Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Methods and materials
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(2):731-741. doi:10.7150/ijbs.64956 This issue Cite
Research Paper
ATPase inhibitory factor 1 protects the heart from acute myocardial ischemia/reperfusion injury through activating AMPK signaling pathway
Jia-Wei Wu, Hao Hu, Jin-sheng Hua, Li-Kun Ma ![]()
Department of Cardiology, The First Affiliated Hospital of USTC, Division of Life Science and Medicine, University of Science and Technology of China, Hefei, 230001, China
Received 2021-7-17; Accepted 2021-11-24; Published 2022-1-1
Abstract

Rationale: Myocardial ischemia/reperfusion (I/R) injury is a common clinic scenario that occurs in the context of reperfusion therapy for acute myocardial infarction (AMI). The mitochondrial F1Fo-ATPase inhibitory factor 1 (IF1) blocks the reversal of the F1Fo-ATP synthase to prevent detrimental consumption of cellular ATP and associated demise. In the present study, we study the role and mechanism of IF1 in myocardial I/R injury.
Methods: Mice were ligated the left anterior descending coronary artery to build the I/R model in vivo. Rat hearts were isolated and perfused with constant pressure according to Langendorff. Also, neonatal cardiomyocytes hypoxia-reoxygenation (H/R) model was also used. Myocardial infarction area, cardiac function, cellular function, and cell viability was conducted and compared.
Results: Our data revealed that IF1 is upregulated in hearts after I/R and cardiomyocytes with hypoxia/re-oxygenation (H/R). IF1 delivered with adenovirus and adeno-associated virus serotype 9 (AAV9) ameliorated cardiac dysfunction and pathological development induced by I/R ex vivo and in vivo. Mechanistically, IF1 stimulates glucose uptake and glycolysis activity and stimulates AMPK activation during in vivo basal and I/R and in vitro OGD/R conditions, and activation of AMPK by IF1 is responsible for its cardioprotective effects against H/R-induced injury.
Conclusions: These results suggest that increased IF1 in the I/R heart confer cardioprotective effects via activating AMPK signaling. Therefore, IF1 can be used as a potential therapeutic target for the treatment of pathological ischemic injury and heart failure.
Keywords: ATPase inhibitory factor 1, ischemia/reperfusion injury, heart, AMPK
Introduction
Acute myocardial infarction (AMI) caused by coronary occlusion is the main cause of disability and death worldwide. After AMI, a large number of dead cardiomyocytes are replaced by fibrous scar tissue, leading to ventricular remodeling and heart failure (HF) 1, 2. In order to rescue ischemic myocardium and limit infarct size, timely and effective reperfusion is the current treatment option for patients with AMI. However, this leads to additional injury, known as ischemia/reperfusion (I/R) injury 3, 4. Therefore, it is necessary to identify the methods to protect cardiomyocytes from I/R injury in the early stage of reperfusion and to elucidate the potential protective mechanisms to promote the development of new therapeutic methods for ischemic heart disease.
The efficiency of the mitochondrial F1Fo-ATP synthase that generates large amounts of ATP guaranteed energy homeostasis of mammalian cells. ATPase inhibitory factor 1 (IF1) is a protein that binds with F1F0-ATP synthase 5-7. It was reported that when F1F0-ATP synthase reverses its function and hydrolyzes ATP, IF1 binds and inhibits ATP synthase with a large drop in the inner membrane potential 8, 9. Thus, IF1 prevents total ATP depletion and consequent cell death during hypoxia or starvation, a function which is essential under pathologic conditions, such as cardiac ischemia 10, 11. This is consistent with the report that endogenous IF1 overexpression is involved in the reprograming of energy metabolism to enhance glycolysis by limiting ATP production by F1F0-ATP synthase in both cell culture and tissue-specific mouse models 12, 13. This promotes the production of mitochondrial reactive oxygen species (ROSs) that modulate various signaling pathways favoring cellular processes 14. Many studies have evaluated the physiologic role and gene expression of endogenous IF1, in spite of this, the underlying molecular events by which IF1 could be protective in I/R injury has not been yet properly addressed.
AMPK is a stress response kinase, which can regulate a variety of important physiological and metabolic processes, including energy homeostasis, cell apoptosis, and cell metabolism 15-18. It has been demonstrated that AMPK can up-regulate cellular antioxidant enzyme systems, thereby reducing oxidant-induced damage in I/R injury 19. Therefore, this study aims to investigate the cardioprotective effect of IF1 on I/R injury and whether AMPK activation is involved in the protective effect of IF1.
Methods and materials
Animals. Adult male C57BL/6 mice from (Shanghai Slac Laboratory Animal Co. Ltd.) were nursed according to the Guidelines for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication, 8th Edition, 2011), and the animal procedure was approved by the Institutional Review Board of The First Affiliated Hospital of USTC.
The mouse I/R injury model. Mice were fasting overnight before surgery. 50 mg/kg pentobarbital sodium was injected intraperitoneally. Injected and fixed in the lateral position on the operating table. The fourth intercostal space was opened to expose the heart, and the left anterior descending artery (LAD) was sutured with silk thread on the inflated balloon. After 30 minutes, LAD was reperfused by deflation and balloon withdrawal. Echocardiography was carried out in each animal after the conduction of myocardial IR with an ultrasound machine (Vevo 2100, USA).
Adenovirus and adeno-associated virus vector construction, infection, and injection
Recombinant adenoviruses expressing mouse IF1 (Ad-IF1) and NC (Ad-NC) were prepared as previously described 20. The AAV9 expressing IF1 (AAV9-IF1) and control (AAV9-Ctrl) were constructed as described previously 21. Surgery and adenovirus delivery was performed as previously described. In short, mice anesthetized with pentobarbital sodium (50 mg/kg, i.p.) underwent thoracotomy. A 27-G needle containing 100 μL of diluted adenovirus (3 x 1010 pfu/mL) or sterile saline was injected into left ventricle. When the solution was injected, the aorta and pulmonary aorta were clamped at the distal end of the syringe for 10 s, and then the chest was closed. The hemodynamics of the heart was studied on the 4th day after adenovirus injection. AAV9 was injected into adult mice by using a 29-gauge insulin syringe (BD, Ultral fine needle) as described previously 21. 6-week-old C57BL/6 mice were injected with 100 μl containing 1x1012 vg AAV9-NC or 200 μl containing 3 × 1012 vg AAV9-IF1 viruses using a 29-gauge insulin syringe (BD, Ultral fine needle) through tail vein. 3 weeks after AAV injection, hearts were treated with I/R injury in vivo (30min/24h). The heart function, infarct size, LDH level, and serum cTnT level were measured after 24 hours.
Isolation, culture, and adenoviral infection of neonatal rat cardiomyocytes (NRCMs)
NRCMs were isolated from neonatal S-D rat hearts. After 2 h of culture to achieve myocytes attachment, adenovirus was added at a multiplicity of infection (MOI) of 100 for 2 h. All experiments were performed after 36 h of infection.
I/R injury model in Langendorff-perfused mouse hearts
After anesthetizing the mice with pentobarbital sodium (50 mg/kg i.p.), the hearts were removed quickly and perfused with Krebs-Henseleit solution at 37℃ using a Langendorff apparatus as previously described 20. The development pressure of LV (LVDP), LVEDP and the maximum rate of pressure development or decay with time (+dP/dtmax) were evaluated with the PowerLab system (AD Instrument) by powerlab system (AD instrument). At the end of reperfusion, the heart was quickly removed and frozen in liquid nitrogen for further analysis.
TUNEL assay
Fixed cultured cells were incubated with a TUNEL stain (In situ Cell Death Detection Kit, POD, Roche Diagnostics GmbH, Mannheim, Germany) and co-stained with Hoechst 33342 and visualized using a Leica SP5 confocal microscopy system. The images were analyzed using ImageJ software.
Infarct size measurement
The isolated mice hearts subjected to 30 min of ischemia followed by 1h of reperfusion ex vivo and mice hearts subjected to 30 min of ischemia followed by 24 h of reperfusion in vivo were frozen, and the LV tissue was cut into 5 slices. The slices were incubated in 1% TTC for 20 min Infarct size was calculated using Image-J software, and the infarct area was expressed as a percentage of area at risk (ARR). Cardiac function was evaluated by echocardiography using VEVO 2100 (Visual Sonics) under anesthesia with 1.5% isoflurane. For fibrosis measurement, Masson's Trichrome staining was performed.
Immunoblotting analysis
The LV tissue and the cardiomyocytes were lysed by RIPA buffer with protein inhibitors mixture as described previously 22. The protein fractions were analyzed by standard immunoblotting when transferred to polyvinylidene fluoride membrane (BioRad) and probed with relevant antibodies. Relative level of proteins was determined by antibodies against IF1 (1:2000), b-actin (1:4000), phospho-AMPK (Thr172, 1:1000), AMPK (1:2000), and Cleaved-caspase 3 (1:1000). Antibodies were purchased from Cell Signaling Technology.
Statistical analysis
Data were expressed as means ± SEM and performed with Graphpad Prism software (version 8.0). For multiple comparisons, ANOVA or repeated ANOVA followed by the LSD post-hoc test was used. A P-value of p < 0.05 was considered statistically significant.
Results
IF1 is downregulated during I/R and H/R
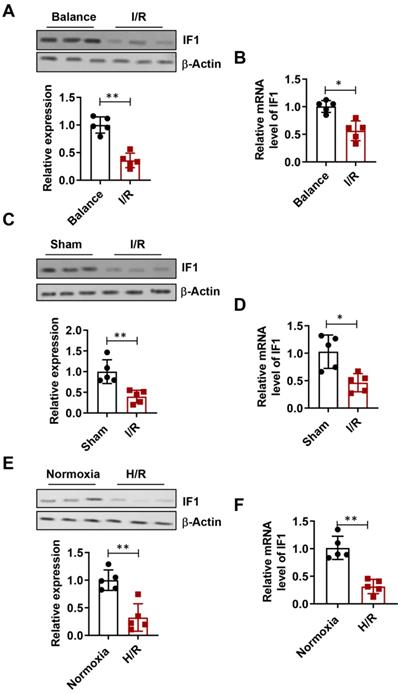
The expression level of IF1 in isolated perfused I/R (30 min/1 h) hearts (Fig. 1A), I/R (30min/24h) hearts (Fig. 1C), simulated I/R (30 min/1 h) cardiomyocytes (Fig. 1E) were analyzed. IF1 protein levels were downregulated by I/R injury in hearts. Similar expression pattern of IF1 mRNA was observed in the I/R cardiomyocytes and hearts (Fig. 1B, D and F). These results suggest a possible involvement of IF1 in I/R injury.
IF1 protein expression is downregulated during I/R and H/R. A, Immunoblotting analysis of IF1 protein in isolated perfused I/R (30/45 min) hearts. β-Actin was used as an internal reference. B, Q-PCR analysis of IF1 mRNA in isolated perfused I/R (30/45 min) hearts; C, Immunoblotting analysis of IF1 protein in I/R (30min/24h) hearts; D, Q-PCR analysis of IF1 mRNA in I/R (30min/24h) hearts; E, Immunoblotting analysis of IF1 protein in simulated I/R (20/30 min) cardiomyocytes. F, Q-PCR analysis of IF1 mRNA in simulated I/R (20/30 min) cardiomyocytes. n = 5 each. *P <0.05, **P<0.01.
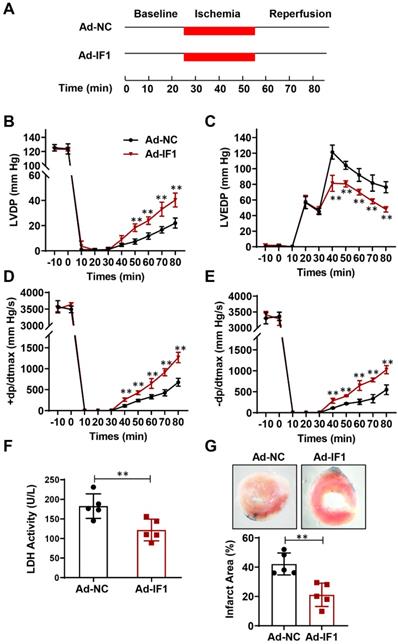
Effect of IF1 overexpression with adenovirus encoding IF1 on cardiac function and LDH release during I/R ex vivo
We then studied the role of IF1 in cardiac I/R injury with adenovirus-mediated IF1 overexpression (Ad-IF1) (Fig. 2A). Western blot analysis showed that IF1 was significantly overexpressed in the hearts of mice infected with Ad-IF1 (Fig. S1A). There are no significant differences were observed in LVDP (Fig. 2B), LVEDP (Fig. 2C), ± dP/dt (Fig. 2D and E) between the Ad-NC control and Ad-IF1 groups during the pre-ischaemic phase in Langendorff perfused mice hearts, while I/R-induced contractile dysfunction (Fig. 2B-E) in Ad-NC hearts were significantly improved by Ad-IF1. Meanwhile, I/R-induced increases of LDH activity in coronary perfusate after 45 min of reperfusion as well as infarct size after 2 h of reperfusion (Fig. 2F and G) were significantly decreased by Ad-IF1. These results suggest that a higher level of IF1 is needed for cardiac repair after I/R injury, and the change of IF1 in a certain level does not affect basic myocardial function.
Effect of IF1 overexpression with adenovirus encoding IF1 on cardiac function and LDH release during I/R ex vivo. A, Schematic of I/R stimulation and IF1 overexpression with adenovirus. B, Time course of LVDP (B), LVEDP (C), and +dP/dtmax (D) and 2dP/dtmax (E); n = 5 each. F, Fold changes of lactate dehydrogenase (LDH) activities in Ad-IF1 group compared with the Ad-NC group. n = 4-5. G, Representative images and analysis of the infarct size in isolated I/R (30 min/2 h) hearts; n = 5 each. **P<0.01.
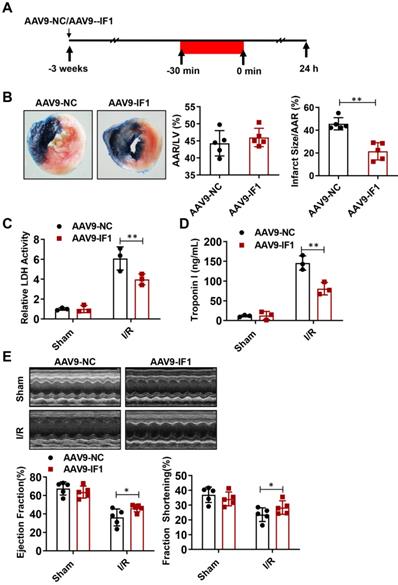
Effect of IF1 overexpression with AAV9 vector encoding IF1 on infarct size and cardiac function during myocardial I/R (mice)
To determine whether IF1 contributes to the recovery of myocardial I/R injury, we overexpressed IF1 in the heart by tail vein injection of AAV9-IF1 at 3 weeks before the myocardial I/R (Fig. 3A). Western blot analysis showed that IF1 was significantly overexpressed in the hearts of mice infected with AAV-IF1 (Fig. S1B). We first analyzed whether AAV9-IF1 decrease infarct size at day 1 post-I/R. The I/R-insulted hearts with AAV9-NC and AAV9-IF1 infected have similar AAR to LV (Figure 3B), while the infarct area (Fig. 3B), serum LDH activity (Fig. 3C), and serum Troponin I level (Fig. 3D) were all significantly reduced in the AAV9-IF1 group compared with these in the AAV9-NC group. We next analyzed the long-term protective effects of AAV9-IF1 in the I/R hearts. The mice injected with AAV9-NC and AAV9-IF1 showed comparable LVEF and LVFS with these in the Sham groups, whereas the AAV9-IF1 treatment significantly improved these parameters worsened in the I/R control group at the 4-week after I/R (Fig. 3E). Notably, the fibrotic area was significantly smaller in AAV9-IF1-infected hearts than in controls at 4 weeks post I/R (Figure S3A). Therefore, IF1 may function as an endogenous protective factor in the heart against I/R injury.
Effect of IF1 overexpression with AAV9 vector encoding IF1 on infarct size and cardiac function during myocardial I/R (mice). A, Schematic of in vivo I/R stimulation and IF1 overexpression with AAV9. B, Representative and quantification of 2, 3, 5-triphenyte-trazoliumchloride (TTC)/Evans blue staining for infarct size in the hearts at 24 hours post-I/R. n = 5. C, Fold changes of LDH activities in AAV9-IF1 group compared with the AAV9-NC group. n = 3. D, Serum cTnI ELISA analysis in AAV9-IF1 group compared with the AAV9-NC group. n = 3. E, left ventricle (LV) ejection fraction (LVEF), and LV fractional shortening (LVFS) measured by echocardiography (Echo). n = 5. *P < 0.05, **P < 0.01.
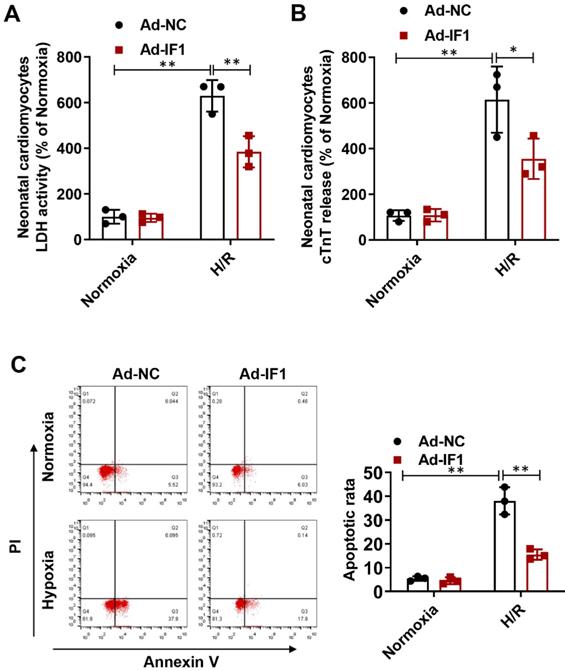
IF1 is involved in the response to H/R-induced injury in cardiomyocytes
To further explore whether the protective effects of IF1 against I/R injury is related to cardiomyocytes, we performed H/R injury model on NRCMs. We found the H/R injury-increased LDH release (Fig. 4A) at the 1 hour of reperfusion was decreased by Ad-IF1. Concomitantly, Ad-IF1 significantly decreased the cTnT release induced by H/R injury (Fig. 4B). Similar results were found by Annexin V/PI staining, that is, the proportion of the right quadrant cells was increased in the H/R-treated NRCMs and recovered with Ad-IF1 (Fig. 4C). Moreover, IF1 knockdown aggravated H/R-induced cardiomyocytes injury, as evidenced by increased LDH release, cTnT release, and increased number of apoptotic cells (Fig. S2). Therefore, IF1 is an endogenous protective factor for cardiomyocytes against I/R injury.
IF1 is involved in the response to H/R-induced injury (cTnT and LDH release, apoptosis) in cardiomyocytes. A, LDH released from NRCMs at the 1 hour of reperfusion. B, cTnT released from NRCMs induced by H/R injury. C, Representative flow cytometric assessment of apoptosis via Annexin V/PI staining and Flow cytometric quantification. n = 3. *P <0.05, **P<0.01.
IF1 stimulates glucose uptake and glycolysis activity
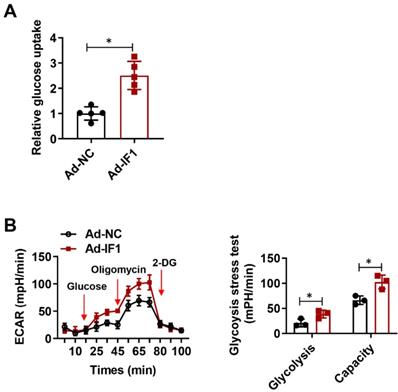
IF1 is reported to exert its cardioprotective effects in the I/R injury whereas ATP wastage accounts for a large proportion of the bio-energetic damage, but whether by overexpression IF1 to target the F1Fo-ATPase is protective or not in I/R injury has not been yet properly addressed. To determine whether IF1 could save energy consumption after I/R injury, we performed glucose uptake assay and glycolytic and mitochondrial respiration measurement with Seahorse Extracellular Flux Analyzer, with ECAR to reflect aerobic glycolysis. IF1 overexpression remarkably promotes glucose uptake (Fig. 5A), increased glycolysis and glycolytic capacity, which was reflected by ECAR measurement (Fig. 5B).
IF1 stimulates glucose uptake and glycolysis activity. A, Glucose uptake assay. n = 3. B, Glycolytic and mitochondrial respiration measurement with Seahorse Extracellular Flux Analyzer, with ECAR to reflect aerobic glycolysis. n = 5. *P <0.05.
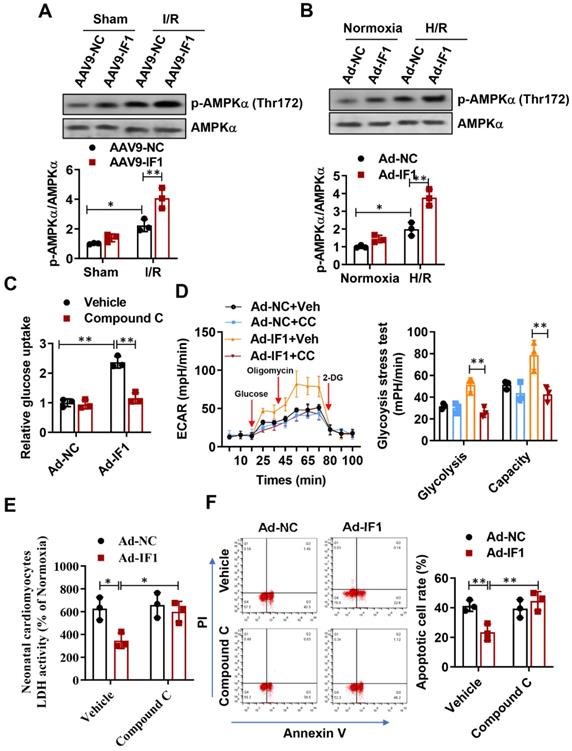
IF1 stimulated AMPK activation during in vivo basal and I/R and in vitro OGD/R conditions, and activation of AMPK by IF1 is involved in its cardioprotection against H/R-induced injury
AMPK activation in the heart is protective against cardiac ischemic injury 23-25. Thus, the activating phosphorylation levels of AMPK at Thr-172 in the heart were assessed. Phosphorylation levels of AMPK at Thr-172 in the heart were significantly increased after I/R, while systemic delivery of IF1 by AAV9 resulted in a more increase in AMPK phosphorylation in the ischemic heart following I/R injury (Fig. 6A). Furthermore, we also observed this phenomenon on H/R-induced injured NRCMs, where H/R-induced injury increased AMPK phosphorylation in the cardiomyocytes and treatment of cardiac myocytes with IF1 by adenovirus enhanced the phosphorylation of AMPK (Fig. 6B). Next, we examined whether AMPK phosphorylation mediate the regulation of IF1 on glucose uptake and glycolysis activity. In cardiomyocytes, pretreatment of compound C markedly decreased IF1-induced AMPK phosphorylation (Fig. S1C). Our data indicated that Ad-IF1 increased glycolysis and glycolytic capacity, while pretreatment of AMPK inhibitor Compound C abolished these effects (Fig. 6C and D). To further determine whether the cardioprotective effects of IF1 on cardiomyocyte is mediated by AMPK, we induced H/R injury in the NRCMs and infected with Ad-IF1 with or without treatment of Compound C and measured LDH release and cell apoptosis. Ad-IF1 decreased H/R injury-increased LDH release and cardiomyocytes apoptosis at the 1 hour of reperfusion while Compound C abolished these cardioprotective effects (Fig. 6E, F and Fig. S4).
IF1 confer cardioprotection via stimulating AMPK activation. A, Western blot analysis of phosphorylation levels of AMPK at Thr-172 in the heart. B, Western blot analysis of phosphorylation levels of AMPK at Thr-172 in cardiomyocytes. C, Glucose uptake assay and glycolytic and mitochondrial respiration (D) measurement with Seahorse Extracellular Flux Analyzer. E, LDH released from NRCMs at the 1 hour of reperfusion. F, Representative flow cytometric assessment of apoptosis via Annexin V/PI staining and Flow cytometric quantification. n = 3 each. *P <0.05, **P<0.01.
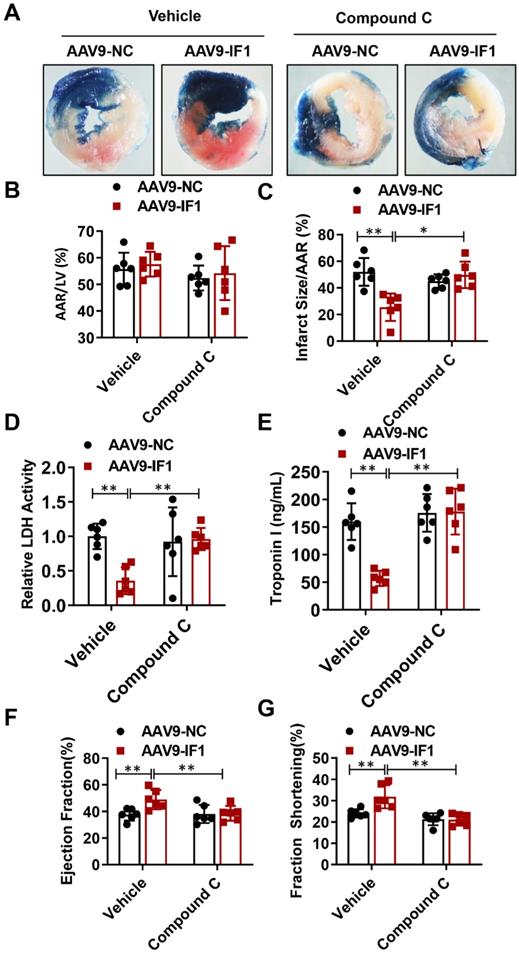
Compound C abrogated IF1-afforded cardioprotection in vivo
To further determine whether IF1 contributes to the alleviation of myocardial I/R injury through AMPK signaling pathway, we overexpressed IF1 in the heart and treat the I/R mice at the beginning of reperfusion with Compound C. Although AAV9-NC and AAV9-IF1 groups showed comparable AAR to LV after I/R (Fig. 7A, B), the infarct area (Fig. 7C), serum LDH activity (Fig. 7D) and serum Troponin I level (Fig. 7E) were significantly attenuated in the AAV9-IF1 group compared with these in the AAV9-NC group without Compound C, while these effects were all abrogated by Compound C treatment. At 4 weeks post-I/R, treatment with Compound C almost completely abolished IF1-improved cardiac function, as evidenced by reduced LVEF and LVFS (Fig. 7D-7E). Meanwhile, cardiac fibrosis was mitigated by AAV-IF1, which was reversed with Compound C treatment, as evaluated by Masson's Trichrome staining (Fig. S3B). Therefore, AMPK mediates the cardioprotective effects of IF1 against I/R injury in vivo.
Compound C abrogated IF1-afforded cardioprotection. A-C, Representative images, and analysis of the infarct size in isolated I/R (30 min/2 h) hearts. D, serum LDH activity analysis. E, Serum Troponin I level analysis. F-G, left ventricle (LV) ejection fraction (LVEF) and LV fractional shortening (LVFS) measured by echocardiography (Echo). n = 6. *P <0.05, **P<0.01.
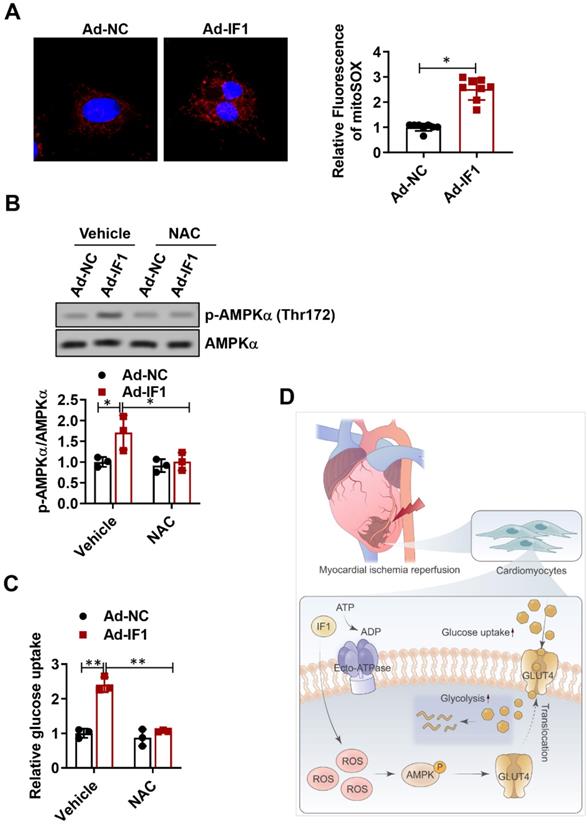
IF1 activates ROS as an AMPK upstream pathway
ROS has been shown to be associated with AMPK activation (30, 31). Thus, we attempted to determine whether the release of ROS is involved in IF1-induced AMPK phosphorylation. Following infection of NRCMs for 48h with Ad-IF1, there was an increased intensity of staining with DCF-DA, a ROS-sensitive dye (Fig. 8A). Moreover, ROS chelation using NAC, a ROS scavenger, resulted in decreased AMPK phosphorylation (Fig. 8B). In addition, NAC blocked IF1-mediated glucose uptake (Fig. 8C), demonstrating that IF1 increases AMPK phosphorylation through ROS generation.
IF1 activates ROS as an AMPK upstream pathway. A, following infection of NRCMs for 48h with Ad-IF1, ROS were stained with an ROS-sensitive dye DCF-DA. B, Western blot analysis of phosphorylation levels of AMPK. C, NAC blocked IF1-mediated glucose uptake. n = 3. D, The graphic abstract of the study. *P <0.05, **P<0.01.
Discussion
In the present study, we study the role and mechanism of IF1 in myocardial I/R injury. First, our data indicated that IF1 protein expression is downregulated during I/R and H/R. Second, IF1 delivered with adenovirus and AAV9 protected against cardiac dysfunction and pathological development induced by I/R ex vivo and in vivo. Third, IF1 stimulates glucose uptake and glycolysis activity and stimulates AMPK activation during in vivo basal and I/R and in vitro OGD/R conditions. Finally, we found activation of AMPK by IF1 is involved in its cardioprotection against H/R-induced injury. These results indicate that upregulation of IF1 in the I/R heart exerts its cardioprotection by activating AMPK signaling pathway.
It has been reported that the main function of IF1 is to reduce the decrease of ATP by inhibiting the hydrolysis activity of ATP synthase under myocardial ischemia 7-9, 26. Inhibiting ATP hydrolysis has been proposed as a potential way for the treatment of various pathological conditions, including cardiac I/R injury 10, 27. In preclinical models, IF1 mimetic compounds have been shown to enhance the cardiac performance of isolated hearts subjected to myocardial ischemia/reperfusion 28-30. Inhibition of ATP hydrolysis has been proposed as a potential clinical strategy for the treatment of various pathological conditions, including myocardial ischemia/reperfusion injury [10, 26]. IF1 mimics, such as bms-199264 and bms-250685, have been shown to enhance the cardiac performance of isolated hearts subjected to myocardial ischemia/reperfusion [27-29]. Before identifying IF1 as a therapeutic target for heart disease, it is necessary to obtain the functional role of IF1 in pathological heart. In this study, we further explored the protective effect of IF1 delivered by adenovirus and AAV9 and found that IF1 overexpression protects the heart from I/R-induced cardiac dysfunction and pathological development in vitro and in vivo.
There are many literatures that activation of pro-survival kinases, such as ERK and AMPK, can achieve strong cardiac protection in myocardial reperfusion 4, 20, 31. Activation of myocardial AMPK has been shown to display salutary effects on ischemic heart disease and glucose homeostasis 32-36, we, therefore, hypothesized that the AMPK signaling pathway is primarily involved in the protective effects of IF1 as an underlying mechanism. At the cellular level, AMPK activation was found to result from ROS production induced by IF1 treatment. ROS-induced AMPK activation might be involved in the observed hypoglycemic effects of IF1. It is widely accepted that AMPK phosphorylation increases glucose uptake through the enhanced GLUT4 translocation from the cytoplasmic fraction to the plasma membrane 37-40; therefore, AMPK activation induced by IF1 could increase glucose uptake in these mice, ultimately eliciting glucose-lowering effects. This was confirmed by our in vitro results that IF1 significantly increased glucose uptake in NRCMs. Glucose uptake was attenuated after the inactivation of the AMPK pathway using AMPK-specific silencing by the AMPK inhibitor compound C. Thus, the IF1/AMPK signaling axis may contribute to the cardioprotective effects. Collectively, the therapeutic approaches to enhance IF1 production can be valuable for the prevention or treatment of I/R injury. A previous study suggests that in mammalian neurons mitochondria adapt to respiratory stress by upregulating IF1, which exerts a protective role by coordinating pro-survival cell mitophagy and bioenergetics resilience41. Therefore, it is of great interest to investigate whether IF1 confer cardioprotective effects via activating mitophagy.
In conclusion, in the present study, we showed that IF1 is downregulated in mouse hearts under I/R stress, IF1 delivered by adenovirus and AAV9 protected the hearts from I/R injury. Mechanistically, we demonstrated that activation of AMPK by IF1 is involved in the cardioprotection of IF1 against I/R injury (Fig. 8D). These results suggest that IF1 delivery may serve as a potential therapeutic strategy in treating pathological cardiac ischemic injury and heart failure.
Supplementary Material
Supplementary figures.
Acknowledgements
The authors would like to thank Yanju Zhen for their technical assistance and for providing expert opinions and feedback.
Author Contributions
L.K.M. designed the study and edited the manuscript. J.W.W. helped with the study design, designed, and performed experiments, analyzed, and interpreted data, and wrote the manuscript; H.H. and H.J.S. performed experiments, analyzed data.
Funding
The present study is supported by the Natural Science Foundation of Anhui Province (2008085QH354), the National Natural Science Foundation of China (81870192), and the National Key Research and Development Program of China (2021YFA0804904).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Prabhu SD, Frangogiannis NG. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circulation research. 2016;119:91-112
2. Ivey MJ, Kuwabara JT, Pai JT. et al. Resident fibroblast expansion during cardiac growth and remodeling. Journal of molecular and cellular cardiology. 2018;114:161-174
3. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121-35
4. Davidson SM, Ferdinandy P, Andreadou I. et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. Journal of the American College of Cardiology. 2019;73:89-99
5. Cabezon E, Montgomery MG, Leslie AG. et al. The structure of bovine F1-ATPase in complex with its regulatory protein IF1. Nature structural biology. 2003;10:744-50
6. Cabezon E, Runswick MJ, Leslie AG. et al. The structure of bovine IF(1), the regulatory subunit of mitochondrial F-ATPase. The EMBO journal. 2001;20:6990-6
7. Gledhill JR, Montgomery MG, Leslie AG. et al. How the regulatory protein, IF(1), inhibits F(1)-ATPase from bovine mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:15671-6
8. Campanella M, Parker N, Tan CH. et al. IF(1): setting the pace of the F(1)F(o)-ATP synthase. Trends in biochemical sciences. 2009;34:343-50
9. Pullman ME, Monroy GC. A Naturally Occurring Inhibitor of Mitochondrial Adenosine Triphosphatase. The Journal of biological chemistry. 1963;238:3762-9
10. Campanella M, Casswell E, Chong S. et al. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell metabolism. 2008;8:13-25
11. Rouslin W, Broge CW. Mechanisms of ATP conservation during ischemia in slow and fast heart rate hearts. The American journal of physiology. 1993;264:C209-16
12. Esparza-Molto PB, Cuezva JM. The Role of Mitochondrial H(+)-ATP Synthase in Cancer. Frontiers in oncology. 2018;8:53
13. Sanchez-Cenizo L, Formentini L, Aldea M. et al. Up-regulation of the ATPase inhibitory factor 1 (IF1) of the mitochondrial H+-ATP synthase in human tumors mediates the metabolic shift of cancer cells to a Warburg phenotype. The Journal of biological chemistry. 2010;285:25308-13
14. Formentini L, Sanchez-Arago M, Sanchez-Cenizo L. et al. The mitochondrial ATPase inhibitory factor 1 triggers a ROS-mediated retrograde prosurvival and proliferative response. Mol Cell. 2012;45:731-42
15. Madhavi YV, Gaikwad N, Yerra VG. et al. Targeting AMPK in Diabetes and Diabetic Complications: Energy Homeostasis, Autophagy and Mitochondrial Health. Current medicinal chemistry. 2019;26:5207-5229
16. Wang W, Xiao ZD, Li X. et al. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nature cell biology. 2015;17:490-9
17. Jia J, Bissa B, Brecht L. et al. AMPK, a Regulator of Metabolism and Autophagy, Is Activated by Lysosomal Damage via a Novel Galectin-Directed Ubiquitin Signal Transduction System. Molecular cell. 2020;77:951-969 e9
18. Zhao P, Sun X, Chaggan C. et al. An AMPK-caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science. 2020;367:652-660
19. Rabinovitch RC, Samborska B, Faubert B. et al. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell reports. 2017;21:1-9
20. Chen Y, Liu J, Zheng Y. et al. Uncoupling protein 3 mediates H(2)O(2) preconditioning-afforded cardioprotection through the inhibition of MPTP opening. Cardiovascular research. 2015;105:192-202
21. Xie C, Zhang YP, Song L. et al. Genome editing with CRISPR/Cas9 in postnatal mice corrects PRKAG2 cardiac syndrome. Cell research. 2016;26:1099-1111
22. Bi W, Wang J, Jiang Y. et al. Neurotrophin-3 contributes to benefits of human embryonic stem cell-derived cardiovascular progenitor cells against reperfused myocardial infarction. Stem cells translational medicine. 2021;10:756-772
23. Qi D, Young LH. AMPK: energy sensor and survival mechanism in the ischemic heart. Trends in endocrinology and metabolism: TEM. 2015;26:422-9
24. Chen X, Li X, Zhang W. et al. Activation of AMPK inhibits inflammatory response during hypoxia and reoxygenation through modulating JNK-mediated NF-kappaB pathway. Metabolism: clinical and experimental. 2018;83:256-270
25. Ding R, Wu W, Sun Z. et al. AMP-activated protein kinase: An attractive therapeutic target for ischemia-reperfusion injury. European journal of pharmacology. 2020;888:173484
26. Sah JF, Kumar C, Mohanty P. pH dependent conformational changes modulate functional activity of the mitochondrial ATPase inhibitor protein. Biochemical and biophysical research communications. 1993;194:1521-8
27. Formentini L, Pereira MP, Sanchez-Cenizo L. et al. In vivo inhibition of the mitochondrial H+-ATP synthase in neurons promotes metabolic preconditioning. The EMBO journal. 2014;33:762-78
28. Grover GJ, Atwal KS, Sleph PG. et al. Excessive ATP hydrolysis in ischemic myocardium by mitochondrial F1F0-ATPase: effect of selective pharmacological inhibition of mitochondrial ATPase hydrolase activity. American journal of physiology Heart and circulatory physiology. 2004;287:H1747-55
29. Grover GJ, Malm J. Pharmacological profile of the selective mitochondrial F1F0 ATP hydrolase inhibitor BMS-199264 in myocardial ischemia. Cardiovascular therapeutics. 2008;26:287-96
30. Grover GJ, Marone PA, Koetzner L. et al. Energetic signalling in the control of mitochondrial F1F0 ATP synthase activity in health and disease. The international journal of biochemistry & cell biology. 2008;40:2698-701
31. Hausenloy DJ, Yellon DM. Reperfusion injury salvage kinase signalling: taking a RISK for cardioprotection. Heart failure reviews. 2007;12:217-34
32. Kambara T, Ohashi K, Shibata R. et al. CTRP9 protein protects against myocardial injury following ischemia-reperfusion through AMP-activated protein kinase (AMPK)-dependent mechanism. The Journal of biological chemistry. 2012;287:18965-73
33. Li J, Minczuk K, Massey JC. et al. Metformin Improves Cardiac Metabolism and Function, and Prevents Left Ventricular Hypertrophy in Spontaneously Hypertensive Rats. Journal of the American Heart Association. 2020;9:e015154
34. Steneberg P, Lindahl E, Dahl U. et al. PAN-AMPK activator O304 improves glucose homeostasis and microvascular perfusion in mice and type 2 diabetes patients. JCI insight. 2018;3(12):e99114
35. Nam DH, Kim E, Benham A. et al. Transient activation of AMPK preceding left ventricular pressure overload reduces adverse remodeling and preserves left ventricular function. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2019;33:711-721
36. Potenza MA, Sgarra L, Nacci C. et al. Activation of AMPK/SIRT1 axis is required for adiponectin-mediated preconditioning on myocardial ischemia-reperfusion (I/R) injury in rats. PloS one. 2019;14:e0210654
37. Habegger KM, Hoffman NJ, Ridenour CM. et al. AMPK enhances insulin-stimulated GLUT4 regulation via lowering membrane cholesterol. Endocrinology. 2012;153:2130-41
38. Rao XS, Cong XX, Gao XK. et al. AMPK-mediated phosphorylation enhances the auto-inhibition of TBC1D17 to promote Rab5-dependent glucose uptake. Cell death and differentiation. 2021
39. Ducommun S, Wang HY, Sakamoto K. et al. Thr649Ala-AS160 knock-in mutation does not impair contraction/AICAR-induced glucose transport in mouse muscle. American journal of physiology Endocrinology and metabolism. 2012;302:E1036-43
40. Blanco CL, Gastaldelli A, Anzueto DG. et al. Effects of intravenous AICAR (5-aminoimidazole-4-carboximide riboside) administration on insulin signaling and resistance in premature baboons, Papio sp. PloS one. 2018;13:e0208757
41. Lefebvre V, Du Q, Baird S. et al. Genome-wide RNAi screen identifies ATPase inhibitory factor 1 (ATPIF1) as essential for PARK2 recruitment and mitophagy. Autophagy. 2013;9:1770-9
Author contact
![]() Corresponding author: Li-Kun Ma, Department of Cardiology, The First Affiliated Hospital of USTC, Division of Life Science and Medicine, University of Science and Technology of China, Hefei, 230001, China. E-mail: lkmaedu.cn
Corresponding author: Li-Kun Ma, Department of Cardiology, The First Affiliated Hospital of USTC, Division of Life Science and Medicine, University of Science and Technology of China, Hefei, 230001, China. E-mail: lkmaedu.cn