Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(1):426-440. doi:10.7150/ijbs.64640 This issue Cite
Research Paper
Engineering of human mesenchymal stem cells resistant to multiple natural killer subtypes
Dejin Zheng1,2, Xiaoyan Wang1,2, Zhenwu Zhang1,2,3, Enqin Li1,2, Cheungkwan Yeung1,2, Roma Borkar1,2, Guihui Qin1,2, Yaojiong Wu4, Ren-He Xu1,2 ![]()
1. Center of Reproduction, Development & Aging, and Institute of Translational Medicine, Faculty of Health Sciences, University of Macau, Taipa, Macau, China.
2. Ministry of Education Frontiers Science Center for Precision Oncology, University of Macau, Taipa, Macau, China.
3. School of Life Science and Technology, ShanghaiTech University, Shanghai, China.
4. The Shenzhen Key Laboratory of Health Sciences and Technology, International Graduate School at Shenzhen, Tsinghua University, Shenzhen, China.
Received 2021-7-6; Accepted 2021-10-25; Published 2022-1-1
Abstract

Mesenchymal stem cells (MSCs) as a therapeutic promise are often quickly cleared by innate immune cells of the host including natural killer (NK) cells. Efforts have been made to generate immune-escaping human embryonic stem cells (hESCs) where T cell immunity is evaded by defecting β-2-microglobulin (B2M), a common unit for human leukocyte antigen (HLA) class I, and NK cells are inhibited via ectopic expression of HLA-E or -G. However, NK subtypes vary among recipients and even at different pathologic statuses. It is necessary to dissect and optimize the efficacy of the immune-escaping cells against NK subtypes. Here, we first generated B2M knockout hESCs and differentiated them to MSCs (EMSCs) and found that NK resistance occurred with B2M-/- EMSCs expressing HLA-E and -G only when they were transduced via an inducible lentiviral system in a dose-dependent manner but not when they were inserted into a safe harbor. HLA-E and -G expressed at high levels together in transduced EMSCs inhibited three major NK subtypes, including NKG2A+/LILRB1+, NKG2A+/LILRB1-, and NKG2A-/LILRB1+, which was further potentiated by IFN-γ priming. Thus, this study engineers MSCs with resistance to multiple NK subtypes and underscores that dosage matters when a transgene is used to confer a novel effect to host cells, especially for therapeutic cells to evade immune rejection.
Keywords: Human embryonic stem cells, mesenchymal stem cells, natural killer cells, innate immunity, immune rejection
Introduction
Mesenchymal stem cells (MSCs) exert promising therapeutic effects on many disease models mainly through secreted factors and exosomes for immunomodulation and regeneration, and in a rarer chance via direct differentiation. However, their outcomes in clinical trials often fluctuate [1], which might partially result from clearance of allogeneic MSCs infused either systemically or locally, due to both innate immunity immediately and adaptive immunity at a later time [2-4]. T cell-mediated adaptive immunity plays an essential role in the clearance of allogeneic MSCs [2], which is triggered by allogeneic peptides presented on the cell surface by human leukocyte antigens (HLAs) [5]. Many strategies have been developed to reduce the immunogenicity of MSCs. Classical HLA class I (HLA-Ia) is the primary target of manipulation due to its important role in MSC immunogenicity. β-2-microglobulin (B2M) knockout MSCs were generated from umbilical cord MSCs and MSCs differentiated from induced pluripotent stem cells (iPSCs). B2M-/- MSCs became more resistant to MSC-primed CD8+ T cell-mediated lysis even after IFN-γ priming [6-8]. In addition, B2M-/- MSCs also had better therapeutic effects for treating myocardial infarction and ischemic hindlimb compared to wild-type (WT) MSCs [7, 8].
However, since HLA-Ia molecules are the classical ligands for the killer-cell immunoglobulin-like receptors (KIRs) on natural killer (NK) cells, a major type of innate immune cells [9], removal of HLA-Ia from any cells including MSCs sensitizes the cells to NK cytotoxicity [6]. To solve this problem, MSCs were derived from HLA-Ia pseudo-homozygous iPSCs with monoallelic deletion of HLA-A, B, and C genes to avoid NK cytotoxicity [10]. However, this strategy still requires HLA matching to evade adaptive immunity. Unlike HLA-Ia, non-classical HLA class I molecules (HLA-Ib) such as HLA-E or HLA-G barely activate adaptive immunity, while still protecting cells from NK cell-mediated lysis through interacting with NK inhibitory receptors including natural killer group 2 member A (NKG2A)/CD94 and leukocyte immunoglobulin-like receptor subfamily B member 1 (LILRB1) [11, 12].
Strategies based on HLA-Ib have been developed to evade NK immunity in HLA-null human pluripotent stem cells (hPSCs), including embryonic stem cells (ESCs) and iPSCs, which have led to the generation of so-called universal hPSCs [13]. For example, Gornalusse, et al., inserted a B2M-HLA-E fusion gene into the B2M loci of hESCs using an adeno-associated virus vector. The resultant hESCs and their derived hematopoietic cells can escape the allogeneic responses and NK cell-mediated killing in vitro and in vivo [14]. Moreover, HLA-G expression in HLA-Ia-depleted hESCs also protect against NK cell-mediated lysis [15]. Co-expression of B2M-fused HLA-G1 and -G5 proteins in B2M deficient hESCs has potent inhibitory effects on both T and NK cells [16]. Nevertheless, it awaits to be tested whether such strategies remain effective in hPSC-derived MSCs. Moreover, NK cells are heterozygous with a donor-dependent profile of their receptor expression [17], and the reported approaches only inhibit limited NK subtypes. For example, cells bearing HLA-E or -G alone only escape NKG2A+ or LILRB1+ NK subtypes, respectively [14, 16]. Therefore, it is necessary to develop MSCs with combined resistance to multiple NK subtypes.
In this study, we first knocked out B2M, a common unit for all class I HLAs, in hESCs and then inserted a single copy of engineered HLA-E and -G together in the safe harbor locus AAVS1 in the genome of the B2M-/- hESCs, which were then differentiated to MSCs (EMSCs). Unexpectedly, the genetically manipulated EMSCs were still sensitive to human NK cytotoxicity. However, when HLA-E or -G were expressed via a doxycycline (DOX)-inducible lentiviral system, NK resistance was achieved with high doses of DOX, suggesting that NK cytotoxicity can be escaped only when ectopically expressed immunosuppressive molecules reach a threshold. Based on these, we generated EMSCs with the capability to resist multiple NK subtypes including NKG2A+/LILRB1+, NKG2A+/LILRB1-, and NKG2A-/LILRB1+ NK cells.
Results
B2M-/- EMSCs are sensitized to NK cytotoxicity
To develop EMSCs that can evade T cell immunity, we first targeted B2M gene [18] in H1 hESC line using the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) technology with a pair of single guide (sg) RNAs specifically for B2M and obtained two B2M-/- cell clones with frame-shift mutations in exon 1, i.e., -13/+2 and -11/-22, respectively, based on genotyping (Fig. 1A). As expected, western blotting detected B2M expression in the WT control but not the two B2M-/- clones. On the other hand, the total HLA class I expression was detected in the WT control and, to a lesser level, in the B2M-/- clones (Fig. 1B). Flow cytometry confirmed that both B2M and HLA class I were present on the surface of the WT control but not of the B2M-/- hESCs (Fig. 1C). The genome-editing didn't affect the pluripotency of the hESCs based on assays for expression of pluripotency marker genes (Fig. S1A) and teratoma formation (Fig. S1B). Normal karyotypes were also preserved in the genetically manipulated hESC clones (Fig. S1C).
B2M-/- EMSCs are sensitized to NK cytotoxicity. (A) Schematic graph and genotyping results for B2M-/- hESC clones #1 and 2 compared to WT control. (B) Western blotting for HLA class I and B2M expression in WT and B2M-/- hESCs. GAPDH was tested as an internal control. (C) Flow cytometry analysis for HLA class I and B2M expression on the surface of WT and B2M-/- hESCs. (D) Western blotting for HLA class I and B2M expression in WT and B2M-/- EMSCs. GAPDH was tested as an internal control. (E) Flow cytometry analysis for B2M and HLA class I expression on the surface of WT and B2M-/- EMSCs treated with (IFN-γ-primed) or without (naïve) 10 ng/ml IFN-γ. (F) Summary of the medium fluorescence intensities for the above results. N = 3, ****P < 0.0001 for (1) versus (3), ####P < 0.0001 for (1) versus (5), &&&&P < 0.0001 for (2) versus (4), $$$$P < 0.0001 for (2) versus (6), ++++P < 0.0001 for (2) versus (1) EMSCs per ordinary one-way ANOVA followed by Tukey's multiple comparison test. (G) NK-92MI cell-mediated lysis against WT and B2M-/- EMSCs (naïve). N = 3, *P < 0.05, ***P < 0.001 for (2) versus (1), #P < 0.05 for (3) versus (1) per two-way ANOVA followed by Dunnett's multiple comparison test. (H) NK-92MI cell-mediated lysis against WT and B2M-/- EMSCs (IFN-γ-primed). N = 3, ***P < 0.001, ****P < 0.0001 for (2) versus (1); #P < 0.05, ####P < 0.0001 for (3) versus (1) per two-way ANOVA followed by Dunnett's multiple comparison test.
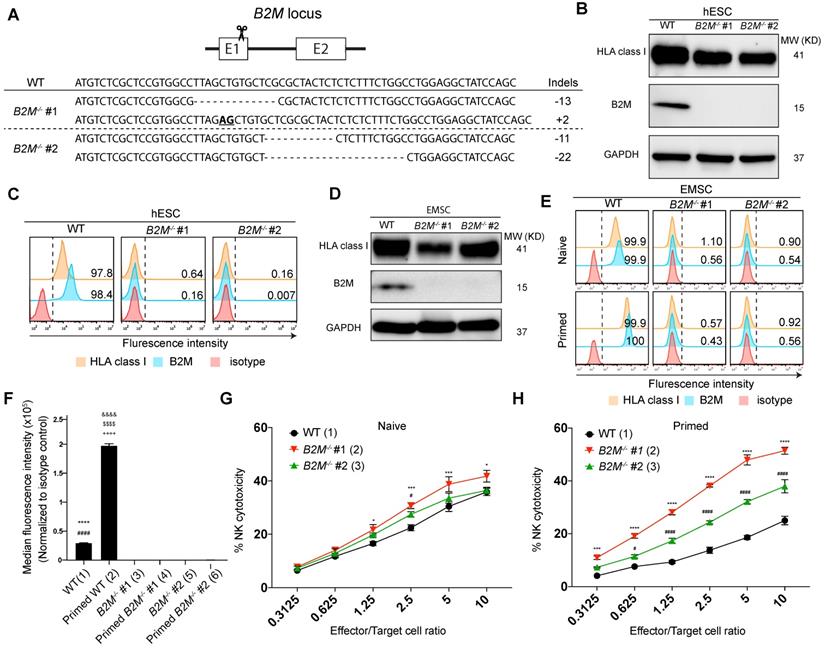
Further, we differentiated B2M-/- hESCs to MSCs using an established protocol [19, 20] (Fig. S2A). The resultant EMSCs, like the WT control, met the minimum criteria for MSCs based on their positivity of CD90, CD44, CD105, and CD73 and negativity of CD34, CD45, CD11b, CD19, and HLA-DR (Fig. S2B) and their capability of trilineage differentiation to adipocytes, osteoblasts, and chondrocytes in vitro (Fig. S2C). Consistently, B2M expression was detected in the WT but not B2M-/- EMSCs using western blotting, while the total HLA class I expression was detected in both WT and B2M-/- EMSCs (Fig. 1D). Moreover, both B2M and HLA class I were present on the surface of the WT but not the B2M-/- EMSCs no matter whether the cells were primed with or without 10 ng/ml interferon-gamma (IFN-γ) (Figs. 1E and F).
To test whether the absence of HLA class I on the cell surface would sensitize the EMSCs to NK cells, we set up NK cytotoxicity assay in vitro using NK-92MI cells as effectors [21]. B2M-/- EMSCs, as target cells, were more susceptive than WT EMSCs to the NK cell-mediated killing (Fig. 1G). It has been known that IFN-γ priming increases the resistance of MSCs to NK cells through elevated expression of HLA class I molecules [22]. However, IFN-γ treatment increased the surface expression of HLA class I only in the WT EMSCs but not in the B2M-/- EMSCs (Figs. 1E and F). Consistently, following IFN-γ treatment, the WT but not the B2M-/- EMSCs had reduced sensitivity to the NK cytotoxicity compared to the unprimed controls (Figs. 1G and H). These results suggest that the absence of HLA class I on the cell surface sensitizes EMSCs to NK cytotoxicity.
HLA-E and -G fused with B2M are inserted in a safe harbor locus in B2M-/- hESCs
To reduce the sensitivity of B2M-/- EMSCs to NK cytotoxicity, we expressed two HLA-Ib genes HLA-E and -G ectopically in the AAVS1 site, a safe harbor locus, in human cells via recombinase-mediated cassette exchange (RMCE) [23]. A cassette including floxed PGK promoter-driven puΔTK was inserted into AAVS1 in the B2M-/- hESCs to establish a master hESC line for RMCE (Fig. 2A). One single cell-derived clone was selected to verify the integration of the cassette into the AAVS1 site. Polymerase chain reaction (PCR) results showed that the clone was heterozygous with the cassette integrated into one allele of the AAVS1 site as the borders between the insert and its flanking regions were amplified but the full insert was too long (around 2.5 kb) to be amplified (Fig. 2B). Both HLA-Ia and -Ib require the subunit B2M for assembly and presence on the cell surface [24]. Thus, we fused B2M with HLA-E or -G using a linker containing 4 repeats of gly-gly-gly-gly-ser, namely (G4S)4, to generate single-chain dimers (E-SCD or G-SCD) [25] (Fig. 2C), which were further linked with a neomycin-resistant gene NeoR via the P2A peptide [26], together forming an RMCE exchange donor vector for both HLA-E and -G expression (Fig. 2D).
B2M-fused HLA-E and -G are inserted in a safe harbor locus in B2M-/- hESCs. (A) Schematic graph for generation of RMCE master line in B2M-/- hESCs. A RMCE cassette containing floxed PGK promoter driven puΔTK is inserted into the AAVS1 locus using TALENs. (B) PCR confirmation of stable integration of the RMCE cassette in the AAVS1 site of master hESCs. PCR products were amplified from genomic DNA in WT and master hESCs using primes as indicated in A. (C) Schematic graph for a single-chain dimer (SCD) of HLA-E (E-SCD) or -G (G-SCD), which fused B2M with HLA-E or -G using a linker (G4S)4. (D) Schematic graph for knock-in of a cassette including CAG promoter-driven E-SCD, G-SCD, and NeoR from the RMCE donor vector into the AAVS1 locus of B2M-/- hESCs via Cre-mediated RMCE. (E) PCR confirmation of the integration of E-SCD, G-SCD, and NeoR in the AAVS1 site of B-E+G+ hESCs. PCR products were amplified from the genomic DNA of master and B-E+G+ hESCs using primes as indicated in D. (F) Flow cytometry analysis for HLA-E and -G expression on the surface of WT, master, and B-E+G+ hESCs (with or without IFN-γ treatment).
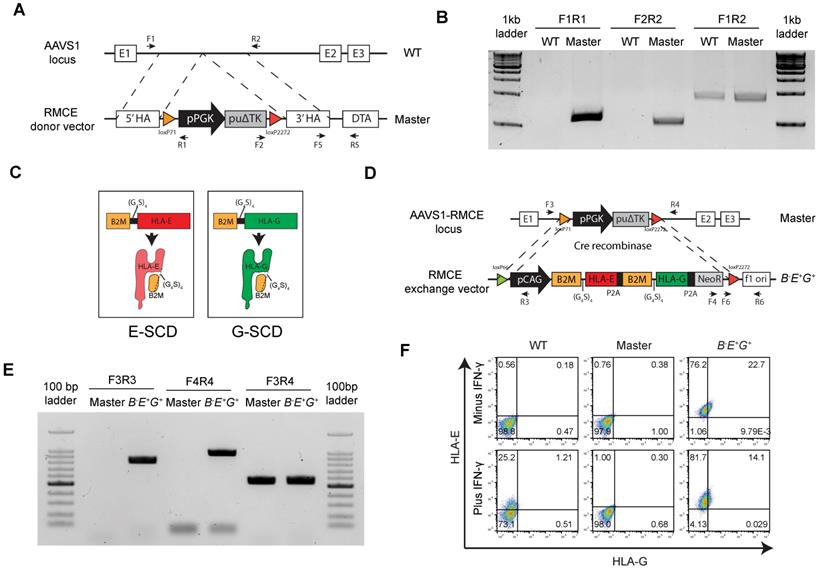
Next, we replaced the PGK promoter-driven puΔTK in the cassette in the master hESCs with the donor vector through Cre recombinase-mediated RMCE (Fig. 2D). One single-cell clone was selected and verified via PCR for the successful exchange in the master hESC line based on the detection of the flanking sequences of the transgenes HLA-E and -G, hence named B-E+G+ hESCs (Fig. 2E). Further, both HLA-E and -G were detected on the surface of B-E+G+ hESCs compared to the original master cells, and IFN-γ priming did not affect the surface expression of both HLA-E and -G (Fig. 2F). The pluripotency of the cells was verified via flow cytometry (Fig. S3A) and teratoma formation (Fig. S3B). PCR analysis further assured no integration of either the RMCE (Fig. S3C) or RMCE exchange donor vectors (Fig. S3D) in any other region in the genome. Thus, we introduced a single copy of HLA-E and -G into the AAVS1 site of B2M-/- hESCs successfully.
Site-specifically expressed HLA-E, but not HLA-G, increases NK resistance of only IFN-γ-primed B2M-/- EMSCs
To test the NK resistance of the RMCE-derived B-E+G+ hESCs, we differentiated them and the master hESCs as a control to MSCs, named B-E+G+ and master EMSCs, respectively, which both expressed typical MSC markers at similar levels based on flow cytometry analysis (Fig. S4A) and retained the capability of trilineage differentiation in vitro (Fig. S4B). The expression levels of E-SCD and G-SCD in the B-E+G+ EMSCs treated with or without IFN-γ were confirmed using quantitative PCR (qPCR) (Fig. 3A). Through flow cytometry, HLA-E and -G were detected on the surface of only the B-E+G+ EMSCs but not the master EMSCs (Fig. 3B), suggesting successful generation of B-E+G+ EMSCs. Interestingly, IFN-γ treatment increased the surface expression of both HLA-E and -G on the B-E+G+ EMSCs (Fig. 3B), which might result from an increased supply of high-affinity immunopeptides for HLA-E and -G assembly after IFN-γ treatment [27, 28].
Site-specifically expressed HLA-E, but not HLA-G, increases NK resistance of IFN-γ-primed B2M-/- EMSCs. (A) qPCR analysis for E-SCD and G-SCD transcript levels in master and B-E+G+ EMSCs with (primed) or without (naïve) IFN-γ treatment. N = 3, ****P < 0.0001 for B-E+G+ versus master EMSCs per two-tailed unpaired t test. (B) Flow cytometry analysis for HLA-E and -G expression on the surface of master and B-E+G+ EMSCs with or without IFN-γ treatment. (C) NK-92MI cell-mediated lysis of master and B-E+G+ EMSCs (naïve). N = 3. (D) NK-92MI cell-mediated lysis of master and B-E+G+ EMSCs (IFN-γ-primed). N = 3. **P < 0.01, ***P < 0.001, ****P < 0.0001 for B-E+G+ versus master EMSCs per two-way ANOVA followed by Sidak's multiple comparison test. (E) Flow cytometry analysis for NKG2A and LILRB1 expression on the surface of LILRB1- and NKG2A- NK-92MI cells. (F) LILRB1- NK-92MI cell-mediated lysis of master and B-E+G+ EMSCs (naïve). N = 3. (G) LILRB1- NK-92MI cell-mediated lysis of master and B-E+G+ EMSCs (IFN-γ-primed). N = 3, ***P < 0.001, ****P < 0.0001 for B-E+G+ versus master EMSCs per two-way ANOVA followed by Sidak's multiple comparison test. (H) NKG2A- NK-92MI cell-mediated lysis of master and B-E+G+ EMSCs (naïve). N = 3. (I) NKG2A- NK-92MI cell-mediated lysis of master and B-E+G+ EMSCs (IFN-γ-primed). N = 3.
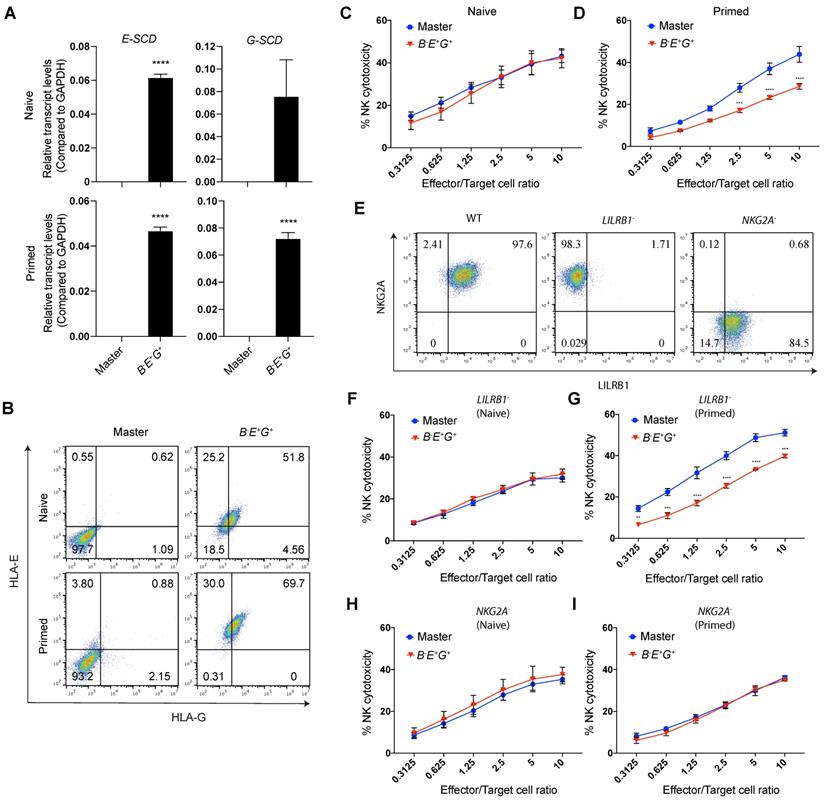
HLA-E and -G can inhibit NK cytotoxicity by binding to two major inhibitory receptors NKG2A/CD94 and LILRB1, respectively, on NK cells [11, 12]. Thus, we tested whether B-E+G+ EMSCs can escape NK killing by co-culturing them with NK-92MI cells. Unexpectedly, ectopic HLA-E and -G couldn't protect the cells from the NK cytotoxicity (Fig. 3C) unless they were primed with IFN-γ, in which around 20-50% NK inhibition was observed compared to the control master EMSCs (Fig. 3D). This result suggests that the site-specific expression of a single copy of HLA-E and -G is not sufficient to reduce the NK sensitivity of unprimed B2M-/- EMSCs, whereas IFN-γ priming assists the ectopic HLA-E or -G in executing the NK-inhibitory effect.
To test the contribution of HLA-E and -G in B-E+G+ EMSCs to the NK inhibition, we generated LILRB1- and NKG2A- subtypes from NK-92MI cells using CRISPR/Cas9 (Figs. S5A) and confirmed the knockouts via flow cytometry (Fig. 3E). To examine the functionality of LILRB1- and NKG2A- NK-92MI cells, we tested their cytotoxicity against K562 cells negative for both HLA-E and -G [29]. Compared to the WT control, LILRB1- and NKG2A- NK-92MI cells showed similar cytotoxicity against K562 cells (Fig. S5B), suggesting that knockout of LILRB1 or NKG2A didn't compromise the general cytotoxic activity of the NK cell line. Interestingly, IFN-γ-induced NK inhibition only happened in B-E+G+ EMSCs incubated with LILRB1- but not NKG2A- NK-92MI cells (Figs. 3F-I), suggesting the protective role of HLA-E in B-E+G+ EMSCs after IFN-γ priming. Moreover, expression of genes involved in other NK-inhibitory mechanisms independent of B2M or HLA-Ia, including PTGS2, IDO1, TGFB1, CD274, CD47, and PVR, was similar between the master and B-E+G+ EMSCs no matter whether primed with IFN-γ or not, although many of the genes were upregulated by IFN-γ priming in both master and B-E+G+ EMSCs (Fig. S4C). This result implicates that the genetic manipulations didn't affect the expression of the irrelevant NK-regulatory genes.
Lentivirus-transduced HLA-E and -G render dose-dependent NK resistance to B2M-/- EMSCs even without priming
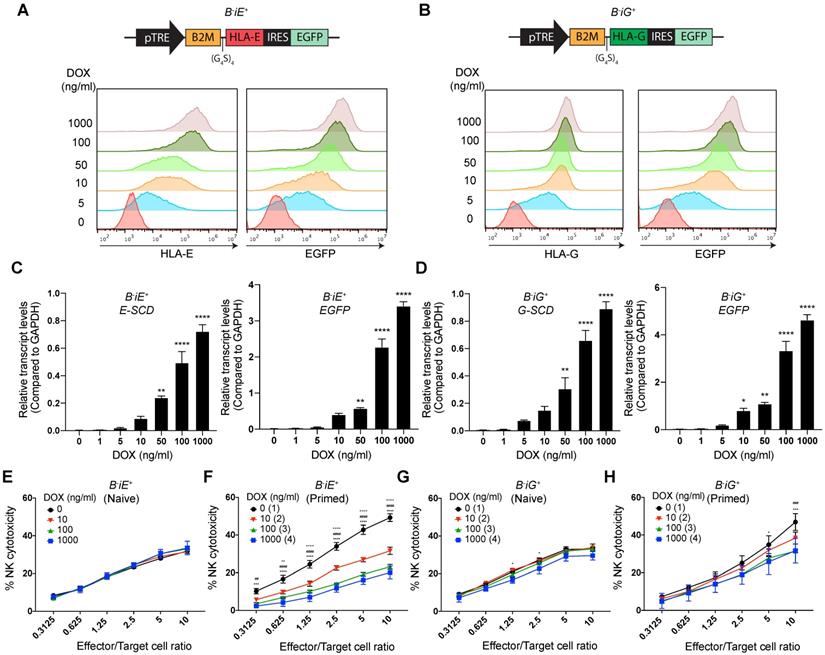
Based on the results above, we wondered whether increased expression of HLA-E and -G can render such protection or not. To achieve gradient expression of both molecules, we employed a Tet-on based lentiviral vector to co-express enhanced green fluorescent protein (EGFP) and an E-SCD or G-SCD in the B2M-/- EMSCs under the control of DOX, named B-iE+ and B-iG+ EMSCs, respectively (Figs. 4A and B). Flow cytometry and qPCR assays demonstrate DOX-induced expression of HLA-E or E-SCD, HLA-G or G-SCD, and EGFP in B-iE+ and B-iG+ EMSCs were in a dose-dependent manner (Figs. 4A-D). Surprisingly, inhibited NK cytotoxicity was not observed in naïve B-iE+ and B-iG+ EMSCs (Figs. 4E and G). Only after IFN-γ priming, the NK-inhibitory effects of the ectopic HLA-E and -G showed a clear parallel relationship with the DOX dose (Figs. 4F and H).
HLA-E or -G inducibly expressed in B2M-/- EMSCs resist NK cytotoxicity dose-dependently. (A) Schematic graph of the construct for inducible E-SCD expression and flow cytometry analysis for HLA-E and EGFP expression in B-iE+ EMSCs treated with DOX. (B) Schematic graph of the construct for inducible G-SCD expression and flow cytometry analysis for HLA-G and EGFP expression in B-iG+ EMSCs treated with DOX. (C) qPCR analysis of E-SCD and EGFP transcripts in B-iE+ EMSCs under treatment with various concentrations of DOX. N = 3. **P < 0.01 and ****P < 0.0001 for DOX-treated versus vehicle-treated B-iE+ EMSCs per ordinary one-way ANOVA followed by Dunnette's multiple comparison test. (D) qPCR analysis of G-SCD and EGFP transcripts in B-iG+ EMSCs under treatment with DOX. N = 3. *P < 0.05, **P < 0.01, and ****P < 0.0001 for DOX-treated versus vehicle-treated B-iG+ EMSCs per ordinary one-way ANOVA followed by Dunnette's multiple comparison test. (E) NK-92MI cell-mediated lysis of B-iE+ EMSCs (naïve) treated with DOX. N = 3. (F) NK-92MI cell-mediated lysis of B-iE+ EMSCs (IFN-γ-primed) treated with DOX. N = 3. ++P < 0.01 and ++++P < 0.0001 for (2) versus (1); ##P < 0.01 and ####P < 0.0001 for (3) versus (1); ***P < 0.001 and ****P < 0.0001 for (4) versus (1) per two-way ANOVA followed by Dunnett's multiple comparison test. (G) NK-92MI cell-mediated lysis of B-iG+ EMSCs (naïve) treated with DOX. N = 3. *P < 0.05 for (4) versus (1) per two-way ANOVA followed by Dunnett's multiple comparison test. (H) NK-92MI cell-mediated lysis of B-iG+ EMSCs (IFN-γ-primed) treated with DOX. N = 3. ###P < 0.001 for (3) versus (1); *P < 0.05 and ***P < 0.001 for (4) versus (1) per two-way ANOVA followed by Dunnett's multiple comparison test.
To achieve constitutive high-level expression of HLA-E and -G, we transduced B2M-/- EMSCs with a lentiviral vector containing a cassette for co-expression of a puromycin-resistant gene PuroR with luciferase (Luc) (as a control), E-SCD or G-SCD (Fig. 5A), resulting in lenti-B-L+, -B-E+, -B-G+, and -B-E+G+ EMSCs, respectively, as confirmed via qPCR (Fig. 5B). The expression of E-SCD in the lenti-B-E+ and lenti-B-E+G+ EMSCs was 10-fold, and the expression of G-SCD in the lenti-B-G+ and lenti-B-E+G+ EMSCs was 2.5-fold, of that in the B-E+G+ EMSCs generated via RMCE (Fig. 5C). The cell surface presence of HLA-E and -G was also confirmed using flow cytometry, which was further enhanced after IFN-γ priming (Fig. 5D).
EMSC ectopically expressing both HLA-E and -G resist NK cytotoxicity more than each alone. (A) Schematic graphs of lentiviral transfer vectors overexpressing luciferase, E-SCD, and G-SCD, respectively. (B) qPCR analysis of E-SCD and G-SCD transcripts in lenti-B-L+, -B-E+, -B-G+, and -B-E+G+ EMSCs. N = 3. ****P<0.0001 for (2) versus (1); $$$$P < 0.0001 for (3) versus (1); ####P < 0.0001 for (4) versus (1) per ordinary one-way ANOVA followed by Dunnette's multiple comparison test. (C) qPCR analysis of relative transcript levels of E-SCD or G-SCD in lenti-B-E+, -B-G+, and -B-E+G+ EMSCs compared to B-E+G+ EMSCs. N = 3. ****P < 0.0001 for (2) versus (1); $P < 0.05 and $$$$P < 0.0001 for (3) versus (1); #P < 0.05 and ####P < 0.0001 for (4) versus (1) per ordinary one-way ANOVA followed by Dunnette's multiple comparison test. (D) Flow cytometry analysis for HLA-E and -G expression on the surface of lenti-B-L+, -B-E+, -B-G+, and -B-E+G+ EMSCs. (E) NK-92MI cell-mediated lysis of lenti-B-L+, -B-E+, -B-G+, and -B-E+G+ EMSCs (naïve). N = 3, ***P < 0.001 and ****P < 0.0001 for (3) versus (1); ###P < 0.001 and ####P < 0.0001 for (4) versus (1) per two-way ANOVA followed by Dunnett's multiple comparison test. (F) NK-92MI cell-mediated lysis of lenti-B-L+, -B-E+, -B-G+, and -B-E+G+ EMSCs (IFN-γ-primed). N = 3, ++++P < 0.0001 for (2) versus (1); ***P < 0.001, and ****P < 0.0001 for (3) versus (1); ####P < 0.0001 for (4) versus (1) per two-way ANOVA followed by Dunnett's multiple comparison test. (G) LILRB1- NK-92MI cell-mediated lysis of lenti-B-L+, -B-E+, -B-G+, and -B-E+G+ EMSCs (naïve). N = 3. (H) LILRB1- NK-92MI cell-mediated lysis of lenti-B-L+, -B-E+, -B-G+, and -B-E+G+ EMSCs (IFN-γ-primed). N = 3, +P < 0.05, +++P < 0.001, and ++++P < 0.0001 for (2) versus (1); *P < 0.05 for (3) versus (1); ##P < 0.01, ###P < 0.001, and ####P < 0.0001 for (4) versus (1) per two-way ANOVA followed by Dunnett's multiple comparison test. (I) NKG2A- NK-92MI cell-mediated lysis of lenti-B-L+, -B-E+, -B-G+, and -B-E+G+ EMSCs (naïve). N = 3, *P < 0.05, **P < 0.01, and ***P < 0.001 for (3) versus (1); ##P < 0.01, ###P < 0.001 for (4) versus (1) per two-way ANOVA followed by Dunnett's multiple comparison test. (J) NKG2A- NK-92MI cell-mediated lysis of lenti-B-L+, -B-E+, -B-G+, and -B-E+G+ EMSCs (IFN-γ-primed). N = 3, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 for (3) versus (1); ###P < 0.001 and ####P < 0.0001 for (4) versus (1) per two-way ANOVA followed by Dunnett's multiple comparison test.

To test the inhibitory effect of EMSCs on diverse NK subtypes, we used WT, LILRB1-, and NKG2A- NK-92MI cells to represent the three subtypes of primary NK cells: NKG2A+/LILRB1+, NKG2A+/LILRB1-, and NKG2A-/LILRB1+, respectively. The inhibitory effect on WT NK-92MI cells was observed the highest in lenti-B-E+G+, intermediate with lenti-B-G+, and the lowest with lenti-B-E+, compared to lenti-B-L+ EMSCs control (Fig. 5E). Again, IFN-γ priming enhanced the inhibitory effect of lenti-B-E+ and lenti-B-G+, while lenti-B-E+G+ EMSCs still being the highest in NK inhibition (Fig. 5F). Consistently, lenti-B-G+ and lenti-B-E+ EMSCs lost resistance to LILRB1- and NKG2A- NK-92MI cells, respectively, and only lenti-B-E+G+ EMSCs remained resistant to both NK subtypes especially following IFN-γ priming (Figs. 5G-J).
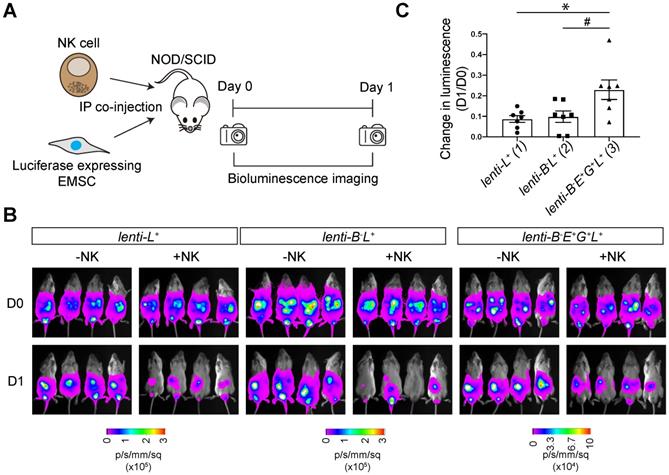
To track transplanted cells in vivo, we transduced WT and lenti-B-E+G+ EMSCs with a lentiviral vector containing a cassette for EF1α promoter-driven co-expression of NeoR linked with luc, resulting in lenti-L+ and lenti-B-E+G+L+ EMSCs, respectively. We tested NK resistance of the EMSCs in vivo by co-injecting NOD/SCID mice intraperitoneally (i.p.) with NK-92MI cells and lenti-L+, lenti-B-L+, or lenti-B-E+G+L+ EMSCs (Fig. 6A). The bioluminescent signal in groups without NK-92MI cell co-injection was retained one day after the injection (Fig. 6B). Co-injection of NK-92MI cells reduced the bioluminescent signal, which was more obvious in mice injected with lenti-L+ and lenti-B-L+ EMSCs than in mice treated with lenti-B-E+G+L+ EMSCs (Figs. 6B and C), suggesting that lenti-B-E+G+L+ EMSCs resist NK cytotoxicity more than lenti-L+ and lenti-B-L+ EMSCs. Overall, these experiments demonstrate that a high-level of HLA-E and -G expression reduces NK cytotoxicity on B2M-/- EMSCs in vivo.
HLA-E and -G protect B2M-/- EMSCs from NK cell-mediated lysis in vivo. (A) Schematic graph of NK cell-mediated lysis of EMSCs in vivo. (B) In vivo luminescence changes after co-injection of NK-92MI cells with lenti-L+, lenti-B-L+, and lenti-B-E+G+L+ EMSCs, N = 7. (C) Bioluminescence changes between day 0 (prior to NK-92MI cell injection) and day 1 after injection of NK-92MI cells with lenti-L+, lenti-B-L+, and lenti-B-E+G+L+ EMSCs, N = 7. *P < 0.05 for (3) versus (1); #P < 0.05 for (3) versus (2) per one-way ANOVA followed by Sidak's multiple comparison test.
Quality control of genetically manipulated EMSCs
First, we conducted Sanger sequencing of top 6 potential off-target sites of B2M sgRNAs in lenti-L+ and lenti-B-E+G+ EMSCs and found no mutation in any of the sites (Figs. S6A and B). Next, since genome editing often affects the expression of TP53, a tumor suppressor gene critical for maintaining genetic stability [30, 31], we tested its expression and found that both total and phosphorylated p53 levels were similar between B2M-/- and WT hESCs per western blotting following X-ray irradiation (Fig. S7), suggesting that B2M knockout didn't compromise TP53 expression and function. Interestingly, total and phosphorylated p53 were absent in B2M-/- and WT EMSCs even after X-ray irradiation (Fig. S7), implying that TP53 expression is repressed at this developmental stage.
To examine whether B2M knockout or lentiviral expression of HLA-E and -G affects the paracrine functions of EMSCs, we checked the cytokine profiles of lenti-L+, lenti-B-L+, and lenti-B-E+G+ EMSCs. Surprisingly, we found that secretion of interleukin (IL)-6 and -8 reduced dramatically in lenti-B-L+ EMSCs compared to lenti-L+ EMSCs but recovered in lenti-B-E+G+ EMSCs (Figs. S8A and B). This result indicates that B2M might assist the production of these cytokines in EMSCs through a not-yet-known mechanism, whereas overexpression of the B2M-fused HLA-E and -G can rescue the B2M knockout effect on the paracrine activity.
It has been reported that ectopic expression of either HLA-E or -G in tumor and virus-infected cells can evoke cytotoxic T lymphocyte (CTL) response [32, 33]. To address this concern, we performed CTL assays and found that lenti-B-E+G+ EMSCs didn't trigger the proliferation and activation of CD8+ T cells under both naïve and primed states compared to lenti-L+ and lenti-B-L+ EMSCs (Figs. S9A-D). These results suggest that ectopic expression of HLA-E and -G in EMSCs does not evoke CTL response, probably due to the immunosuppressive property of MSCs via secretion of PD-L1, PD-L2, CTLA-4, PGE2, IDO, and TGFβ1 to counteract T cell responses [34-39].
Discussion
Although MSCs possess potent immunosuppressive capability by regulating both innate and adaptive immunity through cell-cell contact and secretion of soluble factors, they are not immune privileged as they, like other cell types, also elicit a humoral and cellular immune response in vivo. Following systemic infusion of human MSCs in immunocompromised mice, mouse MSCs in syngeneic mice, and rat MSCs in allogeneic rats, the majority of the cells die within 48 h after the transplantation [40]. HLAs are the major factor to induce the T cell response [41], and depleting HLAs negates the T cell immunity but activates NK immunity [42]. In this study, we found that EMSCs depleted of B2M became more sensitive to NK lysis than the WT control, and priming EMSCs with IFN-γ exacerbates the sensitivity. Site-specific knock-in of HLA-E and -G only showed a limited protective effect against NK cytotoxicity, whereas lentiviral transduction of HLA-E and -G dose-dependently reduced the NK sensitivity of B2M-/- EMSCs.
It has been reported that B2M-/- MSCs can escape the CTL-mediated killing in vitro [6-8]. Moreover, B2M-/- allogeneic MSCs showed better therapeutic effects than WT MSCs on myocardial infarction and hindlimb ischemia partly due to escaping from CD8+ T cell-mediated immune rejection in vivo [7, 8]. Nonetheless, B2M-/- MSCs exhibited increased NK sensitivity [6], and small interfering RNA (siRNA) against HLA class I also increased the susceptibility of MSCs to autologous NK cells [3]. These results are consistent with our findings and can be explained by the “missing-self” effect of the HLA absence on the cell surface [42].
It has been known that IFN-γ treatment of MSCs inhibits the NK cytotoxicity against MSCs by increasing the expression of HLA class I and soluble factors, e.g., PGE2 and IDO [3, 22]. Unexpectedly, our results showed that IFN-γ treatment increased NK cytotoxicity to B2M-/- EMSCs, which might be explained by the increased expression of NK-activating genes such as ICAM1, PVR, and Nectin 2 in MSCs after IFN-γ treatment [22] and the absence of the NK-inhibitory ligands HLA class I on the cell surface.
Both HLA-E and -G can protect allogeneic [14, 15] and xenogeneic cells [43-45] from NK cytotoxicity, respectively. Thus, Russel and coworkers reported that HLA-E knock-in protects B2M-/- hESC-derived CD45+ hematopoietic cells from primary NK-mediated lysis [14]. However, we only observed such protection in IFN-γ-primed but not naïve B2M-/- EMSCs with site-specific insertion of HLA-E (Fig. 3). This result might result from differences between the present and previous studies in terms of the promoters (the CAG promoter versus the endogenous B2M promoter) used to drive HLA-E expression, the effectors (the NK-92MI cell line versus primary NK cells), and target cells (EMSCs versus hematopoietic cells). HLA-G knock-in into the B2M locus has also been reported to inhibit NK-92 cell-mediated cytotoxicity on cardiomyocytes differentiated from B2M-/- hESCs [16], which, however, failed to occur with B2M-/- EMSCs (Fig. 3). This might also be caused by reasons similar to the above, except that similar effector cells were used in both studies.
The major inhibitory receptors on NK cells for HLA-E and -G are NKG2A/CD94 and LILRB1 [11, 12], respectively. NKG2A- and LILRB1- NK subpopulations vary in different individuals [17]. We found that B2M-/- EMSCs with co-expression of HLA-E and -G showed robust resistance to NKG2A- and LILRB1- NK cells, while HLA-E and -G expression alone cannot provide resistance to NKG2A- and LILRB1- NK cells, respectively. This finding is consistent with a previous report that HLA-E provides limited protection to B2M-/- CD45+ cells against NK cells with low NKG2A expression [14]. We have further clarified that HLA-G alone is insufficient for fully overcoming NK cytotoxicity, which is different from a previous study [15]. Consistently, co-expression of HLA-E and -G protected B2M-/- EMSCs in vivo against WT NK-92MI cells representing NKG2A+/LILRB1+ NK cells. Thus, co-expression of HLA-E and -G renders B2M-/- EMSCs resistant to broader NK subtypes, including NKG2A+/LILRB1+, NKG2A+/LILRB1-, and NKG2A-/LILRB1+, than expression of HLA-E or -G each alone. In addition, EMSCs express high levels of PD-L1 [46] and CD47 (Fig. S4C), which may make EMSCs also resistant to PD-1+/SIRPα+ NK cells [47, 48] and macrophages [49, 50], respectively.
Interestingly, naïve EMSCs with high-level expression of HLA-E presented no inhibitory effects on NK cells. In contrast, IFN-γ-primed EMSCs with a relatively low-level of HLA-E expression revealed significant inhibition against NK cell-mediated lysis. Peptides loaded by HLA-E have dramatic effects on the interaction of HLA-E with corresponding receptors. For example, HLA-E loaded with HLA-A2 peptide has more surface expression than HLA-E loaded with HLA-B7 peptide [27]. HLA-E loaded with HLA-G peptide has higher affinity for CD94/NKG2A than HLA-E loaded with peptides from HLA-Ia [51]. However, HLA-E loaded with heat shock protein 60 peptide loses the capability of interacting with NKG2A/CD94 and cannot inhibit NKG2A+ NK cells [51]. Based on these reports, IFN-γ-primed EMSCs might generate more high-affinity peptides loaded by HLA-E than naïve EMSCs.
It is worth noting that MSCs as well as other therapeutic cell types also encounter clearance in the host by the instant blood-mediated inflammatory reaction including coagulation, complement reaction, and phagocyte activation [52] and the lung or precapillary entrapment following systemic infusion [53-56]. Furthermore, although HLA class II antigens are not expressed in naïve MSCs, they can be expressed in MSCs primed with cytokines like IFN-γ, undergoing hypoxia or following differentiation [57-61]. Consistently, HLA class II molecules were detected on the surface of IFN-γ-primed, but not naïve, EMSCs, which were not affected by co-culture with NK cells. Interestingly, both lenti-B-L+ and lenti-B-E+G+ EMSCs had much higher expression of HLA class II on their surface than lenti-L+ EMSCs (Figs. S10A and B). These results suggest that HLA class II expression might be regulated by multiple factors including B2M and upregulated following B2M knockout, which cannot be reversed by HLA-E and -G overexpression. Thus, it is also necessary to block the HLA class II expression in B2M-/- cells to generate fully immunoresistant hPSCs by knocking out genes such as CIITA, RFXANK, or HLA class II directly [62-64].
On the other hand, it is critical to evaluate other biological activities of B2M, CIITA, etc., and their loss of functions in targeted EMSCs and their progeny. Genetic manipulations are a double-edged sword as they can create novel biological functions of a cell and also damage the genomic integrity of the cell. We didn't detect any mutation in the top 6 potential off-target sites of B2M sgRNAs in lenti-B-E+G+ EMSCs. Nevertheless, for future therapeutic application, whole-genome sequencing is required to identify any mutated genes and reduce the risk of tumorigenesis if an oncogene is triggered or a tumor-suppressor gene is disabled. To safeguard the use of hPSC progeny, a suicide gene such as iCaspase-9 is installed in the genome for quick clearance of targeted cells once they are becoming malignant or infected [65]. Nevertheless, the ideal cells for therapy are those without any genomic manipulation.
In conclusion, this study demonstrates that ectopic expression of HLA-E and -G in B2M-/- EMSCs reverses enhanced NK sensitivity of the cells in a synergistic and dose-dependent manner. In addition, we suggest that dose-dependence should be considered when using HLA-E and -G to resist NK cytotoxicity. Moreover, IFN-γ priming increases, instead of, decreases NK sensitivity of B2M-/- EMSCs, whereas HLA-E and -G have more substantial protective effects on IFN-γ-primed B2M-/- EMSCs than naïve control cells, which may help the cells survive better in inflammatory diseases. The generation of NK-resistant MSCs from B2M-/- hESCs shall greatly enhance their therapeutic efficacy via increased survival from both innate and adaptive immunity. More studies are warranted to evaluate the efficacy and biosafety of the cells in conditions with high diversity of NK cell populations in patients.
Methods
Cell culture
The experiments carried out in this project were in accordance with the National Institutes of Health Guidelines on Human Stem Cell Research. The protocols were approved by the University of Macau Panel of Research Ethics. H1 hESCs (Wicell, WA01) were maintained in monolayer on Matrigel (Corning, 354230) in E8 medium (Thermo, A1517001), 293T cells (ATCC, CRL-3216) in high glucose DMEM medium (Thermo, 11965092) with 10% FBS (Thermo, 16000044), K562 cells (ATCC, CCL-243) in RPMI1640 medium (Thermo, 11875093) with 10% FBS, NK-92MI cells (ATCC, CRL-2408) in αMEM (Thermo, 32561037) with inositol (Sigma, I5125, 0.2 mM), folic acid (Sigma, F8758, 0.02 mM), 2-mercaptoethanol (Thermo, 21985023, 0.1 mM), 12.5% horse serum (Thermo, 26050088) and 12.5% FBS, and MSCs in αMEM (Thermo, 12571063) with 20% FBS.
MSC differentiation
MSC differentiation was performed as previously described [19, 20]. In brief, hESCs were dissociated with TrypLE (Thermo, 12605010) and plated in the density of 90,000 cells/cm2 in E8 medium supplemented with ROCK inhibitor Y27632 (Stemgent, 04-0012, 10 μM) for 24 h. Cells were treated with Essential 6 (E6) medium (Thermo, A1516401) supplemented with sodium heparin (Sigma, H3149, 22.5 ng/ml), bFGF (Thermo, PHG0261, 10 ng/ml), CHIR99021 (Selleck, S1263, 1 μM), SB431542 (Stemgent, 04-0010, 10 μM) and dorsomorphin (R&D, 3093, 1 μM). Cells were split at a 1:6 ratio when they became confluent. Fifteen days later, cells were cultured in MSC medium for continued differentiation to MSCs for 5 more days. Following routine characterization for MSC markers and tri-lineage differentiation [66], the resulting cells were designated as EMSCs at passage 0 (p0). EMSCs within 10 passages were used in this study.
Plasmid construction
Lentiviral transfer vectors for single-chain dimers of HLA-E (E-SCD) and HLA-G (G-SCD)
Firstly, B2M-linker was amplified from cDNA of H1 hESCs using primers B2M-cDNA-F/R and B2M-G4S-F/R carrying a (G4S)4 linker and Esp3I ends; HLA-E fragments were amplified from lv236-HLA-E (Genecopoeia) using primers HLA-E1-F/R and HLA-E2-F/R carrying Esp3I ends, and lv236-destination vector was constructed by inserting Esp3I cloning sites (annealed from Esp3I-anneal1-F/R) into KpnI- and NotI-digested lv236-mCherry (Genecopoeia). Then, B2M-linker, HLA-E fragments, and lv236-destination vector were assembled through Golden Gate Cloning [67]. Similarly, HLA-G fragment with Esp3I ends was amplified from lv236-HLA-G1 using primers HLA-G-F/R. Then, B2M-linker, HLA-G fragment, and lv236-destination vector were assembled as above. The resultant lv236-E-SCD and lv236-G-SCD were verified using Sanger sequencing. Q5 DNA polymerase for PCR and restriction enzymes for DNA digestions were all from NEB for plasmid constructions.
RMCE donor vector
The left and right homologous arms (5' HA and 3' HA, respectively) amplified from Puro-Cas9 donor plasmid (Addgene # 58409, a gift of Danwei Huangfu) [68] were inserted into KpnI- or EcoRV-digested plasmid PGKdtabpA (Addgene #13440, a gift of Philippe Soriano) [69] to obtain PGK-HA-DTA-pA. In addition, the lox71-PGK-puΔTK-lox2272 fragment was digested from pLCA.66/2272 (Addgene #22733, a gift of Mark Magnuson) [70] and inserted into ClaI- and BstBI-digested PGK-HA-DTA-pA plasmid. The RMCE donor vector AAVS1-lox71-PGK-puΔTK-lox2272 was verified using Sanger sequencing.
E-SCD and G-SCD RMCE exchange vector
The E-SCD-P2A fragment with Esp3I ends was amplified from RMCE-E-SCD-P2A-NeoR (constructed elsewhere), the G-SCD fragment with Esp3I ends was amplified from the lv236-G-SCD, and the P2A-NeoR fragment with Esp3I ends was amplified from the pCDH-iC9-P2A-NeoR plasmid [65]. The RMCE destination vector was constructed by inserting Esp3I sites into PacI- and BsrGI-digested RMCE-mCherry vector (constructed elsewhere). Then, the E-SCD-P2A, G-SCD, and P2A-NeoR fragments were assembled with the RMCE destination through Golden Gate Cloning. The RMCE exchange vector RMCE-E-SCD-G-SCD-NeoR was verified using Sanger sequencing.
Lentiviral transfer vectors for luciferase
The luciferase gene was amplified from the pGL3-control plasmid (Promega) and inserted into NotI- and EcoRI-digested pCDH-CAG-MSC-T2A-PuroR (Clontech) or AgeI- and BamHI-digested lenti-SpCas9 neo (Addgene #104996, a gift of Brett Stringer) [71] through Gibson assembly. The resultant pCDH-CAG-Luc-T2A-PuroR and plenti-EF1-Luc-P2A-NeoR were verified using Sanger sequencing.
Lentiviral transfer vector for inducible expression of E-SCD and G-SCD
E-SCD and G-SCD amplified from the lv236-E-SCD and lv236-G-SCD, respectively, were inserted into BamHI- and NdeI-digested pTight-TYR-EGFP plasmid (constructed elsewhere) through Gibson assembly. The resultant pTight-E-SCD-IRES-EGFP and pTight-G-SCD-IRES-EGFP were verified using Sanger sequencing.
Lentiviral packaging and transduction
293T cells were transfected with lentiviral transfer vectors, pCMVR8.74 (Addgene #22036, a gift of Didier Trono), and pMD2.G (Addgene #12259, a gift of Didier Trono) at a ratio of 3:2:1 using the Lipofectamine 3000 transfection reagent (Thermo, L30000015). The supernatant containing lentiviral particles was collected 48 and 72 h after transfection. EMSCs were transduced with corresponding lentiviral particles supplemented with polybrene reagent (Sigma, TR1003, 10 μg/ml). 48 h later, cells were treated with the corresponding antibiotic puromycin (Sigma, P9620, 500 ng/ml) or G418 (Thermo, 10131027, 500 µg/ml) for two weeks to enrich transduced cells.
Genome editing
B2M knockout
The guide sequences were designed according to the B2M gene using the online CRISPR design tool (crispr.mit.edu) as follows CGCGAGCACAGCTAAGGCCA and ACTCTCTCTTTCTGGCCTGG. Corresponding annealed oligos B2M-sg1-F/R and B2M-sg2-F/R (Table S1) were cloned into BbsI-digested pSpCas9n(BB)-2A-GFP (PX461) and pSpCas9n(BB)-2A-Puro (PX462) V2.0 (Addgene: #48140 and #62987, gifts of Feng Zhang) [72] to generate B2M targeting plasmids PX461-sgB2M1 and PX462-sgB2M2.
HESCs were transfected with B2M targeting plasmids using the Lipofectamine 3000 reagent. GFP+ cells were enriched using FACSAria III (BD Biosciences). Single-cell clones were obtained using the Incucyte Live Cell Analysis Imaging System (Sartorius). Two individual clones were selected, referred to as H1 B2M-/- #1 and #2, respectively. For genotyping, genomic DNA was extracted from both H1 B2M-/- #1 and #2 hESCs using the TIANamp Genomic DNA Extraction kit (Tiangen, DP304). Amplicons were obtained using Q5 DNA polymerase, cloned into T vectors, and verified via Sanger sequencing.
RMCE master line
H1 B2M-/- #2 hESCs were transfected with AAVS1-TALEN-L and AAVS1-TALEN-R (Addgene, gifts of Danwei Huangfu) [68] and the RMCE donor vector AAVS1-lox71-PGK-puΔTK-lox2272 using the Lipofectamine 3000 reagent. After puromycin selection, single-cell clones were obtained using the Incucyte system leading to the establishment of a RMCE master line. Site-specific integration of puΔTK in master hESCs was confirmed through PCR. Random integration was excluded through PCR.
E-SCD and G-SCD exchange
The master hESCs were transfected with pCAG-Cre:GFP (Addgene #13776, a gift of Connie Cepko) [73] and the exchange vector RMCE-E-SCD-G-SCD-NeoR (1:1) using the Lipofectamine 3000 reagent. After selection with G418, single-cell clones were obtained using the Incucyte system. Site-specific exchange of E-SCD and G-SCD in B-E+G+ hESCs was confirmed through PCR. Random integration was excluded through PCR.
LILRB1 and NKG2A knockout
The guide sequences were designed according to the sequences of LILRB1 (gene id: 10859) and NKG2A (KLRC1, gene id: 3821) using the online CRISPR design tool as follows TGTACCACCACCTGCGACTC and AACAACTATCGTTACCACAG. Corresponding annealed oligos LILRB1-sg-F/R and NKG2A-sg-F/R (Table S1) were cloned into Esp3I-digested LentiCRISPRv2 (Addgene: #52961, a gift of Feng Zhang) [74]. NK-92MI cells were transduced with the corresponding lentiviral particles. Puromycin-selected cells and WT controls were stained with anti-LILRB1 and -NKG2A antibodies (Table S2). LILRB1- and NKG2A- NK-92MI cells were enriched using FACSAria III.
Teratoma assay
All animal procedures were conducted under the guidelines and animal use protocol (#UMARE-011-2019) approved by the University of Macau subpanel on animal research ethics. hESCs (~1 million) were suspended in 100 μl of cold Matrigel-DPBS (1:1) and injected subcutaneously into inguinal flanks of 4-6 weeks old NOD/SCID mice. Eight weeks after injection, mice were euthanized, and teratomas were dissected and analyzed using hematoxylin and eosin staining on the paraffin-embedded sections.
Karyotyping
Cells were treated with KaryoMAX colcemid (Thermo, 15212012), followed by 25% acetic acid and 75% methanol fixation. The fixed cells were sent to Global Medical Laboratory Center (Wuhan, China) for G-band karyotyping.
Western blotting
Cells were lysed in the radioimmunoprecipitation assay (RIPA) buffer (Thermo, 89900) supplemented with proteinase inhibitor (Thermo, 87786). 50 μg proteins per sample per lane were separated on a SDS-PAGE gel and then transferred to polyvinylidene difluoride (PVDF) membrane (Bio-Rad, 1620177). Membranes were blocked with 5% milk in phosphate-buffered saline-0.1% Tween 20 (Sigma, P7949) and then incubated with primary antibodies overnight at 4 °C, followed by corresponding secondary antibodies for 1 h at room temperature (Table S2). Detected signals were visualized with enhanced chemiluminescence and imaged using ChemiDoc Imager (Bio-Rad).
Flow cytometry
Cells were harvested using TrypLE to obtain single-cell suspensions. For cell surface staining, cells were incubated with primary antibodies for 30 min on ice, followed by corresponding secondary antibodies for another 30 min on ice. For intracellular staining, cells were fixed with 4% paraformaldehyde for 15 min and permeabilized with 90% ice-cold methanol for 1 h or overnight at -20 °C. Cells were then blocked with 3% bovine serum albumin (Beyotime, ST023) for 1 h at room temperature, followed by incubation with primary antibodies for 1 h on ice and corresponding secondary antibodies for another 1 h on ice. Data were collected using the Cytoflex cytometer (Beckman Coulter) and analyzed using the FlowJo software (Treestar). Antibody information was listed in Table S2.
Real-time qPCR
Total RNA was extracted using Trizol reagent (Thermo, 15596026) according to the manufacturer's instructions. Reverse transcription was performed using the PrimeScript RT Reagent Kit with gDNA Eraser (Takara, RR047A). qPCR was carried out using iTaq Universal SYBR Green Supermix (Bio-rad, 1725124) with gene-specific primers (Table S3) in CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Gene expression levels were normalized to that of GAPDH.
Trilineage differentiation
MSCs were differentiated into adipocytes, osteoblasts, and chondrocytes using the corresponding StemPro differentiation medium (Thermo, A1007001, A1007201, and A1007101). In brief, cells were seeded into 24-well plates and cultured in the corresponding differentiation medium. For adipogenesis, cells were fixed and stained with Oil Red O (Sigma, O0625) at 40 days after differentiation. For osteogenesis and chondrogenesis, cells were fixed and stained with Alizarin Red S (Sigma, A5533) and Alcian Blue (Sigma, 109-09), respectively, at 21 days after differentiation. Staining was visualized under a microscope (Axio observer, Carl Zeiss).
NK cytotoxicity assay in vitro
Ten thousand Calcein-AM (ebioscience, 65-0853-78, 5 µM)-labeled target cells (MSCs or K562 cells) were seeded in 100 μl per well in a flat-bottom 96-well plate. Effector cells (NK-92MI) were added in 100 μl at different effector to target cell ratios and incubated for 4 h at 37 °C. All groups were set up in triplicate. Target cells without NK cells were used for spontaneous release and target cells lysed with 1% Triton X-100 (Sigma, T8787) for maximum release. 100 μl supernatant was collected and measured using Victor X5 microplate reader (PerkinElmer) with excitation filter at 485 nm and emission filter at 535 nm. Percent lysis was calculated using the following formula:

NK cytotoxicity assay in vivo
NOD/SCID mice were injected i.p. with 2.5×105 luciferase-expressing EMSCs. Four hours after EMSCs injection, 2.5×106 NK-92MI cells were injected i.p. per mouse. Bioluminescence imaging was performed using In-Vivo Xtreme imaging system (Bruker) at days 0 and 1 for pre-NK-92MI and post-NK-92MI luciferase signals, respectively. The average change in the luminescence of NK-92MI non-injected mice was used as control.
Statistics
Statistical analysis was carried out with Prism 9 software (GraphPad). All data shown in the study are presented as mean ± S.E.M. Statistically significant results were considered when P < 0.05. Two-tailed, unpaired t tests, ordinary one-way ANOVA tests, and two-way ANOVA tests were used to analyze the data in this study.
Abbreviations
MSC: mesenchymal stem cell; HLA: human leukocyte antigen; HLA-Ia: classical HLA class I; iPSC: induced pluripotent stem cell; WT: wild-type; KIR: killer-cell immunoglobulin-like receptor; NK: natural killer; HLA-Ib: non-classical HLA class I; NKG2A: natural killer group 2, member A; LILRB1: leukocyte immunoglobulin-like receptor subfamily B member 1; hPSC: human pluripotent stem cell; ESC: embryonic stem cell; hESC: human embryonic stem cell; B2M: β-2-microglobulin; EMSC: hESC-derived MSC; DOX: doxycycline; CRISPR: clustered regularly interspaced short palindromic repeats; Cas9: CRISPR-associated protein 9; IFN-γ: interferon-gamma; RMCE: recombinase-mediated cassette exchange; PCR: polymerase chain reaction; E-SCD: HLA-E single-chain dimer; G-SCD: HLA-G single-chain dimer; qPCR: quantitative PCR; PTGS2: prostaglandin-endoperoxide synthase 2; IDO: indoleamine 2,3-dioxygenase; TGFβ1: transforming growth factor beta 1; PVR: poliovirus receptor; EGFP: enhanced green fluorescent protein; Luc: luciferase; i.p.: intraperitoneally; IL: interleukin; CTL: cytotoxic T lymphocyte; PD-L1/2: programmed death-ligand 1/2; CTLA-4: cytotoxic T-lymphocyte-associated protein 4; PGE2: prostaglandin E2; siRNA: small interfering RNA; ICAM1: intercellular adhesion molecule 1; PD-1: programmed cell death protein 1; SIRPα: signal-regulatory protein alpha; CIITA: MHC class II transactivator; RFXANK: regulatory factor x associated ankyrin containing protein.
Supplementary Material
Supplementary methods, figures and tables.
Acknowledgements
We thank all the core facilities in the Faculty of Health Sciences, especially the Animal Facility and the Bioimaging and Stem Cell Core, for their excellent services. This work was supported with the University of Macau Research Committee funds MYRG #2016-00070-FHS and #2017-00124-FHS, Macau Science and Technology Development Fund (FDCT) #095/2017/A1 and 0112-2018-A3, and FDCT-National Natural Science Foundation of China joint grant #0008-2019-AFJ to R.X. The authors thank the council members of the Macau Society for Stem Cell Research for inspiring discussion.
Author Contributions
D.Z. and R.X. conceived of and designed the research. D.Z., X.W., Z.Z., E.L., C.Y., R.B., and G.Q. performed the experiments. D.Z., Y.W., and R.X. analyzed all the data and wrote the manuscript. R.X. gave the final approval of the manuscript.
Competing Interests
R.X. is a founder of ImStem Biotechnology, Inc., a stem cell company. The other authors declare no competing financial interests.
References
1. Levy O, Kuai R, Siren EMJ, Bhere D, Milton Y, Nissar N. et al. Shattering barriers toward clinically meaningful MSC therapies. Sci Adv. 2020;6:eaba6884
2. Eliopoulos N, Stagg J, Lejeune L, Pommey S, Galipeau J. Allogeneic marrow stromal cells are immune rejected by MHC class I- and class II-mismatched recipient mice. Blood. 2005;106:4057-65
3. Crop MJ, Korevaar SS, de Kuiper R, Ijzermans JN, van Besouw NM, Baan CC. et al. Human mesenchymal stem cells are susceptible to lysis by CD8(+) T cells and NK cells. Cell transplantation. 2011;20:1547-59
4. Kotobuki N, Katsube Y, Katou Y, Tadokoro M, Hirose M, Ohgushi H. In vivo Survival and Osteogenic Differentiation of Allogeneic Rat Bone Marrow Mesenchymal Stem Cells (MSCs). Cell Transplantation. 2008;17:705-12
5. Le Blanc K, Tammik C, Rosendahl K, Zetterberg E, Ringden O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol. 2003;31:890-6
6. Zha S, Tay JC, Zhu S, Li Z, Du Z, Wang S. Generation of Mesenchymal Stromal Cells with Low Immunogenicity from Human PBMC-Derived beta2 Microglobulin Knockout Induced Pluripotent Stem Cells. Cell Transplant. 2020;29:963689720965529
7. Shao L, Zhang Y, Pan X, Liu B, Liang C, Zhang Y. et al. Knockout of beta-2 microglobulin enhances cardiac repair by modulating exosome imprinting and inhibiting stem cell-induced immune rejection. Cell Mol Life Sci. 2020;77:937-52
8. Zhang Y, Wang Y, Shao L, Pan X, Liang C, Liu B. et al. Knockout of beta-2 microglobulin reduces stem cell-induced immune rejection and enhances ischaemic hindlimb repair via exosome/miR-24/Bim pathway. J Cell Mol Med. 2020;24:695-710
9. Masuda K, Kawamoto H. Possible NK cell-mediated immune responses against iPSC-derived cells in allogeneic transplantation settings. Inflamm Regen. 2021;41:2
10. Kwon D, Ahn H-J, Han M-J, Ji M, Ahn J, Seo K-W. et al. Human Leukocyte Antigen Class I Pseudo-Homozygous Mesenchymal Stem Cells Derived from Human Induced Pluripotent Stem Cells. Stem cell reviews and reports. 2020;16:792-808
11. Lee N, Llano M, Carretero M, Ishitani A, Navarro F, Lopez-Botet M. et al. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci U S A. 1998;95:5199-204
12. Favier B, Lemaoult J, Lesport E, Carosella ED. ILT2/HLA-G interaction impairs NK-cell functions through the inhibition of the late but not the early events of the NK-cell activating synapse. FASEB J. 2010;24:689-99
13. Zheng D, Wang X, Xu RH. Concise Review: One Stone for Multiple Birds: Generating Universally Compatible Human Embryonic Stem Cells. Stem Cells. 2016;34:2269-75
14. Gornalusse GG, Hirata RK, Funk SE, Riolobos L, Lopes VS, Manske G. et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat Biotechnol. 2017;35:765-72
15. Han X, Wang M, Duan S, Franco PJ, Kenty JH, Hedrick P. et al. Generation of hypoimmunogenic human pluripotent stem cells. Proc Natl Acad Sci U S A. 2019;116:10441-6
16. Shi L, Li W, Liu Y, Chen Z, Hui Y, Hao P. et al. Generation of hypoimmunogenic human pluripotent stem cells via expression of membrane-bound and secreted beta2m-HLA-G fusion proteins. Stem Cells. 2020;38:1423-37
17. Smith SL, Kennedy PR, Stacey KB, Worboys JD, Yarwood A, Seo S. et al. Diversity of peripheral blood human NK cells identified by single-cell RNA sequencing. Blood Adv. 2020;4:1388-406
18. Yang J, Park JW, Zheng D, Xu RH. Universal Corneal Epithelial-Like Cells Derived from Human Embryonic Stem Cells for Cellularization of a Corneal Scaffold. Transl Vis Sci Technol. 2018;7:23
19. Stebbins MJ, Gastfriend BD, Canfield SG, Lee MS, Richards D, Faubion MG. et al. Human pluripotent stem cell-derived brain pericyte-like cells induce blood-brain barrier properties. Sci Adv. 2019;5:eaau7375
20. Menendez L, Kulik MJ, Page AT, Park SS, Lauderdale JD, Cunningham ML. et al. Directed differentiation of human pluripotent cells to neural crest stem cells. Nat Protoc. 2013;8:203-12
21. Neri S, Mariani E, Meneghetti A, Cattini L, Facchini A. Calcein-acetyoxymethyl cytotoxicity assay: standardization of a method allowing additional analyses on recovered effector cells and supernatants. Clin Diagn Lab Immunol. 2001;8:1131-5
22. Spaggiari GM, Capobianco A, Becchetti S, Mingari MC, Moretta L. Mesenchymal stem cell-natural killer cell interactions: evidence that activated NK cells are capable of killing MSCs, whereas MSCs can inhibit IL-2-induced NK-cell proliferation. Blood. 2006;107:1484-90
23. Ordovas L, Boon R, Pistoni M, Chen Y, Wolfs E, Guo W. et al. Efficient Recombinase-Mediated Cassette Exchange in hPSCs to Study the Hepatocyte Lineage Reveals AAVS1 Locus-Mediated Transgene Inhibition. Stem Cell Reports. 2015;5:918-31
24. Lopez de Castro JA, Barbosa JA, Krangel MS, Biro PA, Strominger JL. Structural analysis of the functional sites of class I HLA antigens. Immunol Rev. 1985;85:149-68
25. Hansen T, Yu YY, Fremont DH. Preparation of stable single-chain trimers engineered with peptide, beta2 microglobulin, and MHC heavy chain. Curr Protoc Immunol. 2009 Chapter 17: Unit17 5
26. Liu Z, Chen O, Wall JBJ, Zheng M, Zhou Y, Wang L. et al. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci Rep. 2017;7:2193
27. Forte P, Baumann BC, Weiss EH, Seebach JD. HLA-E expression on porcine cells: protection from human NK cytotoxicity depends on peptide loading. Am J Transplant. 2005;5:2085-93
28. Diehl M, Munz C, Keilholz W, Stevanovic S, Holmes N, Loke YW. et al. Nonclassical HLA-G molecules are classical peptide presenters. Curr Biol. 1996;6:305-14
29. Hirano N, Takahashi T, Takahashi T, Ohtake S, Hirashima K, Emi N. et al. Expression of costimulatory molecules in human leukemias. Leukemia. 1996;10:1168-76
30. Brady CA, Attardi LD. p53 at a glance. J Cell Sci. 2010;123:2527-32
31. Enache OM, Rendo V, Abdusamad M, Lam D, Davison D, Pal S. et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat Genet. 2020;52:662-8
32. Lenfant F, Pizzato N, Liang S, Davrinche C, Le Bouteiller P, Horuzsko A. Induction of HLA-G-restricted human cytomegalovirus pp65 (UL83)-specific cytotoxic T lymphocytes in HLA-G transgenic mice. J Gen Virol. 2003;84:307-17
33. Joosten SA, Sullivan LC, Ottenhoff TH. Characteristics of HLA-E Restricted T-Cell Responses and Their Role in Infectious Diseases. J Immunol Res. 2016;2016:2695396
34. Davies LC, Heldring N, Kadri N, Le Blanc K. Mesenchymal Stromal Cell Secretion of Programmed Death-1 Ligands Regulates T Cell Mediated Immunosuppression. Stem Cells. 2017;35:766-76
35. Gaber T, Schonbeck K, Hoff H, Tran CL, Strehl C, Lang A. et al. CTLA-4 Mediates Inhibitory Function of Mesenchymal Stem/Stromal Cells. Int J Mol Sci. 2018;19:2312
36. Fu QL, Chow YY, Sun SJ, Zeng QX, Li HB, Shi JB. et al. Mesenchymal stem cells derived from human induced pluripotent stem cells modulate T-cell phenotypes in allergic rhinitis. Allergy. 2012;67:1215-22
37. Wang D, Feng X, Lu L, Konkel JE, Zhang H, Chen Z. et al. A CD8 T cell/indoleamine 2,3-dioxygenase axis is required for mesenchymal stem cell suppression of human systemic lupus erythematosus. Arthritis Rheumatol. 2014;66:2234-45
38. Böttcher M, Hofmann AD, Bruns H, Haibach M, Loschinski R, Saul D. et al. Mesenchymal Stromal Cells Disrupt mTOR-Signaling and Aerobic Glycolysis During T-Cell Activation. Stem Cells. 2016;34:516-21
39. Li M, Sun X, Kuang X, Liao Y, Li H, Luo D. Mesenchymal stem cells suppress CD8+ T cell-mediated activation by suppressing natural killer group 2, member D protein receptor expression and secretion of prostaglandin E2, indoleamine 2, 3-dioxygenase and transforming growth factor-beta. Clin Exp Immunol. 2014;178:516-24
40. Ankrum JA, Ong JF, Karp JM. Mesenchymal stem cells: immune evasive, not immune privileged. Nat Biotechnol. 2014;32:252-60
41. Ayala Garcia MA, Gonzalez Yebra B, Lopez Flores AL, Guani Guerra E. The major histocompatibility complex in transplantation. J Transplant. 2012;2012:842141
42. Karre K. Natural killer cell recognition of missing self. Nat Immunol. 2008;9:477-80
43. Yue Y, Xu W, Kan Y, Zhao HY, Zhou Y, Song X. et al. Extensive germline genome engineering in pigs. Nat Biomed Eng. 2021;5:134-43
44. Forte P, Pazmany L, Matter-Reissmann UB, Stussi G, Schneider MK, Seebach JD. HLA-G inhibits rolling adhesion of activated human NK cells on porcine endothelial cells. J Immunol. 2001;167:6002-8
45. Lilienfeld BG, Crew MD, Forte P, Baumann BC, Seebach JD. Transgenic expression of HLA-E single chain trimer protects porcine endothelial cells against human natural killer cell-mediated cytotoxicity. Xenotransplantation. 2007;14:126-34
46. Wang X, Lazorchak AS, Song L, Li E, Zhang Z, Jiang B. et al. Immune modulatory mesenchymal stem cells derived from human embryonic stem cells through a trophoblast-like stage. Stem Cells. 2016;34:380-91
47. Pesce S, Greppi M, Grossi F, Del Zotto G, Moretta L, Sivori S. et al. PD/1-PD-Ls Checkpoint: Insight on the Potential Role of NK Cells. Front Immunol. 2019;10:1242
48. Deuse T, Hu X, Agbor-Enoh S, Jang MK, Alawi M, Saygi C. et al. The SIRPalpha-CD47 immune checkpoint in NK cells. J Exp Med. 2021;218:e20200839
49. Kono Y, Saito H, Miyauchi W, Shimizu S, Murakami Y, Shishido Y. et al. Increased PD-1-positive macrophages in the tissue of gastric cancer are closely associated with poor prognosis in gastric cancer patients. BMC Cancer. 2020;20:175
50. Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS. et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A. 2012;109:6662-7
51. Michaelsson J, Teixeira de Matos C, Achour A, Lanier LL, Karre K, Soderstrom K. A signal peptide derived from hsp60 binds HLA-E and interferes with CD94/NKG2A recognition. J Exp Med. 2002;196:1403-14
52. Moll G, Rasmusson-Duprez I, von Bahr L, Connolly-Andersen AM, Elgue G, Funke L. et al. Are therapeutic human mesenchymal stromal cells compatible with human blood? Stem Cells. 2012;30:1565-74
53. Toma C, Wagner WR, Bowry S, Schwartz A, Villanueva F. Fate of culture-expanded mesenchymal stem cells in the microvasculature: in vivo observations of cell kinetics. Circ Res. 2009;104:398-402
54. Eggenhofer E, Benseler V, Kroemer A, Popp FC, Geissler EK, Schlitt HJ. et al. Mesenchymal stem cells are short-lived and do not migrate beyond the lungs after intravenous infusion. Front Immunol. 2012;3:297
55. Mao AS, Ozkale B, Shah NJ, Vining KH, Descombes T, Zhang L. et al. Programmable microencapsulation for enhanced mesenchymal stem cell persistence and immunomodulation. Proc Natl Acad Sci U S A. 2019;116:15392-7
56. Lee RH, Pulin AA, Seo MJ, Kota DJ, Ylostalo J, Larson BL. et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell. 2009;5:54-63
57. Schu S, Nosov M, O'Flynn L, Shaw G, Treacy O, Barry F. et al. Immunogenicity of allogeneic mesenchymal stem cells. J Cell Mol Med. 2012;16:2094-103
58. Abu-El-Rub E, Sareen N, Sequiera G, Ammar HI, Yan W, ShamsEldeen AM. et al. Hypoxia-induced increase in Sug1 leads to poor post-transplantation survival of allogeneic mesenchymal stem cells. The FASEB Journal. 2020;34:12860-76
59. Huang XP, Sun Z, Miyagi Y, McDonald Kinkaid H, Zhang L, Weisel RD. et al. Differentiation of allogeneic mesenchymal stem cells induces immunogenicity and limits their long-term benefits for myocardial repair. Circulation. 2010;122:2419-29
60. Dhingra S, Li P, Huang XP, Guo J, Wu J, Mihic A. et al. Preserving prostaglandin E2 level prevents rejection of implanted allogeneic mesenchymal stem cells and restores postinfarction ventricular function. Circulation. 2013;128:S69-78
61. Abu-El-Rub E, Sequiera GL, Sareen N, Yan W, Moudgil M, Sabbir MG. et al. Hypoxia-induced 26S proteasome dysfunction increases immunogenicity of mesenchymal stem cells. Cell Death Dis. 2019;10:90
62. Chen H, Li Y, Lin X, Cui D, Cui C, Li H. et al. Functional disruption of human leukocyte antigen II in human embryonic stem cell. Biol Res. 2015;48:59
63. Colunga A, Hirata R, Russell D. Generation of HLA Class II Deficient Human Embryonic Stem Cells By AAV Mediated Knockout of RFXANK. Molecular Therapy. 2014;22:S14-S
64. Crivello P, Ahci M, Maassen F, Wossidlo N, Arrieta-Bolanos E, Heinold A. et al. Multiple Knockout of Classical HLA Class II beta-Chains by CRISPR/Cas9 Genome Editing Driven by a Single Guide RNA. J Immunol. 2019;202:1895-903
65. Wang X, Jiang B, Sun H, Zheng D, Zhang Z, Yan L. et al. Noninvasive application of mesenchymal stem cell spheres derived from hESC accelerates wound healing in a CXCL12-CXCR4 axis-dependent manner. Theranostics. 2019;9:6112-28
66. Jiang B, Yan L, Wang X, Li E, Murphy K, Vaccaro K. et al. Concise Review: Mesenchymal Stem Cells Derived from Human Pluripotent Cells, an Unlimited and Quality-Controllable Source for Therapeutic Applications. Stem Cells. 2019;37:572-81
67. Engler C, Kandzia R, Marillonnet S. A one pot, one step, precision cloning method with high throughput capability. PLoS One. 2008;3:e3647
68. Gonzalez F, Zhu Z, Shi ZD, Lelli K, Verma N, Li QV. et al. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell Stem Cell. 2014;15:215-26
69. Soriano P. The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development. 1997;124:2691-700
70. Chen SX, Osipovich AB, Ustione A, Potter LA, Hipkens S, Gangula R. et al. Quantification of factors influencing fluorescent protein expression using RMCE to generate an allelic series in the ROSA26 locus in mice. Dis Model Mech. 2011;4:537-47
71. Stringer BW, Day BW, D'Souza RCJ, Jamieson PR, Ensbey KS, Bruce ZC. et al. A reference collection of patient-derived cell line and xenograft models of proneural, classical and mesenchymal glioblastoma. Sci Rep. 2019;9:4902
72. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281-308
73. Matsuda T, Cepko CL. Controlled expression of transgenes introduced by in vivo electroporation. Proc Natl Acad Sci U S A. 2007;104:1027-32
74. Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783-4
Author contact
![]() Corresponding author: Ren-He Xu, Faculty of Health Sciences, University of Macau, Taipa, Macau, China. E-mail: renhexuedu.mo.
Corresponding author: Ren-He Xu, Faculty of Health Sciences, University of Macau, Taipa, Macau, China. E-mail: renhexuedu.mo.