Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Markers of pEMT in SCC
Regulatory mechanism of pEMT in...
pEMT and collective migration in...
pEMT and SCC stemness
pEMT and therapeutic resistance...
Conclusions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(12):3036-3047. doi:10.7150/ijbs.61566 This issue Cite
Review
Partial EMT in Squamous Cell Carcinoma: A Snapshot
Chengcheng Liao1,2, Qian Wang2,4, Jiaxing An3, Qian Long1,2, Hui Wang3, Meiling Xiang1, Mingli Xiang1, Yujie Zhao1,2, Yulin Liu1,2, Jianguo Liu1,2 ![]() , Xiaoyan Guan1
, Xiaoyan Guan1 ![]()
1. Department of Orthodontics II, Affiliated Stomatological Hospital of Zunyi Medical University, Zunyi 563000, China
2. Oral Disease Research Key Laboratory of Guizhou Tertiary Institution, School of Stomatology, Zunyi Medical University, Zunyi 563006, China
3. Department of Gastroenterology, Affiliated Hospital of Zunyi Medical University, Zunyi 563000, China
4. Microbial Resources and Drug Development Key Laboratory of Guizhou Tertiary Institution, Life Sciences Institute, Zunyi Medical University, Zunyi 563006, China
Received 2021-4-12; Accepted 2021-6-25; Published 2021-7-13
Abstract

In the process of cancer EMT, some subgroups of cancer cells simultaneously exhibit both mesenchymal and epithelial characteristics, a phenomenon termed partial EMT (pEMT). pEMT is a plastic state in which cells coexpress epithelial and mesenchymal markers. In squamous cell carcinoma (SCC), pEMT is regulated, and the phenotype is maintained via the HIPPO pathway, NOTCH pathway and TGF-β pathways and by microRNAs, lncRNAs and the cancer microenvironment (CME); thus, SCC exhibits aggressive tumorigenic properties and high stemness, which leads collective migration and therapy resistance. Few studies have reported therapeutic interventions to address cells that have undergone pEMT, and this approach may be an effective way to inhibit the plasticity, drug resistance and metastatic potential of SCC.
Keywords: Partial EMT, Squamous Cell Carcinoma, FAT1, HIPPO, NOTCH, TGF-β, Collective migration, Stemness, Therapeutic resistance
Introduction
Epithelial-mesenchymal transition (EMT), which refers to the biological process through which epithelial cells transform into cells with a mesenchymal phenotype and plays an important role in cancer progression, embryonic development, inflammation, tissue reconstruction, and fibrosis [1]. EMT endows tumors with malignant properties, including aggressive behaviors, metastasis, cancer stem cell (CSC) activity, and therapeutic resistance [2]. EMT was historically considered a binary switch, and any partial state was regarded as a metastable state or transient snapshot acquired during the EMT process. Transcriptional and epigenetic characteristics determine the potential gene regulatory networks, transcription factors and signaling pathways that control these different EMT transition states [3]. However, many studies have shown that cancer cells can stably acquire one or more partial EMT (pEMT) phenotypes, and that they can exhibi a mixture of epithelial and mesenchymal characteristics at the molecular and/or morphological level [4, 5, 6, 7, 8, 9, 10, 11]. pEMT is also known as the hybrid epithelial/mesenchymal (E/M), intermediate EMT, intermediate mesenchymal, incomplete EMT, semimesenchymal, and EMT-like phenotype [12].
The pEMT phenotype plays an important role in the tumor progression, process of organ branching morphogenesis, diabetic kidney disease and wound healing [13, 14, 15, 16]. Cells in the pEMT state show greater tumor-initiating potential, therapeutic resistance and apoptosis resistance than purely epithelial or mesenchymal cells [17, 18]. Tumorigenesis depends on single cells in the pEMT state, and cannot be replicated by mixing epithelial and mesenchymal phenotype cells [19]. Cells with a pEMT phenotype cells exhibit loss of apical-basal polarity and have better motility, while maintaining adhesion characteristics with neighboring cells and acquiring mesenchymal-like characteristics [20]; thus, they can assemble and move together. If these cell clusters reach the bloodstream intact, circulating tumor cell clusters (CTCs) clusters that can migrate collectively are generated [18]. Small numbers of CTCs have the highest plasticity, are believed to possess CSC characteristics and the ability to initiate tumors, can form new lesions and are fatal in tumor patients [21]. These results indicate that the pEMT phenotype cells may be the most suitable for metastasis. The feedback loop between EMT and immunosuppression promotes tumor progression [22, 23], but how pEMT affects tumor immunity remains undiscussed [24].
Squamous cell carcinoma (SCC) accounts for most esophageal cancers (upper esophagus) [25], more than 90% of cervical cancers [26], 90% of head and neck cancers [27], 30% of non-small-cell lung cancers (NSCLCs) [28] and 20% of skin cancers [29]. Head and neck squamous cell carcinoma (HNSCC), esophageal squamous cell carcinoma (ESCC), NSCLC and cutaneous squamous cell carcinoma (cSCC) are prone to metastasis, recurrence and invasion and have low 5-year survival rates [30, 31, 32, 33]. HNSCC is prone to cervical lymph node and lung metastasis, and its 5-year survival rate is only 43% [32]. The 5-year survival rate for esophageal cancer is less than 13% [30]. NSCLC accounts for 85% of lung cancers; approximately 40% are unresectable, and the 5-year overall survival (OS) rate is approximately 15.9% [34, 35]. NSCLC includes a variety of cancer types, such as lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), and lung large cell carcinoma. The largest subgroups of NSCLC are LUAD and LUSC, which are significantly different at both the transcriptome and cell control network levels [36]. However, many current NSCLC studies have not distinguished the difference between LUAD and LUSC. In this review, the description of NSCLC in this review is focused more on LUSC than on LUAD. Many types of SCC have a poor response to treatment and are prone to metastasis, and thus, they are life-threatening. Therapeutic targeting of the pEMT maintenance mechanism and phenotype seems to be the best strategy to overcome the poor prognosis of SCC.
Markers of pEMT in SCC
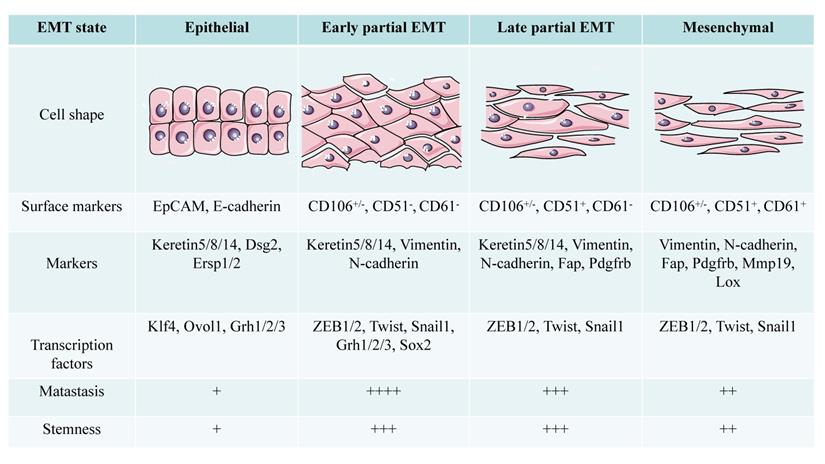
pEMT is usually evidenced by simultaneous expression of epithelial and mesenchymal protein markers in a single cell [37]. A detailed single-cell analysis of more than 6000 cells in 18 cases of HNSCC found that some of the cells showed pEMT properties; Vimentin and integrin-α5 were upregulated, although epithelial markers were not downregulated [38]. Immunohistochemical detection of E-cadherin and Vimentin expression in 200 oral squamous cell carcinoma (OSCC) patients confirmed that evidence of non-EMT, pEMT, and full EMT was present among the clinical samples, accounting for 49.5% (99), 43.5% (87), and 7.5% (14) of cases, respectively [39]. According to the expression of ESCC extracellular adhesion proteins, Vimentin and the adhesion and junction components Claudin1 and Claudin7, ESCC cells may be divided into the epithelial, full EMT and pEMT subgroups [40]. In another study, according to the expression level and localization of E-cadherin, Vimentin and N-cadherin, approximately 43% of the ESCC primary tumors and 53% of ESCC metastatic lymph nodes were classified as having a pEMT phenotype [41]. The pEMT phenotype can also be induced in SCC cell lines. In the p16-positive cervical carcinoma cell line CERV196, β-catenin levels are increased under epidermal growth factor (EGF) induction, while the expression levels of vimentin and E-cadherin change only slightly, which indicates a pEMT phenotype [42]. In addition, overexpression of Bcl-2 in the OSCC cell line HSC-3 induces the expression of N-cadherin but cannot completely eliminate E-cadherin expression or induce increased expression of typical EMT-related transcription factors (EMT-TFs) [43].
Pastushenko et al. [44] divided the pEMT state into early and late stages based on the expression patterns of the tumor cell surface markers CD106, CD61, and CD51. Epithelial tumor cells express epithelial cell adhesion molecule (EpCAM), early pEMT is characterized by lack of EpCAM expression and a CD106+/-/CD51-/CD61- phenotype, late pEMT status is characterized by a CD106+/-/CD51+/CD61- phenotype, and full EMT tumor cells exhibit a CD106+/-/CD51+/CD61+ phenotype [20, 44]. The pEMT subgroup in the SCC model established by Ievgenia et al. [45] is negative for CD106, CD61, and CD51 or expresses only CD106. Expression of epithelial markers such as E-cadherin and EpCAM is lost in the early stage of EMT, while keratin 5/8/14 expression is retained in pEMT and disappears completely in the later stage of EMT [44]. N-cadherin and Vimentin are highly upregulated in the early hybrid state, and their levels are maintained in late EMT. The expression of fibroblast activation protein (FAP), cadherin 11, complexes that regulate transcription of the collagen XXIV gene (Col24a1), lysyl oxidase-like 1 (LOXL1), matrix metalloproteinase-19 (MMP19), platelet-derived growth factor receptor (PDGFR) a/b or paired related homeobox 1 (PRRX1) increases in late EMT [44].
The conversion state of EMT shows different morphological and functional characteristics with different markers in SCC. Compared with cells in epithelial and mesenchymal state, partial EMT cells have stronger metastatic ability and stemness (+: low to ++++: high).
Regulatory mechanism of pEMT in SCC
pEMT may be a transient state in the EMT process, but the pEMT state can be stabilized or maintained through several mechanisms [46]. pEMT is controlled by signaling pathways, TFs, epigenetic regulators, phenotype stabilizing factors (PSFs) and posttranslational modifications [47].
pEMT-TFs in SCC
TFs bind to and inhibit the transcription of genes encoding adhesion junction and tight junction molecules, thereby triggering EMT. These TFs include Snail1/2, zinc finger E-box binding homeobox (ZEB) 1/2, Twist and lymphoid enhancer-binding factor 1 (LEF-1) [48]. pEMT, which is characterized by Twist-induced expression of ZEB1, Vimentin and Podoplanin (PDPN) but without the absence of E-cadherin expression, mediates the acquisition of invasive characteristics by cSCC [49]. In clinical samples of NSCLC, the frequency of Snail2 and E-cadherin expression seems to control EMT phenotypic plasticity. Specific expression patterns of E-cadherin and Snail2 distinguish NSCLC with different phenotypic characteristics and are related to prognosis [50]. In the model of early pEMT in SCC established by Ievgenia et al. [45], pEMT is induced by the expression of ZEB1 and sex determining region Y-box2 (SOX2). Snail1, Twist and ZEB1/2 are highly upregulated in the early hybrid state, and their levels are maintained in late EMT [44]. Another study suggested that early hybrid EMT is initiated by the expression of ZEB1, P63, Twist and LIM-protein 2 and that Smad2 promotes late EMT [51].
FAT1/HIPPO pathway-mediated regulation of pEMT in SCC
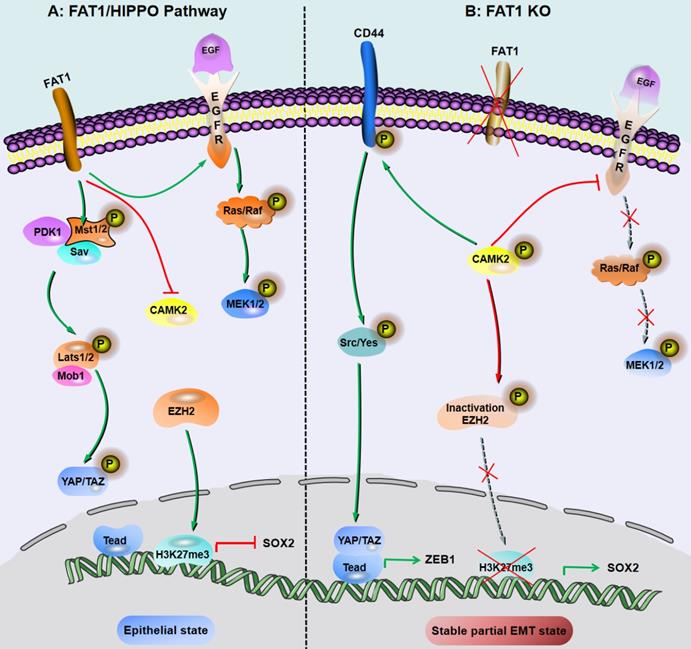
FAT1 is a transmembrane protein involved in the regulation of EMT, cell growth and actin dynamics and plays a key role in tumorigenesis and development [52]. FAT1 inhibits tumor progression by activating Hippo signaling [53]. The Hippo core complex controls the transport of YAP1/TAZ proteins to the nucleus, and abnormal upregulation of YAP1/TAZ expression or their nuclear localization occurs in SCC, which promotes tumor progression and metastasis [54]. FAT1 deletion promotes the acquisition of pEMT status in mouse and human SCC, thereby increasing tumor stemness and metastasis. Depletion of FAT1 functionally activates the CAMK2/CD44/SRC axis, promotes YAP1 nuclear translocation-mediated ZEB1 expression, and stimulates the mesenchymal state of SCC [36]. Moreover, depletion of FAT1 inactivates EZH2 and promotes the expression of SOX2, thereby maintaining the epithelial state [45]. The results of a recent study also showed that the ZIP4-miR-373-LATS2-ZEB1/YAP1-ITGA3 signaling axis has a significant impact on pancreatic cancer metastasis and pEMT phenotype acquisition [55].
Inhibition of FAT1 can activate HIPPO-mediated partial EMT in SCC. A: Under the background of function of FAT1 and amplification or overexpression of EGFR, which is involved in the molecular mechanisms of SCC development. B: The loss of Fat1 accelerates tumor initiation and malignant progression, and promotes the partial EMT phenotype. The loss of FAT1 function activates the CAMK2-CD44-SRC axis, promotes YAP1 nuclear translocation and ZEB1 expression, and stimulates the mesenchymal state. This loss of function also inactivates EZH2 and promotes the expression of SOX2, thereby maintaining the epithelial state.
NOTCH/Jagged pathway-mediated regulation of pEMT in SCC
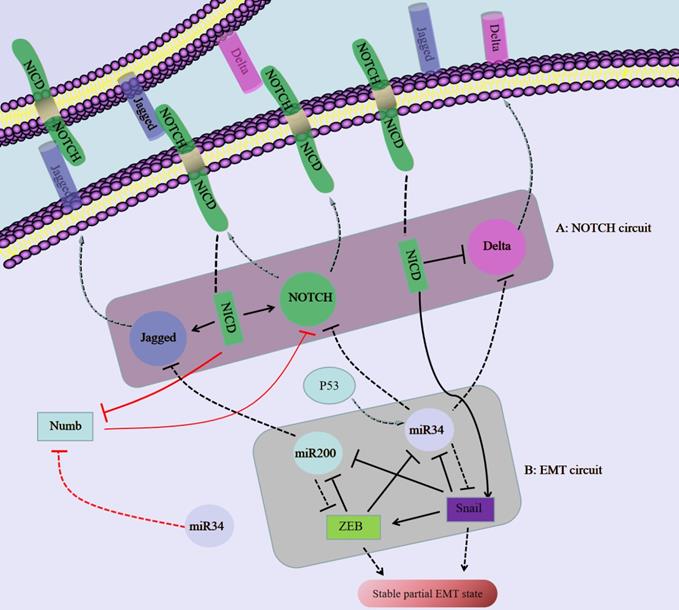
A ligand (Delta-like 1/3/4, Jagged-1/2) binds to a receptor (NOTCH1-4) to activate Notch signaling, thereby initiating the intercellular communication system [56]. Ligand binding induces conformational changes in Notch, resulting in exposure of the S2 site, which is sequentially cleaved by the A Disintegrin and Metalloproteinase (ADAM) family of proteases and the γ-secretase complex, thereby releasing the Notch intracellular domain (NICD), which then translocates to the nucleus [57]. In the nucleus, NICD binds to the transcription factor CBF-1/suppressor of hairless/Lag1 (CSL) and regulates gene expression [57]. The Notch signaling pathway can not only activate cell proliferation and antagonize apoptosis, but it can also participate in crosstalk with a variety of TFs to promote the occurrence of EMT, thereby enhancing cell activity, invasion and metastasis in vivo [58].
Numb, which regulates the endocytosis of adhesion molecules (such as E-cadherin), is important for epithelial cell-cell and cell-matrix interactions [59]. In addition, Numb inhibits Notch intercellular signaling and suppresses complete EMT by stabilizing the pEMT phenotype, which promotes mass migration of NSCLC cells [49]. In addition, Numb is associated with poor survival rates and increased lung cancer aggressiveness [60]. EMT induction in a given cell to increases the levels of Notch ligands and can activate Notch signaling in neighboring cells; in turn Notch-Delta and Notch-Jagged signaling then induces EMT [60]. Notch/Jagged signaling but not Notch/Delta signaling can cause aggregation of pEMT cells and maintain the population of cells with a pEMT phenotype [60]. In addition, the pEMT phenotype is strongly correlated with CSC attributes and increased Notch-Jagged signaling [60]. Jagged signaling is often related to the maintenance of CSC populations [61], and Notch-Jagged signaling-induced stemness may be caused by the pEMT phenotype [60].
TGF-β-mediated regulation of pEMT in SCC
Transforming growth factor β (TGF-β) is a main driver of EMT. TGF-β is a secreted cytokine that can regulate cell proliferation, migration and differentiation of many different types of cells [62]. In the process of TGF-β-induced EMT, the key effectors are transcriptional inhibitors of E-cadherin, such as Snail1/2, ZEB1/2 and Twist [62]. In scleroderma-affected skin, TGF-β-induced pEMT-like changes are characterized by the induction of Snail1 without loss of E-cadherin. Similarly, HaCaT cells (human skin keratinocytes) under continuous TGF-β stimulation exhibit pEMT characteristics [63, 64].
pEMT depends on the TGF-β pathway and is involved in lymph node metastasis in NSCLC patients [65]. In addition, TGF-β causes proliferation arrest and changes in epithelial morphology in benign and malignant HaCaT cells. The epithelial connexin ZO-1 and E-cadherin are downregulated in response to TGF-β in benign and malignant HaCaT cells but do not induce mesenchymal markers, which suggests a pEMT response [66]. In SCC cells (SiHa and FaDu), acidosis-induced TGF-β activation can promote pEMT and fatty acid metabolism [67]. TGF-β/Smad participates in crosstalk with the Wnt, Notch, Hippo, Hedgehog, PI3K-Akt, NF-κB, and JAK/STAT signaling pathways [68]. TGF-β drives Notch1-mediated EMT to generate ESCC tumor-initiating cells with high CD44 expression and inhibits Notch3 via ZEB1 expression, thereby preventing cell differentiation and allowing pEMT to progression [69].
Nuclear factor E2-related factor 2 (NRF2) can prevent adequate EMT during wound healing [70], activates pEMT and maximizes the presentation of the pEMT phenotype [71]. TGF-β transcription activates P21, thereby stabilizing NRF2, which significantly promotes glutathione metabolism and reduces the effectiveness of SCC treatment [72].
The regulatory network composed of NOTCH/Jagged and miR34/miR200 regulates the plasticity of partial EMT in SCC. A: Notch binds to an external ligand (Delta/jagged), resulting in the cleavage and release of Notch that produces NICD. NICD translocates to the nucleus, activates Notch and Jagged in transcription, and inhibits Delta at the same time. B: The EMT circuit contains miR-34, miR-200 and Snail, ZEB, which inhibit each other.
Epigenetic regulators of pEMT-TFs in SCC
Epigenetic regulatory factors (acetylation, methylation and noncoding RNAs) and posttranslational modifications (ubiquitination, sumoylation and phosphorylation) regulate the expression of EMT-TFs, thereby regulating pEMT and subsequent metastasis, stemness and therapeutic resistance [38].
miR-200 is considered a marker of cancer cells and a determinant of the epithelial phenotype [73]. miR-200 directly targets the mRNA of the E-cadherin transcriptional repressor ZEB1/2 [73]. Activation of p53 downregulates Snail expression by inducing miR-34a/b/c gene expression [74]. The miR-200/ZEB axis is driven by the miR-34/Snail axis to form a three-component stable loop; miR-200high/ZEBlow, miR-200low/ZEBhigh, and miR-200medium/ZEBmedium cells have epithelial, full EMT and pEMT phenotypes, respectively [75]. Moreover, Sukanta et al. [76] analyzed the cell fate transitions among epithelial, pEMT and mesenchymal states and confirmed that these transitions are mediated by the miR-200/ZEB mutual inhibition feedback loop, which is driven by the expression level of Snail. In addition to regulating E-cadherin and Snail, miR-200 and miR-34 also inhibit Jagged and Notch/Delta, respectively [77, 78, 79]. These regulatory mechanisms of miR-200/miR-34 can affect the pEMT status in SCC.
miR-151a promotes the contact and barrier properties of endothelial cells and promotes endothelial cell movement and angiogenesis by inducing Snail2, which is frequently amplified in solid tumors, including lung tumors [80, 81, 82]. As an oncomiR in the pathogenesis of NSCLC, miR-151a promotes tumor cell growth by regulating the expression of E-cadherin, Fibronectin and Slug, among others. In addition, as a direct target of miR-151a, E-cadherin can inhibit the migration of NSCLC cells and the transition to a mesenchymal-like cell phenotype, which suggests that the miR-151a-mediated induction of E-cadherin inhibition is the main mechanism by which miR-151a enhances pEMT in NSCLC [83].
Based on a comparison of pEMT cells with non-pEMT cells by single-cell sequencing, Snail2 is the only activated EMT-TF in pEMT cells [38]. Similarly, in the three-dimensional Madin-Darby canine kidney (MDCK) tubule formation system, Snail2 has also been shown to be a key regulator of pEMT processes in vivo [84]. Laminin subunit beta 3 (LAMB3) and PDPNs are thought to be pEMT markers and to be related to cancer metastasis. The expression of the lncRNA MYOSLID in HNSCC is closely related to that of Slug, LAMB3 and PDPN. In addition, knockout of MYOSLID significantly reduces the expression levels of Snail 2, LAMB3 and PDPN but has no effect on E-cadherin and Vimentin expression. The lncRNA MYOSLID promotes invasion and metastasis by regulating the pEMT process in HNSCC [85].
Cancer microenvironment-mediated regulation of pEMT
The cancer microenvironment (CME) surrounding tumor cells contributes to the emergence, stabilization and regulation of the pEMT phenotype, thereby promoting tumor progression [86]. The CME reflects the heterogeneity, spatial organization and complex fusion of tumor cells, fibroblasts, endothelial cells, immune cells and other mesenchymal cells in the surrounding extracellular matrix (ECM). Cancer-associated fibroblasts (CAFs) directly interact with cancer cells to promote pEMT [87, 88]. The local distribution of CAFs in the CME differs significantly between patients with local relapse and those without relapse. While those with relapse accumulate more CAFs, cancer cells adjacent to CAFs express both E-cadherin and Vimentin [88], which is similar to the findings in the study by Wang et al. [89]. pEMT cells are located at the primary tumor front in HNSCC and are near CAFs [38]. The paracrine interaction between CAFs and malignant cells promotes the pEMT program at the tumor front in HNSCC and plays a potential role in tumor invasion [38]. In cSCC and breast tumors, different EMT populations are distributed in different tumor areas and are related to specific microenvironments [44]. With the development of EMT in tumor cells, the composition of different matrix components changes; most noticeably, the immune infiltration of monocytes and macrophages increases significantly, and the density of blood and lymphatic vessels increases [44]. The reduction in macrophages in vivo increases the proportion of EpCAM+ tumor epithelial cells and cells in the early mixed EMT state and prevents further progression of EMT to a fully mesenchymal state [44].
Hypoxia is a powerful driving force for the disruption of normal tissue homeostasis and tumor-stromal interactions [90]. The most characteristic hypoxia response pathway is mediated by hypoxia inducible factor-1 (HIF-1) [91]. Hypoxic ESCC cells express high levels of HIF-1A and EIF5A2. The two-way regulation between HIF-1A and EIF5A2 plays an important role in ESCC metastasis, invasion, angiogenesis, and pEMT phenotypic coexpression of E-cadherin and Vimentin [92]. In addition, periodic or intermittent hypoxia may stabilize the pEMT phenotype via HIF-1A stabilization and/or crosstalk between NRF2 and HIF-1A [93].
Acidosis triggers the upregulation/activation of TGF-β2 and the partial EMT phenotype in a Tsp-1-dependent manner in SCC.
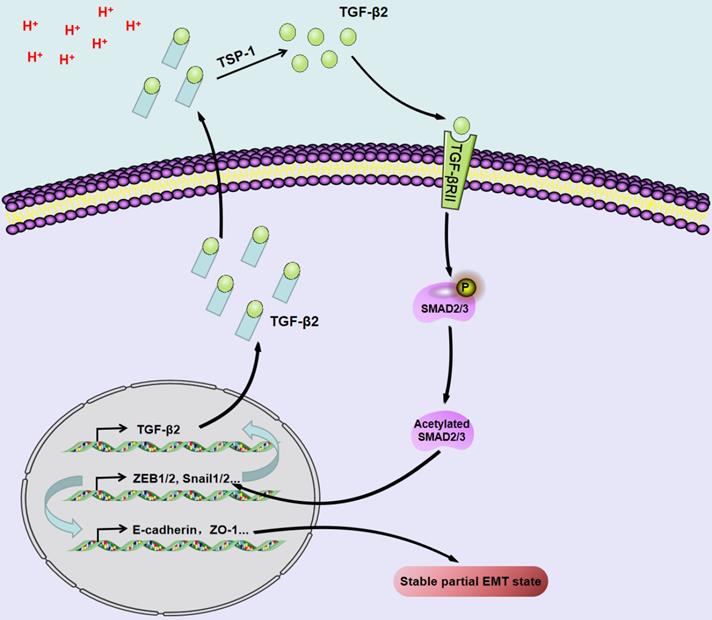
The extracellular pH of most solid tumors is acidic due to the high lactate production rate and poor perfusion. Acidosis induces genomic instability, promotes local tumor invasion and metastasis, and inhibits antitumor immunity and therapeutic resistance, thereby promoting tumor progression [94]. Acidosis triggers the upregulation/activation of TGF-β2 in a TSP-1-dependent manner and promotes pEMT in SCC through TGF-βRI and subsequent phosphorylation of Smad2/3 [69].
pEMT and collective migration in SCC
Metastasis is a process by which localized cancer becomes a systemic disease; cancer spreads due to the migration of individual cells of the primary tumor. However, new evidence found in many types of cancers, such as breast cancer, lung cancer, and mesenchymal tumors, indicates that tumor metastasis can also occur via the spread of large, cohesive cell populations that accumulate in adjacent tissues [95]. Collective migration does not require complete EMT. Cell clusters maintain intercellular connections and have some epithelial features; the acquisition of interstitial features allows the cells to migrate as a cluster. In contrast to the complete loss of cell-cell adhesion, which occurs during single-cell migration, during collective migration, cell clusters maintain cell-cell connections primarily through E-cadherin, gap junctions, and surface adhesion proteins of the immunoglobulin family [96]. Collective migration is related to metastasis, and the clinical effect of collective tumor cell migration is worse than that of single-cell migration, as collective migration confers greater potential for metastasis and proliferation as well as higher therapeutic resistance [95, 97].
A higher pEMT score is related to HNSCC lymph node metastasis and higher lymph node staging [38, 98], and pEMT is associated with a greater metastasis rate of SCC cells [45, 88]. In HNSCC, Snail induces Claudin-11-mediated Src activation and then suppresses RhoA activity at intercellular junctions through p190RhoGAP, maintains stable cell-cell contacts and induces collective migration [99]. In addition, increased Src activity stabilizes E-cadherin-based connections and collective migration of HNSCC cells [100]. In addition, Snail-mediated epithelial phenotype maintenance is important for the collective migration of SCC cells during EMT [99, 101, 102, 103], which suggests that the pEMT phenotype may affect the migration pattern in SCC.
Cells in migrating clusters are usually organized into two groups: leader cells and follower cells. Leader cells are responsible for sensing the microenvironment, generating traction to move other members of the group and reshaping the matrix through proteolysis to create a path through which the group can navigate [104]. Fourteen differentially expressed mutations were found between the leader and follower subgroups isolated from NSCLC. The functional characteristics of the two phenotype-specific candidate mutations indicated that ARP3 enhances collective invasion by promoting the phenotype of leader cells and that KDM5B inhibits chain-like cooperative behavior in follower cells [105], in contrast to pEMT plasticity [106-107], supporting the relationship between pEMT, collective migration, and leader/follower cell genetic and phenotypic differences.
pEMT and SCC stemness
EMT is associated with an increase in tumor stemness [108-109, 110]. However, complete EMT may reduce the tumor initiation potential [111, 112, 113, 114]. Some studies have reported that the stemness of CSC is most likely to be maintained in the pEMT state rather than in the pure epithelial or mesenchymal state [115, 116]. Clustered cancer cells have been reported to have greater potential to become CSC compared with individual cancer cells[117-118]. A coupled EMT-stemness network showed that acquisition of the pEMT phenotype can increase tumor stemness [119]. Moreover, research has shown that pEMT can greatly promote the stemness of SCC [120]. At least two factors may lead to enhanced stem cell properties of pEMT cells: pEMT cells have the ability to self-renewal ability [109, 121-122] and can generate hybrid subgroups of epithelial and mesenchymal cells [109, 123-124]. pEMT cells can be considered conceptually similar to adult stem cells in tissues, and this view is supported by the pEMT phenotype found in subpopulations of breast CSC [109, 125-126]. However, stemness does not always monotonically increase during EMT. When cells begin to undergo EMT, stemness may initially increase, and when cells cross the pEMT threshold to acquire a complete mesenchymal phenotype, their stemness may decrease [109].
The mechanism by which pEMT regulates stemness in SCC is still largely unknown. However, some evidence has helped us understand the connection between pEMT and stemness. In NSCLC, TGF-β1 promotes the expression of CD133 in pEMT cells, which in turn leads to the conversion of non-stem cells to CSC [127]. The OvoL family transcription factor Shavenbaby (Svb) is the downstream target of the Wnt and EGFR pathways, and this protein mediates their activity with regard to stem cell survival and proliferation. In addition, systemic steroid hormones produced by the ovaries regulate the conversion between Svb inhibitors and activators. Therefore, the Svb axis allows the internal integration of local signal cues and interorgan communication to regulate the proliferation and differentiation of stem cells and has a wide range of roles in adult stem cells and cancer stem cells [128]. A recent study showed that the interaction of EMT-TFs (Snail, Zeb1/2) and the epithelial stabilizing factor Svb can regulate stemness and pEMT [129]. In the SCC model established by Ievgenia et al.[45], the expression of CD44 and SOX2 mediated by changes in the HIPPO pathway is an important mechanism for the emergence and maintenance of pEMT. However, CD44 and SOX2 are one of the key proteins that regulate tumor SCC stemness [130]. Based on these studies, while pEMT promotes stemness in SCC, the maintenance of the pEMT phenotype may depend to a large extent on the promotion of tumor cell stemness by some pathways.
pEMT and therapeutic resistance in SCC
NSCLC patients usually benefit from treatment with epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs), such as gefitinib and erlotinib. However, EGFR resistance is likely to occur and is at least partially mediated by EMT [131, 132, 133, 134, 135, 136]. The frequency of Vimentin and E-cadherin coexpression in erlotinib-resistant NSCLC cell lines is significantly increased compared with that in the parental cell lines. In NSCLC-resistant cells, the pEMT phenotype, collective cell migration and increased stemness are associated with erlotinib resistance [137]. CTCs isolated from NSCLC patients exhibit both PD-1 ligand 1 (PD-L1) positivity and the pEMT phenotype, which may represent the molecular background of immune escape in NSCLC [138]. The pEMT phenotype may lead to unique and broad drug resistance to a variety of cancer therapies. Therapies targeting CSC in the pEMT state are expected to prevent metastasis and treatment resistance in OSCC [139]. In an OSCC cell line, the epithelial cell subset (CD44high/EpCAMhigh/CD24low) is sensitive to cisplatin, paclitaxel and salinomycin; the mesenchymal cell subset (CD44high/EpCAMlow/CD24low) is sensitive to cisplatin and salinomycin; and the pEMT subgroups (CD44high/EpCAMlow/CD24high) are resistant to all three drugs [139].
Drug resistance is often associated with the EMT process and CSC. Increasing evidence shows that traditional therapies often fail to eradicate cancer cells that have been activated by EMT programs and that have become CSC, thereby allowing CSC-mediated clinical recurrence [140]. Like EMT, the coexistence of pEMT and CSC is associated with poor prognosis and therapeutic resistance in cancer patients [115]. Similar to normal stem cells, most CSC grow slowly, which is one of the reason why CSC are resistant to chemotherapy [141]. It is reasonable to believe that pEMT cells acquire stronger drug resistance because of their stronger stemness. However, more direct and detailed studies are needed to determine the role of pEMT cells in the treatment of drug resistance.
Conclusions
Through EMT studies on breast, pancreatic and ovarian cancer, the existence of pEMT was confirmed [142]. pEMT is not a metastable transient state acquired during EMT but a stable phenotype. In addition, the functional role of pEMT may vary depending on the type of tumor, the state of spread, and the degree of metastasis and colonization [142]. At present, the research on pEMT in SCC has just started, but some phenomena have been observed. Through pEMT, SCC cells acquire a stronger tumor initiation ability than that of epithelial and mesenchymal phenotype cells and undergo collective migration, thereby promoting invasion and metastasis into SCC patients' lymphatic and circulatory systems. After pEMT phenotype cells reach a suitable niche, the epithelial tumor phenotype can be completely or partially restored by mesenchymal-epithelial transition (MET) [96]. A stochastic dynamics study, which used the dimension reduction approach of landscape (DRL) method to study the gene regulatory network interacting with metabolism and EMT, found a wide range of parameters that can produce four stable states, corresponding to epithelial (E), abnormal metabolism (A), pEMT (H) and mesenchymal (M) cell state. Further calculations quantified the transition path between these states and regarded it as a biological path. Cells tend to follow the sequence during EMT or MET. For EMT, cells in the E state need to enter the A state first , and then enter the M state, while for the MET, before the cells in the M state reaching the E state, cells are likely to enter the H state first.[143]. Therefore, tumor cells in the mesenchymal state can still transform into the pEMT phenotype when the microenvironment changes. The driving factors (internal and external) of pEMT in SCC cannot be fully determined. Changes in some signaling pathways (the HIPPO, NOTCH, and TGF-β pathways), EMT-TFs, the CME and noncoding RNAs can produce or/and maintain the pEMT phenotype in SCC. PSFs such as GRHL2, OVOL2 and miR-145 are also considered to be related to the maintenance of pEMT [144]; however, the importance of PSFs for pEMT in SCC has not been reported.
Although we have noted the markers of each stage of EMT in SCC, which is very important for the study and separation of pEMT subgroups, there is no clear consensus on the current definition of pEMT based on the coexpression of selected mesenchymal and epithelial markers in SCC. Therefore, determining the model of epithelial and mesenchymal marker coexpression to standardize the characterization of pEMT remains a main challenge in this field. In addition, developing strategies to design treatments for pEMT subgroups is a direction worthy of consideration. The drug resistance and plasticity of pEMT subgroups, the tendency of SCC to metastasize and the poor prognosis of SCC patients with metastasis emphasize that traditional treatment methods cannot effectively overcome the poor prognosis conferred by pEMT.
Acknowledgements
Author Contributions
Xiaoyan Guan, Chengcheng Liao and Jianguo Liu had the idea for the article. Literature search was performed by Chengcheng Liao, Guanyi Wang and Enming Kang. The manuscript was written by Yibin Jia and Qian Wang, Jiaxing An, Qian Long and Hui Wang critically revised the work. Meiling Xiang, Mingli Xiang, Yujie Zhao and Yulin Liu design and draw the figures in this review. All authors read and approved the final manuscript.
Funding
This study was supported by the the Graduate Research Fund of Guizhou Province (Qian-Jiao-He YJSCXJH [2019]087); the Sixth Talent Foundation in Guizhou province (rcjd2019-9); the Youth Science and Technology Talents Growth Project of Guizhou Education Department (Qian-Jiao-He KY ZI [2018]236); Outstanding Young Talent Project of Zunyi Medical University (17zy-002) (F-801); National Natural Science Foundation of China (82060205); and The Project to Cultivate Young Scientific and Technological Talents in Colleges and Universities of Guizhou Province (Qian Jiao He KY [2021] 215).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Singh M, Yelle N, Venugopal C, Singh SK. EMT: Mechanisms and therapeutic implications. Pharmacol Ther. 2018;182:80-94
2. Lu W, Kang Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev Cell. 2019;49:361-74
3. Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S. et al. Identification of the tumour transition states occurring during EMT. Nature. 2018;556:463-468
4. Huang RY, Wong MK, Tan TZ, Kuay KT, Ng AH, Chung VY, Chu YS. et al. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death Dis. 2013;4:e915
5. Zhang J, Tian XJ, Zhang H, Teng Y, Li R, Bai F. et al. TGF-β-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci Signal. 2014;7:ra91
6. Hong T, Watanabe K, Ta CH, Villarreal-Ponce A, Nie Q, Dai X. An Ovol2-Zeb1 Mutual Inhibitory Circuit Governs Bidirectional and Multi-step Transition between Epithelial and Mesenchymal States. PLoS Comput Biol. 2015;11:e1004569
7. Jolly MK, Tripathi SC, Jia D, Mooney SM, Celiktas M, Hanash SM. et al. Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget. 2016;7:27067-84
8. Bierie B, Pierce SE, Kroeger C, Stover DG, Pattabiraman DR, Thiru P. et al. Integrin-β4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc Natl Acad Sci U S A. 2017;114:E2337-46
9. Burger GA, Danen EHJ, Beltman JB. Deciphering Epithelial-Mesenchymal Transition Regulatory Networks in Cancer through Computational Approaches. Front Oncol. 2017;7:162
10. Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166:21-45
11. Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL. et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet. 2017;49:708-18
12. Grigore AD, Jolly MK, Jia D, Farach-Carson MC, Levine H. Tumor Budding: The Name is EMT. Partial EMT. J Clin Med. 2016;5:51
13. Jolly MK, Ward C, Eapen MS, Myers S, Hallgren O, Levine H. et al. Epithelial-mesenchymal transition, a spectrum of states: Role in lung development, homeostasis, and disease. Dev Dyn. 2018;247:346-58
14. Bhatia S, Monkman J, Toh AKL, Nagaraj SH, Thompson EW. Targeting epithelial-mesenchymal plasticity in cancer: clinical and preclinical advances in therapy and monitoring. Biochem J. 2017;474:3269-306
15. Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia. 2010;15:117-34
16. Li Y, Hu Q, Li C, Liang K, Xiang Y, Hsiao H. et al. PTEN-induced partial epithelial-mesenchymal transition drives diabetic kidney disease. J Clin Invest. 2019;129:1129-51
17. Saxena K, Jolly MK, Balamurugan K. Hypoxia, partial EMT and collective migration: Emerging culprits in metastasis. Transl Oncol. 2020;13:100845
18. Jolly MK, Boareto M, Huang B, Jia D, Lu M, Ben-Jacob E. et al. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front Oncol. 2015;5:155
19. Kröger C, Afeyan A, Mraz J, Eaton EN, Reinhardt F, Khodor YL. et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc Natl Acad Sci U S A. 2019;116:7353-62
20. Pastushenko I, Blanpain C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019;29:212-26
21. Tayoun T, Faugeroux V, Oulhen M, Aberlenc A, Pawlikowska P, Farace F. CTC-Derived Models: A Window into the Seeding Capacity of Circulating Tumor Cells (CTCs). Cells. 2019;8:1145
22. Taki M, Abiko K, Ukita M, Murakami R, Yamanoi K, Yamaguchi K. et al. Tumor Immune Microenvironment during Epithelial-Mesenchymal Transition [published online ahead of print, 2021 Apr 7]. Clin Cancer Res. 2021; clincanres.4459. 2020
23. Dodagatta-Marri E, Meyer DS, Reeves MQ, Paniagua R, To MD, Binnewies M. et al. α-PD-1 therapy elevates Treg/Th balance and increases tumor cell pSmad3 that are both targeted by α-TGFβ antibody to promote durable rejection and immunity in squamous cell carcinomas. J Immunother Cancer. 2019;7:62
24. Jia D, Li X, Bocci F, Tripathi S, Deng Y, Jolly MK. et al. Quantifying Cancer Epithelial-Mesenchymal Plasticity and its Association with Stemness and Immune Response. J Clin Med. 2019;8:725
25. Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet. 2013;381:400-12
26. Li B, Sun H, Zhang S, Wang X, Guo Q. The utility of APT and IVIM in the diagnosis and differentiation of squamous cell carcinoma of the cervix: A pilot study. Magn Reson Imaging. 2019;63:105-13
27. Sanderson RJ, Ironside JAD. Squamous cell carcinomas of the head and neck. BMJ. 2002;325:822-7
28. Rooney M, Devarakonda S, Govindan R. Genomics of squamous cell lung cancer. Oncologist. 2013;18:707-16
29. Stratigos A, Garbe C, Lebbe C, Malvehy J, del Marmol V, Pehamberger H. et al. Diagnosis and treatment of invasive squamous cell carcinoma of the skin: European consensus-based interdisciplinary guideline. Eur J Cancer. 2015;51:1989-2007
30. Hu X, Wu D, He X, Zhao H, He Z, Lin J. et al. circGSK3β promotes metastasis in esophageal squamous cell carcinoma by augmenting β-catenin signaling. Mol Cancer. 2019;18:160
31. Ahadiat O, Higgins S, Sutton A, Ly A, Wysong A. SLNB in cutaneous SCC: A review of the current state of literature and the direction for the future. J Surg Oncol. 2017;116:344-50
32. Liao C, An J, Tan Z, Xu F, Liu J, Wang Q. Changes in Protein Glycosylation in Head and Neck Squamous Cell Carcinoma. J Cancer. 2021;12:1455-66
33. Dezube AR, Jaklitsch MT. Minimizing residual occult nodal metastasis in NSCLC: recent advances, current status and controversies. Expert Rev Anticancer Ther. 2020;20:117-30
34. Goldstraw P, Ball D, Jett JR, Le Chevalier T, Lim E, Nicholson AG. et al. Non-small-cell lung cancer. Lancet. 2011;378:1727-40
35. Parsons A, Daley A, Begh R, Aveyard P. Influence of smoking cessation after diagnosis of early stage lung cancer on prognosis: systematic review of observational studies with meta-analysis. BMJ. 2010;340:b5569
36. Relli V, Trerotola M, Guerra E, Alberti S. Abandoning the Notion of Non-Small Cell Lung Cancer. Trends Mol Med. 2019;25:585-94
37. Sinha D, Saha P, Samanta A, Bishayee A. Emerging Concepts of Hybrid Epithelial-to-Mesenchymal Transition in Cancer Progression. Biomolecules. 2020;10:1561
38. Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S. et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell. 2017;171:1611-24 e24
39. Wangmo C, Charoen N, Jantharapattana K, Dechaphunkul A, Thongsuksai P. Epithelial-Mesenchymal Transition Predicts Survival in Oral Squamous Cell Carcinoma. Pathol Oncol Res. 2020;26:1511-8
40. Usami Y, Satake S, Nakayama F, Matsumoto M, Ohnuma K, Komori T. et al. Snail-associated epithelial-mesenchymal transition promotes oesophageal squamous cell carcinoma motility and progression. J Pathol. 2008;215:330-9
41. Wen J, Luo KJ, Liu QW, Wang G, Zhang MF, Xie XY. et al. The epithelial-mesenchymal transition phenotype of metastatic lymph nodes impacts the prognosis of esophageal squamous cell carcinoma patients. Oncotarget. 2016;7:37581-8
42. Umbreit C, Flanjak J, Weiss C, Erben P, Aderhold C, Faber A. et al. Incomplete Epithelial- Mesenchymal Transition in p16-positive Squamous Cell Carcinoma Cells Correlates with β-Catenin Expression. Anticancer Res. 2014;34:7061-9
43. Wen J, Luo KJ, Liu QW, Wang G, Zhang MF, Xie XY. et al. The epithelial-mesenchymal transition phenotype of metastatic lymph nodes impacts the prognosis of esophageal squamous cell carcinoma patients. Oncotarget. 2016;7:37581-8
44. Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S. et al. Identification of the tumour transition states occurring during EMT. Nature. 2018;556:463-8
45. Pastushenko I, Mauri F, Song Y, de Cock F, Meeusen B, Swedlund B. et al. Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis. Nature. 2021;589:448-55
46. Norgard RJ, Stanger BZ. Isolation and Identification of EMT Subtypes. Methods Mol Biol. 2021;2179:315-26
47. Sinha D, Saha P, Samanta A, Bishayee A. Emerging Concepts of Hybrid Epithelial-to-Mesenchymal Transition in Cancer Progression. Biomolecules. 2020;10:1561
48. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014;7:re8
49. Toll A, Masferrer E, Hernández-Ruiz ME, Ferrandiz-Pulido C, Yébenes M, Jaka A. et al. Epithelial to mesenchymal transition markers are associated with an increased metastatic risk in primary cutaneous squamous cell carcinomas but are attenuated in lymph node metastases. J Dermatol Sci. 2013;72:93-102
50. Andriani F, Bertolini G, Facchinetti F, Baldoli E, Moro M, Casalini P. et al. Conversion to stem-cell state in response to microenvironmental cues is regulated by balance between epithelial and mesenchymal features in lung cancer cells. Mol Oncol. 2016;10:253-71
51. Pastushenko I., Blanpain C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019;29:212-26
52. Hu X, Zhai Y, Kong P, Cui H, Yan T, Yang J. et al. FAT1 prevents epithelial mesenchymal transition (EMT) via MAPK/ERK signaling pathway in esophageal squamous cell cancer. Cancer Lett. 2017;397:83-93
53. Katoh M. Function and cancer genomics of FAT family genes (review). Int J Oncol. 2012;41:1913-8
54. Maehama T, Nishio M, Otani J, Mak TW, Suzuki A. The role of Hippo-YAP signaling in squamous cell carcinomas. Cancer Sci. 2021;112:51-60
55. Liu M, Zhang Y, Yang J, Zhan H, Zhou Z, Jiang Y. et al. Zinc-Dependent Regulation of ZEB1 and YAP1 Coactivation Promotes Epithelial-Mesenchymal Transition Plasticity and Metastasis in Pancreatic Cancer. Gastroenterology. 2021;160:1771-83.e1
56. Capaccione KM, Pine SR. The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis. 2013;34:1420-30
57. Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling-are we there yet? Nat Rev Drug Discov. 2014;13:357-78
58. Yuan X, Wu H, Han N, Xu H, Chu Q, Yu S. et al. Notch signaling and EMT in non-small cell lung cancer: biological significance and therapeutic application. J Hematol Oncol. 2014;7:87
59. Chatterjee SJ, Halaoui R, Deagle RC, Rejon C, McCaffrey L. Numb regulates cell tension required for mammary duct elongation. Biol Open. 2019;8:bio042341
60. Bocci F, Jolly MK, Tripathi SC, Aguilar M, Hanash SM, Levine H. et al. Numb prevents a complete epithelial-mesenchymal transition by modulating Notch signalling. J R Soc Interface. 2017;14:20170512
61. Espinoza I, Miele L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett. 2013;341:41-5
62. Hao Y, Baker D, Ten Dijke P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int J Mol Sci. 2019;20:2767
63. Liarte S, Bernabé-García Á, Nicolás FJ. Human Skin Keratinocytes on Sustained TGF-β Stimulation Reveal Partial EMT Features and Weaken Growth Arrest Responses. Cells. 2020;9:255
64. Hahn JM, McFarland KL, Combs KA, Supp DM. Partial epithelial-mesenchymal transition in keloid scars: regulation of keloid keratinocyte gene expression by transforming growth factor-β1. Burns Trauma. 2016;4:30
65. Emprou C, Le Van Quyen P, Jégu J, Prim N, Weingertner N, Guérin E. et al. SNAI2 and TWIST1 in lymph node progression in early stages of NSCLC patients. Cancer Med. 2018;7:3278-91
66. Räsänen K, Vaheri A. TGF-beta1 causes epithelial-mesenchymal transition in HaCaT derivatives, but induces expression of COX-2 and migration only in benign, not in malignant keratinocytes. J Dermatol Sci. 2010;58:97-104
67. Corbet C, Bastien E, Santiago de Jesus JP, Dierge E, Martherus R, Vander Linden C. et al. TGFβ2-induced formation of lipid droplets supports acidosis-driven EMT and the metastatic spreading of cancer cells. Nat Commun. 2020;11:454
68. Luo K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb Perspect Biol. 2017;9:a022137
69. Natsuizaka M, Whelan KA, Kagawa S, Tanaka K, Giroux V, Chandramouleeswaran PM. et al. Interplay between Notch1 and Notch3 promotes EMT and tumor initiation in squamous cell carcinoma. Nat Commun. 2017;8:1758
70. Riahi R, Long M, Yang Y, Dean Z, Zhang DD, Slepian MJ. et al. Single cell gene expression analysis in injury-induced collective cell migration. Integr Biol (Camb). 2014;6:192-202
71. Bocci F, Tripathi SC, George JT, Casabar J, Mercedes S V, Wong P. et al. NRF2 activates a partial Epithelial-Mesenchymal Transition and is maximally present in a hybrid Epithelial/Mesenchymal phenotype. Integr Biol (Camb). 2019;11:251-63
72. Oshimori N, Oristian D, Fuchs E. TGF-β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell. 2015;160:963-76
73. Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894-907
74. Siemens H, Jackstadt R, Hünten S, Kaller M, Menssen A, Götz U. et al. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle. 2011;10:4256-71
75. Lu M, Jolly MK, Levine H, Onuchic JN, Ben-Jacob E. MicroRNA-based regulation of epithelial-hybrid-mesenchymal fate determination. Proc Natl Acad Sci U S A. 2013;110:18144-9
76. Sarkar S, Sinha SK, Levine H, Jolly MK, Dutta PS. Anticipating critical transitions in epithelial-hybrid-mesenchymal cell-fate determination. Proc Natl Acad Sci U S A. 2019;116:26343-52
77. Brabletz S, Bajdak K, Meidhof S, Burk U, Niedermann G, Firat E. et al. The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J. 2011;30:770-82
78. de Antonellis P, Medaglia C, Cusanelli E, Andolfo I, Liguori L, De Vita G. et al. MiR-34a targeting of Notch ligand delta-like 1 impairs CD15+/CD133+ tumor-propagating cells and supports neural differentiation in medulloblastoma. PLoS One. 2011;6:e24584
79. Bu P, Chen KY, Chen JH, Wang L, Walters J, Shin YJ. et al. A microRNA miR-34a-regulated bimodal switch targets Notch in colon cancer stem cells. Cell Stem Cell. 2013;12:602-15
80. Wong MP, Fung LF, Wang E, Chow WS, Chiu SW, Lam WK. et al. Chromosomal aberrations of primary lung adenocarcinomas in nonsmokers. Cancer. 2003;97:1263-70
81. Lips EH, van Eijk R, de Graaf EJ, Oosting J, de Miranda NF, Karsten T. et al. Integrating chromosomal aberrations and gene expression profiles to dissect rectal tumorigenesis. BMC Cancer. 2008;8:314
82. Ding J, Huang S, Wu S, Zhao Y, Liang L, Yan M. et al. Gain of miR-151 on chromosome 8q24.3 facilitates tumour cell migration and spreading through downregulating RhoGDIA. Nat Cell Biol. 2010;12:390-9
83. Daugaard I, Sanders KJ, Idica A, Vittayarukskul K, Hamdorf M, Krog JD. et al. miR-151a induces partial EMT by regulating E-cadherin in NSCLC cells. Oncogenesis. 2017;6:e366
84. Leroy P, Mostov KE. Slug is required for cell survival during partial epithelial-mesenchymal transition of HGF-induced tubulogenesis. Mol Biol Cell. 2007;18:1943-52
85. Xiong HG, Li H, Xiao Y, Yang QC, Yang LL, Chen L. et al. Long noncoding RNA MYOSLID promotes invasion and metastasis by modulating the partial epithelial-mesenchymal transition program in head and neck squamous cell carcinoma. J Exp Clin Cancer Res. 2019;38:278
86. Bhatia S, Wang P, Toh A, Thompson EW. New insights into the role of phenotypic plasticity and EMT in driving cancer progression. Front. Mol. Biosci. 2020;7:71
87. Ligorio M, Sil S, Malagon-Lopez J, Nieman LT, Misale S, Di Pilato M. et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell. 2019;178:160-75.e27
88. Okuyama K, Suzuki K, Yanamoto S, Naruse T, Tsuchihashi H, Yamashita S. et al. Anaplastic transition within the cancer microenvironment in early-stage oral tongue squamous cell carcinoma is associated with local recurrence. Int J Oncol. 2018;53:1713-20
89. Wang C, Huang H, Huang Z, Wang A, Chen X, Huang L. et al. Tumor budding correlates with poor prognosis and epithelial-mesenchymal transition in tongue squamous cell carcinoma. J Oral Pathol Med. 2011;40:545-51
90. Harris AL. Hypoxia-a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38-47
91. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721-32
92. Li Y, Fu L, Li JB, Qin Y, Zeng TT, Zhou J. et al. Increased expression of EIF5A2, via hypoxia or gene amplification, contributes to metastasis and angiogenesis of esophageal squamous cell carcinoma. Gastroenterology. 2014;146:1701-13 e1709
93. Saxena K, Jolly MK, Balamurugan K. Hypoxia, partial EMT and collective migration: emerging culprits in metastasis. Tranl. Oncol. 2020;13:100845
94. Ibrahim-Hashim A, Estrella V. Acidosis and cancer: from mechanism to neutralization. Cancer Metastasis Rev. 2019;38:149-55
95. Yang Y, Zheng H, Zhan Y, Fan S. An emerging tumor invasion mechanism about the collective cell migration. Am J Transl Res. 2019;11:5301-12
96. Aggarwal V, Montoya CA, Donnenberg VS, Sant S. Interplay between tumor microenvironment and partial EMT as the driver of tumor progression. iScience. 2021;24:102113
97. Cheung KJ, Ewald AJ. A collective route to metastasis: Seeding by tumor cell clusters. Science. 2016;352:167-9
98. Parikh AS, Puram SV, Faquin WC, Richmon JD, Emerick KS, Deschler DG. et al. Immunohistochemical quantification of partial-EMT in oral cavity squamous cell carcinoma primary tumors is associated with nodal metastasis. Oral Oncol. 2019;99:104458
99. Li CF, Chen JY, Ho YH, Hsu WH, Wu LC, Lan HY. et al. Snail-induced claudin-11 prompts collective migration for tumour progression. Nat Cell Biol. 2019;21:251-62
100. Veracini L, Grall D, Schaub S, Beghelli-de la Forest Divonne S, Etienne-Grimaldi MC, Milano G. et al. Elevated Src family kinase activity stabilizes E-cadherin-based junctions and collective movement of head and neck squamous cell carcinomas. Oncotarget. 2015;6:7570-83
101. Rizqiawan A, Tobiume K, Okui G, Yamamoto K, Shigeishi H, Ono S. et al. Autocrine galectin-1 promotes collective cell migration of squamous cell carcinoma cells through up-regulation of distinct integrins. Biochem Biophys Res Commun. 2013;441:904-10
102. Tanaka F, Rizqiawan A, Higashikawa K, Tobiume K, Okui G, Shigeishi H. et al. Snail promotes Cyr61 secretion to prime collective cell migration and form invasive tumor nests in squamous cell carcinoma. Cancer Lett. 2013;329:243-52
103. Okui G, Tobiume K, Rizqiawan A, Yamamoto K, Shigeishi H, Ono S. et al. AKT primes snail-induced EMT concomitantly with the collective migration of squamous cell carcinoma cells. J Cell Biochem. 2013;114:2039-49
104. Khalil AA, Friedl P. Determinants of leader cells in collective cell migration. Integr Biol (Camb). 2010;2:568-74
105. Zoeller EL, Pedro B, Konen J, Dwivedi B, Rupji M, Sundararaman N. et al. Genetic heterogeneity within collective invasion packs drives leader and follower cell phenotypes. J Cell Sci. 2019;132:jcs231514
106. Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S. et al. Identification of the tumour transition states occurring during EMT. Nature. 2018;556:463-8
107. Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell. 2013;155:1639-51
108. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704-15
109. Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008;3:e2888
110. Tanabe S, Quader S, Cabral H, Ono R. Interplay of EMT and CSC in Cancer and the Potential Therapeutic Strategies. Front Pharmacol. 2020;11:904
111. Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S. et al. Identifification of the tumour transition states occurring during EMT. Nature. 2018;556:463-8
112. Ruscetti M, Quach B, Dadashian EL, Mulholland DJ, Wu H. Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis. Cancer Res. 2015;75:2749-59
113. Celià-Terrassa T, Meca-Cortés O, Mateo F, Martínez de Paz A, Rubio N, Arnal-Estapé A. et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest. 2012;122:1849-68
114. Grosse-Wilde A, Fouquier d'Hérouël A, McIntosh E, Ertaylan G, Skupin A, Kuestner RE. et al. Stemness of the hybrid Epithelial/Mesenchymal State in Breast Cancer and Its Association with Poor Survival. PLoS One. 2015;10:e0126522
115. Jolly MK, Somarelli JA, Sheth M, Biddle A, Tripathi SC, Armstrong AJ. et al. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol Ther. 2019;194:161-84
116. Biddle A, Liang X, Gammon L, Fazil B, Harper LJ, Emich H. et al. Cancer stem cells in squamous cell carcinoma switch between two distinct phenotypes that are preferentially migratory or proliferative. Cancer Res. 2011;71:5317-26
117. Kai K, D'Costa S, Yoon BI, Brody AR, Sills RC, Kim Y. Characterization of side population cells in human malignant mesothelioma cell lines. Lung Cancer. 2010;70:146-51
118. Yeung TM, Gandhi SC, Wilding JL, Muschel R, Bodmer WF. Cancer stem cells from colorectal cancer-derived cell lines. Proc Natl Acad Sci U S A. 2010;107:3722-7
119. Pasani S, Sahoo S, Jolly MK. Hybrid E/M Phenotype(s) and Stemness: A Mechanistic Connection Embedded in Network Topology. J Clin Med. 2020;10:60
120. Andriani F, Bertolini G, Facchinetti F, Baldoli E, Moro M, Casalini P. et al. Conversion to stem-cell state in response to microenvironmental cues is regulated by balance between epithelial and mesenchymal features in lung cancer cells. Mol Oncol. 2016;10:253-71
121. Vega S, Morales AV, Ocaña OH, Valdés F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004;18:1131-43
122. Comaills V, Kabeche L, Morris R, Buisson R, Yu M, Madden MW. et al. Genomic Instability Is Induced by Persistent Proliferation of Cells Undergoing Epithelial-to-Mesenchymal Transition. Cell Rep. 2016;17:2632-47
123. Ruscetti M, Dadashian EL, Guo W, Quach B, Mulholland DJ, Park JW. et al. HDAC inhibition impedes epithelial-mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene. 2016;35:3781-95
124. Tripathi S, Chakraborty P, Levine H, Jolly MK. A mechanism for epithelial-mesenchymal heterogeneity in a population of cancer cells. PLoS Comput Biol. 2020;16:e1007619
125. Colacino JA, Azizi E, Brooks MD, Harouaka R, Fouladdel S, McDermott SP. et al. Heterogeneity of Human Breast Stem and Progenitor Cells as Revealed by Transcriptional Profiling. Stem Cell Reports. 2018;10:1596-609
126. Thong T, Wang Y, Brooks MD, Lee CT, Scott C, Balzano L. et al. Hybrid Stem Cell States: Insights Into the Relationship Between Mammary Development and Breast Cancer Using Single-Cell Transcriptomics. Front Cell Dev Biol. 2020;8:288
127. Andriani F, Bertolini G, Facchinetti F, Baldoli E, Moro M, Casalini P. et al. Conversion to stem-cell state in response to microenvironmental cues is regulated by balance between epithelial and mesenchymal features in lung cancer cells. Mol Oncol. 2016;10:253-71
128. Al Hayek S, Alsawadi A, Kambris Z, Boquete JP, Bohère J, Immarigeon C. et al. Steroid-dependent switch of OvoL/Shavenbaby controls self-renewal versus differentiation of intestinal stem cells. EMBO J. 2021;40:e104347
129. Mancheno-Ferris A, Polesello C, Payre F. Les facteurs OvoL - Des régulateurs clés de la plasticité épithélium-mésenchyme et des cellules souches [OvoL factors: a family of key regulators of epithelium mesenchyme plasticity and stem cells]. Med Sci (Paris). 2020;36:61-6
130. Yu SS, Cirillo N. The molecular markers of cancer stem cells in head and neck tumors. J Cell Physiol. 2020;235:65-73
131. Thomson S, Buck E, Petti F, Griffin G, Brown E, Ramnarine N. et al. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005;65:9455-62
132. Shien K, Toyooka S, Yamamoto H, Soh J, Jida M, Thu KL. et al. Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell-like properties in cancer cells. Cancer Res. 2013;73:3051-61
133. Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L. et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005;11:8686-98
134. Thomson S, Petti F, Sujka-Kwok I, Epstein D, Haley JD. Kinase switching in mesenchymal-like non-small cell lung cancer lines contributes to EGFR inhibitor resistance through pathway redundancy. Clin Exp Metastasis. 2008;25:843-54
135. Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M. et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19:279-90
136. Li L, Han R, Xiao H, Lin C, Wang Y, Liu H. et al. Metformin sensitizes EGFR-TKI-Resistant human lung cancer cells in vitro and in vivo through inhibition of IL-6 signaling and EMT reversal. Clin Cancer Res. 2014;20:2714-26
137. Fustaino V, Presutti D, Colombo T, Cardinali B, Papoff G, Brandi R. et al. Characterization of epithelial-mesenchymal transition intermediate/hybrid phenotypes associated to resistance to EGFR inhibitors in non-small cell lung cancer cell lines. Oncotarget. 2017;8:103340-63
138. Raimondi C, Carpino G, Nicolazzo C, Gradilone A, Gianni W, Gelibter A. et al. PD-L1 and epithelial-mesenchymal transition in circulating tumor cells from non-small cell lung cancer patients: A molecular shield to evade immune system? Oncoimmunology. 2017;6:e1315488
139. Biddle A, Gammon L, Liang X, Costea DE, Mackenzie IC. Phenotypic Plasticity Determines Cancer Stem Cell Therapeutic Resistance in Oral Squamous Cell Carcinoma. EBioMedicine. 2016;4:138-45
140. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611-29
141. Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10:717-28
142. Bakir B, Chiarella AM, Pitarresi JR, Rustgi AK. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020;30:764-76
143. Kang X, Li C. A Dimension Reduction Approach for Energy Landscape: Identifying Intermediate States in Metabolism-EMT Network. Adv Sci (Weinh). 2021;8:2003133
144. Watanabe K, Villarreal-Ponce A, Sun P, Salmans ML, Fallahi M, Andersen B. et al. Mammary morphogenesis and regeneration require the inhibition of EMT at terminal end buds by Ovol2 transcriptional repressor. Dev Cell. 2014;29:59-74
Author contact
![]() Corresponding authors: Xiaoyan Guan; E-mail: 1278279125com; Tel.: 86-18089620011; Jianguo Liu; E-mail: jgl_zmucom; Tel.: 86-13087891001
Corresponding authors: Xiaoyan Guan; E-mail: 1278279125com; Tel.: 86-18089620011; Jianguo Liu; E-mail: jgl_zmucom; Tel.: 86-13087891001