Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Material and Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(12):3255-3267. doi:10.7150/ijbs.63125 This issue Cite
Research Paper
Discovery and Validation of Nitroxoline as a Novel STAT3 Inhibitor in Drug-resistant Urothelial Bladder Cancer
Wenfeng Lin1, Jingkai Sun1,2, Takuya Sadahira1, Naijin Xu1,3, Koichiro Wada1, Chunxiao Liu2, Motoo Araki1, Abai Xu2, Masami Watanabe1,4, Yasutomo Nasu1, Peng Huang1,2,5 ![]()
1. Department of Urology, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama, Japan
2. Department of Urology, Zhujiang Hospital, Southern Medical University, Guangzhou, China
3. Department of Urology, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine, Hangzhou, China
4. Center for Innovative Clinical Medicine, Okayama University Hospital, Okayama, Japan
5. Okayama Medical Innovation Center, Okayama University, Okayama, Japan
Received 2021-5-25; Accepted 2021-7-6; Published 2021-7-25
Abstract

Repeated cycles of first-line chemotherapy drugs such as doxorubicin (DOX) and cisplatin (CIS) trigger frequent chemoresistance in recurrent urothelial bladder cancer (UBC). Nitroxoline (NTX), an antibiotic to treat urinary tract infections, has been recently repurposed for cancer treatment. Here we aimed to investigate whether NTX suppresses drug-resistant UBC and its molecular mechanism. The drug-resistant cell lines T24/DOX and T24/CIS were established by continual exposure of parental cell line T24 to DOX and CIS, respectively. T24/DOX and T24/CIS cells were resistant to DOX and CIS, respectively, but they were sensitive to NTX time- and dose-dependently. Overexpressions of STAT3 and P-glycoprotein (P-gp) were identified in T24/DOX and T24/CIS, which could be reversed by NTX. Western blot revealed that NTX downregulated p-STAT3, c-Myc, Cyclin D1, CDK4, CDK6, Bcl-xL, Mcl-1, and Survivin, which were further confirmed by Stattic, a selective STAT3 inhibitor. In vivo, NTX exhibited the significant anti-tumor effect in T24/DOX and T24/CIS tumor-bearing mice. These results suggested that NTX-induced P-gp reversal, G0/G1 arrest, and apoptosis in drug-resistant UBC were mediated by inhibition of STAT3 signaling. Our findings repurpose NTX as a novel STAT3 inhibitor to induce P-gp reversal, G0/G1 arrest, and apoptosis in drug-resistant UBC.
Keywords: Urothelial bladder cancer, doxorubicin, cisplatin, chemoresistance, nitroxoline, STAT3.
Introduction
Urothelial bladder cancer (UBC) is a malignant tumor originating primarily from the transitional cells of bladder urothelium [1]. The 2021 Cancer Statistics [2] reported an estimation of 83,730 new UBC cases and 17,200 UBC-specific deaths in America. Moreover, 50%-70% patients with non-muscle-invasive bladder cancer (NMIBC) will suffer a relapse following transurethral resection, while approximately 30% will progress into muscle-invasive bladder cancer (MIBC), mainly causing UBC-specific deaths [3]. Both doxorubicin (DOX/ADM/ADR) and cisplatin (CIS/DDP/CDDP) have been the first-line chemotherapy drugs effectively against UBC, but the high rates of DOX- and CIS- chemoresistance in recurrent UBC remain a major barrier to improve the prognosis of patients [4, 5]. It still maintains a challenge to explore novel effective chemotherapeutic agents for the treatment of drug-resistant UBC.
Multidrug resistance (MDR), a clinical obstacle to the long-term success in chemotherapy, represents the cross-resistance of tumor cells to various chemotherapeutic agents with different structures and functions [6]. One of the main reasons for MDR phenotype is the overexpressed 170-kDa transmembrane glycoprotein, P-glycoprotein (P-gp). It is encoded by the ATP-binding cassette B1 (ABCB1) gene, with an alternative name as multidrug resistance 1 (MDR1) [7]. As a drug efflux pump, P-gp extrudes its substrates such as DOX out of tumor cells, thus resulting in a reduction of intracellular drug concentration [8]. P-gp has also been reported to reduce the tumor sensitivity to the non-P-gp substrates such as CIS, while inhibition of P-gp has the potential to increase CIS sensitivity [9, 10]. Signal transducer and activators of transcription 3 (STAT3) acts as a crucial transcription factor to regulate its downstream target genes, which subsequently involves in diverse biological processes such as cell proliferation, survival, and chemoresistance [11]. Overexpression and activation of STAT3 correlate with the poor prognosis of UBC [12, 13], and STAT3 requires the phosphorylation at the residues of Tyr705 (p-STAT3 (Y705)) and Ser727 (p-STAT3 (S727)) for its maximal activation [14]. The chromatin immunoprecipitation (ChIP) assay revealed the potential binding of STAT3 to MDR1 gene promoter region, and increasing evidence suggests that inhibiting STAT3 activation effectively downregulates the expression of P-gp [15, 16]. Based on the above research evidence, the development of potent inhibitors targeting STAT3 signaling may be a promising therapeutic strategy against drug-resistant UBC. Although multiple inhibitors targeting STAT3 in cancer have been identified in preclinical and early phase clinical trials, the potential low efficacy or adverse effects may limit their clinical transformation [17].
Drug repurposing refers to discovering novel indications of the currently marketed drugs which have been applied to treat other diseases [18]. Nitroxoline (NTX), an antibiotic used clinically to treat urinary tract infections, has been repurposed for the treatment of multiple tumors such as UBC [19-21], pancreatic cancer [22], and glioma [23]. Several studies have reported its anti-tumor activity against UBC since Shim and coworkers firstly identified NTX as an antiangiogenic agent in UBC through inhibition of methionine aminopeptidase-2 (MetAP2) [19-21]. In an orthotopic mouse model, NTX was demonstrated to exhibit anti-UBC effect with favorable safety profile and pharmacokinetic properties [21]. Our previous study has also revealed that NTX suppresses the progression of UBC by reversing epithelial-mesenchymal transition (EMT) and enhancing anti-tumor immunity [20]. Prior studies have noted that P-gp expression is identified in pre-chemotherapy UBC tissue samples, which increases with MDR after chemotherapy [24, 25].
However, it has not been reported whether NTX and its analogues suppress the growth of drug-resistant UBC. And the mechanism whether NTX inhibits STAT3 signaling and reverses P-gp overexpression, remains to be explored at the molecular level. Drug repurposing of NTX in drug-resistant UBC may contribute to the following advantages including its well-known drug properties (pharmacokinetics, efficacy, toxicity and drug interactions), developing a putative agent with fewer funds and shorter period [21]. Both in vitro and in vivo experiments were performed to investigate whether NTX could reverse P-gp overexpression and effectively inhibit the growth of drug-resistant UBC cell lines through STAT3 signaling regulation.
Material and Methods
Chemicals and antibodies
The chemicals including DOX and CIS were purchased from MedChemExpress (MCE, New Jersey, USA). Both DOX and CIS were dissolved in phosphate buffered saline (PBS), followed by ultrasonic bath and 0.22-µm filtration. Nitroxoline (NTX) was provided by Jiangsu Asieris Pharmaceuticals in China. NTX was treated as PBS dissolution with 0.22-µm filtration. Stattic, a selective inhibitor of STAT3 activation and dimerization, was obtained from Tocris Bioscience (#2978, Bristol, UK), which was treated as dimethyl sulfoxide (DMSO) dissolution with a 20-mM stock solution. Primary antibodies against β-Actin (#4970), STAT3 (#12640), p-STAT3 (Y705) (#9145), p-STAT3 (S727) (#9134), MDR1/P-gp (#13342), c-Myc (#5605), Cyclin D1 (#2978), CDK4 (#2906), CDK6 (#3136), Bcl-xL (#2764), Mcl-1 (#4572), and Survivin (#2808) were provided by Cell Signaling Technology (CST, USA). Anti-rabbit IgG (#7074) and anti-mouse IgG (#7076) were the corresponding secondary antibodies.
Cell culture of drug-resistant UBC cell lines
Human UBC cell line T24 was purchased from the American Type Culture Collection (ATCC, USA). DOX-resistant bladder cancer cell line, T24/DOX, and CIS-resistant bladder cancer cell line, T24/CIS, were established from our laboratory based on continual exposure of parental cell line T24 to the culture media with DOX and CIS, respectively [26-28]. T24, T24/DOX, and T24/CIS cells were incubated in DMEM medium containing 10% FBS, 1% penicillin-streptomycin (Invitrogen, USA). To maintain the drug-resistant characteristics, T24/DOX cells were cultured with 1 μM DOX while T24/CIS cells cultured with 10 μM CIS prior to experiment.
Cell viability assay
T24, T24/DOX, and T24/CIS (2 × 103 /well) in 96-well plates were allowed to attach overnight. For drug sensitivity analysis, T24 and T24/DOX cells were treated with DOX (0, 0.1, 1, 10, 20, and 100 μM) for 24 h, while T24 and T24/CIS cells underwent 24 h treatment with CIS (0, 10, 20, 40, and 80 μM). To determine the effect of NTX on cell proliferation, T24, T24/DOX, and T24/CIS cells were exposed to NTX (0, 2.5, 5, 10, 20, and 40 μM) for 24, 48, and 72 h. At the indicated time points, culture media were removed and the cells were washed gently with PBS. The prepared XTT working solution (Cell Proliferation Kit II, #11465015001, Sigma-Aldrich) was added to the cells, which were subsequently incubated for 4-24 h at 37 ℃. Finally, the optical density (OD) levels were determined by the Model 680 Microplate Reader (Bio-Rad, USA).
Hoechst 33342 staining
T24, T24/DOX, and T24/CIS (2 × 105 /well) in 6-well plates firstly underwent 24 h culture. Next, after 48 h treatment with 0 and 40 μM NTX, cells were stained at room temperature with 5 µg/ml Hoechst 33342 (#H3570, ThermoFisher Scientific, USA) for 20 min. The occurrence of apoptotic cells was observed under fluorescence microscope.
Cell cycle distribution analysis
T24, T24/DOX, and T24/CIS (2 × 105 /well) were seeded in 6-well plates. After 24 h incubation with NTX (0, 10, 20, or 40 µM), cells were rinsed twice with PBS and fixed in precooling 70% ethanol for over 18 h at 4℃. Subsequently, they were rinsed twice with PBS and stain buffer (BD Biosciences, #554656). Following by 15 min staining in PI/RNase buffer (BD Biosciences, #550825), the analysis was performed by MACSQuant Analyzer 10 and MACSQuantifyTM Software 2.6.
Apoptosis examination
T24, T24/DOX, and T24/CIS (1.5 × 105 /well) in 6-well plates underwent 48 h incubation of NTX. After twice rinsing, they underwent 1X Annexin V binding buffer resuspension to obtain a density of 1 × 106 cells/ml. In the dark, cell suspension (100 μl) underwent 15 min treatment with FITC Annexin V (5 µl) and PI (5 µl) (BD Biosciences, #556547). Finally, apoptosis was examined by MACSQuant Analyzer 10 and analyzed by MACSQuantifyTM Software 2.6.
Western blot analysis
Total protein extraction was conducted with M-PERTM Mammalian Protein Extraction Reagent (#78501, ThermoFisher Scientific, USA) and then quantified by Bradford protein assay. The same amount of protein (10 μg/well) was separated by 10% or 12% Mini-PROTEAN Gels and then transferred to 0.2 µm PVDF Transfer Packs (#1704156, Bio-Rad, USA) on the Trans-Blot Turbo transfer system (#170-4155, Bio-Rad, USA). After 1.5 h bovine serum albumin or non-fat milk blocking, and 4°C overnight incubation with primary antibodies, membranes underwent TBST washing. At room temperature, the secondary antibody was diluted to treat membranes for 1.5 h. The ECL detection reagent (#RPN2232, GE Healthcare) and ChemiDoc Imaging System (Bio-Rad, USA) were used to visualize proteins.
Subcutaneous xenograft models of T24/DOX and T24/CIS
Male BALB/c nude mice (five-week-old) kept in specific pathogen-free (SPF) conditions were from Japan SLC, Inc. (Shizuoka, Japan). The mice experimentation was conducted in compliance with the Animal Care and Use Committee, Okayama University.
T24/DOX cells (1×106) or T24/CIS cells (1×106) in 100 µl resuspension, mixing Matrigel (BD Biosciences, USA) and Hank's buffered saline solution (HBSS) at 1:1, were used for subcutaneous inoculation to mouse right flank. The mice bearing about 100 mm3 T24/DOX tumor received two-week oral administration of NTX (40 mg/kg) or PBS (n = 6 per group) once per day. It was the same with T24/CIS tumor-bearing mice. Both mouse body weight and tumor growth were measured twice a week. Surgical resection, weighing, and photography of T24/DOX and T24/CIS tumors were conducted at the indicated endpoint. The formula of volume = 0.52 × length × width2, was applied to calculate tumor growth. T-PER Tissue Protein Extraction Reagent (#78510, ThermoFisher Scientific, USA) was used for extracting tumor tissue protein, which was subsequently analyzed by western blot. For histological examination, T24/DOX and T24/CIS tumors underwent 10% formaldehyde fixation, paraffin imbedding, and 4 μm section cut. Hematoxylin and eosin (H&E) were applied to stain the sections, followed by observing NTX-induced morphological alterations.
Statistical analysis
GraphPad Prism 8.3 was used to perform statistical analyses including half maximal inhibitory concentration (IC50) calculation and statistical charts creation. Between-group difference was assessed by Student's t-test or one-way analysis of variance (ANOVA), with data expressing as mean ± standard deviation (SD).
Results
Characterization of DOX-resistant and CIS-resistant bladder cancer cell lines
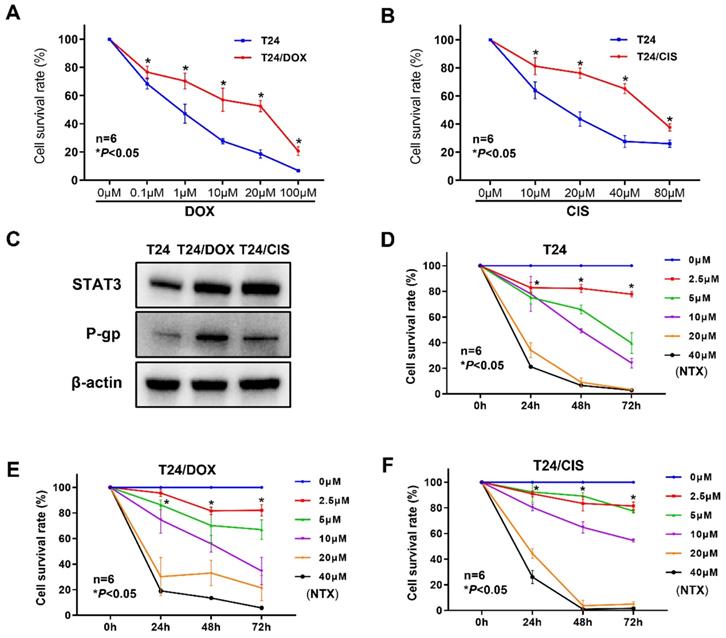
To confirm whether bladder cancer cell lines acquired DOX or CIS resistance, the viability of T24/DOX or T24/CIS cells was determined by XTT assay following the indicated treatment for 24 h. Although DOX dose-dependently inhibited the proliferation of T24/DOX cells, the sensitivity to DOX was lower than that of parental T24 cells (Figure 1A). For DOX, the IC50 value in T24 cells was 0.71 μM [95% confidence interval (95%CI), 0.56-0.89], while that in T24/DOX cells was 10.93 μM (95%CI, 6.88-17.55), with a 15.4-fold change in IC50 (Table 1). Similarly, CIS dose-dependently inhibited the proliferation of T24/CIS cells, but the sensitivity to CIS was lower than that of parental T24 cells (Figure 1B). For CIS, the IC50 value in T24 cells was 16.27 μM (95%CI, 13.29-19.11), while that in T24/CIS cells was 58.04 μM (95%CI, 51.11-67.55), with a 3.6-fold change in IC50 (Table 1).
T24/DOX and T24/CIS cells were resistant to DOX and CIS, respectively, but sensitive to NTX. (A) T24 and T24/DOX cells were treated with different concentrations of DOX (0, 0.1, 1, 10, 20, or 100 μM) for 24 h, followed by the evaluation of cell viability using XTT assay. (B) T24 and T24/CIS cells were treated with different concentrations of CIS (0, 10, 20, 40, or 80 μM) for 24 h, followed by the evaluation of cell viability using XTT assay. Data are expressed as the mean ± SD (n=6). *P < 0.05 vs T24 parental cells. (C) The expression levels of STAT3 and P-gp in T24, T24/DOX, and T24/CIS cells were analyzed by western blot analysis. β-actin was used as a loading control. (D) T24, (E) T24/DOX, and (F) T24/CIS cells were exposed to different concentrations of NTX (0, 2.5, 5, 10, 20, and 40 μM) for 24, 48, and 72 h. The cell viability was determined by XTT assay. Data are shown as the mean ± SD (n=6). *P < 0.05 vs the control group.
IC50 values* of DOX, CIS, or NTX determined in T24, T24/DOX and T24/CIS cells.
| T24 | T24/DOX | T24/Cis | ||||
|---|---|---|---|---|---|---|
| IC50 (μM) | 95%CI | IC50 (μM) | 95%CI | IC50 (μM) | 95%CI | |
| DOX (24 h) | 0.71 | 0.56-0.89 | 10.93 | 6.88-17.55 | - | - |
| CIS (24 h) | 16.27 | 13.29-19.11 | - | - | 58.04 | 51.11-67.55 |
| NTX (24 h) | 15.22 | 12.77-18.19 | 14.91 | 12.99-17.02 | 19.72 | 18.11-21.53 |
| NTX (48 h) | 7.85 | 7.15-8.61 | 10.69 | 9.31-12.27 | 11.20 | 10.17-12.30 |
| NTX (72 h) | 4.48 | 4.17-4.82 | 7.32 | 6.55-8.16 | 9.15 | 8.09-10.27 |
*IC50 values represent the concentrations of DOX, CIS, or NTX producing 50% cell growth inhibition. Abbreviations: IC50, half maximal inhibitory concentration; 95%CI: 95% confidence interval; DOX: doxorubicin; CIS: cisplatin; NTX: nitroxoline.
We next analyzed the expressions of STAT3 and P-gp by western blot. In Figure 1C, the levels of STAT3 and P-gp were upregulated in T24/DOX and T24/CIS cells compared with T24 parental cells.
NTX inhibits the proliferation of DOX-resistant and CIS-resistant bladder cancer cells
The dose- and time-dependent viability inhibitions were observed in T24 parental cells (Figure 1D), and drug-resistant cell lines T24/DOX (Figure 1E), T24/CIS (Figure 1F). Moreover, for 48 h treatment of NTX, the IC50 values in T24/DOX (10.69 μM) and T24/CIS (11.20 μM) were slightly higher than that in T24 cells (7.85 μM), but IC50 values of NTX were not significantly different between T24 parental cells and drug-resistant cells (Table 1).
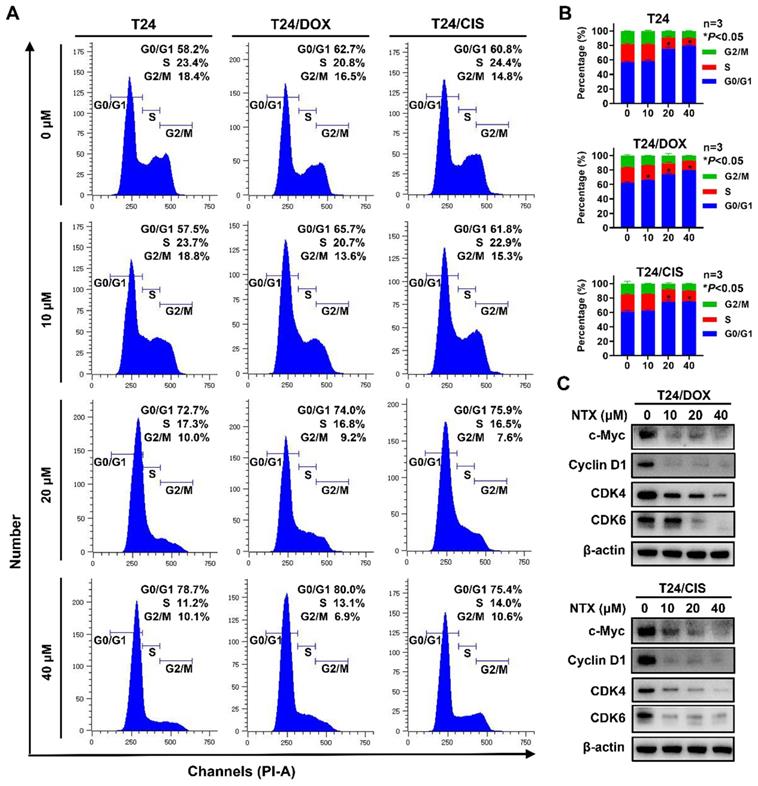
NTX triggers G0/G1 phase arrest in DOX-resistant and CIS-resistant bladder cancer cells
To examine whether NTX inhibited the cell cycle progression, T24, T24/DOX, and T24/CIS cells were treated with different concentrations of NTX for 24 h, followed by flow cytometric analysis (Figure 2A). As drug concentration increased, a higher percentage of cells stayed at G0/G1 phase, while a lower percentage of cells stayed at the phases of S and G2/M in T24, T24/DOX, and T24/CIS cells (Figure 2B). NTX negatively regulated the G0/G1-related protein expressions (c-Myc, Cyclin D1, CDK4, and CDK6) in T24/DOX and T24/CIS cells (Figure 2C).
NTX triggers G0/G1 phase cell cycle arrest in T24, T24/DOX, and T24/CIS cells. (A) T24, T24/DOX, and T24/CIS cells were treated with the indicated concentrations of NTX (0, 10, 20, and 40 μM) for 24 h. The harvested cells were incubated with PI/RNase, followed by flow cytometric analysis of cell cycle distribution. (B) The proportions of cells in each phase (G0/G1, S, and G2/M) are presented in the histograms. Data are expressed as the mean ± SD from three independent experiments. *P < 0.05 vs the control group. (C) T24/DOX and T24/CIS cells were treated with NTX (0, 10, 20, and 40 μM) for 48 h. Proteins were extracted and western blot was used to analyze the expressions of c-Myc, Cyclin D1, CDK4, and CDK6. β-actin was used as a loading control.
NTX promotes apoptosis of DOX-resistant and CIS-resistant bladder cancer cells
T24, T24/DOX, and T24/CIS cells were treated with NTX for 48 h, followed by flow cytometry (Figure 3A). The apoptotic rates of T24 cells in NTX groups were (14.65±1.47)%, (38.26±7.21)%, and (58.35±0.43)% of the total cells, compared with only (5.96±0.58)% in control group (Figure 3B). As NTX concentration increased, the apoptotic rates of T24/DOX cells were (4.42±0.92)%, (16.52±0.48)%, (30.84±0.17)%, and (53.37±2.67)%, while the rates of T24/CIS cells were (6.05±0.14)%, (14.49±1.89)%, (53.19±3.68)%, and (62.36±2.51)%, respectively (Figure 3B). Consistent with the apoptosis trend detected by flow cytometry, Hoechst staining results indicated a higher proportion of apoptotic T24, T24/DOX, and T24/CIS cells in NTX-treated groups, exhibiting such typical morphological changes as chromatin condensation and nuclear fragmentations (Figure 3C).
NTX induces apoptosis of T24, T24/DOX, and T24/CIS cells. (A) T24, T24/DOX, and T24/CIS cells were treated with the indicated concentrations of NTX (0, 10, 20, and 40 μM) for 48 h. The cells were stained with Annexin V-FITC/PI and flow cytometry was used to analyze the apoptotic rates. (B) The percentages of apoptotic cells are shown in the histograms. Data are expressed as the mean ± SD from three independent experiments. *P < 0.05, ***P < 0.001 vs the control group. (C) The indicated concentrations of NTX (0 and 40 μM) were used to treat T24, T24/DOX, and T24/CIS cells for 48 h. Hoechst 33342 was then used to stain the cell nuclei (blue), followed by fluorescence microscopy. Scale bar = 100 μm. (D) T24/DOX and T24/CIS cells were treated with NTX (0, 10, 20, and 40 μM) for 48 h, followed by western blot to assess the expressions of Bcl-xL, Mcl-1, and Survivin. β-actin was used as a loading control.
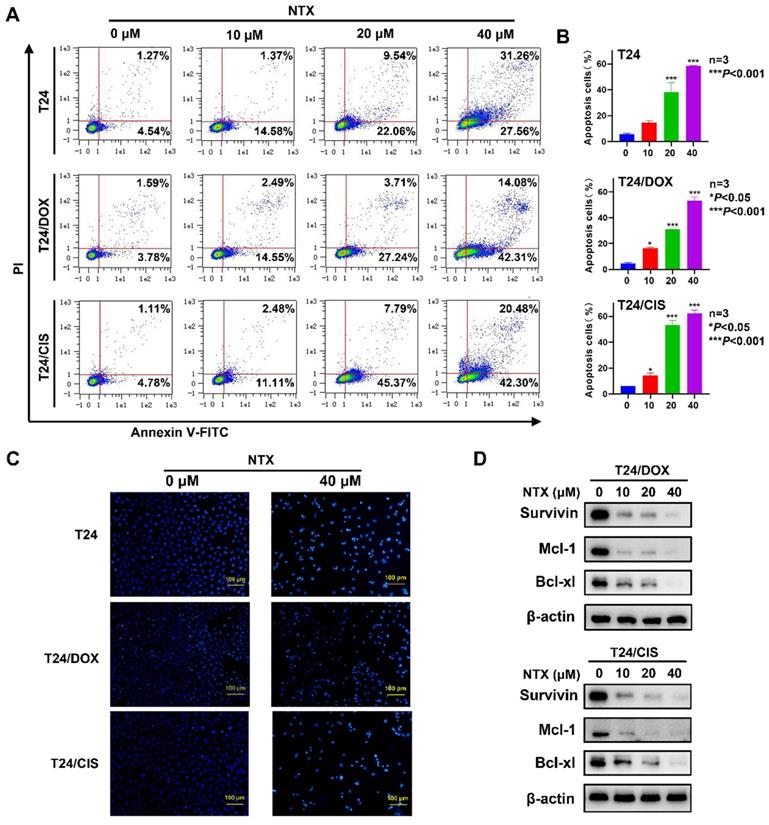
Western blot analysis revealed that the levels of Bcl-xL, Mcl-1, and Survivin decreased following NTX treatment (Figure 3D). Our results indicated that NTX promoted the apoptosis of both DOX-resistant and CIS-resistant T24 cells.
NTX suppresses the STAT3 signaling and P-gp in T24/DOX and T24/CIS cells
Western blot was applied to analyze whether NTX treatment regulated the expressions of proteins associated with STAT3 signaling and P-gp. Figure 4A illustrates that NTX significantly decreased the levels of STAT3, p-STAT3 (Y705), p-STAT3 (S727), and P-gp dose-dependently.
NTX suppresses the STAT3 signaling pathway in T24/DOX and T24/CIS cells. (A) Western blot was used to assess the expressions of STAT3, p-STAT3 (Y705), p-STAT3 (S727), and P-gp in T24, T24/DOX, and T24/CIS cells after they were treated with NTX (0, 10, 20, and 40 μM) for 48 h. (B) T24/DOX and T24/CIS cells were exposed to 10 μM NTX in the presence or absence of 4 μM Stattic for 24 h. Western blot was then performed to detect the expressions of STAT3, p-STAT3 (Y705), P-gp, Cyclin D1, and Mcl-1.
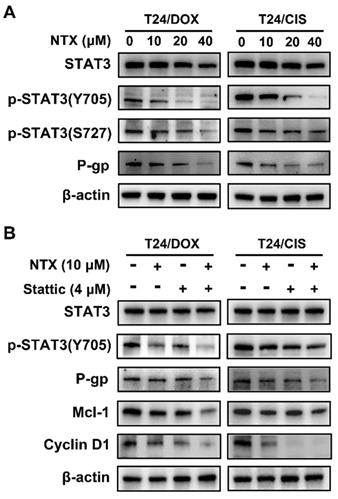
To further verify whether NTX induced apoptosis and G0/G1 arrest through the STAT3 signaling, T24/DOX and T24/CIS cells were incubated with NTX (10 μM) or Stattic (4 μM) for 24 h. Western blot analysis demonstrated that NTX (10 μM, 24h or 48 h) or Stattic (4 μM, 24h) alone did not significantly inhibit STAT3 expression, but higher concentrations of NTX (20 and 40 μM) decreased the levels of STAT3 (Figure 4A, B). The combination of NTX and Stattic synergistically downregulated the expressions of p-STAT3 (Y705), P-gp, Cyclin D1, and Mcl-1 (Figure 4B). These findings suggested that NTX could inhibit P-gp, promote apoptosis and G0/G1 arrest in DOX-resistant and CIS-resistant T24 cell lines via the STAT3 pathway.
NTX exhibits the anti-tumor effect in T24/DOX and T24/CIS tumor-bearing mice
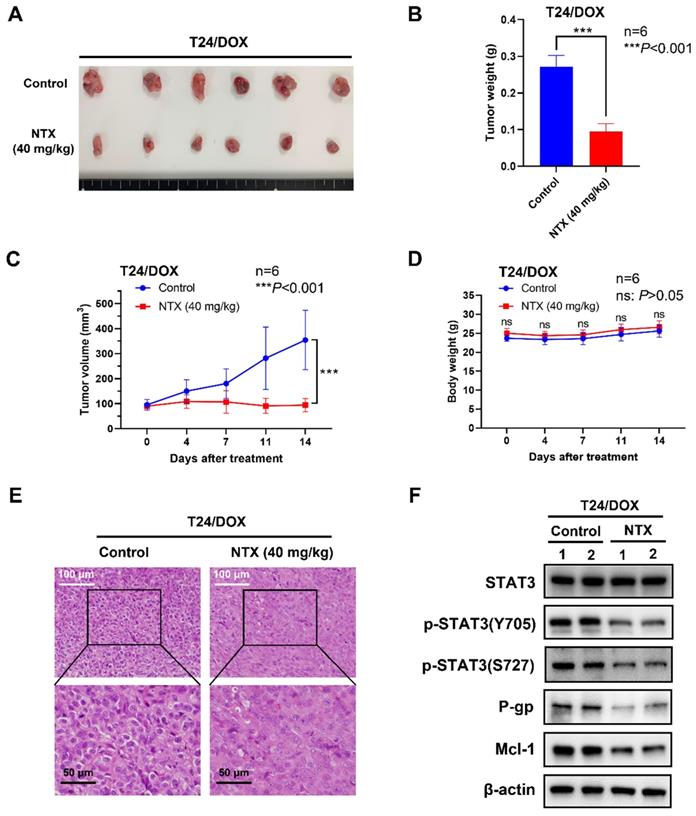
To determine NTX-induced anti-tumor effect in vivo, we established the T24/DOX and T24/CIS tumor xenograft models, followed by oral administration of NTX (40 mg/kg) or PBS (vehicle group). The representative changes of tumors were shown in Figure 5A, 6A, showing a significantly lower tumor size in NTX group than that in control group. The average tumor weights were (0.272±0.031) g for vehicle group and (0.095±0.022) g for NTX group in T24/DOX model (Figure 5B), while those were (0.420±0.121) g for vehicle group and (0.192±0.072) g for NTX group in T24/CIS model (Figure 6B). Similar for T24/DOX and T24/CIS models, the tumors grew slowly in NTX group when compared with the vehicle group (Figure 5C, 6C). No significant differences were found in the mean body weights of T24/DOX or T24/CIS tumor-bearing mice between NTX group and the corresponding control group (Figure 5D, 6D), indicating a low toxicity of NTX in vivo.
NTX exhibits the anti-tumor effect on T24/DOX in vivo. (A) T24/DOX tumors were excised from mice and photographed after two-week treatment. (B) The average tumor weight of NTX treatment group significantly decreased compared with that of vehicle group. (C) The growth curves of T24/DOX tumors at the indicated days following NTX treatment. (D) No significant difference was found in the body weight of T24/DOX tumor-bearing mice between treatment group and vehicle group. Data are expressed as the mean ± SD. ***P < 0.001, ns: P > 0.05, vs the vehicle group. (E) H&E staining was performed to observe the morphological alterations in T24/DOX tumors after NTX treatment. White scale bar = 100 μm; Black scale bar = 50 μm. (F) Proteins were extracted from T24/DOX tumors and western blot was used to detect the expressions of STAT3, p-STAT3 (Y705), p-STAT3 (S727), P-gp, and Mcl-1.
NTX exhibits the anti-tumor effect on T24/CIS in vivo. (A) T24/CIS tumors were excised from mice and photographed after two-week treatment. (B) The average tumor weight of NTX treatment group significantly decreased compared with that of vehicle group. (C) The growth curves of T24/CIS tumors at the indicated days following NTX treatment. (D) No significant difference was found in the body weight of T24/CIS tumor-bearing mice between treatment group and vehicle group. Data are expressed as the mean ± SD. **P < 0.01, ***P < 0.001, ns: P > 0.05, vs the vehicle group. (E) H&E staining was performed to observe the morphological alterations in T24/CIS tumors after NTX treatment. White scale bar = 100 μm; Black scale bar = 50 μm. (F) Proteins were extracted from T24/CIS tumors and western blot was used to detect the expressions of STAT3, p-STAT3 (Y705), p-STAT3 (S727), P-gp, and Mcl-1.
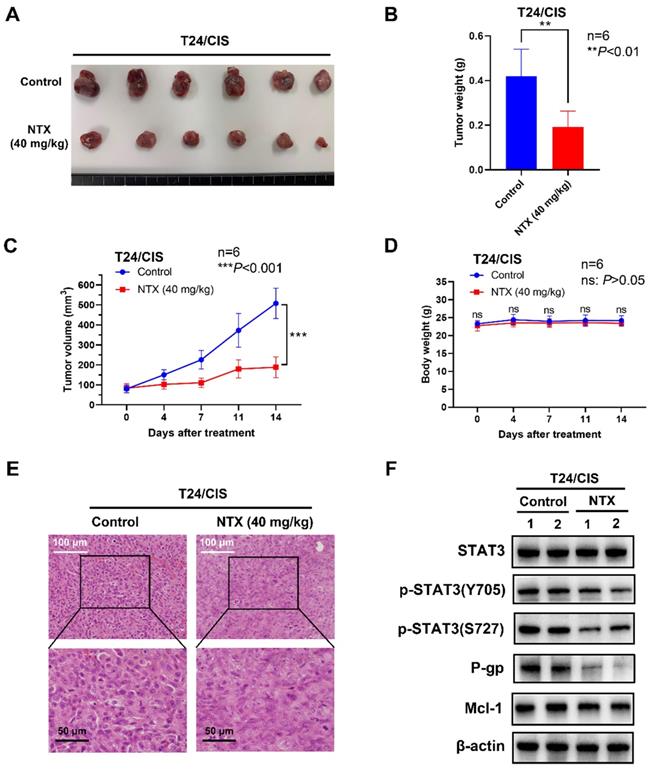
The morphological changes of H&E staining were shown in Figure 5E, 6E. The tumor cells in vehicle group were intact with deeply stained, large and abnormal nuclei, while those in NTX group presented with disappeared nuclei and tumor necrosis. Western blot analysis of T24/DOX and T24/CIS tumor tissues revealed that the expressions of p-STAT3 (Y705), p-STAT3 (S727), P-gp, and Mcl-1 were reduced by NTX treatment as compared to vehicle group, but there was no significant difference in STAT3 (Figure 5F, 6F).
Discussion
The high rates of recurrence and progression require repeated cycles of chemotherapy for a long-term duration, and subsequently trigger frequent MDR in UBC, leading to poor prognosis and heavy economic burden [3, 5]. In this study, NTX was firstly discovered as a STAT3 signaling inhibitor to trigger G0/G1 cell cycle arrest, apoptosis, and reverse P-gp overexpression in drug-resistant UBC. T24/DOX and T24/CIS cells have been well-established for decades to apply for basic research on UBC drug resistance to DOX or CIS [26-28]. Following the confirmation of drug-resistant characteristics, we identified the overexpressions of STAT3 and P-gp in T24/DOX and T24/CIS cells. Moreover, we demonstrated by XTT assay that T24/DOX and T24/CIS cells were resistant to DOX and CIS, respectively, while both of them were sensitive to NTX time- and dose-dependently.
Recent studies have reported NTX arrests glioma and myeloma cells at G0/G1 stage [23, 29]. Flow cytometric analysis also indicated NTX treatment led to the G0/G1 accumulation of DOX- and CIS-resistant T24 cells. Promoting cell cycle progression is a major carcinogenic mechanism of the proto-oncogene c-Myc, which potentially binds to Cyclin D1 promoter and upregulates its expression [30]. As an allosteric regulator, Cyclin D1 forms a complex with cyclin-dependent kinase 4/6 (CDK4/6), and active complex (Cyclin D1-CDK4/CDK6) drives cell cycle transition from G1 to S phase [31]. To explain the mechanism on NTX-induced G0/G1 arrest in T24/DOX and T24/CIS cells, we next demonstrated that NTX treatment downregulated c-Myc, Cyclin D1, CDK4, and CDK6.
Typical cellular changes, such as chromatin condensation, nuclear fragmentation, and even cell shrinkage, often accompany with the process of programmed cell death, apoptosis [32]. Consistently, we observed these typical morphological changes in T24, T24/DOX, and T24/CIS cells following NTX treatment, which were further confirmed by flow cytometric analysis. Mitochondria-mediated apoptosis is crucially regulated by the B-cell lymphoma-2 (Bcl-2) family members, among which the anti-apoptotic ones such as myeloid cell leukemia 1 (Mcl-1), B-cell lymphoma-extra large (Bcl-xL) are well-validated anticancer targets [33]. As a member of the inhibitor of apoptosis (IAP) family, Survivin is overexpressed in multiple cancers, and its upregulation in cancer is associated with chemoresistance and radioresistance [34]. The anti-apoptotic protein expressions of Bcl-xL, Mcl-1, and Survivin could be negatively regulated by NTX treatment in T24/DOX and T24/CIS cells.
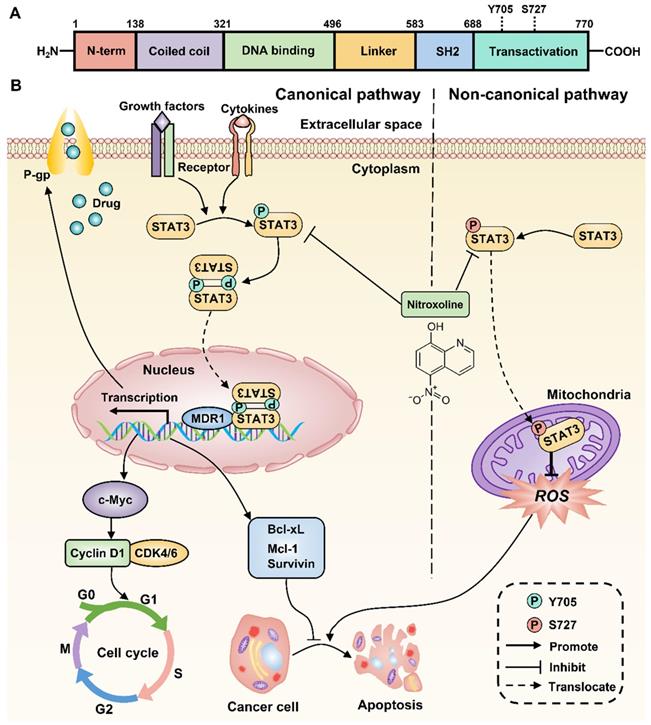
Increasing evidence suggests the critical role of STAT3 in chemoresistance, proliferation, and apoptosis of multiple tumor cells [11]. Previous studies have also reported that STAT3 overexpression occurs in drug-resistant UBC cell lines and associates with poor prognosis of UBC patients [12, 13]. Therefore, inhibition of STAT3 signaling is a promising therapeutic strategy to decrease chemoresistance and induce apoptosis of drug-resistant UBC. Consistently, the expressions of STAT3 and P-gp were upregulated in T24/DOX and T24/CIS cells compared with T24 parental cells. After membrane surface receptors are stimulated by cytokines or growth factors, STAT3 can be activated to signal through both canonical and non-canonical pathways [35]. For the canonical pathway, the subsequent phosphorylation of STAT3 at Y705 residue will homodimerize or heterodimerize through their SH2 domains (Figure 7A). Following nuclear translocation, STAT3 dimers can bind to promoter elements of target genes including the MDR1 gene, the cell cycle regulatory genes such as c-Myc, Cyclin D1, and the anti-apoptotic genes such as Survivin, Mcl-1, Bcl-xL, and further modulate their transcription [36]. For the non-canonical pathway, the phosphorylation of STAT3 at S727 residue is required for maximal STAT3 activation [14]. The subsequent translocation of p-STAT3 (S727) to the mitochondria promotes tumor cell proliferation by reducing the production of reactive oxygen species (ROS), a mediator of cell apoptosis [37]. In this study, NTX was identified as a potent STAT3 inhibitor to downregulate the expressions of STAT3, p-STAT3 (Y705), and p-STAT3 (S727) in T24/DOX and T24/CIS cells. The molecular-level explanation for NTX causing P-gp reversal, G0/G1 arrest, and apoptosis in T24/DOX and T24/CIS cells, is presented in Figure 7B. NTX treatment downregulated the expressions of STAT3 and p-STAT3 (Y705), which subsequently decreased the translocation of STAT3 dimers into the nucleus. Then, the downstream targets, including MDR1, c-Myc, Cyclin D1, Survivin, Mcl-1, and Bcl-xL, were downregulated, which could reverse MDR, trigger G0/G1 arrest, and induce apoptosis, respectively. Moreover, the downregulated p-STAT3 (S727) could promote ROS generation, which further mediated the apoptosis of T24/DOX and T24/CIS cells. According to the recent reports, there is still a huge controversy on whether p-STAT3 (S727) regulates p-STAT3 (Y705) negatively or positively [38-40], which needs further experimental confirmation. NTX and Stattic, a selective inhibitor of STAT3 activation, dimerization, and nuclear translocation [41], synergistically decreased p-STAT3 (Y705), P-gp, and the anti-apoptotic member Mcl-1, which further verified NTX-mediated STAT3 inhibition in drug-resistant UBC.
Proposed mechanism for NTX-induced P-gp reversal, G0/G1 arrest and apoptosis in drug-resistant T24 cells. The molecular structure of STAT3 is classic in STAT family, consisting of N-term Domain for cooperative DNA binding, Coiled-coil Domain for STAT3 recruitment to a receptor, DNA binding Domain, Linker Domain, SH2 Domain for STAT3 dimerization, and Transactivation Domain for transcription activation. (B) STAT3 can be activated to signal through both canonical and non-canonical pathways. For the canonical pathway, NTX inhibits STAT3 phosphorylation at Y705 residue and then decreases the translocation of STAT3 dimers to the nucleus in T24/DOX and T24/CIS cells. The subsequent downregulation of target genes including MDR1 gene, the cell cycle regulatory genes such as c-Myc, Cyclin D1, and the anti-apoptotic genes such as Survivin, Mcl-1, Bcl-xL, will reverse P-gp, trigger G0/G1 arrest, and induce apoptosis, respectively. For the non-canonical pathway, NTX-downregulated STAT3 phosphorylation at S727 residue reduces the translocation of p-STAT3 (S727) to the mitochondria and finally induces cell apoptosis by the increased generation of reactive oxygen species (ROS).
The two-week NTX treatment (40 mg/kg/day) also significantly in vivo inhibited T24/DOX and T24/CIS tumor growth. The dosage was selected based on the recommended dose for urinary tract infections [42], the guide for dose conversion between animals and human [43], and animal experiments in previous studies [20, 21]. The mean body weights of T24/DOX or T24/CIS tumor-bearing mice exhibited no between-group difference, which also indicated its favorable safety profile. Interestingly, by H&E staining, we also observed the disappeared nuclei and tumor necrosis in NTX group.
To our knowledge, this is the first report to discover NTX as a STAT3 inhibitor for drug-resistant UBC therapy. However, it should be noted that single agent therapy for UBC potentially leads to chemotherapy failure with limited therapeutic efficacy and drug resistance [44]. It remains to be investigated whether NTX or its analogues combined with DOX or CIS can enhance the anti-tumor effect, promote the extrusion of drugs into extracellular space by drug efflux pumps, and even delay the development of drug resistance in UBC without significantly increasing toxicity. We also noticed that previous clinical trials focused on the therapeutic effect of NTX (APL-1202) on NMIBC, while a multicenter study (NCT04813107, Phase I/II) on the combination of NTX with tislelizumab, a monoclonal antibody against programmed cell death protein 1 (PD-1) [45] will be started to assess their efficacy and safety in cisplatin-ineligible MIBC patients. Our previous work found that NTX decreases the levels of myeloid-derived suppressor cells (MDSCs) in UBC-bearing murine model [20]. Interestingly, Takeyama et al. recently reported the elevated MDSCs infiltration in CIS-resistant UBC microenvironment [46]. Further studies are expected to verify the combined efficacy of NTX and PD-1 antibody, and whether NTX could potently inhibit drug-resistant UBC through targeting MDSCs.
Conclusions
Here we reported that NTX as a STAT3 inhibitor exhibits the significant efficacy against drug-resistant UBC. Our observations demonstrated that NTX induces P-gp reversal, G0/G1 arrest, and apoptosis in drug-resistant UBC via suppression of the STAT3 signaling pathway. Our findings could provide a molecular-level basis in repurposing NTX with clinical implications against drug-resistant UBC by targeting STAT3 signaling.
Abbreviations
UBC: urothelial bladder cancer; NTX: nitroxoline; MIBC: muscle-invasive bladder cancer; DOX/ADM/ADR: doxorubicin; CIS/DDP/CDDP: cisplatin; P-gp: P-glycoprotein; MDR: multidrug resistance; ABCB1: ATP-binding cassette B1; MDR1 gene: multidrug resistance 1 gene; ChIP: chromatin immunoprecipitation; STAT3: Signal transducer and activators of transcription 3; MetAP2: methionine aminopeptidase-2; PD-1: programmed cell death protein 1; H&E staining: hematoxylin and eosin staining; IC50: half maximal inhibitory concentration; 95%CI: 95% confidence interval; CDK4/6: cyclin-dependent kinase 4/6; Mcl-1: myeloid cell leukemia 1; ROS: reactive oxygen species; MDSCs: myeloid-derived suppressor cells; ANOVA: one-way analysis of variance; Bcl-xL: B-cell lymphoma-extra large.
Acknowledgements
We extended our thanks to Shunai Li (Okayama University) and Hideo Ueki (Okayama University) for their help in experimental technique.
Funding
This research was funded by the Ministry of Education, Culture, Sports, Science and Technology of Japan (grant No. 17K11138, 21K09371). Dr. Wenfeng Lin was supported by the China Scholarship Council.
Author Contributions
All authors read and approved the final manuscript. PH and YN conceived and supervised the study. WL and JS contributed to the study design and conducted most of the experiments. TS, NX, and KW interpreted data and performed the statistical analysis. WL prepared the manuscript, while CL, MA, AX, and MW revised the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Oliveira MC, Caires HR, Oliveira MJ, Fraga A, Vasconcelos MH, Ribeiro R. Urinary Biomarkers in Bladder Cancer: Where Do We Stand and Potential Role of Extracellular Vesicles. Cancers (Basel). 2020 12
2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71:7-33
3. Patel VG, Oh WK, Galsky MD. Treatment of muscle-invasive and advanced bladder cancer in 2020. CA Cancer J Clin. 2020;70:404-23
4. Gomez De Liano A, Duran I. The continuing role of chemotherapy in the management of advanced urothelial cancer. Ther Adv Urol. 2018;10:455-80
5. Mollaei M, Hassan ZM, Khorshidi F, Langroudi L. Chemotherapeutic drugs: Cell death- and resistance-related signaling pathways. Are they really as smart as the tumor cells? Transl Oncol. 2021;14:101056
6. Wang F, Li D, Zheng Z, Kin Wah To K, Chen Z, Zhong M. et al. Reversal of ABCB1-related multidrug resistance by ERK5-IN-1. J Exp Clin Cancer Res. 2020;39:50
7. Wei X, Song M, Li W, Huang J, Yang G, Wang Y. Multifunctional nanoplatforms co-delivering combinatorial dual-drug for eliminating cancer multidrug resistance. Theranostics. 2021;11:6334-54
8. Choi YH, Yu AM. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr Pharm Des. 2014;20:793-807
9. Bukowski K, Kciuk M, Kontek R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int J Mol Sci. 2020 21
10. He C, Sun Z, Hoffman RM, Yang Z, Jiang Y, Wang L. et al. P-Glycoprotein Overexpression Is Associated With Cisplatin Resistance in Human Osteosarcoma. Anticancer Res. 2019;39:1711-8
11. Loh CY, Arya A, Naema AF, Wong WF, Sethi G, Looi CY. Signal Transducer and Activator of Transcription (STATs) Proteins in Cancer and Inflammation: Functions and Therapeutic Implication. Front Oncol. 2019;9:48
12. Chen Z, Chen X, Xie R, Huang M, Dong W, Han J. et al. DANCR Promotes Metastasis and Proliferation in Bladder Cancer Cells by Enhancing IL-11-STAT3 Signaling and CCND1 Expression. Mol Ther. 2019;27:326-41
13. Gatta LB, Melocchi L, Bugatti M, Missale F, Lonardi S, Zanetti B. et al. Hyper-Activation of STAT3 Sustains Progression of Non-Papillary Basal-Type Bladder Cancer via FOSL1 Regulome. Cancers (Basel). 2019 11
14. Yang J, Kunimoto H, Katayama B, Zhao H, Shiromizu T, Wang L. et al. Phospho-Ser727 triggers a multistep inactivation of STAT3 by rapid dissociation of pY705-SH2 through C-terminal tail modulation. Int Immunol. 2020;32:73-88
15. Ji L, Liu X, Zhang S, Tang S, Yang S, Li S. et al. The Novel Triazolonaphthalimide Derivative LSS-11 Synergizes the Anti-Proliferative Effect of Paclitaxel via STAT3-Dependent MDR1 and MRP1 Downregulation in Chemoresistant Lung Cancer Cells. Molecules. 2017 22
16. Zhang X, Xiao W, Wang L, Tian Z, Zhang J. Deactivation of signal transducer and activator of transcription 3 reverses chemotherapeutics resistance of leukemia cells via down-regulating P-gp. PLoS One. 2011;6:e20965
17. Thilakasiri PS, Dmello RS, Nero TL, Parker MW, Ernst M, Chand AL. Repurposing of drugs as STAT3 inhibitors for cancer therapy. Semin Cancer Biol. 2021;68:31-46
18. Talevi A, Bellera CL. Challenges and opportunities with drug repurposing: finding strategies to find alternative uses of therapeutics. Expert Opin Drug Discov. 2020;15:397-401
19. Shim JS, Matsui Y, Bhat S, Nacev BA, Xu J, Bhang HE. et al. Effect of nitroxoline on angiogenesis and growth of human bladder cancer. J Natl Cancer Inst. 2010;102:1855-73
20. Xu N, Lin W, Sun J, Sadahira T, Xu A, Watanabe M. et al. Nitroxoline inhibits bladder cancer progression by reversing EMT process and enhancing anti-tumor immunity. J Cancer. 2020;11:6633-41
21. Zhang QI, Wang S, Yang D, Pan K, Li L, Yuan S. Preclinical pharmacodynamic evaluation of antibiotic nitroxoline for anticancer drug repurposing. Oncol Lett. 2016;11:3265-72
22. Veschi S, De Lellis L, Florio R, Lanuti P, Massucci A, Tinari N. et al. Effects of repurposed drug candidates nitroxoline and nelfinavir as single agents or in combination with erlotinib in pancreatic cancer cells. J Exp Clin Cancer Res. 2018;37:236
23. Lazovic J, Guo L, Nakashima J, Mirsadraei L, Yong W, Kim HJ. et al. Nitroxoline induces apoptosis and slows glioma growth in vivo. Neuro Oncol. 2015;17:53-62
24. Mari A, D'Andrea D, Abufaraj M, Foerster B, Kimura S, Shariat SF. Genetic determinants for chemo- and radiotherapy resistance in bladder cancer. Transl Androl Urol. 2017;6:1081-9
25. Vallo S, Michaelis M, Rothweiler F, Bartsch G, Gust KM, Limbart DM. et al. Drug-Resistant Urothelial Cancer Cell Lines Display Diverse Sensitivity Profiles to Potential Second-Line Therapeutics. Transl Oncol. 2015;8:210-6
26. Byun SS, Kim SW, Choi H, Lee C, Lee E. Augmentation of cisplatin sensitivity in cisplatin-resistant human bladder cancer cells by modulating glutathione concentrations and glutathione-related enzyme activities. BJU Int. 2005;95:1086-90
27. Kim WT, Kim J, Yan C, Jeong P, Choi SY, Lee OJ. et al. S100A9 and EGFR gene signatures predict disease progression in muscle invasive bladder cancer patients after chemotherapy. Ann Oncol. 2014;25:974-9
28. Tanaka M, Koul D, Davies MA, Liebert M, Steck PA, Grossman HB. MMAC1/PTEN inhibits cell growth and induces chemosensitivity to doxorubicin in human bladder cancer cells. Oncogene. 2000;19:5406-12
29. Li G, Zheng YH, Xu L, Feng J, Tang HL, Luo C. et al. BRD4 inhibitor nitroxoline enhances the sensitivity of multiple myeloma cells to bortezomib in vitro and in vivo by promoting mitochondrial pathway-mediated cell apoptosis. Ther Adv Hematol. 2020;11:2040620720932686
30. Garcia-Gutierrez L, Delgado MD, Leon J. MYC Oncogene Contributions to Release of Cell Cycle Brakes. Genes (Basel). 2019 10
31. Reinius MAV, Smyth E. Anti-cancer therapy with cyclin-dependent kinase inhibitors: impact and challenges. Expert Rev Mol Med. 2021;23:e6
32. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29:347-64
33. Hata AN, Engelman JA, Faber AC. The BCL2 Family: Key Mediators of the Apoptotic Response to Targeted Anticancer Therapeutics. Cancer Discov. 2015;5:475-87
34. Rathore R, McCallum JE, Varghese E, Florea AM, Busselberg D. Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis. 2017;22:898-919
35. Zhang ZH, Li MY, Wang Z, Zuo HX, Wang JY, Xing Y. et al. Convallatoxin promotes apoptosis and inhibits proliferation and angiogenesis through crosstalk between JAK2/STAT3 (T705) and mTOR/STAT3 (S727) signaling pathways in colorectal cancer. Phytomedicine. 2020;68:153172
36. Laudisi F, Cherubini F, Monteleone G, Stolfi C. STAT3 Interactors as Potential Therapeutic Targets for Cancer Treatment. Int J Mol Sci. 2018 19
37. Meier JA, Hyun M, Cantwell M, Raza A, Mertens C, Raje V. et al. Stress-induced dynamic regulation of mitochondrial STAT3 and its association with cyclophilin D reduce mitochondrial ROS production. Sci Signal. 2017 10
38. Huang G, Yan H, Ye S, Tong C, Ying QL. STAT3 phosphorylation at tyrosine 705 and serine 727 differentially regulates mouse ESC fates. Stem Cells. 2014;32:1149-60
39. Mandal T, Bhowmik A, Chatterjee A, Chatterjee U, Chatterjee S, Ghosh MK. Reduced phosphorylation of Stat3 at Ser-727 mediated by casein kinase 2 - protein phosphatase 2A enhances Stat3 Tyr-705 induced tumorigenic potential of glioma cells. Cell Signal. 2014;26:1725-34
40. Wakahara R, Kunimoto H, Tanino K, Kojima H, Inoue A, Shintaku H. et al. Phospho-Ser727 of STAT3 regulates STAT3 activity by enhancing dephosphorylation of phospho-Tyr705 largely through TC45. Genes Cells. 2012;17:132-45
41. Jin J, Li Y, Zhao Q, Chen Y, Fu S, Wu J. Coordinated regulation of immune contexture: crosstalk between STAT3 and immune cells during breast cancer progression. Cell Commun Signal. 2021;19:50
42. Wagenlehner FM, Munch F, Pilatz A, Barmann B, Weidner W, Wagenlehner CM. et al. Urinary concentrations and antibacterial activities of nitroxoline at 250 milligrams versus trimethoprim at 200 milligrams against uropathogens in healthy volunteers. Antimicrob Agents Chemother. 2014;58:713-21
43. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7:27-31
44. Alfarouk KO, Stock CM, Taylor S, Walsh M, Muddathir AK, Verduzco D. et al. Resistance to cancer chemotherapy: failure in drug response from ADME to P-gp. Cancer Cell Int. 2015;15:71
45. Ye D, Liu J, Zhou A, Zou Q, Li H, Fu C. et al. Tislelizumab in Asian patients with previously treated locally advanced or metastatic urothelial carcinoma. Cancer Sci. 2021;112:305-13
46. Takeyama Y, Kato M, Tamada S, Azuma Y, Shimizu Y, Iguchi T. et al. Myeloid-derived suppressor cells are essential partners for immune checkpoint inhibitors in the treatment of cisplatin-resistant bladder cancer. Cancer Lett. 2020;479:89-99
Author contact
![]() Corresponding author: Peng Huang, M.D., Ph.D., Department of Urology, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama 700-8558, Japan. Tel: + 81-86-235-7997; Fax: +81-86-235-7884. Email: huangpeng509com
Corresponding author: Peng Huang, M.D., Ph.D., Department of Urology, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama 700-8558, Japan. Tel: + 81-86-235-7997; Fax: +81-86-235-7884. Email: huangpeng509com