Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
The structure of the PXR protein
PXR undergoes epigenetic...
PXR as sensor and effector of...
PXR confers anti-cancer drug...
Application and disadvantages of...
Conclusions and perspectives
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(2):742-759. doi:10.7150/ijbs.68724 This issue Cite
Review
Insights into the critical role of the PXR in preventing carcinogenesis and chemotherapeutic drug resistance
Xiaxia Niu1, Ting Wu1, Gege Li1, Xinsheng Gu2, Yanan Tian3 ![]() , Hongmei Cui1
, Hongmei Cui1 ![]()
1. Institute of Toxicology, School of Public Health, Lanzhou University, 730000, Lanzhou, China.
2. Department of Pharmacology, College of Basic Medical Sciences, Hubei University of Medicine, Shiyan 442000, Hubei, China.
3. Department of Veterinary Physiology and Pharmacology, Texas A&M University, USA.
Received 2021-11-4; Accepted 2021-11-21; Published 2022-1-1
Abstract

Pregnane x receptor (PXR) as a nuclear receptor is well-established in drug metabolism, however, it has pleiotropic functions in regulating inflammatory responses, glucose metabolism, and protects normal cells against carcinogenesis. Most studies focus on its transcriptional regulation, however, PXR can regulate gene expression at the translational level. Emerging evidences have shown that PXR has a broad protein-protein interaction network, by which is implicated in the cross signaling pathways. Furthermore, the interactions between PXR and some critical proteins (e.g., p53, Tip60, p300/CBP-associated factor) in DNA damage pathway highlight its potential roles in this field. A thorough understanding of how PXR maintains genome stability and prevents carcinogenesis will help clinical diagnosis and finally benefit patients. Meanwhile, due to the regulation of CYP450 enzymes CYP3A4 and multidrug resistance protein 1 (MDR1), PXR contributes to chemotherapeutic drug resistance. It is worthy of note that the co-factor of PXR such as RXRα, also has contributions to this process, which makes the PXR-mediated drug resistance more complicated. Although single nucleotide polymorphisms (SNPs) vary between individuals, the amino acid substitution on exon of PXR finally affects PXR transcriptional activity. In this review, we have summarized the updated mechanisms that PXR protects the human body against carcinogenesis, and major contributions of PXR with its co-factors have made on multidrug resistance. Furthermore, we have also reviewed the current promising antagonist and their clinic applications in reversing chemoresistance. We believe our review will bring insight into PXR-targeted cancer therapy, enlighten the future study direction, and provide substantial evidence for the clinic in future.
Keywords: Pregnane nuclear receptor (PXR), post-translational modifications, protein-protein interactions, chemotherapeutic drug resistance
Introduction
Pregnane x receptor (PXR, SXR, NR1I2) is one of the important nuclear receptors (NRs), which is expressed mainly in the liver, intestine, and colon. Kliewer et al. discovered the full-length cDNA of mouse PXR in 1997, and human PXR (hPXR) was cloned simultaneously [1]. PXR plays important roles in disposing of endogenous and xenobiotic compounds and maintains metabolizing homeostasis, therefore, PXR protects the liver against toxic insult from both endogenous and xenobiotic compounds [2, 3]. Once xenobiotics as ligand bind to PXR, PXR forms a heterodimer with RXRα and then translocate to nucleus, recognizing the conserved sequence on the promoter region of downstream genes and triggering transcriptional regulation. The PXR responsive elements on the promoter region of target genes (such as CYP3A4 and CYP2B6) are some of the short repeats (AGGTCA), including direct repeats (DR) DR-3, DR-4, DR-5, and the eversion repeats (ER) ER-6 and ER-8 [4-6] (Figure 1). It is estimated that CYP3A4 is responsible for the metabolism of over 50% of drugs in use today, many of which are either metabolically activated and/or detoxified through this enzyme [2].
Schematic of PXR' functional domains contain DNA binding domain, hinge region, and ligand binding domain. Xenobiotics (including agonist and antagonist) bind to the ligand binding domain, and then PXR forms a heterodimer with RXRα and translocate to the nucleus, recognizing the consensus sequence on the promoter region of target genes to initiate transcriptional regulation of CYP450 phase I, II, III enzymes. Antagonists can be used to overcome chemoresistance in lab while applied in clinic with caution.
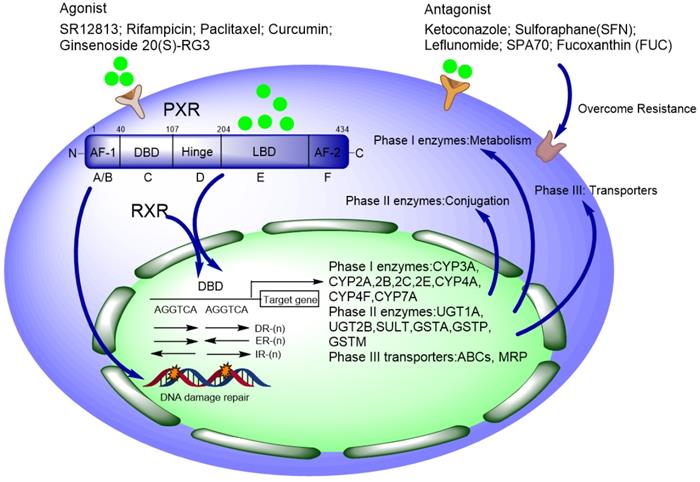
Normally, DNA methylation on the PXR promoter is much higher in some cell lines (like Caco-2, HT29, HCT116, and SW48), resulting in silenced PXR expression and finally transcriptionally repressed downstream target genes [7]. Besides, the transcription activity of PXR is also affected by some auxiliary co-activators and co-repressors, which have been reviewed in [8, 9]. Agonists of PXR include rifampicin (RIF), SR12813, paclitaxel (PTX), et al. [10-12]. The ligand-bound activity is thought to make conformational changes to activate PXR by disassociation of co-repressors, and simultaneously recruitment of co-activators [8, 9, 13, 14]. These co-factors comprehensively determine how PXR is activated, thereby affecting PXR-regulated response to xenobiotics [14].
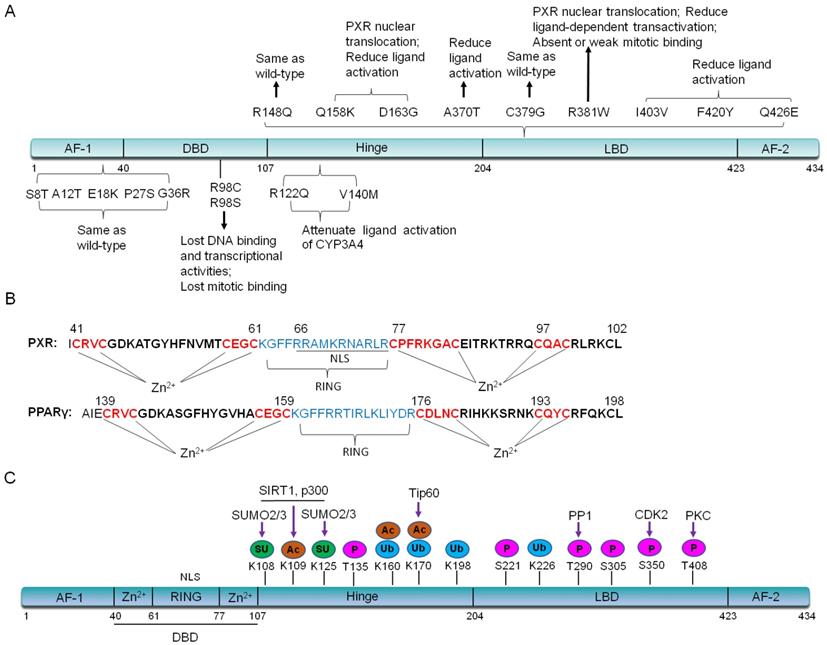
De mattia et al. have summarized important single nucleotide polymorphisms (SNPs) variants on PXR exons [15-18]. In the activating function (AF) -1 and DNA binding domain (DBD), SNPs such as G23C (S8T), G34A (A12T), G52A (E18K), C79T (P27S), G106A (G36R) and C292T (R98C), have no effects on PXR localization whereas affect its ligand-bound capability. C79T (P27S) occurs on exon 2, which is population specific in African Americans with a frequency ranging from 15-20%. This allele has no significant effect on the hepatic CYP3A4 expression [16, 19]. C292T (R98C) makes PXR can not bind to promoter region, thus will lose the transcriptional regulation on downstream target genes [17, 20]. In the PXR hinge region, the variant G365A (R122Q) and G418A (V140M), potentially decrease affinity of the PXR-DNA binding and attenuate ligand (Such as RIF and corticosterone) activation of the CYP3A4 reporter plasmid in an in vitro transient transfection assay [15, 16]. It is worthy of note that nine SNPs variants of PXR have been found in the LBD region, including G443A (R148Q), C472A (Q158K), A488G (D163G), G1108A (A370T), T1135G (C379G), C1141T (R381W), A1207G (I403V), T1258A (F420Y), and C1276G (Q426E) [17]. The G443A (R148Q) variant has no effect on PXR transcriptional activity [20]. The C1141T (R381W) and A1207G (I403V) variants also have shown varying reduction in PXR-mediated transactivation [20]. In COS-1 cells, in vitro transient transfection with the Q158K, D163G, and R381W variants of PXR, demonstrates that these three variants lead to PXR nuclear translocation as well as the reduced ligand-dependent transactivation. D163G makes PXR completely lose transactivation activity in LS174 T cells, reduce CYP3A4 induction by corticosterone, whereas enhances promoter-dependent induction by RIF [21]. The rs1054190-TT genotype, which locates at the 3'UTR of PXR, reduces mRNA expression of PXR, and can be used as a marker of poor prognosis [22]. Currently, PXR SNPs (rs3732359, rs3732360 and rs3814058) have been linked to the decreased percentage in nadir haemoglobin, suggesting increased docetaxel-associated toxicity in nasopharyngeal carcinoma patients and PXR (rs3814058C>T) polymorphism increases the risk of lung cancer in smokers [23, 24]. We have summarized these important SNPs and their functions in Figure 2A. Due to the specificity of PXR SNPs, further research is needed for the precision clinical medicine.
(A). Important PXR SNPs on exons and their functions. (B). The DBD region of PXR contains a bidirectional nuclear localization sequence (NLS) and zinc finger I (CX2CX13CX2C) and zinc finger II (CX6CX9CX2C) in DBD domain, which is quite similar with that of PPARγ. In contrast, PPARγ has zinc finger I (CX2CX13CX2C) and zinc finger II (CX3CX9CX2C). The RING domain of PPARγ plays E3 ligase activity to degrade NF-κB. (C). PXR protein structure can be altered by post-translational modifications. P: Phosphorylation; SU: SUMOylation; Ac: Acetylation; Ub: Ubiquitination.
Most studies focus on PXR's role as a transcriptional factor, whereas emerging evidences suggest that PXR can regulate gene expression at translational levels. Recently, the protein-protein interactions centered by PXR have raised much attention, especially the interactions between PXR and histone deacetylate transferase, histone arginine methyltransferase, kinase and phosphatase, and ubiquitination enzyme, indicating that PXR is implicated in posttranslational modifications (e.g., acetylation, phosphorylation, and SUMOylation, ubiquitination) and ultimately affects its transcriptional regulation and metabolic detoxification process. Protein-protein interactions also have provided a novel insight that PXR protects against toxic insult and plays important roles in the prevention of carcinogenesis. A thorough understanding of the regulation and function of PXR in carcinogenesis will help improve the diagnosis and treatment of cancer in the clinic.
Additionally, due to modulation of PXR on metabolic detoxification, inflammation, and DNA damage repair, the cell tolerance to genotoxic agents is increased, therefore, PXR also contributes to chemotherapeutic drug resistance. So far, numerous PXR antagonists have been invented and applied in clinical cancer therapy with caution to overcome drug resistance.
In this review, we have summarized the post-translational modifications of PXR, and have put much emphasis on the protective mechanisms of PXR against carcinogenesis. Additionally, we have discussed about the current understanding of PXR-induced drug resistance mechanisms to chemotherapy. We will focus on the new shreds of evidence about interaction protein network centered by PXR, including some progresses on the PXR involved DNA damage signaling.
The structure of the PXR protein
The full-length PXR contains 434 amino acids [25, 26]. The DNA binding domain (DBD) is more conserved between humans and other species. There is a bidirectional nuclear localization sequence (NLS) and two zinc finger structures: CX2CX13CX2C (zinc finger I) and CX5CX9CX2C (zinc finger II) in the DBD region, which is quite similar as that of peroxisome proliferator-activated receptor γ (PPARγ) [27] (Figure 2B). Each zinc finger structure consists of four cysteine residuals that chelate a zinc ion [13, 27, 28]. The ligand binding domain (LBD) of PXR is composed of a seven-member α-helical sandwich, assembled in three layers with an antiparallel five-strand β sheet spherical structure, which accommodates more ligands in this region [29]. The amino acid sequence of LBD in human PXR is only 75%-80% homology with that of rats [30]. Compared with other nuclear receptors, the LBD of PXR is wider and more flexible, which makes it easier to bind with many hydrophobic compounds [31]. The LBD serves as the docking site for ligands and contains dimerization motifs and transcriptional activation function domains (AF-2) helix. The binding of a ligand to the LBD results in a conformational change in the AF-2 helix, and this change allows the nuclear receptor to interact with accessory proteins and to regulate the expression of target genes [32, 33].
PXR undergoes epigenetic modifications through protein-protein interaction
PXR may be modified by acetylation, phosphorylation, ubiquitination, and SUMOylation through protein-protein interactions (Figure 2C). In the following part, we will highlight the recent studies about the interactions between PXR and histone methyltransferase, histone acetylate transferase, and E3 ubiquitin ligase. The interaction network-centered by PXR will uncover the multifunctional property of PXR in different signaling pathways.
The interactions between PXR and histone methyltransferase, histone acetyltransferase (HAT)/ histone deacetylate transferase (HDAC)
Protein arginin methyltransferases (PRMT1) is a major histone methyltransferase that associates with PXR. PXR agonist RIF recruits PRMT1 to the promoter region of CYP3A4, and in the meantime, methylates arginine 3 of histone H4. It has been proved that PRMT1 is indispensable for PXR-regulated CYP3A4 expression. Interestingly, the interaction between PXR and PRMT1 appears to have a mutual effect on both PRMT1 localization and PXR epigenetic modifications [34].
The acetylation status of PXR is quite important during its activation [35]. The lysine (K) 109 in the hinge region on PXR is the major site to be acetylated by E1A binding protein p300, and the acetylation of K109 represses PXR transcriptional activity by reducing DNA-bound capability, further inhibits the formation of heterodimer with RXRα [36]. Besides, steroid receptor co-activators (SRCs) facilitate PXR transcriptional modulation by recruiting endogenous histone acetyltransferases (HAT) to loosen tensed chromosome structure, thus lead to nucleosome remodeling and covalent modifications on histone tails, such as histones H3K4 methylation, H3K9 acetylation, H4K20 acetylation, and phosphorylation of the linker histone H1. Hence, acetylation has been seen as a promotional signal for transcriptional response [37]. A recent study has demonstrated that PXR can form a complex with histone acetylate transferase Tip60 and augments the latter protein's enzyme activity, thus regulates colon cancer cell migration and invasion [38], suggesting that Tip60 might be one of co-activators of PXR [35, 39].
Normally, silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) forms a repressor complex with histone de-acetylase (HDAC) 1 and nuclear receptor corepressor (NCoR), which inhibits PXR activity. Under this situation, HDAC activity creates a repressive chromatin environment [40, 41]. The interaction between PXR and SMRT occurs within the LBD of PXR and the nuclear receptor (NR)-interacting domain of SMRT. Deletion of the PXR's AF-2 helix enhances SMRT binding and abolishes ligand-dependent dissociation of SMRT. Overexpression of SMRT abrogates PXR's transactivation of the CYP3A4, whereas silencing of SMRT enhances the CYP3A4 promoter reporter signal [42]. PXR strongly represses vitamin D3 activation of the CYP24A1, in which PXR indirectly interacts with SMRT and prevents dissociation of SMRT from the CYP24A1 promoter [43]. HDAC3-SMRT complex as co-repressor mainly regulates acetylation of ligand-dependent PXR [44]. Treatment with TSA (deacetylase inhibitor) dramatically elevates acetylation level of PXR. Surprisingly, PXR agonist RIF can dampen the TSA-induced acetylation of PXR. TSA also alters the subcellular localization of PXR in primary hepatocytes which is isolated from C57BL6 mice [44]. Pharmacologically, inhibition of HDAC3-SMRT co-repressor complex activity, together with TSA will lead to a synergistic transactivation of PXR.
NAD-dependent deacetylase protein Sirtuin 1 (SIRT1) belongs to the HDAC3 class, which is responsible for major deacetylation modifications on PXR protein. Research has showed that fasting-activated SIRT1 can bind with PXR and deacetylate PXR on lysine (K) 109 to promote PXR-mediated lipogenesis or lipid accumulation. Furthermore, SIRT1 opposes PXR coactivation by abolishing peroxisome proliferator-activated receptor γ coactivator 1 alpha (PGC-1a) expression, whereas PGC-1a increases PXR expression and transactivation function [35, 45].
The interaction between PXR and Ubiquitination enzyme
So far, there are a lot of studies have reported interactions between nuclear receptors and E3 ubiquitin ligases [27, 46]. Accordingly, the interaction may affect PXR transcriptional activity, which has been reviewed in ref. [46]. For example, androgen receptor (AR) belongs to the nuclear receptor superfamily and its activation is critical for prostate cancer development and progression. Updated research has demonstrated that the RNA helicase DEAH-box (DHX) 15 stabilizes E3 ligase Siah2 and enhances Siah2 activity, thus ubiquitinates and degrades AR, whereas enhanced AR transcriptional activity contributes to prostate cancer progression [47]. Hu et al. have reported that celastrol as an agonist of nuclear receptor Nur77, binds and promotes Nur77 translocation from the nucleus to mitochondria. In mitochondria, Nur77 interacts with tumor necrosis factor receptor-associated factor 2 (TRAF2), further stabilizes TRAF2. The stabilization of TRAF2 contributes to K63-linked Nur77 ubiquitination, inducing autophagy under inflammatory conditions [48]. Another orphan nuclear receptor small heterodimer partner (SHP) is a negative regulator of bile acid and lipid metabolism in the liver. SHP physically occupies the transactivation domain of x-box-binding protein 1 (XBP1s), thereby preventing the ubiquitination and degradation of XBP1s by the E3 ligase complex cullin3-SPOP (speckle-type POZ protein), which has provided novel evidence that SHP governs ER homeostasis [49].
However, there are little evidence about the interaction between PXR and E3 ligase. One study has demonstrated that PXR interacts with ring-B-box-coiled-coil protein interacting with protein kinase C-1 (RBCK1) and ubiquitin protein ligase E3 component n-recognin 5 (UBR5), by which PXR is degraded by RBCK1 and UBR5 and finally altered xenobiotic's metabolism [50, 51]. Domain mapping has revealed that the full-length of E3 ligase RBCK1 binds with PXR including UBL domain, novel zinc finger domain, and RING 1/2 domain. RBCK1 down-regulates the expression of PXR, and the down-regulation is inhibited by the application of MG132 [50]. Using mass spectrometry (MS) analysis and genome-wide siRNA screen, Ong. et al have reported that the E3 ligase UBR5 and tyrosine-phosphorylation-regulated kinase 2 (DYRK2) regulate PXR protein stability. PXR has been shown to be a substrate for DYRK2, and DYRK2-dependent phosphorylation of PXR facilitates itself subsequent ubiquitination by UBR5 [51]. Another study has demonstrated that the interaction between PXR and E3 ligase c-terminus of Hsp70-interacting protein (CHIP) exists in both the cytoplasm and nucleus. Threonine (T) 408 on PXR is phosphorylated by protein kinase C (PKC) and then recruits CHIP, to modulate cancer cell autophagy [52]. PXR ubiquitination can be increased by protease inhibitor MG132, facilitated by the activation of the protein kinase A (PKA) signaling pathway [39]. Interestingly, PXR can be heavily mono-ubiquitinated on K109, K160, K170, K198, and K226, which sets PXR in a silent state. The ubiquitination on K198 and K226 is abrogated with treatment with TSA whereas the formation of K48-linked ubiquitin chains at K170 strongly promotes the degradation of ligand-activated PXR [53]. These evidences supports that the acetylation status of PXR affects its ubiquitination modification.
Interestingly, the DBD region of PXR is composed of two zinc-RING structures, which is similar as that in PPARγ (Figure 2A), leading us to think about whether the zinc-RING pocket could grant E3 ligase property to PXR since PPARγ has been proved as an E3 ligase to degrade NF-κB [27]. However, until now, none of the studies has been reported yet.
The PXR protein contains a SIM (SUMO-interacting motifs) consensus amino acid sequence. The PIASy-mediated SUMOylation of PXR inhibits ubiquitin-mediated degradation by the 26S proteasome and further leads to transcriptional repression of PXR during pathological states [44, 53]. SUMOylation site of PXR has been found on K108 of PXR [39]. K170 has been seen as a primary site of ubiquitination of PXR whereas SUMOylation of K108 and K128 abolishes ubiquitination on K170. Obviously, SUMOylation and ubiquitination of the PXR both determine its biologic function in response to xenobiotics [53]. Surprisingly, the acetylation status of PXR enhances SUMOylation, whereas deacetylation modification of PXR by HDAC3-SMRT may inhibit its SUMOylation. Acetylated PXR interacts with HDAC3 and forced overexpression of SMRT further increases PXR-HDAC3 interactions [44].
PXR contains numerous serine (S) / threonine (T) amino acids and can be phosphorylated and implicated in cellular stress response
Structurally, there are numerous serine (S) and threonine (T) amino acids in the PXR protein structure, which are easily to be phosphorylated, suggesting that the main function of PXR can be finely tuned by its phosphorylation. PKC can directly phosphorylate the T408 of PXR, which decreases PXR protein stability, followed by recruiting E3 ligase CHIP and chaperon HSP90 to modulate autophagic degradation [52]. The activation of the PKA signaling pathway also increases the ubiquitination of PXR [39]. The amino acid residues S8, S57, S208, S05, S350, and T408 are critical for the biological activity of the PXR protein. S57 and T408 mutationsabolish ligand-inducible PXR activity. Mutations in the PXR LBD domain at S305, S350, and T408 decrease the ability of PXR to form a heterodimer with RXRα. Mutation at position S208, S305, S350, and T408 alters the recruitment of co-factors. T408 mutation potentially affects the subcellular localization of the PXR. These data suggest that phosphorylation at specific amino acid residues can profoundly manipulate PXR biological activity [54]. Besides, the phosphorylation modification at T135 and S221 of PXR decreases affinity with RXRα and co-activator SRC1 while augments the interaction with SMRT and NCoR [55, 56]. Phosphatase PP1 interacts with PXR and phosphorylates T290, thus further affects PXR cellular localization and transcriptional regulation of downstream target CYP3A4, UGT1A1 [57]. Activated PXR recruits PP2Cα and interacts each other, leading to dephosphorylation of serum/glucocorticoid regulated kinase 2 (SGK2) at T193. Dephosphorylated SGK2 conversely augments PXR-mediated transactivation of gluconeogenic genes in human liver cells, thereby enhancing gluconeogenesis [58]. In one of the recent studies, fasting-activated PXR interacts with vaccinia virus-related kinase 1 (VRK1) and phosphorylates S350 of PXR, further inhibits the sulfotransferase (SULT1E1) activity, modulating glucose metabolism [59]. Phosphomimic mutation at T290 (T290D) makes PXR protein remain in the cytoplasm whereas phosphorylation at S350 may alter the binding with RXRα and its co-activators, attenuating transactivation of UGT1A1 by roscovitine [60].
Interestingly, we have also checked the protein structure of PXR, and there are three consensus S/TQ motifs, which suggest that PXR could be the substrate of ATM/ATR and phosphorylated by ATM/ATR, which may contribute to DNA damage and repair, DNA replication signaling (Figure 3). The consensus S/TQ motif is the most critical structure of the substrate of ATM/ATR [61]. PXR might play very important roles in DNA damage and DNA replication signaling.
PXR protein structure contains 3 S/TQ motifs (Green color), suggesting PXR possibly is one of the substrates of ATM/ATR and is implicated in DNA damage response signaling.

PXR as sensor and effector of xenobiotics, regulates metabolizing detoxification, glucose metabolism, and protects against carcinogenesis
As the sensor and effector of xenobiotics, PXR binds with agonists, then the corepressor complex will be released from PXR, subsequently, PXR forms a heterodimer with RXRα and then translocates to nuclear, transcriptionally regulates downstream gene's expression [62]. In addition to its role in xenobiotic metabolism, PXR is implicated in regulating inflammatory responses, glucose metabolism, and protects normal cells against carcinogenesis [62-70]. Most importantly, protein-protein interactions centered by PXR define specificity in signal transduction crosstalk, and influence every aspect of cellular function. In this part, we will highlight the possible mechanisms that PXR protects the normal cell against carcinogenesis.
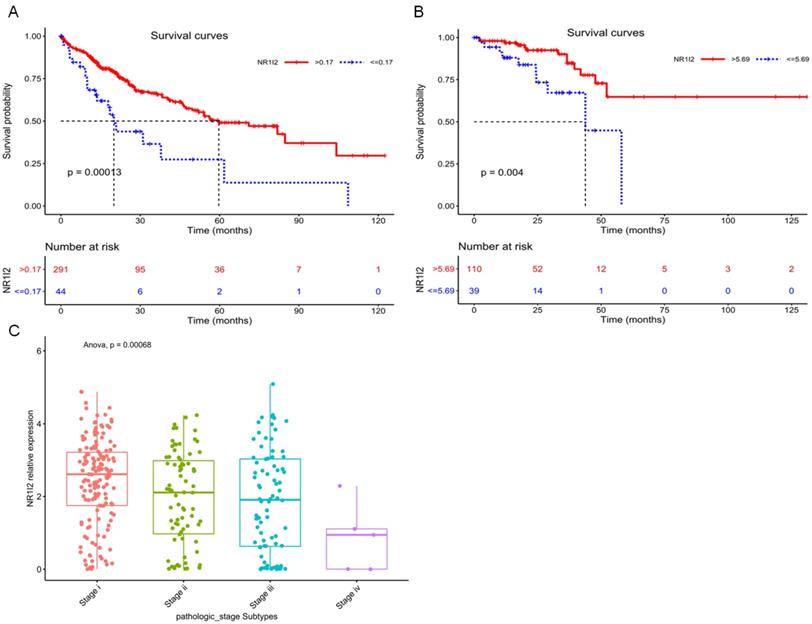
Current research evidences have shown that the high expression of PXR is positively correlated with the specific survival rate of prostate cancer patients, suggesting PXR can be used as an indicator of a good prognosis for prostate cancer [71]. By utilizing the cancer genome atlas (TCGA), we have analyzed the relationship between survival probability and mRNA expression level of PXR in liver hepatocellular carcinoma (LIHC) patients, as well as in rectum adenocarcinoma (READ) patients. Result shows that the high mRNA expression of the PXR is positively correlated with the survival probability of LICH and READ cancer patients (Figure 4A and B), whereas negatively correlated with tumor stage and grade in LICH samples (Figure 4C), suggesting that PXR could be a protective factor in liver hepatocellular carcinoma and rectum adenocarcinoma.
Kaplan-Meier curves for survival analysis of Liver hepatocellular carcinoma (LIHC) patients and Rectum adenocarcinoma (READ) patients based on PXR (NR1I2) mRNA expression from TCGA database. A Survival analysis based on the best cutoff threshold (0.17) of PXR mRNA expression in LIHC patients (335 samples) (Red line: >0.17, blue line: < = 0.17). B Survival analysis based on the best cutoff threshold (5.69) of PXR mRNA expression in READ patients (149 samples) (Red line: >5.69, blue line: < = 5.69). C PXR mRNA expression in different pathologic stages of LIHC patients (Stage I 154 samples, Stage II 78 samples, Stage III 80 samples, Stage IV 5 samples) (Anova, p = 0.00068).
PXR promotes cancer cell apoptosis, inhibits proliferation, and regulates cell cycle
It has been shown that PXR is induced by PTX or cisplatin (CDDP) in a ligand-dependent manner in endometrial cancer and ovarian cancer cells [72, 73]. A lot of studies have shown that PXR extensively inhibits cancer cell proliferation or induces damaged cells undergoing apoptosis to prevent carcinogenesis. In colon cancer, PXR arrests the colon cancer cell cycle at G0/G1 phase, which is mediated by upregulated p21 and downregulated E2F/RB signaling, therefore, promoting apoptosis and inhibiting the proliferation of human colon cancer cells [66]. In breast cancer and cervical cancer, PXR is activated by its ligand RIF, leading to cell cycle arrest at G2/M phase and inhibiting cell growth and proliferation [74, 75]. In addition, PXR also reduces Benzoapyrene (BaP) -induced genotoxicity [76, 77]. However, there are also other studies have reported that activated-PXR mediates drug-induced hepatoxicity [78, 79]. Therefore, PXR is a strictly context-dependent receptor, and how it plays its function depends on the post-translational modifications and cross-talk between signaling pathways [9].
PXR activation induces metabolic detoxification enzymes to clear genotoxicity mediated by xenobiotics
So far, many CYP450 enzymes have been reported to be regulated by PXR, including Phase I metabolizing enzymes CYP2B6, CYP2C9, CYP2C19, CYP3A4, and CYP3A5; Phase II conjugating enzymes including UDP-glucuronsy transferase (UGT1A1), glutathione S-Transferase (GST), favoring detoxification of xenobiotics; some drug transporters including multidrug resistance protein 1 (MDR1/P-gp/ABCB1), multidrug resistance related protein 2 (MRP2/ABCC2) [80, 81] (Figure 1).
Most of the PXR-regulated drug metabolism enzymes and transporters detoxify xenobiotics and protect normal cells against carcinogenesis. Naspinski et al. have identified that PXR protects HepG2 cells from BaP-induced DNA damage by inducing CYP450 enzymes GSTA1, GSTA2, GSTM1, UGT1A6, and Breast cancer resistance protein (BCRP, ABCG2) gene expression. Additionally, the total GST enzymatic activity which favors the metabolic detoxification of BaP is found to be significantly induced in the presence of PXR [3]. However, PXR-mediated GST induction is independent of the nuclear factor-erythroid 2 p45-related factor 2 (Nrf2)/Kelchlike Ech-associated protein 1 (Keap1) pathway, although Nrf2 and Keap1 have been shown to regulate GST expression positively and negatively, respectively [82]. In line with Naspinski's finding, ginsenoside 20(S)-Rg3 as a functional PXR agonist significantly decreases BaP-induced DNA damage by increasing the gene expression of an important phase II detoxifying enzyme NAD(P)H: quinine oxidoreductase1 (NQO1), which is associated with the activation of Akt/Nrf2 pathway, suggesting that PXR serves as a cytoprotective factor against environmental carcinogens [77].
PXR represses immune/inflammatory responses by inhibition of NF-κB signaling pathway
Inflammation always has been viewed as the major factor to promote carcinogenesis. PXR plays a critical role in suppressing the NF-κB-regulated signaling. Moreover, the PXR null mice display more severe inflammation in their small bowels [83, 84]. Zhou et al. have shown that nuclear factor-κB (NF-κB) activation inhibits PXR/SXR and its primary target gene CYP3A by interrupting the formation of the PXR·RXRα complex, in which NF-κB p65 directly interacts with the DNA-binding domain of RXRα, thus inhibiting the transactivation regulation by the PXR·RXRα complex [2]. PXR agonist pregnenolone-16α-carbonitrile (PCN) alleviates inflammatory bowel disease (IBD) symptoms, whereas mice lacking PXR expression has not been affected. Activation of PXR suppresses the expression of the NF-κB-mediated inflammatory response [85]. In coordination with these findings, rifaximin, a semi-systemic rifamycin-derived antibiotic, as an intestine-specific human PXR agonist, also represses NF-κB signaling and displays a preventive and therapeutic role for the IBD model [85]. Therefore, rifaximin may be a promising anticancer tool due to PXR-mediated inhibition of inflammation factors [86]. Additionally, curcumin activates human PXR and significantly reduces the histological signs of colonic inflammation by induction of MDR1 expression. These data also suggest that curcumin or curcumin-like derivatives could be further developed as intestine-specific PXR activators [87]. Solomonsterol A, a newly-reported PXR agonist, also prevents colitis through the reduction of NF-κB in PXR-humanized mice. These studies indicate that PXR agonists are very promising in the therapy of inflammation-driven cancer [88]. Another putative mouse PXR agonist chrysin has been reported to protect against colitis by inhibiting NF-κB signal [89]. It is worth to note that long-term treatment of PXR agonist to the patients inhibits the inflammatory response in the liver. Activation of the inflammatory response in hepatocytes renders modifications of SUMOylation on PXR, and SUMOylated PXR conversely further represses the immune response in hepatocytes [90].
Furthermore, many studies have reported that PXR expressed in human CD4, CD8, CD19, and CD14 cells. Upon immune stress, activated PXR inhibits the T cell proliferation, decreases the expression of CD25 and interferon γ (IFN-γ), as well as phosphorylation of NF-κb and MEK1/2 [91, 92].
PXR may promote DNA damage repair through protein-protein interaction
It has been shown that PXR protects cells against liver injury/DNA damage induced by carcinogens and anti-cancer drugs, such as polycyclic aromatic hydrocarbons (PAHs) and their metabolites, aflatoxin B [93, 94], alkylating agents (cyclophosphamide, nimustine, lomustine) [95], anthracyclines (doxorubicin, daunorubicin and adriamycin) [96] and platinum-based compounds (loplatin, oxaliplatin, cisplatin) [97]. PXR interrupts the PAHS-AhR pathway and enhances CYP450 phase II enzyme activity, protecting liver cells from BaP-induced DNA damages [3, 76].
Treatment of HepG2 cells with RIF not only blocks the G0/G1 transition of the cells and inhibits cell growth, but also protects the cells from DNA damage induced by UVB [77, 98, 99]. The PXR agonist ginsenoside 20(S)-RG3 activates the phosphatidylinositol 3-kinase (PI3k)/Akt/Nrf2 pathway to attenuate the DNA damage induced by BaP in human dermal fibroblasts. After inhibiting the expression of PXR by siRNA, the protective effect of ginsenoside 20(S)-RG3 has been abolished [77]. The tumor suppressor gene p53 is activated when carcinogens induce DNA damage, blocking the cycle of colon cancer cells. PXR interacted with p53, and further represses its downstream genes to regulate tumor formation [100, 101].
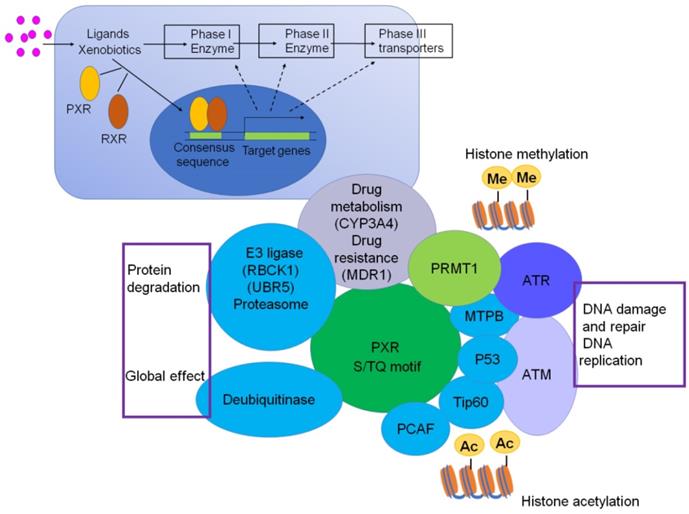
Until now, emerging evidences have shown that PXR is implicated in protein-protein network in response to DNA damage signaling. Cui et al. have demonstrated that PXR directly interacts with AhR, and prevents the liver cancer cells from BaP-induced DNA damage through inhibiting AhR-CYP1A1 transactivation [76]. Recent research has demonstrated that PXR could interact with Tip60 without ligand and augment the acetylation activity of Tip60 [38]. The histone acetyltransferase Tip60 closely associates with genomic instability and cancer progress. Low expression of Tip60 compromises DNA repair efficiency either under normal conditions or genotoxic stress. Further study has revealed that Tip60 regulates homology recombination (HR) to promote DNA repair [102]. Additionally, Tip60 accelerates the DNA damage repair process through acetylation of ATM/ATR kinase in response to DNA damage stress [103-105]. Mechanically, PXR should bear huge potential in promoting DNA damage response and repair signaling, and consequently plays protective roles in counteracting carcinogenesis, which is worth to be further investigated (Figure 5).
PXR centered protein-protein interactions. PXR protein structurally interacts with p53, Tip60, PCAF, MTBP, which are critical proteins to sense ATM/ATR signaling and regulate DNA damage and repair, DNA replication. PXR also interacts with PRMT1 and Tip60, thereby implicated for regulation of histone methylation and acetylation. The interaction between PXR and E3 ligase and DUB enzymes are responsible for protein degradation and ubiquitination, which might be a global effect to influence cell function. The transcriptional regulation of PXR to CYP3A4 and MDR1 makes it contribute to chemotherapeutic drug resistance.
PXR confers anti-cancer drug resistance in the clinic
Currently, a significant barrier to effective cancer therapy is the development of drug resistance upon prolonged use. The mechanisms of drug resistance are quite complicated. The possible mechanisms include decreased drug inflow [106], increased drug efflux [106], activation of the drug detoxification system, alteration of drug targets, initiation of DNA repair reprogramming [107], regulation of cell death pathways [108], and inducing changes of microenvironments [109].
Nowadays, most chemotherapeutic regimens typically are genotoxic agents, which are designed either to damage DNA of tumor cells or to prevent the damaged DNA being repaired. Normally, DNA damage induced by genotoxic agents in normal cells can be repaired efficiently or tolerated to maintain genomic stability. Defect in DNA damage repair makes DNA lesions can't be removed timely, therefore, more and more damaged DNA accumulated and mutated, finally developed cancer. In contrast, increased repair or tolerance of DNA lesions may grant cancer cell the ability to survive in genotoxic environments upon clinical chemotherapy, thus generate resistance to DNA repair agents. DNA repair status may explain intrinsically or acquired drug resistance [110].
Due to facilitating MDR1/CYP3A4-mediated drug metabolisms [111], and promoting DNA damage repair, PXR presents huge tolerance to genotoxic agents, thus contributes to drug resistance. So far, PXR has been reported to confer paclitaxel and docetaxel resistance [65, 112], cisplatin resistance [113, 114], tamoxifen resistance [115, 116], doxorubicin resistance [117] and sorafenib resistance [118, 119] (Figure 6).
PXR confers to chemotherapeutic drug resistance including Sorafenib resistance, Paclitaxel and docetaxel resistance, Cisplatin resistance, Tamoxifen resistance, Doxorubicin/Adriamycin resistance, Irinotecan resistance through regulation of CYP450 metabolic enzymes and protein-protein interactions and transcriptional modulation. Ac: Acetylation.
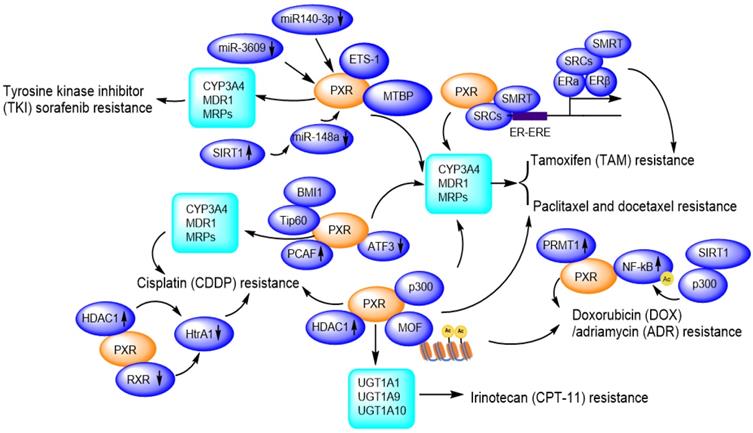
PXR contributes to paclitaxel and docetaxel resistance
In lung cancer cells A549, NCI-H358, HCC827, NCI-H1650, and NCI-H1299, upon paclitaxel treatment, the PXR mRNA and protein expression levels are elevated. The PXR expression levels are positively correlated with those of P-gp. The co-expression profile of PXR and P-gp in the peripheral blood mononuclear cells of non-small cell lung cancer (NSCLC) patients has been used as biomarker for predicting paclitaxel resistance [120]. The PXR agonist SR12813 significantly increases the resistance of MDA-MB-231 cells to docetaxel, suggesting that PXR is involved in the docetaxel resistance of human breast cancer [65].
Previously, literature has reported that PXR compromises p53 function upon DNA damage response pathway [101]. Coordinately, p53 is inactivated in most drug-resistant cell lines. Studies have been reported that in contrast to parental cells, p53 functionally inactive in all PTX-resistant sublines which results from two distinct point mutations in codons 236 and 239 of the DNA binding domain. This observation suggests that p53-loss-of-function may facilitate the development of paclitaxel resistance and restoring p53 tumor suppressor function, might be a promising approach to reverse chemoresistance [121, 122].
PXR contributes to tyrosine kinase inhibitor sorafenib resistance
Hepatocellular carcinoma (HCC) is a highly heterogeneous tumor, and small molecule tyrosine kinase inhibitors (TKIs) including erlotinib, gefitinib, nilotinib, sorafenib, vandetanib, have shown dramatic therapeutic effects in cancer therapy, however, the drug resistance is an important obstacle to the successful use in the clinic. These TKIs are able to induce the expression of P-gp, which is mediated by PXR, conferring acquired TKI resistance in cancer cells [123]. Sorafenib binds to and activates PXR, and promotes the expression of CYP3A4 and MDR1, facilitating the clearance or elimination of sorafenib, then HCC cells gain sorafenib-resistance to promote proliferation and invasion/migration [119]. LINE-1 ORF-1p (LINE-1 ORF-1p is encoded by the human pro-oncogene LINE-1) interacts with PXR and enhances its cytoplasmic/nuclear translocation, and further mediates the resistance to and clearance of sorafenib in HCC cells by inducing CYP3A4 and MDR1 [118] . Transcription factor E26 transformation specific sequence 1 (ETS-1) also binds with PXR and promotes PXR transactivation, thereby confers the sorafenib resistance in HCC tumor models. ETS-1 blockade enhances the anti-tumor capacity of sorafenib by withdrawing PXR activation [124]. Conversely, miR-140-3p and miR-3609 inhibit PXR/CYP3A4/MDR1 signal, and finally result in the decelerating clearance of sorafenib in HCC cells [125, 126]. Rhamnetin as an inhibitor of SIRT1, enhances miR-148a transcription thus inhibits PXR/CYP3A4/MDR1 signal, slowing down the metabolic detoxification of sorafenib in HCC cells and enhancing the sensitivity of HCC cells to sorafenib [127].
In addition to the regulation of PXR/CYP3A4/MDR1 pathway, sorafenib alone can activate p53 in a concentration-dependent manner [128]. Additionally, most of HCC patients who clinically acquired resistant to sorafenib also accompany with increased P-gp expression, enhanced glycolytic metabolism, and increased nuclear factor kappa B (NF-κB) activity [129]. Literature has also reported that the MDM2 binding protein (MTBP) interacts with the PXR and accumulates PXR in the nucleus, thereby facilitating PXR-CYP3A4 transactivation, which leads to the resistance of HCC cells to sorafenib. In this way, MTBP could work synergistically with PXR and might be a promising target to reverse sorafenib resistance of HCC cells [130].
PXR contributes to cisplatin resistance
Cisplatin is a platinum compound applied for killing cancer cells and patients easily developed cisplatin resistance in clinic. In Skov-3 and OVCAR-8 ovarian cancer cells, cisplatin treatment strongly activates PXR-mediated transcription through binding with the MDR1/CYP3A4 responsive element [72, 131]. Downregulation of PXR suppresses the augmented MDR1/CYP3A4 expression and significantly inhibits cell growth and apoptosis in the presence of cisplatin [72]. PXR antagonist ketoconazole induces higher caspase-3 activity, whereas phenethyl isothiocyanate (PEITC) accumulates intracellular platinum level by suppressing ABCC2 protein expression, therefore, enhances the antitumor activity of cisplatin [113]. Another PXR antagonist leflunomide significantly enhances caspase-3 activation in cisplatin-resistant HepG2 cells [113, 114].
A HAT gene, p300/CBP-associated factor (PCAF), as co-activator of PXR, overexpressed in cisplatin-resistant cells and endows an antiapoptotic phenotype by enhancing E2F1 expression [132]. A member of the polycomb repressor complex 1 (BMI1, a proto-oncogene) recruits another HAT gene Tip60, promoting acetylation of histone H2A and H3, upregulating MDR1 expression, and contributes to acquired cross-resistance against paclitaxel, doxorubicin, as well as other chemotherapeutic drugs [133, 134].
PXR contributes to doxorubicin (DOX) / adriamycin (ADR) resistance
Cytotoxicity experiment has revealed that the resistance of the osteosarcoma cell lines to etoposide is correlated with PXR protein expression levels and activation of CYP3A4 [135]. Evidences have shown that resistance to DOX can be acquired through NF-κB activation while the drug resistance to PTX can be developed through the toll-like receptor 4 (TLR4)-NF-κB pathway [117]. PRMT1 as an arginine methyltransferase, interacts with PXR in MCF7/ADR cells, and this interaction could be disrupted by PRMT1 inhibitor 1 (AMI-1). AMI-1 significantly suppresses the expression of MDR1 in MCF7/ADR cells and increases cells sensitivity to adriamycin. In the term of acquired resistance, PRMT1 may be an important co-activator of PXR in regulating MDR1 gene and PRMT1 inhibitor may be promising for overcoming ADR resistance [136].
PXR contributes to tamoxifen (TAM) resistance
PXR also is involved in the tamoxifen resistance of human breast cancer [65]. An outstanding activation of PXR and persistent expression of PI3k/p-Akt has been observed, together with an elevated expression of CYP3A4 and MDR1 [115, 116]. Additionally, TAM-resistant MCF-7 cells (TAMR-MCF-7) express higher levels of MRP2 than parental MCF-7 cells. LY294002 (PI3k inhibitor) significantly reduces both the PXR activity and MRP2 expression in TAMR-MCF-7 cells, suggesting that PI3k plays important roles in activating PXR-MRP2 signaling and these observations might bear clinical implications during long-term TAM therapy-acquired chemoresistance [115].
PXR contributes to irinotecan resistance
The resistance of the lung cancer cells to irinotecan (CPT-11) and its active metabolite SN38 is associated with the elevated expression of PXR downstream genes UGT1A1 and UGT1A10 [137]. Elevated expression of UGT1A1 and UGT1A10 enhances metabolism of SN-38, resulting in a reduced concentration of SN-38 in the lung cancer cells [138]. Activation of PXR confers the resistance of colorectal cancer cells LS174T, SW480 and SW620 to irinotecan, which is mediated by the increased expression of UGT1A1, UGT1A9 and UGT1A10. The resistance can be reversed by PXR repression [138].
The PXR genetic polymorphisms affect chemoresistance
As we mentioned above, the SNPs of PXR may alter its transcriptional regulation of CYP3A4, and MDR1, the ligand-bound capability, and cellular localization. There is no wonder that SNPs also influence pharmacokinetic profile and drug sensitivity. De Mattia et al. summarized the role of PXR polymorphisms in toxicity, efficacy, and pharmacokinetics of chemotherapeutic drugs [15]. The haplotype formed by rs2276707 (39823C>T) and rs3814058 (42961T>C) linked with a lower doxorubicin clearance. An allele in rs10934498 (10051G>A) associated with decreased exposition and increased degradation of SN-38. 2654T>C polymorphism have been shown to be associated with reduced intestinal ABCB1 expression whereas the IVS617C>T polymorphism is related to intestinal CYP3A inducibility, leading to an accelerated clearance of azithromycin [19, 139]. However, since SNPs vary between different people, different gender, and different study subject, the results lack independent validation. In future, new sequencing technology might provide better genetic characterizations of PXR to thoroughly understand the variability in drug sensitivity and drug resistance among individuals.
Application and disadvantages of PXR antagonists in clinic
A lot of studies have further verified the key role of PXR antagonists in overcoming resistance.
Ketoconazole is a weak antagonist of PXR, which binds to the AF-2 of PXR instead of LBD [140]. It efficiently antagonizes RIF-induced PXR activation, and inhibits the activity of CYP3A enzyme as well as MDR-1, MRP2 expression in ovarian cancer, liver cancer, and colon cancer cells. It dramatically enhances drug sensitivity such as paclitaxel and cisplatin, inhibits cell proliferation, and improves the effect of drugs [141-143]. In paclitaxel-resistant SKOV-3 cells, ketoconazole significantly inhibits cell growth and proliferation, and promotes the apoptosis of drug-resistant cells [72]. This effect has also been further verified in combination with cisplatin [113]. Indeed, ketoconazole combination with paclitaxel or cisplatin inhibits cell growth and promotes apoptosis, increases the sensitivity of endometrial cancer cells to chemotherapeutic drugs [144]. However, low-dose ketoconazole cannot directly inhibit the expression of PXR, only high-dose and long-term can inhibit the expression of PXR [16]. This is consistent with the results of another study, which claimed that ketoconazole as a PXR antagonist has limitations, and cannot be applied to clinical due to its high toxicity [140]. Because ketoconazole is more toxic and its clinical application is very limited, researchers are also committed to develop some of its analogs to overcome the effect of drug resistance, and find that the toxicity of FLB12 is less than that of ketoconazole, counteracting the resistance of LS174T colon cancer cells to SN-38 [142].
Fucoxanthin (FUC) is a carotenoid bearing anti-inflammatory and anti-cancer effects [145]. It is reported that fucoxanthin can be used as a PXR antagonist, not only promotes apoptosis of prostate cancer cells (PC-3, DU145 and LNCaP) through G0/G1 phases arrest, but also enhances the activation of caspase-3 in cisplatin-resistant HepG2 cells [114, 146]. The dietary carotenoid fucoxanthin (FUC) can overcome DOX resistance in breast cancer cells (MCF-7/ADR), hepatocytes (HepG-2/ADR), ovarian cells (Skov-3/ADR) through inhibiting RIF-induced PXR, MDR1, and CYP3A4 expression at both the mRNA level and protein level. Therefore, fucoxanthin might be very promising to overcome PXR-mediated drug resistance [146].
Leflunomide as PXR antagonist is clinically used for anti-rheumatic treatment. It has an IC50 of 6.8μM, which can significantly enhance the activation of caspase-3 in parental and CDDP-resistant HepG2 cells [147].
Sulforaphane (SFN) is the first naturally-developed PXR antagonist in vitro. It is directly bound with PXR and inhibits PXR-mediated expression of CYP3A4 in human primary liver cancer cells [148]. SFN arrests cancer cells in the G2/M phase, inhibits cell growth, and further inhibits the formation of DNA adducts, thus enhances the therapeutic effect of cancer [149]. SFN metabolites sulforaphane-cysteine (SFN-Cys) or sulforaphane-N-acetyl-cysteine (SFN-NAC) increases the binding of phosphorylated extracellular regulated protein kinases 1/2 (ERK1/2) to α-tubulin, thus alternates the microtubule morphology and disturbs the microtubule assembly [150]. Another study has reported that SFN induces apoptosis by sustained activation of ERK1/2 in NSCLC cells [151]. SFN metabolites phosphorylate ERK1/2, leading to upregulation of 26S proteasome and Hsp70, as well as downregulation of βIII-tubulin, X-linked inhibitor of apoptosis protein (XIAP), Tau, Stathmin1, and α-tubulin, ultimately develops microtubule catastrophe, then reverses PTX resistance in NSCLC cells [152].
SPA70 is a newly developed antagonist of PXR by Chen's lab [153]. It acts on the LBD region of PXR and blocks activation of PXR in the liver cancer cell HepG2 in a concentration-dependent manner. It suppresses the expression of multiple PXR target genes such as CYP3A4 and MDR1. SPA70 is a powerful and specific antagonist of PXR with low toxicity, which is an analogue of ketoconazole [153]. Structurally, SPA70 contains a 2,5-dimethoxyphenyl component with the 5-methoxy group replacing the 5-methyl group in SJB7 (PXR agonist) [153]. Nonetheless, SPA70 can work as agonist to mutant PXR (a mutation residue W299A in the ligand domain), since W299A transition is closely related to the AF-2 region of PXR contact [154]. In such case, W299 amino acid is quite important for PXR activation, and the agonistic effect of CITCO also requires W299 [155]. Recently, the researchers have modified the four different positions of SPA70, the 4-(tert-butyl) phenyl group, the methoxy group, the hydrogen group, and the 5-methyl-1H-1,2,3-triazole ring, and have produced 81 analogs of SPA70 totally, and the binding ability of these compounds with PXR has been altered and investigated. Interestingly, although these compounds have similar structures, their functions and effects on cancer cells are quite different [156].
Conclusions and perspectives
Since Kliewer et al. initiated PXR study in 1997, so far, it has been well studied in drug metabolisms [1]. However, how PXR acts in tumorigenesis still needs further clarification. PXR per se is a context-dependent gene since its functional regulation occurs in multiple levels: firstly, it has high or low hypermethylation in the promoter region, which makes its gene expression silenced or overactivated [7]; secondly, PXR plays its function as a heterodimer with RXRα, however, RXRα is also co-factors of many other nuclear receptors and can be affected by these nuclear receptors [26, 157]; thirdly, PXR activation needs some auxiliary co-activators or co-repressors, most of which may modify PXR in post-translational modifications, thereby influence the performance of PXR [33]; fourthly, the protein structure of PXR is enriched of serine and threonine amino acid, which makes PXR possibly be one of the substrates of multiple kinases and further employs with phosphorylation signaling transmission [55, 56]; fifthly, the outcome of PXR binding with or without ligand varies because the process may recruit E3 ligase or co-activators or co-repressors, thus finally affects the transactivation effect of PXR [45, 52, 70]. Collectively, PXR displays quite a complicated performance in response of xenobiotics. Further understanding the biological regulation of PXR will help clinical application or diagnosis.
Although PXR is a context-dependent gene, it exhibits as a protective factor in tumorigenesis, as shown by survival analysis in LICH and READ samples according to PXR mRNA expression (Figure 4). PXR manipulates drug metabolism to clear the toxicity of xenobiotics, furthermore, it represses inflammation via NF-κB inhibition. The current study has resonated that PXR represses NF-κB activation whereas NF-κB activation disrupts the heterodimer of PXR-RXRα, and further inhibits PXR binding to CYP3A4 promoter [2, 83, 84]. Interestingly, we found a quite similar zinc ring domain structure between PPARγ and PXR [27], which highlighted a possibility that PXR may bear similar E3 ligase activity as PPARγ to degrade NF-κB in protein level, however, this possibility needs further examined.
Although most researches focus on PXR as the transcriptional factor, PXR also regulates cross signaling through protein-protein interactions, which occurs at the protein translational or post-translational level [14]. Till date, the PXR-mediated protein-protein interactions are involved in glucose and lipid metabolism, bile acid/cholesterol metabolism, inflammation regulation, ER stress, autophagy, drug metabolism, liver injury, type II diabetes, steroid/endocrine homeostasis, and carcinogenesis [62, 80]. Indeed, by applying yeast two-hybrid and GST-pulldown assays, as well as an endogenous co-IP experiment, it has been found that PXR protein structurally has interactions with many proteins, which have been reviewed in ref. [33]. Strikingly, PXR can interact with many critical proteins in DNA damage signaling, including p53, Tip60, PCAF, MTBP (Figure 5), suggesting PXR is involved in DNA damage and repair network. There are several classical S/TQ motifs on the PXR protein structure, substantiating the fact that PXR physically might be the substrate of ATM/ATR kinase and play undefined roles in the DNA damage response pathway.
Due to the regulation of CYP3A4 and MDR1 gene expression, there is no any disputation that PXR contributes to chemoresistance in cancer therapy. PXR structurally binds to the promoter region of MDR1, MRP2 and BCRP, thus inducing anti-cancer drug resistance [158-160]. Consistently, PXR activation coordinates with the expression of these drug-resistance related proteins, and inhibition of PXR by an antagonist could reverse and sensitize the response to anti-cancer drugs in patients. In the meantime, the fact that PXR promotes DNA damage response and repair provides another explanation that PXR confers to drug resistance. During carcinogenesis, PXR protects the human body against xenobiotics-induced DNA damage and maintains genome stability; however, once cancer cell gained drug resistance, the capability of promotion DNA damage repair compromises the killing effect of the anti-cancer drug to cells. At this point, PXR and tumor suppressors, such as p53, et al. work like a two-side sword in cancer therapy. Nonetheless, the mechanisms that PXR confreres multidrug resistance are not limited to transcriptional upregulation of MDR1, MRP2 and BCRP, as evidenced that SFN metabolites overcome paclitaxel resistance through microtubular disruption. The finding provokes our attention that PXR governs the receptor interaction with mitotic chromatin and the nuclear localization signal (NLS) (R66-76R) region of PXR is essential for this process [28]. It is worth to note that some SNPs also influence mitotic binding of PXR. Indeed, the mechanisms that PXR-conferring multidrug resistance are quite complicated, especially its co-activators SRCs, co-repressors HDCA/SMRT/NCoR, and heterodimer partner RXRα, SNPs, governing post-translational modification-mediated drug resistance, which bears significant implications and emerged a plethora of clinical ramifications. Expanded knowledge unraveling the mechanisms of PXR-mediated drug resistance may delineate new interventive procedure for more comprehensive treatments.
Antagonist of PXR bears huge potential to overcome drug resistance. So far, many small molecules have been developed and shown potency in this field. However, the toxic effects of these drugs during administration cannot be avoided [161, 162]. At present, scientists have conducted in-depth studies on the structure and metabolic pathways of PXR, using current molecular technology, computing software to develop the desired antagonist counteracting PXR-mediated chemotherapy resistance [162]. Future research directions will focus on low- or non-toxic, more efficient, safer, and more specific PXR reversal agents to enhance the sensitivity of chemotherapy drugs, to explore the clinical therapeutic effects of these antagonists, and hope these inventions can solve these clinical drug resistance obstacles. Furthermore, rational drug combination therapy has yielded substantial effects in particular malignancy cases. Future evaluations are warranted in providing opportunities for the effective clinical manipulation of these PXR-mediated molecular processes.
Acknowledgements
Funding: This work was supported by the initiative fund for faculty development from Lanzhou University (Grant # 561119203).
Author Contributions: X.N. drafted the raw manuscript, T.W., G.L., X.G., H.C. and Y.T edited the final version. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement: Not Applicable.
Informed Consent Statement: Not Applicable.
Data Availability Statement: Not Applicable.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA. et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73-82
2. Gu X, Ke S, Liu D, Sheng T, Thomas PE, Rabson AB. et al. Role of NF-kappaB in regulation of PXR-mediated gene expression: a mechanism for the suppression of cytochrome P-450 3A4 by proinflammatory agents. J Biol Chem. 2006;281:17882-17889
3. Naspinski C, Gu XS, Zhou GD, Mertens-Talcott SU, Donnelly KC, Tian YA. Pregnane X receptor protects HepG2 cells from BaP-induced DNA damage. Toxicological Sciences. 2008;104:67-73
4. Orans J, Teotico DG, Redinbo MR. The nuclear xenobiotic receptor pregnane X receptor: recent insights and new challenges. Mol Endocrinol. 2005;19:2891-2900
5. Umesono K, Murakami KK, Thompson CC, Evans RM. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell. 1991;65:1255-1266
6. Casabar RC, Das PC, Dekrey GK, Gardiner CS, Cao Y, Rose RL. et al. Endosulfan induces CYP2B6 and CYP3A4 by activating the pregnane X receptor. Toxicol Appl Pharmacol. 2010;245:335-343
7. Habano W, Gamo T, Terashima J, Sugai T, Otsuka K, Wakabayashi G. et al. Involvement of promoter methylation in the regulation of Pregnane X receptor in colon cancer cells. BMC Cancer. 2011;11:81
8. Pavek P. Pregnane X Receptor (PXR)-Mediated Gene Repression and Cross-Talk of PXR with Other Nuclear Receptors via Coactivator Interactions. Front Pharmacol. 2016;7:456
9. Pondugula SR, Pavek P, Mani S. Pregnane X Receptor and Cancer: Context-Specificity is Key. Nucl Receptor Res. 2016 3
10. Li TG, Chiang JYL. Mechanism of rifampicin and pregnane X receptor inhibition of human cholesterol 7 alpha-hydroxylase gene transcription. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2005;288:G74-G84
11. Terc J, Hansen A, Alston L, Hirota SA. Pregnane X receptor agonists enhance intestinal epithelial wound healing and repair of the intestinal barrier following the induction of experimental colitis. Eur J Pharm Sci. 2014;55:12-9
12. Harmsen S, Meijerman I, Febus CL, Maas-Bakker RF, Beijnen JH, Schellens JHM. PXR-mediated induction of P-glycoprotein by anticancer drugs in a human colon adenocarcinoma-derived cell line. Cancer Chemotherapy and Pharmacology. 2010;66:765-771
13. Umesono K, Evans RM. Determinants of target gene specificity for steroid/thyroid hormone receptors. Cell. 1989;57:1139-1146
14. Tian Y. Epigenetic regulation of pregnane X receptor activity. Drug Metab Rev. 2013;45:166-172
15. De Mattia E, Cecchin E, Roncato R, Toffoli G. Pregnane X receptor, constitutive androstane receptor and hepatocyte nuclear factors as emerging players in cancer precision medicine. Pharmacogenomics. 2016;17:1547-1571
16. Fuchs I, Hafner-Blumenstiel V, Markert C, Burhenne J, Weiss J, Haefeli WE. et al. Effect of the CYP3A inhibitor ketoconazole on the PXR-mediated induction of CYP3A activity. Eur J Clin Pharmacol. 2013;69:507-513
17. Rana M, Devi S, Gourinath S, Goswami R, Tyagi RK. A comprehensive analysis and functional characterization of naturally occurring non-synonymous variants of nuclear receptor PXR. Biochim Biophys Acta. 2016;1859:1183-1197
18. Skandalaki A, Sarantis P, Theocharis S. Pregnane X Receptor (PXR) Polymorphisms and Cancer Treatment. Biomolecules. 2021 11.1142
19. Zhang J, Kuehl P, Green ED, Touchman JW, Watkins PB, Daly A. et al. The human pregnane X receptor: genomic structure and identification and functional characterization of natural allelic variants. Pharmacogenetics. 2001;11:555-572
20. Satoru K, Kouichi K, Yoshiro S, Shogo O, Ryuichi H, Kazuo K. et al. Functional characterization of four naturally occurring variants of human pregnane X receptor (PXR): one variant causes dramatic loss of both DNA binding activity and the transactivation of the CYP3A4 promoter/enhancer region. Drug Metab Dispos. 2004;32:149-154
21. Hustert E, Zibat A, Presecan-Siedel E, Eiselt R, Mueller R, Fuss C. et al. Natural protein variants of pregnane X receptor with altered transactivation activity toward CYP3A4. Drug Metab Dispos. 2001;29:1454-1459
22. De Mattia E, Polesel J, Roncato R, Labriet A, Bignucolo A, Dreussi E. et al. Germline Polymorphisms in the Nuclear Receptors PXR and VDR as Novel Prognostic Markers in Metastatic Colorectal Cancer Patients Treated With FOLFIRI. Front Oncol. 2019;9:1312
23. Chew SC, Lim J, Singh O, Chen X, Tan EH, Lee EJ. et al. Pharmacogenetic effects of regulatory nuclear receptors (PXR, CAR, RXRalpha and HNF4alpha) on docetaxel disposition in Chinese nasopharyngeal cancer patients. Eur J Clin Pharmacol. 2014;70:155-166
24. Zhang L, Qiu F, Lu X, Li Y, Fang W, Zhang L. et al. A functional polymorphism in the 3'-UTR of PXR interacts with smoking to increase lung cancer risk in southern and eastern Chinese smoker. Int J Mol Sci. 2014;15:17457-17468
25. Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841-850
26. Falkner KC, Pinaire JA, Xiao GH, Geoghegan TE, Prough RA. Structure and function of the human nuclear xenobiotic receptor PXR. Molecular Pharmacology. 2001;60:611-619
27. Hou Y, Moreau F, Chadee K. PPARγ is an E3 ligase that induces the degradation of NFκB/p65. Nat Commun. 2012;3:1300
28. Rana M, Dash AK, Ponnusamy K, Tyagi RK. Nuclear localization signal region in nuclear receptor PXR governs the receptor association with mitotic chromatin. Chromosome Research. 2018;26:255-276
29. Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP. et al. The human nuclear xenobiotic receptor PXR: Structural determinants of directed promiscuity. Science. 2001;292:2329-2333
30. Zhang Z, Burch PE, Cooney AJ, Lanz RB, Pereira FA, Wu J. et al. Genomic analysis of the nuclear receptor family: new insights into structure, regulation, and evolution from the rat genome. Genome Res. 2004;14:580-590
31. Ngan CH, Beglov D, Rudnitskaya AN, Kozakov D, Waxman DJ, Vajda S. The structural basis of pregnane X receptor binding promiscuity. Biochemistry. 2009;48:11572-11581
32. Bourguet W, Germain P, Gronemeyer H. Nuclear receptor ligand-binding domains: three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol Sci. 2000;21:381-388
33. Oladimeji P, Cui H, Zhang C, Chen T. Regulation of PXR and CAR by protein-protein interaction and signaling crosstalk. Expert Opin Drug Metab Toxicol. 2016;12:997-1010
34. Xie Y, Ke S, Ouyang N, He J, Xie W, Bedford MT. et al. Epigenetic regulation of transcriptional activity of pregnane X receptor by protein arginine methyltransferase 1. J Biol Chem. 2009;284:9199-9205
35. Biswas A, Pasquel D, Tyagi RK, Mani S. Acetylation of pregnane X receptor protein determines selective function independent of ligand activation. Biochem Biophys Res Commun. 2011;406:371-376
36. Pasquel D, Doricakova A, Li H, Kortagere S, Krasowski MD, Biswas A. et al. Acetylation of lysine 109 modulates pregnane X receptor DNA binding and transcriptional activity. Biochim Biophys Acta. 2016;1859:1155-1169
37. Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405-1428
38. Bakshi K, Ranjitha B, Dubey S, Jagannadham J, Jaiswal B, Gupta A. Novel complex of HAT protein TIP60 and nuclear receptor PXR promotes cell migration and adhesion. Sci Rep. 2017;7:3635
39. Staudinger JL, Xu C, Biswas A, Mani S. Post-translational modification of pregnane x receptor. Pharmacol Res. 2011;64:4-10
40. Nagy L, Kao HY, Chakravarti D, Lin RJ, Hassig CA, Ayer DE. et al. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell. 1997;89:373-380
41. Li H, Leo C, Schroen DJ, Chen JD. Characterization of receptor interaction and transcriptional repression by the corepressor SMRT. Mol Endocrinol. 1997;11:2025-2037
42. Johnson DR, Li CW, Chen LY, Ghosh JC, Chen JD. Regulation and binding of pregnane X receptor by nuclear receptor corepressor silencing mediator of retinoid and thyroid hormone receptors (SMRT). Mol Pharmacol. 2006;69:99-108
43. Konno Y, Kodama S, Moore R, Kamiya N, Negishi M. Nuclear xenobiotic receptor pregnane X receptor locks corepressor silencing mediator for retinoid and thyroid hormone receptors (SMRT) onto the CYP24A1 promoter to attenuate vitamin D3 activation. Mol Pharmacol. 2009;75:265-271
44. Cui W, Sun M, Zhang S, Shen X, Galeva N, Williams TD. et al. A SUMO-acetyl switch in PXR biology. Biochim Biophys Acta. 2016;1859:1170-1182
45. Buler M, Aatsinki SM, Skoumal R, Hakkola J. Energy sensing factors PGC-1alpha and SIRT1 modulate PXR expression and function. Biochem Pharmacol. 2011;82:2008-2015
46. Helzer KT, Hooper C, Miyamoto S, Alarid ET. Ubiquitylation of nuclear receptors: new linkages and therapeutic implications. J Mol Endocrinol. 2015;54:R151-67
47. Jing Y, Nguyen MM, Wang D, Pascal LE, Guo W, Xu Y. et al. DHX15 promotes prostate cancer progression by stimulating Siah2-mediated ubiquitination of androgen receptor. Oncogene. 2018;37:638-650
48. Hu M, Luo Q, Alitongbieke G, Chong S, Xu C, Xie L. et al. Celastrol-Induced Nur77 Interaction with TRAF2 Alleviates Inflammation by Promoting Mitochondrial Ubiquitination and Autophagy. Mol Cell. 2017;66:141-53.e6
49. Sun S, Kelekar S, Kliewer SA, Mangelsdorf DJ. The orphan nuclear receptor SHP regulates ER stress response by inhibiting XBP1s degradation. Genes Dev. 2019;33:1083-1094
50. Rana R, Coulter S, Kinyamu H, Goldstein JA. RBCK1, an E3 ubiquitin ligase, interacts with and ubiquinates the human pregnane X receptor. Drug Metab Dispos. 2013;41:398-405
51. Ong SS, Goktug AN, Elias A, Wu J, Saunders D, Chen T. Stability of the human pregnane X receptor is regulated by E3 ligase UBR5 and serine/threonine kinase DYRK2. Biochem J. 2014;459:193-203
52. Sugatani J, Noguchi Y, Hattori Y, Yamaguchi M, Yamazaki Y, Ikari A. Threonine-408 Regulates the Stability of Human Pregnane X Receptor through Its Phosphorylation and the CHIP/Chaperone-Autophagy Pathway. Drug Metab Dispos. 2016;44:137-150
53. Cui W, Sun M, Galeva N, Williams TD, Azuma Y, Staudinger JL. SUMOylation and Ubiquitylation Circuitry Controls Pregnane X Receptor Biology in Hepatocytes. Drug Metab Dispos. 2015;43:1316-1325
54. Lichti-Kaiser K, Brobst D, Xu C, Staudinger JL. A systematic analysis of predicted phosphorylation sites within the human pregnane X receptor protein. J Pharmacol Exp Ther. 2009;331:65-76
55. Cui W, Shen X, Agbas E, Tompkins B, Cameron-Carter H, Staudinger JL. Phosphorylation Modulates the Coregulatory Protein Exchange of the Nuclear Receptor Pregnane X Receptor. J Pharmacol Exp Ther. 2020;373:370-380
56. Pondugula SR, Dong H, Chen T. Phosphorylation and protein-protein interactions in PXR-mediated CYP3A repression. Expert Opin Drug Metab Toxicol. 2009;5:861-873
57. Sugatani J, Hattori Y, Noguchi Y, Yamaguchi M, Yamazaki Y, Ikari A. Threonine-290 regulates nuclear translocation of the human pregnane X receptor through its phosphorylation/dephosphorylation by Ca2+/calmodulin-dependent protein kinase II and protein phosphatase 1. Drug Metab Dispos. 2014;42:1708-1718
58. Gotoh S, Negishi M. Statin-activated nuclear receptor PXR promotes SGK2 dephosphorylation by scaffolding PP2C to induce hepatic gluconeogenesis. Sci Rep. 2015;5:14076
59. Hu H, Yokobori K, Negishi M. PXR phosphorylated at Ser350 transduces a glucose signal to repress the estrogen sulfotransferase gene in human liver cells and fasting signal in mouse livers. Biochem Pharmacol. 2020;180:114197
60. Sugatani J, Uchida T, Kurosawa M, Yamaguchi M, Yamazaki Y, Ikari A. et al. Regulation of pregnane X receptor (PXR) function and UGT1A1 gene expression by posttranslational modification of PXR protein. Drug Metab Dispos. 2012;40:2031-2040
61. Chen C, Zhang L, Huang NJ, Huang B, Kornbluth S. Suppression of DNA-damage checkpoint signaling by Rsk-mediated phosphorylation of Mre11. Proc Natl Acad Sci U S A. 2013;110:20605-20610
62. Ma X, Chen J, Tian Y. Pregnane X receptor as the "sensor and effector" in regulating epigenome. J Cell Physiol. 2015;230:752-7
63. Ma X, Idle JR, Gonzalez FJ. The pregnane X receptor: from bench to bedside. Expert Opin Drug Metab Toxicol. 2008;4:895-908
64. Hassani-Nezhad-Gashti F, Rysa J, Kummu O, Napankangas J, Buler M, Karpale M. et al. Activation of nuclear receptor PXR impairs glucose tolerance and dysregulates GLUT2 expression and subcellular localization in liver. Biochemical Pharmacology. 2018;148:253-264
65. Qiao EQ, Yang HJ. Effect of pregnane X receptor expression on drug resistance in breast cancer. Oncology Letters. 2014;7:1191-1196
66. Ouyang N, Ke S, Eagleton N, Xie Y, Chen G, Laffins B. et al. Pregnane X receptor suppresses proliferation and tumourigenicity of colon cancer cells. British Journal of Cancer. 2010;102:1753-1761
67. Pondugula SR, Mani S. Pregnane xenobiotic receptor in cancer pathogenesis and therapeutic response. Cancer Letters. 2013;328:1-9
68. Wahli W. A gut feeling of the PXR, PPAR and NF-kappaB connection. J Intern Med. 2008;263:613-619
69. Moreau A, Vilarem MJ, Maurel P, Pascussi JM. Xenoreceptors CAR and PXR activation and consequences on lipid metabolism, glucose homeostasis, and inflammatory response. Mol Pharm. 2008;5:35-41
70. Spruiell K, Richardson RM, Cullen JM, Awumey EM, Gonzalez FJ, Gyamfi MA. Role of pregnane X receptor in obesity and glucose homeostasis in male mice. J Biol Chem. 2014;289:3244-3261
71. Fujimura T, Takahashi S, Urano T, Tanaka T, Zhang W, Azuma K. et al. Clinical significance of steroid and xenobiotic receptor and its targeted gene CYP3A4 in human prostate cancer. Cancer Sci. 2012;103:176-180
72. Masuyama H, Nakamura K, Nobumoto E, Hiramatsu Y. Inhibition of pregnane X receptor pathway contributes to the cell growth inhibition and apoptosis of anticancer agents in ovarian cancer cells. International Journal of Oncology. 2016;49:1211-1220
73. Masuyama H, Hiramatsu Y, Kodama J, Kudo T. Expression and potential roles of pregnane X receptor in endometrial cancer. J Clin Endocrinol Metab. 2003;88:4446-4454
74. Niu YD, Wang ZL, Huang HH, Zhong SP, Cai WF, Xie YM. et al. Activated pregnane X receptor inhibits cervical cancer cell proliferation and tumorigenicity by inducing G2/M cell-cycle arrest. Cancer Letters. 2014;347:88-97
75. Verma S, Tabb MM, Blumberg B. Activation of the steroid and xenobiotic receptor, SXR, induces apoptosis in breast cancer cells. Bmc Cancer. 2009;9:3
76. Cui H, Gu X, Chen J, Xie Y, Ke S, Wu J. et al. Pregnane X receptor regulates the AhR/Cyp1A1 pathway and protects liver cells from benzo-[α]-pyrene-induced DNA damage. Toxicol Lett. 2017;275:67-76
77. Poon PY, Kwok HH, Yue PY, Yang MS, Mak NK, Wong CK. et al. Cytoprotective effect of 20S-Rg3 on benzo[a]pyrene-induced DNA damage. Drug Metab Dispos. 2012;40:120-129
78. Wang YM, Chai SC, Brewer CT, Chen T. Pregnane X receptor and drug-induced liver injury. Expert Opin Drug Metab Toxicol. 2014;10:1521-1532
79. Shizu R, Abe T, Benoki S, Takahashi M, Kodama S, Miayata M. et al. PXR stimulates growth factor-mediated hepatocyte proliferation by cross-talk with the FOXO transcription factor. Biochem J. 2016;473:257-266
80. Wang YM, Ong SS, Chai SC, Chen T. Role of CAR and PXR in xenobiotic sensing and metabolism. Expert Opin Drug Metab Toxicol. 2012;8:803-817
81. Xie W, Uppal H, Saini SPS, Mu Y, Little JM, Radominska-Pandya A. et al. Orphan nuclear receptor-mediated xenobiotic regulation in drug metabolism. Drug Discovery Today. 2004;9:442-449
82. Gong HB, Singh SV, Singh SP, Mu Y, Lee JH, Saini SPS. et al. Orphan nuclear receptor pregnane X receptor sensitizes oxidative stress responses in transgenic mice and cancerous cells. Molecular Endocrinology. 2006;20:279-290
83. Zhou C, Tabb MM, Nelson EL, Grün F, Verma S, Sadatrafiei A. et al. Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J Clin Invest. 2006;116:2280-2289
84. Xie W, Tian Y. Xenobiotic receptor meets NF-kappaB, a collision in the small bowel. Cell Metab. 2006;4:177-178
85. Cheng J, Shah YM, Gonzalez FJ. Pregnane X receptor as a target for treatment of inflammatory bowel disorders. Trends Pharmacol Sci. 2012;33:323-330
86. Esposito G, Gigli S, Seguella L, Nobile N, D'Alessandro A, Pesce M. et al. Rifaximin, a non-absorbable antibiotic, inhibits the release of pro-angiogenic mediators in colon cancer cells through a pregnane X receptor-dependent pathway. Int J Oncol. 2016;49:639-645
87. Nones K, Dommels YE, Martell S, Butts C, McNabb WC, Park ZA. et al. The effects of dietary curcumin and rutin on colonic inflammation and gene expression in multidrug resistance gene-deficient (mdr1a-/-) mice, a model of inflammatory bowel diseases. Br J Nutr. 2009;101:169-181
88. Sepe V, Ummarino R, D'Auria MV, Mencarelli A, D'Amore C, Renga B. et al. Total synthesis and pharmacological characterization of solomonsterol A, a potent marine pregnane-X-receptor agonist endowed with anti-inflammatory activity. J Med Chem. 2011;54:4590-4599
89. Dou W, Zhang J, Zhang E, Sun A, Ding L, Chou G. et al. Chrysin ameliorates chemically induced colitis in the mouse through modulation of a PXR/NF-kappaB signaling pathway. J Pharmacol Exp Ther. 2013;345:473-482
90. Hu G, Xu C, Staudinger JL. Pregnane X receptor is SUMOylated to repress the inflammatory response. J Pharmacol Exp Ther. 2010;335:342-350
91. Dubrac S, Elentner A, Ebner S, Horejs-Hoeck J, Schmuth M. Modulation of T lymphocyte function by the pregnane X receptor. J Immunol. 2010;184:2949-2957
92. Schote AB, Turner JD, Schiltz J, Muller CP. Nuclear receptors in human immune cells: expression and correlations. Mol Immunol. 2007;44:1436-1445
93. Schoket B. DNA damage in humans exposed to environmental and dietary polycyclic aromatic hydrocarbons. Mutation Research-Fundamental and Molecular Mechanisms of Mutagenesis. 1999;424:143-153
94. Shen H, Liu J, Wang Y, Lian H, Wang J, Xing L. et al. Aflatoxin G1-induced oxidative stress causes DNA damage and triggers apoptosis through MAPK signaling pathway in A549 cells. Food Chem Toxicol. 2013;62:661-669
95. Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes & Development. 2004;18:1272-1282
96. Carvalho H, Garrido LM, Furlan RLA, Padilla G, Agnoletto M, Guecheva T. et al. DNA damage induced by the anthracycline cosmomycin D in DNA repair-deficient cells. Cancer Chemotherapy and Pharmacology. 2010;65:989-994
97. Rocha CRR, Silva MM, Quinet A, Cabral-Neto JB, Menck CFM. DNA repair pathways and cisplatin resistance: an intimate relationship. Clinics. 2018 73
98. Zucchini-Pascal N, de Sousa G, Pizzol J, Rahmani R. Pregnane X receptor activation protects rat hepatocytes against deoxycholic acid-induced apoptosis. Liver Int. 2010;30:284-297
99. Zhuang W, Jia Z, Feng H, Chen J, Wang H, Guo Y. et al. The mechanism of the G0/G1 cell cycle phase arrest induced by activation of PXR in human cells. Biomed Pharmacother. 2011;65:467-473
100. Muller M. Transcriptional control of hepatocanalicular transporter gene expression. Semin Liver Dis. 2000;20:323-337
101. Robbins D, Cherian M, Wu J, Chen T. Human pregnane X receptor compromises the function of p53 and promotes malignant transformation. Cell Death Discov. 2016;2:16023
102. Bassi C, Li YT, Khu K, Mateo F, Baniasadi PS, Elia A. et al. The acetyltransferase Tip60 contributes to mammary tumorigenesis by modulating DNA repair. Cell Death Differ. 2016;23:1198-1208
103. Sun Y, Xu Y, Roy K, Price BD. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol Cell Biol. 2007;27:8502-8509
104. Cui H, Guo M, Xu D, Ding ZC, Zhou G, Ding HF. et al. The stress-responsive gene ATF3 regulates the histone acetyltransferase Tip60. Nat Commun. 2015;6:6752
105. Cui H, Li X, Han C, Wang QE, Wang H, Ding HF. et al. The Stress-responsive Gene ATF3 Mediates Dichotomous UV Responses by Regulating the Tip60 and p53 Proteins. J Biol Chem. 2016;291:10847-10857
106. Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219-234
107. Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nature Reviews Cancer. 2012;12:587-598
108. Assaraf YG, Brozovic A, Goncalves AC, Jurkovicova D, Line A, Machuqueiro M. et al. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resistance Updates. 2019;46:100645
109. Wojtkowiak JW, Verduzco D, Schramm KJ, Gillies RJ. Drug Resistance and Cellular Adaptation to Tumor Acidic pH Microenvironment. Molecular Pharmaceutics. 2011;8:2032-2038
110. Fox M, Roberts JJ. Drug resistance and DNA repair. Cancer Metastasis Rev. 1987;6:261-281
111. Jiang H, Chen K, He J, Pan F, Li J, Chen J. et al. Association of pregnane X receptor with multidrug resistance-related protein 3 and its role in human colon cancer chemoresistance. J Gastrointest Surg. 2009;13:1831-1838
112. Chen Y, Huang W, Chen F, Hu G, Li F, Li J. et al. Pregnane X receptors regulate CYP2C8 and P-glycoprotein to impact on the resistance of NSCLC cells to Taxol. Cancer Med. 2016;5:3564-3571
113. Yasuda M, Kishimoto S, Amano M, Fukushima S. Comparison of Pregnane X Receptor Antagonists for Enhancing the Antitumor Effect of Cisplatin. Anticancer Research. 2019;39:4749-4755
114. Yasuda M, Kishimoto S, Amano M, Fukushima S. The Involvement of Pregnane X Receptor-regulated Pathways in the Antitumor Activity of Cisplatin. Anticancer Res. 2019;39:3601-3608
115. Choi HK, Yang JW, Roh SH, Han CY, Kang KW. Induction of multidrug resistance associated protein 2 in tamoxifen-resistant breast cancer cells. Endocrine-Related Cancer. 2007;14:293-303
116. Chen Y, Tang Y, Chen S, Nie D. Regulation of drug resistance by human pregnane X receptor in breast cancer. Cancer Biol Ther. 2009;8:1265-1272
117. Xu F, Wang F, Yang T, Sheng Y, Zhong T, Chen Y. Differential drug resistance acquisition to doxorubicin and paclitaxel in breast cancer cells. Cancer Cell Int. 2014;14:142
118. Chen Y, Zeng Q, Liu X, Fu J, Zeng Z, Zhao Z. et al. LINE-1 ORF-1p enhances the transcription factor activity of pregnenolone X receptor and promotes sorafenib resistance in hepatocellular carcinoma cells. Cancer Manag Res. 2018;10:4421-4438
119. Feng F, Jiang Q, Cao S, Cao Y, Li R, Shen L. et al. Pregnane X receptor mediates sorafenib resistance in advanced hepatocellular carcinoma. Biochim Biophys Acta Gen Subj. 2018;1862:1017-1030
120. Kong QN, Han ZL, Zuo XL, Wei HJ, Huang WQ. Co-expression of pregnane X receptor and ATP-binding cassette sub-family B member 1 in peripheral blood: A prospective indicator for drug resistance prediction in non-small cell lung cancer. Oncology Letters. 2016;11:3033-3039
121. Giannakakou P, Poy G, Zhan Z, Knutsen T, Blagosklonny MV, Fojo T. Paclitaxel selects for mutant or pseudo-null p53 in drug resistance associated with tubulin mutations in human cancer. Oncogene. 2000;19:3078-3085
122. Parmakhtiar B, Burger RA, Kim JH, Fruehauf JP. HIF Inactivation of p53 in Ovarian Cancer Can Be Reversed by Topotecan, Restoring Cisplatin and Paclitaxel Sensitivity. Mol Cancer Res. 2019;17:1675-1686
123. Harmsen S, Meijerman I, Maas-Bakker RF, Beijnen JH, Schellens JH. PXR-mediated P-glycoprotein induction by small molecule tyrosine kinase inhibitors. Eur J Pharm Sci. 2013;48:644-649
124. Shao Z, Li Y, Dai W, Jia H, Zhang Y, Jiang Q. et al. ETS-1 induces Sorafenib-resistance in hepatocellular carcinoma cells via regulating transcription factor activity of PXR. Pharmacol Res. 2018;135:188-200
125. Li J, Zhao J, Wang H, Li X, Liu A, Qin Q. et al. MicroRNA-140-3p enhances the sensitivity of hepatocellular carcinoma cells to sorafenib by targeting pregnenolone X receptor. Onco Targets Ther. 2018;11:5885-94
126. Shao QP, Wei C, Yang J, Zhang WZ. miR-3609 Decelerates the Clearance of Sorafenib in Hepatocellular Carcinoma Cells by Targeting EPAS-1 and Reducing the Activation of the Pregnane X Receptor Pathway. Onco Targets Ther. 2020;13:7213-27
127. Li B, Feng F, Jia H, Jiang Q, Cao S, Wei L. et al. Rhamnetin decelerates the elimination and enhances the antitumor effect of the molecular-targeting agent sorafenib in hepatocellular carcinoma cells via the miR-148a/PXR axis. Food Funct. 2021;12:2404-17
128. Vatsyayan R, Singhal J, Nagaprashantha LD, Awasthi S, Singhal SS. Nutlin-3 enhances sorafenib efficacy in renal cell carcinoma. Mol Carcinog. 2013;52:39-48
129. Liang Y, Zheng T, Song R, Wang J, Yin D, Wang L. et al. Hypoxia-mediated sorafenib resistance can be overcome by EF24 through Von Hippel-Lindau tumor suppressor-dependent HIF-1α inhibition in hepatocellular carcinoma. Hepatology. 2013;57:1847-57
130. Jiang Q, Ma Y, Han J, Chu J, Ma X, Shen L. et al. MDM2 Binding Protein Induces the Resistance of Hepatocellular Carcinoma Cells to Molecular Targeting Agents via Enhancing the Transcription Factor Activity of the Pregnane X Receptor. Front Oncol. 2021;11:715193
131. Masuyama H, Suwaki N, Tateishi Y, Nakatsukasa H, Segawa T, Hiramatsu Y. The pregnane X receptor regulates gene expression in a ligand- and promoter-selective fashion. Molecular Endocrinology. 2005;19:1170-80
132. Hirano G, Izumi H, Kidani A, Yasuniwa Y, Han B, Kusaba H. et al. Enhanced expression of PCAF endows apoptosis resistance in cisplatin-resistant cells. Mol Cancer Res. 2010;8:864-72
133. Banerjee Mustafi S, Chakraborty PK, Naz S, Dwivedi SK, Street M, Basak R. et al. MDR1 mediated chemoresistance: BMI1 and TIP60 in action. Biochim Biophys Acta. 2016;1859:983-93
134. Cui H, Arnst K, Miller DD, Li W. Recent Advances in Elucidating Paclitaxel Resistance Mechanisms in Non-small Cell Lung Cancer and Strategies to Overcome Drug Resistance. Curr Med Chem. 2020;27:6573-95
135. Mensah-Osman EJ, Thomas DG, Tabb MM, Larios JM, Hughes DP, Giordano TJ. et al. Expression levels and activation of a PXR variant are directly related to drug resistance in osteosarcoma cell lines. Cancer. 2007;109:957-65
136. Li T, Kong AN, Ma Z, Liu H, Liu P, Xiao Y. et al. Protein arginine methyltransferase 1 may be involved in pregnane x receptor-activated overexpression of multidrug resistance 1 gene during acquired multidrug resistant. Oncotarget. 2016;7:20236-48
137. Oguri T, Takahashi T, Miyazaki M, Isobe T, Kohn N, Mackenzie PI. et al. UGT1A10 is responsible for SN-38 glucuronidation and its expression in human lung cancers. Anticancer Research. 2004;24:2893-6
138. Raynal C, Pascussi JM, Legueline G, Breuker C, Kantar J, Lallemant B. et al. Pregnane x Receptor (PXR) expression in colorectal cancer cells restricts irinotecan chemosensitivity through enhanced SN-38 glucuronidation. Molecular Cancer. 2010;9:46
139. Sandanaraj E, Lal S, Selvarajan V, Ooi LL, Wong ZW, Wong NS. et al. PXR pharmacogenetics: association of haplotypes with hepatic CYP3A4 and ABCB1 messenger RNA expression and doxorubicin clearance in Asian breast cancer patients. Clin Cancer Res. 2008;14:7116-26
140. Huang H, Wang H, Sinz M, Zoeckler M, Staudinger J, Redinbo MR. et al. Inhibition of drug metabolism by blocking the activation of nuclear receptors by ketoconazole. Oncogene. 2007;26:258-68
141. Gupta D, Venkatesh M, Wang HW, Kim S, Sinz M, Goldberg GL. et al. Expanding the roles for pregnane X receptor in cancer: Proliferation and drug resistance in ovarian cancer. Clinical Cancer Research. 2008;14:5332-40
142. Venkatesh M, Wang HW, Cayer J, Leroux M, Salvail D, Das B. et al. In vivo and In vitro Characterization of a First-in-Class Novel Azole Analog That Targets Pregnane X Receptor Activation. Molecular Pharmacology. 2011;80:124-35
143. Li H, Redinbo MR, Venkatesh M, Ekins S, Chaudhry A, Bloch N. et al. Novel Yeast-based Strategy Unveils Antagonist Binding Regions on the Nuclear Xenobiotic Receptor PXR. Journal of Biological Chemistry. 2013;288:13655-68
144. Masuyama H, Nakatsukasa H, Takamoto N, Hiramatsu Y. Down-regulation of pregnane X receptor contributes to cell growth inhibition and apoptosis by anticancer agents in endometrial cancer cells. Mol Pharmacol. 2007;72:1045-53
145. Shiratori K, Ohgami K, Ilieva I, Jin XH, Koyama Y, Miyashita K. et al. Effects of fucoxanthin on lipopolysaccharide-induced inflammation in vitro and in vivo. Exp Eye Res. 2005;81:422-8
146. Liu CL, Lim YP, Hu ML. Fucoxanthin Attenuates Rifampin-Induced Cytochrome P450 3A4 (CYP3A4) and Multiple Drug Resistance 1 (MDR1) Gene Expression Through Pregnane X Receptor (PXR)-Mediated Pathways in Human Hepatoma HepG2 and Colon Adenocarcinoma LS174T Cells. Marine Drugs. 2012;10:242-57
147. Ekins S, Kholodovych V, Ai N, Sinz M, Gal J, Gera L. et al. Computational discovery of novel low micromolar human pregnane X receptor antagonists. Molecular Pharmacology. 2008;74:662-72
148. Poulton EJ, Levy L, Lampe JW, Shen DD, Tracy J, Shuhart MC. et al. Sulforaphane is not an effective antagonist of the human pregnane X-receptor in vivo. Toxicol Appl Pharmacol. 2013;266:122-31
149. Wang LG, Liu DL, Ahmed T, Chung FL, Conaway C, Chiao JW. Targeting cell cycle machinery as a molecular mechanism of sulforaphane in prostate cancer prevention. International Journal of Oncology. 2004;24:187-92
150. Zhou Y, Yang G, Tian H, Hu Y, Wu S, Geng Y. et al. Sulforaphane metabolites cause apoptosis via microtubule disruption in cancer. Endocr Relat Cancer. 2018;25:255-68
151. Geng Y, Zhou Y, Wu S, Hu Y, Lin K, Wang Y. et al. Sulforaphane Induced Apoptosis via Promotion of Mitochondrial Fusion and ERK1/2-Mediated 26S Proteasome Degradation of Novel Pro-survival Bim and Upregulation of Bax in Human Non-Small Cell Lung Cancer Cells. J Cancer. 2017;8:2456-70
152. Wang Y, Zhou Y, Zheng Z, Li J, Yan Y, Wu W. Sulforaphane metabolites reduce resistance to paclitaxel via microtubule disruption. Cell Death Dis. 2018;9:1134
153. Lin WW, Wang YM, Chai SC, Lv LL, Zheng J, Wu J. et al. SPA70 is a potent antagonist of human pregnane X receptor. Nature Communications. 2017;8:741
154. Huber AD, Wright WC, Lin WW, Majumder K, Low JA, Wu J. et al. Mutation of a single amino acid of pregnane X receptor switches an antagonist to agonist by altering AF-2 helix positioning. Cellular and Molecular Life Sciences. 2021;78:317-35
155. Lin W, Bwayi M, Wu J, Li Y, Chai SC, Huber AD. et al. CITCO Directly Binds to and Activates Human Pregnane X Receptor. Mol Pharmacol. 2020;97:180-90
156. Li Y, Lin W, Wright WC, Chai SC, Wu J, Chen T. Building a Chemical Toolbox for Human Pregnane X Receptor Research: Discovery of Agonists, Inverse Agonists, and Antagonists Among Analogs Based on the Unique Chemical Scaffold of SPA70. J Med Chem. 2021;64:1733-1761
157. Wang W, Zhao M, Cui L, Ren Y, Zhang J, Chen J. et al. Characterization of a novel HDAC/RXR/HtrA1 signaling axis as a novel target to overcome cisplatin resistance in human non-small cell lung cancer. Mol Cancer. 2020;19:134
158. Geick A, Eichelbaum M, Burk O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. Journal of Biological Chemistry. 2001;276:14581-7
159. Kast HR, Goodwin B, Tarr PT, Jones SA, Anisfeld AM, Stoltz CM. et al. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J Biol Chem. 2002;277:2908-15
160. To KKW, Zhan Z, Bates SE. Aberrant promoter methylation of the ABCG2 gene in renal carcinoma. Molecular and Cellular Biology. 2006;26:8572-85
161. Buchman CD, Chai SC, Chen T. A current structural perspective on PXR and CAR in drug metabolism. Expert Opin Drug Metab Toxicol. 2018;14:635-47
162. Chai SC, Wright WC, Chen T. Strategies for developing pregnane X receptor antagonists: Implications from metabolism to cancer. Med Res Rev. 2020;40:1061-83
Author contact
![]() Corresponding authors: Yanan Tian, Department of Veterinary Physiology and Pharmacology, College of Veterinary Medicine, Texas A&M University, College Station, TX 77843, USA; Email: ytiantamu.edu. Hongmei Cui, Institute of Toxicology, School of Public Health, Lanzhou University, 730000, Lanzhou, China; Email: Cuihmedu.cn.
Corresponding authors: Yanan Tian, Department of Veterinary Physiology and Pharmacology, College of Veterinary Medicine, Texas A&M University, College Station, TX 77843, USA; Email: ytiantamu.edu. Hongmei Cui, Institute of Toxicology, School of Public Health, Lanzhou University, 730000, Lanzhou, China; Email: Cuihmedu.cn.