Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
HPV status and chemoradiotherapy
EGFR/PI3K/AKT/mTOR pathway might...
Targeting P53 is an effective...
HPV-associated disease trend to...
Epidrugs have a long way to go...
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(4):1104-1118. doi:10.7150/ijbs.58077 This issue Cite
Review
Therapeutic strategies of different HPV status in Head and Neck Squamous Cell Carcinoma
Yingming Sun1, Zhe Wang2,3 ![]() , Sufang Qiu4, Ruoyu Wang2,3
, Sufang Qiu4, Ruoyu Wang2,3 ![]()
1. Department of Radiation and Medical Oncology, Affiliated Sanming First Hospital of Fujian Medical University, Sanming 365001, P. R. China.
2. Department of Medical Oncology, Affiliated Zhongshan Hospital of Dalian University, Dalian 116001, P. R. China.
3. The Key Laboratory of Biomarker High Throughput Screening and Target Translation of Breast and Gastrointestinal Tumor, Dalian University, Dalian 116001, P. R. China.
4. Department of Radiation Oncology, Fujian Medical University Cancer Hospital & Fujian Cancer Hospital; Fujian Provincial Key Laboratory of Translational Cancer Medicine, Fuzhou 350014, P.R. China.
Received 2021-1-11; Accepted 2021-2-9; Published 2021-3-10
Abstract

Head and neck squamous cell carcinoma (HNSCC) is the 9th most common malignant tumor in the world. Based on the etiology, HNSCC has two main subtypes: human papillomavirus (HPV) -related and HPV-unrelated. HPV-positive HNSCC is more sensitive to treatment with favorable survival. Due to the different biological behaviors, individual therapy is necessary and urgently required to deduce the therapeutic intensity of HPV-positive disease and look for a more effective and toxicity-acceptable regimen for HPV-negative disease. EGFR amplification and PI3K/AKT/mTOR pathway aberrant activation are quite common in HPV-positive HNSCC. Besides, HPV infection alters immune cell infiltrating in HNSCC and encompasses a diverse and heterogeneous landscape with more immune infiltration. On the other hand, the chance of HPV-negative cancers harboring mutation on the P53 gene is significantly higher than that of HPV-positive disease. This review focuses on the updated preclinical and clinical data of HPV-positive and HPV-negative HNSCC and discusses the therapeutic strategies of different HPV status in HNSCC.
Keywords: HPV, HNSCC, PI3K, P53, immunotherapy
Introduction
Head and neck squamous cell carcinoma, including those of the lip and oral cavity, nasal cavity, paranasal sinuses, oropharynx, larynx, and nasopharynx, is the 9th most common malignant tumor in the world, representing 6% of all cancer cases and up to 2% of all cancer-related deaths [1, 2]. HNSCC is a biologically diverse and genetically heterogeneous disease. Smoking, betel nut, and alcohol consumption are the traditional high-risk factors for HNSCC [3-7]. During the past three decades, people realized human papillomavirus (HPV) [1, 8, 9] and Epstein Barr Virus (EBV) [10-12] are associated with the development of HNSCC.
Based on the etiology, HNSCC has two main subtypes: human papillomavirus-related (HPV-positive) and human papillomavirus-unrelated (HPV-negative). The HPV-positive disease occurs predominantly in the oropharynx [13]. In contrast, HPV-negative disease is driven by chemical mutagenesis associated with tobacco and alcohol exposure and origins in anatomic sites [14]. HPV infection transforms cells in tonsillar crypts and boosts carcinogenesis in the oropharynx [15]. HPV-positive HNSCC tending to arise in a younger patient population is more sensitive to treatment with more favorable survival. Overall survival rates at 3 years are estimated at 82% in locally advanced HPV-positive HNSCC compared to 57% for locally advanced HPV-negative HNSCC [1, 9, 16-19]. More preclinical and clinical data recommends a different therapeutic strategy for HPV-positive and HPV-negative HNSCC as two clinically distinct diseases [20]. Recently, many clinical trials design for HNSCC based on HPV status (Table 1). As for the different biological behaviors, individual therapy is necessary and urgently required for deducing the therapeutic intensity of HPV-positive disease and looks for a more effective and toxicity-acceptable regimen for HPV-negative disease [19, 21]. This review summarizes the up-to-date research and describes the emerging therapeutic strategies on HPV biased HNSCCs.
Clinical trials based on HPV status in NHSCC
| Clinical trial number | Project name |
|---|---|
| HPV+ | |
| NCT03946358 | Combination of UCPVax Vaccine and Atezolizumab for the treatment of Human Papillomavirus Positive Cancers (VolATIL) |
| NCT04260126 | Study of PDS0101 and Pembrolizumab combination I/O in subjects with HPV16 + recurrent and/or metastatic HNSCC |
| NCT02048020 | Paclitaxel and Carboplatin before radiation therapy with Paclitaxel in treating HPV-positive patients with Stage III-IV Oropharynx, Hypopharynx, or Larynx Cancer |
| NCT04444869 | Testing less intensive radiation with chemotherapy to treat low-risk patients with HPV-positive Oropharyngeal Cancer |
| NCT03829722 | Radiotherapy, Carboplatin/Paclitaxel and Nivolumab for high risk HPV-related Head and Neck Cancer |
| NCT03618134 | Stereotactic Body Radiation Therapy and Durvalumab with or without Tremelimumab before surgery in treating participants with Human Papillomavirus positive Oropharyngeal Squamous Cell Cancer |
| NCT04630353 | A Study HB-201 in patients with newly diagnosed HPV16+ Oropharynx or Locally Advanced Cervical Cancer |
| NCT01721525 | Induction Chemotherapy with Afatinib, Ribavirin, and weekly Carboplatin/Paclitaxel for Stage IVA/IVB HPV associated Oropharynx Squamous Cell Cancer (OPSCC) |
| NCT03715946 | Adjuvant de-escalated radiation + Adjuvant Nivolumab for Intermediate-high risk P16+ Oropharynx Cancer |
| NCT00257738 | 0804 GCC: MAGE-A3/HPV 16 Vaccine for Squamous Cell Carcinoma of the Head and Neck |
| NCT03396718 | De-escalation of Adjuvant Radio (Chemo) Therapy for HPV-positive Head-neck Squamous Cell Carcinomas |
| NCT03578406 | HPV-E6-specific anti-PD1 TCR-T cells in the treatment of HPV-positive NHSCC or Cervical Cancer |
| NCT04534205 | A clinical trial investigating the safety, tolerability, and therapeutic effects of BNT113 in combination with Pembrolizumab versus Pembrolizumab alone for patients with a form of Head and Neck Cancer positive for Human Papilloma Virus 16 and expressing the protein PD-L1 |
| NCT04489212 | Study of Mucosal Sparing Adjuvant Radiotherapy after Surgical Exploration in HPV+ Head and Neck Cancer of Unknown Primaries |
| NCT03260023 | Phase Ib/II of TG4001 and Avelumab in HPV16 positive R/M Cancers including Oropharyngeal SCCHN |
| NCT03978689 | A Phase 1 Study in patients with HPV+ recurrent/metastatic Head and Neck Squamous Cell Carcinoma |
| NCT04369937 | HPV-16 Vaccination and Pembrolizumab Plus Cisplatin for “Intermediate Risk” HPV-16-associated Head and Neck Squamous Cell Carcinoma |
| NCT04252248 | Decitabine treatment in HPV-induced anogenital and Head and Neck Cancer patients after radiotherapy or as novel late salvage |
| NCT03942380 | Cell-free tumor DNA in Head and Neck Cancer patients |
| NCT02163057 | Study of HPV specific immunotherapy in patients with HPV associated Head and Neck Squamous Cell Carcinoma |
| HPV- | |
| NCT03944915 | De-escalation therapy for Human Papillomavirus negative Disease |
| NCT04220749 | Radiotherapy vs. Trans-Oral Surgery for HPV-negative Oropharyngeal Squamous Cell Carcinoma |
| NCT03356223 | Evaluation of ABEMACICLIB monotherapy in patients with locally advanced/metastatic Head and Neck Cancer after failure of Platinum and Cetuximab or anti-EGFR-based therapy and harboring an Homozygous Deletion of CDKN2A, and/or an amplification of CCND1 and/or of CDK6 |
| NCT03389477 | Los Tres Paso: Neoadjuvant Palbociclib Monotherapy, Concurrent Chemoradiation Therapy, Adjuvant Palbociclib Monotherapy in patients with p16INK4a negative, HPV-unrelated Head and Neck Squamous Cell Carcinoma |
| NCT03635164 | Radiotherapy with Durvalumab prior to surgical resection for HPV negative Squamous Cell Carcinoma |
| NCT04169074 | Modulation of the tumor microenvironment by Abemaciclib in operable HPV-Negative Head and Neck Cancer (HNC) |
| NCT03673735 | Maintenance Immune Check-point Inhibitor following post-operative Chemo-radiation in subjects with HPV-negative HNSCC |
| NCT03624231 | Feasibility & Efficacy of Durvalumab+Tremelimumab+RT and Durvalumab+RT in Non-resect. Locally Advanced HPVnegativ HNSCC |
| NCT04247282 | Anti-PD-L1/TGF-beta Trap (M7824) alone and in combination with TriAd Vaccine and N-803 for Resectable Head and Neck Squamous Cell Carcinoma not associated with Human Papillomavirus Infection |
HPV status and chemoradiotherapy
HPV has prognostic but not predictive effectiveness of chemoradiotherapy
Radiotherapy and chemotherapy are critical therapeutic ways to treat HNSCC, and they are even the gold standard for patients with local advanced stage or recurrence and metastasis disease. RTOG 0129, the most cited trial, reported that HPV-positive patients had better 3-year rates of overall survival (82.4%, vs 57.1%; p<0.001) [22-25]. Many scholars concluded that the clinical outcomes after chemoradiotherapy were similar between HPV+ and HPV- cohort [16, 17, 20, 25]. Retrospective sub-analyses in randomized trials failed to conclude a benefit from HPV status: in the DAHANCA 5 study, nimorazole with radiotherapy has more effect in the p16-positive cohort [26]. However, the TROG 0202 phase III trial presented a trend favoring the tirapazamine arm in p16-negative patients [27, 28]. In conclusion, the present data in HNSCC suggest that HPV (p16) has prognostic but not predictive effectiveness of chemoradiotherapy.
HPV-associate disease may not receive aggressive chemoradiotherapy
The clinical significance of HPV status indicates the prognosis but cannot change the clinical decision-making. According to the guidelines of NCCN 2020, surgery or radiotherapy alone can be considered for HPV-positive oropharyngeal carcinoma in the T1-2 N0 stage, surgery, radiotherapy, or concurrent radiotherapy and chemotherapy can be regarded as in T1-2N1 (lymph nodes smaller than 3cm), while tumors in a later stage can consider surgery, concurrent radiotherapy, and chemotherapy or sequential radiotherapy and chemotherapy after induced chemotherapy. For radical radiotherapy, the equivalent biological dose (EQD2) is still required to 70Gy or above (National Comprehensive Cancer Network. Head and Neck Cancers (Version 1.2021). https://www.nccn.org. Accessed December 27, 2020).
However, because of the treatment sensitivity of HPV-positive oropharyngeal carcinoma, appropriately reducing the treatment intensity would be an ideal strategy [29]. Therefore, in recent years, studies on intensity reduction therapy for HPV-positive oropharyngeal carcinoma emerge one after another. In 2018, ECOG1308 suggested that local advanced HPV-positive oropharyngeal cancer with complete remission after induced chemotherapy can safely reduce the radiation dose to 54Gy [30]. OPTIMA is a phase II trial to explore different radiotherapy doses according to recurrence risk. Risk-stratified dose and volume de-escalated RT/CRT are associated with favorable outcomes and less acute and chronic toxicity for HPV-positive oropharyngeal cancer [31]. In 2020, ASCO also updated the data of ECOG 3311. The moderate-risk group were randomized and received standard adjuvant dose (60 Gy) radiotherapy reduction therapy (50 Gy). 2 years PFS was similar in the two groups [32].
Reducing the intensity of chemotherapy is another strategy for HPV-positive disease. RTOG 1016 indicates that cetuximab has no merit to improve prognosis [33]. In the same period, two other studies with a similar design, De-ESCALaTE [34] and TROG 12.01 [35], also achieved negative results, suggesting that cisplatin should still be used as the standard treatment. Many ongoing trials are going on to uncover the results; however, the treatment of HPV negative HNSCC in clinical practice needs more exploration.
EGFR/PI3K/AKT/mTOR pathway might be potentially targeted to HPV-associated HNSCC
EGFR/PI3K/AKT/mTOR pathway aberrant activation frequently occurs in HPV-positive HNSCC
As described above, HPV status contributes less to conventional treatment. As we know, cancer derives through the accumulation of genetic and epigenetic alteration in genes involved in a variety of signaling pathways and precipitates the cancer-associated phenotypes [29, 36].
EGFR blockade is not a good choice for HPV-positive HNSCC
About 70% of HNSCC has an epidermal growth factor receptor (EGFR) overexpression. Hwang et al. reported that E5 forms a complex with the EGFR and triggers the EGFR pathway activation [37]. Also, E5 binds the 16 kDa subunit C of the V-H+-ATPase inhibits the degradation of EGFR. c-Cbl as a ubiquitin ligase associates with the activated EGFR and targets it for degradation was hijacked by E5 [38-40]. To sum up, E5 is essential to trigger the EGFR pathway.
Decades ago, scholars initiated to block EGFR [41-45] on HNSCC. Monoclonal antibodies and inhibitors of EGFR had been developed, including cetuximab, afatinib, and panitumumab. A retrospective analysis from the SPECTRUM trial [46] indicated that panitumumab's therapeutic benefit was restricted to the p16 negative patients. A small sample from the PRISM trial suggested that patients with p16 negative trends benefit from panitumumab [47]. Afatinib, the 2nd generation of EGFR-TKI, is more pronounced in p16 negative tumors [48, 49]. The EXTREME trial was the soundest in HNSCC recurrent/metastatic disease and showed that patients with p16 positive would have more benefit from cetuximab [50, 51]. However, more studies regarding anti-EGFR, including the PARTNER study [52] and some other small sample trials, showed that HPV status failed to differentiate the EGFR blockade [48, 53, 54]. In 2015, Seiwert et al. analyzed the TCGA HNSCC samples and showed fewer EGFR aberrations in HPV-negative tumors in genomic analyses [55]. Thus, many scholars agree that EGFR expression or amplification may not be a predictive factor for EGFR blockade therapy.
HPV trigger PI3K/AKT/mTOR pathway in various ways
The heterogeneity of EGFR blockade therapy, in part, because of the downstream driver gene mutations, remains an incomplete understanding. PI3K mutation is highlighted in HPV-associated HNSCC. In the TCGA database, 27.8% PI3K mutated in HPV positive cases [55]. Vivian et al. analyzed the GWAS data from 151 HNSCC cases and determined the PI3K was the most frequently mutated (30.5%), and in HPV-associated tumors, only PIK3R1 (453_454insN), PIK3CA (E542K), and PIK3CA (H1047L) were identified [56]. PI3K pathogenic mutation is relevant to advanced disease, suggesting that PI3K fuels the progress of HPV-associated HNSCC [56-59]. However, the PIK3CA mutations are not the only genetic alterations that maintain activation of PI3K and downstream targets, including AKT and mTOR, in HNSCC [60-62]. Indeed, 80-90% of HNSCC gain an abnormal activation of the PI3K/AKT/mTOR pathway, indicating that multiple steps of genetic and epigenetic alteration may involve the carcinogenesis which PI3K drives. Downstream signaling genes of PI3K, AKT2, mTOR, TSC1, and TSC2 were less mutated (<2%) [13, 55, 56, 59, 61].
Aside from the PI3K pathway's stromal mutations, many studies reveal that E6 can target phosphorylase and activate the pathway. HPV E6 oncoprotein contains a PDZ-binding domain and inactivates PTEN [62-65], leading to fuel pAkt [66, 67]. Also, E6 and BPV1 interact with acidic LxxLL motifs and activate mTOR [68]. E6‐E6AP complex binds and degrades the TSC2 [69]. E6 can degrade the NHERF-1 and activate the PI3K/Akt pathway [70]. The E7 protein is known as Rb disruption and results in dysfunction of cell cycle checkpoints that promotes carcinogenesis [71-73]. Loss-of-function of Rb induces AKT activation, and E7 can directly phosphorylate Akt at Thr 308 and Ser 473 [74, 75]. Also, E7 can bind to the ubiquitous and conserved serine/threonine phosphatase, which is crucial to protect AKT dephosphorylation and sustain AKT activation [76] (Figure 1).
EGFR/PI3K/AKT/mTOR pathway might be potentially targeted to HPV-associated HNSCC. HPV E5, E6, E7 can activate PI3K/AKT/mTOR in various ways. The heterogeneity of the EGFR blockade is attributed to, somehow, the downstream driver gene mutation. About 80-90% of HPV-associated HNSCC has PI3K/AKT/mTOR pathway activation. Monotherapy of PI3K/AKT/mTOR is less effective. PI3K and MAPK pathways conduct in parallel. Combine PI3K with MAPK inhibitors in patients where genetic alterations coexist in both pathways that may induce a synthetic lethal.

PI3K inhibitors are promising but still have a long way to go
Many pan-class I PI3K inhibitors have been developed and tested. The safety profile of inhibitors is acceptable [77]. Single-agent PI3K inhibitor does not present a potent effect like EGFR-TKI and works on the selected tumor [77-80]. FDA only approves alpelisib for breast cancer in PI3K mutated breast cancer and paxalisib for DIPG. Alpelisib monotherapy is unavailable in all breast cancer, and it needs to combine with fulvestrant for its therapeutic effect [81, 82]. Paxalisib prolongs 5 months overall survival (OS) than TMZ, independent of PI3K mutation status [83, 84]. In HNSCC, only BKM120 monotherapy was applied in the clinical trial (NCT01737450).
Herein, finding a reasonable mode of combined therapy is the best choice for targeting PI3K. As the same as PI3K inhibitor, mTOR inhibitors often present a short-term efficacy [85]. Preclinical and clinical studies indicate mTOR blockade results in PI3K and Akt's reactivation via various negative feedback blockade [86-88]. Dactolisib, a PI3K/mTOR dual inhibitor, exhibits a therapeutic effect superior to everolimus in the mouse model. However, excessive toxicity in patients restrains Dactolisib for pharmaceutical use [89, 90]. MAPK pathway and interpathway inhibition should be explored.
Synthetic lethal with PI3K inhibitors may be highlighted for HPV-positive HNSCC
It is well known that PI3K and MAPK pathways conduct in parallel. Combine PI3K with MAPK inhibitors in patients whose genetic alterations in both pathways coexist [58, 91-93]. Scholars note that PIK3CA mutations often coexist with KRAS and BRAF mutations [94]. A parallel oncogenic pathway activation abrogates the effects of PI3K/Akt/mTOR inhibitors [77, 79]. Vemurafenib and dabrafenib are two successful drugs targeting BRAF; however, they present a very modest effect and only last 6 months PFS [95-98]. Vitro drug screening shows that PI3K pathway aberrant activation may contribute to BRAF inhibitor resistance [99]. As the same, the MEK inhibitor presents the same phenotype. Drugs or RNA interfere, targeting PI3K rescues the resistance to BRAF and MEK inhibitors. An animal model double-agent of PI3K and MEK pathway inhibitor shows a strong therapeutic effect [94, 100, 101]. Unfortunately, a Phase Ib study of the pan-PI3K inhibitor Buparlisib combined with the MEK1/2 inhibitor Trametinib meets the problems of frequent dose interruptions and reductions for toxicity ease before Phase II trial [102]. In 2018, novel MEK inhibitor pimasertib and PI3K/mTOR inhibitor voxtalisib also failed due to long-term tolerability and limited anti-tumor activity [103]. EMR 20006-012 is another trial to evaluate pimasertib (MEK inhibitor) with SAR245409 (PI3K inhibitor) and terminates early due to low ORR and high rate of discontinuation [104]. MEK inhibitor trametinib and the AKT inhibitor GSK2141795 combination strategy lunched in cervical cancer; AML also terminated due to the high rate of discontinuation or lack of clinical efficacy [105, 106]. It seems that Ras mutation status does not affect the efficacy of the dual blockade of PI3K and MAPK.
It has been reported that activation might be a prognostic marker for radiotherapy in HNSCC and indicates PI3K pathway inhibition would potentially exert a synergistic effect with radiotherapy [62]. In 2011, NCCTG N057K showed a well-tolerance of the combination of everolimus and radiotherapy; however, the group updated their result in 2015, and everolimus did not change the clinical outcome [107]. A phase Ib clinical trial indicates a novel PI3K inhibitor Alpelisib at 250 mg/d combined with cetuximab and IMRT is tolerable and presents a clinical efficacy in local advanced HNSCC [108]. Furthermore, some PI3K inhibitors, such as voxtalisib [103, 109], nelfinavir [110, 111], and buparlisib [112], also present a favorable safety profile with radiotherapy, but the clinical efficacy needs further investigation (Table 2).
Clinical trials basing on PI3K/AKT/mTOR pathway in NHSCC
| Clinical trial number | Project name |
|---|---|
| NCT03740100 | Single-arm study with Bimiralisib in patients with HNSCC Harboring NOTCH1 loss of function mutations |
| NCT02822482 | Copanlisib in association with Cetuximab in patients with recurrent and/or Metastatic Head and Neck Squamous Cell Carcinomas harboring a PI3KCA mutation/amplification and/or a PTEN loss |
| NCT03795610 | Window of Opportunity Study of IPI-549 in patients with locally advanced HPV+ and HPV- Head and Neck Squamous Cell Carcinoma |
| NCT04193293 | A Study of Duvelisib in combination with Pembrolizumab in Head and Neck Cancer |
| NCT03356587 | A Biomarker-driven, Open Label, Single Arm, Multicentre Phase II Study of Abemaciclib in patients with recurrent or metastatic Head and Neck Squamous Cell Carcinoma who failed to Platinum-based Therapy |
| NCT01602315 | A Phase Ib/II Study of BYL719 and Cetuximab in Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma |
| NCT02537223 | Phase I Study of BYL719 in combination with Cisplatin and Radiotherapy in patients with Squamous Cell Head and Neck Cancer |
| NCT02573493 | Nab-Paclitaxel and Cisplatin or Nab-paclitaxel as induction therapy for locally advanced Squamous Cell Carcinoma of the Head and Neck (HNSCC) |
| NCT03896412 | Detection of Circulating Tumor DNA in p16- Locally Advanced Head Neck Squamous Cell Carcinoma |
| NCT02051751 | A study to evaluate the potential benefit of the addition of BYL719 to Paclitaxel in the Treatment of Breast Cancer and Head-and-neck Cancer |
| NCT03022409 | A Study to investigate biomarker effects of pre-surgical Treatment with DNA Damage Repair (DDR) agents in patients with Head and Neck Squamous Cell Carcinoma (HNSCC). |
| NCT01204099 | Study of PX-866 and Docetaxel in Solid Tumors |
| NCT01252628 | Phase 1 and 2 Study of PX-866 and Cetuximab |
| NCT02113878 | Phase Ib Study of BKM120 with Cisplatin and XRT in high risk locally advanced Squamous Cell Cancer of Head and Neck |
| NCT02277184 | Ficlatuzumab, Cisplatin and IMRT in locally advanced Head and Neck Squamous Cell Carcinoma |
| NCT01816984 | PI3K Inhibitor BKM120 and Cetuximab in treating patients with recurrent or metastatic Head and Neck Cancer |
| NCT02644122 | SF1126 in recurrent or progressive SCCHN and mutations in PIK3CA Gene and/or PI-3 Kinase Pathway Genes |
| NCT03292250 | Korean Cancer Study Group: Translational bIomarker Driven UMbrella Project for Head and Neck (TRIUMPH), Esophageal Squamous Cell Carcinoma- Part 1 (HNSCC)] |
| NCT02298595 | Cetuximab, Cisplatin and BYL719 for HPV-associated Oropharyngeal Squamous Cell Carcinoma |
| NCT01349933 | Akt Inhibitor MK2206 in treating patients with recurrent or metastatic Head and Neck Cancer |
| NCT01195922 | Rapamycin Therapy in Head and Neck Squamous Cell Carcinoma |
| NCT01172769 | Efficacy Study of Temsirolimus to Treat Head and Neck Cancer |
| NCT03740100 | Single-arm Study with Bimiralisib in patients with HNSCC Harboring NOTCH1 loss of function mutations |
| NCT01111058 | Everolimus versus Placebo in Head and Neck Cancer |
| NCT01051791 | Phase II Study of RAD001 Head and Neck Cancer |
| NCT01016769 | Temsirolimus + weekly Paclitaxel + Carboplatin for recurrent or metastatic Head and Neck Squamous Cell Cancer (HNSCC) |
| NCT01313390 | Everolimus and Docetaxel in treating patients with recurrent, locally advanced, or metastatic Head and Neck Cancer |
| NCT03065062 | Study of the CDK4/6 Inhibitor Palbociclib (PD-0332991) in combination with the PI3K/mTOR Inhibitor Gedatolisib (PF-05212384) for patients with Advanced Squamous Cell Lung, Pancreatic, Head & Neck and Other Solid Tumors |
Targeting P53 is an effective strategy to fight against HPV-negative HNSCC
Directly targeting P53 is good but hard to fight
Many trials have proved a worse clinical outcome in HPV-negative HNSCC. In RTOG 0129 trial, 433 patients with oropharyngeal cancer received cisplatin with radiotherapy. Compared with HPV-positive patients, the OS of HPV-positive tumor patients was remarkably worse (the 8-year survival rate was 71% vs 30%, HR 0.34, 95%CI 0.22-0.52). All the present data indicate that patients with HPV-negative tumors should be considered high-risk, and intensive, multimodal therapy is needed to avoid compromising their survival [22, 23, 25].
Genomic alterations are very common in HNSCC. HPV-negative cancers harbor significantly more mutations in the P53 gene than HPV-positive disease; meanwhile, loss-of-function variants in P53 are almost low to none in HPV-associated HNSCC [13, 55]. In HPV-positive HNSCC, E6 promotes the MDM2-independent degradation of P53. Inactivation variants of P53 driven by tobacco may not be the same as the degradation of P53 by HPV E6 [113, 114]. Therefore, HPV-positive HNSCC may contain full-of-function p53.
As a transcription factor, P53 plays a crucial role in downstream target gene transcription. Under the stress of DNA damage, proto-oncogene activation, hypoxia, and microtubule damage, P53 is activated in the process of signal transduction. As a result, cellular behaviors, such as cell cycle arrest, apoptosis, aging, and angiogenesis inhibition, are performed to keep the benignancy of the cells'. P53 repairs the abnormal chromosome distribution in cells after DNA damage and mitosis [113, 115, 116]. However, the anti-tumor route of targeting the P53 strategy is not easy in the past 50 years.
Decades ago, to overcome the loss function of mutated P53, people tried to reintroduce wild type P53 into the tumor via adenoviruses as vectors, and the virus can then selectively target and kill cancer cells. Adenovirus is safe and should be an ideal carrier for gene therapy as it can infect both mitoses and quiescent cells, and the genome keeps episomal and does not integrate into the host cells [117, 118]. Studies have shown that the introduction of the Ad-p53 gene into P53 mutated HNSCC cells can increase tumor cells' radiosensitivity [119]. Moreover, the Ad-p53 virus is active in vivo and compromise tumor growth in the HNSCC animal model. INGN 201 is the first candidate of Ad-p53 for the Phase I clinical trial. The study recruited 33 patients with recurrent HNSCC and received INGN 201 intratumor injection. In the resectable cohort, 27% of HNSCC patients remained disease-free, with a median follow-up period of 18 months after surgery. Of the 17 unresectable diseases, in 2 cases had PR, 6 cases kept an SD, and 9 cases evaluated as PD. Multiple direct intratumor injection courses of INGN 201 were well tolerated, and no dose-limiting toxicity or serious adverse events were observed [120-122]. ONYX-015 is a chimeric virus that consists of 2 species of C adenovirus genomes, serotypes 5 (Ad5) and serotype 2 (Ad2). ONYX-015 was designed to efficiently replicate in and lyse p53-deficient cells while not affect cells with wild-type p53 [123]. In a Phase II trial in recurrent or refractory HNSCC patients to evaluate the safety and effect of ONYX-015, in the hyper-dose group, 1 patient had CR, 4 patients had SD, and 2 patients had PD, there was no severe AEs observed [124, 125]. Besides, the effect of ONYX-015 was dependent on the mutant p53 status.
Loss-of-function P53 mutation is mainly located in the DNA binding domain, which effectively prevents the mutant p53 from binding to the target gene's response elements. p53-reactivating small molecules, including CP-31398 and PRIMA-1, are developed and tested in HNSCC [126]. Roh et al. report that both CP-31398 and PRIMA-1 can attenuate HNSCC cells' proliferation and exert a synergetic effect with chemotherapy agents [127]. RITA, another p53-Reactivating small molecule, directly disrupts the interaction between p53 and MDM2 and presents a potent anti-tumor impact on HNSCC, and enhances the sensitivity of cisplatin in HNSCC cells [128]. However, the cell often obtains the resistance profile after directly targeting P53 treatment very soon. Adenoviral therapeutic strategies, such as Ad-p53 and ONYX-015, and small molecular compounds, have progressed to clinical trials in the HNSCC but have shown a very mild activity [113, 122, 126, 127]. More and more trials list in Table 2 are ongoing to figure out the best candidate or partner of P53 RAs.
DNA damage responders and P53 mutation may exert synthetic lethality in HPV-negative HNSCC
Due to the PARP inhibitor's great success, scholars move their focus on hunting the synthetic lethal genes of P53. There is a complicated synthetic lethal relationship between mutated p53 and its corresponding target genes in all kinds of tumors. Based on the various research results of p53 synthetic lethality, the corresponding synthetic lethal relationship can be explained from two aspects: periodic regulatory genes and aperiodic regulatory genes [129, 130].
It is well known that p53 activation can lead to cell cycle arrest and initiate DNA repair in response to DNA damage. Key protein inhibitors that regulate DNA damage response and cell cycle progression have the potential to be synthetic lethal partners [115]. As shown in Figure 2, targeting ataxia-telangiectasia mutant (ATM), ATM-Rad3-associated (ATR), DNA-dependent protein kinase (DNA-PK), checkpoint kinase-1/2 (Chk1/2), and Wee1 kinase shows up-and-coming prospects to overcome mutant P53 HNSCC [130].
Targeting P53 is an effective strategy to fight against HPV-negative HNSCC. HPV-negative cancers harbor a mutation in the P53 gene as a result of tobacco and alcohol consumption. DNA damage occurs in p53 wild-type cells; the cell cycle checkpoints of G1 and G2/M are activated, which prevents the accumulation of DNA damage and may induce senescence. DNA damage repair carries out cell cycle arrest and initiates by acting on G1 and G2/M cell cycle checkpoints. In p53 mutated tumor cells, due to the lack of G1 checkpoint regulated by p53, it is more dependent on the G2-M cell cycle checkpoint when DNA is damaged. When p53 mutated cells are injured by chemotherapy or radiotherapy, blocking the G2/M cell cycle checkpoint push the unrepaired chromosome into M phase and result in a mitosis catastrophe.
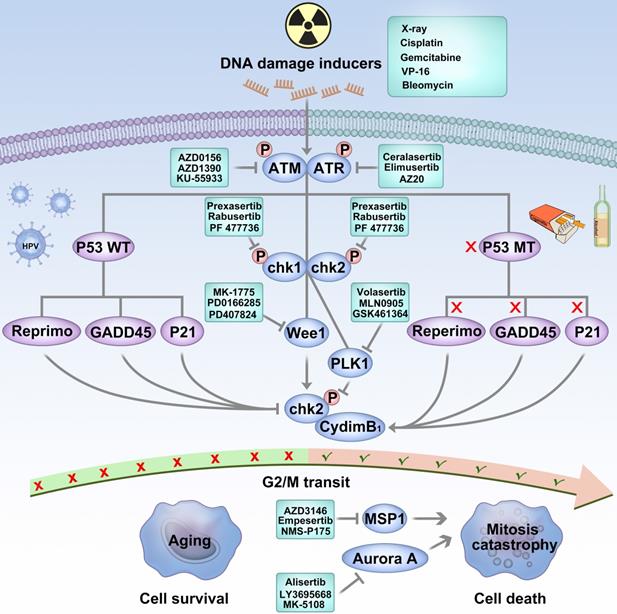
ATM and ATR are members of the phosphatidylinositol 3-kinase-related kinase (PIKK) family of serine/threonine protein kinases. ATM and ATR are two critical factors to initiate the DNA damage response as a DNA damage sensor. ATM and ATR act as primary regulators of double-strand breaks and DNA replication stress, respectively. These two kinases perform functionally overlapping but non-redundant activities [131, 132]. We have a strong rationale to explore ATR inhibitors' anti-tumor efficacy in p53-mutated tumors within certain clinical settings. The dual loss of ATR and p53 function in adult mice is demonstrated to lead to defective hair follicle and tissue regeneration [133]. In cancer cells treated with DNA-damaging agents, synergistic effects between ATR and TP53 have also been observed. Dual inhibitory of TP53 and ATR comes down in global loss of DNA damage checkpoints and exert a synthetic lethality [134]. As a DBS response sensor, the preclinical data has presented a potent synergistic effect when an ATM inhibitor combines with radiotherapy in P53 mutated cells [135]. Currently, the small-molecule inhibitors of ATR, ATM are developed and progresses to I/II phase clinical trial.
As major regulators of the ATM/ATR pathway, Chk1 and Chk2 kinases are functionally overlapping activated in response to DNA replication stress, cell cycle progression, chromatin remodeling, and apoptosis [136, 137]. Basic research indicates that Chk1/2 faithfully regulates DNA repair and replication when P53 deficiency occurs [134]. Gadhikar et al. used Chk1/2 inhibitor AZD7762 in mutant p53 OSCC cells. Due to the out-of-control G2/M checkpoint, AZD7762 and cisplatin pushed the cell into a mitotic catastrophe [138]. Several early phase clinical trials on Chk1/2 inhibitors have been conducted in HNSCC.
When DNA damage occurred in P53 wild type cells, the cell cycle checkpoints of G1 and G2/M are activated and prevents the accumulation of DNA damage. In P53 mutated cells, due to the dysfunction of the G/1 checkpoint, the G2/M cell cycle checkpoint is crucial when DNA is damaged [115, 116]. In mutated p53 cells, DNA damage repair is predominantly regulated by Wee1. It can mediate the activation of the G2/M cell cycle checkpoint, inhibit the phosphorylation of cyclin-dependent kinase 1 (Cyclin-dependent kinase 1, CDK1), and block the progression of the tumor cell cycle. Thus, the Wee1 kinase inhibitory may sensitize p53 mutant cancer cells to DNA-damaging therapy [139, 140]. Many articles have proved that AZD1775, a selective and potent wee1 inhibitor, abrogated the G2 checkpoint and selectively sensitized p53 mutant cancer cells to DNA-damaging inducers, such as gemcitabine [141], cisplatin [142], and X-ray [143]. Considering these findings, AZD1775 has been tested in a phase I/II clinical trial in patients with advanced solid tumors and showed well-tolerance and promising therapeutic effects [144, 145]. Osman et al. used AZD1775 in HPV-negative HNSCC cells and reversed the cisplatin resistance [138]. Intriguingly, Tanaka et al. reported that AZD1775 monotherapy potentiates the cisplatin response of HPV-positive HNSCC cells. Unlike HPV-negative OSCC cells, AZD1775 induces apoptosis triggered by selective cleavage of the antiapoptotic proteins MCl-1 and XIAP [146]. Busch et al. revealed that the wee1 and chk1 blockade enhanced the radiation sensitivity in HPV-positive cells, and FOXMI could be a catalyst between AZD1775 and X-ray HPV-positive cells [147, 148]. However, most present evidence based on basic research requires further clinical trials to investigate the safety and effect of AZD1775 further. Table 3 listed all the clinical trials targeting P53 and DNA damage responder inhibitors in HNSCC.
Clinical trials targeting P53 and DNA damage responder inhibitor in NHSCC
| Number | Project name |
|---|---|
| NCT02842125 | Safety and Efficacy of Intra-Arterial and intra-tumoral Ad-p53 with Capecitabine (Xeloda) or Anti-PD-1 in Liver Metastases of Solid Tumors and recurrent Head and Neck Squamous Cell Cancer |
| NCT03544723 | Safety and efficacy of p53 Gene Therapy combined with Immune Checkpoint Inhibitors in Solid Tumors. |
| NCT00003257 | Gene Therapy in treating patients with recurrent Head and Neck Cancer |
| NCT02432963 | Vaccine Therapy and Pembrolizumab in treating patients with Solid Tumors that have failed prior therapy |
| NCT00017173 | S0011, Gene Therapy & Surgery Followed by Chemo & RT in Newly Diagnosed Cancer of the Mouth or Throat |
| NCT00404339 | Vaccine Therapy in treating patients with Head and Neck Cancer |
| NCT00041613 | Study to compare the Overall Survival of patients receiving INGN 201 (Study Drug) with patients receiving Methotrexate |
| NCT00041626 | Effectiveness and Safety of INGN 201 in combination with Chemotherapy versus Chemotherapy Alone |
| NCT02567422 | M6620, Cisplatin and Radiation Therapy in treating patients with locally advanced Head and Neck Squamous Cell Carcinoma |
| NCT04576091 | Testing the Addition of an Anti-cancer Drug, BAY1895344, with Radiation Therapy to the usual Pembrolizumab Treatment for Recurrent Head and Neck Cancer |
| NCT03022409 | A Study to Investigate Biomarker effects of pre-surgical treatment with DNA Damage Repair (DDR) agents in patients with Head and Neck Squamous Cell Carcinoma (HNSCC) |
| NCT01275183 | Pilot Study of Raltegravir and Cisplatin in Squamous Cell Carcinoma of Head and Neck |
| NCT01115790 | A Phase 1 study in participants with Advanced Cancer |
| NCT02797964 | A Phase 1/2 trial of SRA737 in subjects with Advanced Cancer |
| NCT02508246 | WEE1 Inhibitor MK-1775, Docetaxel, and Cisplatin before surgery in treating patients with Borderline Resectable Stage III-IVB Squamous Cell Carcinoma of the Head and Neck |
| NCT02585973 | Dose-escalating AZD1775 + Concurrent Radiation + Cisplatin for Intermediate/High Risk HNSCC |
| NCT03028766 | WEE1 inhibitor with Cisplatin and Radiotherapy: A trial in Head and Neck Cancer |
| NCT02196168 | Cisplatin with or without WEE1 inhibitor MK-1775 in treating patients with Recurrent or Metastatic Head and Neck Cancer |
HPV-associated disease trend to obtain more benefit from immunotherapy
Landscape of microenvironment of different HPV-status HNSCC
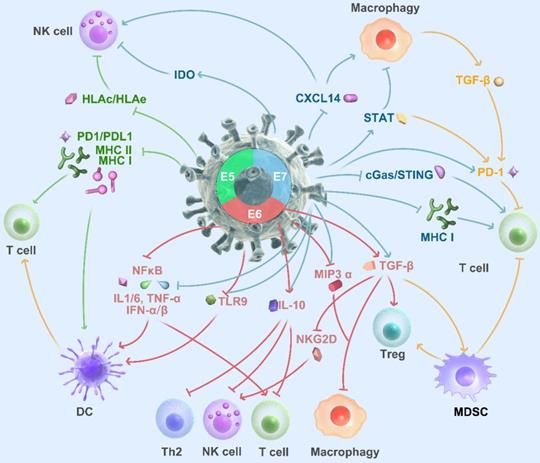
Tumor-infiltrating lymphocytes are associated with improved prognosis. HNSCC arises from squamous epithelium associated with the tongue's tonsils and base and is deemed to have more immune cells infiltrated within the tumor microenvironment. HPV infection alters immune cell, which infiltrates in HNSCC to encompass diverse and heterogeneous landscapes [36].
HPV viral factors, E5, E6, and E7, play a crucial role in generating an immunosuppressive microenvironment that promotes tumor progression. Oncoprotein E5 blocks HLA-C and HLA-E from tumor stroma to interact with MHC I on the cancer cell, impairing T cell and NK cells' activity. [149]. Moreover, E5 attenuates MHC class II's expression and stability by blocking peptide loading and transportation. By interfering with MHC, E5 may severely impair antigen processing and T cell activation [150]. In the past 20 years, scholars get extensive knowledge about E6 and E7. The mechanism of E6 and E7 to regulate the tumor microenvironment is not linear. Briefly, in 2012, Vandermark et al. reported that E6 and E7 proteins alter the NF-kB pathway in tumor cells, impair the innate immunosystem, and evade supervision [151]. E6 and E7 can interact with keratinocytes and inhibit macrophage infiltration by decreasing the secretion of MIP-3α [152]. IL-10 is a double-edged sword, and it can exert both pro-tumor and anti-tumor effects, including hiding the MHC, repression of DCs, and activation of NK and T cells. Poved et al. reported, HPV E6 and E7 can bind to the promoter of IL10 and enhance the expression of IL10. What is more, IL10 can promote the expression of E6 and E7 that create positive feedback for an immunosuppressive environment [153, 154]. Toll-like receptor 9 (TLR9) expresses on the surface of dendritic cells, macrophages, natural killer cells, and initiates the signals that cytokines' production is needed for innate and acquired immunity. HPV E7 induces a transcriptional repressor complex on the TLR9 promotor and abolishes its expression and function. HPV E6 also blocks TLR9 by NFκB pathway [155-157]. Transforming growth factor beta (TGF-β) is a multifunctional cytokine, which can regulate the inflammation process of tumor, polarize the macrophagy and stimulate the myeloid-derived suppressor cell (MDSC) [158]. E6 and E7 enable to activate the TGF-β promoter throughout the Sp1 recognition sequence [159]. E5 and E7 play different role in PD1/PDL1 axis. Briefly, E5 mediates resistance to PD-L1 Blockade by hiding the MHC [160]. Chaoqi Liu exogenously expressed E7 on PC3 cells and led to a PDL1 increase [161]. Furthermore, HPV E7 also can regulate IDO [162], CXCL14 [163], and c-GAS-STING [164] to generate an immunosuppressive microenviroment.
Mandal et al. reported in an analysis of TCGA found that HPV-positive HNSCCs had more significant immune infiltration and had higher T cell levels with granzyme and perforin [165]. Tregs and NK cells were also frequently infiltrated in HPV positive tissues and associated with a favorable prognosis [166, 167]. Wood et al. identified increased expression of genes encoding PD1, CTLA-4, and TIM-3 in HPV-positive HNSCC [168], which suggests patients with HPV-positive status may obtain more benefit from immune checkpoints blockade (Figure 3).
HPV infection alters immune cell infiltrates in HNSCC. HPV E5, ER6, E7 module HNSCC microenvironment. Briefly, E5 interacts with HLA and MHCII on tumor cells to inhibit the DC and T cell activation; E6, E7 impair the NFκB pathway and regulates the immune cell infiltrate; IL-10 creates a loop with E6 and E7; E7 also regulates CXCL14 and c-GAS/STING pathway to modulate the immune cell infiltration.
Promising but not sure, HPV-positive HNSCC could obtain more benefit from immunotherapy
Promising clinical trial results led to pembrolizumab's FDA approval for treating refractory or metastatic HNSCC in 2016. Keynote 012 study was the first clinical trial to evaluate the relationship between HPV status and immune checkpoints blockade response. 60 patients with PD-L1+ (> 1%) were treated with pembrolizumab. ORR was 24% (95% CI, 13-40%) in those patients with HPV positive, and 16% in HPV negative patients (95% CI, 10-23%, P > 0.05) [169].
In the phase III Checkmate 141 study, nivolumab obtained a better OS than standard treatment (median OS: 7.5 months vs 5.1 months, HR: 0.70; 97.73% CI, 0.51-0.96; P=0.01), with an approximately 19% increase for 1-year survival. In the Checkmate 141 subgroup analysis, patients with p16 positive tumors tend to obtain more benefit from nivolumab than the standard treatment group (median OS: 9.1 months vs. 4.4 months HR: 0.56; 95% CI: 0.32 to 0.99). On the other hand, in patients with p16 negative disease, the median OS was 7.5 months in the nivolumab monotherapy group and 5.8 months in the standard treatment group (HR: 0.73; 95% CI: 0.42 to 1.25) [170].
However, the advantage of HPV status in nivolumab treatment failed to exhibit in pembrolizumab. In phase II KEYNOTE-055 trial, ORR was similar regardless of HPV status, with rates of 16% in HPV-associated disease and 15% in HPV-negative disease. However, medium PFS and OS did not differ based on HPV status [171]. The KEYNOTE-040 trial design is the same model as checkmate 141. The median OS in the pembrolizumab group was 8.4 months, compared with 6.9 months in the standard treatment group. Although the trial failed to reach the final endpoint, the data is still promising. Patients with p16-negative disease in the pembrolizumab group had more prolonged overall survival than the standard treatment group (HR: 0.77; 95%CI: 0.61-0.97), whereas this survival benefit did not present among patients with the p16-positive disease (HR: 0.97; 95%CI: 0.63-1.49) [172-174]. KEYNOTE-048 trial followed up to KEYNOTE-040; the data showed that the treatment with pembrolizumab significantly improved OS compared with the EXTREME regimen in patients with PD-L1 expression (CPS ≥ 1 and CPS ≥ 20 arms). From the date, PD-L1 expression is the only reliable biomarker for HNSCC to receive pembrolizumab. While approximately 21% of patients in this study were p16-positive, they were not analyzed separately as the HPV status may not tell any difference [175].
Atezolizumab is an anti-PD-L1 mAb. In a phase I trial, 32 patients with R/M HNSCC were enrolled. The data were in line with anti-PD1 mAb. Atezolizumab monotherapy had a 22% ORR rate, mPFS of 2.6 months, and mOS of 6 months. However, this early trial's primary date displayed the effect of Atezolizumab, regardless of HPV status or PD-L1 expression level [176]. Durvalumab is another anti-PDL1 and rises a Pacific storm in NSCLC. The Phase III EAGLE trial data shows the HPV-positive did not affect durvalumab [177], and patients with negative were associated with worse OS. Avelumab is a fully human anti-PD-L1. The IgG1 construction can competitively block with PD-1; however, the trial on HNSCC prematurely terminated as recommended because the boundary for futility has been crossed (NCT02952586).
Although many retrospective or bio-informative studies indicated a higher degree of T cells in curium titration in HPV-associated HNSCC, the only nivolumab showed HPV+HNSCC could obtain more benefit from ICI therapy. The present clinical data indicate that HPV status does not support strategic treatment with checkpoint inhibitors.
Epidrugs have a long way to go in NHSCC
The imbalance, mutation, and aberrant expression of epigenetic regulatory factors boost carcinogenesis and maintain the growth and metastasis of HNSCC. Also, the levels of these epigenetic regulators varied with the type of HNSCC. From 2004, the US FDA approved only 6 epidrugs in the clinic: 2 DNMT inhibitors of azacitidine and decitabine; 4 HDAC inhibitors of Vorinostat, Romidepsin, Farydak, and Panobinostat. Many other agents are in the pipeline.
Although scientists have done much work on the epigenetic regulation of HNSCC, rare epidrugs are in clinical trials. Many studies report that HPV-HNSCC has relatively low DNA methylation levels and promotes genomic instability. Meanwhile, HPV+ HNSCC harbors distinctly hypermethylated genomes. The diversity of molecular and epigenetic between HPV+ and HPV- tumors provides a therapeutic strategy that forces the demethylation of genomes of HPV associated NHSCC.
DNMT inhibitors/DNA methylation agents present the cytotoxicity on HPV status bias. Asel Biktasova et al. reported that HPV+ HNSCC cells are sensitive to azacitidine partially due to stabilization of p53 and attenuation of the expression of HPV genes. Moreover, azacytidine is sufficient for suppressing cell invasion and sensitizing the cell to interferons. [178]. However, in 2019, Ricard Mesia posted that azacitidine had an insufficient activity to further study in advanced NPC [179].
CUDC-101 is a small molecule that simultaneously blocks the EGFR, human growth factor receptor 2 (HER2), and histone deacetylase (HDAC) with promising activity in HNSCC. Thomas Galloway et al. reveals that CUDC-101 is tolerable in a phase I trial; however, a high discontinuation rate suggests CUDC-10 needs to alternate the schedules or routes of administration [180, 181]. NCT02178072 is an ongoing clinical trial to evaluate CUDC-10 with Azacitidine. Asel Biktasova et al. reports that single-agent epidrug rarely presents therapeutic effects in NHSCC; most drugs cease the clinical trial before phase II [182]. Oncologists move their efforts to combine epidrugs with radiotherapy or chemotherapy. Panobinostat, an HDAC inhibitor, has been identified as a tolerable drug and exerts the effectiveness of erlotinib in HNSCC [183]. Valproic acid, a drug for epilepsy, has a potent effect on blocking histone deacetylase activity. A phase II clinical study showed that Valproic acid plus cisplatin and cetuximab exhibits a less toxic and more effective than standard first-line regimen in advanced HNSCC [184]. Other HADC inhibitors, including Vorinostat, Belinostat, Panobinostat, present the therapeutic effect in HNSCC without HPV bias. HNSCC Oncologist is looking for an optimal combination with epidrugs, and we believe more results about epidrugs in HNSCC will be posted in the next few years; however, our point of view does not support epidrug could exert the effects on HPV bias.
To sum up, we review the conditional therapy, including chemotherapy and radiotherapy, and indicate that HPV-positive patients may obtain more benefit from traditional treatment. More clinical trials need to be investigated for the optimal dosage for HPV-positive patients. PI3K is a promising target in HPV-positive HNSCC; however, the toxicity of combination therapy with PI3K inhibitor should not be ignored. Preclinical data present a suspected result of directly targeting P53, and synthetic lethality with DNA damage responder inhibitors remains too much to be explored. Immunotherapy is effective for HNSCC regardless of HPV status. At the present stage, as the same before 5 years, the HPV status can predict the prognosis more but cannot change the clinical decision-making. More prospective clinical trials are ongoing, which may answer critical questions over the next three to five years.
Acknowledgements
This study was supported by the National R&D Program of China (2019YFB1311300), The Technology Innovation Foundation of Dalian (2020JJ27SN080), Natural Science Foundation of Fujian Province (No. 2020J01126), National Natural Science Foundation of China (NSFC) (82072986).
Author Contributions
Original draft preparation: Yingming Sun; Review and edit: Yingming Sun, Zhe Wang, Sufang Qiu, Ruoyu Wang; Visualization: Yingming Sun. All authors have read and agreed to the published version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF. et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363:24-35
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7-30
3. Akhtar S, Sheikh AA, Qureshi HU. Chewing areca nut, betel quid, oral snuff, cigarette smoking and the risk of oesophageal squamous-cell carcinoma in South Asians: a multicentre case-control study. Eur J Cancer. 2012;48:655-61
4. Applebaum KM, McClean MD, Nelson HH, Marsit CJ, Christensen BC, Kelsey KT. Smoking modifies the relationship between XRCC1 haplotypes and HPV16-negative head and neck squamous cell carcinoma. Int J Cancer. 2009;124:2690-6
5. Brennan JA, Boyle JO, Koch WM, Goodman SN, Hruban RH, Eby YJ. et al. Association between cigarette smoking and mutation of the p53 gene in squamous-cell carcinoma of the head and neck. N Engl J Med. 1995;332:712-7
6. Lewin F, Norell SE, Johansson H, Gustavsson P, Wennerberg J, Biorklund A. et al. Smoking tobacco, oral snuff, and alcohol in the etiology of squamous cell carcinoma of the head and neck: a population-based case-referent study in Sweden. Cancer. 1998;82:1367-75
7. Zhang ZF, Morgenstern H, Spitz MR, Tashkin DP, Yu GP, Hsu TC. et al. Environmental tobacco smoking, mutagen sensitivity, and head and neck squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. 2000;9:1043-9
8. Bratman SV, Bruce JP, O'Sullivan B, Pugh TJ, Xu W, Yip KW. et al. Human Papillomavirus Genotype Association with Survival in Head and Neck Squamous Cell Carcinoma. JAMA Oncol. 2016;2:823-6
9. Klussmann JP, Weissenborn S, Fuchs PG. Human papillomavirus infection as a risk factor for squamous-cell carcinoma of the head and neck. N Engl J Med. 2001;345:376 author reply 7
10. Kamel AH, el B, Hashish MH, el-Sheikh SM. Epstein-Barr virus in head and neck squamous cell carcinoma. East Mediterr Health J. 2003;9:364-71
11. Karbalaie Niya MH, Safarnezhad Tameshkel F, Keyvani H, Esghaei M, Panahi M, Zamani F. et al. Epstein-Barr virus molecular epidemiology and variants identification in head and neck squamous cell carcinoma. Eur J Cancer Prev. 2020;29:523-30
12. Sisk EA, Bradford CR, Carey TE, Paulino A, Robertson E. Epstein-Barr virus detected in a head and neck squamous cell carcinoma cell line derived from an immunocompromised patient. Arch Otolaryngol Head Neck Surg. 2003;129:1115-24
13. Gillison ML, Chaturvedi AK, Anderson WF, Fakhry C. Epidemiology of Human Papillomavirus-Positive Head and Neck Squamous Cell Carcinoma. J Clin Oncol. 2015;33:3235-42
14. Gillison ML. Human papillomavirus and prognosis of oropharyngeal squamous cell carcinoma: implications for clinical research in head and neck cancers. J Clin Oncol. 2006;24:5623-5
15. Egawa N, Egawa K, Griffin H, Doorbar J. Human Papillomaviruses; Epithelial Tropisms, and the Development of Neoplasia. Viruses. 2015;7:3863-90
16. Wuerdemann N, Wittekindt C, Sharma SJ, Prigge ES, Reuschenbach M, Gattenlohner S. et al. Risk Factors for Overall Survival Outcome in Surgically Treated Human Papillomavirus-Negative and Positive patients with Oropharyngeal Cancer. Oncol Res Treat. 2017;40:320-7
17. Granata R, Miceli R, Orlandi E, Perrone F, Cortelazzi B, Franceschini M. et al. Tumor stage, human papillomavirus and smoking status affect the survival of patients with oropharyngeal cancer: an Italian validation study. Ann Oncol. 2012;23:1832-7
18. Adjei Boakye E, Buchanan P, Hinyard L, Osazuwa-Peters N, Schootman M, Piccirillo JF. Incidence and Risk of Second Primary Malignant Neoplasm After a First Head and Neck Squamous Cell Carcinoma. JAMA Otolaryngol Head Neck Surg. 2018;144:727-37
19. Wai KC, Strohl MP, van Zante A, Ha PK. Molecular Diagnostics in Human Papillomavirus-Related Head and Neck Squamous Cell Carcinoma. Cells. 2020 9
20. Semrau R, Duerbaum H, Temming S, Huebbers C, Stenner M, Drebber U. et al. Prognostic impact of human papillomavirus status, survivin, and epidermal growth factor receptor expression on survival in patients treated with radiochemotherapy for very advanced nonresectable oropharyngeal cancer. Head Neck. 2013;35:1339-44
21. Wang H, Zhang Y, Bai W, Wang B, Wei J, Ji R. et al. Feasibility of Immunohistochemical p16 Staining in the Diagnosis of Human Papillomavirus Infection in patients with Squamous Cell Carcinoma of the Head and Neck: A Systematic Review and Meta-Analysis. Front Oncol. 2020;10:524928
22. Xiao C, Hanlon A, Zhang Q, Movsas B, Ang K, Rosenthal DI. et al. Risk factors for clinician-reported symptom clusters in patients with advanced head and neck cancer in a phase 3 randomized clinical trial: RTOG 0129. Cancer. 2014;120:848-54
23. Galloway TJ, Zhang QE, Nguyen-Tan PF, Rosenthal DI, Soulieres D, Fortin A. et al. Prognostic Value of p16 Status on the Development of a Complete Response in Involved Oropharynx Cancer Neck Nodes After Cisplatin-Based Chemoradiation: A Secondary Analysis of NRG Oncology RTOG 0129. Int J Radiat Oncol Biol Phys. 2016;96:362-71
24. Xiao C, Zhang Q, Nguyen-Tan PF, List M, Weber RS, Ang KK. et al. Quality of Life and Performance Status From a Substudy Conducted within a Prospective Phase 3 Randomized Trial of Concurrent Standard Radiation Versus Accelerated Radiation Plus Cisplatin for Locally Advanced Head and Neck Carcinoma: NRG Oncology RTOG 0129. Int J Radiat Oncol Biol Phys. 2017;97:667-77
25. Fakhry C, Zhang Q, Gillison ML, Nguyen-Tan PF, Rosenthal DI, Weber RS. et al. Validation of NRG oncology/RTOG-0129 risk groups for HPV-positive and HPV-negative oropharyngeal squamous cell cancer: Implications for risk-based therapeutic intensity trials. Cancer. 2019;125:2027-38
26. Lassen P, Overgaard J, Eriksen JG. Expression of EGFR and HPV-associated p16 in oropharyngeal carcinoma: correlation and influence on prognosis after radiotherapy in the randomized DAHANCA 5 and 7 trials. Radiother Oncol. 2013;108:489-94
27. Rischin D, Peters L, Fisher R, Macann A, Denham J, Poulsen M. et al. Tirapazamine, Cisplatin, and Radiation versus Fluorouracil, Cisplatin, and Radiation in patients with locally advanced head and neck cancer: a randomized phase II trial of the Trans-Tasman Radiation Oncology Group (TROG 98.02). J Clin Oncol. 2005;23:79-87
28. Rischin D, Peters LJ, O'Sullivan B, Giralt J, Fisher R, Yuen K. et al. Tirapazamine, cisplatin, and radiation versus cisplatin and radiation for advanced squamous cell carcinoma of the head and neck (TROG 02.02, HeadSTART): a phase III trial of the Trans-Tasman Radiation Oncology Group. J Clin Oncol. 2010;28:2989-95
29. Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. Head and neck squamous cell carcinoma. Nat Rev Dis Primers. 2020;6:92
30. Howard J, Dwivedi RC, Masterson L, Kothari P, Quon H, Holsinger FC. De-intensified adjuvant (chemo)radiotherapy versus standard adjuvant chemoradiotherapy post transoral minimally invasive surgery for resectable HPV-positive oropharyngeal carcinoma. Cochrane Database Syst Rev. 2018;12:CD012939
31. Seiwert TY, Foster CC, Blair EA, Karrison TG, Agrawal N, Melotek JM. et al. OPTIMA: a phase II dose and volume de-escalation trial for human papillomavirus-positive oropharyngeal cancer. Ann Oncol. 2019;30:297-302
32. Ferris RL, Flamand Y, Holsinger FC, Weinstein GS, Quon H, Mehra R. et al. A novel surgeon credentialing and quality assurance process using transoral surgery for oropharyngeal cancer in ECOG-ACRIN Cancer Research Group Trial E3311. Oral Oncol. 2020;110:104797
33. Gillison ML, Trotti AM, Harris J, Eisbruch A, Harari PM, Adelstein DJ. et al. Radiotherapy plus cetuximab or cisplatin in human papillomavirus-positive oropharyngeal cancer (NRG Oncology RTOG 1016): a randomised, multicentre, non-inferiority trial. Lancet. 2019;393:40-50
34. Mehanna H, Robinson M, Hartley A, Kong A, Foran B, Fulton-Lieuw T. et al. Radiotherapy plus cisplatin or cetuximab in low-risk human papillomavirus-positive oropharyngeal cancer (De-ESCALaTE HPV): an open-label randomised controlled phase 3 trial. Lancet. 2019;393:51-60
35. Deschuymer S, Mehanna H, Nuyts S. Toxicity Reduction in the Treatment of HPV Positive Oropharyngeal Cancer: Emerging Combined Modality Approaches. Front Oncol. 2018;8:439
36. Wijetunga NA, Yu Y, Morris LG, Lee N, Riaz N. The head and neck cancer genome in the era of immunotherapy. Oral Oncol. 2020;112:105040
37. Hwang ES, Nottoli T, Dimaio D. The HPV16 E5 protein: expression, detection, and stable complex formation with transmembrane proteins in COS cells. Virology. 1995;211:227-33
38. Crusius K, Rodriguez I, Alonso A. The human papillomavirus type 16 E5 protein modulates ERK1/2 and p38 MAP kinase activation by an EGFR-independent process in stressed human keratinocytes. Virus Genes. 2000;20:65-9
39. Zhang B, Srirangam A, Potter DA, Roman A. HPV16 E5 protein disrupts the c-Cbl-EGFR interaction and EGFR ubiquitination in human foreskin keratinocytes. Oncogene. 2005;24:2585-8
40. Ilahi NE, Bhatti A. Impact of HPV E5 on viral life cycle via EGFR signaling. Microb Pathog. 2020;139:103923
41. Stoehlmacher-Williams J, Obermann L, Ehninger G, Goekkurt E. Polymorphisms of the epidermal growth factor receptor (EGFR) and survival in patients with advanced cancer of the head and neck (HNSCC). Anticancer Res. 2012;32:421-5
42. Nagalakshmi K, Jamil K, Pingali U, Reddy MV, Attili SS. Epidermal growth factor receptor (EGFR) mutations as biomarker for head and neck squamous cell carcinomas (HNSCC). Biomarkers. 2014;19:198-206
43. Fernandez-Mateos J, Seijas-Tamayo R, Mesia R, Taberna M, Pastor Borgonon M, Perez-Ruiz E. et al. Epidermal growth factor receptor (EGFR) pathway polymorphisms as predictive markers of cetuximab toxicity in locally advanced head and neck squamous cell carcinoma (HNSCC) in a Spanish population. Oral Oncol. 2016;63:38-43
44. Koi L, Lock S, Linge A, Thurow C, Hering S, Baumann M. et al. EGFR-amplification plus gene expression profiling predicts response to combined radiotherapy with EGFR-inhibition: A preclinical trial in 10 HNSCC-tumour-xenograft models. Radiother Oncol. 2017;124:496-503
45. Swick AD, Prabakaran PJ, Miller MC, Javaid AM, Fisher MM, Sampene E. et al. Cotargeting mTORC and EGFR Signaling as a Therapeutic Strategy in HNSCC. Mol Cancer Ther. 2017;16:1257-68
46. Vermorken JB, Stohlmacher-Williams J, Davidenko I, Licitra L, Winquist E, Villanueva C. et al. cisplatin and fluorouracil with or without panitumumab in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck (SPECTRUM): an open-label phase 3 randomised trial. Lancet Oncol. 2013;14:697-710
47. Rischin D, Spigel DR, Adkins D, Wein R, Arnold S, Singhal N. et al. PRISM: Phase 2 trial with panitumumab monotherapy as second-line treatment in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. Head Neck. 2016;38(Suppl 1):E1756-61
48. Seiwert TY, Fayette J, Cupissol D, Del Campo JM, Clement PM, Hitt R. et al. A randomized, phase II study of afatinib versus cetuximab in metastatic or recurrent squamous cell carcinoma of the head and neck. Ann Oncol. 2014;25:1813-20
49. Chung CH, Rudek MA, Kang H, Marur S, John P, Tsottles N. et al. A phase I study afatinib/carboplatin/paclitaxel induction chemotherapy followed by standard chemoradiation in HPV-negative or high-risk HPV-positive locally advanced stage III/IVa/IVb head and neck squamous cell carcinoma. Oral Oncol. 2016;53:54-9
50. Rivera F, Garcia-Castano A, Vega N, Vega-Villegas ME, Gutierrez-Sanz L. Cetuximab in metastatic or recurrent head and neck cancer: the EXTREME trial. Expert Rev Anticancer Ther. 2009;9:1421-8
51. Vermorken JB, Psyrri A, Mesia R, Peyrade F, Beier F, de Blas B. et al. Impact of tumor HPV status on outcome in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck receiving chemotherapy with or without cetuximab: retrospective analysis of the phase III EXTREME trial. Ann Oncol. 2014;25:801-7
52. Wirth LJ, Dakhil S, Kornek G, Axelrod R, Adkins D, Pant S. et al. PARTNER: An open-label, randomized, phase 2 study of docetaxel/cisplatin chemotherapy with or without panitumumab as first-line treatment for recurrent or metastatic squamous cell carcinoma of the head and neck. Oral Oncol. 2016;61:31-40
53. Saloura V, Cohen EE, Licitra L, Billan S, Dinis J, Lisby S. et al. An open-label single-arm, phase II trial of zalutumumab, a human monoclonal anti-EGFR antibody, in patients with platinum-refractory squamous cell carcinoma of the head and neck. Cancer Chemother Pharmacol. 2014;73:1227-39
54. Al-Saleh K, El-Sherify M, Safwat R, Elbasmy A, Shete J, Hussein A. et al. Phase II/III Randomized Controlled Trial of Concomitant Hyperfractionated Radiotherapy plus Cetuximab (Anti-EGFR Antibody) or Chemotherapy in Locally Advanced Head and Neck Cancer. Gulf J Oncolog. 2019;1:6-12
55. Seiwert TY, Zuo Z, Keck MK, Khattri A, Pedamallu CS, Stricker T. et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin Cancer Res. 2015;21:632-41
56. Lui VW, Hedberg ML, Li H, Vangara BS, Pendleton K, Zeng Y. et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013;3:761-9
57. Mazumdar T, Byers LA, Ng PK, Mills GB, Peng S, Diao L. et al. A comprehensive evaluation of biomarkers predictive of response to PI3K inhibitors and of resistance mechanisms in head and neck squamous cell carcinoma. Mol Cancer Ther. 2014;13:2738-50
58. Rampias T, Giagini A, Siolos S, Matsuzaki H, Sasaki C, Scorilas A. et al. RAS/PI3K crosstalk and cetuximab resistance in head and neck squamous cell carcinoma. Clin Cancer Res. 2014;20:2933-46
59. Isaacsson Velho PH, Castro G Jr, Chung CH. Targeting the PI3K Pathway in Head and Neck Squamous Cell Carcinoma. Am Soc Clin Oncol Educ Book. 2015:123-8
60. Horn D, Hess J, Freier K, Hoffmann J, Freudlsperger C. Targeting EGFR-PI3K-AKT-mTOR signaling enhances radiosensitivity in head and neck squamous cell carcinoma. Expert Opin Ther Targets. 2015;19:795-805
61. Simpson DR, Mell LK, Cohen EE. Targeting the PI3K/AKT/mTOR pathway in squamous cell carcinoma of the head and neck. Oral Oncol. 2015;51:291-8
62. Pfisterer K, Fusi A, Klinghammer K, Knodler M, Nonnenmacher A, Keilholz U. PI3K/PTEN/AKT/mTOR polymorphisms: association with clinical outcome in patients with head and neck squamous cell carcinoma receiving cetuximab-docetaxel. Head Neck. 2015;37:471-8
63. Stankiewicz E, Prowse DM, Ng M, Cuzick J, Mesher D, Hiscock F. et al. Alternative HER/PTEN/Akt pathway activation in HPV positive and negative penile carcinomas. PLoS One. 2011;6:e17517
64. Benderli Cihan Y. Could PTEN be a biomarker in head and neck cancers subjected to radiotherapy as similar to HPV? J BUON. 2018;23:1560
65. Tang JY, Li DY, He L, Qiu XS, Wang EH, Wu GP. HPV 16 E6/E7 Promote the Glucose Uptake of GLUT1 in Lung Cancer Through Downregulation of TXNIP Due to Inhibition of PTEN Phosphorylation. Front Oncol. 2020;10:559543
66. Howie HL, Koop JI, Weese J, Robinson K, Wipf G, Kim L. et al. Beta-HPV 5 and 8 E6 promote p300 degradation by blocking AKT/p300 association. PLoS Pathog. 2011;7:e1002211
67. Zhang B, Song Y, Sun S, Han R, Hua C, van der Veen S. et al. Human Papillomavirus 11 Early Protein E6 Activates Autophagy by Repressing AKT/mTOR and Erk/mTOR. J Virol. 2019 93
68. Moon MS, Lee CJ, Um SJ, Park JS, Yang JM, Hwang ES. Effect of BPV1 E2-mediated inhibition of E6/E7 expression in HPV16-positive cervical carcinoma cells. Gynecol Oncol. 2001;80:168-75
69. Zheng L, Ding H, Lu Z, Li Y, Pan Y, Ning T. et al. E3 ubiquitin ligase E6AP-mediated TSC2 turnover in the presence and absence of HPV16 E6. Genes Cells. 2008;13:285-94
70. Drews CM, Case S, Vande Pol SB. E6 proteins from high-risk HPV, low-risk HPV, and animal papillomaviruses activate the Wnt/beta-catenin pathway through E6AP-dependent degradation of NHERF1. PLoS Pathog. 2019;15:e1007575
71. Barbosa MS, Edmonds C, Fisher C, Schiller JT, Lowy DR, Vousden KH. The region of the HPV E7 oncoprotein homologous to adenovirus E1a and Sv40 large T antigen contains separate domains for Rb binding and casein kinase II phosphorylation. EMBO J. 1990;9:153-60
72. Stoppler H, Stoppler MC, Adduci A, Koval D, Schlegel R. The serine protease inhibitors TLCK and TPCK react with the RB-binding core of HPV-18 E7 protein and abolish its RB-binding capability. Virology. 1996;217:542-53
73. Wang J, Sampath A, Raychaudhuri P, Bagchi S. Both Rb and E7 are regulated by the ubiquitin proteasome pathway in HPV-containing cervical tumor cells. Oncogene. 2001;20:4740-9
74. Menges CW, Baglia LA, Lapoint R, McCance DJ. Human papillomavirus type 16 E7 up-regulates AKT activity through the retinoblastoma protein. Cancer Res. 2006;66:5555-9
75. Strickland SW, Vande Pol S. The Human Papillomavirus 16 E7 Oncoprotein Attenuates AKT Signaling To Promote Internal Ribosome Entry Site-Dependent Translation and Expression of c-MYC. J Virol. 2016;90:5611-21
76. Zhang L, Wu J, Ling MT, Zhao L, Zhao KN. The role of the PI3K/Akt/mTOR signalling pathway in human cancers induced by infection with human papillomaviruses. Mol Cancer. 2015;14:87
77. Marquard FE, Jucker M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochem Pharmacol. 2020;172:113729
78. Dong G, Chen Z, Li ZY, Yeh NT, Bancroft CC, Van Waes C. Hepatocyte growth factor/scatter factor-induced activation of MEK and PI3K signal pathways contributes to expression of proangiogenic cytokines interleukin-8 and vascular endothelial growth factor in head and neck squamous cell carcinoma. Cancer Res. 2001;61:5911-8
79. Zhang M, Jang H, Nussinov R. PI3K inhibitors: review and new strategies. Chem Sci. 2020;11:5855-65
80. Duncan L, Shay C, Teng Y. PI3K Isoform-Selective Inhibitors in Cancer. Adv Exp Med Biol. 2020;1255:165-73
81. Andre F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS. et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N Engl J Med. 2019;380:1929-40
82. Andre F, Ciruelos EM, Juric D, Loibl S, Campone M, Mayer IA. et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: final overall survival results from SOLAR-1. Ann Oncol. 2020
83. Heffron TP, Ndubaku CO, Salphati L, Alicke B, Cheong J, Drobnick J. et al. Discovery of Clinical Development Candidate GDC-0084, a Brain Penetrant Inhibitor of PI3K and mTOR. ACS Med Chem Lett. 2016;7:351-6
84. Wen PY, Cloughesy TF, Olivero AG, Morrissey KM, Wilson TR, Lu X. et al. First-in-Human Phase I Study to Evaluate the Brain-Penetrant PI3K/mTOR Inhibitor GDC-0084 in patients with Progressive or Recurrent High-Grade Glioma. Clin Cancer Res. 2020;26:1820-8
85. Mirza-Aghazadeh-Attari M, Ekrami EM, Aghdas SAM, Mihanfar A, Hallaj S, Yousefi B. et al. Targeting PI3K/Akt/mTOR signaling pathway by polyphenols: Implication for cancer therapy. Life Sci. 2020;255:117481
86. Yuan R, Kay A, Berg WJ, Lebwohl D. Targeting tumorigenesis: development and use of mTOR inhibitors in cancer therapy. J Hematol Oncol. 2009;2:45
87. Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol. 2009;27:2278-87
88. Liu FY, Zhao ZJ, Li P, Ding X, Zong ZH, Sun CF. Mammalian target of rapamycin (mTOR) is involved in the survival of cells mediated by chemokine receptor 7 through PI3K/Akt in metastatic squamous cell carcinoma of the head and neck. Br J Oral Maxillofac Surg. 2010;48:291-6
89. Massard C, Chi KN, Castellano D, de Bono J, Gravis G, Dirix L. et al. Phase Ib dose-finding study of abiraterone acetate plus buparlisib (BKM120) or dactolisib (BEZ235) in patients with castration-resistant prostate cancer. Eur J Cancer. 2017;76:36-44
90. Wise-Draper TM, Moorthy G, Salkeni MA, Karim NA, Thomas HE, Mercer CA. et al. A Phase Ib Study of the Dual PI3K/mTOR Inhibitor Dactolisib (BEZ235) Combined with Everolimus in patients with Advanced Solid Malignancies. Target Oncol. 2017;12:323-32
91. Siegfried Z, Bonomi S, Ghigna C, Karni R. Regulation of the Ras-MAPK and PI3K-mTOR Signalling Pathways by Alternative Splicing in Cancer. Int J Cell Biol. 2013;2013:568931
92. Kandil E, Tsumagari K, Ma J, Abd Elmageed ZY, Li X, Slakey D. et al. Synergistic inhibition of thyroid cancer by suppressing MAPK/PI3K/AKT pathways. J Surg Res. 2013;184:898-906
93. Khajah MA, Mathew PM, Luqmani YA. Inhibitors of PI3K/ERK1/2/p38 MAPK Show Preferential Activity Against Endocrine-Resistant Breast Cancer Cells. Oncol Res. 2017;25:1283-95
94. Janku F, Lee JJ, Tsimberidou AM, Hong DS, Naing A, Falchook GS. et al. PIK3CA mutations frequently coexist with RAS and BRAF mutations in patients with advanced cancers. PLoS One. 2011;6:e22769
95. Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY. et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N Engl J Med. 2015;373:726-36
96. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations; Adjuvant Pertuzumab and Trastuzumab in Early HER2-Positive Breast Cancer. N Engl J Med. 2018; 379: 1585.
97. Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland A. et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 2017;18:1307-16
98. Subbiah V, Lassen U, Elez E, Italiano A, Curigliano G, Javle M. et al. Dabrafenib plus trametinib in patients with BRAF(V600E)-mutated biliary tract cancer (ROAR): a phase 2, open-label, single-arm, multicentre basket trial. Lancet Oncol. 2020;21:1234-43
99. Perna D, Karreth FA, Rust AG, Perez-Mancera PA, Rashid M, Iorio F. et al. BRAF inhibitor resistance mediated by the AKT pathway in an oncogenic BRAF mouse melanoma model. Proc Natl Acad Sci U S A. 2015;112:E536-45
100. Wee S, Jagani Z, Xiang KX, Loo A, Dorsch M, Yao YM. et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69:4286-93
101. Chen CH, Hsia TC, Yeh MH, Chen TW, Chen YJ, Chen JT. et al. MEK inhibitors induce Akt activation and drug resistance by suppressing negative feedback ERK-mediated HER2 phosphorylation at Thr701. Mol Oncol. 2017;11:1273-87
102. Bedard PL, Tabernero J, Janku F, Wainberg ZA, Paz-Ares L, Vansteenkiste J. et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res. 2015;21:730-8
103. Schram AM, Gandhi L, Mita MM, Damstrup L, Campana F, Hidalgo M. et al. A phase Ib dose-escalation and expansion study of the oral MEK inhibitor pimasertib and PI3K/MTOR inhibitor voxtalisib in patients with advanced solid tumours. Br J Cancer. 2018;119:1471-6
104. Arend RC, Davis AM, Chimiczewski P, O'Malley DM, Provencher D, Vergote I. et al. EMR 20006-012: A phase II randomized double-blind placebo controlled trial comparing the combination of pimasertib (MEK inhibitor) with SAR245409 (PI3K inhibitor) to pimasertib alone in patients with previously treated unresectable borderline or low grade ovarian cancer. Gynecol Oncol. 2020;156:301-7
105. Ragon BK, Odenike O, Baer MR, Stock W, Borthakur G, Patel K. et al. Oral MEK 1/2 Inhibitor Trametinib in Combination with AKT Inhibitor GSK2141795 in patients with Acute Myeloid Leukemia with RAS Mutations: A Phase II Study. Clin Lymphoma Myeloma Leuk. 2019;19:431-40 e13
106. Liu JF, Gray KP, Wright AA, Campos S, Konstantinopoulos PA, Peralta A. et al. Results from a single arm, single stage phase II trial of trametinib and GSK2141795 in persistent or recurrent cervical cancer. Gynecol Oncol. 2019;154:95-101
107. Ma DJ, Galanis E, Anderson SK, Schiff D, Kaufmann TJ, Peller PJ. et al. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro Oncol. 2015;17:1261-9
108. Dunn LA, Riaz N, Fury MG, McBride SM, Michel L, Lee NY. et al. A Phase 1b Study of Cetuximab and BYL719 (Alpelisib) Concurrent with Intensity Modulated Radiation Therapy in Stage III-IVB Head and Neck Squamous Cell Carcinoma. Int J Radiat Oncol Biol Phys. 2020;106:564-70
109. Wen PY, Omuro A, Ahluwalia MS, Fathallah-Shaykh HM, Mohile N, Lager JJ. et al. Phase I dose-escalation study of the PI3K/mTOR inhibitor voxtalisib (SAR245409, XL765) plus temozolomide with or without radiotherapy in patients with high-grade glioma. Neuro Oncol. 2015;17:1275-83
110. Alonso-Basanta M, Fang P, Maity A, Hahn SM, Lustig RA, Dorsey JF. A phase I study of nelfinavir concurrent with temozolomide and radiotherapy in patients with glioblastoma multiforme. J Neurooncol. 2014;116:365-72
111. Lin C, Verma V, Ly QP, Lazenby A, Sasson A, Schwarz JK. et al. Phase I trial of concurrent stereotactic body radiotherapy and nelfinavir for locally advanced borderline or unresectable pancreatic adenocarcinoma. Radiother Oncol. 2019;132:55-62
112. McGowan DR, Skwarski M, Bradley KM, Campo L, Fenwick JD, Gleeson FV. et al. Buparlisib with thoracic radiotherapy and its effect on tumour hypoxia: A phase I study in patients with advanced non-small cell lung carcinoma. Eur J Cancer. 2019;113:87-95
113. Crook T, Tidy JA, Vousden KH. Degradation of p53 can be targeted by HPV E6 sequences distinct from those required for p53 binding and trans-activation. Cell. 1991;67:547-56
114. Band V, Dalal S, Delmolino L, Androphy EJ. Enhanced degradation of p53 protein in HPV-6 and BPV-1 E6-immortalized human mammary epithelial cells. EMBO J. 1993;12:1847-52
115. Bradner JE. Cancer: An essential passenger with p53. Nature. 2015;520:626-7
116. Bieging KT, Attardi LD. Cancer: A piece of the p53 puzzle. Nature. 2015;520:37-8
117. Graat HC, Carette JE, Schagen FH, Vassilev LT, Gerritsen WR, Kaspers GJ. et al. Enhanced tumor cell kill by combined treatment with a small-molecule antagonist of mouse double minute 2 and adenoviruses encoding p53. Mol Cancer Ther. 2007;6:1552-61
118. Ma G, Kawamura K, Li Q, Suzuki N, Liang M, Namba M. et al. cytotoxicity of adenoviruses expressing the wild-type p53 gene to esophageal carcinoma cells is linked with the CAR expression level and indirectly with the endogenous p53 status. Cancer Gene Ther. 2009;16:832-40
119. Perri F, Pacelli R, Della Vittoria Scarpati G, Cella L, Giuliano M, Caponigro F. et al. Radioresistance in head and neck squamous cell carcinoma: Biological bases and therapeutic implications. Head Neck. 2015;37:763-70
120. Merritt JA, Roth JA, Logothetis CJ. Clinical evaluation of adenoviral-mediated p53 gene transfer: review of INGN 201 studies. Semin Oncol. 2001;28:105-14
121. Swisher SG, Roth JA, Komaki R, Gu J, Lee JJ, Hicks M. et al. Induction of p53-regulated genes and tumor regression in lung cancer patients after intratumoral delivery of adenoviral p53 (INGN 201) and radiation therapy. Clin Cancer Res. 2003;9:93-101
122. Wolf JK, Bodurka DC, Gano JB, Deavers M, Ramondetta L, Ramirez PT. et al. A phase I study of Adp53 (INGN 201; ADVEXIN) for patients with platinum- and paclitaxel-resistant epithelial ovarian cancer. Gynecol Oncol. 2004;94:442-8
123. Nemunaitis J, Ganly I, Khuri F, Arseneau J, Kuhn J, McCarty T. et al. Selective replication and oncolysis in p53 mutant tumors with ONYX-015, an E1B-55kD gene-deleted adenovirus, in patients with advanced head and neck cancer: a phase II trial. Cancer Res. 2000;60:6359-66
124. Kirn DH. A Phase II Trial of Intratumoral Injection with a Selectively Replicating Adenovirus (ONYX-015) in patients with Recurrent, Refractory Squamous Cell Carcinoma of the Head and Neck. Methods Mol Med. 2000;35:559-74
125. Nemunaitis J, Khuri F, Ganly I, Arseneau J, Posner M, Vokes E. et al. Phase II trial of intratumoral administration of ONYX-015, a replication-selective adenovirus, in patients with refractory head and neck cancer. J Clin Oncol. 2001;19:289-98
126. Rao CV, Patlolla JM, Qian L, Zhang Y, Brewer M, Mohammed A. et al. Chemopreventive effects of the p53-modulating agents CP-31398 and Prima-1 in tobacco carcinogen-induced lung tumorigenesis in A/J mice. Neoplasia. 2013;15:1018-27
127. Roh JL, Kang SK, Minn I, Califano JA, Sidransky D, Koch WM. p53-Reactivating small molecules induce apoptosis and enhance chemotherapeutic cytotoxicity in head and neck squamous cell carcinoma. Oral Oncol. 2011;47:8-15
128. Roh JL, Ko JH, Moon SJ, Ryu CH, Choi JY, Koch WM. The p53-reactivating small-molecule RITA enhances cisplatin-induced cytotoxicity and apoptosis in head and neck cancer. Cancer Lett. 2012;325:35-41
129. Morandell S, Yaffe MB. Exploiting synthetic lethal interactions between DNA damage signaling, checkpoint control, and p53 for targeted cancer therapy. Prog Mol Biol Transl Sci. 2012;110:289-314
130. Tongyang L, Haiqiang G, Meiyan Z, Yingze H, Shuting J, Ying L. et al. [Synthetic lethal genes to mutant p53]. Yi Chuan. 2015;37:321-6
131. Bao S, Tibbetts RS, Brumbaugh KM, Fang Y, Richardson DA, Ali A. et al. ATR/ATM-mediated phosphorylation of human Rad17 is required for genotoxic stress responses. Nature. 2001;411:969-74
132. Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605-11
133. Ruzankina Y, Schoppy DW, Asare A, Clark CE, Vonderheide RH, Brown EJ. Tissue regenerative delays and synthetic lethality in adult mice after combined deletion of Atr and Trp53. Nat Genet. 2009;41:1144-9
134. Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007;11:175-89
135. Hammond EM, Muschel RJ. Radiation and ATM inhibition: the heart of the matter. J Clin Invest. 2014;124:3289-91
136. Zeng Y, Forbes KC, Wu Z, Moreno S, Piwnica-Worms H, Enoch T. Replication checkpoint requires phosphorylation of the phosphatase Cdc25 by Cds1 or Chk1. Nature. 1998;395:507-10
137. Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410:842-7
138. Gadhikar MA, Sciuto MR, Alves MV, Pickering CR, Osman AA, Neskey DM. et al. Chk1/2 inhibition overcomes the cisplatin resistance of head and neck cancer cells secondary to the loss of functional p53. Mol Cancer Ther. 2013;12:1860-73
139. Geenen JJJ, Schellens JHM. Molecular Pathways: Targeting the Protein Kinase Wee1 in Cancer. Clin Cancer Res. 2017;23:4540-4
140. Ghelli Luserna di Rora A, Cerchione C, Martinelli G, Simonetti G. A WEE1 family business: regulation of mitosis, cancer progression, and therapeutic target. J Hematol Oncol. 2020;13:126
141. Meng X, Bi J, Li Y, Yang S, Zhang Y, Li M. et al. AZD1775 Increases Sensitivity to Olaparib and Gemcitabine in Cancer Cells with p53 Mutations. Cancers (Basel). 2018 10
142. Matheson CJ, Venkataraman S, Amani V, Harris PS, Backos DS, Donson AM. et al. A WEE1 Inhibitor Analog of AZD1775 Maintains Synergy with Cisplatin and Demonstrates Reduced Single-Agent Cytotoxicity in Medulloblastoma Cells. ACS Chem Biol. 2016;11:921-30
143. Cuneo KC, Morgan MA, Sahai V, Schipper MJ, Parsels LA, Parsels JD. et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination with Gemcitabine and Radiation for patients with Locally Advanced Pancreatic Cancer. J Clin Oncol. 2019;37:2643-50
144. Leijen S, van Geel RM, Sonke GS, de Jong D, Rosenberg EH, Marchetti S. et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in patients with TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy within 3 Months. J Clin Oncol. 2016;34:4354-61
145. Cole KA, Pal S, Kudgus RA, Ijaz H, Liu X, Minard CG. et al. Phase I Clinical Trial of the Wee1 Inhibitor Adavosertib (AZD1775) with Irinotecan in Children with Relapsed Solid Tumors: A COG Phase I Consortium Report (ADVL1312). Clin Cancer Res. 2020;26:1213-9
146. Tanaka N, Patel AA, Wang J, Frederick MJ, Kalu NN, Zhao M. et al. Wee-1 Kinase Inhibition Sensitizes High-Risk HPV+ HNSCC to Apoptosis Accompanied by Downregulation of MCl-1 and XIAP Antiapoptotic Proteins. Clin Cancer Res. 2015;21:4831-44
147. Busch CJ, Kroger MS, Jensen J, Kriegs M, Gatzemeier F, Petersen C. et al. G2-checkpoint targeting and radiosensitization of HPV/p16-positive HNSCC cells through the inhibition of Chk1 and Wee1. Radiother Oncol. 2017;122:260-6
148. Diab A, Gem H, Swanger J, Kim HY, Smith K, Zou G. et al. FOXM1 drives HPV+ HNSCC sensitivity to WEE1 inhibition. Proc Natl Acad Sci U S A. 2020;117:28287-96
149. Ashrafi GH, Haghshenas MR, Marchetti B, O'Brien PM, Campo MS. E5 protein of human papillomavirus type 16 selectively downregulates surface HLA class I. Int J Cancer. 2005;113:276-83
150. Zhang B, Li P, Wang E, Brahmi Z, Dunn KW, Blum JS. et al. The E5 protein of human papillomavirus type 16 perturbs MHC class II antigen maturation in human foreskin keratinocytes treated with interferon-gamma. Virology. 2003;310:100-8
151. Vandermark ER, Deluca KA, Gardner CR, Marker DF, Schreiner CN, Strickland DA. et al. Human papillomavirus type 16 E6 and E 7 proteins alter NF-kB in cultured cervical epithelial cells and inhibition of NF-kB promotes cell growth and immortalization. Virology. 2012;425:53-60
152. Guess JC, McCance DJ. Decreased migration of Langerhans precursor-like cells in response to human keratinocytes expressing human papillomavirus type 16 E6/E7 is related to reduced macrophage inflammatory protein-3alpha production. J Virol. 2005;79:14852-62
153. Torres-Poveda K, Bahena-Roman M, Madrid-Gonzalez C, Burguete-Garcia AI, Bermudez-Morales VH, Peralta-Zaragoza O. et al. Role of IL-10 and TGF-beta1 in local immunosuppression in HPV-associated cervical neoplasia. World J Clin Oncol. 2014;5:753-63
154. Berti FCB, Pereira APL, Cebinelli GCM, Trugilo KP, Brajao de Oliveira K. The role of interleukin 10 in human papilloma virus infection and progression to cervical carcinoma. Cytokine Growth Factor Rev. 2017;34:1-13
155. Hasan UA, Bates E, Takeshita F, Biliato A, Accardi R, Bouvard V. et al. TLR9 expression and function is abolished by the cervical cancer-associated human papillomavirus type 16. J Immunol. 2007;178:3186-97
156. Hasan UA, Zannetti C, Parroche P, Goutagny N, Malfroy M, Roblot G. et al. The human papillomavirus type 16 E7 oncoprotein induces a transcriptional repressor complex on the Toll-like receptor 9 promoter. J Exp Med. 2013;210:1369-87
157. Richards KH, Wasson CW, Watherston O, Doble R, Eric Blair G, Wittmann M. et al. The human papillomavirus (HPV) E7 protein antagonises an Imiquimod-induced inflammatory pathway in primary human keratinocytes. Sci Rep. 2015;5:12922
158. Liu M, Kuo F, Capistrano KJ, Kang D, Nixon BG, Shi W. et al. TGF-beta suppresses type 2 immunity to cancer. Nature. 2020;587:115-20
159. Peralta-Zaragoza O, Bermudez-Morales V, Gutierrez-Xicotencatl L, Alcocer-Gonzalez J, Recillas-Targa F, Madrid-Marina V. E6 and E7 oncoproteins from human papillomavirus type 16 induce activation of human transforming growth factor beta1 promoter throughout Sp1 recognition sequence. Viral Immunol. 2006;19:468-80
160. Miyauchi S, Sanders PD, Guram K, Kim SS, Paolini F, Venuti A. et al. HPV16 E5 Mediates Resistance to PD-L1 Blockade and Can Be Targeted with Rimantadine in Head and Neck Cancer. Cancer Research. 2020;80:732-46
161. Liu C, Lu J, Tian H, Du W, Zhao L, Feng J. et al. Increased expression of PD-L1 by the human papillomavirus 16 E7 oncoprotein inhibits anticancer immunity. Mol Med Rep. 2016
162. Mittal D, Kassianos AJ, Tran LS, Bergot AS, Gosmann C, Hofmann J. et al. Indoleamine 2,3-Dioxygenase Activity Contributes to Local Immune Suppression in the Skin Expressing Human Papillomavirus Oncoprotein E7. J Invest Dermatol. 2013;133:2686-94
163. Cicchini L, Westrich JA, Xu T, Vermeer DW, Berger JN, Clambey ET. et al. Suppression of Antitumor Immune Responses by Human Papillomavirus through Epigenetic Downregulation of CXCL14. Mbio. 2016 7
164. Lau L, Gray EE, Brunette RL, Stetson DB. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science. 2015;350:568-71
165. Mandal R, Senbabaoglu Y, Desrichard A, Havel JJ, Dalin MG, Riaz N. et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight. 2016;1:e89829
166. Sridharan V, Margalit DN, Lynch SA, Severgnini M, Zhou J, Chau NG. et al. Definitive chemoradiation alters the immunologic landscape and immune checkpoints in head and neck cancer. Br J Cancer. 2016;115:252-60
167. Cillo AR, Kurten CHL, Tabib T, Qi Z, Onkar S, Wang T. et al. Immune Landscape of Viral- and Carcinogen-Driven Head and Neck Cancer. Immunity. 2020;52:183-99 e9
168. Wood O, Woo J, Seumois G, Savelyeva N, McCann KJ, Singh D. et al. Gene expression analysis of TIL rich HPV-driven head and neck tumors reveals a distinct B-cell signature when compared to HPV independent tumors. Oncotarget. 2016;7:56781-97
169. Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP. et al. safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17:956-65
170. Harrington KJ, Ferris RL, Blumenschein G Jr, Colevas AD, Fayette J, Licitra L. et al. Nivolumab versus standard, single-agent therapy of investigator's choice in recurrent or metastatic squamous cell carcinoma of the head and neck (CheckMate 141): health-related quality-of-life results from a randomised, phase 3 trial. Lancet Oncol. 2017;18:1104-15
171. Bauml J, Seiwert TY, Pfister DG, Worden F, Liu SV, Gilbert J. et al. Pembrolizumab for Platinum- and Cetuximab-Refractory Head and Neck Cancer: Results From a Single-Arm, Phase II Study. J Clin Oncol. 2017;35:1542-9
172. Bins S, Mathijssen RHJ. p16 status and choice of chemotherapy in the KEYNOTE-040 study. Lancet. 2019;394:1322-3
173. Cohen EEW, Soulieres D, Le Tourneau C, Dinis J, Licitra L, Ahn MJ. et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): a randomised, open-label, phase 3 study. Lancet. 2019;393:156-67
174. Harrington KJ, Soulieres D, Le Tourneau C, Dinis J, Licitra LF, Ahn MJ. et al. Quality of Life with Pembrolizumab for Recurrent/Metastatic Head and Neck Squamous Cell Carcinoma: KEYNOTE-040. J Natl Cancer Inst. 2020
175. Burtness B, Harrington KJ, Greil R, Soulieres D, Tahara M, de Castro G Jr. et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet. 2019;394:1915-28
176. Colevas AD, Bahleda R, Braiteh F, Balmanoukian A, Brana I, Chau NG. et al. Safety and clinical activity of atezolizumab in head and neck cancer: results from a phase I trial. Ann Oncol. 2018;29:2247-53
177. Ferris RL, Haddad R, Even C, Tahara M, Dvorkin M, Ciuleanu TE. et al. Durvalumab with or without tremelimumab in patients with recurrent or metastatic head and neck squamous cell carcinoma: EAGLE, a randomized, open-label phase III study. Ann Oncol. 2020;31:942-50
178. Biktasova A, Hajek M, Sewell A, Gary C, Bellinger G, Deshpande HA. et al. Demethylation Therapy as a Targeted Treatment for Human Papillomavirus-associated Head and Neck Cancer. Clin Cancer Res. 2017;23:7276-87
179. Mesia R, Bossi P, Hansen AR, Hsieh CY, Licitra LF, Tan EH. et al. Phase II study of CC-486 (oral azacitidine) in previously treated patients with locally advanced or metastatic nasopharyngeal carcinoma. Eur J Cancer. 2019;123:138-45
180. Shimizu T, LoRusso PM, Papadopoulos KP, Patnaik A, Beeram M, Smith LS. et al. Phase I first-in-human study of CUDC-101, a multitargeted inhibitor of HDACs, EGFR, and HER2 in patients with advanced solid tumors. Clin Cancer Res. 2014;20:5032-40
181. Galloway TJ, Wirth LJ, Colevas AD, Gilbert J, Bauman JE, Saba NF. et al. A Phase I Study of CUDC-101, a Multitarget Inhibitor of HDACs, EGFR, and HER2, in Combination with Chemoradiation in patients with Head and Neck Squamous Cell Carcinoma. Clin Cancer Res. 2015;21:1566-73
182. Bais MV. Impact of Epigenetic Regulation on Head and Neck Squamous Cell Carcinoma. J Dent Res. 2019;98:268-76
183. Gray JE, Haura E, Chiappori A, Tanvetyanon T, Williams CC, Pinder-Schenck M. et al. A phase I, pharmacokinetic, and pharmacodynamic study of panobinostat, an HDAC inhibitor, combined with erlotinib in patients with advanced aerodigestive tract tumors. Clin Cancer Res. 2014;20:1644-55
184. Caponigro F, Di Gennaro E, Ionna F, Longo F, Aversa C, Pavone E. et al. Phase II clinical study of valproic acid plus cisplatin and cetuximab in recurrent and/or metastatic squamous cell carcinoma of Head and Neck-V-CHANCE trial. BMC Cancer. 2016;16:918
Author contact
![]() Corresponding authors: Zhe Wang PhD, MD. Department of Medical Oncology, Affiliated Zhongshan Hospital of Dalian University, Dalian 116001, China, Tel +86-411-6289-3627, E-mail: wangzheedu.cn; Ruoyu Wang, M.D., PhD, Department of Medical Oncology Affiliated Zhongshan Hospital of Dalian University. Dalian 116001, China, E-mail: wangruoyu1963com.
Corresponding authors: Zhe Wang PhD, MD. Department of Medical Oncology, Affiliated Zhongshan Hospital of Dalian University, Dalian 116001, China, Tel +86-411-6289-3627, E-mail: wangzheedu.cn; Ruoyu Wang, M.D., PhD, Department of Medical Oncology Affiliated Zhongshan Hospital of Dalian University. Dalian 116001, China, E-mail: wangruoyu1963com.