Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(14):3898-3910. doi:10.7150/ijbs.65488 This issue Cite
Research Paper
ADAM9 functions as a transcriptional regulator to drive angiogenesis in esophageal squamous cell carcinoma
Yu-Sen Lin1,2, Ting-Ting Kuo3, Chia-Chien Lo3, Wei-Chung Cheng4, Wei-Chao Chang3, Guan-Chin Tseng5, Shih-Ting Bai3, Yu-Kai Huang4, Chih-Ying Hsieh3, Han-Shui Hsu6,7, Yi-Fan Jiang8, Chen-Yuan Lin9,10, Liang-Chuan Lai11, Xing-Guo Li4, Yuh-Pyng Sher1,3,4,12 ![]()
1. Graduate Institute of Clinical Medical Science, China Medical University, Taichung 404, Taiwan.
2. Division of Thoracic Surgery, China Medical University Hospital, Taichung 404, Taiwan.
3. Center for Molecular Medicine, China Medical University Hospital, Taichung 404, Taiwan.
4. Graduate Institute of Biomedical Sciences, China Medical University, Taichung 404, Taiwan.
5. Department of Anatomic Pathology, Nantou Hospital of the Ministry of Health and Welfare, Nantou 540, Taiwan.
6. Division of Thoracic Surgery, Department of Surgery, Taipei Veterans General Hospital, Taipei 112, Taiwan.
7. Institute of Emergency and Care Medicine, School of Medicine, National Yang-Ming University, Taipei 112, Taiwan.
8. Graduate Institute of Molecular and Comparative Pathobiology, School of Veterinary Medicine, National Taiwan University, Taipei 106, Taiwan.
9. School of Pharmacy, China Medical University, Taichung 404, Taiwan.
10. Division of Hematology and Oncology, China Medical University Hospital, Taichung 404, Taiwan.
11. Graduate Institute of Physiology, College of Medicine, National Taiwan University, Taipei 100, Taiwan.
12. Chinese Medicine Research Center, China Medical University, Taichung 404, Taiwan.
Received 2021-7-29; Accepted 2021-8-29; Published 2021-9-7
Abstract

Hypoxia and angiogenesis play key roles in the pathogenesis of esophageal squamous cell carcinoma (ESCC), but regulators linking these two pathways to drive tumor progression remain elusive. Here we provide evidence of ADAM9's novel function in ESCC progression. Increasing expression of ADAM9 was correlated with poor clinical outcomes in ESCC patients. Suppression of ADAM9 function diminished ESCC cell migration and in vivo metastasis in ESCC xenograft mouse models. Using cellular fractionation and imaging, we found a fraction of ADAM9 was present in the nucleus and was uniquely associated with gene loci known to be linked to the angiogenesis pathway demonstrated by genome-wide ChIP-seq. Mechanistically, nuclear ADAM9, triggered by hypoxia-induced translocation, functions as a transcriptional repressor by binding to promoters of genes involved in the negative regulation of angiogenesis, and thereby promotes tumor angiogenesis in plasminogen/plasmin pathway. Moreover, ADAM9 suppresses plasminogen activator inhibitor-1 gene transcription by interacting with its transcription factors at the promoter. Our findings uncover a novel regulatory mechanism of ADAM9 as a transcriptional regulator in angiogenesis and highlight ADAM9 as a promising therapeutic target for ESCC treatment.
Keywords: ESCC, ADAM9, PAI-1, angiogenesis, transcriptional regulator
Introduction
Esophageal cancer is one of the most fatal types of malignancies, with rapid tumor mass growth and frequently a poor prognosis [1]. The histological type of this disease is disproportionate between Asian and Caucasian patients [2]. In contrast to Western countries where esophageal adenocarcinoma predominates, esophageal squamous cell carcinoma (ESCC) occurs more commonly in Asian populations, with, for example, a ratio of 10-15:1 squamous cell carcinoma to adenocarcinoma in Taiwan [3]. The 5-year survival of ESCC is rarely more than 25% even after curative-intent resection [4]. More than 50% of tumors recur within one year after surgery due to locoregional recurrence and distal metastases [5]. The molecular mechanism associated with ESCC progression is unclear and limits improvements in the clinical therapy of ESCC patients.
Hypoxia-associated biomarkers are associated with poor outcomes in esophageal cancer [6]. Under hypoxic conditions, angiogenic factors are induced that promote tumor angiogenesis, which is a critical factor for the progression of solid tumors [7]. The plasminogen activating system can increase local proteolysis for a proangiogenic effect, thereby promoting tumorigenesis. For example, tissue plasminogen activator (tPA) is required for the growth, invasion, and angiogenesis of pancreatic cancer, and urokinase plasminogen activator (uPA) is a strong prognostic factor in adenocarcinoma of the esophagus [8, 9]. Plasminogen activator inhibitor type 1 (PAI-1) can bind to plasminogen activators (tPA and uPA) to influence the plasminogen activator pathway and prevent the generation of plasmin [10]. Based on this, PAI-1 was considered to have antitumorigenic effects. However, a paradoxical association between PAI-1 and an unfavorable clinical outcome was reported, where PAI-1-mediated tumor angiogenesis is considered regardless of angiogenic stimuli in malignant pleural mesothelioma [11]. Although PAI-1 has been reported as a potential marker for the malignancy of ESCC [12], the role of PAI-1 in angiogenesis relating to plasminogen activator-based pathways in ESCC remains unclear.
Tumor metastasis involves degradation of the extracellular matrix and requires several proteolytic enzymes, such as matrix metalloproteinases, and a disintegrin and metalloproteases (ADAMs) either from tumor cells or surrounding stromal cells in ESCC [13, 14]. ADAM9, a type I transmembrane protein of the ADAM family, is involved in cell adhesion and migration via its disintegrin domain for adhesion with integrin and its metalloproteinase domain for ectodomain shedding to release a number of molecules with important roles in tumorigenesis and angiogenesis [15]. ADAM9 is linked with tumor progression in different types of cancer. For example, ADAM9 promotes lung cancer metastasis to brain by a plasminogen activator-based pathway and increases angiogenesis [16, 17]. In ESCC, up-regulated DNA methyltransferase 1 contributes to tumor growth through ADAM9-mediated epidermal growth factor receptor (EGFR)-AKT signaling [18]. However, the role of ADAM9 in ESCC tumorigenesis has not been fully elucidated. In this study, we showed that ADAM9 enhances the plasminogen activator-based pathway for ESCC progression. Notably, ADAM9 can translocate to the nucleus and occupy chromatin for the inhibition of transcription of genes in negative regulation of angiogenesis such as PAI-1 and PGK1, thereby promoting angiogenesis in ESCC.
Materials and Methods
Patient information
Formalin-fixed paraffin-embedded ESCC samples were obtained from 213 patients who received curative esophagectomy between August 2000 and September 2009 at China Medical University Hospital in compliance with protocols approved by the hospital's institutional review board (DMR98-IRB-059). The informed consent was obtained for experimentation with human subjects. Clinical staging and clinicopathological TNM classification were determined according to criteria proposed by the American Joint Committee on Cancer, 6th edition.
ESCC tumor animal models
SCID mice (8 weeks of age) were used as models of ESCC distal metastasis by intracardial injection with control and ADAM9 knockdown CE146T or KYSE170 cells (5×104). All experimental procedures were approved by the Institutional Animal Care and Use Committee of China Medical University and Hospital. After two months, the mice were sacrificed and tumors were excised and weighed.
Transmission electron microscopy
For immunogold EM analysis of ADAM9, samples were performed as previously described [19]. Briefly, the ESCC cells were fixed by 4% paraformaldehyde and 0.25% glutaraldehyde in 0.1 M phosphate buffer (pH 7.3). After rinsing with 0.1 M phosphate buffer (pH 7.3), the cell pellets were post-fixed in 4% low melting agar. The agar samples were dehydrated with ascending concentrations of ethanol, then infiltrated and embedded with Spurr's resin at 65 degrees for 16 hours. 100-nm-thick sections were stained with anti-ADAM9 antibody (AF939, R&D, 1:500) and 12-nm-gold anti-goat antibody (1:20). The stained specimens were imaged with the FEI Tecnai G2 TF20 Super TWIN microscope operating at 120 kV.
Immunofluorescence and Proximity Ligation Assay
The experiments were conducted as previously described [20]. Briefly, for immunofluorescence, cells were cultured for 24 h on coverslips, fixed with 4% paraformaldehyde pH 7.4 in PBS for 15 min at room temperature, and blocked with 0.1% tween-PBS and 5% BSA for 1 h at 37 °C. Incubations with primary antibodies were performed overnight at 4 °C. After incubation with secondary antibodies for 1 h at 37 °C, cells were stained with DAPI (4',6'-diamidino-2-phénylindole) and mounted using Vectashield Hardset mounting medium (Vector Laboratories, H-1400). PLA assays were conducted using Duolink™ In situ Red Starter Kit (#DUO92101, Sigma-Aldrich) according to the recommendations. The primary antibodies against ADAM9 (AF939; R&D), TP53 (#2524; CST), HIF1α (#610959; BD), and USF1 (sc-390027; santa cruz) were used.
Statistical analysis
Comparisons between the analyzed parameters were performed using Student's t-test or two-way ANOVA for continuous variables. All statistical tests were two-sided. Survival curves were obtained by the Kaplan-Meier method. Statistical significance was set for all tests at P < 0.05.
Results
ADAM9 overexpression is associated with poor outcomes of ESCC patients
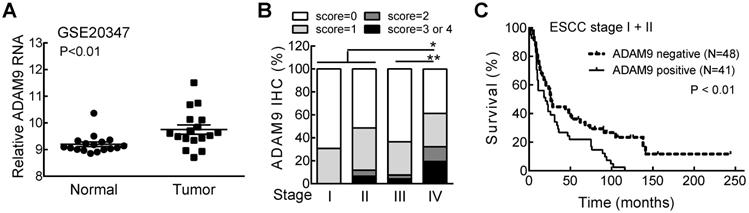
We first investigated the relationship of ADAM9 with the clinicopathological parameters of ESCC patients, and found significantly higher levels of ADAM9 mRNA in ESCC tumors than in normal esophageal tissues from the Gene Expression Omnibus (GEO) databases (GSE20347) (Figure 1A). Next, we examined the ADAM9 proteins in paraffin-embedded ESCC specimens by immunohistochemical (IHC) staining with anti-ADAM9 antibody [16]. Different staining levels of ADAM9 in ESCC tissues were scored from 0 to 4 by a pathologist, and 44% of 213 specimens were scored greater than 0 as positive ADAM9 staining (Table S1). In stage IV patients, the percentage of positive ADAM9 staining (61%) and the staining intensity represented by the IHC score were significantly higher than in all other stages (Figure 1B). Among early stage ESCC patients (stage I and II), those with positive ADAM9 staining had a shorter survival time compared with the ADAM9-negative group (Figure 1C), but that trend was not observed in late stages (stage III and IV, Figure S1), probably due to the very short survival time of late stage-patients. These data reveal that ADAM9 is correlated with tumor progression in ESCC.
ADAM9 is overexpressed in patients with advanced ESCC and correlated with poor outcome. (A) The RNA levels of ADAM9 in the specimens from ESCC patients in the GSE20347 dataset. N = 17, each group. (B) The distribution of ADAM9 IHC scores in tumor specimens from 4 stages of ESCC. Wilcoxon rank sum test. *, P < 0.05; **, P < 0.01. (C) Kaplan-Meier survival curve of ESCC patients with stage I and II grouped by ADAM9 IHC staining status (positive or negative).
ADAM9 suppression reduces the migration of ESCC cells and metastasis of ESCC tumor xenografts
To investigate the role of ADAM9 in ESCC progression, ADAM9 knockdown cells were generated using lentiviral shRNAs targeting different regions of ADAM9. No obvious change in cell proliferation rate was detected in control versus ADAM9 knockdown cells by MTT assays over a period of 3 days (Figure S2A-B). However, we found that control cells migrated longer distances than ADAM9 knockdown cells when tracking each cell's movement with time-lapse detection (Figure 2A and Figure S2C). Moreover, compared to control cells, ADAM9 knockdown ESCC cells significantly decreased the invasion ability in a transwell assay (Figure 2B and Figure S2D). Thus, ADAM9 promotes the invasion and migration ability of ESCC cells.
ADAM9 knockdown reduces ESCC cell growth and metastasis in animal models. (A) Videomicroscopy of individual cells is quantitated to determine the distance and direction of cellular migration of CE146T and KYSE170 cells. Black indicates migration to the left and red indicates migration to the right. Bar chart data are mean ± SD. (B) Crystal violet staining of control and ADAM9 knockdown cells in the transwell assays that migrated with Matrigel after 2 days. One of three independent assays performed in triplicate is shown. (C) Control or ADAM9 knockdown CE146T cells were inoculated into the left cardiac ventricle of 6~8-week-old SCID mice (N=5 in each group). The mice were sacrificed on day 64. The metastatic nodules were collected from the whole body of each mouse and weighed for analysis. (D) Intracardiac injection of control or ADAM9 knockdown KYSE170 cells into 10-week-old SCID mice (N=8 in each group). The mice were sacrificed on day 48. Metastatic tumors were weighed for analysis. Student's t test. *, P < 0.05. **, P < 0.01.
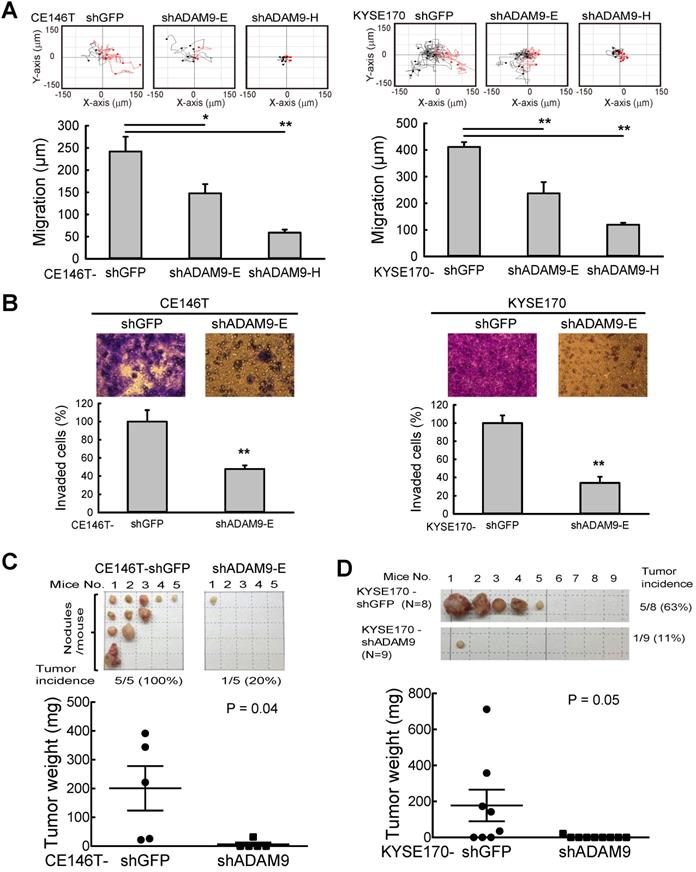
To further examine the metastatic effect of ADAM9, control and ADAM9 knockdown CE146T cells were intracardially injected into SCID mice for evaluating distal metastasis. In the control group, in which mice were injected with the control CE146T cells, all mice (100%) developed several distal metastatic tumors within 64 days after cancer cell injection, whereas only one mouse (20%) had one small distal metastatic tumor in the ADAM9 knockdown group (Figure 2C). The metastatic nodules in the control group grew in the neck, ribs, kidney, and subcutaneous regions. Similar results were observed in mice injected with control and ADAM9 knockdown KYSE170 cells (Figure 2D). Although only one distant metastatic tumor was found in each mouse, the tumor incidence was 63% (5 of 8) in the control group, markedly higher than 11% (1 of 9) in the ADAM9 knockdown group. Notably, the tumor size was significantly bigger in the control group than in the ADAM9 knockdown group. These data indicate that ADAM9 promotes tumor metastases in ESCC.
ADAM9 regulates plasminogen activator-based pathway in ESCCs
To further investigate how ADAM9 can promote the pathogenesis of ESCC, we generated stable cell clones using a CRISPR/Cas9 system to knock out (KO) ADAM9 and demonstrated that cell proliferation was reduced in ADAM9 KO KYSE170 cells (Figure S3A). The reduced cell proliferation in ADAM9 KO cells but not in ADAM9 KD cells is likely due to the complete reduction of ADAM9 expression in KO cells. Next, we explored ADAM9-regulated genes by comparing transcriptomes between control and ADAM9 KO cells. Notably, we found genes in the plasminogen activator system (PLAT and SERPINE1) in the top three functional enrichment analysis (Figure 3A). Given that the plasminogen activation system plays a central role in the regulation of a variety of pathological processes including malignancy, chemoresistance, coagulation, and angiogenesis [21, 22], we further examined whether ADAM9 regulated the plasminogen activation system for the pathogenesis of ESCC cells.
ADAM9 promotes the plasminogen activator-based pathway in ESCC. (A) Gene ontology-based functional analysis of ADAM9-regulated genes using differentially expressed genes in the control and ADAM9 KO KYSE170 cells. (B) tPA activity measured in the two-day culture conditioned media. Two individual clones of ADAM9 KO (#17 and #29) KYSE170 cells were analyzed. (C) Western blot analysis of indicated proteins in ADAM9 KO KYSE170 cells transiently transfected with plasmids of vector (V), ADAM9 wild type (WT), and E348A mutant. (D - F) The measured tPA activity (D), cell morphology; scale bar, 200 μm (E), and migration distance (F) of ADAM9 KO cells transiently transfected with plasmids of ADAM9 WT and E348A mutant. Bar chart data are mean ± SD. Student's t test. *, P < 0.05. **, P < 0.01. (G) The correlation of ADAM9 and PLAT or SERPINE1 RNA expression in ESCC patients from TCGA dataset.
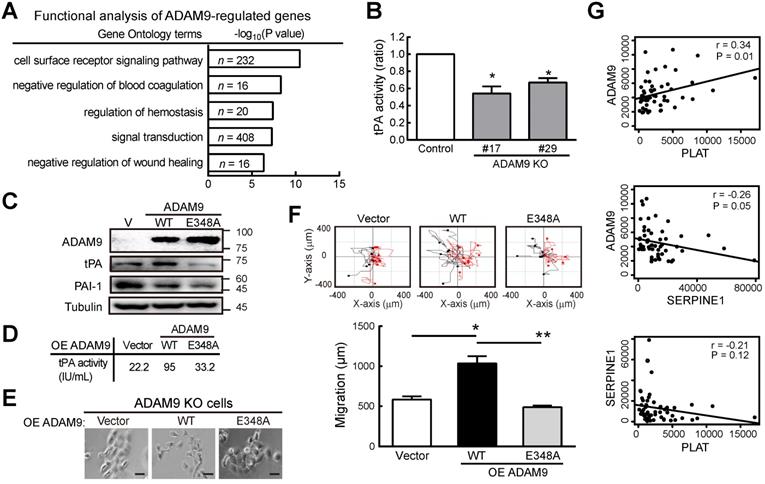
First, we demonstrated a decreased tPA activity in ADAM9 KO KYSE170 cells compared to control cells (Figure 3B). Accordingly, we found decreased RNA levels of tPA (encoded by PLAT) but increased PAI-1 (encoded by SERPINE1) in ADAM9 knockdown cells compared with control cells (Figure S3B). Moreover, the secretion of tPA proteins into the culture media was also significantly decreased in ADAM9 knockdown TE and KYSE170 cells (Figure S3C). To further investigate whether the plasminogen activator-based pathway was regulated by the protease activity of ADAM9 in ESCC, we introduced ectopic ADAM9 wild-type (WT) or catalytic mutant E348A [23], which is defective in the protease activity, into ADAM9 KO cells. Compared to WT, the ADAM9 protease mutant E348A decreased tPA expression (Figure 3C) and reduced the tPA activity by nearly 3-fold (Figure 3D). In terms of cell morphology and motility, we found that overexpression of ADAM9 WT showed more cell scattering, a dispersion of compact cells (Figure 3E), and increased cell migration ability (Figure 3F), whereas expression of the E348A mutant in ADAM9 KO cells retained the cell contact and migration characteristics of vector control cells.
To investigate whether the role of ADAM9 for promoting the plasminogen activator-based pathway is clinically relevant, we measured the correlation of ADAM9 and PLAT or SERPINE1 expression in ESCC specimens from The Cancer Genome Atlas (TCGA) dataset. In patients with high ADAM9 expression (above the mean), a positive correlation of ADAM9 and PLAT RNA levels was detected (r = 0.34), whereas a negative correlation of ADAM9 and SERPINE1 levels was detected (r = - 0.26) (Figure 3G). Notably, we found a poor correlation of PLAT and SERPINE1 RNA (r = -0.21; P = 0.12), suggesting that the correlation of ADAM9 and PLAT or SERPINE1 was not caused by a negative correlation between PLAT and SERPINE1 (Figure 3G). Taken together, these results show that ADAM9 can promote the pathogenesis of ESCC by activating the plasminogen activator-based pathway.
Nuclear ADAM9 is increased in ESCC under hypoxia
IHC staining of ESCC specimens demonstrated that ADAM9 was more highly expressed in esophageal tumor cells, as compared to adjacent normal esophageal tissue (Figure 4A). Notably, ADAM9-positive staining was clearly detected in the nuclei of ESCC specimens despite that ADAM9 is a membrane protein. To validate this observation, we performed cellular fractionation and showed that ADAM9 proteins were indeed detected in the nuclear fraction of ESCC cells, using two different antibodies against the N-terminal and C-terminal domains of ADAM9, the signals of which were markedly suppressed in ADAM9 knockdown cells (Figure S4A). Similar results were also observed in control and ADAM9 KO KYSE170 cells (Figure 4B). Furthermore, we performed transmission electron microscopy (TEM) with immunogold labeling and validated the localization of ADAM9 in the membrane and nuclei of KYSE170 cells (Figure 4C) whereas no signals were detected in ADAM9 KO cells (Figure S4B). Notably, ADAM9 E348A protease mutant showed much lower nuclear ADAM9 levels compared to WT by Western blot (Figure 4D) and confocal microscopy (Figure S4C).
ADAM9 protein is detected in the nuclei of ESCC cells. (A) Representative image of ADAM9 IHC staining in tumor and adjacent normal tissue in ESCC specimens. Scale bar, 25 μm. (B) Nuclear localization of ADAM9 in control and ADAM9 KO KYSE170 cells. C, control; KO, ADAM9 KO. Short or long, short or long exposure. (C) Presence of ADAM9 in cell membrane and nucleus of KYSE170 cells using transmission electron microscopy. ADAM9 is detected using 15 nm gold particles. Arrows indicate the positive signals for ADAM9 detection (top). The highlighted square areas are shown (right). (D) Cellular fractionation followed by western blot analysis in ADAM9 KO KYSE170 cells transiently transfected with plasmids of ADAM9. W, wild type and E, E348A mutant. Representative images of total proteins and nuclear proteins using the same antibody staining from the same blots. Nuclear ADAM9 protein levels were calculated from three independent assays. (E) Western blot analysis in ADAM9 KO KYSE170 cells transiently transfected with plasmids of ADAM9 WT and 4A mutant. One and two-fold of plasmid amount of ADAM9 4A mutant were used. Nuclear ADAM9 protein levels were calculated from three independent assays. (F) Migration ability of ADAM9 KO KYSE170 cells transiently transfected with plasmids of ADAM9 WT and 4A mutant. (G) Western blot analysis of KYSE170 cells treated with normoxic or hypoxic conditions. The amount of ADAM9 protein relative to the normoxia group was shown. (H) Fluorescence immunohistochemistry of ADAM9 and HIF1α in KYSE170 cells with confocal microscopy. Hypoxia, 1% O2. Scale bars, 20 µm.
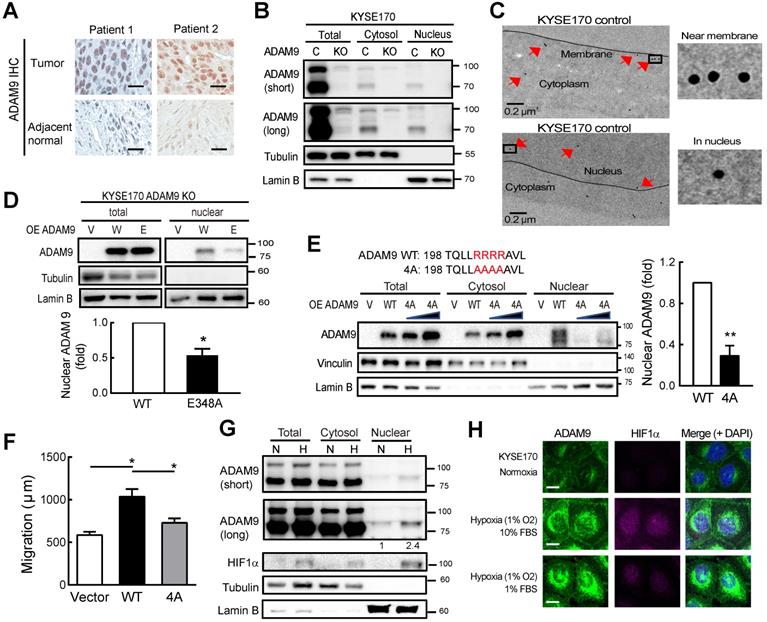
NLS (nuclear localization signal) is known to contain three to five basic residues that are required for nuclear translocation and interaction with importin β1. Using two independent prediction programs, cNLS Mapper program [24] and NucPred program [25], we found one putative NLS containing 4 arginine residues R202 to R205 adjacent to metalloprotease domain in the N-terminal of ADAM9. By replacing 4 arginine sites to alanine in ADAM9 NLS (4A), the nuclear translocation of ADAM9 was decreased compared with ADAM9 WT (Figure 4E). Moreover, the migration ability was also disrupted in cells expressing with ADAM9 4A compared with ADAM9 WT (Figure 4F).
A high level of hypoxia inducible factor 1 subunit alpha (HIF1α was detected in the esophageal tissue of ESCC patients and correlated with clinical TNM stage and poor outcomes [26]. We then investigated whether hypoxia could induce nuclear translocation of ADAM9 in ESCC. Cellular fractionation revealed that nuclear ADAM9 was increased under hypoxic conditions (Figure 4G) or induced by CoCl2 treatment in a time-dependent manner (Figure S4D). Moreover, as shown by confocal fluorescence, co-localization of ADAM9 with DAPI (4′,6-diamidino-2-phenylindole) in the nucleus was increased by hypoxia (achieved by culturing in 1% O2), as well as under stressed conditions of low serum with hypoxia (Figure 4H). Taken together, ADAM9 proteins can translocate to the nucleus of ESCC cells, especially under hypoxia.
Nuclear ADAM9 regulates genes involved in angiogenesis in ESCC
Next, to investigate whether nuclear ADAM9 can bind to DNA for genetic regulation, we performed chromatin immunoprecipitation (ChIP) using an antibody that can pull down ADAM9 in ESCC cells (Figure S5A). A genome-wide survey by ChIP-seq was carried out to determine the binding profile of nuclear ADAM9 by comparing control and ADAM9 KO KYSE170 cells. The top 10 specific peaks of ChIP-seq were identified (Table S2). Gene ontology-based functional analysis reveal that these top hits are involved in functions, such as regulation of angiogenesis and plasminogen activation (Figure 5A). Notably, SERPINE1 gene (encoding PAI-1) is listed in all these functions (Figure 5A). By deep analysis of the binding regions from the ChIP-seq profile, ADAM9 occupied the SERPINE1 promoter from position -736 to 293, with a width of 1030 bp in control cells but no signals in ADAM9 KO KYSE170 cells (Figure 5B). ADAM9's occupation of the promoter of SERPINE1 was confirmed using ChIP-quantitative PCR (ChIP-qPCR), which showed enrichment over an extended region (from site 2 to 6) in control compared to ADAM9 KO KYSE170 cells (Figure 5C). Similar results were observed in control (shGFP) TE cells compared with ADAM9 knockdown TE cells (Figure S5B).
Nuclear ADAM9 protein occupies DNA and regulates gene expression to promote angiogenesis in ESCC cells. (A) Gene ontology-based functional analysis of target genes occupied by ADAM9. (B) Peaks of ADAM9 occupancy were identified in the SERPINE1 promoter by ADAM9 ChIP-sequencing in KYSE170 control and ADAM9 KO cells. The SERPINE1 gene location is shown at the bottom. (C) ChIP-qPCR enrichment analysis with primer sets (1 to 6) in the SERPINE1 promoter region in control and ADAM9 KO KYSE170 cells using ADAM9 antibody and control IgG. Data are mean ± SD. (D) RT-qPCR of ADAM9, SERPINE1, and PGK1 in control and ADAM9 knockout KYSE170 cells. (E) Western blot analysis of angiostatin proteins in control and ADAM9 KO KYSE170 cells. Cells were cultured in starvation for 3 days. (F) Cellular fractionation followed by western blot analysis of control and ADAM9 KO KYSE170 cells treated with 5 μM Brefeldin A at different time points. (G) Representative images of tumor tissues processed for IHC staining of PGK1 and CD31 from metastatic KYSE170 tumor animal models. Scale bar, 50 μm. CD31 staining was quantified as the percentage of positively stained area (bottom). Bar chart data are mean ± SD. Student's t test. *, P < 0.05. **, P < 0.01.
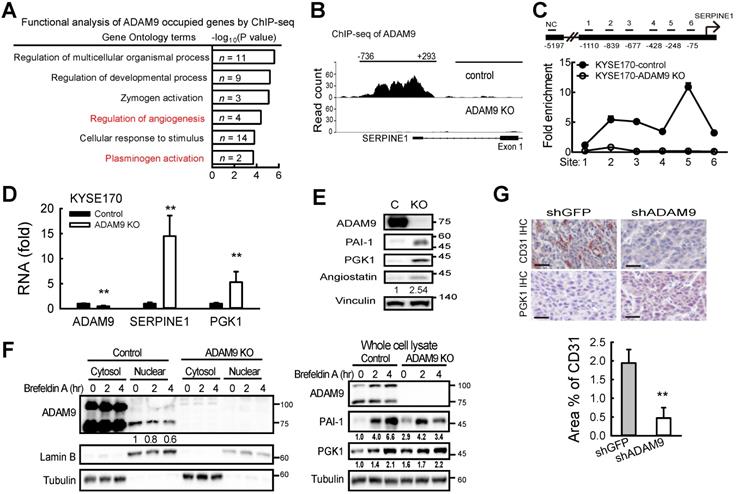
Of the two genes, SERPINE1 and PGK1 (encoding phosphoglycerate kinase 1), identified for their roles in regulating plasminogen activation (Figure 5A), we found that ADAM9 also occupied the PGK1 promoter (Figure S5C). PGK1 has been reported to act as a disulfide reductase to stimulate proangiogenic plasmin to release the blood vessel inhibitor angiostatin, which blocks angiogenesis [27]. Importantly, we found that the levels of SERPINE1 and PGK1 mRNAs were increased in ADAM9 KO and knockdown cells (Figure 5D and Figure S5D), as well as increased protein levels of PAI-1, PGK1, and angiostatin in ADAM9 KO cells compared with control cells (Figure 5E). Conversely, ectopic expression of ADAM9 decreased PGK1 expression in KYSE170-ADAM9 KO cells (Figure S5E).
Next, we asked whether suppression of ADAM9 nuclear translocation affects PAI-1 and PGK1 expression. To this end, we treated control and ADAM9 KO KYSE170 cells with brefeldin A (BFA), an inhibitor of intracellular protein transport. The levels of nuclear ADAM9 were decreased and the total PAI-1 and PGK1 proteins were increased in the control cells in a time-dependent manner (Figure 5F). In ADAM9 KO cells, PAI-1 initially increased after 2 h of BFA treatment but then declined to a level equivalent to that at the zero timepoint whereas PGK1 levels remained constant (Figure 5F). We speculated that the negative association between the amounts of nuclear ADAM9 and the PAI-1 or PGK1 proteins may be due to the transcriptional repression by nuclear ADAM9.
Next, to validate ADAM9 function in angiogenesis, we demonstrated that endothelial marker CD31 was higher in control tumors than in ADAM9 knockdown KYSE170 tumors whereas IHC staining of PGK1 was lower in control tumors than ADAM9 knockdown KYSE170 tumors (Figure 5G). Moreover, angiogenesis (tube formation) was reduced in human umbilical vein endothelial cells (HUVECs) cultured in conditioned media from ADAM9 KO cells (Figure S5F). Conditioned media from ADAM9 KO cells with overexpression of ADAM9 WT strongly restored angiogenesis in HUVECs whereas conditioned media from ADAM9 KO cells with overexpression of the E348A mutant only slightly restored angiogenesis (Figure S5G). Taken together, these results reveal that nuclear ADAM9 functions as a transcriptional repressor by binding to promoters of genes involved in the negative regulation of angiogenesis, and thereby promotes tumor angiogenesis.
ADAM9 suppresses SERPINE1 transcription by interacting with known transcription factors for SERPINE1 gene regulation in ESCC
Next, we investigated whether ADAM9 influenced SERPINE1 gene transcription by measuring SERPINE1 promoter activity. Two repressor elements (-764 to -628 and -266 to -188) have been previously identified in the SERPINE1 promoter, but the identity of potential repressors remains unknown [28]. We linked the 5'-flanking regions of the SERPINE1 promoter at positions -1200, -600, and -300, respectively, with the luciferase gene to measure the promoter activity in control and ADAM9 KO cells (Figure 6A). SERPINE1 promoter activity was greater in ADAM9 KO cells compared to control cells for all three promoter constructs, but the two shorter constructs (-600 and -300) showed greater promoter activity than the longest construct (-1200) (Figure 6A). This is likely due to the presence of an unknown repressor sequence located within the region of -1200 to -600 bp. Similar observations of SERPINE1 promoter activity were found in ADAM9 knockdown ESCC cells (Figure S6A). Moreover, in ADAM9 KO KYSE170 cells, overexpression of ADAM9 WT strongly reduced the promoter activity of SERPINE1 compared with vector controls (Figure 6B). Although SERPINE1 promoter contains a hypoxia-responsive element [29], its activity remains low under stressed condition of hypoxia with starvation in ADAM9-expressing shGFP cells (Figure S6B). In contrast, ADAM9 deficiency resulted in activation of SERPINE promoter under stressed conditions (Figure S6B). These results suggest that ADAM9 plays an important role in the negative regulation of SERPINE1 gene expression.
Nuclear ADAM9 proteins interact with transcription factors HIF1α and USF1 in ESCC cells. (A) SERPINE1 promoter activity in control and ADAM9 KO KYSE170 cells. A schematic diagram of SERPINE1 promoter constructs from sites -1200, -600, or -300 driving luciferase expression (top). The two previously reported repressor sites are marked in red. Relative luciferase activity was shown by Firefly/Renilla luciferase ratio in the dual-luciferase assay. (B) The transcriptional activity of the indicated promoters of SERPINE1 in ADAM9 KO KYSE170 cells with overexpression of ADAM9 WT. (C) Identification of potential transcription factors involved in ADAM9-mediated transcriptional suppression of SERPINE1 in KYSE170 cells. Schematics of transcriptional responses of SERPINE1 regulated by the interaction of ADAM9 and transcription factor (TF) (top). A9, ADAM9; Pol, RNA polymerase. The shRNA of TF was transiently transfected in control and ADAM9 KO cells and then cells were subjected to assay for the luciferase activity (bottom). Error bar, ±s.d. (D-F) PLA detection of ADAM9 and transcription factors in KYSE170 cells. Duolink PLA analysis of ADAM9/p53 complexes (D); ADAM9/HIF1α complexes (E); ADAM9/USF1 complexes (F). Representative images of PLA (red) and nuclei (DAPI, blue). Quantification of PLA was analyzed from 3 fields. Scale bars, 10 µm. Statistical analysis was performed with a t-test. (G) KYSE170 cells were ChIP and re-ChIP using indicated transcription factor and ADAM9 antibodies sequentially or in reverse order. Chromatin samples were analyzed by qPCR at the transcription factor binding sites. A t-test was compared with the NC1 group. *, p < 0.05; **, p < 0.01.
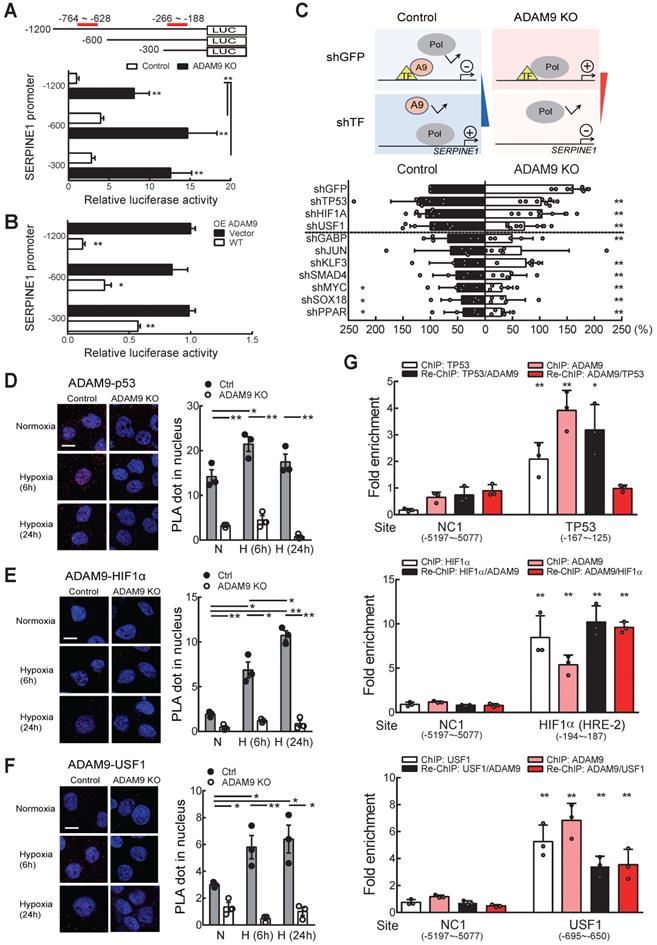
Because ADAM9 has no DNA binding domain, we speculate that it may be recruited by known transcription factors and then suppress their transactivation. To screen for the potential ADAM9-interacting transcription factors, we evaluated the promoter activity in control and ADAM9 KO cells by transiently knocking down known transcription factors involved in SERPINE1 gene regulation. We reason that the transcriptional inhibitory effect of ADAM9 for SERPINE1 gene would be reversed upon additional knockdown of ADAM9-interacting transcription factors in control cells based on the assumption that ADAM9 may not be able to be recruited by these transcription factors to the promoter region. In contrast, in the absence of ADAM9 (in ADAM9 KO cells), these transcription factors may increase the transactivation of SERPINE1 promoter and this would be reversed upon further knockdown of transcription factors (Figure 6C, top). Consistent with our prediction, we found that knockdown of transcription factor TP53, HIF1α, and upstream transcription factor 1 (USF1) in control cells showed elevated transactivation of SERPINE1 promoter whereas the promoter activity in ADAM9 KO cells was reduced (Figure 6C, bottom). The other transcription factors we screened may not be related to ADAM9 transcriptional regulation in SERPINE1 promoter based on our observations that their knockdown non-selectively decreased the promoter activity in control and ADAM9 KO cells (Figure 6C, bottom). To validate whether ADAM9 can interact with these transcription factors in a physiologically relevant manner, we conducted a sensitive proximity ligation assay (PLA), which can detect the close proximity of two molecules in the cells using two primary antibodies. We found that PLA signals of ADAM9 and TP53 were elevated in the whole cell and the nucleus of control cells during hypoxia (Figure 6D and Figure S6C). Similar phenomenon was detected in PLA signals of ADAM9 and HIF1α (Figure 6E and Figure S6C), as well as those of ADAM9 and USF1 (Figure 6F and Figure S6C). Re-ChIP analysis of sequential immunoprecipitation of two chromatin-binding proteins can be used to study co-occupancy of two factors on a specific DNA sequence [30]. In single ChIP analysis, either individual known transcription factor or ADAM9 bound well to the SERPINE1 promoter (Figure 6G). In re-ChIP analysis, we showed that the three transcription factors and ADAM9 were co-enriched on their binding sites at SERPINE1 promoter (Figure 6G). Similar levels of co-enrichment in HIF1α (first IP)/ADAM9 (second IP) and ADAM9 (first IP)/HIF1α (second IP) indicated that HIF1α and ADAM9 co-occupied the SERPINE1 promoter. Notably, a higher level of enrichment is seen with TP53/ADAM9 than ADAM9/TP53, suggesting that these two proteins show partial co-occupancy: TP53 can co-bind with ADAM9, but ADAM9 may not always co-bind with TP53 to the SERPINE1 promoter. Co-occupancy of USF1 and ADAM9 was also detected with reciprocal re-ChIP at known USF1 binding site (-695 ~ -650) of SERPINE1 promoter. Taken together, these results suggest that ADAM9 proteins interact with selective transcription factors to dampen transcription of SERPINE1 gene in ESCC.
Discussion
Here we have demonstrated that a high level of ADAM9 is correlated with poor outcomes in ESCC patients. Silencing of ADAM9 reduced tumorigenic characteristics of ESCC in vitro and in vivo, such as invasive and metastatic ability, and distant metastasis. We propose that ADAM9 coordinates plasminogen activator-based network for ESCC progression through two major functions of ADAM9: protease activity for increasing tPA expression/activity and nuclear translocation of ADAM9 for suppressing PAI-1 and PGK1 expression (PAI-1 for reducing plasmin and PGK1 for increasing angiostatin), two inhibitory factors involved in plasminogen activator-mediated angiogenesis. Thus, we revealed a novel role of ADAM9 in promoting angiogenesis through a previously unappreciated mechanism of transcriptional repression of genes participating in the negative regulation of angiogenesis (Figure 7).
Schematic diagram showing a dual role of ADAM9 in angiogenesis regulation through its protease activity and transcriptional regulation in nucleus. The protease function of ADAM9 increases PLAT expression for extracellular matrix degradation and proangiogenesis. In hypoxia, nuclear translocation of ADAM9 is increased and nuclear ADAM9 occupies the promoters of SERPINE1 and PGK1, leading to transcriptional repression of SERPINE1 and PGK1 for reducing angiostatin formation and thus promoting angiogenesis.
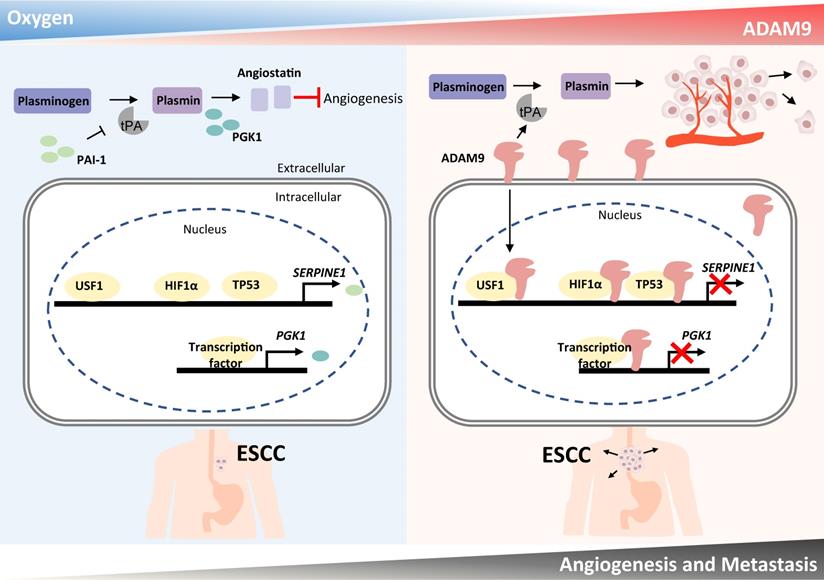
The sheddase function of ADAM9 has been considered critical for promoting cell proliferation and migration in ESCC through yielding soluble EGFR ligands, thereby activating the EGFR-AKT pathway [18]. Here we demonstrated that the sheddase activity of ADAM9 is also involved in increasing tPA protein expression and activity. Importantly, protease-defective ADAM9 can reduce its nuclear translocation. We found that nuclear ADAM9 can occupy the SERPINE1 promoter over nearly 1 kb and suppress SERPINE1 transcription. We provide evidence suggesting that ADAM9 is likely to interact with several transcription factors previously known for regulation of SERPINE1 expression, such as p53, HIF1α, and USF1. These transcription factors play important roles in tumorigenesis and interplay with each other to regulate target gene expression. They serve as positive regulators by binding to SERPINE1 promoter to activate PAI-1 gene expression under various contexts. Beyond the role as a TF, the tumor suppressor p53 binds to PAI-1 mRNA 3'-UTR and stabilizes PAI-1 mRNA, leading to enhanced PAI-1 expression [31]. The stress sensor USF1 not only binds to and transactivates TP53 promoter [32], but also stabilizes p53 proteins to coordinate p53 function in cell fate decision [33]. Loss of USF1 drives p53 degradation and accelerates gastric carcinogenesis [34]. p53 influences HIF-1α by suppressing its hypoxic induction and target activation, while p53 loss of function enhances HIF-1α in tumors [35]. Conversely, severe and prolonged hypoxic stress upregulates p53 for cell fate decision [36]. Upregulation of HIF-1α activates crucial cancer hallmarks such as angiogenesis leading to poor cancer patient prognosis. Altogether, p53, HIF1α, and USF1 portray a complicated regulation network on multiple target genes, including PAI-1, suggesting that PAI-1 level needs to be critically regulated in maintaining cell function. Our study uncovers ADAM9 as a new player of PAI-1 regulation network, presumably through protein-protein interaction between ADAM9 and the above mentioned transcription factors. However, whether and how the interaction of ADAM9 with these transcription factors regulate other genes or pathways during cancer progression need to be clarified in future studies.
Previous studies showed positive nuclear staining of ADAM9 and its association with lethal phenotypic progression of prostate cancer [37], but the role and regulation of nuclear ADAM9 has not been elucidated. Our finding reveals that ADAM9 can be induced upon hypoxia to translocate to the nucleus, where nuclear ADAM9 acts as a transcriptional repressor to regulate expression of key angiogenesis genes in ESCC cells. Another member of the ADAM family, ADAM10, has been reported to be transported into the nucleus and contributes to the pathogenesis and progression of human prostate cancer [38]. Although the overall sequence similarity between ADAM9 and ADAM10 is about 25%, the putative nuclear localization sequence (NLS, aa 199 to 206) in ADAM9 is highly similar (87.5%) to the NLS region (aa 207 to 214) in ADAM10. Whether the putative NLS of ADAM9 mediates protein nuclear translocation in a similar manner as that of ADAM10 is worth further investigation.
Angiogenesis contributes to the aggressive characteristics of ESCC [39]. The plasminogen activator-based pathway promotes tumor-associated angiogenesis via protease activity [9]. Although PAI-1 has been considered as an inhibitor of cancer development, the proangiogenic and promigratory effects of PAI-1 promote tumor growth at physiological concentration by stimulating plasmin-mediated proteolysis [40]. Several studies have revealed that elevated levels of PAI-1 in tumor tissue are predictors of poor patient outcomes in the subset of breast cancer patients with lymph node-negative disease [41]. However, genetic ablation of PAI-1 had no effects on tumor development or metastasis [42]. Given that a typical rapid decline in PAI-1 levels was detected prior to the onset of DNA synthesis [43] and PAI-1 reduction leading to escape from senescence-associated proliferative arrest in primary fibroblasts [44], PAI-1 may have a negative influence on cell growth. Indeed, at supraphysiological levels, PAI-1-mediated inhibition of tumor vascularization was observed in either host cells or tumor cells [45], suggesting that high levels of PAI-1 reduce angiogenesis. Our current work provides new insights into how ADAM9 regulates tumor-associated angiogenesis in a context-dependent manner. Under stressed conditions, nuclear ADAM9 may function as a transcriptional repressor to maintain PAI-1 at a low level. Consistent with this concept, our data show that stressed conditions enhance a strong induction of PAI-1 expression only in ADAM9-deficient cells, but not in ADAM9-abundant cells. Taken together, we reveal a novel role of ADAM9 as a stress-induced transcriptional regulator to undermine PAI-1 expression to promote tumor vascularization.
Abbreviations
ADAM9: a disintegrin and metalloprotease 9; ChIP: chromatin immunoprecipitation; EGFR: epidermal growth factor receptor; ESCC: esophageal squamous cell carcinoma; HIF1α: hypoxia inducible factor 1 subunit alpha; IHC: immunohistochemistry; KO: knockout; PAI-1: plasminogen activator inhibitor type I; PGK1: phosphoglycerate kinase 1; PLA: proximity ligation assay; SERPINE1: serpin family E member 1; TIC: tumor-initiating cell; tPA: tissue plasminogen activator; TCGA: The Cancer Genome Atlas; WT: wild type.
Supplementary Material
Supplementary methods, figures and tables.
Acknowledgements
This work was supported by grants from the Ministry of Science and Technology (MOST 105-2314-B-039-034-MY3, MOST 108-2314-B-039-054-MY3 to Y-P.S.; MOST 107-2314-B-039-051-MY2 to C-Y.L.), National Health Research Institutes (NHRI-EX109-10706BI to Y-P.S.), and China Medical University (DMR-109-203; CMU110-MF-32). This study was supported by the Chinese Medicine Research Center, China Medical University, from the Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education in Taiwan.
Ethics approval and consent to participate
Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of China Medical University. Ethical approval for data extraction of patients with ESCC was approved by the hospital's institutional review board of China Medical University Hospital.
Author Contributions
Y-S.L., and Y-P.S. conceived and designed the experiments. Y-S.L., T-T.K., C-C.L., W-C.Chang, Y-K.H., S-T.B., and C-Y.H performed the experiments. G-C.T., T-T.K., L-C. L., and W-C.Cheng analyzed the data. C-Y.L., H-S.H., and Y-F.J. contributed reagents, materials, and/or analysis tools. Y-S.L., C-Y.L., L-C. L., X-G. L., and Y-P.S. wrote the paper. All authors reviewed the manuscript.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen W, Zheng R, Baade PD. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115-32
2. Torre LA, Bray F, Siegel RL. et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87-108
3. Lu CL, Lang HC, Luo JC. et al. Increasing trend of the incidence of esophageal squamous cell carcinoma, but not adenocarcinoma, in Taiwan. Cancer Causes Control. 2010;21:269-74
4. Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349:2241-52
5. Abate E, DeMeester SR, Zehetner J. et al. Recurrence after esophagectomy for adenocarcinoma: defining optimal follow-up intervals and testing. Journal of the American College of Surgeons. 2010;210:428-35
6. Peerlings J, Van De Voorde L, Mitea C. et al. Hypoxia and hypoxia response-associated molecular markers in esophageal cancer: A systematic review. Methods. 2017;130:51-62
7. Leo C, Giaccia AJ, Denko NC. The hypoxic tumor microenvironment and gene expression. Semin Radiat Oncol. 2004;14:207-14
8. Nekarda H, Schlegel P, Schmitt M. et al. Strong prognostic impact of tumor-associated urokinase-type plasminogen activator in completely resected adenocarcinoma of the esophagus. Clin Cancer Res. 1998;4:1755-63
9. Diaz VM, Planaguma J, Thomson TM. et al. Tissue plasminogen activator is required for the growth, invasion, and angiogenesis of pancreatic tumor cells. Gastroenterology. 2002;122:806-19
10. McMahon B, Kwaan HC. The plasminogen activator system and cancer. Pathophysiol Haemost Thromb. 2008;36:184-94
11. Takayama Y, Hattori N, Hamada H. et al. Inhibition of PAI-1 Limits Tumor Angiogenesis Regardless of Angiogenic Stimuli in Malignant Pleural Mesothelioma. Cancer Res. 2016;76:3285-94
12. Sakakibara T, Hibi K, Kodera Y. et al. Plasminogen activator inhibitor-1 as a potential marker for the malignancy of esophageal squamous cell carcinoma. Clin Cancer Res. 2004;10:1375-8
13. Groblewska M, Siewko M, Mroczko B. et al. The role of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) in the development of esophageal cancer. Folia Histochem Cytobiol. 2012;50:12-9
14. Ma B, Zhang HY, Bai X. et al. ADAM10 mediates the cell invasion and metastasis of human esophageal squamous cell carcinoma via regulation of E-cadherin activity. Oncol Rep. 2016;35:2785-94
15. Chou CW, Huang YK, Kuo TT. et al. An Overview of ADAM9: Structure, Activation, and Regulation in Human Diseases. Int J Mol Sci. 2020 21
16. Lin CY, Chen HJ, Huang CC. et al. ADAM9 promotes lung cancer metastases to brain by a plasminogen activator-based pathway. Cancer Res. 2014;74:5229-43
17. Lin CY, Cho CF, Bai ST. et al. ADAM9 promotes lung cancer progression through vascular remodeling by VEGFA, ANGPT2, and PLAT. Sci Rep. 2017;7:15108
18. Liu R, Gu J, Jiang P. et al. DNMT1-microRNA126 epigenetic circuit contributes to esophageal squamous cell carcinoma growth via ADAM9-EGFR-AKT signaling. Clin Cancer Res. 2015;21:854-63
19. Jiang YF, Lin SS, Chen JM. et al. Electron tomographic analysis reveals ultrastructural features of mitochondrial cristae architecture which reflect energetic state and aging. Sci Rep. 2017;7:45474
20. Lin CC, Huang YK, Cho CF. et al. Targeting positive feedback between BASP1 and EGFR as a therapeutic strategy for lung cancer progression. Theranostics. 2020;10:10925-39
21. Bharadwaj AG, Holloway RW, Miller VA. et al. Plasmin and Plasminogen System in the Tumor Microenvironment: Implications for Cancer Diagnosis, Prognosis, and Therapy. Cancers (Basel). 2021 13
22. Napolitano F, Montuori N. The Role of the Plasminogen Activation System in Angioedema: Novel Insights on the Pathogenesis. J Clin Med. 2021 10
23. Maretzky T, Swendeman S, Mogollon E. et al. Characterization of the catalytic properties of the membrane-anchored metalloproteinase ADAM9 in cell-based assays. Biochem J. 2017;474:1467-79
24. Kosugi S, Hasebe M, Tomita M. et al. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci U S A. 2009;106:10171-6
25. Brameier M, Krings A, MacCallum RM. NucPred-predicting nuclear localization of proteins. Bioinformatics. 2007;23:1159-60
26. Sun G, Hu W, Lu Y. et al. A meta-analysis of HIF-1alpha and esophageal squamous cell carcinoma (ESCC) risk. Pathol Oncol Res. 2013;19:685-93
27. Lay AJ, Jiang XM, Kisker O. et al. Phosphoglycerate kinase acts in tumour angiogenesis as a disulphide reductase. Nature. 2000;408:869-73
28. Johnson MR, Bruzdzinski CJ, Winograd SS. et al. Regulatory sequences and protein-binding sites involved in the expression of the rat plasminogen activator inhibitor-1 gene. J Biol Chem. 1992;267:12202-10
29. Fink T, Kazlauskas A, Poellinger L. et al. Identification of a tightly regulated hypoxia-response element in the promoter of human plasminogen activator inhibitor-1. Blood. 2002;99:2077-83
30. Truax AD, Greer SF. ChIP and Re-ChIP assays: investigating interactions between regulatory proteins, histone modifications, and the DNA sequences to which they bind. Methods Mol Biol. 2012;809:175-88
31. Shetty S, Shetty P, Idell S. et al. Regulation of plasminogen activator inhibitor-1 expression by tumor suppressor protein p53. J Biol Chem. 2008;283:19570-80
32. Reisman D, Rotter V. The helix-loop-helix containing transcription factor USF binds to and transactivates the promoter of the p53 tumor suppressor gene. Nucleic Acids Res. 1993;21:345-50
33. Bouafia A, Corre S, Gilot D. et al. p53 requires the stress sensor USF1 to direct appropriate cell fate decision. PLoS Genet. 2014;10:e1004309
34. Costa L, Corre S, Michel V. et al. USF1 defect drives p53 degradation during Helicobacter pylori infection and accelerates gastric carcinogenesis. Gut. 2020;69:1582-91
35. Sermeus A, Michiels C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011;2:e164
36. Parandavar E, Yazdanparast R. Differential impact of various reactive oxygen species (ROS) on HIF-1alpha/p53 direct interaction in SK-N-MC neuroblastoma cells. Cell Biosci. 2017;7:52
37. Pen CC, Liu CM, Lin CC. et al. Combined Dynamic Alterations in Urinary VEGF Levels and Tissue ADAM9 Expression as Markers for Lethal Phenotypic Progression of Prostate Cancer. Chin J Physiol. 2012;55:390-7
38. Arima T, Enokida H, Kubo H. et al. Nuclear translocation of ADAM-10 contributes to the pathogenesis and progression of human prostate cancer. Cancer science. 2007;98:1720-6
39. Kubota Y, Kaneko K, Konishi K. et al. The onset of angiogenesis in a multistep process of esophageal squamous cell carcinoma. Front Biosci (Landmark Ed). 2009;14:3872-8
40. Devy L, Blacher S, Grignet-Debrus C. et al. The pro- or antiangiogenic effect of plasminogen activator inhibitor 1 is dose dependent. FASEB J. 2002;16:147-54
41. Duffy MJ, McGowan PM, Harbeck N. et al. uPA and PAI-1 as biomarkers in breast cancer: validated for clinical use in level-of-evidence-1 studies. Breast Cancer Res. 2014;16:428
42. Almholt K, Nielsen BS, Frandsen TL. et al. Metastasis of transgenic breast cancer in plasminogen activator inhibitor-1 gene-deficient mice. Oncogene. 2003;22:4389-97
43. Qi L, Allen RR, Lu Q. et al. PAI-1 transcriptional regulation during the G0 -> G1 transition in human epidermal keratinocytes. J Cell Biochem. 2006;99:495-507
44. Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8:877-84
45. Bajou K, Maillard C, Jost M. et al. Host-derived plasminogen activator inhibitor-1 (PAI-1) concentration is critical for in vivo tumoral angiogenesis and growth. Oncogene. 2004;23:6986-90
Author contact
![]() Corresponding author: Yuh-Pyng Sher, Graduate Institute of Biomedical Sciences, China Medical University, Taichung 404, Taiwan, Phone: 886-4-22052121 ext. 7819, Fax: 886-4-2233-3496, E-mail: ypshercmu.edu.tw.
Corresponding author: Yuh-Pyng Sher, Graduate Institute of Biomedical Sciences, China Medical University, Taichung 404, Taiwan, Phone: 886-4-22052121 ext. 7819, Fax: 886-4-2233-3496, E-mail: ypshercmu.edu.tw.