Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Methods and Materials
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(4):1153-1167. doi:10.7150/ijbs.58365 This issue Cite
Research Paper
MicroRNA-223 restricts liver fibrosis by inhibiting the TAZ-IHH-GLI2 and PDGF signaling pathways via the crosstalk of multiple liver cell types
Xiaolin Wang1,2, Wonhyo Seo1, Seol Hee Park1, Yaojie Fu1, Seonghwan Hwang1, Robim M. Rodrigues1, Dechun Feng1, Bin Gao1 ![]() , Yong He1
, Yong He1 ![]()
1. Laboratory of Liver Diseases, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, MD 20892, USA.
2. Department of Infectious Diseases, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, 200025, China.
Received 2021-1-18; Accepted 2021-3-1; Published 2021-3-11
Abstract

Background & Aims: Liver fibrosis is a common consequence of chronic liver injury and is characterized by the accumulation of extracellular matrix mainly generated from activated hepatic stellate cells (HSCs). At present, the mechanisms underlying liver fibrogenesis remain obscure and effective pharmacological therapies are lacking. Neutrophil-specific microRNA-223 (miR-223) plays an important role in controlling the development of various liver diseases; however, its role in HSC activation and liver fibrosis remains unclear.
Methods: Liver fibrosis was induced by chronic carbon tetrachloride (CCl4) injection of miR-223 knockout (miR-223KO) mice and littermate wild-type controls. MiR-223 was overexpressed in cultured HSCs to determine its function and targets during HSC activation and proliferation. The expression of miR-223 and pri-miR-223 was examined in primary HSCs isolated from CCl4-treated mice and in cultured HSCs. The communication between HSCs and neutrophils was studied by performing in vitro co-culture experiments.
Results: Genetic deletion of miR-223 exacerbated chronic CCl4-induced liver fibrosis. Administration of miR-223 inhibited liver fibrosis by inhibiting the transcriptional coactivator with PDZ-binding motif (TAZ)-Indian hedgehog (IHH)-GLI Family Zinc Finger 2 (GLI2) pathway via the crosstalk between hepatocytes and HSCs. Overexpression of miR-223 also directly attenuated Gli2 as well as platelet-derived growth factor receptor α/β (Pdgfra/b) expression in HSCs, thereby suppressing HSC activation and proliferation. The expression of pri-miR-223 and miR-223 was downregulated during HSC activation in vitro. Expression of pri-miR-223 was also decreased in activated HSCs in vivo in fibrotic livers but mature miR-223 expression was not reduced. Finally, in co-culture experiments, activated HSCs were able to take up miR-223-enriched extracellular vesicles from neutrophils, resulting in elevation of miR-223.
Conclusion: MiR-223 restricts liver fibrosis by targeting multiple genes in hepatocytes and HSCs, providing potential therapeutic targets for the treatment of liver fibrosis.
Keywords: Liver fibrosis, HSC, miR-223, Gli2, PDGFRα/β
Introduction
Hepatic fibrosis is a dynamic process characterized by the net accumulation of extracellular matrix (ECM) and myofibroblast-rich scar tissue, caused by chronic liver injury, including chronic viral infection, alcoholic liver disease, nonalcoholic steatohepatitis (NASH), and autoimmune hepatitis [1]. Of note, liver fibrosis can progress to liver cirrhosis and liver cancer but its mechanisms remain obscure and effective pharmacological therapies are lacking [2].
Activation of hepatic stellated cells (HSCs) is the key step in the development of liver fibrosis [3]. Following liver injury or culture in vitro, HSCs transdifferentiate from a quiescent phenotype into ECM-producing myofibroblasts, serving as the major source of ECM production in liver fibrosis [4]. A variety of fibrogenic mediators and their downstream signaling pathways have been identified to drive HSC activation, such as transforming growth factor β (TGFβ), platelet-derived growth factor (PDGF), connective tissue growth factor, and the Hedgehog pathway [1]. Immune cell-mediated liver inflammation is well known to play an important role in regulating HSC activation. For example, macrophages contribute to liver fibrogenesis by secreting TGFβ, which is the most potent fibrogenic cytokine for HSC activation [5]. Neutrophils likely promote liver fibrosis via production of ROS but may also promote liver fibrosis resolution by transferring microRNA into macrophages [6-9]. Moreover, natural killer cells have been reported to kill activated HSCs, thereby inhibiting liver fibrosis [10]. Up till now, there are no clinically effective ways to inhibit HSC activation and reduce liver fibrosis. Therefore, understanding of the regulatory mechanisms involved in HSC activation could provide new insights for the treatment of liver fibrosis.
MicroRNAs (miRNAs), which are endogenous, noncoding, single-stranded RNAs of ~22 nucleotides in length, play a key role in controlling multiple physiological processes by negatively regulating target gene expression through complementary binding with the 3' untranslated regions (UTRs) [11-13]. Accumulating studies have demonstrated the important roles of miRNAs in the progression of liver diseases [13,14]. Moreover, miRNAs can be packaged and released in extracellular vehicles (EVs), serving as a messenger for cell-to-cell communication [13]. MiR223 is a myeloid cell-specific miRNA which is expressed at the highest level in neutrophils and is specifically enriched in neutrophil/myeloid cell-derived EVs [15]. Importantly, over the past ten years, miR-223 has attracted great attention as an important regulator in liver pathology [13,16]. Notably, hepatic miR-223 expression was significantly increased in patients with liver fibrosis and in mice with CCl4 or bile duct ligation (BDL)-induced liver fibrosis [17]. Calvente et al. have demonstrated that neutrophil-derived EVs can transfer miR-223 from neutrophils to hepatic macrophages and covert macrophages into a restorative phenotype that mitigates fibrogenesis [18]. In addition, our previous study demonstrated that overexpression of miR-223 ameliorated NASH-related liver fibrosis by inhibiting several targets in hepatocytes [15]. However, the functions of miR-223 in HSCs remain largely unknown.
In the current study, we demonstrated that miR-223 played a protective role in liver fibrosis by targeting several genes in hepatocytes and HSCs. The downregulation of miR-223 is accompanied by the activation of HSCs, while neutrophil-derived EVs can transfer miR-223 into HSCs, thus mitigating liver fibrosis.
Methods and Materials
Mice
MiR-223-/- (miR-223KO) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). MiR-223KO (miR-223-/y) (the miR-223 locus is on the X chromosome) and WT (miR-223+/y) littermate controls were generated as previously described [19]. All animal experiments were approved by the National Institute on Alcohol Abuse and Alcoholism Animal Care and Use Committee.
For chronic CCl4-induced liver fibrosis, mice were injected intraperitoneally with 2 ml/kg body weight of 10% CCl4 (Sigma-Aldrich, dissolved in olive oil) twice a week for 4 weeks or 8 weeks. All mice were sacrificed 48 h after the last injection. Serum samples and liver tissues were collected at the indicated time points. For acute CCl4-induced acute liver injury, mice were injected intraperitoneally with 2 ml/kg body weight of 10% CCl4 and mice were sacrificed 24 h, 48 h or 72h after the injection.
For adenovirus (Ad)-miR-223 injection experiment, mice were treated with CCl4 for 4 weeks followed by injection (via the tail vein) of adenovirus expressing mouse miR-223 (Ad-miR-223) (Vector Biolabs, Malvern, PA) or green fluorescence protein (Ad-Gfp) (Vector Biolabs) as a control. After another 2 weeks of CCl4 treatment, the mice were sacrificed.
Primary HSC isolation and flow Cytometry Cell Sorter (FACS) sorting
Mice were anesthetized with sodium pentobarbital (30 mg/kg intraperitoneally), the portal vein was cannulated and the liver was perfused with EGTA (ethylene glycol-bis (β-aminoethyl ether)-N,N,N',N'-tetraacetic acid) solution (5.4 mM KCl, 0.44 mM KH2PO4,140mM NaCl, 0.34 mM Na2HPO4, 0.5 mM EGTA, and 25 mM Tricine, pH 7.2) and with 0.075% type I collagenase (Worthington, Lakewood, NJ) in Hank's Balanced Salt Solution (Sigma, St. Louis, MO). Liver tissue was further digested with 0.009% collagenase at 37 °C agitation for 20 minutes. Primary hepatocytes were separated after centrifugation at 50 g for 5 minutes and the supernatant was further centrifuged at 400 g for 10 minutes at 4 °C. After centrifuge, the cell pellet was suspended in 11.5% OptiPrep and loaded carefully with Hank's Balanced Salt Solution. After centrifugation at 1500 g for 20 minutes at 4 °C, HSCs were collected from the interface fraction and washed with Hank's Balanced Salt Solution. The finally collected HSCs were cultured in RPMI-1640 medium containing 10% fetal bovine serum, 10% horse serum and penicillin-streptomycin.
For the analysis of in vivo miR-223 expression in HSCs, HSCs were isolated from olive oil and CCl4-treated mice (4 weeks of CCl4 treatment). FACS was conducted following density-gradient centrifugation. HSCs were sorted based on their positive auto fluorescent signals [20].
Culture of LX-2 cells and miR-223 mimics transfection
Human hepatic stellate cell line LX-2 cells (a kind gift from Dr. Scott Friedman, Mount Sinai) were cultured in DMEM with 10% fetal bovine serum, and penicillin-streptomycin.
MiRNA transfection was performed in primary mouse HSCs and LX-2 cells. Briefly, miR-223 mimics or nonspecific miRNA mimics (NS-miRNA) (Thermo fisher Scientific, Waltham, MA) were transfected into cells at 1 nM using Lipofectamine RNAiMAX transfection reagent (Thermo fisher Scientific, Waltham, MA) following the manufacture's instruction. In some experiments, cells were treated with PDGFBB (PeproTech, Cranbury, NJ; 10 ng/ml) for 20 min after transfection.
Tissue processing, histological analysis and immunohistochemistry
Formalin fixed liver samples were processed and 4-µm-thick paraffin sections were stained with Sirius Red (collagen/fibrosis) (Sigma, St. Louis, MO). For immunohistochemistry, after heat-induced epitope retrieval, paraffin-embedded sections were incubated in 3% H2O2, and blocked for another 60 mins in 3% normal serum buffer. Sections were incubated with primary antibodies overnight at 4 °C. Incubation with the secondary antibody against rabbit was performed by SignalStain® Boost IHC Detection Reagent (Cell Signaling Technology, Danvers, MA). DAB Peroxidase Substrate Kit (Vector 2 Laboratories, Inc., Burlingame, CA) were used to visualize the staining according to the manufacturer's instructions. Immunochemistry double staining was performed by using ImmPRESS Duet Double staining Polymer Kit (Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions. The primary antibodies used are listed below: anti-TAZ antibody was purchased from Sigma-Aldrich (Biocare Medical, LLC, Concord, CA); anti-PDGFRα, anti-PDGFRβ, anti-alpha-SMA and anti-Ki67 antibodies were purchased from Cell Signaling Technology (Danvers, MA); anti-IHH was purchased from Novus Biologicals (Centennial, CO) and anti-GLI2 antibody was purchased from Proteintech (Rosemont, IL); anti-myeloperoxidase (MPO) antibody was purchased from Biocare Medical (Concord, CA).The images were taken with an Olympus camera DP72.
BrdU immunostaining
The rate of cell proliferation was determined by using BrdU immunostaining Kit (BD Biosciences, San Jose, CA) according to the manufacturer's instructions. Primary HSCs were incubated with BrdU (10 μM) for 6 hours before staining. The positively stained HSC nuclei were counted, and the mean values were analyzed.
Cell counting kit 8 (CCK-8) assay
CCK8 assay was performed using CCK-8 cell counting kit (Vitascientific, MD) according to the manufacturer's instructions. Briefly, cells were seeded into 96-well plates, 10 μl CCK-8 was added to each well and optical density (OD) was measured at 450 nm after incubation for 1.5 hours at 37 °C.
Co-culture of HSCs and neutrophils
Transwell plates with 0.4 µm pore size polycarbonate membrane inserts (Corning, Glendale, Arizona) were used to set up the co-culture experiments according to the manufacturer's instructions. Primary mouse HSCs were isolated from miR-223KO mice and cultured in the lower chamber for 1 day. Bone marrow neutrophils were isolated by using the mouse neutrophil isolation kit (MiltenyiBiotec, Ashburn, CA) according to the manufacturer's protocol. Neutrophils were added to the upper chamber. After co-culture for 6 hours, the HSCs were collected and subjected to RT-qPCR analysis of miR-223.
Total RNA isolation and quantitative reverse transcription PCR (RT-qPCR)
Total RNA was extracted from liver tissues or cells using TRIzol reagents (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. RNA was then reverse transcribed into cDNA using a High-capacity cDNA Reverse Transcription kit (Invitrogen, Carlsbad, CA). The expression levels of mRNA were measured by RT-qPCR by using QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems, Foster City, CA). The mRNA levels of 18s were used as an internal control. The relative expression of mRNA was determined by 2-∆∆Ct method. The primers used for RT-qPCR are listed in Supporting Table S1.
For miRNA detection, total RNA was extracted from liver tissues and cells by using TRIzol reagent (Invitrogen, Carlsbad, CA), and then the mature miRNA strand cDNA was synthesized using TaqMan® MicroRNA Reverse Transcription Kit (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. MiRNA was amplified by using TaqMan® MicroRNA Assays (Invitrogen) and TaqMan® Universal PCR Master Mix (Invitrogen) according to the manufacturer's instructions. The fold-change for miRNA relative to snoRNA202 or spiked-in cel-miR-39 (Qiagen, Valencia, CA) was calculated by the formula 2-∆∆Ct.
Western blotting
Cells and liver tissues were lysed or homogenized in RIPA buffer containing a cocktail of protease inhibitors (Santa Cruz, CA) according to the manufacturer's instruction. Protein extracts were loaded onto 12% acrylamide gels (Bio-Rad) and transferred onto nitrocellulose membranes. Protein bands were visualized with ECL-chemiluminescent kit (GE Healthcare, Piscataway, NJ). The antibodies against anti-PDGFRα, PDGFRβ, ERK, AKT, phosphorylated ERK (Thr202/Tyr204) and phosphorylated AKT (Ser473) were purchased from Cell Signaling Technology (Danvers, MA). GLI2 and GLI3 antibodies were purchased from Proteintech (Rosemont, IL). The antibodies against β-actin was purchased from Abcam (Cambridge, MA).
Isolation of EVs and EV uptake assay
EVs were isolated from culture medium of neutrophils or hepatocytes by using ExoQuick-TC solution (System Biosciences, Palo Alto, CA) according to the manufacturer's instructions. For EV uptake assay, the EVs isolated from neutrophils were labeled with DiD red-fluorescent dye (Thermo Fisher Scientific, Inc, Waltham, MA) by incubating with 1 mL DiD red-fluorescent dye solution (1:200 dilution) for 15 min. The labeled EV solution was then centrifuged at 100,000 g for 70 min at 4 °C and re-suspended in PBS and rotated at 4 °C for overnight. The EV fractions were incubated with HSCs for 24 hours and subjected to immunofluorescent staining. PBS was used as a negative control. The images were obtained by using LSM 710 confocal microscope (Zeiss, Thornwood, NY).
Statistical analysis
Data are expressed as the means ± SEM for each group and were analyzed using GraphPad Prism software (GraphPad Software, La Jolla, CA). To compare values obtained from three or more groups, a one-way ANOVA was used, followed by Tukey post-hoc test. To compare values obtained from two groups, the Student t test was performed. P values of <0.05 were considered significant.
Results
MiR-223 deletion exacerbates CCl4-induced liver fibrosis
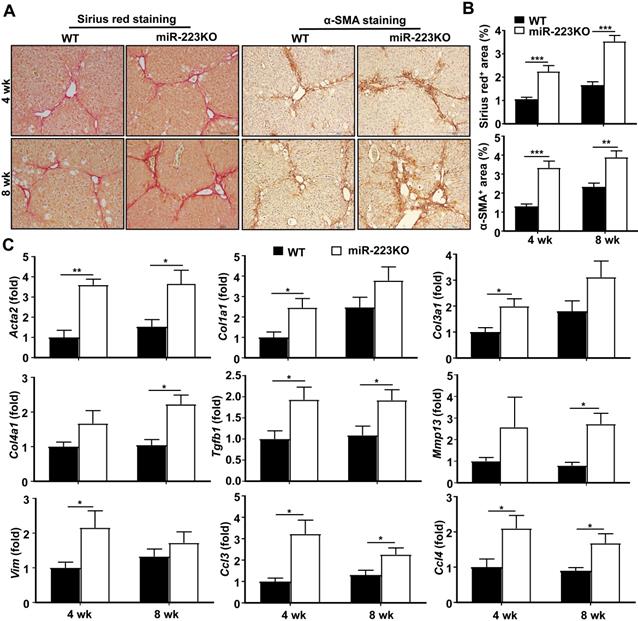
Our previous work revealed that miR-223KO mice are more susceptible to NASH-related liver fibrosis [19]. To further define the functional role of miR-223 in the development of liver fibrosis, miR-223KO mice and their WT littermate control mice were injected with CCl4 for 4 weeks or 8 weeks. As demonstrated in Fig. 1A and 1B, miR-223KO mice showed more severe liver fibrosis as evidenced by Sirius red staining and α-SMA staining. Moreover, serum ALT levels were higher in miR-223KO mice compared to WT mice (Supplementary Fig. S1A). Consistently, ALT and AST levels were also higher in miR-223KO mice compared with WT mice in acute liver injury induced by a single injection of CCl4 (Supplementary Fig. S1B). Furthermore, hepatic mRNA levels of fibrogenic genes (Acta2, Col1a1, Col3a1, Col4a1, Tgfb1, Mmp13 and Vim) were significantly upregulated in miR-223KO mice compared with WT mice, suggesting that miR-223 deletion accelerated liver fibrosis (Fig. 1C). Increased liver inflammation was further confirmed by the upregulation of Ccl3 and Ccl4 in miR-223KO mice, whereas Ccl2 and Il6 levels had an increasing trend although no statistical significance was reached between WT and miR-223KO mice (Fig. 1C and Supplementary Fig. S1C).
MiR-223KO mice are more susceptible to CCl4-induced liver fibrosis. WT and miR-223KO mice were treated with CCl4 twice a week for 4 weeks or 8 weeks. (A) Representative images of Sirius red and α-SMA staining are shown. (B) Fibrotic area per field was quantified. (C) RT-qPCR analyses of the expression profiles of the genes related to fibrogenesis and inflammation in the liver from WT and miR-223KO mice. Values represent means ± SEM (n=5-8/group). *P< 0.05, **P< 0.01, ***P< 0.001.
MiR-223 attenuates liver fibrosis by inhibiting the TAZ-IHH-GLI2 signaling pathway via the crosstalk between hepatocytes and HSCs
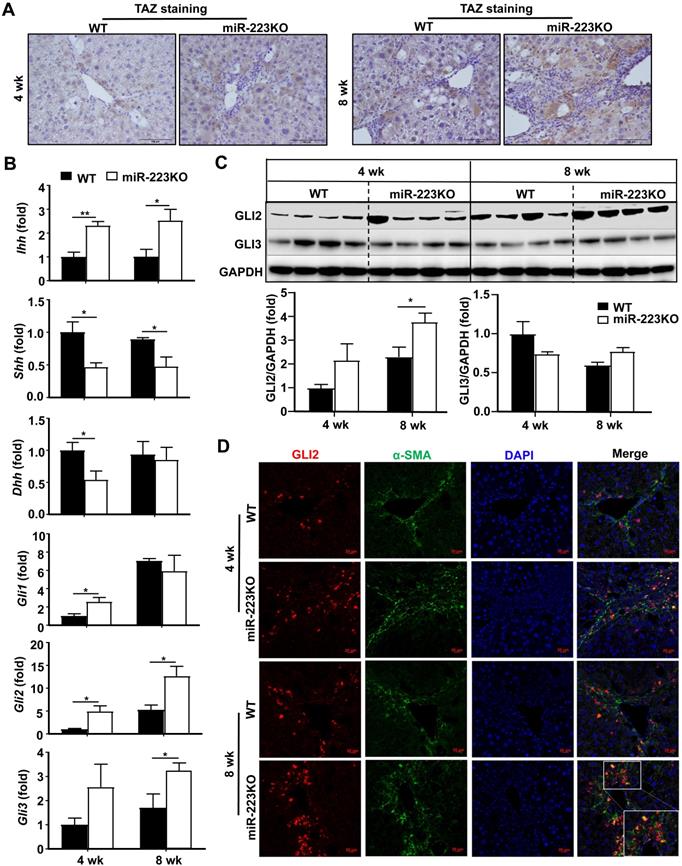
We have previously demonstrated that transcriptional coactivator with PDZ-binding motif (TAZ) is the direct target of miR-223 in hepatocytes in NASH-related liver fibrosis [19]. These findings prompted us to investigate whether TAZ in hepatocytes is also the target of miR-223 in CCl4-induced liver fibrosis. As shown in Fig. 2A and Supplementary Fig. S2A, TAZ expression in hepatocytes was markedly increased in miR-223KO mice compared to WT mice after chronic CCl4 injection. TAZ has been reported to promote liver fibrosis by inducing Indian hedgehog (IHH) in hepatocytes [21], which is a secretory factor that activates the Hedgehog signaling pathway in HSCs and mediates steatosis-to-NASH progression [21]. In the presence of Hedgehog ligand, including IHH, Sonic hedgehog (SHH) and Desert hedgehog (DHH), the signal transducer Smoothened is activated, which promotes the accumulation and nuclear translocation of transcription factor GLI family including GLI1, GLI2, and GLI3, finally activating HSCs [1,22]. To further clarify whether miR-223 attenuates liver fibrosis via TAZ-Hedgehog pathway in CCl4-induced liver fibrosis, we analyzed the expression of Hedgehog signaling-related factors in the liver of WT and miR-223KO mice. Indeed, as illustrated in Fig. 2B, miR-223KO mice exhibited higher hepatic mRNA levels of Ihh than WT mice; however, hepatic Shh and Dhh mRNA levels were downregulated in miR-223KO mice compared with WT mice. Other hedgehog signaling-related gene (e.g Smo, Ptch1) levels showed no significant difference between WT and miR-223KO mice (Supplementary Fig. S2B). Immunohistochemistry staining also revealed that the greater hepatic IHH expression in miR-223KO mice than that in WT mice after chronic CCl4 treatment (Supplementary Fig. S2C). Additionally, the expression of transcription factor GLI family especially Gli2 was remarkably elevated in miR-223KO mice. Elevation of GLI2 protein in miR-223KO mice was also observed by western blot analysis (Fig. 2C). To further determine which cell type expressed GLI2 in the liver, we performed α-SMA and GLI2 immunofluorescence double staining in the livers of WT and miR-223KO mice. Notably, GLI2 expression was highly elevated in the fibrotic area with co-staining of α-SMA in miR-223KO mice compared with WT mice (Fig. 2D). Taken together, these results strongly suggest that miR-223 inhibits paracrine IHH production by targeting TAZ in hepatocytes, leading to inhibition of Hedgehog signaling pathway in HSCs.
MiR-223KO mice are more susceptible to CCl4-induced liver fibrosis due to enhanced secretion of the TAZ-targeting Indian hedgehog (IHH) by hepatocytes. WT and miR-223KO mice were treated with CCl4 twice a week for 4 weeks or 8 weeks. (A) Representative images of TAZ staining are shown. (B) RT-qPCR analyses of the genes related to Hedgehog signaling pathway in the livers from CCl4-treated WT and miR-223KO mice. (C) Liver tissues were subjected to western blot analyses of GLI2 and GLI3 (Upper panel). The blots were quantified (lower panel). (D) Liver tissue samples were subjected to α-SMA and GLI2 immunofluorescence double staining. Representative images of GLI2 (red), α-SMA (green), and nuclei (blue) in the livers from WT and miR-223KO mice are shown. Values represent means ± SEM (n=5-8/group). *P< 0.05, **P< 0.01.
Gli2 is a target of miR-223 in HSCs
The above data suggest that the increased GLI2 expression in the HSCs from miR-223KO mice is due to the enhanced stimulation of IHH produced by hepatocytes. Next we investigated the effect of miR-223 overexpression in hepatocytes on CCl4-induced liver fibrosis in vivo. Mice were injected with CCl4 for 4 weeks followed by Gfp- or miR-223 expressing adenovirus injection for 2 weeks. As we previously reported, overexpression of miR-223 led to decreased fibrogenic gene expression and attenuated CCl4-induced liver fibrosis [23]. In the current study, we demonstrated that TAZ and IHH protein levels in hepatocytes were markedly decreased in mice injected with miR-223-expressing adenovirus (Ad-miR-223) (Fig. 3A and 3B). In addition, hepatic expressions of Gli2, Gli3 and IHH target gene, osteopontin (Opn) [24], were downregulated after administration of Ad-miR-223 (Fig. 3C). Consistently, immunofluorescence staining revealed that injection of Ad-miR-223 reduced GLI2 expression in HSCs in CCl4-induced liver fibrosis (Fig. 3D).
Overexpression of miR-223 attenuates TAZ and GLI2 expression in the liver and directly inhibits Gli2 in HSCs. (A-D) C57BL/6J mice were injected with CCl4 for 6 weeks and were given intravenous injection of Ad-Gfp or Ad-miR-223 at the end of the 4th week. (n=6/group). The liver tissues were collected at the 6th week. (A-B) Representative images of TAZ staining and IHH staining are shown. Quantification of the TAZ+ and IHH+ area per field was performed and is shown. (C) RT-qPCR analyses of Gli2, Gli3 and Opn in liver tissues. (D) Representative images of GLI2 (red), α-SMA (green), and nuclei (blue) in the livers are shown. (E) Using a miRNA database (http://www.targetscan.org/), putative binding sites of miR-223 were predicted in the 3'-UTR of Gli2 mRNA in mice. The dashed line represents complementary base pairs between miR-223 and Gli2 3'-UTR. (F) Primary mouse HSCs were transfected with miR-223 mimics and nonspecific miRNA mimics (NS) for 24 hours. The mRNA levels of Gli2 and Gli3 were analyzed by RT-qPCR (G) Primary hepatocytes were transfected with Ad-miR-223 for 24 hours followed by H2O2 (500 µM, 5h) treatment. The expression of miR-223 in hepatocytes and hepatocyte-derived EVs was analyzed by RT-qPCR. Values represent means ± SEM. In panels A-D, n=6/group; Panels F-G are from three independent experiments. *P< 0.05.
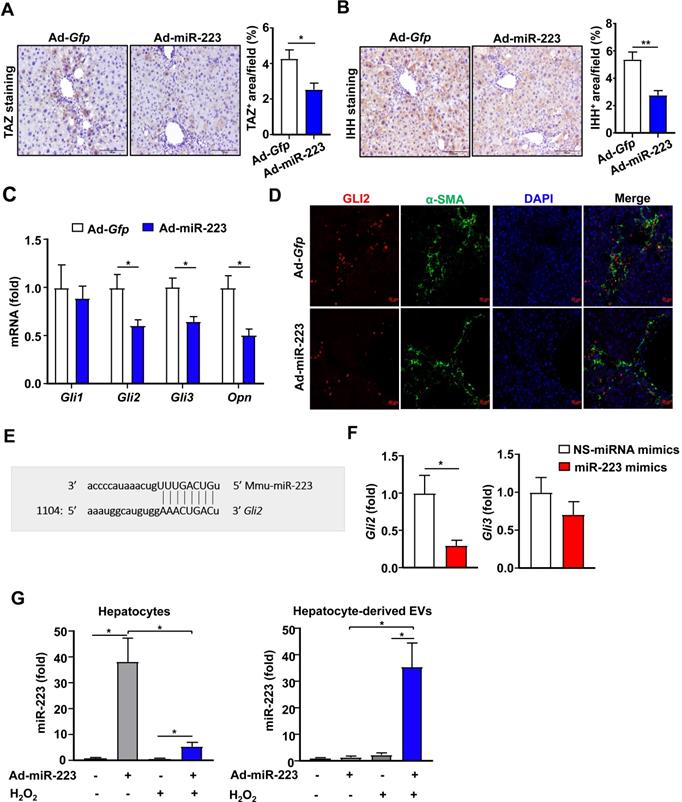
Next, we wondered whether GLI2 can be directly regulated by miR-223 in HSCs. To verify this hypothesis, we conducted bioinformatic analysis by using targetscan (http://www.targetscan.org/). As a result, miR-223 was predicted to directly bind the 3'-UTR of Gli2 (Fig. 3E). To determine the function of miR-223 in regulating GLI2 expression, we overexpressed miR-223 in primary mouse HSCs via the transfection of miR-223 mimics. As shown in Fig. 3F, overexpression of miR-223 significantly suppressed Gli2 but not Gli3 expression in primary HSCs.
Given that adenovirus are vehicles which mainly deliver genes to hepatocytes, we speculated that overexpressed miR-223 in hepatocyte can be transferred into HSCs by hepatocyte-derived EVs. To address this issue, we examined the expression of miR-223 in hepatocyte-derived EVs after transduction of Ad-miR-223 in vitro. Since oxidative stress plays a vital role in CCl4-induced toxicity, we treated hepatocytes with hydrogen peroxide (H2O2), which is an inducer of oxidative stress in vitro [25]. As shown in Fig. 3G, miR-223 expression in hepatocytes was dramatically elevated after treatment with Ad-miR-223; however, after treatment with H2O2, such miR-223 expression in hepatocytes was markedly reduced while miR-223 express was highly elevated in hepatocyte-derived EVs. These data suggest that oxidative stress can stimulate hepatocytes to release miR-223 enriched EVs.
MiR-223 ameliorates liver fibrosis by targeting PDGFRα/β in HSCs
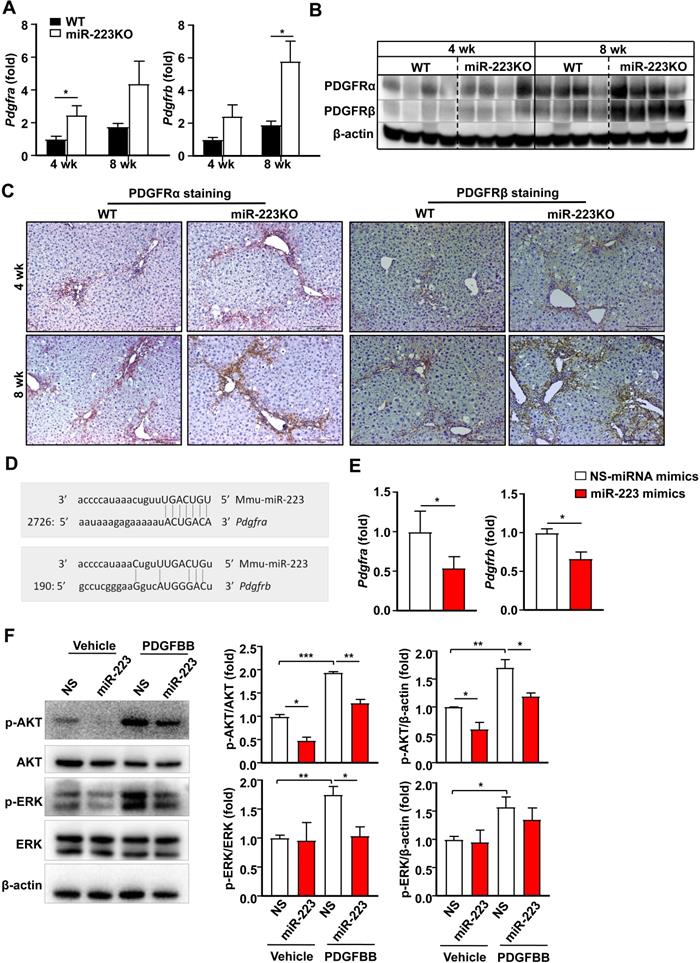
PDGF signaling plays a critical role in promoting HSC proliferation and migration [26,27]. MiR-223 has been reported to directly target PDGF receptor β (PDGFRβ) in vascular smooth muscle cells [28]. We speculated that miR-233 may interfere with the PDGF signaling pathway by modulating PDGFR expression in HSCs. To address this hypothesis, we first analyzed the expression of PDGFRα and PDGFRβ in the livers of chronic CCl4-treated WT and miR-223KO mice. As illustrated in Fig. 4A, B and Supplementary Fig. S3A, hepatic PDGF receptor α (PDGFRα) and PDGFRβ mRNA and protein levels were significantly elevated in chronic CCl4-treated miR-223KO mice compared to chronic CCl4-treated WT mice. Immunohistochemistry staining further identified a greater expression of PDGFRα and PDGFRβ in the fibrotic regions of chronic CCl4-treated miR-223KO mice than in WT mice (Fig. 4C and Supplementary Fig. S3B). To further investigate whether miR-223 can regulate PDGFRα/β expression, we performed bioinformatics analysis and identified the binding site for miR-223 in the 3'-UTR of Pdgfra and Pdgfrb (Fig. 4D). The direct target of PDGFRα and PDGFRβ by miR-223 was further supported by RT-qPCR data showing that overexpression of miR-223 significantly downregulated the expressions of PDGFRα and PDGFRβ in HSCs (Fig. 4E). PDGF-BB, a ligand for PDGF signaling, binds to PDGF receptors and induces phosphorylation of ERK and AKT, thereby leading to HSC proliferation, activation and migration [3]. As illustrated in Fig. 4F, PDGF-BB treatment induced the phosphorylation of ERK and AKT, such phosphorylation was blocked by overexpression of miR-223 in primary HSCs. These findings support the notion that miR-223 inhibits the activation of PDGF signaling pathway in HSCs by targeting PDGFRα/β.
MiR-223 inhibits PDGF signaling pathway in HSCs by targeting Pdgfra and Pdgfrb. (A-C) WT and miR-223KO mice were treated with CCl4 twice a week for 4 weeks or 8 weeks. (A) RT-qPCR analyses of Pdgfra and Pdgfrb. (B) Liver tissues were subjected to western blot analyses of PDGFRα and PDGFRβ. (C) Representative images of PDGFRα and PDGFRβ staining are shown. (D) Using a miRNA database (http://www.targetscan.org/), putative binding sites of miR-223 were predicted in the 3'-UTR of mouse Pdgfra and Pdbgrb mRNAs. (E) Primary mouse HSCs were transfected with miR-223 mimics and nonspecific miRNA mimics (NS), and the mRNA levels of Pdgfra and Pdgfrb were detected by RT-qPCR analyses. (F) Primary mouse HSCs were transfected with miR-223 mimics and nonspecific miRNA mimics (NS) for 24 hours followed by treatment with vehicle or PDGFBB (10 ng/ml) for 20 min. Total cell lysates were subjected to western blot analyses of PDGF signaling pathway. The blots were quantified (right panel). Values represent means ± SEM. In panels A-C, n=5-8/group; Panels E-F from three independent experiments. *P< 0.05, **P< 0.01, ***P< 0.001.
MiR-223 directly inhibits HSC activation and proliferation
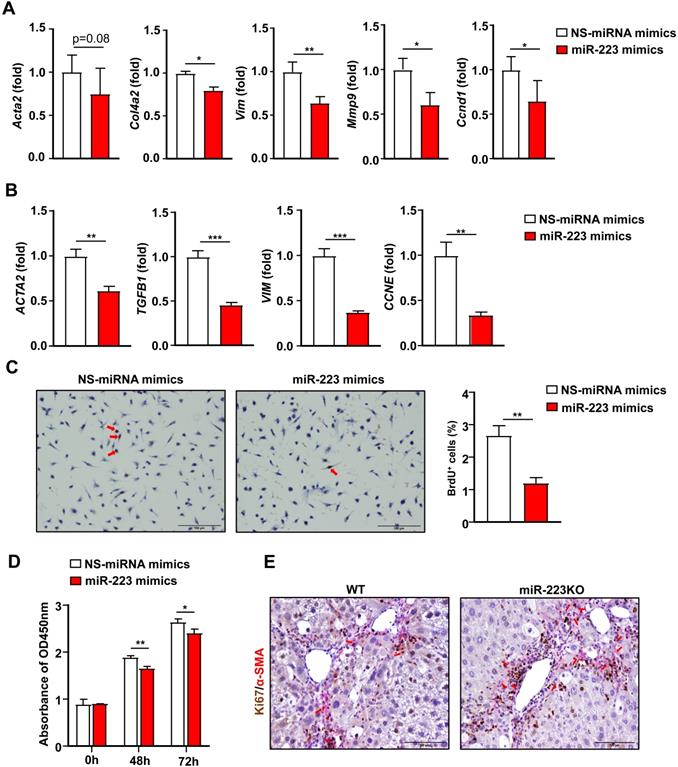
Activation of HSCs is the central event in liver fibrosis [3], thus we focused on the effects of miR-223 on HSCs for the rest of our study. We have previously demonstrated that primary HSCs from miR-223KO mice expressed higher levels of fibrotic genes [19], indicating that miR-223 deficiency promotes fibrogenesis in HSCs. In addition, the above data also show that miR-223 directly inhibits GLI2, PDGFRα and PDGFRβ in HSCs. Collectively, this prompted us to hypothesize that miR-223 may directly inhibit HSC activation. To test this hypothesis, we overexpressed miR-223 in primary mouse HSCs and human HSC cell line LX-2 cells via the transfection of miR-223 mimics. As illustrated in Fig. 5A, overexpression of miR-223 downregulated the expression of Acta2, Col4a2, Vim and Mmp9. Consistently, in LX-2 cells, overexpression of miR-223 leads to decreased expression of ACTA2, TGFB1, and VIM (Fig. 5B). Interestingly, the cell cycle genes Ccnd1 and CCNE were also downregulated by miR-223 in primary HSCs and LX-2 cells, respectively (Fig. 5A and 5B). These results promoted us to determine the function of miR-223 in the proliferation of HSCs. We performed BrdU staining in primary HSCs and observed the reduced number of BrdU positive cells in HSCs after overexpression of miR-223 (Fig. 5C). Besides, CCK-8 assay further revealed that overexpression of miR-223 significantly suppressed the proliferation of LX-2 cells (Fig. 5D). To further confirm the role of miR-223 in HSC proliferation in vivo, we performed α-SMA and Ki67 immunofluorescence double staining in the livers of WT and miR-223KO mice after 8 weeks CCl4 injection. Notably, more Ki67 and α-SMA double positive cells were detected in miR-223KO mice compared with WT mice, suggesting that miR-223 deletion enhances the proliferation of HSCs in liver fibrosis (Fig. 5E).
Overexpression of miR-223 suppresses HSC activation and proliferation. (A) Primary mouse HSCs were transfected with miR-223 mimics and nonspecific miRNA mimics (NS) for 24 hours. The expression of the genes related to fibrogenesis and cell proliferation was analyzed by RT-qPCR. (B) Human HSC cell line LX-2 cells were transfected with miR-223 mimics and nonspecific miRNA mimics (NS) for 48 hours. The expression of genes involved in fibrogenesis and cell proliferation was analyzed by RT-qPCR. (C) Primary mouse HSCs were transfected with miR-223 mimics and nonspecific miRNA mimics (NS) for 24 hours. Cells were incubated with BrdU (10 uM) for 6 hours before staining. Representative images of BrdU staining are shown in the left panel. The number of BrdU+ HSCs was counted in the right panel. (D) LX-2 cells were transfected with miR-223 mimics and nonspecific miRNA mimics (NS) for 48 hours and 72 hours followed by CCK8 assay. (E) Liver tissue samples from 8-week CCl4-treated WT and miR-223KO mice were subjected to α-SMA and Ki67 immunofluorescence staining. Representative images of α-SMA (pink) and Ki67 (brown) in the liver from WT and miR-223KO mice are shown. Values represent means ± SEM. Panels A-D from three independent experiments. In panel E, n=5-8/group. *P< 0.05, **P< 0.01, ***P< 0.001.
Regulation of miR-223 expression in HSCs during the progression of liver fibrosis
Our previous data show that hepatic miR-223 expression is increased in a NASH-related fibrosis mouse model induced by high fat diet feeding [19]. Therefore, we hypothesized that miR-223 expression might be altered during the pathogenesis of liver fibrosis. Similar to the result of NASH-related liver fibrosis [19], hepatic miR-223 expression was significantly increased after chronic CCl4 treatment (Fig. 6A). To determine whether miR-223 expression in HSCs was altered in vivo during fibrosis, we isolated primary HSCs from mice treated with and without CCl4, followed by analysis of miR-223 expression. As demonstrated in Fig. 6B, as expected, Acta2 and Col1a1 mRNA levels were dramatically elevated in activated HSCs from CCl4-treated mice compared with quiescent HSCs from control mice; however, miR-223 expression was comparable between activated HSCs and quiescent HSCs. To our surprise, pri-miR-223, which is the precursor of miR-223 [29], was significantly decreased in activated HSCs from CCl4-treated mice compared to quiescent HSCs from control mice.
Regulation of miR-223 expression in vivo and in vitro during HSC activation. (A) C57BL/6J mice were injected with CCl4 for 4 weeks or 8 weeks. Hepatic miR-223 was measured by RT-qPCR. (B) C57BL/6J mice were injected with CCl4 for 4 weeks. Primary HSCs were isolated from CCl4-treated mice and control mice. Acta2, Col1a1, miR-223 and pri-223 expression levels were measured by RT-qPCR. (C) Primary HSCs were isolated and cultured for 1 day, 3 days, 5 days and 7 days, the expression of Acta2, Col1a1, miR-223 and pri-miR-223 was analyzed by RT-qPCR at indicated time points. Values represent means ± SEM. In panel A, n=5-8/group. Panels B-C from three independent experiments. *P< 0.05, **P< 0.01, ***P< 0.001.
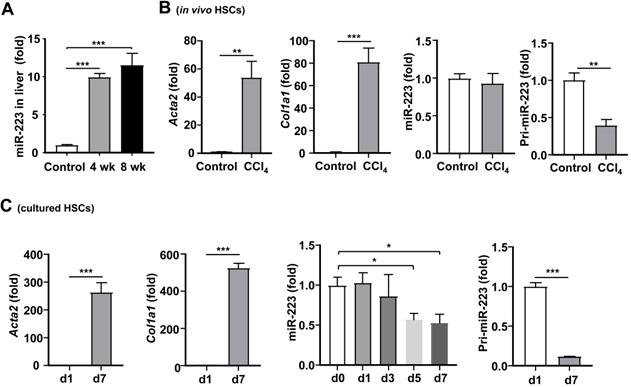
To further examine the effect of HSC activation on miR-223 expression, we examined the expression of miR-223 at different time points during HSC activation in vitro. As illustrated in Fig. 6C, compared to freshly isolated quiescent HSCs, miR-223 expression was decreased in activated HSCs on day 5 and day 7. The expression of Acta2 and Col1a1 mRNAs was highly elevated in activated HSCs, confirming the HSC activation. Furthermore, pri-miR-223 expression was also reduced in activated HSCs compared to quiescent HSCs.
Activated HSCs take up neutrophil-derived EVs, thus elevating miR-223
The above data show that the biosynthesis of miR-223 is decreased in activated HSCs in vivo and in vitro, but mature miR-223 remains unchanged in activated HSCs in vivo, suggesting the possibility that other types of cells may compensate the reduction of miR-223 levels in activated HSCs in vivo by transferring miR-223.
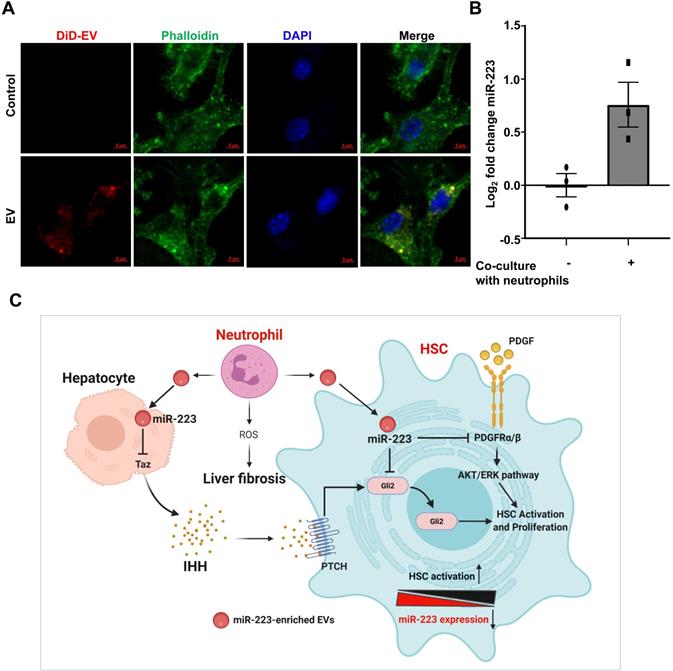
In chronic CCl4-induced liver fibrosis model, CCl4 injection induces hepatocyte death, followed by infiltration of inflammatory cells, such as neutrophils [30]. We detected significant neutrophil infiltration in fibrotic areas after chronic CCl4 treatment as evidenced by MPO (a neutrophil marker) staining in Supplementary Fig. S4. Since neutrophils express miR-223 at very high levels and can transfer miR-223 into hepatocytes [15] and macrophages [9], we hypothesized that during liver fibrosis, miR-223 can be transferred from neighboring neutrophils into HSCs. To address this question, we tested whether HSCs can take up neutrophil-derived miR-223 enriched EVs. As illustrated in Fig. 7A, after co-cultured with fluorescence (DiD)-labelled neutrophil-derived EVs, HSCs had significant amount of fluorescence staining, suggesting that HSCs can take up neutrophil-derived EVs. Furthermore, we co-cultured WT neutrophils with miR-223 KO HSCs isolated from miR-223KO mice, and observed detectable miR-223 expression in HSCs (Fig. 7B). Since miR-223 deficient HSCs do not have miR-223, we concluded that the detected miR-223 was due to its transfer from neutrophils into HSCs.
Activated HSCs take up neutrophil-derived EVs, thus elevating miR-223. (A) EVs derived from neutrophils were fluorescently labeled with the cell membrane dye DiD prior to its incubation with HSCs. The HSCs were incubated with fluorescently labeled-EVs for 24 hours. Representative images of DiD immunofluorescence (red), F-actin (green) and nuclei (blue) are shown. (B) Primary HSCs were isolated from miR-223KO mice. The expression of miR-223 was measured in HSCs after co-culture with bone marrow neutrophils for 6 hours. Values represent means ± SEM from three independent experiments. (C) Schematic representation of the mechanism by which miR-223 inhibits HSC activation and proliferation. MiR-223 protects against liver fibrosis by regulating multiple targets in different hepatic cells. In hepatocytes, miR-223 inhibits TAZ expression, leading to decreased IHH secretion from hepatocytes, which serves as the ligand for Hedgehog signaling in HSCs. In HSCs, miR-223 directly suppresses Gli2 and Pdgfra/b expression and eventually suppresses HSC activation and proliferation. During the activation of HSCs, miR-223 expression is decreased, which accelerates HSC activation. Neutrophils can deliver miR-223 into HSCs by EVs, which can compensate the decreased miR-223 biosynthesis in HSCs in vivo, thereby inhibiting liver fibrosis. (Image created with BioRender.com).
Discussion
To our knowledge, the current study demonstrated for the first time that miR-223 directly inhibits HSC activation and proliferation by targeting Gli2 and Pdgfra/b, thus ameliorating liver fibrosis. Furthermore, results from our mechanistic studies suggest that pri-miR-223 and miR-223 expression are downregulated during HSC activation in vitro, and such downregulation may accelerate HSC activation. However, despite of pri-miR-223 downregulation, miR-223 expression was not decreased in activated HSCs in vivo from fibrotic livers. In vitro co-culture experiments demonstrated that HSCs can take up neutrophil-derived miR-223-enriched EVs. We have integrated all of these findings together into a model depicting how miR-223 restricts liver fibrosis via the potential crosstalk of several types of cells in the liver (Fig. 7C).
The anti-fibrotic role of miR-223 has been reported in NASH-related fibrosis [15,19,31]. However, the functional roles of miR-223 in CCl4-induced liver fibrosis and in HSCs have not been fully illustrated. Our data clearly revealed that miR-223KO mice had greater liver fibrosis as evidenced by stronger Sirius red and α-SMA immunostaining and increased fibrogenesis genes expression, suggesting that miR-223 plays a protective role in CCl4-induced liver fibrosis. Our finding is in line with the previous report by Calvente et al. which demonstrated that miR-223 deficiency impaired the spontaneous resolution of liver fibrosis caused by CCl4 administration [9]. However, Schueller et al. reported no differences in the degree of liver injury and fibrosis between WT and miR-223KO mice in the chronic CCl4 mouse model [17]. The reasons for the discrepancy among these reports may be due to the difference in animal facility, the source of mice, different WT control mice used, and different doses of CCl4 treatment. In our current study, we used littermate WT mice as the control group; while non-littermate WT control mice were used in other studies [17].
Till date, various targets of miR-223 have been identified in hepatocytes, neutrophils and macrophages [13]; however, the targets of miR-223 in HSCs have not been explored. In the current study, we have identified several targets of miR-223 in HSCs. First, we demonstrated that Gli2 is a direct target of miR-223 in HSCs in vivo and in vitro. GLI2, which is a transcriptional activator of Hh signaling pathway [22], was significantly elevated in miR-223KO mice and downregulated by Ad-miR-223 administration after CCl4 challenge. Inhibition of GLI2 may contribute to the anti-fibrotic functions of miR-223. Second, we also found that Pdgfra and Pdgfrb are targets of miR-223 in HSCs and overexpression of miR-223 in HSCs blunted PDGF downstream ERK and AKT signaling, thus inhibiting HSC proliferation. PDGR signaling is one of the most important pathways which regulates activation and proliferation of HSCs during hepatic fibrosis [26,27]. It has been reported that PDGFRα promotes HSC proliferation and migration [26,32], while PDGFRβ induces the activation and transdifferentiation of HSCs into myofibroblasts [33]. Taken together, miR-223 suppresses the activation and proliferation of HSCs by regulating Hh and PDGF signaling pathway.
In addition to the targets of miR-223 in HSCs, our data also suggest that miR-223 limits liver fibrosis partially by targeting TAZ in hepatocytes. TAZ has been implicated in promoting liver fibrosis, particularly in the steatosis-to-NASH conversion via the induction of IHH in hepatocytes, a secretory factor that activates HSCs [21]. Of note, overexpression of miR-223 in hepatocytes ameliorates CCl4-induced liver fibrosis along with the hepatic downregulation of TAZ and IHH. However, the mechanism by which hepatic Shh and Dhh mRNA levels are decreased in miR-223KO mice compared with WT mice after chronic CCl4 injection remains unclear. Further studies are needed to investigate whether SHH and DHH can be regulated by miR-223. Collectively, miR-223 ameliorates liver fibrosis by regulating multiple targets in HSCs and hepatocytes.
In mouse models of liver fibrosis induced by BDL or chronic CCl4 injection, hepatic miR-223 levels were found to be elevated [17]. In line with previous reports, we also observed the increased hepatic miR-223 expression after chronic CCl4 treatment. One of the major reasons for elevated miR-223 expression in the fibrotic lives is probably due to the infiltration of neutrophils that express much higher miR-223 than hepatocytes [34]. Interestingly, miR-223 expression was markedly downregulated in activated HSCs in vitro but was not reduced in activated HSCs in vivo from CCl4-induced fibrotic livers despite of downregulation of pri-miR-223 in both in vitro and in vivo activated HSCs. Levels of mature miR-223 in the cells are controlled by biogenesis, secretion and uptake of miR-223 via the EVs. Because the biogenesis (pri-miR-223) was reduced, the unchanged miR-223 levels in activated HSCs in vivo were probably due to reduced secretion and/or increased uptake of miR-223. At the present, we do not have data whether secretion of miR-223 was reduced in activated HSCs, however, several lines of evidence suggest that activated HSCs may have increased uptake of miR-223 in the fibrotic livers in vivo. First, neutrophils, which expressed miR-223 at the highest levels among all types of cells, are accumulated in the fibrotic regions, and contact activated HSCs. Second, previous studies have reported that neutrophils can transfer miR-223 into macrophages [9] and hepatocytes [15] via the EVs. In the current study, we found that neutrophil-derived EVs can be taken up by HSCs, leading to elevated miR-223 levels in HSCs. The mechanism of EV uptake by HSCs is a challenging question. Several endocytic pathways have been implicated in EV uptake, including endocytosis, micropinocytosis, phagocytosis, and lipid raft-mediated internalization [35]. Different cell types likely have different underlying mechanisms for EV uptake. For example, low-density lipoprotein receptor on hepatocytes has been shown to promote selective uptake of miR-223-enriched EVs from neutrophils in obese mice [15]. Besides, several stimuli such as TGFβ may also be involved in regulating EV uptake [36]. Further studies are warranted to elucidate the mechanisms of EV uptake by HSCs in the pathogenesis of liver fibrosis.
Hepatic neutrophil infiltration is a hallmark for liver inflammation in various types of liver diseases. However, the mechanisms by which neutrophils contribute to liver fibrosis remain obscure. One previous study showed that impaired neutrophil recruitment does not impact on CCl4-induced liver fibrosis [37]. While Calvente et al reported that neutrophil depletion impaired resolution from both CCl4- and MCD-induced liver fibrosis [9]. To better understand the function of neutrophils in liver fibrosis, some studies have focused on the communication between neutrophils and HSCs [6]. Zhou et al. demonstrated that activated HSCs prolong the survival of neutrophils, which further promotes liver fibrosis via the production of reactive oxygen species (ROS) [6]. However, our data suggest a protective role of neutrophils in liver fibrosis by delivering exosomal miR-223 into HSCs, and reveal an important mechanism responsible for neutrophils in regulating liver fibrosis, which will help understand the complex roles of neutrophils in the progression from chronic liver inflammation to fibrosis. Further studies are needed to clarify the specific functions of miR-223 in neutrophils in liver fibrosis by using neutrophil-specific miR-223 knockout mice.
Due to an important role of miR-223 in inhibiting HSC activation and liver fibrosis, elevation of miR-223 levels may open new avenues for the treatment of liver fibrosis in the future. Indeed, Calvente et al have reported the therapeutic effect of the synthetic miR-223 analog miR-223-3p in the progression of chronic inflammation and fibrosis in NASH [31]. Restoration of hepatic miR-223 ameliorates steatosis and fibrosis in HFD-fed Ldlr KO mice [15]. In addition, the therapeutic effect of adenovirus-mediated overexpression of miR-223 was also recently demonstrated in liver fibrosis induced by chronic CCl4 treatment [23]. In the current study, we demonstrated that overexpression of miR-223 in hepatocytes can also ameliorate CCl4-induced liver fibrosis by secreting TAZ-targeting IHH and by releasing miR-223-enriched EVs. Since HSCs are in close contact with hepatocytes in vivo, we predicted that adenovirus-mediated overexpression of miR-223 in hepatocytes may be an effective approach to treat liver fibrosis not only via the direct inhibition of fibrogenic genes in hepatocytes but also via the production of miR-223-enriched EVs that can be transferred into HSCs. Finally, due to the high stability and rapid biodistribution of EVs in the liver [38], it will be interesting to explore the therapeutic effect of miR-223-loaded EVs in liver fibrosis.
Abbreviations
ALT: alanine aminotransferase; BDL: bile duct ligation; CCl4: carbon tetrachloride; DHH: Desert hedgehog; ECM: extracellular matrix; EVs: extracellular vesicles; GLI2: GLI Family Zinc Finger 2; H2O2: hydrogen peroxide; Hh: Hedgehog; miRNAs: microRNAs; HSCs: hepatic stellate cells; KO: knockout; MPO: myeloperoxidase; NASH: nonalcoholic steatohepatitis; Opn: osteopontin; PDGF: platelet-derived growth factor; ROS: reactive oxygen species; RT-qPCR: reverse transcription quantitative PCR; SHH: Sonic hedgehog; TAZ: PDZ-binding motif; TGFβ: transforming growth factor β; UTRs: untranslated regions; WT: wild-type.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Financial support
This work was supported by the intramural program of NIAAA, NIH (B.G.). Dr. Xiaolin Wang was supported by a Fellowship from Ruijin Hospital when she performed experiments at the NIAAA during 2019-2021.
Author contribution
XW and YH designed and conducted the experiments, XW wrote the paper; WS, SHP, YF, SH, RMR and DF conducted some experiments; YH and BG supervised the whole project and edited the paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nature reviews Gastroenterology & hepatology. 2017;14:397-411
2. Mehal WZ, Iredale J, Friedman SL. Scraping fibrosis: expressway to the core of fibrosis. Nature medicine. 2011;17:552-553
3. Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Advanced drug delivery reviews. 2017;121:27-42
4. Mederacke I, Hsu CC, Troeger JS. et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nature communications. 2013;4:2823
5. Koyama Y, Brenner DA. Liver inflammation and fibrosis. The Journal of clinical investigation. 2017;127:55-64
6. Zhou Z, Xu MJ, Cai Y. et al. Neutrophil-Hepatic Stellate Cell Interactions Promote Fibrosis in Experimental Steatohepatitis. Cellular and molecular gastroenterology and hepatology. 2018;5:399-413
7. Cho Y, Szabo G. Two faces of neutrophils in liver disease development and progression. Hepatology. 2020
8. Liu K, Wang FS, Xu R. Neutrophils in liver diseases: pathogenesis and therapeutic targets. Cellular & molecular immunology. 2021;18:38-44
9. Calvente CJ, Tameda M, Johnson CD. et al. Neutrophils contribute to spontaneous resolution of liver inflammation and fibrosis via microRNA-223. The Journal of clinical investigation. 2019;129:4091-4109
10. Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130:435-452
11. Pritchard CC, Cheng HH, Tewari M. MicroRNA profiling: approaches and considerations. Nature reviews Genetics. 2012;13:358-369
12. Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nature reviews Genetics. 2010;11:597-610
13. Wang X, He Y, Mackowiak B, Gao B. MicroRNAs as regulators, biomarkers and therapeutic targets in liver diseases. Gut. 2020
14. Szabo G, Bala S. MicroRNAs in liver disease. Nature reviews Gastroenterology & hepatology. 2013;10:542-552
15. He Y, Rodrigues RM, Wang X. et al. Neutrophil-to-hepatocyte communication via LDLR-dependent miR-223-enriched extracellular vesicle transfer ameliorates nonalcoholic steatohepatitis. The Journal of clinical investigation. 2020
16. Ye D, Zhang T, Lou G, Liu Y. Role of miR-223 in the pathophysiology of liver diseases. Experimental & molecular medicine. 2018;50:128
17. Schueller F, Roy S, Loosen SH. et al. miR-223 represents a biomarker in acute and chronic liver injury. Clinical science. 2017;131:1971-1987
18. Calvente CJ, Tameda M, Johnson CD. et al. Neutrophils contribute to spontaneous resolution of liver inflammation and fibrosis via microRNA-223. The Journal of clinical investigation. 2019;130:4091-4109
19. He Y, Hwang S, Cai Y. et al. MicroRNA-223 Ameliorates Nonalcoholic Steatohepatitis and Cancer by Targeting Multiple Inflammatory and Oncogenic Genes in Hepatocytes. Hepatology. 2019;70:1150-1167
20. Mederacke I, Dapito DH, Affo S, Uchinami H, Schwabe RF. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nature protocols. 2015;10:305-315
21. Wang X, Zheng Z, Caviglia JM. et al. Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2016;24:848-862
22. Machado MV, Diehl AM. Hedgehog signalling in liver pathophysiology. Journal of hepatology. 2018;68:550-562
23. Hou X, Yin S, Ren R. et al. Myeloid cell-specific IL-6 signaling promotes miR-223-enriched exosome production to attenuate NAFLD-associated fibrosis. Hepatology. 2020 doi: 10.1002/hep.31658
24. Syn WK, Choi SS, Liaskou E. et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology. 2011;53:106-115
25. Hwang S, Wang X, Rodrigues RM. et al. Protective and Detrimental Roles of p38alpha Mitogen-Activated Protein Kinase in Different Stages of Nonalcoholic Fatty Liver Disease. Hepatology. 2020;72:873-891
26. Kikuchi A, Pradhan-Sundd T, Singh S, Nagarajan S, Loizos N, Monga SP. Platelet-Derived Growth Factor Receptor alpha Contributes to Human Hepatic Stellate Cell Proliferation and Migration. The American journal of pathology. 2017;187:2273-2287
27. Yu J, Moon A, Kim HR. Both platelet-derived growth factor receptor (PDGFR)-alpha and PDGFR-beta promote murine fibroblast cell migration. Biochemical and biophysical research communications. 2001;282:697-700
28. Zeng Z, Xia L, Fan X. et al. Platelet-derived miR-223 promotes a phenotypic switch in arterial injury repair. The Journal of clinical investigation. 2019;129:1372-1386
29. Treiber T, Treiber N, Meister G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nature reviews Molecular cell biology. 2019;20:5-20
30. Louis H, Van Laethem JL, Wu W. et al. Interleukin-10 controls neutrophilic infiltration, hepatocyte proliferation, and liver fibrosis induced by carbon tetrachloride in mice. Hepatology. 1998;28:1607-1615
31. Jimenez Calvente C, Del Pilar H, Tameda M, Johnson CD, Feldstein AE. MicroRNA 223 3p Negatively Regulates the NLRP3 Inflammasome in Acute and Chronic Liver Injury. Molecular therapy: the journal of the American Society of Gene Therapy. 2020;28:653-663
32. Kikuchi A, Singh S, Poddar M. et al. Hepatic Stellate Cell-Specific Platelet-Derived Growth Factor Receptor-alpha Loss Reduces Fibrosis and Promotes Repair after Hepatocellular Injury. The American journal of pathology. 2020;190:2080-2094
33. Kocabayoglu P, Lade A, Lee YA. et al. beta-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. Journal of hepatology. 2015;63:141-147
34. Li M, He Y, Zhou Z. et al. MicroRNA-223 ameliorates alcoholic liver injury by inhibiting the IL-6-p47(phox)-oxidative stress pathway in neutrophils. Gut. 2017;66:705-715
35. Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. Journal of extracellular vesicles. 2014 3
36. de la Cuesta F, Passalacqua I, Rodor J, Bhushan R, Denby L, Baker AH. Extracellular vesicle cross-talk between pulmonary artery smooth muscle cells and endothelium during excessive TGF-beta signalling: implications for PAH vascular remodelling. Cell Commun Signal. 2019;17:143
37. Moles A, Murphy L, Wilson CL. et al. A TLR2/S100A9/CXCL-2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. Journal of hepatology. 2014;60:782-791
38. Szabo G, Momen-Heravi F. Extracellular vesicles in liver disease and potential as biomarkers and therapeutic targets. Nature reviews Gastroenterology & hepatology. 2017;14:455-466
Author contact
![]() Corresponding authors: Yong He, Ph.D., Laboratory of Liver Diseases, NIAAA/NIH, 5625 Fishers Lane, Bethesda, MD 20892; Tel: 301-480-7478. E-mail: yong.hegov; Bin Gao, M.D., Ph.D., Laboratory of Liver Diseases, NIAAA/NIH, 5625 Fishers Lane, Bethesda, MD 20892; Tel: 301-443-3998. E-mail: bgaonih.gov.
Corresponding authors: Yong He, Ph.D., Laboratory of Liver Diseases, NIAAA/NIH, 5625 Fishers Lane, Bethesda, MD 20892; Tel: 301-480-7478. E-mail: yong.hegov; Bin Gao, M.D., Ph.D., Laboratory of Liver Diseases, NIAAA/NIH, 5625 Fishers Lane, Bethesda, MD 20892; Tel: 301-443-3998. E-mail: bgaonih.gov.