer: evidence and implications for consideration of risks and benefits of hormone replacement therapy. J Womens Health. 1999;8(3):347-57
29. Green PS, Simpkins JW. Neuroprotective effects of estrogens: potential mechanisms of action. Int J Dev Neurosci. 2000;18(4-5):347-58
30. Engler-Chiurazzi EB, Brown CM, Povroznik JM. et al. Estrogens as neuroprotectants: Estrogenic actions in the context of cognitive aging and brain injury. Prog Neurobiol. 2017;157:188-211
31. Del Gaudio P, Sansone F, Mencherini T. et al. Nanospray drying as a novel tool to improve technological properties of soy isoflavone extracts. Planta Med. 2017;83(5):426-33
32. Li L, Xue J, Liu R. et al. Neuroprotective effects of genistein-3'-sodium sulfonate on focal cerebral ischemia in rats. Neurosci Lett. 2017;646:43-8
33. Xue J, Shui R, Huang J. et al. Genistein's effect on inflammatory cytokines levels in cerebral ischemia/reperfusion injury. Chinese J Exp Trad Med Formulae. 2013;19(22):191-5 Chinese
34. Aryanpour R, Pasbakhsh P, Zibara K. et al. Progesterone therapy induces an M1 to M2 switch in microglia phenotype and suppresses NLRP3 inflammasome in a cuprizone-induced demyelination mouse model. Int Immunopharmacol. 2017;51:131-139
35. Roshni T, Wang R, Wang J. et al. 17β -estradiol regulates microglia activation and polarization in the hippocampus following global cerebral ischemia. Oxid Med Cell Longev. 2018;2018:4248526
36. Longa EZ, Weinstein PR, Carlson S. et al. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84-91
37. Garcia JH, Wagner S, Liu KF. et al. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats: statistical validation. Stroke. 1995;26(4):627-34
38. Yoshida IY. The effects of early exercise on brain damage and recovery after focal cerebral infarction in rats. Acta Physiol (Oxf). 2011;201(2):275-87
39. Zhang Q, Lu Y, Bian H. et al. Activation of the α7 nicotinic receptor promotes lipopolysaccharide-induced conversion of M1 microglia to M2. Am J Transl Res. 2017;9(3):971-85
40. Lee RH, Vazquez G. Evidence for a prosurvival role of alpha-7 nicotinic acetylcholine receptor in alternatively (M2)-activated macrophages. Physiol Rep. 2013;1:e189
41. Liu C, Liu S, Yang Y. et al. The effect of genistein sodium sulfonate on the expression of MMP-3 and MMP-9 protein following focal cerebral ischemia-reperfusion injury in rats. Journal of Gannan Medical University. 2018;38(10):959-63 Chinese
42. Taylor RA, Sansing LH. Microglial Responses after Ischemic Stroke and Intracerebral Hemorrhage. Clin Dev Immunol. 2013;2013:746068
43. Langdon KD, Maclellan CL, Corbett D. Prolonged, 24-h delayed peripheral inflammation increases short- and long-term functional impairment and histopathological damage after focal ischemia in the rat. J Cereb Blood Flow Metab. 2010;30(8):1450-9
44. Morrison HW, Filosa JA. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J Neuroinflammation. 2013;10:4
45. Fernández-Arjona M, Grondona JM, Fernández-Llebrez P. et al. Microglial morphometric parameters correlate with the expression level of il-1β, and allow identifying different activated morphotypes. Front Cell Neurosci. 2019;13:472
46. Torres-Platas SG, Comeau S, Rachalski A. et al. Morphometric characterization of microglial phenotypes in human cerebral cortex. J Neuroinflammation. 2014;11:12
47. Zhou T, Huang Z, Sun X. et al. Microglia polarization with m1/m2 phenotype changes in rd1 mouse model of retinal degeneration. Front Neuroanat. 2017;11:77
48. Hendrickx D, van Eden CG, Schuurman KG. et al. Staining of HLA-DR, Iba1 and CD68 in human microglia reveals partially overlapping expression depending on cellular morphology and pathology. J Neuroimmunol. 2017;309:12-22
49. Chistiakov DA, Killingsworth MC, Myasoedova VA. et al. CD68/macrosialin: not just a histochemical marker. Lab Invest. 2017;97(1):4-13
50. Donat CK, Scott G, Gentleman SM. et al. Microglial activation in traumatic brain injury. Front Aging Neurosci. 2017;9:208
51. Walker DG, Lue LF. Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther. 2015;7(1):56
52. Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1:a000034
Author contact
![]() Corresponding authors: Zhihua Huang (zh.huangedu.cn) and Cheng Huang (huangc6a2013edu.cn). Key Laboratory of Prevention and treatment of cardiovascular and cerebrovascular diseases of Ministry of Education, Gannan Medical University, Ganzhou 341000, China.
Corresponding authors: Zhihua Huang (zh.huangedu.cn) and Cheng Huang (huangc6a2013edu.cn). Key Laboratory of Prevention and treatment of cardiovascular and cerebrovascular diseases of Ministry of Education, Gannan Medical University, Ganzhou 341000, China.