Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Infection of SARS-CoV-2
3. Mechanism of the cytokine...
4. Clinical outcome and...
5. Therapy of COVID-19 disease
6. Summary
Acknowledgements
References
Author Biography

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(2):459-472. doi:10.7150/ijbs.59272 This issue Cite
Review
Cytokine storm in COVID-19: from viral infection to immune responses, diagnosis and therapy
Yizhou Jiang1#, Limor Rubin, MD2#, Tangming Peng1, Linlin Liu1, Xingan Xing1, Philip Lazarovici, PhD3, Wenhua Zheng, MD, PhD1 ![]()
1. Center of Reproduction, Development & Aging, Faculty of Health Sciences, University of Macau, Taipa, Macau SAR, China.
2. Allergy and Clinical Immunology Unit, Department of Medicine, Hadassah-Hebrew University Medical Center, Jerusalem, Israel.
3. School of Pharmacy Institute for Drug Research, Faculty of Medicine, The Hebrew University of Jerusalem, Jerusalem 91120, Israel.
#Co-first equal contributing authors
Received 2021-2-10; Accepted 2021-8-5; Published 2022-1-1
Abstract

The COVID-19 outbreak is emerging as a significant public health challenge. Excessive production of proinflammatory cytokines, also known as cytokine storm, is a severe clinical syndrome known to develop as a complication of infectious or inflammatory diseases. Clinical evidence suggests that the occurrence of cytokine storm in severe acute respiratory syndrome secondary to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection is closely associated with the rapid deterioration and high mortality of severe cases. In this review, we aim to summarize the mechanism of SARS-CoV-2 infection and the subsequent immunological events related to excessive cytokine production and inflammatory responses associated with ACE2-AngII signaling. An overview of the diagnosis and an update on current therapeutic regimens and vaccinations is also provided.
Keywords: cytokine storm, COVID-19, SARS-CoV-2, immune response, cytokines, diagnosis, therapy, vaccination
1. Introduction
In the past two decades, before the emergence of COVID-19, coronaviruses were known for two massive pandemics: severe acute respiratory syndrome (SARS) in 2003 and Middle East respiratory syndrome (MERS) in 2012 [1-3]. The COVID-19 we faced last and this year, is a highly infectious and pathogenic epidemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection [4]. Despite quarantine measures taken, the virus spread rapidly worldwide without specific effective therapies, but the pandemic has been halted thanks to up-to-date vaccines in countries with national vaccination programs, e.g., Israel [5]. By June 2021, SARS-CoV-2 has caused more than 170 million infections and more than 3.7 million deaths worldwide [6]. Although most COVID-19 patients have mild symptoms and a good prognosis, some patients may develop serious complications [7]. Medical evidence suggests that acute respiratory distress syndrome (ARDS) caused by cytokine storm is a major cause of mortality [8, 9]. Cytokine storm is the result of overproduction of cytokines triggered by a variety of infectious and noninfectious diseases or even some therapeutic interventions [10]. The excessive cytokines can trigger an uncontrolled immune response, damage lung tissue, and develop into multiorgan failure [10, 11]. Therefore, suppression of the cytokine storm is critical for reducing mortality in severe COVID-19 patients. This review summarizes the mechanism of cytokine storm and tissue damage induced by SARS-CoV-2 infection and reviews diagnosis and current therapies with emphasis on vaccination.
2. Infection of SARS-CoV-2
SARS-CoV-2 was first isolated from broncho-alveolar lavage fluid of patients in December 2019 [12]. The virus was then sequenced, and phylogenetic analysis showed that SARS-CoV-2 belongs to the genus Betacoronavirus and shares 79% and 50% identity with SARS-CoV and MERS-CoV, respectively [13, 14]. Of note, at the whole-genome level, SARS-CoV-2 shares high similarity (nearly 90%) with two bat coronaviruses, bat-SL-CoVZC45 and bat-SL-CoVZXC21, indicating its possible bat origin [13, 14]. The sequence identity between SARS-CoV-2 and these two bat coronaviruses is greater than 90% in five gene regions (E, M, 7, N, and 14). However, the S gene and its corresponding S protein were an exception, with approximately 75% and 80% identity, respectively. The S protein is a viral protein that mediates binding to the receptor and fusion with the cell membrane, making it a key determinant of host infection and transmission [15]. It can be divided functionally into an N-terminal S1 subunit responsible for receptor recognition and binding, and a membrane-bound C-terminal S2 region responsible for cell membrane fusion [13, 15], both of which are essential for viral entry via the endocytic pathway and infection into host cells [16]. The S1 domain is highly variable among different coronaviruses, whereas the S2 domain is more conserved [15, 17]. Although the S2 domain of SARS-CoV-2 is identical (approximately 93% similarity) to that of bat-SL-CoVZC45 and bat-SL-CoVZXC21, the S1 domain is quite different (approximately 68% similarity) [13]. In addition, the S1 domain of SARS-CoV-2 is similar to that of SARS-CoV, although variations have been found in several key residues, suggesting that they may bind the same receptor [13].
Angiotensin-converting enzyme 2 (ACE2) has been shown to be the cell entry receptor of SARS-CoV [18]. Therefore, attempts were made to verify whether it is also the cell entry receptor of SARS-CoV-2. Zhou et al. used SARS-CoV-2 to infect HeLa cells expressing ACE2 protein from humans or other animals, including Chinese horseshoe bats, civet cats, pigs, and mice [14]. The results showed significantly efficient virus entry into cells expressing ACE2 except mouse ACE2, whereas cells without ACE2 expression were not infected. In addition to the cell receptor ACE2, other essential cellular proteins such as the transmembrane protease serine 2 (TMPRSS2) and the endosomal cysteine proteases cathepsin B and L (CatB/L), which have S-protein priming, are involved in SARS-CoV infection [19, 20]. Hoffmann et al. also found that inhibition of TMPRSS2 activity by camostat mesylate or inhibition of CatB/L by ammonium chloride could only partially block cell entry of SARS-CoV-2 in the presence of the activity of another enzyme [21]. Co-treatment of camostat mesylate and E-64d, another CatB/L inhibitor, completely blocked viral infection. These findings suggest that both TMPRSS2 and CatB/L are involved in S-protein priming of SARS-CoV-2. Interestingly, TMPRSS2 appears to have a more dominant role than CatB/L in virus entry, reminiscent of other viral infections. Since camostat is already in clinical use for the treatment of chronic pancreatitis, it is tempting to suggest its potential use for the treatment of COVID-19 disease. Ammonium chloride is also used as an autophagy inhibitor. Since it is still controversial whether and how autophagy is involved in SARS-CoV-2 infection [16], further work is needed to clarify the involvement of autophagy in the intracellular transport of SARS-CoV-2 after its binding and fusion with the target cell membrane.
Binding of the spike protein of SARS-CoV-2 to ACE2 triggers entry of the virus into the cell, but other proteins may also be involved, such as the neuropilin-1 receptor (NRP-1) [22], a transmembrane receptor that lacks a cytosolic protein kinase domain and has high expression in the respiratory and olfactory epithelium. Recent studies have shown that the SARS-CoV-2 spike S1 protein can bind to the b1b2 domain of NRP-1. This interaction occurs via a polybasic amino acid sequence Arg-Arg-Ala-Arg C-terminal sequence on S1 (682RRAR685) that is not conserved in SARS and MERS and is referred to as the "C-end rule" (CendR) motif, which significantly potentiates its entry into cells upon binding to the cell surface receptors neuropilin-1 (NRP-1) and neuropilin-2 (NRP-2) [23]. While NRP-1 significantly potentiates SARS-CoV-2 infectivity, an effect blocked by a monoclonal neutralizing antibody to NRP-1, a SARS-CoV-2 mutant with an altered furin' cleavage site was not dependent on NRP-1 for infectivity. Pathological analysis of human COVID-19 autopsies revealed SARS-CoV-2-infected cells, including olfactory neurons positive for NRP-1A in the nasal cavity. In addition, significant upregulation of NRP-1 was found in biological samples from COVID-19 patients compared to healthy controls. Collectively, these data suggest the involvement of NRP-1 in SARS-CoV-2 cell infectivity and define an additional potential target for antiviral intervention in pulmonary and neurological manifestations of COVID-19.
The mechanism of viral entry of SARS-CoV-2 via ACE2 could also involve the neutral amino acid transporter B0AT1, also known as SLC6A19. ACE2 may act as a chaperone for membrane trafficking of B0AT1, thereby regulating the uptake of neutral amino acids into intestinal cells [24]. A recent study presenting the cryo-electron microscopic structures of ACE2 showed that ACE2 and B0AT1 form a dimer or heterodimer [25]. Residues 697 to 716 of the C-terminal segment of ACE2, where proteases such as TMPRSS2 that cleave ACE2 and facilitate viral entry, are involved in dimerization. Therefore, B0AT1 can suppress SARS-CoV-2 infection by blocking the access of TMPRSS2 to the cleavage site on ACE2. It is worth noting that the expression and distribution of B0AT1 is mainly restricted to kidney and intestine, which is lower than that of ACE2 [26]. Moreover, ACE2 may be able to form homodimers in the absence of B0AT1 because B0AT1 is not involved in dimerization. B0AT1 may play a partial role in the enteric infection of SARS-CoV-2, and this idea should be strengthened by further studies. Figure 1 summarizes the mechanism of viral entry.
Mechanism of SARS-CoV-2 viral entry. SARS-CoV-2 uses angiotensin-converting enzyme 2 (ACE2) as its cellular receptor. The cell entry of SARS-CoV-2 is also dependent on transmembrane protease serine 2 (TMPRSS2), Neuropilin-1 (NRP1) and cathepsin B and L (CatB/L).
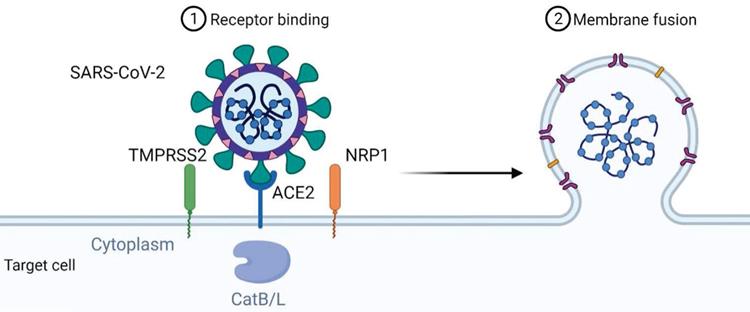
3. Mechanism of the cytokine storm triggered by SARS-CoV-2 infection
The immune system is vital for the host to fight viral infections. However, excessive immune responses can lead to pathologies [27]. Viral entry can trigger a series of immune responses that can further lead to cytokine storm and eventually acute respiratory distress syndrome (ARDS) and death. Both clinical and animal studies have shown that the cytokine storm plays a key role in the pathogenesis of SARS and MERS [28]. In clinical studies, higher concentrations of proinflammatory cytokines were found in plasma in critical patients than in mild to moderate patients, suggesting that the cytokine storm is also directly related to the severity of COVID-19 disease [29]. The mechanism(s) by which SARS-CoV-2 infection induces cytokine overproduction is not yet fully understood. In the present review, we will mainly focus on the role of ACE2-AngII-related signaling on the cytokine storm induced by SARS-CoV-2 infection.
The ACE2 protein has been found to be most abundant in lung alveolar epithelial cells and small intestinal enterocytes. In addition, ACE2 protein is also highly expressed in endothelial cells and smooth muscle cells of arteries and endothelial cells of veins in all organs studied [30]. Analysis of autopsy specimens from SARS patients indicated that SARS-CoV primarily infected epithelial cells of the respiratory tract, consistent with expression levels of ACE2 [31]. However, despite the lack of ACE2 expression, immune cells, including T lymphocytes, monocytes, and macrophages, were also infected and destroyed by SARS-CoV [30, 31]. Furthermore, although SARS-CoV viral particles were detected in other cell types such as epithelial cells of the digestive tract, kidney, and neurons in the brain, many organs with ACE2 expression remained uninfected [30, 31]. These discrepancies support the notion that cell entry of SARS-CoV is not solely dependent on ACE2.
As mentioned above, although cell subsets expressing ACE2 are likely targets of SARS-CoV-2, viral entry also depends on other cellular factors such as TMPRSS2. Therefore, cells that co-express ACE2 and TMPRSS2 may be at a higher risk of direct infection with SARS-CoV-2. A recent study that analyzed new and existing single-cell RNA sequencing datasets found co-expressing ACE2+/ TMPRSS2+ cell types, including lung-type pneumocytes II, nasal secretory goblet cells, and ileal absorptive enterocytes, and that ACE2 is a human interferon-stimulated gene (ISG) in vitro [32]. SARS-CoV-2 has been shown to actively and productively infect cardiomyocytes [33] and myocardial damage correlated with outcome in autopsies showing myocarditis with presence of SARS-CoV-2 viral RNA in hearts from COVID-19 patients [34]. Lymphocytopenia is a hallmark of COVID-19 disease [35, 36]. Both SARS-CoV and MERS-CoV can infect T cells [31, 37]. SARS-CoV-2 has also been found to infect T lymphocytes through S protein-mediated membrane fusion [38]. Similar to MERS-CoV, SARS-CoV-2 is unable to replicate in T cells. In addition, T lymphocytes have been reported to be more sensitive to SARS-CoV-2 than to SARS-CoV infection. Because T cells have very low ACE2 mRNA expression, SARS-CoV-2 could infect T cells via a different receptor, such as CD147 [39]. Extensive T-cell apoptosis was found in COVID-19 patients, in direct correlation with increased expression of pro-apoptotic molecules [40, 41]. The question of whether SARS-CoV-2 virus directly causes T-cell apoptosis requires further investigation.
After entry into the host cell, the virus may undergo rapid replication, associated with cell apoptosis, pyroptosis, and production of proinflammatory cytokines and chemokines [42]. Most COVID-19 patients were found to have lymphopenia, a decrease in lymphocyte count associated with increased mortality [35, 36]. It was hypothesized that lymphopenia is induced by several factors: i. Lymphocyte infiltration and sequestration in the lungs or other tissues such as the gastrointestinal tract and lymphoid tissues; ii. SARS-CoV-2 direct infection of lymphocytes, as some lymphocytes express ACE2 and SARS-CoV-2 could also infect lymphocytes through other pathways; iii. Excess production of pro-inflammatory cytokines including IL-6, which may contribute to additional lymphocyte reduction [35,38, 43]. Moreover, the excess pro-inflammatory cytokines and chemokines could further attract neutrophils and monocytes, leading to excessive infiltration of inflammatory cells enhancing the lung injury [27]. Indeed, biopsy and autopsy studies have shown lung damage with infiltration of inflammatory immune cells, such as lymphocytes and macrophages, into the lungs of COVID-19 patients [44, 45].
ACE2 functions as a negative regulator of the renin-angiotensin system, which is important for blood pressure regulation and maintenance of fluid and electrolyte balance [46-49]. ACE2 balances the activity of angiotensin-converting enzyme (ACE), the homolog of ACE2, and inhibits the production of angiotensin II (AngII) [50]. In addition to its cardiovascular modulatory effects, AngII is considered a proinflammatory cytokine [51, 52]. AngII activates nuclear factor-κB (NF-κB) through its action on both AngII receptors AT1 and AT2 and mediates inflammatory responses by stimulating the expression of proinflammatory cytokines, chemokines, and adhesion molecules [51, 52]. In addition, the renin-angiotensin system plays a critical role in severe acute respiratory failure, including SARS-CoV-2 infection. This conclusion is supported by studies showing that acid aspiration causes reduced ACE2 expression and increased AngII levels, and ACE2 knockout mice developed increased lung failure in the acute lung injury model, compared with wild-type mice [53]. Moreover, injection of human recombinant ACE2 protein reduced the severity of lung injury caused by acid treatment and ACE2 deficiency [53]. Similar results were also obtained in lung injury induced by viral infections. Reduced ACE2 expression in the lung and increased angiotensin levels in serum II were found after H5N1 infection with avian influenza in a mouse model [54]. Importantly, SARS-CoV infection and SARS-CoV spike protein caused reduced ACE2 surface expression [50]. The lung injury induced by SARS-CoV spike protein is prevented by ACE2 knockout and by inhibition of AngII receptor type 1 (AT1R) by a specific inhibitor. Therefore, ACE2 has a protective role towards AngII-induced inflammatory response during SARS infection and lung injury caused by other factors. These findings strongly suggest that ACE2 downregulation is critical for SARS-CoV infection-induced lung injury.
After binding to their cellular receptors, viruses frequently use endocytosis machinery to invade host cells [55, 56]. Therefore, the downregulation of ACE2 at the cell surface after SARS-CoV infection may be due to the internalization of ACE2. Indeed, flow cytometry and immunostaining showed that the receptor-binding domain of the S protein of SARS-CoV was internalized along with ACE2 [57]. Shedding of the ACE2 ectodomain may be another mechanism responsible for SARS-CoV infection-induced ACE2 downregulation [58]. TACE (TNFα-converting enzyme) is a metalloprotease that releases soluble regulatory factors from cells [59]. It has been previously reported that TACE (ADAM 17, a disintegrin and metalloprotease 17) is required for phorbol-12-myristate-13-acetate (PMA)-induced ACE2 shedding [59, 60]. TNFα and IL-1β, another proinflammatory cytokine, were able to induce ACE2 shedding [61]. Similarly, the spike protein of SARS-CoV-induced ACE2 shedding was dependent on TACE, a biological process that directly correlates with TNFα production [58]. These cumulative findings suggest an essential role of proinflammatory cytokines, particularly TNFα in SARS-CoV infection-induced-ACE2 shedding. Interestingly, the shed form of ACE2 was catalytically active and could function in the renin-angiotensin system. However, it has not yet been established that ACE2 shedding is a key step in SARS-CoV-induced lung injury [58]. Moreover, in addition to its effect on TNFα release and ACE2 shedding, TACE also cleaves other cellular substrates and releases corresponding molecules such as EGFR ligands and soluble IL-6Rα (sIL-6Rα) [62, 63]. Both TNFα and EGFR ligands are capable of activating NF-κB, the major regulator of inflammation [64-66]. sIL-6Rα can form a complex with IL-6 and activate another important inflammatory regulatory protein, Signal Transducer and Activator of Transcription 3 (STAT3) [63]. NF-κB and STAT3 produce a synergistic effect on IL-6 transcription in nonimmune cells, a process described as IL-6 amplification mechanism [63]. In addition, SARS-CoV-2 itself can directly activate NF-κB via pattern recognition receptors [64]. The IL-6 amplification leads to an overproduction of pro-inflammatory cytokines and chemokines, including IL-6, and eventually results in a cytokine storm with subsequent ARDS [64]. Interestingly, AngII may increase the activity of TACE, thereby promoting the release of its negative regulator ACE2 [67]. Moreover, AngII also induced the internalization and degradation of ACE2 in an AT1R-dependent manner [68]. These findings represent a positive feedback mechanism in the renin-angiotensin system. Therefore, ACE2 downregulation by SARS-CoV-2 infection leads to increased AngII signaling, thereby triggering the excessive inflammatory response and lung injury.
A recent study investigated the effect of another subset of cytokines (IFN-α2, IFN-γ, IL-4, IL-13, IL-17A, and IL-1β) on ACE2 expression on cultured human primary basal epithelial cells [32]. The results showed that IFN-α2 and IFN-γ led to a significant upregulation of ACE2. Since SARS-CoV-2 mediates the downregulation of surface ACE2 protein by several mechanisms including shedding and internalization, IFNs may act in a different way to maintain a certain level of cellular targets to allow the infection of the virus in the host lung. Although the detailed mechanism of each cytokine is not fully elucidated, these findings further imply a tight correlation between cytokine production and ACE2 expression and function. Notably, it was also found that IFN failed to stimulate ACE2 in mice, indicating that the species difference between humans and mice should be considered in modeling COVID-19 disease [32]. In addition to functioning in the renin-angiotensin system, ACE2 also interacted with other pathways involved in cytokine production, like the plasma kallikrein-kinin system [69] and Ang-(1-7)/ Mas receptor [70]. Moreover, SARS-COV-2 infection could also result in cytokine production by activating complement pathways such as the C3a-C3aR/ C5a-C5aR axis [71]. A complex signaling network as proposed in Figure 2, may regulate the COVID-19-associated cytokine storm.
Proposed mechanisms of COVID-19 -induced cytokine storm. SARS-CoV-2 can directly activate the key inflammation regulator NF-kB via pattern recognition receptors (PPRs). ACE2 downregulation caused by shedding and internalization of ACE2 leads to increased angiotensin II (AngII) levels and hyperactivation of NF-kB, followed by excessive proinflammatory cytokine production. AngII can stimulate both shedding and internalization of ACE2, representing a positive feedback mechanism in the renin-angiotensin system. In addition, AngII can also promote the shedding of molecules that can activate NF-kB including TNFα, sIL-6Rα and EGFR ligands. These positive feedback mechanisms may further enhance the proinflammatory cytokine production and lead to a cytokine storm.
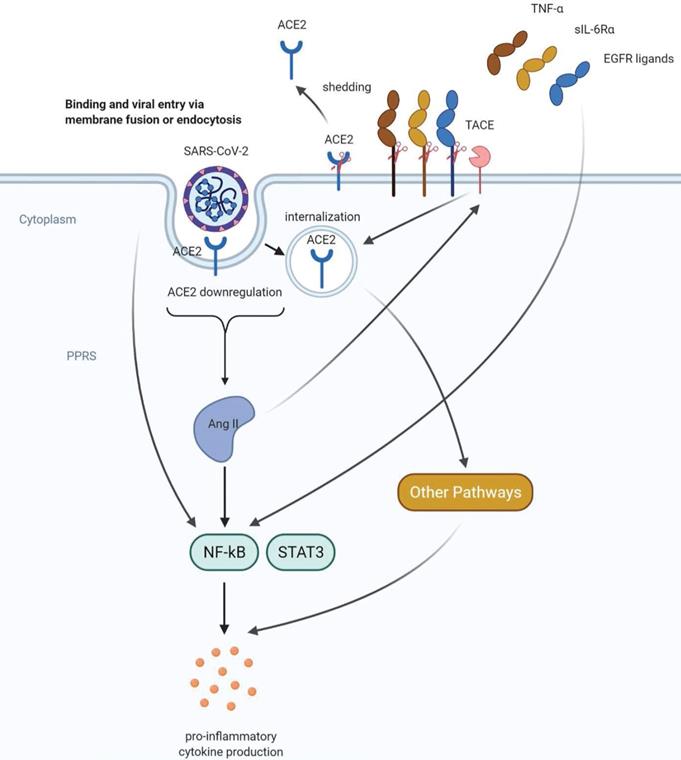
4. Clinical outcome and diagnosis of COVID-19 disease
Similar to most viral respiratory illnesses, COVID-19 symptoms include fever and respiratory symptoms such as cough and dyspnea. Additional symptoms may include loss of smell/taste, myalgia, sore throat, rhinorrhea diarrhea, headaches, nausea/vomiting, extreme fatigue, and chest pain [29]. COVID-19 infection should be suspected over other respiratory viral infections in the two following scenarios: i. Visit/ resident in areas with significant COVID-19 respiratory syndrome outbreaks; ii. Close contact (with no use of standard precautions) with a patient who has confirmed/ suspected of COVID-19 infection. Furthermore, the development of dyspnea and exertional desaturation several days after the initial onset of symptoms is a typical symptom [72]. Important to mention, approximately 40% of patients infected with SARS-CoV-2 infection are found to be asymptomatic [73]. However, they may present with abnormal imaging and laboratory test [74].
Infection with SARS-CoV-2 is classified into three different clinical phases: i. Host viral response phase-infection (flu-like symptoms); ii. Host inflammatory response/ hypercoagulation phase - ARDS/ elevated acute-phase reaction and cytokines; iii. Pulmonary fibrosis formation [75]. As mentioned earlier, ARDS is the major life-threatening complication of COVID-19 infection leading to respiratory failure and the need for mechanical ventilation. Additional complications were reported: cardiac arrhythmia, myocarditis, cardiomyopathy [76], thromboembolic (pulmonary embolism( [77], deep vein thrombosis (DVT) and stroke [78], secondary infection (bacterial/fungal), an inflammatory complication such as cytokine storm [9], Gullian Barre syndrome [79], Kawasaki disease in children [80] multisystem inflammatory syndrome in adults (MIS-A) [81], hemaphagocytic lymphoistiocytosis (HLH) [82] and toxic shock syndrome. Since the identification of SARS-CoV-2 in humans, a multitude of cardiovascular complications including myocardial injury, heart failure, arrhythmias, thromboembolic disease as well as kidney disease have been reported. Clinical outcomes are worse in patients with COVID-19 with cardiovascular disease and risk factors (eg, hypertension, diabetes, and obesity). Acute cardiac injury, inferred from elevations in cardiac troponin levels, is reported in 8% to 62% of patients hospitalized with COVID-19 and is associated with greater disease severity, including the need for mechanical ventilation, and death [83-85].
The preferred diagnostic assay for SARS-CoV-2 is the nucleic acid amplification test (NAAT) using reverse transcription-polymerase chain reaction to detect SARS Cov-2 RNA in a specimen taken from the upper respiratory tract [86]. A single positive test is sufficient for diagnosis, however, if a high clinical suspicion and a negative RT-PCR result, the test should be repeated, lower tract secretion could be examined and a serological test performed. Serological test my also assist in diagnosing asymptomatic patients [87].
5. Therapy of COVID-19 disease
As mentioned above, most COVID-19 complications are related to the inflammatory response and cytokine storm, therefore immunosuppression plays an important role in the management of these patients. One of the first medication proposed for COVID-19 therapy was hydroxychloroquine, an antimalarial drug, due to it's in vitro evidence of suppressing SARS-CoV-2 replication, in addition to its known immunomodulatory effects [88]. Mechanism of action proposed was impairment in the glycosylation terminal of ACE2 and elevation of endosomal pH, resulting in impairment of endosomal mediated viral entrance to the cells [89-91]. Hydroxychloroquine effect was assumed to be potentiated by Azithromycin (macrolide antibiotic) administration, leading to a reduction in the viral load [92]. Unfortunately, large cohort studies performed showed no significant improvement in clinical outcomes in patients receiving hydroxychloroquine, with/ without Azithromycin treatment [93, 94].
For the present, steroids are the mainstay of treatment. Randomized clinical trials (RCT) presented a decrease in mortality rate among COVID-19 patients aged over 60 years old, who received methylprednisolone therapy [95]. RECOVER trial (open label RCT) examined the yield of dexamethasone therapy delivered orally and by intravenous injection. A lower 28-day mortality rate and a shorter hospitalization stay was significantly demonstrated in the treatment subgroup of patients that were in need for oxygen support or mechanical ventilation [96]. Interestingly, the combination of methylprednisolone and dexamethasone therapy increased the risk of thromboembolism, despite achieving a rapid anti-inflammatory effect [97]. As for the hyper-inflammatory disease phase, biological treatment was in need in order to neutralize the effect of elevated plasma pro-inflammatory cytokines including interleukins (IL-2, 6, 7, 10, gCSF, INFγ, TNFα). Hence, Toclizumab was investigated for this purpose. Toclizumab is a monoclonal antibody that targets membrane bound IL-6 receptor, therefore inhibiting soluble IL-6 signal transduction. At first, reduced mortality was attributed to Tocalizumab treatment in intensive care unit patients [98]. However, further studies demonstrated no mortality differences [99] and an increased risk for late onset secondary infections in the treatment group [100]. On the other hand, when testing efficacy of low dose Tocalizumab treatment in a subgroup of patients with moderate to severe COVID-19 infection, opposing results were encountered. Low dose Tocalizumab treatment, as add on therapy to standard of care, prevented disease progression in hospitalized patients with moderate COVID-19 infection [101]. Patients with severe COVID-19 infection, presenting with elevated levels of inflammatory cytokines (including IL-6), and acute phase reactants, improved lung manifestation in follow-up CT imaging under Tocalizumab treatment and did not progress to develop cytokine release syndrome [102]. Recent studies on Tocalizumab treatment conclude lack of efficacy in prevention of intubation and mortality in moderately ill-hospitalized patients [103]. Unless presenting with COVID-19 pneumonia, however survival rates remained unchanged, regardless of treatment [104].
In addition to anti - inflammatory agents, the choice of antiviral medication is also under investigation. Favipiravir, an RNA-dependent, RNA polymerase inhibitor prodrug, used as second-line treatment for influenza infection, was proved to demonstrate in vivo antiviral activity against SARS-CoV-2 and shorten the time for viral clearance in COVID-19 patients [105]. A small RCT demonstrated improved clinical outcome and efficient viral clearance in patients receiving Favipiravir [106] and open-label control study indicated improvement in chest CT findings [107]. Favipiravir entered treatment guidelines in several countries such as India and Russia. However, further ongoing clinical trials are in need in order to confirm these results [107].
Remdesivir is another antiviral medication, used on a compassionate-base for patients hospitalized with COVID-19 infection. Remdesivir is a nucleotide analogue prodrug that inhibit RNA polymerase, and showed to have activity against filoviruses (eg. Ebola) [108] and coronaviruses (es. SARS-CoV and MERS-CoV) [105, 109]. The use of Remdesivir for hospitalized COVID-19 patients with oxygen saturation of 94 % or less, initially showed improved clinical outcomes [110], with parallel studies presenting contradictory results [111]. Further studies indicated superiority to placebo in shortening time to recovery in hospitalized high risk patients that required oxygen support and diagnosed early during illness (>10 days) [112]. Remdesivir is an important treatment option and its advantage value in combination with immunomodulators and monoclonal antibodies remains to be studied [113]. For example, combination treatment with Baricitinib (JAK inhibitor), accelerated clinical improvement and recovery time in patients receiving high-flow oxygen or noninvasive ventilation [114].
Another aspect of COVID-19 therapy includes prevention of thrombotic micro-and macro-vascular complications with the use of prophylactic or therapeutic anticoagulation therapy. Meta-analysis described the occurrence of VTE (venous thromboembolism) in 31 % of ICU admitted COVID-19 patients despite prophylactic and/or therapeutic anticoagulation, emphasizing the need for personalized dose adjustment and anti-Xa coagulation factor monitoring [115]. COVID-19 coagulopathy scoring system is proposed to asses high-risk patients that could benefit from a therapeutic-dose of anticoagulant and perhaps a combination with anti-platelets agents may be beneficial [116]. In patients presenting with what appears to be a typical cardiac syndrome, COVID-19 infection should be in the differential diagnosis during the current pandemic, even in the absence of fever or cough. One should have a low threshold to assess for cardiogenic shock in the setting of acute systolic heart failure related to COVID-19. If inotropic support fails in these patients, intra-aortic balloon pump (IABP) device is the first line of mechanical circulatory support, because it requires the least maintenance from medical support staff. When patients on veno-venous extracorporeal membrane oxygenation (VV-ECMO) for respiratory support develop superimposed cardiogenic shock, the addition of an arterial conduit at relatively low blood flow rates may provide the necessary circulatory support without inducing left ventricular distension. Rescue of patients even with profound cardiogenic or mixed shock may be possible with temporary hemodynamic support at centers with availability of such devices. COVID-19 infection can cause decompensation of underlying heart failure, and may lead to mixed shock. Invasive hemodynamic monitoring, if feasible, may be helpful to manage the cardiac component of shock in such cases. Medications that prolong the QT interval are being considered for COVID-19 patients and may require closer monitoring in patients with underlying structural heart disease [117].
As no definite therapy was proved for COVID-19 infection, convalescent plasma therapy is under investigation for rescue therapy in critical ill patients. A 44 % reduction in mortality rate in COVID ‐19 patients receiving convalescent plasma was demonstrated, in addition to improvement in viral clearance. However, it is important to emphasize reservation on this data which is of low quality and indicated high variability among different studies [118]. It appears that high-titer convalescent plasma is efficient to prevent disease progression when administered early in disease course to older adults with mild disease [119]. In addition immunocompromised patients, especially B cell depleted, may benefit from combined therapy with convalescent plasma and prolonged antiviral treatment [120]. Following initial therapy with convalescent plasma, monoclonal neutralizing antibody products targeting different epitopes on the spike protein, entered clinical trials for therapeutic and prophylactic use [121]. Neutralizing antibodies proved to reduce viral load especially when administered before initiation of the immune response by COVID-19 infection [122, 123].
As the global pandemic expanded, serological testing and research was vital for COVID-19 prevention and for the development of an efficient vaccination. Most immunocompetent patients will develop an immune response after exposure to SARS-CoV-2'infection and serum antibodies will be detected 1-3 weeks post symptoms onset. SARS-CoV-2 antibodies target two major antigens: Spike glycoprotein (antigen S) present on the viral envelope and nucleocapsid phosphoprotein (antigen N) which interacts with RNA. Different antibody assays are used worldwide that can be classified as binding and neutralizing antibody assays. Binding antibodies are IgA, IgM, IgG type antibodies that recognize SARS-CoV-2 antigens and their expression levels can be measured in serum/ plasma/ saliva, using ELISA method. Neutralizing antibodies are detected in functional laboratory tests analyzing the ability of antibodies to neutralize the virus by incubating live SARS-CoV-2 virus/ recombinant virus with patient serum/ plasma. A strong positive correlation was found between IgG binding antibodies level and neutralizing antibodies level [124]. Furthermore, neutralizing antibodies titer was directly correlated to disease severity [125]. However, SARS-CoV-2 antibodies levels in the serum decline overtime, with a presumed half-life of 36 days [126]. Adaptive immunity essential for efficient vaccine development and T cell response (CD4 and CD8) was recognized, and was directly correlated to anti-SARS-CoV-2 immunoglobulins level [127].
On January 2020 the genetic sequence of SARS-CoV-2 become available. This data accelerated vaccine development. Vaccination development employed different vaccines platforms: live attenuated vaccines, recombinant proteins, virus-like particles, inactivated/ activated vector vaccines, and new platforms of DNA and RNA vaccines. These products are under different developments phases from preclinical research to clinical trials and distribution, following FDA licensing [128]. As of June 2021, there are 287 candidate vaccines, 102 undergoing clinical phase and 185 in pre-clinical phase [129]. Initially, two major vaccines were under clinical investigation, both targeting SARS-CoV-2 antigen S. The ChAdOx1 nCoV-19 AstraZeneca viral (adenovirus) vector vaccine, expected to generate neutralizing antibodies towards SARS-CoV-2 in 91 % of patients after the first dose, and 100 % after the second dose and elicited a profound T cell response with an acceptable safety profile [130]. The other, Moderna vaccine based on mRNA-1273, achieved 50 % neutralizing antibodies after the first dose and 100 % after the second dose and also presented a satisfying safety profile [131]. However, Pfizer-BioNTech BNT162b2 mRNA vaccine encoding SARS-CoV-2 full-length spike protein was the first to receive FDA approval following its phase 3 clinical trials. It indicated that a two-dose regimen caused 95 % efficacy in disease prevention and a good safety profile in adults patients [132] and adolescents [133]. Israel, representing the current leading country with the highest percentage of vaccinated population, initiated a mass vaccination campaign of the BNT162b2 mRNA vaccine and demonstrated high efficacy in prevention of symptomatic COVID-19 and its serious outcomes: hospitalization, severe illness and death [134, 135]. Phase 3 clinical trial results of Moderna mRNA-1273 were published, revealing 94 % efficacy in disease prevention with favorable safety profile [136] and antibody persistence for 6 months following the second dose [137]. Sputnik V, Gam-COVID-Vac phase 3 clinical trial indicated 91.6 % efficacy [138]. In addition, phase 3 clinical trial demonstrated safety and efficacy of single dose Ad26.COV2.S vaccine [139]. ChAdOx1 nCoV-19 AstraZeneca vaccine' phase 2-3 trial revealed similar immunogenicity across all age groups following a boost dose, with better tolerance among older adults [140, 141]. Reports of thrombosis and thrombocytopenia following ChAdOx1 nCoV-19 AstraZeneca vaccine raised great concern [142], with a proposed mechanism of antibodies to PF4, unrelated to heparin use [143]. However, analysis of adverse events reports exhibited similar risk for venous thromboembolism and thrombocytopenia in vaccinated patients in comparison to the general population. However, the prolonged safety profile, dose adjustment in subpopulation of children, and lasting immunity especially in immunocompromised patients, remain to be investigated.
As mentioned above, vaccines demonstrated 70-95 % efficacy in protection against mild to severe COVID-19 symptoms and almost total mortality prevention. However, numerous new variants of SARS-CoV-2 have emerged representing a possible threat to the control of the COVID-19 pandemic. During the process of SARS-CoV-2 RNA replication in the human host, mutations occur, a virus with a single or multiple mutations is referred to as a "variant" of the original virus. These variants could differ by distinct properties such as binding affinity to the cell receptor, change in the replication rate, increased efficacy of transmission and/or virulence. In the early 2020, the first SARS-CoV-2 variant emerged, exhibiting a D614G substitution in the spike protein [144] . Over a period of six months, the D614G mutation replaced the initial SARS-CoV-2 strain and by June 2020, became the dominant form of the virus circulating globally, due to its high replication rate and/or transmission, by assembling a more functional S protein into the virion [145]. At present, four major variants are of concern: i. B.1.1.7 (known as 20I/ 501Y.V1 or VOC 202012/ 01 or α-variant) first noted in the UK in December 2020, and characterized by 17 non-synonymous mutations, along with 8 S protein mutations and including the D614G mutation; ii. B.1.351 (known as 20H/ 501Y.V2 or VOC 202012/ 02 or β-variant) first identified in South Africa in late 2020, that has multiple mutations in the S protein including K417N, E484K, and N501Y; iii. P.1 (known as 20J/ 501Y.V3 or γ-variant) initially found in travelers that came to Japan from Brazil, that has multiple mutations within the receptor binding domain K417T, E484K, and N501Y; and iv. B.1.617.2 (known as δ-variant) found in India in late 2020, consisting of D111D, G142D, L452R, E484Q, D614G, and P681R mutations in the S protein and containing within the receptor binding domain three mutations including L452R and E484Q, along with P681R in the furin cleavage site. In all these variants, the mutations in the receptor-binding domain of the spike (S) protein were found to be associated with increased reinfection, escaping binding by neutralizing antibodies of the patient and increased affinity for the human ACE2 receptor [146, 147].
Immunity following COVID-19 infection or vaccination is both humoral, B cell depended and cellular, T cell immunity. SARS-CoV-2 S protein consist of multiple epitopes recognized by T cells, resulting in the production of variable antibodies and a diversity in T cell response [148]. Hence, host translation of the S protein following mRNA vaccination induce an immunogenic humoral and cellular response that can overcomes some of the pivotal SARS-CoV-2 variants activities. Regarding the therapeutic effects on the mutant D614G variant, studies showed that the Pfizer-BioNTech BNT162b2 vaccine which was based on the original D614 sequence had a 1.7-2.0 decrease in neutralization, making it less effective [149, 150]. The Moderna mRNA-1273 vaccine showed similar neutralization when compared to the original SARS-CoV-2 strain [151]. The Moderna and Novavax vaccines exhibited only a moderate reduction in neutralization of the SARS-CoV-2 α-variant compared to the original strain [152] and Pfizer-BioNTech and Moderna vaccines showed no change in S477N, but decreased neutralization of E484K SARS-CoV-2 α-variant [146]. In addition, Pfizer-BioNtech and Moderna vaccines showed decreased neutralization towards SARS-CoV-2 α, β and δ-variants, while NVX-CoV2373 vaccine showed good neutralization towards SARS-CoV-2 β-variant [146].
It is clearer now, that adaptive mutations of SARS-CoV-2 could alter its pathogenic potential, and at the same time would raise difficulties on drug and vaccine development. From a clinical point of view, the SARS-CoV-2 variants demonstrating high disease transmission and severity including hospitalization and death, characterized by significant decrease in antibody neutralization and decreased effectiveness of diagnosis and treatment, represent a big challenge. Control measures remain the key for prevention of viral transmission, hence reduction in the opportunity to viral mutation and development of variants. This aspect requires increased efforts to control SARS-CoV-2 spread by developing novel diagnostic kits and increasing research to ascertain the efficacy of the different vaccines and treatments against the new variants. Global mass vaccination programs offer a great promise in controlling this historical pandemic. Unfortunately, in countries such as India and Brazil, the COVID-19 outbreak shows new signs of acceleration, despite the fact that long-delayed vaccination efforts finally gaining steam. We anticipate that COVID-19 vaccines will be administered in every country, to overcome this pandemic and to protect and promote human health. Although COVID-19 vaccines proved to be highly effective in disease prevention, a small percentage of the fully immunized population could still be infected with COVID-19 disease. Several hurdles remain with vaccine administration such as immunocompromised patients, diverse ethnicities and children that have not been satisfactorily represented in vaccine trials. COVID-19 vaccine hesitancy has been also an important aspect that physicians encounter due to large amount of false and misleading information published in the social media [153, 154]. It is also important to address that lack of vaccination and associated comorbidities that predispose individuals to get infected with the SARS-CoV-2 δ-variant [155].
6. Summary
The immune responses triggered upon viral infections are essential for hosts to eliminate the pathogens. However, the infection of SARS-CoV-2 could cause excessive inflammatory responses, including amplified pro-inflammatory cytokine production and tissue damage, resulting in fatal ARDS. In spite of lacking specific anti-SARS-CoV-2 therapy, suppressing the cytokine storm with corticosteroids is a common approach, which greatly improves the survival rate of COVID-19 patients. Existing drugs that could block the cellular pathways of infection and the excessive inflammatory molecular response, could be also considered for off-label therapy of SARS-CoV-2 disease. Recent studies have suggested that SARS-CoV-2 shares similar cell entry mechanism with SARS-CoV. However, SARS-CoV-2 is much more contagious and may infect humans or animals through novel routes [38, 156]. Further studies are still needed to fully elucidate the mechanism behind the cytokine storm induced by SARS-CoV-2 infection and thus providing new targets for therapeutic interventions. The high contagiousness and mortality of COVID-19 and the great challenge to induce long term immunity pose an extraordinary threat for the global health. The emergence of vaccines is the most promising control measure in this global pandemic. The emerging variants not only result in increased transmission, morbidity and mortality but also have the ability to evade detection by diagnostic tests, exhibit decreased susceptibility to treatment including antivirals, monoclonal antibodies and convalescent plasma, and possess the ability to cause reinfection in previously recovered and vaccinated individuals. Children and immunocompromised individuals are at increased risk of developing multisystem inflammatory syndrome. The longer the virus propagates the higher are the chances of mutations. Vaccine hesitancy cases are often undercounted and fully vaccinated populations should still practice preventative measures. Future studies are pending regarding the lasting immunity and efficacy with the emergence of resistant COVID-19 variants. Vaccination towards COVID-19 wild type and variants and revaccination to increase the duration of vaccine-induced immunity represents the present cornerstone of the control of the global COVID-19 pandemic.
Acknowledgements
This research was supported by National Natural Science Foundation of China (File No. 31771128 and 32070969), The Science and Technology Development Fund, Macau SAR (File No. 0127/2019/A3, 0044/2019/AGJ and 0113/2018/A3), University of Macau (File No. MYRG2018-- 00134-FHS and MYRG2020-- 00158-FSH). Philip Lazarovici holds the Jacob Gitlin Chair in Physiology and is affiliated with, and partially supported by, the Grass Center for Drug Design and Synthesis of Novel Therapeutics, David R. Bloom Center of Pharmacy, and the Adolph and Klara Brettler Medical Research Center at the Hebrew University of Jerusalem, Israel.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S. et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967-76
2. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367:1814-20
3. de Wit E, van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14:523-34
4. Coronaviridae Study Group of the International Committee on Taxonomy of V. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5:536-44
5. Data OWi. Coronavirus (COVID-19) Vaccinations. https://ourworldindata.org/covid-vaccination
6. WHO. Coronavirus disease (COVID-19) Weekly Epidemiological Update and Weekly Operational Update. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports
7. Chen NS, Zhou M, Dong X, Qu JM, Gong FY, Han Y. et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507-13
8. Ruan QR, Yang K, Wang WX, Jiang LY, Song JX. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020;46:846-8
9. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033-4
10. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the Eye of the Cytokine Storm. Microbiol Mol Biol Rev. 2012;76:16-32
11. Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med. 2020;383:2255-73
12. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J. et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N Engl J Med. 2020
13. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565-74
14. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270 -+
15. Lu G, Wang Q, Gao GF. Bat-to-human: spike features determining 'host jump' of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 2015;23:468-78
16. Yang N, Shen HM. Targeting the Endocytic Pathway and Autophagy Process as a Novel Therapeutic Strategy in COVID-19. Int J Biol Sci. 2020;16:1724-31
17. Masters PS. The molecular biology of coronaviruses. Advances In Virus Research, Vol 66. 2006;66:193 -+
18. Li WH, Moore MJ, Vasilieva N, Sui JH, Wong SK, Berne MA. et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450-4
19. Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J Virol. 2010;84:12658-64
20. Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci U S A. 2005;102:11876-81
21. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S. et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271 -+
22. Cantuti-Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S. et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020
23. Daly JL, Simonetti B, Klein K, Chen KE, Williamson MK, Anton-Plagaro C. et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science. 2020
24. Kowalczuk S, Broer A, Tietze N, Vanslambrouck JM, Rasko JEJ, Broer S. A protein complex in the brush-border membrane explains a Hartnup disorder allele. FASEB J. 2008;22:2880-7
25. Yan RH, Zhang YY, Li YN, Xia L, Guo YY, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444 -+
26. Kleta R, Romeo E, Ristic Z, Ohura T, Stuart C, Arcos-Burgos M. et al. Mutations in SLC6A19, encoding B(0)AT1, cause Hartnup disorder. Nat Genet. 2004;36:999-1002
27. Ye Q, Wang B, Mao J. The pathogenesis and treatment of the `Cytokine Storm' in COVID-19. J Infect. 2020;80:607-13
28. Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. 2017;39:529-39
29. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497-506
30. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631-7
31. Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y. et al. Multiple organ infection and the pathogenesis of SARS. J Exp Med. 2005;202:415-24
32. Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN. et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell. 2020;181:1016 -+
33. Yang J, Chen T, Zhou Y. Mediators of SARS-CoV-2 entry are preferentially enriched in cardiomyocytes. Hereditas. 2021;158:4
34. Roshdy A, Zaher S, Fayed H, Coghlan JG. COVID-19 and the Heart: A Systematic Review of Cardiac Autopsies. Front Cardiovasc Med. 2020;7:626975
35. Huang I, Pranata R. Lymphopenia in severe coronavirus disease-2019 (COVID-19): systematic review and meta-analysis. Journal Of Intensive Care. 2020 8
36. Zhao Q, Meng M, Kumar R, Wu Y, Huang J, Deng Y. et al. Lymphopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: A systemic review and meta-analysis. Int J Infect Dis. 2020;96:131-5
37. Chu H, Zhou J, Wong BH, Li C, Chan JF, Cheng ZS. et al. Middle East Respiratory Syndrome Coronavirus Efficiently Infects Human Primary T Lymphocytes and Activates the Extrinsic and Intrinsic Apoptosis Pathways. J Infect Dis. 2016;213:904-14
38. Wang XL, Xu W, Hu GW, Xia S, Sun ZP, Liu ZZ. et al. SARS-CoV-2 infects T lymphocytes through its spike protein-mediated membrane fusion. Cell Mol Immunol. 2020
39. Wang K, Chen W, Zhou Y-S, Lian J-Q, Zhang Z, Du P. et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. BioRxiv. 2020
40. Adamo S, Chevrier S, Cervia C, Zurbuchen Y, Raeber ME, Yang L. et al. Lymphopenia-induced T cell proliferation is a hallmark of severe COVID-19. bioRxiv. 2020. 2020 08.04.236521
41. Chen ZY, Wherry EJ. T cell responses in patients with COVID-19. Nat Rev Immunol. 2020;20:529-36
42. Fu YJ, Cheng YX, Wu YT. Understanding SARS-CoV-2-Mediated Inflammatory Responses: From Mechanisms to Potential Therapeutic Tools. Virol Sin. 2020
43. Lin L, Lu LF, Cao W, Li TS. Hypothesis for potential pathogenesis of SARS-CoV-2 infection-a review of immune changes in patients with viral pneumonia. Emerging Microbes & Infections. 2020;9:727-32
44. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C. et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8:420-2
45. Tian SF, Hu WD, Niu L, Liu H, Xu HB, Xiao SY. Pulmonary Pathology of Early-Phase 2019 Novel Coronavirus (COVID-19) Pneumonia in Two Patients With Lung Cancer. J Thorac Oncol. 2020;15:700-4
46. Skeggs LT, Dorer FE, Levine M, Lentz KE, Kahn JR. The biochemistry of the renin-angiotensin system. Adv Exp Med Biol. 1980;130:1-27
47. Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N. et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87:E1-E9
48. Boehm M, Nabel EG. Angiotensin-converting enzyme 2-A new cardiac regulator. New Engl J Med. 2002;347:1795-7
49. Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE. et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822-8
50. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B. et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875-9
51. Sadoshima J. Cytokine actions of angiotensin II. Circ Res. 2000;86:1187-9
52. Brasier AR, Recinos A 3rd, Eledrisi MS. Vascular inflammation and the renin-angiotensin system. Arterioscler Thromb Vasc Biol. 2002;22:1257-66
53. Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B. et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112-6
54. Zou Z, Yan YW, Shu YL, Gao RB, Sun Y, Li X. et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat Commun. 2014 5
55. Pelkmans L, Helenius A. Insider information: what viruses tell us about endocytosis. Curr Opin Cell Biol. 2003;15:414-22
56. Marsh M, Helenius A. Virus entry: Open sesame. Cell. 2006;124:729-40
57. Wang S, Guo F, Liu K, Wang H, Rao S, Yang P. et al. Endocytosis of the receptor-binding domain of SARS-CoV spike protein together with virus receptor ACE2. Virus Res. 2008;136:8-15
58. Haga S, Yamamoto N, Nakai-Murakami C, Osawa Y, Tokunaga K, Sata T. et al. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc Natl Acad Sci U S A. 2008;105:7809-14
59. Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF. et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729-33
60. Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI. et al. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem. 2005;280:30113-9
61. Jia HP, Look DC, Tan P, Shi L, Hickey M, Gakhar L. et al. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. American Journal Of Physiology-Lung Cellular And Molecular Physiology. 2009;297:L84-L96
62. Eguchi S, Kawai T, Scalia R, Rizzo V. Understanding Angiotensin II Type 1 Receptor Signaling in Vascular Pathophysiology. Hypertension. 2018;71:804-10
63. Murakami M, Kamimura D, Hirano T. Pleiotropy and Specificity: Insights from the Interleukin 6 Family of Cytokines. Immunity. 2019;50:812-31
64. Hirano T, Murakami M. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release Syndrome. Immunity. 2020;52:731-3
65. Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7-11
66. Aggarwal BB, Vijayalekshmi RV, Sung B. Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin Cancer Res. 2009;15:425-30
67. Patel VB, Clarke N, Wang ZC, Fan D, Parajuli N, Basu R. et al. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: A positive feedback mechanism in the RAS. J Mol Cell Cardiol. 2014;66:167-76
68. Deshotels MR, Xia HJ, Sriramula S, Lazartigues E, Filipeanu CM. Angiotensin II Mediates Angiotensin Converting Enzyme Type 2 Internalization and Degradation Through an Angiotensin II Type I Receptor-Dependent Mechanism (vol 64, pg 1368, 2014). Hypertension. 2014;64:E8-E
69. Chung MK, Karnik S, Saef J, Bergmann C, Barnard J, Lederman MM. et al. SARS-CoV-2 and ACE2: The biology and clinical data settling the ARB and ACEI controversy. EBioMedicine. 2020;58:102907
70. Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong JC, Turner AJ. et al. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ Res. 2020;126:1456-74
71. Mahmudpour M, Roozbeh J, Keshavarz M, Farrokhi S, Nabipour I. COVID-19 cytokine storm: The anger of inflammation. Cytokine. 2020 133
72. Cohen PA, Hall LE, John JN, Rapoport AB. The Early Natural History of SARS-CoV-2 Infection: Clinical Observations From an Urban, Ambulatory COVID-19 Clinic. Mayo Clin Proc. 2020;95:1124-6
73. Lavezzo E, Franchin E, Ciavarella C, Cuomo-Dannenburg G, Barzon L, Del Vecchio C. et al. Suppression of a SARS-CoV-2 outbreak in the Italian municipality of Vo'. Nature. 2020;584:425-9
74. Hu Z, Song C, Xu C, Jin G, Chen Y, Xu X. et al. Clinical characteristics of 24 asymptomatic infections with COVID-19 screened among close contacts in Nanjing, China. Sci China Life Sci. 2020;63:706-11
75. Polak SB, Van Gool IC, Cohen D, von der Thusen JH, van Paassen J. A systematic review of pathological findings in COVID-19: a pathophysiological timeline and possible mechanisms of disease progression. Mod Pathol. 2020
76. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J. et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA. 2020
77. Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM. et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145-7
78. Oxley TJ, Mocco J, Majidi S, Kellner CP, Shoirah H, Singh IP. et al. Large-Vessel Stroke as a Presenting Feature of Covid-19 in the Young. N Engl J Med. 2020;382:e60
79. Toscano G, Palmerini F, Ravaglia S, Ruiz L, Invernizzi P, Cuzzoni MG. et al. Guillain-Barre Syndrome Associated with SARS-CoV-2. N Engl J Med. 2020;382:2574-6
80. Shaigany S, Gnirke M, Guttmann A, Chong H, Meehan S, Raabe V. et al. An adult with Kawasaki-like multisystem inflammatory syndrome associated with COVID-19. Lancet. 2020;396:e8-e10
81. Morris SB, Schwartz NG, Patel P, Abbo L, Beauchamps L, Balan S. et al. Case Series of Multisystem Inflammatory Syndrome in Adults Associated with SARS-CoV-2 Infection-United Kingdom and United States, March-August 2020. Mmwr-Morbidity And Mortality Weekly Report. 2020;69:1450-6
82. Soy M, Atagunduz P, Atagunduz I, Sucak GT. Hemophagocytic lymphohistiocytosis: a review inspired by the COVID-19 pandemic. Rheumatol Int. 2021;41:7-18
83. Sandoval Y, Januzzi JL Jr, Jaffe AS. Cardiac Troponin for Assessment of Myocardial Injury in COVID-19: JACC Review Topic of the Week. J Am Coll Cardiol. 2020;76:1244-58
84. Guo T, Fan Y, Chen M, Wu X, Zhang L, He T. et al. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020;5:811-8
85. Touyz RM, Boyd MO, Guzik T, Padmanabhan S, McCallum L, Delles C. et al. Cardiovascular and renal risk factors and complications associated with COVID-19. CJC Open. 2021
86. Patel A, Jernigan DB, nCo VCDCRT. Initial Public Health Response and Interim Clinical Guidance for the 2019 Novel Coronavirus Outbreak-United States, December 31, 2019-February 4, 2020. MMWR Morb Mortal Wkly Rep. 2020;69:140-6
87. Long QX, Liu BZ, Deng HJ, Wu GC, Deng K, Chen YK. et al. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat Med. 2020;26:845-8
88. Yao X, Ye F, Zhang M, Cui C, Huang B, Niu P. et al. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Clin Infect Dis. 2020;71:732-9
89. Savarino A, Boelaert JR, Cassone A, Majori G, Cauda R. Effects of chloroquine on viral infections: an old drug against today's diseases? Lancet Infect Dis. 2003;3:722-7
90. Liu J, Cao R, Xu M, Wang X, Zhang H, Hu H. et al. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020;6:16
91. Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ. et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14:1435-55
92. Gautret P, Lagier JC, Parola P, Hoang VT, Meddeb L, Mailhe M. et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int J Antimicrob Agents. 2020;56:105949
93. Cavalcanti AB, Zampieri FG, Rosa RG, Azevedo LCP, Veiga VC, Avezum A. et al. Hydroxychloroquine with or without Azithromycin in Mild-to-Moderate Covid-19. N Engl J Med. 2020
94. Geleris J, Sun Y, Platt J, Zucker J, Baldwin M, Hripcsak G. et al. Observational Study of Hydroxychloroquine in Hospitalized Patients with Covid-19. N Engl J Med. 2020;382:2411-8
95. Jeronimo CMP, Farias MEL, Val FFA, Sampaio VS, Alexandre MAA, Melo GC. et al. Methylprednisolone as Adjunctive Therapy for Patients Hospitalized With COVID-19 (Metcovid): A Randomised, Double-Blind, Phase IIb, Placebo-Controlled Trial. Clin Infect Dis. 2020
96. Group RC, Horby P, Lim WS, Emberson JR, Mafham M, Bell JL. et al. Dexamethasone in Hospitalized Patients with Covid-19-Preliminary Report. N Engl J Med. 2020
97. Mareev VY, Orlova YA, Pavlikova EP, Matskeplishvili ST, Krasnova TN, Malahov PS. et al. [Steroid pulse-therapy in patients With coronAvirus Pneumonia (COVID-19), sYstemic inFlammation And Risk of vEnous thRombosis and thromboembolism (WAYFARER Study)]. Kardiologiia. 2020;60:15-29
98. Ip A, Berry DA, Hansen E, Goy AH, Pecora AL, Sinclaire BA. et al. Hydroxychloroquine and tocilizumab therapy in COVID-19 patients-An observational study. PLoS ONE. 2020;15:e0237693
99. Maeda T, Obata R, Rizk DD, Kuno T. The association of interleukin-6 value, interleukin inhibitors, and outcomes of patients with COVID-19 in New York City. J Med Virol. 2020
100. Pettit NN, Nguyen CT, Mutlu GM, Wu D, Kimmig L, Pitrak D. et al. Late onset infectious complications and safety of tocilizumab in the management of COVID-19. J Med Virol. 2020
101. Potere N, Di Nisio M, Rizzo G, La Vella M, Polilli E, Agostinone A. et al. Low-Dose Subcutaneous Tocilizumab to Prevent Disease Progression in Patients with Moderate COVID-19 Pneumonia and Hyperinflammation. Int J Infect Dis. 2020
102. Mastroianni A, Greco S, Apuzzo G, De Santis S, Oriolo C, Zanolini A. et al. Subcutaneous tocilizumab treatment in patients with severe COVID-19-related cytokine release syndrome: An observational cohort study. EClinicalMedicine. 2020;24:100410
103. Stone JH, Frigault MJ, Serling-Boyd NJ, Fernandes AD, Harvey L, Foulkes AS. et al. Efficacy of Tocilizumab in Patients Hospitalized with Covid-19. N Engl J Med. 2020;383:2333-44
104. Salama C, Han J, Yau L, Reiss WG, Kramer B, Neidhart JD. et al. Tocilizumab in Patients Hospitalized with Covid-19 Pneumonia. N Engl J Med. 2021;384:20-30
105. Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M. et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269-71
106. Ivashchenko AA, Dmitriev KA, Vostokova NV, Azarova VN, Blinow AA, Egorova AN. et al. AVIFAVIR for Treatment of Patients with Moderate COVID-19: Interim Results of a Phase II/III Multicenter Randomized Clinical Trial. Clin Infect Dis. 2020
107. Cai Q, Yang M, Liu D, Chen J, Shu D, Xia J. et al. Experimental Treatment with Favipiravir for COVID-19: An Open-Label Control Study. Engineering (Beijing). 2020;6:1192-8
108. Mulangu S, Dodd LE, Davey RT Jr, Tshiani Mbaya O, Proschan M, Mukadi D. et al. A Randomized, Controlled Trial of Ebola Virus Disease Therapeutics. N Engl J Med. 2019;381:2293-303
109. de Wit E, Feldmann F, Cronin J, Jordan R, Okumura A, Thomas T. et al. Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc Natl Acad Sci U S A. 2020;117:6771-6
110. Grein J, Ohmagari N, Shin D, Diaz G, Asperges E, Castagna A. et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N Engl J Med. 2020;382:2327-36
111. Wang Y, Zhang D, Du G, Du R, Zhao J, Jin Y. et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395:1569-78
112. Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC. et al. Remdesivir for the Treatment of Covid-19-Final Report. N Engl J Med. 2020;383:1813-26
113. Young B, Tan TT, Leo YS. The place for remdesivir in COVID-19 treatment. Lancet Infect Dis. 2021;21:20-1
114. Kalil AC, Patterson TF, Mehta AK, Tomashek KM, Wolfe CR, Ghazaryan V. et al. Baricitinib plus Remdesivir for Hospitalized Adults with Covid-19. N Engl J Med. 2021;384:795-807
115. Hasan SS, Radford S, Kow CS, Zaidi STR. Venous thromboembolism in critically ill COVID-19 patients receiving prophylactic or therapeutic anticoagulation: a systematic review and meta-analysis. J Thromb Thrombolysis. 2020
116. Hadid T, Kafri Z, Al-Katib A. Coagulation and anticoagulation in COVID-19. Blood Rev. 2021;47:100761
117. Fried JA, Ramasubbu K, Bhatt R, Topkara VK, Clerkin KJ, Horn E. et al. The Variety of Cardiovascular Presentations of COVID-19. Circulation. 2020;141:1930-6
118. Sarkar S, Soni KD, Khanna P. Convalescent plasma is a clutch at straws in COVID-19 management! A systematic review and meta-analysis. J Med Virol. 2020
119. Libster R, Perez Marc G, Wappner D, Coviello S, Bianchi A, Braem V. et al. Early High-Titer Plasma Therapy to Prevent Severe Covid-19 in Older Adults. N Engl J Med. 2021;384:610-8
120. Malsy J, Veletzky L, Heide J, Hennigs A, Gil-Ibanez I, Stein A. et al. Sustained response after remdesivir and convalescent plasma therapy in a B-cell depleted patient with protracted COVID-19. Clin Infect Dis. 2020
121. Marovich M, Mascola JR, Cohen MS. Monoclonal Antibodies for Prevention and Treatment of COVID-19. JAMA. 2020;324:131-2
122. Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R. et al. REGN-COV2, a Neutralizing Antibody Cocktail, in Outpatients with Covid-19. N Engl J Med. 2021;384:238-51
123. Chen P, Nirula A, Heller B, Gottlieb RL, Boscia J, Morris J. et al. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with Covid-19. N Engl J Med. 2021;384:229-37
124. To KK, Tsang OT, Leung WS, Tam AR, Wu TC, Lung DC. et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: an observational cohort study. Lancet Infect Dis. 2020;20:565-74
125. Ko JH, Joo EJ, Park SJ, Baek JY, Kim WD, Jee J. et al. Neutralizing Antibody Production in Asymptomatic and Mild COVID-19 Patients, in Comparison with Pneumonic COVID-19 Patients. J Clin Med. 2020 9
126. Ibarrondo FJ, Fulcher JA, Goodman-Meza D, Elliott J, Hofmann C, Hausner MA. et al. Rapid Decay of Anti-SARS-CoV-2 Antibodies in Persons with Mild Covid-19. N Engl J Med. 2020;383:1085-7
127. Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR. et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell. 2020;181:1489-501 e15
128. Krammer F. SARS-CoV-2 vaccines in development. Nature. 2020;586:516-27
129. WHO. COVID-19 vaccine tracker and landscape. https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines
130. Folegatti PM, Ewer KJ, Aley PK, Angus B, Becker S, Belij-Rammerstorfer S. et al. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: a preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet. 2020;396:467-78
131. Jackson LA, Anderson EJ, Rouphael NG, Roberts PC, Makhene M, Coler RN. et al. An mRNA Vaccine against SARS-CoV-2-Preliminary Report. N Engl J Med. 2020
132. Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S. et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med. 2020;383:2603-15
133. Frenck RW Jr, Klein NP, Kitchin N, Gurtman A, Absalon J, Lockhart S. et al. Safety, Immunogenicity, and Efficacy of the BNT162b2 Covid-19 Vaccine in Adolescents. N Engl J Med. 2021
134. Haas EJ, Angulo FJ, McLaughlin JM, Anis E, Singer SR, Khan F. et al. Impact and effectiveness of mRNA BNT162b2 vaccine against SARS-CoV-2 infections and COVID-19 cases, hospitalisations, and deaths following a nationwide vaccination campaign in Israel: an observational study using national surveillance data. Lancet. 2021;397:1819-29
135. Abu Jabal K, Ben-Amram H, Beiruti K, Batheesh Y, Sussan C, Zarka S. et al. Impact of age, ethnicity, sex and prior infection status on immunogenicity following a single dose of the BNT162b2 mRNA COVID-19 vaccine: real-world evidence from healthcare workers, Israel, December 2020 to January 2021. Euro Surveill. 2021 26
136. Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R. et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N Engl J Med. 2021;384:403-16
137. Doria-Rose N, Suthar MS, Makowski M, O'Connell S, McDermott AB, Flach B. et al. Antibody Persistence through 6 Months after the Second Dose of mRNA-1273 Vaccine for Covid-19. N Engl J Med. 2021;384:2259-61
138. Logunov DY, Dolzhikova IV, Shcheblyakov DV, Tukhvatulin AI, Zubkova OV, Dzharullaeva AS. et al. Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: an interim analysis of a randomised controlled phase 3 trial in Russia. Lancet. 2021;397:671-81
139. Sadoff J, Gray G, Vandebosch A, Cardenas V, Shukarev G, Grinsztejn B. et al. Safety and Efficacy of Single-Dose Ad26.COV2.S Vaccine against Covid-19. N Engl J Med. 2021;384:2187-201
140. Ramasamy MN, Minassian AM, Ewer KJ, Flaxman AL, Folegatti PM, Owens DR. et al. Safety and immunogenicity of ChAdOx1 nCoV-19 vaccine administered in a prime-boost regimen in young and old adults (COV002): a single-blind, randomised, controlled, phase 2/3 trial. Lancet. 2021;396:1979-93
141. Voysey M, Costa Clemens SA, Madhi SA, Weckx LY, Folegatti PM, Aley PK. et al. Single-dose administration and the influence of the timing of the booster dose on immunogenicity and efficacy of ChAdOx1 nCoV-19 (AZD1222) vaccine: a pooled analysis of four randomised trials. Lancet. 2021;397:881-91
142. Greinacher A, Thiele T, Warkentin TE, Weisser K, Kyrle PA, Eichinger S. Thrombotic Thrombocytopenia after ChAdOx1 nCov-19 Vaccination. N Engl J Med. 2021;384:2092-101
143. Scully M, Singh D, Lown R, Poles A, Solomon T, Levi M. et al. Pathologic Antibodies to Platelet Factor 4 after ChAdOx1 nCoV-19 Vaccination. N Engl J Med. 2021;384:2202-11
144. Pachetti M, Marini B, Benedetti F, Giudici F, Mauro E, Storici P. et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Transl Med. 2020;18:179
145. Zhang L, Jackson CB, Mou H, Ojha A, Peng H, Quinlan BD. et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat Commun. 2020;11:6013
146. Forchette L, Sebastian W, Liu T. A Comprehensive Review of COVID-19 Virology, Vaccines, Variants, and Therapeutics. Curr Med Sci. 2021
147. Nonaka CKV, Franco MM, Graf T, de Lorenzo Barcia CA, de Avila Mendonca RN, de Sousa KAF. et al. Genomic Evidence of SARS-CoV-2 Reinfection Involving E484K Spike Mutation, Brazil. Emerg Infect Dis. 2021;27:1522-4
148. Nelde A, Bilich T, Heitmann JS, Maringer Y, Salih HR, Roerden M. et al. SARS-CoV-2-derived peptides define heterologous and COVID-19-induced T cell recognition. Nat Immunol. 2021;22:74-85
149. Plante JA, Liu Y, Liu J, Xia H, Johnson BA, Lokugamage KG. et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature. 2021;592:116-21
150. Zou J, Xie X, Fontes-Garfias CR, Swanson KA, Kanevsky I, Tompkins K. et al. The effect of SARS-CoV-2 D614G mutation on BNT162b2 vaccine-elicited neutralization. NPJ Vaccines. 2021;6:44
151. Planas D, Bruel T, Grzelak L, Guivel-Benhassine F, Staropoli I, Porrot F. et al. Sensitivity of infectious SARS-CoV-2 B.1.1.7 and B.1.351 variants to neutralizing antibodies. Nat Med. 2021;27:917-24
152. Supasa P, Zhou D, Dejnirattisai W, Liu C, Mentzer AJ, Ginn HM. et al. Reduced neutralization of SARS-CoV-2 B.1.1.7 variant by convalescent and vaccine sera. Cell. 2021;184:2201-11 e7
153. Turner PJ, Larson H, Dube E, Fisher A. Vaccine Hesitancy: Drivers and how the Allergy Community can help. J Allergy Clin Immunol Pract. 2021
154. Troiano G, Nardi A. Vaccine hesitancy in the era of COVID-19. Public Health. 2021;194:245-51
155. Vaishya R, Sibal A, Sharma H, Singh SK. Lack of vaccination and associated comorbidities predispose to the need for intensive care in individuals infected with the delta variant-A case cohort study from a tertiary care hospital in New Delhi, India. Diabetes Metab Syndr. 2021;15:102203
156. Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A. et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci U S A. 2020;117:11727-34
Author Biography
 Wenhua Zheng is a professor at University of Macau, Faculty of Health Sciences. Main research fields: neuropharmacology; aging, neuronal degenerative disorders including Alzheimer's disease and degenerative retinal diseases; molecular mechanisms of neuronal cell death; drug development.
Wenhua Zheng is a professor at University of Macau, Faculty of Health Sciences. Main research fields: neuropharmacology; aging, neuronal degenerative disorders including Alzheimer's disease and degenerative retinal diseases; molecular mechanisms of neuronal cell death; drug development.
 Philip Lazarovici obtained his PhD degree in pharmacology and toxicology 1980 from The Hebrew University, post graduated 1984 on neurobiology at the Weizmann Institute of Science, Israel and was a visiting scientist from 1984-1988 in the Section of Growth Factors at the National Institutes of Child Health and Human Development , NIH, Bethesda, USA. He was a Fogorty visiting scientist at NIH (1990-1998), Drexel University (2004-2011) and Temple University (2012-2015), Philadelphia, USA. Present position: Professor Emeritus, Jacob Gitlin Chair in Physiology, School of Pharmacy, The Hebrew University of Jerusalem, Israel. Main research fields: neuronal and non-neuronal pharmacological activities of nerve growth factor, trkA, p75NTR and α9β1-integrin signaling and role in neuroprotection, angiogenesis and tumor development; toxins, lead compounds, drug discovery and development, cell therapy, stem cells, neurological disorders.
Philip Lazarovici obtained his PhD degree in pharmacology and toxicology 1980 from The Hebrew University, post graduated 1984 on neurobiology at the Weizmann Institute of Science, Israel and was a visiting scientist from 1984-1988 in the Section of Growth Factors at the National Institutes of Child Health and Human Development , NIH, Bethesda, USA. He was a Fogorty visiting scientist at NIH (1990-1998), Drexel University (2004-2011) and Temple University (2012-2015), Philadelphia, USA. Present position: Professor Emeritus, Jacob Gitlin Chair in Physiology, School of Pharmacy, The Hebrew University of Jerusalem, Israel. Main research fields: neuronal and non-neuronal pharmacological activities of nerve growth factor, trkA, p75NTR and α9β1-integrin signaling and role in neuroprotection, angiogenesis and tumor development; toxins, lead compounds, drug discovery and development, cell therapy, stem cells, neurological disorders.
 Yizhou Jiang obtained his bachelor's degree from Southern University of Science and Technology (SUSTech) in 2017. He is currently a SUSTech-University of Macau joint Ph. D. student. His major research interests are aging and neurobiology.
Yizhou Jiang obtained his bachelor's degree from Southern University of Science and Technology (SUSTech) in 2017. He is currently a SUSTech-University of Macau joint Ph. D. student. His major research interests are aging and neurobiology.
 Limor Rubin obtained her M.D. degree in 2015 from the Hebrew University of Jerusalem. Thereafter, she completed her residency in Internal Medicine at Hadassah-Hebrew University and from 2020, she is performing a fellowship in Allergy and Clinical Immunology. Present position: Senior Physician, Internal Medicine Department B, Hadassah-Hebrew University Medical Center, Jerusalem, Israel. Main research fields: internal medicine, respiratory diseases, allergy, asthma, common variable immunodeficiency, COVID -19 immune responses, diagnosis and therapy.
Limor Rubin obtained her M.D. degree in 2015 from the Hebrew University of Jerusalem. Thereafter, she completed her residency in Internal Medicine at Hadassah-Hebrew University and from 2020, she is performing a fellowship in Allergy and Clinical Immunology. Present position: Senior Physician, Internal Medicine Department B, Hadassah-Hebrew University Medical Center, Jerusalem, Israel. Main research fields: internal medicine, respiratory diseases, allergy, asthma, common variable immunodeficiency, COVID -19 immune responses, diagnosis and therapy.
![]() Corresponding author: Prof. Wenhua Zheng, Faculty of Health Sciences, University of Macau, Room 3057, Building E12, Avenida de Universidade, Taipa, Macau. China. Tel: +853-88224919; Email: wenhuazhengmo
Corresponding author: Prof. Wenhua Zheng, Faculty of Health Sciences, University of Macau, Room 3057, Building E12, Avenida de Universidade, Taipa, Macau. China. Tel: +853-88224919; Email: wenhuazhengmo