Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Methods
Results
Discussion
Limitations
Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(9):2240-2251. doi:10.7150/ijbs.60473 This issue Cite
Research Paper
Inhibition of de novo pyrimidine synthesis augments Gemcitabine induced growth inhibition in an immunocompetent model of pancreatic cancer
Thuy Phan, PhD1, Vu H. Nguyen, PhD2, Ralf Buettner, PhD2, Corey Morales, BA2, Lifeng Yang, PhD3, Paul Wong, BS1, Weiman Tsai, BA2, Marcela d'Alincourt Salazar2, Ziv Gil, MD4, Don J Diamond, PhD2, Joshua D. Rabinowitz, MD3, Steven Rosen, MD2, Laleh G. Melstrom, MD MSCI1 ![]()
1. Department of Surgery, City of Hope National Medical Center, Duarte, CA 91010.
2. Department of Hematology, City of Hope National Medical Center, Duarte, CA 91010.
3. Lewis Sigler Institute for Integrative Genomics and Department of Chemistry, Princeton University, Princeton, NJ 08544.
4. Rambam Medical Center, Israel.
Received 2021-3-28; Accepted 2021-5-18; Published 2021-6-1
Abstract

Leflunomide (Lef) is an agent used in autoimmune disorders that interferes with DNA synthesis. De Novo pyrimidine synthesis is a mechanism of Gemcitabine (Gem) resistance in pancreatic cancer. This study aims to assess the efficacy and changes in the tumor microenvironment of Lef monotherapy and in combination with Gem, in a syngeneic mouse model of pancreatic cancer.
Methods: MTS proliferation assays were conducted to assess growth inhibition by Gem (0-20 nM), Lef (0-40 uM) and Gem+Lef in KPC (KrasLSL.G12D/+;p53R172H/+; PdxCretg/+) cells in vitro. An in vivo heterotopic KPC model was used and cohorts were treated with: PBS (control), Gem (75 mg/kg/q3d), Lef (40 mg/kg/d), or Gem+Lef. At d28 post-treatment, tumor burden, proliferation index (Ki67), and vascularity (CD31) were measured. Changes in the frequency of peripheral and intratumoral immune cell subsets were evaluated via FACS. Liquid chromatography-mass spectrometry was used for metabolomics profiling.
Results: Lef inhibits KPC cell growth and synergizes with Gem in vitro (P<0.05; Combination Index 0.44 (<1 indicates synergy). In vivo, Lef alone and in combination with Gem delays KPC tumor progression (P<0.001). CTLA-4+T cells are also significantly decreased in tumors treated with Lef, Gem or in combination (Gem+Lef) compared to controls (P<0.05). Combination therapy also decreased the Ki67 and vascularity (P<0.01). Leflunomide inhibits de novo pyrimidine synthesis both in vitro (p<0.0001) and in vivo (p<0.05).
Conclusions: In this study, we demonstrated that Gem+Lef inhibits pancreatic cancer growth, decrease T cell exhaustion, vascularity and as proof of principle inhibits de novo pyrimidine synthesis. Further characterization of changes in adaptive immunity are necessary to characterize the mechanism of tumor growth inhibition and facilitate translation to a clinical trial.
Keywords: pancreatic cancer, leflunomide, de novo pyrimidine synthesis
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is frequently an incurable disease with less than 9% five year survival [1]. Due to the lack of symptoms the disease has often metastasized by the time of diagnosis. Less than 20% of patients are candidates for tumor resection [2]. Thus most patients undergo palliative systemic therapy. Therapeutic options for patients with metastatic PDAC that have either become resistant to first line chemotherapy or that were non-responders, remain limited. Gemcitabine (Gem) in combination with nab-paclitaxel remains a frequently utilized cytotoxic therapy, as the alternative regimen FOLIFRINOX is difficult to tolerate. Gem is often the only systemic therapy that is tolerated by patients that are more frail and with more advanced disease [3]. Chemoresistance can be related to changes in the tumor environment, drug metabolism and drug efflux [4]. Metabolic reprogramming, a known mechanism of chemoresistance leading to cancer cell proliferation and survival, can arise in response to genotoxic stress [5]. How genotoxic stress leads to metabolic reprogramming has not been clearly defined. Genotoxic chemotherapy agents can induce the de novo pyrimidine synthesis pathway to increase the availability of nucleotides essential for DNA repair [6]. Inhibition of the pyrimidine synthesis pathway can sensitize cancer cells to genotoxic chemotherapy agents [7]. Leflunomide (LEF), an agent with a long history of safety and efficacy in the treatment and prevention of autoimmune disorders and allograft rejection, targets de novo pyrimidine synthesis via inhibition of dihydroorotate dehydrogenase (DHODH) [8, 9]. DHODH is the rate-limiting enzyme in the synthesis chain of uridine and is a critical enzyme in this pathway [10]. Although LEF has also been shown to modulate T cell responses in autoimmune diseases, the role of LEF on the immune microenvironment in solid tumors has not been clearly defined. Pre-clinical data show that LEF has potent anti-neoplastic effects in multiple myeloma, oral squamous cell carcinoma, renal cell carcinoma, melanoma and non-small cell carcinoma [11-15]. In this study we aim to assess the effect of LEF with Gem in an immune competent model of pancreatic cancer and to study the immune tumor microenvironment and changes in de novo pyrimidine biosynthesis.
Methods
Cell culture and Animal Model
The KPC PDAC cell line was obtained from Ximbio (UK). Briefly, this cell line was derived from pancreatic tumors of LSL-Kras-G12D, LSL-p53-R172H, and Pdx-1-Cre mice (KPC) mice. KPC cells were grown at 37 °C, 5% CO2 atmosphere in complete endotoxin-free DMEM media (Corning, USA) containing high glucose and glutamine supplemented with 10% FBS (Gibco, USA) and 1% penicillin and streptomycin (Gibco, USA). 6-week-old female C57BL6 mice were purchased from Jackson Lab and maintained under specific pathogen-free conditions at the City of Hope (COH) Animal Research Center. Animals were handled according to Institutional Animal Care and Use Committee (IACUC) guidelines under the approved protocol #16067.
Reagents
Gemcitabine (G4177500MG, LC Laboratories) was reconstituted in water or 0.9% saline for in vitro or in vivo experiments, respectively. The drug was freshly made and injected intraperitoneally into the mice at the dose of 75 mg/kg. Teriflunomide (S4169, Selleckchem) and Leflunomide (J65917ME, Alfa Aesar) were reconstituted in DMSO or CMC/Tween 80 for in vitro or in vivo experiment, respectively. On the day of treatment, the drug was prepared and administered via oral gavage into the mice at a dose of 40 mg/kg.
Cell proliferation assay and Combination experiment
The growth inhibition of the KPC cell line was determined using the 3-(4,5-diethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfopheyl)-2H-tetrazolium (MTS) assay (CellTiter 96 Aqueous One Solution, Promega, USA). Cells (1×104 cells/100 µl/well) were seeded in 96-well tissue culture plates. After 24hrs, cells were incubated with Teriflunomide (2.5-80 µM) and/or Gem (2.5-80 nM) as single agents or in combination at a constant ratio of Teriflunomide:Gem = 1000:1. After 72 hrs of treatment, cell proliferation was evaluated using an MTS assay according to the manufacturer's instructions. Briefly, 20 μl of MTS reagent was added to each well, and the plates were incubated for 2hrs. Absorbance at 492 nm was measured with a microplate reader (Filtermax F3). Results were represented as the means ± standard deviation of the mean (SD) from triplicate wells. For potential synergistic or additive effects in a drug combination, combination index (CI) values were calculated using CompuSyn software (Cambridge, UK). In order to assess potential synergy or additive effects in drug combinations, the CI is calculated by Chou-Talalay method [16]. This provides the theoretical basis for the CI-isobologram equation that allows quantitative determination of drug interactions. Based on these algorithms, Compusyn software by Chou was applied to assed the drug synergism, addition and antagonism effects at ED50, ED75, ED90 and ED95; which are the effective doses (EDs) at which 50, 75, 90 and 95% of cells are killed. Drug synergism, addition and antagonism effects are defined by CI values of <1.0, 1.0 and >1.0 respectively.
Tumor challenge and therapy
KPC cells (5 × 105) were suspended in PBS and implanted subcutaneously into the right thigh of C57BL6 mice. Three weeks after tumor inoculation, when the tumor sizes reached 50 mm3, the mice were randomized into 4 groups (n=5/group): control, Gem, Leflunomide, and combination. The mice were treated in the following dosing regimens for 4 weeks: (i) intraperitoneal injection of Gem (75 mg/kg) every three days, and (ii) oral gavage of Leflunomide (40 mg/kg) every day. Tumor volume (mm3) was measured once a week with a caliper until the tumor volume exceeded 1000 mm3 or any experimental endpoint, as pre-determined in the IACUC protocol, was reached (V=1/2 × Length × Width × Depth).
Preparation of single cell suspensions from tumor tissues and peripheral blood
At day 28 after the first treatment, animals were euthanized, tumors and blood were harvested. The collected tumor tissues were washed with PBS and minced into 2-3 mm3 pieces using sterile blades. The tissue fragments were then incubated with Collagenase IV (1 mg/ml) and DNAse I (50 ug/ml) for 20 minutes at 37 °C followed by an incubation on ice for 10 minutes to stop the enzymatic digestion. The digested tissue fragments were gently homogenized using a plunger from a 1 ml syringe in serum-free RPMI (Corning, USA), followed by dissociation using a 40 μm cell strainer. The collected tumor cells were washed and re-suspended in PBS prior to further staining. Red blood cells were lysed using RBC lysis buffer (Biolegend, USA). Peripheral blood mononuclear cells (PBMCs) were collected, washed and re-suspended in PBS for further manipulation.
Flow cytometry analysis
For surface staining, single cell suspensions from tumors and PBMCs (1×106) were prepared as described above and incubated with 1 μl Fixable Viability Dye eFlour 506 for 10 min on ice. Next, cells were washed with PBS and incubated with fluorescence-labeled antibodies against target cell surface molecules for 30 minutes in the dark on ice. Next, for intracellular staining, cells were fixed and permeabilized using Fixation/Permeabilization solution (Invitrogen, USA) and then block with anti-mouse CD16/CD32. Cells were stained with intracellular markers (FoxP3 or CTLA-4) in permeabilization buffer for 1 hour. Four different flow cytometry panels have been applied in this study: T cell activation panel using CD3 PerCP-Cy5.5 (45-2C11), CD4 eFlour450 (RM4-5), CD8 FITC (53-6.7), CD69 APC (H1.2F3); T cell exhaustion panel using CD3 PerCP-Cy5.5 (45-2C11), CD4 eFlour450 (RM4-5), CD8 FITC (53-6.7), CTLA-4 PE (UC10-4B9); Treg panel using CD3 PerCP-Cy5.5 (45-2C11), CD4 FITC (RM4-5), CD25 APC (PC61.5), FoxP3 PE (FJK-16s); and MDSC panel using CD45-eFlour450 (30-F11), CD11b PE (M1/70), Ly6G FITC (1A8), Ly6C APC (HK1.4). All labeled antibodies were purchased from eBioscience (USA). At least 10,000 events were analyzed using a FACS Celesta flow cytometer (BD), according to the manufacturer's instructions. Doublets were excluded and alive cells were used for evaluation using FlowJo software (TreeStar).
Immunohistochemistry staining
For immunohistochemistry (IHC) staining, excised tumors from mice were fixed in 10% Neutral Buffered Formalin (NBF), processed and embedded in paraffin. Tumor blocks were then sectioned at a thickness of 5 μm and put on positively charged glass slides. IHC staining was performed on Ventana Discovery Ultra (Ventana Medical Systems, Roche Diagnostics, Indianapolis, USA) IHC automated stainer. The slides were loaded on the machine; deparaffinization, rehydration, endogenous peroxidase activity inhibition and antigen retrieval were first performed. The slides were then incubated with a primary antibody (CD31 and Ki67, Cell Signaling) followed by DISCOVERY anti-Rabbit HQ and DISCOVERY anti-HQ HRP detection system. The IHC tumor sections were visualized with DISCOVERY ChromoMap DAB Kit, counterstained with Hematoxylin (Ventana). The slide images were acquired with Leica Dmi8 Microscope (Leica Microsystems, USA) and analyzed by Image-Pro Premier Software.
Samples preparation for LC/MS Metabolomics
For in vitro experiments, KPC cells were seeded overnight in 6 well-plates using completed DMEM. DMEM with 13C/15N-isotope labeled nutrients [L-glutamine-(amide-15N), Sigma] was prepared to replace 12C/14N nutrients. Cells were treated with 20 nM Gem and 40 µM Leflunomide as single agents or in combination in isotope tracing medium for 4hrs and 24 hrs. Afterward medium was aspirated, immediately washed with PBS and metabolism was quenched with extraction buffer (40:40:20 acetonitrile:methanol:water with 0.5% formic acid). Plates were placed on dry ice for 10 min and neutralized with 15% NH4HCO3. Cell lifter was used to scrape cells from plates, extraction buffer containing cells was transferred into Eppendorf tubes and centrifuged in a benchtop microfuge at maximum speed for 30 min at 4 °C. Supernatants were transferred to LC-MS vials for analysis. For in vivo experiments, tumor tissues were collected and immediately clamped into liquid nitrogen. Tissues were stored in -80 °C until analysis. Frozen tissues were transferred into precooled 2 mL Eppendorf tubes, and pulverized with a cyromill. Around 10 mg of tissue was weighed, and extraction buffer was added (40 µL extraction buffer per mg tissue). The solution was kept on ice for 10 mins, neutralized with NH4HCO3 as described above and centrifuged in a microfuge at a maximum speed for 30 min at 4 °C. Supernatants were transferred to LC-MS vials for analysis.
LC/MS procedures
LC/MS was performed to detect 15N-labeled isotopes of metabolites in the de novo pyrimidine synthesis pathway. LC separation was achieved using a Vanquish UHPLC system (Thermo Fisher Scientific) and an Xbridge BEH Amide column (2.1 mm × 150 mm × 2.5 mm particle size, 130 A ̊pore size; Waters, Milford, MA), column temperature 25°C. Solvent A is 95:5 water:acetonitrile with 20 mM ammonium acetate and 20 mM ammonium hydroxide at pH 9.4, and solvent B is acetonitrile. Flow rate was 150 mL/min. The LC gradient was 0 min, 85% B; 2 min, 85% B; 3 min, 80% B; 5 min, 80% B; 6 min, 75% B; 7 min, 75% B; 8 min, 70% B; 9 min, 70% B; 10 min, 50% B; 12 min, 50% B; 13 min, 25% B; 16 min, 25% B; 18 min, 0% B; 23 min, 0% B; 24 min, 85% B. 25 min, stop run. Injection volume was 5 µL. The Q-Exactive Plus mass spectrometer was operated in negative ion mode scanning from m/z 70-1000 with a resolution at 140,000. Data were analyzed by using El-Maven.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 3 Software (San Diego, CA). P values were calculated by the Student's t-test or two-way ANOVA and were considered significant if P<0.05. The data were expressed as the mean ± standard deviation (SD) in the figures.
Results
Leflunomide enhances the anti-Proliferative Effect of Gemcitabine in KPC cells in vitro
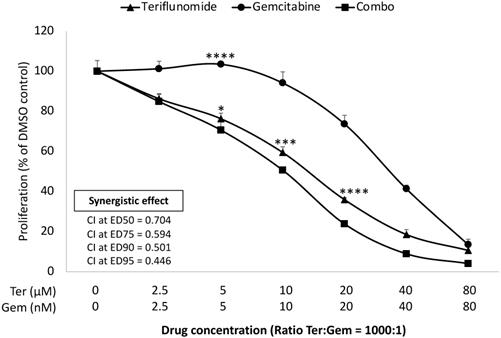
An established mechanism of Gem resistance in pancreatic cancer patients is the induction of de novo pyrimidine synthesis [17]. Leflunomide targets the enzyme DHODH which is important in the pathway of de novo pyrimidine synthesis. The impact of growth inhibition of leflunomide and Gem was assessed in KPC cells lines in vitro using the MTS assay. As Leflunomide in vivo is rapidly converted into its active form Teriflunomide (Teri), we used Teri in this assay [18]. As shown in Figure 1, Teriflunomide alone (2.5-80 µM) suppressed proliferation of KPC cells in a dose-dependent manner. In addition, Teri enhanced the inhibitory effect of Gem (ratio Teri:Gem = 1000:1) in KPC cells (p<0.05 from 0.5 µM Teri and/or 5 nM Gem). Based on the combination index (CI) values of <1, these results indicate that there is demonstrable synergy between Teri and Gem. This data merited further work to be done in the in vivo setting. As Lef has been utilized in the treatment of autoimmune disorders, it was important to evaluate the efficacy of Lef and Gem in an immunocompetent in vivo model.
Leflunomide enhances inhibitory effect in combination with Gem in KPC cell lines in vitro. KPC cells were seeded in 96 well-plate and then treated with Teriflunomide (0-80 µM) and Gem (0-80 nM) for 72 h, as single agents and in combination, at constant ratios (Ter:Gem = 1000:1). MTS assay was performed to measure the cell proliferation. Combination index (CI) values for potential synergistic or additive effects were calculated using CompuSyn software (Cambridge, UK). Drug synergism, addition, and antagonism effects were defined by CI values of <1.0, 1.0, and >1.0, respectively. CI values at ED50, ED75, ED90 and ED95 for combination treatment are shown. Results from one representative experiment are presented as means ± SD, with triplicate determinations. (*) p <0.05, (**) p<0.01, (***) p<0.001, and (****) p< 0.0001 compared to combo.
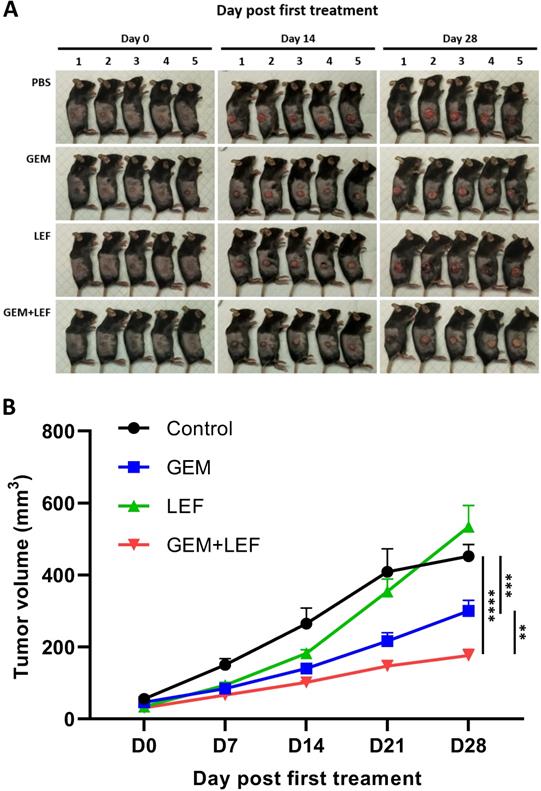
Leflunomide and Gemcitabine suppress tumor growth of KPC cells in an immunocompetent syngeneic flank mouse model
We determined the therapeutic efficacy of Gem, Lef and in combination in a subcutaneous (s.c.) tumor model of KPC cells. When the s.c. tumors reached ≥50 mm3, mice were treated with: PBS (control), Gem, Leflunomide, and combination (Gem+Lef) for 4 weeks (Fig. 2). Although leflunomide attenuated tumor growth from day 7 to day 14, statistical significance was not reached. Similar to early preclinical trials using Gem and Lef in an immunosuppressed mouse model, Gem and combination Gem+Lef significantly halted tumor progression [19]. There was significant growth inhibition by day 21 and persisting through day 28 in Gem+Lef treatment (p<0.05 at day 28). These results report for the first time that Lef (an agent used to mitigate autoimmune conditions) in combination with Gem has anti-tumor growth abilities in an immunocompetent flank model of pancreatic cancer.
Combination of Leflunomide and Gem suppress tumors growth using KPC subcutaneous mouse models. 6-week-old female C57BL6 mice (n = 5/group) were subcutaneously injected into the right thigh with KPC cells (5 × 105 cells/mouse). After 21 days, when the tumor volume reached around 50 mm3, the mice were randomized into 4 groups: control, Gem, Leflunomide, and combination. The mice were treated in the following dosing regimens for 4 weeks: (i) Gem 75mg/kg i.p. every three days, and (ii) Leflunomide 40mg/kg p.o. daily. Tumor volume was measured every 7 days until the end of experiment. Mice were then euthanized when the tumor volume reached 1000mm3. (A) Photos of mice after treatment. (B) Tumor growth curve. Data are presented as means ± SD. (*) p <0.05, (**) p<0.01, and (***) p<0.001 at day 28.
Immune cell profiling of Leflunomide and Gemcitabine treatment in KPC subcutaneous mouse models
In order to assess the role of the immune response in inducing remission following Gem+Lef treatment and as Lef is known to induce changes in the immune cell populations, we investigated the immune profile in treated tumors and PBMCs. First, we sought to examine changes in T cell populations in tumors after 28 days of treatment. As shown in Figure 3A, we first looked at CD4 and CD8 cell populations. There was a significant decrease in expression of markers of T cell exhaustion. All the treated (Gem, Lef, Gem+Lef) tumors showed marked down regulation in the T cell exhaustion marker (CTLA-4+) compared to controls (p<0.05). We chose to look at CTLA-4+ as a marker of exhaustion as uur group has previously studied Leflunomide in multiple myeloma 5TGM1 model in C57BL/KaLwRijHsd mice, which showed significantly differences in only CTLA-4 CD8+ T cells but not other markers of T cell exhaustion (LAG3, PD1, 2B4 and KLRG1) [18].
T cell profiling of Leflunomide and Gem treatment in KPC subcutaneous mouse models. KPC subcutaneous mouse models (50mm3) in C57BL6 mice were treated with Gem, Leflunomide as single agents or in combination as described in the legend of figure 2. (A) At day 28 after the first treatment, tumors were excised and single-cell suspensions were prepared. Cells were then stained with different cell markers CD3, CD4, CD8, CD25, FoxP3, CD69, and CTLA-4. Percentage of CD4+T cells, CD8+T cells from total CD3+ T cells; (CD4+CD25+FoxP3+) Treg cells, (CD8+CD69+) activated T cells, (CD8+CTLA-4+) suppressive T cells from total CD8+ T cells were analyzed using flow cytometry. (B) At day 28 after the first treatment, bloods were collected and PBMC were extracted for further staining. Same procedures were performed for FACS analysis of T cells in PBMCs. Data are presented as means ± SD. (*) p <0.05 compared to control.
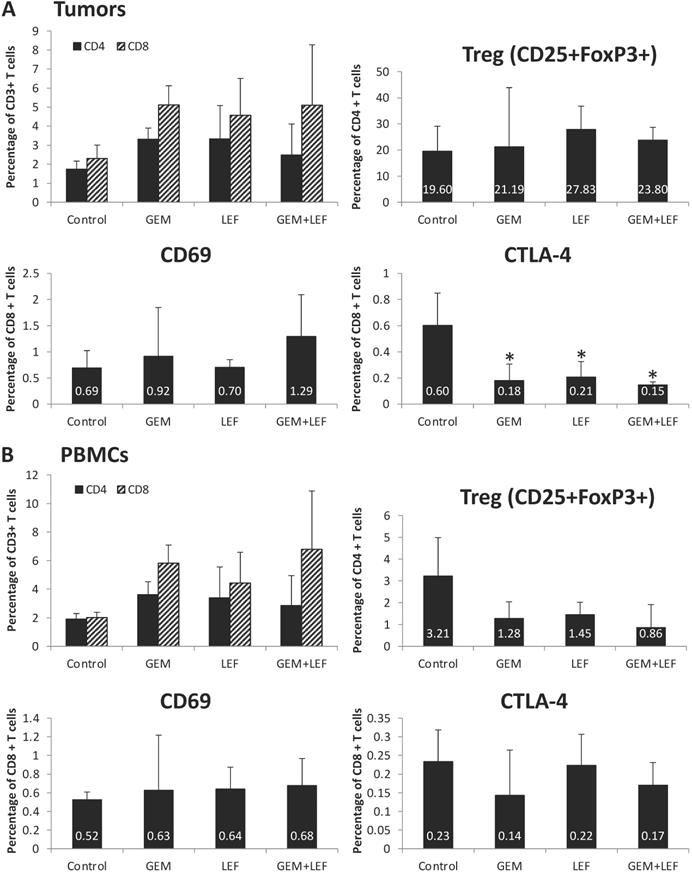
In order to assess changes in the circulating blood, PBMCs were evaluated for populations of T cells. There were no significant changes in markers of T cell activation (CD69+ T cells) or markers of T cell exhaustion (CTLA4+ T cells) based on the treatment group in the PBMC populations (Fig. 3B).
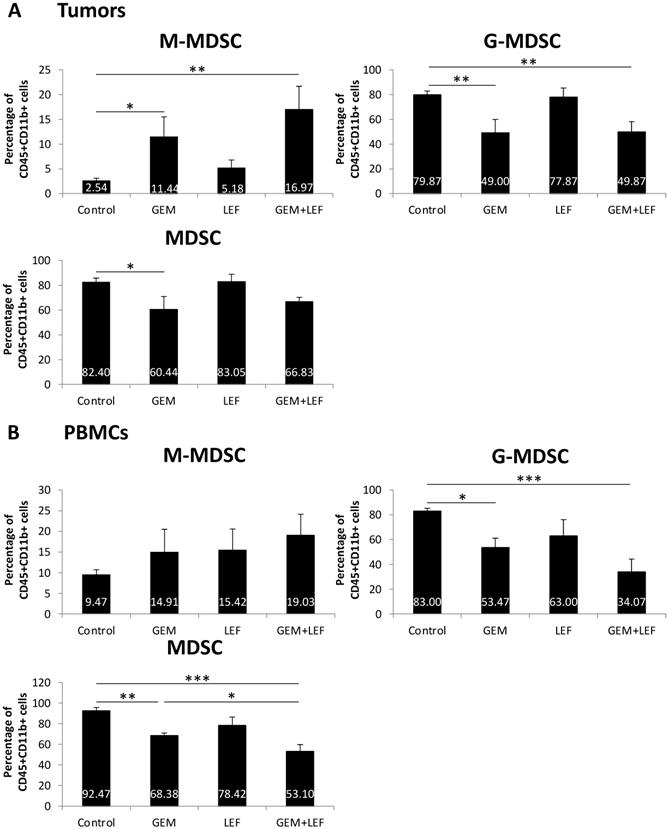
As myeloid-derived suppressor cells (MDSCs) are important in a variety of mechanisms that alter T cell activation or suppression, we studied the population of MDSCs in tumors and in PBMCs in mice treated with Gem, Lef, or Gem+Lef and controls (Fig. 4). We looked at both monocytic-MDSCs (M-MDSCs) and the granulocytic fraction (called granulocytic/polymorphonuclear MDSCs or G-MDSCs). The M-MDSCs are thought to have more immunosuppressive activity.[20, 21] In the tumor models, we found that treatment with Gem alone and Gem+LeF significantly increased the population of M-MDSCs, indicating an increase in the immunosuppressive environment (Fig. 4A, (p<0.05). The G-MDSCs decreased in the treated groups of Gem (p<0.01) and Gem+Lef (p<0.01) but not in Lef alone compared to controls. As there was a greater proportion of G-MDSCs, the overall changes of MDSCs in the tumors were only significantly decreased with Gem alone (p<0.05). In the PBMCs, the findings were similar with a significant decrease in G-MDSCs in the Gem and Gem+Lef treated groups compared to controls (Fig. 4B, p<0.05). Like the tumors there was a significant decrease in overall MDSCs in the Gem and Gem+Lef groups compared to controls (p<0.01 and p<0.001).
Myeloid-Derived Suppressor Cells (MDSC) populations of Leflunomide and Gem treatment in KPC subcutaneous mouse models. On the same experiment as mentioned in figure 3, single-cell suspension from tumors (A) and PBMCs (B) at day 28 after the first treatment were applied for staining with other cell markers for MDSCs: CD45, CD11b, Ly6G and Ly6C. Percentage of (CD11b+Ly6G-Ly6Chi) monocytic (M-MDSC) and (CD11b+Ly6G+Ly6Clo) granulocytic or polymorphonuclear (G-MDSC) from total CD45+CD11b+ cells were analyzed using flow cytometry. Data are presented as means ± SD. (*) p <0.05, (**) p<0.01, (***) p<0.001.
The immune profiling results in the tumors and in the PBMCs can be summarized to indicate that Lef may be enhancing the anti-tumor immunity of Gem treatment via a mechanism that favors the recruitment of activated CD69+CD8+ T cells in the tumors while decreasing the frequency of Tregs, M-MDSCs (systemically) and CTLA-4+CD8+ T cells in the tumor microenvironment. Altogether, Gem+Lef treatment appears to tilt the balance of the host from an immunosuppressive to an immune activated state, thus further delaying tumor progression.
Leflunomide causes anti-angiogenesis effect and enhances the anti-tumor effect of Gemcitabine in KPC subcutaneous mouse models
Based on data that Lef has been shown to inhibit angiogenesis in other solid tumors, CD31staining was performed to evaluate microvessel density. Explanted tumors from mice with flank tumors that had been treated with Gem, Lef, Gem+Lef were stained for CD31 and compared to controls (Fig. 5B). The results for CD31 staining indicate that both the Lef alone and Gem+Lef tumors had significantly less CD31 positive staining indicating inhibition of angiogenesis (P<0.01).
Leflunomide causes anti-angiogenesis effect and enhances the anti-tumor of Gem in KPC subcutaneous mouse models. KPC subcutaneous mouse models (50 mm3) in C57BL6 mice were treated with Gem, Leflunomide as single agents or in combination as described in the legend of figure 2. At day 28 after the first treatment, tumors were excised and immunohistochemistry (IHC) staining was performed with specific antibodies: (B) CD31 (microvessel density) and (C) Ki67 (cell proliferation). Representative photography of H&E (A) and IHC staining of KPC tumor tissues from different experimental groups are shown. Positive staining appears as brown color. Protein expression levels were analyzed by calculating the percentage of integrated optical density (IOD)/area using Image-Pro Premier. Data are presented as means ± SD. (*) p <0.05, (**) p<0.01, and (***) p<0.001.
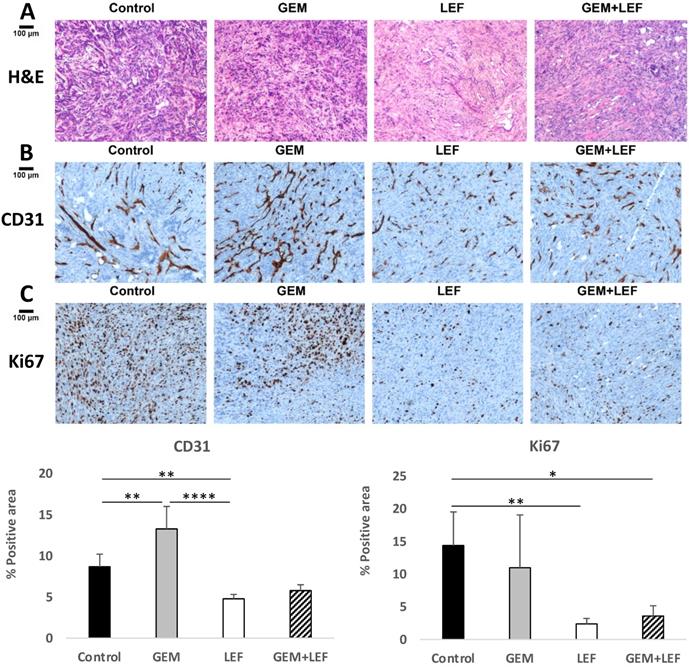
In order to assess if inhibition of angiogenesis also correlated to growth inhibition, tumors were also stained for the proliferation index marker Ki-67. As shown in Figure 5C, Lef alone or Gem+Lef treatment led to significantly fewer proliferating cells (Ki67+ cells) in KPC tumors (P<0.01). These findings indicate that Lef alone and in combination with Gem resulted in both inhibition of angiogenesis and proliferation in this KPC flank model of pancreatic cancer.
Leflunomide inhibits de novo pyrimidine synthesis in KPC cells both in vitro and in vivo
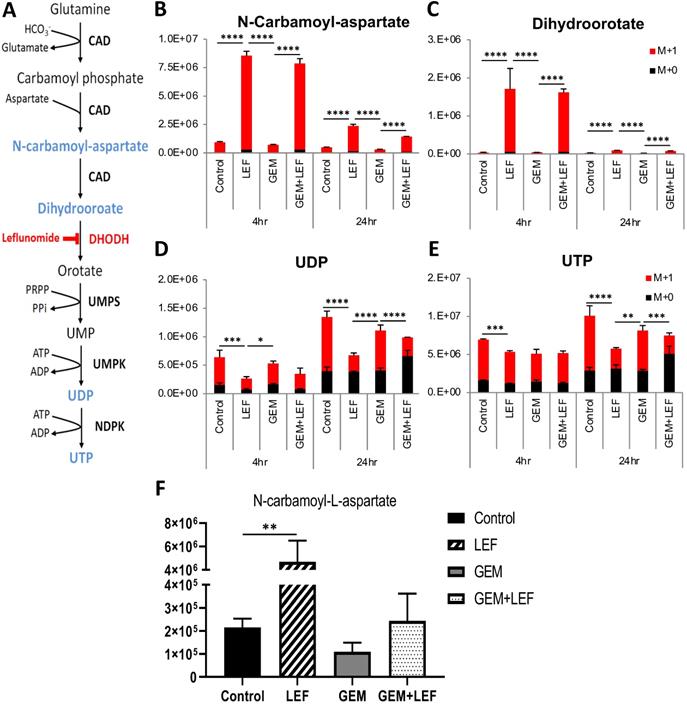
Leflunomide and its active form Teriflunomide (Teri) have been known as a selective inhibitors of the de novo pyrimidine synthesis pathway. Lef inhibits the rate-limiting enzyme DHODH (Fig. 6A). The anti-proliferative effect of Gem+Teri on KPC cells is demonstrated in Figure 1. These findings suggest that Teri enhances the antitumor activity of Gem through the depletion of the nucleoside precursors required for DNA damage repair. To confirm this hypothesis, LC/MS was performed to detect 15N-labeled isotopes of metabolites in the de novo pyrimidine synthesis pathway in KPC cells. The in vitro results showed that Teri alone and Gem+Teri combination therapy significantly increased the abundance of metabolites upstream of DHODH; specifically, there were increased levels of N-carbamoyl-aspartate and dihydroorotate after 4 hrs and 24 hrs of treatment (Fig. 6B & C) p<0.001. Consistently, decreased levels of UDP and UTP were observed in Teri and Gem+Teri combination treated cells (Fig. 6D & E). The in vivo findings were similar. After 28 days of treatment, Lef induced inhibition of the DHODH enzyme was demonstrated by changes in measured metabolites. The metabolomics profile of Lef treated tumors yielded a significant increase in the proportion of N-Carbamoyl-aspartate when compared with the controls or Gem treated tumors (Fig. 6F) p<0.01. Our findings are consistent in that with Lef treatment there is significant inhibition of de novo pyrimidine synthesis in both in vitro and in vivo models and this may account for in part the mechanism of growth inhibition seen in Gem+Lef combination therapy.
Leflunomide inhibits the de novo pyrimidine synthesis pathway in KPC cells in vitro and in vivo. (A) Schematic of the de novo pyrimidine synthesis pathway. KPC cells were treated with Gem 20nM, Leflunomide 40μM as single agents or combination in DMEM supplemented with 200 μM L-Glutamine-(amide-15N), 10% FBS and 1% Penicillin-Streptomycin. After 4hr and 24hr of treatment, cells were quenched with ice-cold solvent (40:40:20 Acetonitrile:Methanol:Water with 0.5% formic acid) and cellular metabolites were extracted. Relative isotopic enrichment of M L-Glutamine-(amide-15N) into Dihydroorotate (B), Carbamoyl-aspartate (C), UDP (D) and UTP (E) was measured by LC/MS-MS. (F) Level of Carbamoyl-aspartate from tumor tissues. Results from one representative experiment are presented as means ± SD, with triplicate determinations. (*) p < 0.05, (**) p < 0.005, (***) p < 0.001, and (****) p < 0.0001 by two-way ANOVA. M+0, all amides unlabeled 14N amide and M+1, all amides labeled 15N.
Discussion
In this study we demonstrate for the first time that Gem + Lef inhibits tumor growth in part via inhibition of angiogenesis, proliferation, de novo pyrimidine synthesis and induces a favorable anti-tumoral immune phenotype in an immunocompetent mouse model.
Pancreatic cancer remains a challenging disease to treat. Patients are often not robust enough to tolerate the more aggressive regimens of Gem with nab-paclitaxel or FOLFIRINOX [22]. Therefore, Gem alone remains a mainstay in systemic therapy. Unfortunately, we know from randomized controlled trials that after several months of therapy, tumor resistance develops and cancer progression ensues [23]. There are several described mechanisms of Gem resistance, which involve the desmoplastic tumor microenvironment, changes in Gem transport, changes in the enzymatic activation of Gem and a host of associated intracellular enzymes and transcription factors [24, 25]. One particular mechanism of Gem resistance is de novo pyrimidine synthesis [19]. Pyrimidine bases are critical for cellular metabolism and growth and are important precursors in DNA (thymine and cytosine) and RNA (uracil and cytosine) biosynthesis [26]. Lef targets the enzyme DHODH which is a rate limiting step in de novo pyrimidine synthesis [27]. Lef binds reversibly to the alpha-helical domain of DHODH and blocks access to this active site inhibiting enzyme activity [27]. Lef has been studied in multiple solid and liquid tumors as an antiproliferative agent [27]. In triple negative breast cancer, inhibition of de novo pyrimidine synthesis sensitizes cells to genotoxic chemotherapy agents by exacerbating DNA damage [7]. Combining treatment with doxorubicin and leflunomide induces regression of TNBC xenografts.
Based on the mechanism of action, the combination of Lef and Gem in an immunocompetent model of pancreatic cancer was the next step in moving this potential therapy to patients. We first evaluated the potential synergy of Gem and Teriflunomide (the active in vitro metabolite Lef) in the KPC mouse pancreatic cell line. Lef is an isoxazole derivative (N-[4-trifluoromethylphenyl]-methylisoxazol-4-carboxamide) that is an oral prodrug that is hydrolyzed during first-pass metabolism in the gut and liver to its singe active metabolite teriflunomide) [26]. The therapeutic effects of leflunomide are primarily mediated via this metabolite [28]. Similar to our published in vitro studies in human pancreatic cancer cell lines, we were able to demonstrate synergy between Gem and Teri and this prompted our in vivo work [29]. As KPC mice harbor p53 and Kras mutations, they have been studied as a well-accepted model in performing preclinical work in the field [30]. Additionally, as Lef is in part an immunomodulator, it was important to perform the in vivo work in an immunocompetent syngeneic model.
Our in vivo findings demonstrate that Gem and Lef together significantly induce growth inhibition of flank implanted KPC cell tumors. The combination of Gem and Lef have been previously studied and demonstrated the synergy with human pancreatic cancer cells implanted in an immunocompromised mouse and in KPC mice [19, 31]. Yu et al demonstrated that mitochondrial fusion was a potential target/regulator of pancreatic cancer growth. PDAC requires mitochondrial oxidative phosphorylation (OXPHOS) for growth. They demonstrated that oral leflunomide promoted mitochondrial fusion which reduced OXPHOS and thus inhibited tumor growth [31]. Shukla et al demonstrated that Gem-resistant cells stabilized hypoxia inducible factor-1 alpha and this lead to increased glycolysis, leading to rapid generation of biosynthetic intermediates to supply the ingredients for cell growth and proliferation [19]. They demonstrated that inhibition of DHODH a key enzyme in the pyrimidine biosynthesis pathway with leflunomide, increased Gem sensitivity in the Gem-R cells [19]. Their findings in conjunction with our in vivo work led us to evaluate the role of Gem+Lef in an immunocompetent model.
Lef is an agent with a long track record of safety that has been used in autoimmune disorders such as rheumatoid arthritis [8]. In autoimmune disease, Lef aims to mitigate the immune response. As inflammation in the tumor microenvironment is an important component of tumor progression, the role of Lef as an immunomodulator has not been adequately assessed [32]. In this study we demonstrate that Lef treated tumors demonstrated decreased CTLA-4 + T cells indicating a decrease in intratumoral T cell exhaustion and perhaps more anti-tumor immunity. Interesting, the combination of Gem+Lef correlated with an increase in the population of the M-MDSCs which are thought to have more immunosuppressive activity. Inversely, G-MDSCs population decreased in the Gem+Lef groups. These immune changes in the tumor microenvironment are potential explanations for the growth inhibitory changes induced by treatment with Gem+Lef and will be further studied in future work.
Angiogenesis in pancreatic cancer is a debated question. There is evidence that the desmoplasia in these tumors can be protective and mitigate more aggressive spread [33]. In contrast, there is literature to say that inhibition of angiogenesis precludes nutrient delivery to tumors and thus can mitigate tumor growth and induce regression [34]. We further examined the tumors for markers of angiogenesis as Lef has been shown to inhibit angiogenesis in other solid tumors (bladder cancer). We found similar findings in our Lef treated tumors with significantly less CD31 staining indicating inhibition of angiogenesis. As the blood supply is a route of nutrition for these tumors, inhibition of angiogenesis is likely a relevant mechanism in Gem+ Lef induced growth inhibition in our PDAC model. The proliferation index was also markedly reduced with the combination of Gem and Lef in these tumors and correlating to the in vivo gross tumor measurement findings.
Lastly, we sought to examine the metabolomics as they pertained to the mechanism of action of Lef. As noted above, DHODH is a rate limiting enzyme in the de novo pyrimidine synthesis pathway. We and others have shown that Lef inhibits this enzyme in human pancreatic cancer cell lines. Here we demonstrate for the first time that Lef inhibits DHODH as measured by upstream metabolite accumulation both in vitro and in vivo in a KPC syngeneic immunocompetent model of pancreatic cancer.
Based on the above evidence, LEF is active in both metabolic and immune contexts. Further work will need to elucidate which mechanism accounts for a greater magnitude of the effect of LEF on pancreatic cancer growth. It is likely that there is significant overlap with inhibition of DHODH impacting both mechanisms.
Limitations
One consideration of utilizing Lef with Gem or with alternative regimens is that in the in vitro and in vivo setting we aren't able to precisely recapitulate the disease process in patients. The thought is that over time de novo pyrimidine synthesis is a mechanism of resistance that develops and that Lef may be used as a maintenance therapy option after a period of time on Gem. Due to the pace of disease progression in these mouse models, it is challenging to recreate this scenario. This will be a focus of study in future work.
Conclusions
In conclusion, our data demonstrates that the combination of Gem and Lef inhibits pancreatic cancer cell growth both in vitro and in an immunocompetent w model. We were also able to demonstrate that combination therapy is associated with decreased CD8+ T cell exhaustion, angiogenesis and proliferation. We were also able to demonstrate that combination therapy changes the metabolomics of cells in vitro and an in vivo such that de novo pyrimidine synthesis with accumulation of upstream metabolites. This may be a mechanism to leverage in treating patients with pancreatic cancer that develop resistance to Gem therapy without significant added toxicity.
Abbreviations
Lef: leflunomide; Gem: Gemcitabine; MTS: (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; KPC: cell line derived from LSL-KRAS-G12, LSL-p53-R172H, and PDx-1-Cre mice); PBS: Phosphate Buffered Solution; FACS: Fluorescence-activated cell sorting (FACS); PDAC: pancreatic ductal adenocarcinoma; FOLFIRINOX: Leucovorin, 5-fluorouracil, Irinotecan, Oxaliplatin; DNA: Deoxyribonucleic acid; DHODH: dihydroorotate dehydrogenase; DMEM: Dulbecco's Modified Eagle Medium; COH: City of Hope; IACUC: Institutional Animal Care and Use Committee; SD: standard deviation; CI: Combination Index; RPMI: Roswell Park Memorial Institute Medium; RBC: red blood cell; PBMC: peripheral blood mononuclear cells; IHC: immunohistochemistry; NBF: neutral buffered formalin; LC/MS: liquid chromatography/mass spectrometry; Teri: Teriflunomide; s.c.: subcutaneous; MDSC: myeloid-derived suppressor cells; M-MDSC: monocytic myeloid-derived suppressor cells; G-MDSC: granulocytic myeloid-derived suppressor cells; RNA: ribonucleic acid; TNBC: triple negative breast cancer; OXPHOS: oxidative phosphorylation.
Acknowledgements
This study was supported in part by Legacy Heritage Fund, the Norman and Sadie Lee Foundation and the City of Hope Chancellors' Fund.
Graphical Abstract Created with BioRender.com with assistance from Supriya Deshpande Ph.D.
Contributions
Conceptualization: TP, LM, SR, RB, VN, MAS, PW, JR, DD; Investigation: CM, WT, LY; Original Writing: LM, TP Review and Editing: SR, RB, MAS, JR, LY; Funding Acquisition: LM, SR.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA: a cancer journal for clinicians. 2021;71:7-33
2. Carpenter E, Nelson S, Bednar F, Cho C, Nathan H, Sahai V. et al. Immunotherapy for pancreatic ductal adenocarcinoma. Journal of surgical oncology. 2021;123:751-9
3. Tempero MA, Malafa MP, Chiorean EG, Czito B, Scaife C, Narang AK. et al. Pancreatic Adenocarcinoma, Version 1.2019. J Natl Compr Canc Netw. 2019;17:202-10
4. Zheng HC. The molecular mechanisms of chemoresistance in cancers. Oncotarget. 2017;8:59950-64
5. Vaidya FU, Sufiyan Chhipa A, Mishra V, Gupta VK, Rawat SG, Kumar A. et al. Molecular and cellular paradigms of multidrug resistance in cancer. Cancer Rep (Hoboken). 2020: e1291.
6. Braun LM, Lagies S, Guenzle J, Fichtner-Feigl S, Wittel UA, Kammerer B. Metabolic Adaptation during nab-Paclitaxel Resistance in Pancreatic Cancer Cell Lines. Cells. 2020 9
7. Brown KK, Spinelli JB, Asara JM, Toker A. Adaptive Reprogramming of De Novo Pyrimidine Synthesis Is a Metabolic Vulnerability in Triple-Negative Breast Cancer. Cancer discovery. 2017;7:391-9
8. Alfaro-Lara R, Espinosa-Ortega HF, Arce-Salinas CA, Precis study group apbtDoIMHCSdP. Systematic review and meta-analysis of the efficacy and safety of leflunomide and methotrexate in the treatment of rheumatoid arthritis. Reumatol Clin. 2019;15:133-9
9. Fragoso YD, Brooks JB. Leflunomide and teriflunomide: altering the metabolism of pyrimidines for the treatment of autoimmune diseases. Expert review of clinical pharmacology. 2015;8:315-20
10. Gaidano V, Houshmand M, Vitale N, Carra G, Morotti A, Tenace V. et al. The Synergism between DHODH Inhibitors and Dipyridamole Leads to Metabolic Lethality in Acute Myeloid Leukemia. Cancers (Basel). 2021 13
11. Baumann P, Mandl-Weber S, Volkl A, Adam C, Bumeder I, Oduncu F. et al. Dihydroorotate dehydrogenase inhibitor A771726 (leflunomide) induces apoptosis and diminishes proliferation of multiple myeloma cells. Molecular cancer therapeutics. 2009;8:366-75
12. Ren A, Fu G, Qiu Y, Cui H. Leflunomide inhibits proliferation and tumorigenesis of oral squamous cell carcinoma. Molecular medicine reports. 2017;16:9125-30
13. Chen Y, Huang Q, Zhou H, Wang Y, Hu X, Li T. Inhibition of canonical WNT/beta-catenin signaling is involved in leflunomide (LEF)-mediated cytotoxic effects on renal carcinoma cells. Oncotarget. 2016;7:50401-16
14. O'Donnell EF, Kopparapu PR, Koch DC, Jang HS, Phillips JL, Tanguay RL. et al. The aryl hydrocarbon receptor mediates leflunomide-induced growth inhibition of melanoma cells. PloS one. 2012;7:e40926
15. Jiang L, Zhang W, Li W, Ling C, Jiang M. Anti-inflammatory drug, leflunomide and its metabolite teriflunomide inhibit NSCLC proliferation in vivo and in vitro. Toxicology letters. 2018;282:154-65
16. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer research. 2010;70:440-6
17. Amrutkar M, Gladhaug IP. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers (Basel). 2017 9
18. Buettner R, Morales C, Caserta E, Troadec E, Gunes EG, Viola D. et al. Leflunomide regulates c-Myc expression in myeloma cells through PIM targeting. Blood Adv. 2019;3:1027-32
19. Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E. et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer cell. 2017;32:392
20. Musolino C, Allegra A, Pioggia G, Gangemi S. Immature myeloid-derived suppressor cells: A bridge between inflammation and cancer (Review). Oncol Rep. 2017;37:671-83
21. Veglia F, Hashimoto A, Dweep H, Sanseviero E, De Leo A, Tcyganov E. et al. Analysis of classical neutrophils and polymorphonuclear myeloid-derived suppressor cells in cancer patients and tumor-bearing mice. J Exp Med. 2021 218
22. Gupta R, Amanam I, Chung V. Current and future therapies for advanced pancreatic cancer. Journal of surgical oncology. 2017;116:25-34
23. Sarvepalli D, Rashid MU, Rahman AU, Ullah W, Hussain I, Hasan B. et al. Gemcitabine: A Review of Chemoresistance in Pancreatic Cancer. Crit Rev Oncog. 2019;24:199-212
24. de Sousa Cavalcante L, Monteiro G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. European journal of pharmacology. 2014;741:8-16
25. Binenbaum Y, Na'ara S, Gil Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug resistance updates: reviews and commentaries in antimicrobial and anticancer chemotherapy. 2015;23:55-68
26. Zhang C, Chu M. Leflunomide: A promising drug with good antitumor potential. Biochem Biophys Res Commun. 2018;496:726-30
27. Madak JT, Bankhead A 3rd, Cuthbertson CR, Showalter HD, Neamati N. Revisiting the role of dihydroorotate dehydrogenase as a therapeutic target for cancer. Pharmacol Ther. 2019;195:111-31
28. Hopkins AM, Moghaddami M, Foster DJ, Proudman SM, Upton RN, Wiese MD. Intracellular CD3+ T Lymphocyte Teriflunomide Concentration Is Poorly Correlated with and Has Greater Variability Than Unbound Plasma Teriflunomide Concentration. Drug Metab Dispos. 2017;45:8-16
29. Buettner R, Morales C, Wu X, Sanchez JF, Li H, Melstrom LG. et al. Leflunomide Synergizes with Gemcitabine in Growth Inhibition of PC Cells and Impairs c-Myc Signaling through PIM Kinase Targeting. Mol Ther Oncolytics. 2019;14:149-58
30. Ariston Gabriel AN, Jiao Q, Yvette U, Yang X, Al-Ameri SA, Du L. et al. Differences between KC and KPC pancreatic ductal adenocarcinoma mice models, in terms of their modeling biology and their clinical relevance. Pancreatology. 2020;20:79-88
31. Yu M, Nguyen ND, Huang Y, Lin D, Fujimoto TN, Molkentine JM. et al. Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer. JCI Insight. 2019 5
32. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860-7
33. Melstrom LG, Salazar MD, Diamond DJ. The pancreatic cancer microenvironment: A true double agent. Journal of surgical oncology. 2017;116:7-15
34. Lafaro KJ, Melstrom LG. The Paradoxical Web of Pancreatic Cancer Tumor Microenvironment. Am J Pathol. 2019;189:44-57
Author contact
![]() Corresponding author: Laleh Melstrom MD, City of Hope National Medical Center, Department of Surgery and Immuno-oncology, 1500 E Duarte Road, Duarte, CA 91010. E-mail: lmelstromorg; Phone: 626 218 0282; Fax: 626 218 1113.
Corresponding author: Laleh Melstrom MD, City of Hope National Medical Center, Department of Surgery and Immuno-oncology, 1500 E Duarte Road, Duarte, CA 91010. E-mail: lmelstromorg; Phone: 626 218 0282; Fax: 626 218 1113.