Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(3):1107-1119. doi:10.7150/ijbs.64285 This issue Cite
Research Paper
The Role of Autophagy in Lamellar Body Formation and Surfactant Production in Type 2 Alveolar Epithelial Cells
Xiaoman Li1,2,3#, Liang Wang4#, Jialin Hao1,2, Qingfeng Zhu1,2, Min Guo1,2, Changjing Wu1*, Sihui Li1, Qiqiang Guo1,2, Qiuhong Ren1, Ning Bai1,2, Fei Yi1,2, Bo Jiang1,2, Wenyu Zhang1, Yanling Feng1,2, Hongde Xu1,2, Han Jiang5, Xiaoyue Zhai1, Guohua Zhang6, Hong-long Ji7, Xuesong Yang8, Dan Zhang9, Jianhua Fu9, Jianjun Chang3, Xiaoyu Song1,2,3 ![]() , Liu Cao1,2
, Liu Cao1,2 ![]()
1. College of Basic Medical Sciences, China Medical University, Shenyang, China.
2. Key Laboratory of Medical Cell Biology, Ministry of Education, China Medical University, Shenyang, China.
3. Institute of Health Sciences, China Medical University, Shenyang, China.
4. Department of Pathology, College of Basic Medical Sciences, China Medical University, Shenyang, China.
5. Department of Vascular Surgery, The First affiliated Hospital, China Medical University, Shenyang, China.
6. Department of Forensic Pathology, School of Forensic Medicine, China Medical University, Shenyang, China.
7. Department of Cellular and Molecular Biology, University of Texas Health Science Center at Tyler, Tyler, USA.
8. Institute of Biomedicine, National Engineering Research Center of Genetic Medicine, Jinan University, Guangzhou, China.
9. Department of Pediatrics, Shengjing Hospital of China Medical University, Shenyang, China.
*Current affiliation: West China Hospital, Sichuan University, China.
#These authors contribute equally to this work.
Received 2021-6-27; Accepted 2021-12-4; Published 2022-1-1
Abstract

The lamellar body (LB), a concentric structure loaded with surfactant proteins and phospholipids, is an organelle specific to type 2 alveolar epithelial cells (AT2). However, the origin of LBs has not been fully elucidated. We have previously reported that autophagy regulates Weibel-Palade bodies (WPBs) formation, and here we demonstrated that autophagy is involved in LB maturation, another lysosome-related organelle. We found that during development, LBs were transformed from autophagic vacuoles containing cytoplasmic contents such as glycogen. Fusion between LBs and autophagosomes was observed in wild-type neonate mice. Moreover, the markers of autophagic activity, microtubule-associated protein 1 light chain 3B (LC3B), largely co-localized on the limiting membrane of the LB. Both autophagy-related gene 7 (Atg7) global knockout and conditional Atg7 knockdown in AT2 cells in mice led to defects in LB maturation and surfactant protein B production. Additionally, changes in autophagic activity altered LB formation and surfactant protein B production. Taken together, these results suggest that autophagy plays a critical role in the regulation of LB formation during development and the maintenance of LB homeostasis during adulthood.
Keywords: autophagy, Atg7, lamellar body, type 2 alveolar cells, LC3B, surfactant protein B
Introduction
Type 2 alveolar epithelial cells (AT2) in the lung tissue play a critical role in retaining the alveoli's integrity and function. AT2 cells contain a unique organelle called the lamellar body (LB), a concentric structure loaded with surfactant proteins and phospholipids [1]. LBs are secreted into the air space by exocytosis, where they unravel into a tubular network (tubular myelin) that is absorbed and spread at the air-liquid interface to form a phospholipid film [2]. The origin of LBs has not been fully elucidated. The well-accepted view that LBs are derived from a multivesicular body (MVB) is supported by evidence from transmission electron microscopy (TEM) studies showing the transition from MVB to LB [3-6]. Autoradiography studies of murine AT2 cells from mouse lungs have shown that although proteins that were metabolically labeled with 3H leucine were visualized in MVBs before delivery to LBs, phospholipids labeled with 3H choline were delivered directly to LBs [7, 8]. Therefore, although surfactant proteins may be supplied by MVBs, phosphatidylcholine is not. Notably, glycogen is an established substrate for surfactant phospholipid production during lung development because glycogen depletion correlated with phospholipid synthesis [9-14].
Surfactant proteins are closely related to the formation of LBs, and when secreted into the alveolar cavity with surfactant phospholipids, they play a pivotal role in the formation and maintenance of the surfactant film [15-17]. Out of four surfactant proteins (SFTPA, SFTPB, SFTPC, and SFTPD), SFTPB and SFTPC are hydrophobic and contribute to the surface tension-lowering activity [18]. Only SFTPB is essential for LB formation, postnatal viability, and breathing [19-22]. SFTPB-deficient mice are not viable, and consistently, infants carrying homozygous SFTPB mutations die of respiratory failure after birth [21, 22]. A previous study has shown that mice may exhibit perinatal lethality when the SFTPB level is reduced to 25% of the normal level [23].
Weibel-Palade bodies (WPBs) of endothelial cells, the melanosomes of melanocytes, the LBs of AT2 cells, and the dense bodies of platelets are all lysosome-related organelles (LROs). Autophagy regulates the formation of several LROs, including WPBs and melanosomes [24, 25]. Hermansky-Pudlak Syndrome (HPS), an autosomal recessive disease, is characterized by clinical manifestations of hypopigmentation, abnormal platelets, and pulmonary fibrosis, which are all coincidentally related to LROs [26]. Cells from lung fibrosis patients suffering from HPS show altered autophagy and abnormal LBs [27, 28]. Taken together, it is plausible to speculate that autophagy is involved in regulating the homeostasis of another LRO, the LB. Although LB formation was compromised by treatment with the autophagic inhibitor 3-methyladenine (3-MA) in a cellular model [8, 15, 29], the detailed mechanism of how autophagy regulates the formation of LBs has not been fully elucidated in an in vivo autophagy-deficient model.
In the present study, we demonstrated that autophagy is an essential physiological process for regulating LB formation and maintaining the proper function of AT2 cells. Mice with global deletion of autophagy-related gene 7 (Atg7) show abnormal lung morphology, impaired autophagic activity, smaller/fewer LBs, and reduced surfactant protein B. Additionally, deletion of Atg7 in AT2 cells using the SPC-specific promotor produced similar effects to those observed with the global knockout of Atg7 at the neonatal stage but led to large and irregular vacuoles (LB-like vacuoles) and degenerative AT2 cells during adulthood. In summary, our results indicate that autophagy plays a critical role in regulating the formation of LBs during development and maintaining LB homeostasis during adulthood in AT2 cells.
Results
Changes in autophagic activity alter LB formation and surfactant protein production
To test whether altered autophagic activity affects LB formation, an autophagy inducer and an inhibitor were used. Rapamycin, which enhances autophagic activity by inhibiting the mechanistic target of rapamycin kinase (mTOR)-UNC-51-like kinase 1 (ULK1) pathway [30, 31], was administrated to 8-week-old WT mice, and lungs were harvested and analyzed 8 hours later. Upon rapamycin treatment, the size of LBs was increased (Figure 1A, B), as demonstrated by the immunofluorescent staining of ATP-binding cassette sub-family A (ABC1), member 3 (ABCA3), which is localized on the limiting membrane of the LB [32]. The size distribution of LBs in control and rapamycin-treated mice is shown in Figure 1C. The number of LBs was also significantly increased upon treatment with rapamycin (Figure 1D). Consistent with the downregulation of mTOR signaling by rapamycin, we observed an increased level of LC3B-II (Figure 1E). Furthermore, the expression level of SFTPB was elevated upon rapamycin treatment (Figure 1E). To further elucidate the role of autophagy in LB formation, we administered 3-MA, which blocks the phosphatidylinositol 3-kinase (PI3K) pathway and inhibits autophagy, or vehicle control to WT mice. 3-MA treatment decreased both the size and number of LBs (Figure 1A-D). Consistently, western blot analysis showed that the SFTPB level was downregulated upon 3-MA treatment, and the extent of autophagy inhibition was determined by measuring the LC3B-II level (Figure 1F).
Changes in autophagic activity alter LB formation and surfactant protein production. (A) Immunofluorescent staining of frozen sections from control (PBS), rapamycin (Rapa), and 3-MA-treated mice. The limiting membrane of LBs was detected using an anti-ABCA3 antibody (scale bar: 5 µm). Representative AT2 cells are shown. The average area (B) and the size distribution (C) of LBs in PBS, Rapa, or 3-MA-treated mice are shown (the area of total 80 LBs in at least 5 random magnification fields per mouse was measured using ImageJ software; n = 3 mice/group). (D) The average number of LBs in Rapa, 3-MA, or PBS-treated mice (at least 25 random AT2 cells were chosen per mouse; n = 3 mice/group). The expression level of SFTPB in mice treated with Rapa (E) or 3-MA (F) (n = 3 mice/group; experiments were duplicated three times; representative results are shown). (G) Primary human AT2 cells treated with control, Rapa, or 3-MA were incubated with LysoTracker to stain LBs (scale bar: 5 µm). The experiments were repeated three times; representative AT2 cells are shown. The average area (H) and size distribution (I) of LBs from Rapa or 3-MA-treated primary human AT2 cells were plotted compared with controls (80 random LBs were measured/group). (J) The average number of LBs in Rapa, 3-MA, or PBS-treated primary human AT2 cells (n ≥25 AT2 cells/group). The expression level of SFTPB in primary human AT2 cells treated with Rapa (K) or 3-MA (L) (experiments were duplicated three times; representative results are shown). *P < 0.05; **P < 0.01; ***P < 0.001.
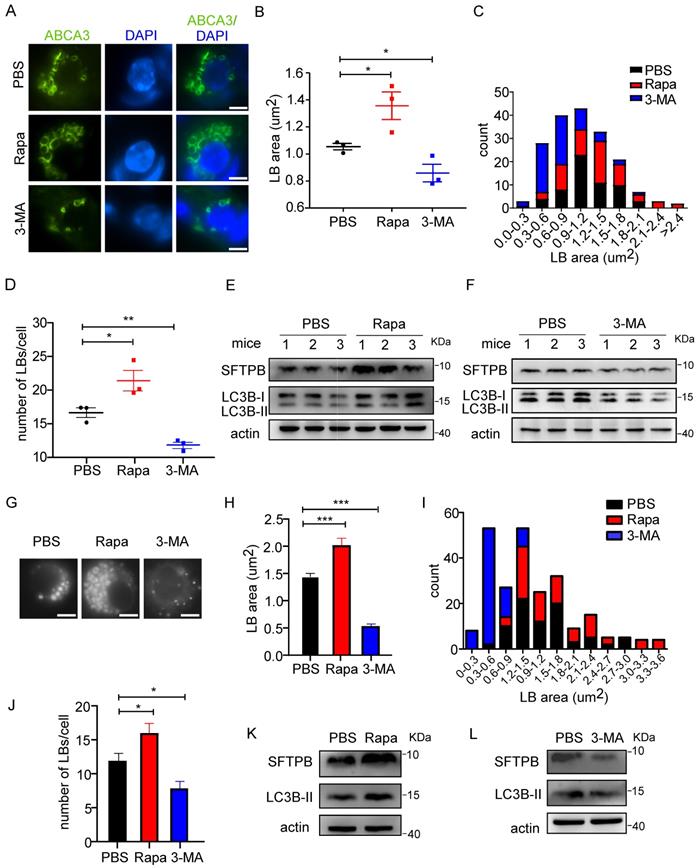
We further studied the effects of autophagy on LBs in isolated primary human AT2 cells. LysoTracker, a fluorescent probe for tracking acidic organelles in live cells, was used to label LBs in AT2 cells. Consistent with the results observed in AT2 cells from mice, rapamycin increased the number and size of LBs (Figure 1G-J) and the level of SFTPB in human AT2 cells (Figure 1K). Conversely, treatment with 3-MA significantly reduced both the number and size of LBs (Figure 1G-J). Moreover, the level of SFTPB was decreased by 3-MA treatment (Figure 1L). Taken together, these results suggest that autophagy regulates LB formation and surfactant protein production in both mice and human AT2 cells.
Atg7-deficient mice show defects in lung development, LB formation, and surfactant biogenesis
ATG7, a component of the ATG conjugation systems, is pivotal to the expansion and closure of autophagosome membranes. Targeted deletion of Atg7 leads to perinatal lethality, which may be due to the non-suckling phenotype in the early neonatal period [33, 34]. Atg7-deficient mice (Atg7 KO) died within 12 h, whereas non-suckling littermate controls died approximately 24 h after birth. Therefore, we analyzed the morphological development of lungs and LB formation in Atg7-/- mice (P1). The gross appearance of Atg7 KO mice indicated their size was smaller than that of wild-type (WT) mice (Figure 2A). The bodyweight of Atg7 KO mice (1.42 g) was slightly lower than that of WT mice (1.58 g). Lungs from Atg7 KO mice and WT littermates were harvested as soon as the pups were found in the breeding cage. Hematoxylin and eosin (H&E) staining showed that Atg7 KO mice had unevenly expanded air sacs and impaired thinning of the alveolar septa. In contrast, the lungs of WT neonates had normal air sac expansion and septal thinning at birth (Figure 2B, C).
Atg7-deficient mice show defects in lung development, LB formation, and surfactant production. (A) Gross appearance of representative neonates. The bodyweight of Atg7-/- mice was slightly but significantly lower than that of wild-type (WT) mice (1.42g vs. 1.58g; n = 5). (B) Histological analysis of lungs from WT control (Ctrl) and Atg7 KO neonates (scale bar: 100 µm). (C) Septal thickness was measured at all points in each visual field (at least five random visual fields in high magnification per mouse were used for measurements; WT, n = 8, Atg7 KO, n = 6). (D) Frozen sections of Ctrl and Atg7 KO neonate lungs were stained with the anti-ABCA3 antibody to detect LBs (scale bar: 5 µm). Representative single cells are shown. The average area (E) and size distribution (F) of LBs were quantified by ImageJ software (80 LBs/mouse in at least five random high magnification fields were measured; WT, n = 5; Atg7 KO, n = 3). (G) The average number of LBs per cell was quantified (at least 25 AT2 cells/mouse were quantified; WT, n = 5; Atg7 KO, n = 3). (H) Electron microscopy analysis shows a marked reduction in the quantity and quality of LBs in Atg7 KO mice compared with controls (* denotes LB; Nu denotes nucleus; Insets are enlarged LBs; scale bar: 1 µm). (I) Quantification of the average area of LBs (n = 4 mice/group). (J) The number of LBs per AT2 cell in each genotype (n = 4 mice/group). (K) The levels of SFTPB and pro-SFTPC in extracts of whole lung tissue from Ctrl and Atg7 KO mice were determined by western blotting. Experiments were repeated more than three times; representative images are shown. (L) Densitometry analysis of SFTPB, pro-SFTPC, p62, and LC3B II/I in Ctrl and Atg7 KO mice (for SFTPB, WT, n = 16, Atg7 KO, n = 13; for pro-SFTPC, p62, and LC3B II/I, WT, n = 9, Atg7 KO, n = 4). (M) Knockdown of ATG7 by adenoviral infection inhibited LB formation. Cells infected with ad-vector have both lysotracker staining and GFP-fluorescence; cells infected with ad-sh-ATG7 have GFP-fluorescence but lack lysotracker staining (scale bar: 5 μm). *P < 0.05; **P < 0.01; ***P < 0.001.
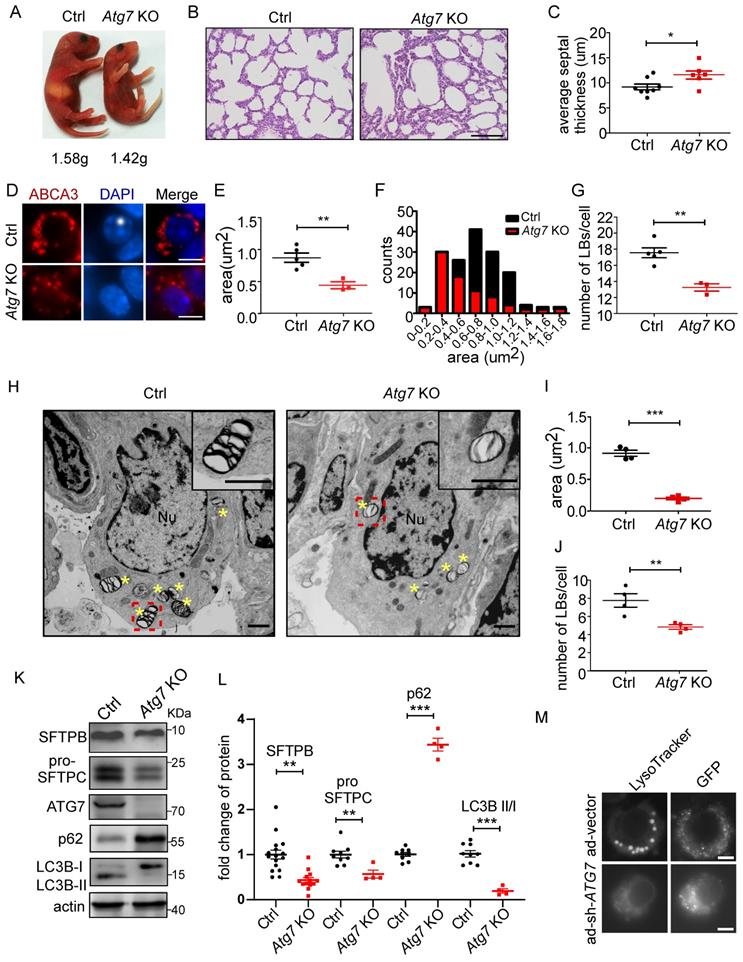
Next, we examined whether the Atg7 deletion in mice compromised the formation of LBs. We found that the LBs in Atg7 KO mice were significantly smaller than those in WT mice (Figure 2D-F and Supplemental Figure 1A-C). Frequently, the hollow circle delineated by ABCA3 staining in WT mice was absent in Atg7 KO mice. Moreover, the number of LBs in Atg7 KO mice was lower than in WT mice (Figure 2G). Similarly, electron microscopy (EM) revealed that the abnormal LBs in Atg7 KO mice contained fewer lamellae than those in WT mice (Figure 2H). EM images also confirmed a reduction in LB size in Atg7 KO mice, compared with the WT controls (Figure 2I). The analysis of random sections showed that the mean number of LBs per AT2 cell was also dramatically reduced (Figure 2J), further supporting the notion that ATG7 may be involved in the growth of LBs.
The surfactant proteins SFTPB and pro-SFTPC were reduced in Atg7 KO mice (Figure 2K and L and Supplemental Figure 2A, B), further supporting the idea that LBs were compromised in Atg7 KO mice. However, SFTPA was unaltered (Supplemental Figure 2C), consistent with previous reports that SFTPA was secreted in an LB-independent manner [35-37]. The SFTPB and SFTPC promoters are reportedly regulated by thyroid transcription factor-1 (TTF-1/Nkx2.1) in AT2 cells [38]. However, TTF-1 expression in Atg7 KO mice was no different from that in WT mice (Supplemental Figure 2D). Next, we assessed autophagic activity by determining the ratio of the expression levels of LC3B-II and LC3B-I and the amount of the autophagy substrate, SQSTM1 (referred to here as p62). In Atg7 KO lungs, the LC3B-II/LC3B-I ratio was reduced compared with the controls (Figure 2K, L), consistent with ATG7 being essential for ATG conjugation systems and autophagosome formation. Western blot analysis also showed the accumulation of p62 in Atg7-deficient lungs compared with the controls (Figure 2K, L). Because AT2 cells constitute only a small percentage of the cell population in the lung, we isolated primary AT2 cells by magnetic-activated cell isolation and subjected them to western blot analysis. Both SFTPB and pro-SFTPC levels were further reduced in the AT2 cells compared with the whole tissue lysate (Supplemental Figure 3A, B). To directly assess the effect of ATG7 on LBs, we infected human primary AT2 cells with adenoviruses carrying sh-ATG7-EGFP to knockdown endogenous ATG7. Consistent with the previously presented results, ablation o of ATG7 compromised LB formation in human primary AT2 cells (Figure 2M, Supplemental Figure 4). Lung surfactant comprises 80% glycerophospholipids, 10% cholesterol, and 10% surfactant proteins. Phosphatidylcholine (PC) and phosphatidylglycerol (PG) are the predominant phospholipids of LBs and make up 70%-80% and 5%-10%, respectively [39]. The primary form of PC is its unsaturated form as dipalmitoyl-phosphatidylcholine (DPPC; 16:0/16:0). To examine whether Atg7 deletion affects the lipid content of LBs, we subjected whole-lung homogenates to quantitative lipid chromatography-tandem mass spectrometry (LC-MS/MS). Among 949 different lipids detected, no significant difference was found in the most abundant lipids, including DPPC and cholesterol. By our criteria, only 14 phosphatidylglycerols were downregulated in the Atg7 KO lung (Supplemental Figure 5 and Supplemental Table 1). Taken together, the present study indicates that Atg7 is critical for lung development, LB formation, and surfactant SFTPB/C production.
The role of autophagy in LB formation
Because the enrichment of glycogen is characteristic of immature AT2 cells [14], we examined the glycogen level in Atg7 KO and control mice. EM revealed that more glycogen-filled AT2 cells were present in P1 Atg7 KO mice than in littermate controls (Figure 3A). Consistently, the glycogen levels, measured by the amount of glucose hydrolyzed from glycogen [40], were higher in Atg7 KO mice than those in littermate controls (Figure 3B). Periodic acid-Schiff (PAS) staining, which stains carbohydrate macromolecules such as glycogen and glycoproteins, was also performed on lung sections. Consistently, the PAS-staining intensity was higher in Atg7 KO mice than in WT controls (Supplemental Figure 6A). Moreover, PAS staining co-stained with ABCA3, suggesting that glycogen mainly accumulated in AT2 cells (Supplemental Figure 6B). Collectively, the abnormally accumulated glycogen in the AT2 cells of Atg7 KO neonate lungs suggests delayed differentiation of AT2 cells.
Autophagic vacuoles are involved in LB formation. (A) EM analysis shows the abnormal glycogen accumulation in Atg7 KO P1 neonate mice (Gly denotes glycogen, scale bar: 1 µm). (B) Glycogen levels in lung homogenates from Atg7 KO mice and littermate controls (mice/group WT, n = 8; KO, n = 6). (C) Electron micrographs of AT2 cells from WT neonatal mice (P1) showing LBs fusing with autophagosomes (* denotes LB; Av denotes autophagic vacuole; scale bar: 200 nm). Electron micrographs of AT2 cells from an E18.5 WT mouse lung with autophagosomes containing cytoplasmic contents (D), glycogen particles (E), and a combination of cytoplasmic contents and undigested glycogen particles (F) (* denotes cytoplasmic contents/organelles, Gly denotes glycogen; scale bar: 500 nm). The lamellar transition from autophagosome could be seen in (G) and (H) (Mt denotes mitochondria, arrowhead denotes the appearance of lamellae; scale bar: 500 nm). (I) Co-localization of ABCA3 and LC3B in WT and Atg7 KO mice (scale bar: 5 µm). (J) Line profile plot of the immunofluorescence (red line) is shown. (K) Pearson correlation coefficient (PCC) for WT and Atg7 KO mice (mice/group WT, n = 4, KO, n = 3) was calculated. *P < 0.05, ***P < 0.001.
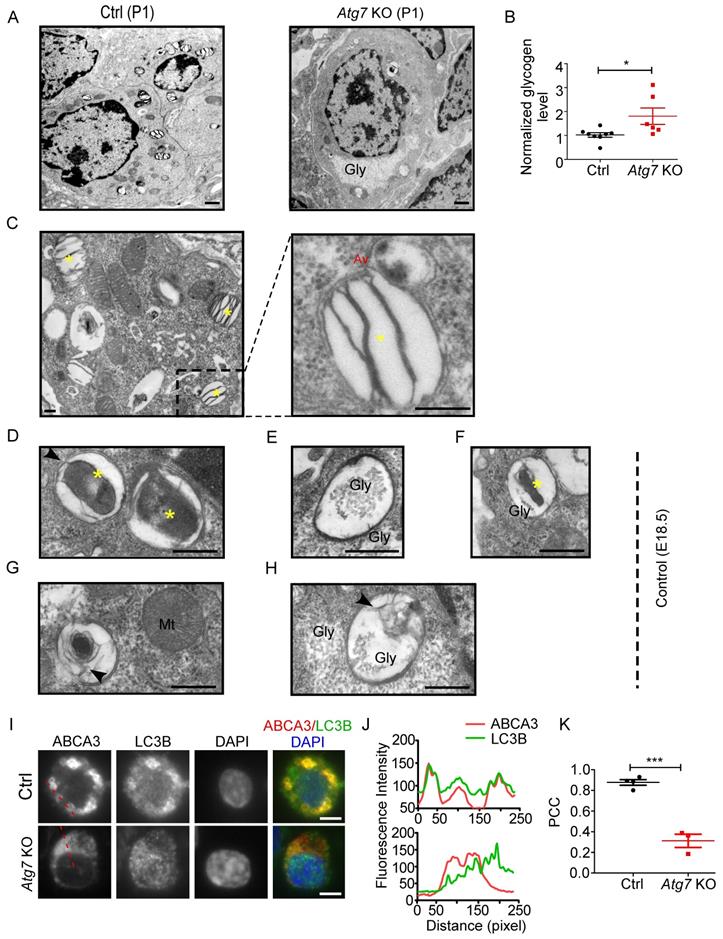
To understand how autophagy is related to LB formation, we analyzed electron micrographs of neonate WT mice (P1). When applying the routine EM fixation procedure, autophagosomes in AT2 cells are characterized by a double membrane enclosing regions of low electron density or an electron-translucent cleft [41]. LBs and autophagosomes were often found in close proximity to each other within AT2 cells. Moreover, we observed multiple instances in which an LB actively fused with an autophagosome in P1 WT neonates (Figure 3C). Because both the glycogen content and the autophagic activity peak prior to term (around E18.5) and fall by birth [42, 43], we also examined the electron micrographs of E18.5 Atg7 KO and WT mice. Interestingly, we readily observed autophagosomes enclosing cytoplasmic contents, including glycogen particles (Figure 3D-F and Supplemental Figure 6C), in the lungs of E18.5 WT mice. The appearance of lamella transitioning from cytoplasmic contents and glycogen was also observed (Figure 3G, 3H). Although glycogen was sequestered in E18.5 Atg7 KO mice, the characteristic autophagic membrane was not observed (Supplemental Figure 6D). Taken together, these data suggest that autophagy may be involved in LB biogenesis during development by engulfing and using cytoplasmic contents such as glycogen. Moreover, autophagy may maintain the LB function by continuous fusion between autophagosomes and LBs.
To further confirm the autophagic origin of LBs, we performed double immunofluorescence using anti-ABCA3 and anti-LC3B antibodies. Interestingly, we found that LC3B and ABCA3 largely colocalized on the LB membrane in WT neonate mice (Figure 3I, J, upper panel). The colocalization of LC3B and ABCA3 was as high as 0.88, as measured by Pearson's correlation coefficient (PCC) (Figure 3K). However, in Atg7 KO mice, LC3B was dispersed (Figure 3I, J, lower panel), and the PCC was 0.31 (Figure 3K). Collectively, these observations suggest that autophagy is essential for LB formation and that the LB membrane, at least in part, is derived from the autophagosome.
Lung development defects and altered LB formation in bronchioalveolar epithelium-specific Atg7 deletion mice
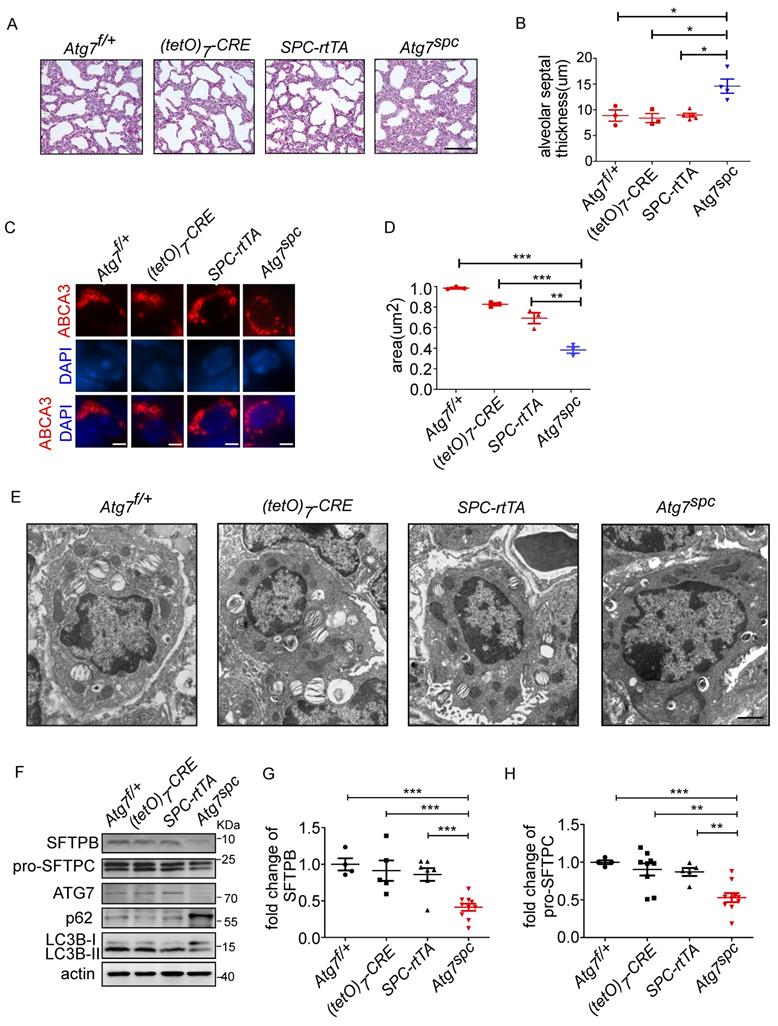
To further elucidate the function of ATG7 in AT2 cells, bronchioalveolar epithelium-specific Atg7 KO mice were generated. Mice harboring floxed Atg7 alleles were mated with SPC-rtTA mice that express the rtTA gene under the control of the SFTPC promoter (hereafter referred to as Atg7SPC mice). Atg7SPC pups did not show neonatal lethality and exhibited no cyanosis or growth retardation. The postnatal growth of Atg7SPC mice was also comparable to that of the control mice.
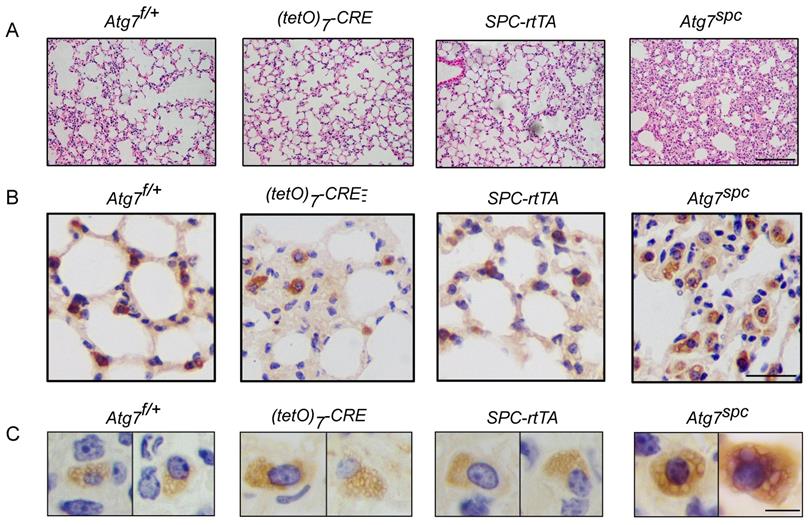
We then analyzed the lungs of Atg7SPC neonates (P1) and littermate mice with the genotype Atg7flox/+, Atg7flox/+:(tetO)7-Cre, and Atg7flox/+:SPC-rtTA. Because it has been reported that treatment of SPC-rtTA mice with doxycycline (DOX) may exert toxicity to alveolar epithelial cells [44], the phenotype of Atg7flox/+:SPC-rtTA mice was carefully compared with littermate controls. However, no apparent abnormality was observed in Atg7flox/+:SPC-rtTA mice under our experimental conditions, except for an enlarged alveolar cavity. H&E staining showed impaired thinning of the alveolar septa in Atg7SPC mice, whereas the lungs of control neonates showed normal septal thinning at birth (Figure 4A). Morphometric analysis revealed an increase in alveolar septal thickness in Atg7SPC mice (Figure 4B). Similar to our observations in Atg7 KO mice, Atg7SPC mice had altered LBs (Figure 4C). Quantitative analysis revealed that the size of LBs in Atg7SPC mice was significantly reduced compared with control mice (Figure 4D). EM also revealed that the abnormal LBs in Atg7SPC mice had fewer lamellae than those in WT mice (Figure 4E). Furthermore, a substantial reduction in ATG7 protein expression was observed in Atg7SPC mice, sufficient to reduce autophagic activity, as evidenced by the increase in p62 and the decrease in the LC3B-II/LC3B-I ratio (Figure 4F). Western blot analysis of SFTPB and pro-SFTPC revealed reduced expression in Atg7SPC mice (Figure 4F-H). Collectively, these findings suggest that the phenotype observed in Atg7 KO lungs is mainly due to the lack of ATG7 in AT2 cells.
Lung dysplasia and abnormal LBs are observed in bronchioalveolar epithelium-specific Atg7 conditional knockout mice. (A) Histological analysis of neonatal lungs from Atg7flox/+, Atg7flox/+:(tetO)7-Cre, Atg7flox/+:SFTPC-rtTA, and Atg7SPC neonates (scale bar: 100 µm). (B) Septal thickness was measured at all points in each visual field, and five visual fields for each animal were used for the measurements (n ≥3 mice/group). (C) Frozen sections of Atg7flox/+, Atg7flox/+:(tetO)7-Cre, Atg7flox/+:SFTPC-rtTA, and Atg7SPC neonates' lungs were stained using anti-ABCA3 antibody to detect LBs (scale bar: 2.5 µm). (D) The average size of LBs in control and Atg7SPC neonate lungs (≥80 LBs in at least 5 ransom high magnification fields were measured/mouse, mice/group n = 3). (E) EM analysis shows robust LBs in the control mice, whereas those in Atg7SPC mice contain few lamellae (scale bar: 1 µm). (F) Western blot analysis of protein lysates prepared from the lungs of Atg7 SPC mice and littermate controls at P1. In Atg7 SPC neonates, LC3B-I failed to convert to LC3B-II, and p62 was accumulated. All experiments were performed at least three times; representative data are shown. A quantitative analysis of SFTPB (G) and pro-SFTPC (H) (mice/group n ≥ 4). *P < 0.05; **P < 0.01; ***P < 0.001.
Postnatal Atg7SPCmice contain large and irregular ABCA3+ vacuoles
To elucidate how ATG7 affects the lung postnatally, we performed microscopy and histological analyses of Atg7SPC mice at P90. Consistent with the observation in P1 mice, H&E staining revealed a thick lung septum (Figure 5A). Immunostaining of lung tissue with anti-ABCA3 antibody showed swelling of AT2 cells in Atg7SPC mice (Figure 5B). Additionally, this analysis showed LBs of uniform size in control mice, whereas the lung tissue from Atg7SPC mice contained large and irregular vacuoles (Figure 5C). Because these vacuoles were ABCA3-positive, we called them “ABCA3+ vacuoles” (LB-like vacuoles). Large LBs were also observed in patients with HPS, which is manifested with lower autophagic activity [28]. Considering autophagosomes constantly fuse with LBs, these enlarged ABCA3+ vacuoles are probably a result of insufficient autophagic activity to maintain normal LBs. Collectively, these data indicate that Atg7 regulates LB formation at the neonatal stage and maintains LB homeostasis at postnatal stages.
Postnatal Atg7SPC mice (P90) contain large and irregular ABCA3+ vacuoles. (A) Histological analysis of lungs from control and Atg7SPC mice (P90, scale bars: 100 µm). Immunostaining of Atg7SPC lungs with anti-ABCA3 antibody shows swollen AT2 cells (scale bar: 20 µm) (B) and large and irregular ABCA3+ vacuoles, compared with littermate controls (scale bars: 5 µm) (C).
Discussion
It is well-established that large amounts of glycogen accumulate in undifferentiated epithelial cells during lung development. The glycogen storage, which provides the substrates for synthesizing surfactant phospholipids and forming LBs, depletes as the pulmonary epithelium matures [6, 9, 10, 22]. In the lungs of Atg7 KO mice, the accumulation of glycogen indicated that the alveolar epithelial cells were defective in using cytoplasmic glycogen in the absence of autophagy. Examining the E18.5 lung, we found autolysosomes containing partially digested glycogen particles, which further confirmed the idea that glycogenolysis occurs through an autophagic process. Moreover, we found extensive fusion between autophagosomes and LBs in control mice (P1), especially in littermate control mice of Atgspc mice. These findings suggest that autophagy is essential for lung development, consistent with the critical role of glycogen autophagy in developing the liver and heart in newborn rats [45].
Currently, it is postulated that the MVB is the precursor of the LB. MVBs play a key role in delivering newly synthesized surfactant proteins and peptides that are internalized/recycled via the endocytic pathway to the LBs. Fluorescence microscopy revealed that autolysosomes could fuse with MVBs, particularly in the case of starvation or drug-induced autophagy [46]. Moreover, the delivery of cytoplasmic components into the LBs was reversibly inhibited by 3-MA [8]. Notably, LBs are similar in nature to the lysosome. Treatment of cells with the lysosomal protease inhibitor leupeptin resulted in the progressive transformation of LBs into electron-dense autophagic vacuoles and the eventual disappearance of LBs [8]. Together with the observation of constant autophagosome-LB fusion, these results support the notion that LB is a specialized autolysosome.
Evidence suggests that activation of the PI3K-AKT-mTOR pathway in pulmonary epithelial cells may play a vital role in the pathogenesis of RDS in infants. The activation of AKT signaling in pulmonary epithelial cells results in transient respiratory difficulties in full-term and pre-term infants [47]. These respiratory difficulties are associated with bronchiolar hyperplasia and maturation defects of pulmonary epithelial cells. Phosphatase and tension homolog deleted on chromosome 10 (PTEN), which inhibits PI3K, is highly expressed in the respiratory epithelium at the terminal sac stage, in which type 1 and type 2 alveolar cells differentiate. Specific deletion of PTEN in alveolar epithelium resulted in RDS, and approximately 90% of neonates died within 2 h after birth [48]. Moreover, it has been reported that PTEN deletion had an adverse effect on LB formation [48]. Although a previous study has demonstrated that the deletion of Ulk1/2, Atg5, or Beclin 1 resulted in compromised lung development, LB formation was not reported to be altered [40, 43, 49]. This discrepancy may be explained by functional differences among individual autophagic genes or different backgrounds. During our manuscript preparation, Mizushima's group showed that LBs were defective in AT2 cells in Atg101- and Fip200-deficient mice [50]. Our results support their notion that autophagic vacuoles can fuse with LBs and autophagy is vital for LB growth. The reason why Atg7spc mice are viable is possibly due to a less critical role of ATG7 as a molecule in the conjunction system compared to upstream autophagic molecules such as FIP200 and ATG101. It cannot be excluded that the deletion efficiency might account for the difference in Atg7 KO and Atg7spc mice in our study.
Taken together, our results provide fundamental insight into the role of autophagy in maintaining the proper function of LBs and AT2 cells. Furthermore, our study considerably improves our understanding of lung diseases, such as RDS and HPS, and will be of great significance in developing therapies for these diseases.
Methods
Mice
Global deletion mice
atg7+/- mice were previously described [33]. atg7-/- mice were generated by mating atg7+/- mice. Newborn pups (P1) were sacrificed after birth.
Generation of bronchioalveolar epithelium-specific Atg7-deficient mice SFTPC-rtTA/(tetO)7-Cre/Atg7flox/flox mice
SFTPC-rtTA transgenic mice, expressing the reverse tetracycline transactivator (rtTA) protein under the control of the human SFTPC promoter, were described previously [51-53]. The SFTPC-rtTA mice line was obtained from Jackson Laboratory with a C57BL/6J background. Transgenic (tetO)7-Cre mice [54], in which rtTA activated Cre expression, were obtained from Jackson Laboratory with a C57BL/6J background. SFTPC-rtTA transgenic mice were mated to tetO-Cre mice to generate SFTPC-rtTA;tetO-Cre mice. The double transgenic mice were further mated with Atg7flox/flox mice (C57BL/6J background) [24] to generate triple-transgenic mice. To induce expression of the Cre transgene, mice were administrated doxycycline in utero. Specifically, dams bearing pups were fed doxycycline in their drinking water (1 mg/mL) at least seven days before delivery.
Histological analysis
Neonate mice were immediately sacrificed after birth by neck removal. Lung tissue was perfused with PBS and fixed in 4% buffered formalin overnight, followed by dehydration, embedding, and sectioning following standard protocols. Sections (5 μm) were mounted on slides for H&E staining. Aerated lung areas and alveolar septal thickness were measured in H&E stained sections using Nikon NIS-Elements BR Analysis software. Septal thickness was measured at all points in each visual field, and five visual fields for each animal were used for the measurement. According to the manufacturer's instructions, sections were also stained with Periodic Acid Schiff stain (PAS, Sigma-Aldrich, St. Louis, MO, #395) to visualize intracellular glycogen. For immunohistochemical staining, tissue sections were deparaffinized and then rehydrated in graded ethanol from 100% to 75%. For antigen retrieval, sections were incubated with 0.1% proteinase K (#19131, Qiagen) for 15 min at room temperature. After washing with PBS, sections were subjected to UltraSensitiveTM SP (Mouse/Rabbit) IHC Kit according to the manufacturer's instructions (MXB KIT-9720, MXB Biotechnologies, China). Brown signals were visualized using DAB (MXB 0031, MXB Biotechnologies, China). Slides without primary antibodies were used as negative controls.
TEM
Mouse lungs were freshly dissected, finely minced into millet size, and fixed in 2.5% glutaraldehyde in PBS at 4 °C. Samples were examined by TEM (JEM-1200EX, JEOL, Japan). Autophagic vacuoles, LBs, and glycogen were identified by visual inspection of EM micrographs using established criteria [13, 29, 55, 56]. To quantify the size of LB in EM, ImageJ software was used. In 10,000 magnification fields, the size of at least 80 LBs/mouse was measured and averaged. To quantify the number of LBs, at least 25 AT2 cells/mouse were quantified.
Isolation of primary AT2 cells from neonate and adult mice
Primary AT2 cells from adult mice were isolated as previously described [57], and primary AT2 cells from neonate mice were isolated similarly with moderate modifications. Neonate mice (P1) were perfused with PBS through the right ventricle of the heart until visually free of blood. The trachea was exposed by clearing surrounding tissue. Dispase was injected into the lung through the trachea by an insulin syringe, and the lung was allowed to collapse for 5 min. Lungs were removed from the animal and put in a tube containing 1mL of dispase. Lungs were minced and incubated on a shaker at room temperature for 45 min. After centrifugation, the isolating procedure followed Sun et al. [57].
Isolation and adenoviral infection of primary human AT2 cells
Primary AT2 cells from human lungs were isolated as previously described [58]. For immunofluorescent staining, isolated cells were seeded into NuncTM Lab-TEKTM Chambered Coverglass (Thermofisher Scientific). 70 nM of LysoTracker (L7528, Thermofisher Scientific) was added two hours before cell collection. Adenoviruses with ATG7 shRNA plasmid constructed into an adenoviral vector (pAdEasy-U6-CMV-EGFP) were purchased from Hanbio Technology Ltd. (Shanghai, China). The shRNA sequence used to target human ATG7 (sh-ATG7) was 5'- GCCTGCTGAGGAGCTCTCCAT -3', and the control sequence was 5'- TTCTCCGAACGTGTCACGTAA -3'. The titter of adenoviruses was 3.16×1010 PFU/mL. The multiplicity of infection was 100:1. Cells were collected for immuno-staining 48 hours after infection.
Glycogen assay
Glycogen assay kits were purchased from Sigma-Aldrich (#MAK016), and the assays were performed according to the manufacturer's instructions. The relative glycogen level was calculated by comparing it with the absorbance from the standard.
Western Blot analysis
Frozen lung tissues were sonicated in RIPA lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 1 mM EDTA) containing protease inhibitor cocktail (Roche Applied Bioscience). Protein content was determined by Bradford assay (Amresco) staining using bovine serum albumin as a standard. Equal amounts of protein were loaded on 10%, 12%, or 15% Tris-glycine gels for electrophoresis. Proteins were wet-transferred to PVDF membranes (Millipore) and then probed with the indicated antibodies. The primary antibodies and dilutions used were as follows: anti-surfactant protein B rabbit polyclonal antibody (1:3,000, #07-614, Millipore), anti-surfactant protein C rabbit polyclonal antibody (1:1000, #AB3786, Millipore), anti-surfactant protein A rabbit polyclonal antibody (1:1000, #AB3420, Millipore), anti-p62 rabbit polyclonal antibody (1:1000, #P0067, Sigma), LC3B rabbit polyclonal antibody (1:1000, #12741, Cell Signaling Technology), anti-ATG7 rabbit polyclonal antibody (1:1000, #A2856, Sigma) and anti-actin mouse polyclonal antibody (1:1000, #TA-09, Zsgb-bio, China). Immunoreactivity was detected using horseradish peroxidase-conjugated secondary antibodies. Chemiluminescence substrates were used (Tiangen, Beijing, China). The images were captured using a MicroChemi 4.2 system (DNR Bio Imaging Systems, Jerusalem, Israel).
Immunofluorescence and confocal microscopy
Lung tissues were fixed with 4% paraformaldehyde for 30 min and cryopreserved by sequential sucrose incubation. Tissues were embedded in OCT medium, and 4 µm sections were cut and mounted on slides. Slides were air-dried and stored at -80 °C. For immunostaining, sections were fixed with 4% paraformaldehyde for 15 mins and permeabilized with 0.5% Triton X-100 for 15 mins. Following permeabilization, sections were blocked with 10% normal horse serum in PBS supplemented with 0.1% Triton X-100 for 1 h at room temperature and then incubated overnight at 4 °C with rabbit polyclonal anti-SFTPB (1:2000, #07-614, Millipore), mouse polyclonal anti-ABCA3 (1:200, #AB24751, Abcam), anti-pro-SFTPC (1:200, #3786, Millipore) or LC3B (1:200, #18725-1-AP, Proteintech). Slides were washed with PBS, incubated with Alexa Fluor-conjugated secondary antibodies (Alexa Fluor-488 or Alexa Fluor-594, Life Technologies), stained with DAPI (Life Technologies). For co-labeling experiments, AT2 cells were isolated from neonate mice (P1), centrifuged using cytospin (Thermo Scientific), and stained with antibodies as needed. Slides were imaged on a Nikon A1R confocal microscope. We conducted fluorescence co-localization analyses using NIH Image J software. Pearson correlation coefficient (PCC) was calculated by Image J scatter J plug-in. The line profile plots were performed as indicated. PCC was calculated from the region of interest.
Rapamycin or 3-MA treatment
Rapamycin (#53123-88-9, BioChemPartner, China) was initially dissolved in 100% DMSO, stored at -20 °C, and further diluted in PBS immediately before use. WT adult mice (eight-week-old) were randomly divided into two groups. Mice were intraperitoneally injected with rapamycin (40 μg/kg) or vehicle. Eight hours later, mice were harvested. Eight-week-old mice were intraperitoneally injected with 20 mg/kg/day 3-MA (#M9281, sigma) or vehicle for five consecutive days. Primary human AT2 cells were treated with rapamycin or vehicle control for eight hours. Primary human AT2 cells were treated with 3-MA (15 mM) or vehicle control for 24 hours. 70 nM of LysoTracker was added two hours before cell collection.
Statistics
Data analyses and the construction of statistical charts were performed using GraphPad Prism 5 software (GraphPad Software, CA, USA). The results were presented as the mean value ( ± SEM). Statistical significance was determined using an independent samples t-test. A value of P < 0.05 was considered statistically significant. For western blots, at least three experiments were performed, and gels were quantified using ImageJ software.
± SEM). Statistical significance was determined using an independent samples t-test. A value of P < 0.05 was considered statistically significant. For western blots, at least three experiments were performed, and gels were quantified using ImageJ software.
Study approval
All animal experiments were approved by the animal experiment review board of the China Medical University (Ethics approval number 16099M). The study of isolation of human primary AT2 cells was approved by the Research Ethical Board of China Medical University [2017]074 for the use of human lungs from distal portions of normal lung tissue from patients undergoing lung resection.
Abbreviations
LB: lamellar body; AT2: alveolar type 2 cell; WPBs: Weibel-Palade bodies; LC3B (MAP1LC3B): microtubule-associated protein 1 light chain 3B; Atg7: autophagy-related 7; MVB: multivesicular body; TEM: transmission electronic microscopy; SFTPB: surfactant protein B; SFTPC: surfactant protein C; LROs: lysosome-related organelles; HPS: Hermansky-Pudlak Syndrome; 3-MA: 3-methyladenine; RDS: respiratory distress syndrome; ABCA3: ATP-binding cassette sub-family A [ABC1]: member 3; mTOR: mechanistic target of rapamycin kinase; ULK1: UNC-51-like kinase 1; Atg7 KO: Atg7 deficient mice; mTOR: mechanistic target of rapamycin kinase; Avs: autophagic vacuoles; PTEN: phosphatase and tension homology.
Supplementary Material
Supplementary methods, figures and table.
Acknowledgements
This study was supported by National Key R&D Program of China under Grant [2016YFC1302400 to LC]; National Science Foundation of China under Grant [81770001 and 82030031 to LC]; the Ministry of Education Innovation Team Development Plan under Grant [IRT17R107 to LC]; Department of Science and Technology of Liaoning Province under Grant [2015225003 to LC]; Science Foundation from Liaoning Province to LC (2019JH2/10300); Shenyang Science and Technology Project (Z18-4-021); Department of Education of Liaoning Province under Grant [LFWK201726 to XL]; Natural Science Foundation of Liaoning to LW [2021-MS-160]; Department of Education of Liaoning Province under Grant [LFWK201725 to XS]; NIH grant to HLJ (HL134828).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Voorhout WF, Weaver TE, Haagsman HP, Geuze HJ, Van Golde LM. Biosynthetic routing of pulmonary surfactant proteins in alveolar type II cells. Microsc Res Tech. 1993;26:366-73
2. Olmeda B, Martinez-Calle M, Perez-Gil J. Pulmonary surfactant metabolism in the alveolar airspace: Biogenesis, extracellular conversions, recycling. Ann Anat. 2017;209:78-92
3. Sorokin SP. A morphologic and cytochemical study on the great alveolar cell. J Histochem Cytochem. 1966;14:884-97
4. Flaks B, Flaks A. Electron-microscope observations on the formation of the cytoplasmic lamellar inclusion bodies in murine pulmonary tumours induced in vitro. J Pathol. 1972;108:211-7
5. Kikkawa Y, Motoyama EK, Gluck L. Study of the lungs of fetal and newborn rabbits. Morphologic, biochemical, and surface physical development. Am J Pathol. 1968;52:177-210
6. Campiche MA, Gautier A, Hernandez EI, Reymond A. An Electron Microscope Study of the Fetal Development of Human Lung. Pediatrics. 1963;32:976-94
7. Chevalier G, Collet AJ. In vivo incorporation of choline- 3 H, leucine- 3 H and galactose- 3 H in alveolar type II pneumocytes in relation to surfactant synthesis. A quantitative radoautographic study in mouse by electron microscopy. Anat Rec. 1972;174:289-310
8. Hariri M, Millane G, Guimond MP, Guay G, Dennis JW, Nabi IR. Biogenesis of multilamellar bodies via autophagy. Mol Biol Cell. 2000;11:255-68
9. Brehier A, Rooney SA. Phosphatidylcholine synthesis and glycogen depletion in fetal mouse lung: developmental changes and the effects of dexamethasone. Exp Lung Res. 1981;2:273-87
10. Rinaudo MT, Curto M, Bruno R, Ponzetto C. Some aspects of carbohydrate metabolism in rat lung during the period of growth. Int J Biochem. 1981;13:571-5
11. Bourbon JR, Rieutort M, Engle MJ, Farrell PM. Utilization of glycogen for phospholipid synthesis in fetal rat lung. Biochim Biophys Acta. 1982;712:382-9
12. Carlson KS, Davies P, Smith BT, Post M. Temporal linkage of glycogen and saturated phosphatidylcholine in fetal lung type II cells. Pediatr Res. 1987;22:79-82
13. Ridsdale R, Post M. Surfactant lipid synthesis and lamellar body formation in glycogen-laden type II cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L743-51
14. Bourbon J, Jost A. Control of glycogen metabolism in the developing fetal lung. Pediatr Res. 1982;16:50-6
15. Weaver TE, Na CL, Stahlman M. Biogenesis of lamellar bodies, lysosome-related organelles involved in storage and secretion of pulmonary surfactant. Semin Cell Dev Biol. 2002;13:263-70
16. Wang WJ, Russo SJ, Mulugeta S, Beers MF. Biosynthesis of surfactant protein C (SP-C). Sorting of SP-C proprotein involves homomeric association via a signal anchor domain. J Biol Chem. 2002;277:19929-37
17. Fisher AB, Dodia C, Ruckert P, Tao JQ, Bates SR. Pathway to lamellar bodies for surfactant protein A. Am J Physiol Lung Cell Mol Physiol. 2010;299:L51-8
18. Weaver TE. Surfactant proteins and SP-D. Am J Respir Cell Mol Biol. 1991;5:4-5
19. Clark JC, Wert SE, Bachurski CJ, Stahlman MT, Stripp BR, Weaver TE. et al. Targeted disruption of the surfactant protein B gene disrupts surfactant homeostasis, causing respiratory failure in newborn mice. Proc Natl Acad Sci U S A. 1995;92:7794-8
20. Nogee LM, de Mello DE, Dehner LP, Colten HR. Brief report: deficiency of pulmonary surfactant protein B in congenital alveolar proteinosis. N Engl J Med. 1993;328:406-10
21. Nogee LM, Wert SE, Proffit SA, Hull WM, Whitsett JA. Allelic heterogeneity in hereditary surfactant protein B (SP-B) deficiency. Am J Respir Crit Care Med. 2000;161:973-81
22. Stahlman MT, Gray MP, Falconieri MW, Whitsett JA, Weaver TE. Lamellar body formation in normal and surfactant protein B-deficient fetal mice. Lab Invest. 2000;80:395-403
23. Melton KR, Nesslein LL, Ikegami M, Tichelaar JW, Clark JC, Whitsett JA. et al. SP-B deficiency causes respiratory failure in adult mice. Am J Physiol Lung Cell Mol Physiol. 2003;285:L543-9
24. Torisu T, Torisu K, Lee IH, Liu J, Malide D, Combs CA. et al. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat Med. 2013;19:1281-7
25. Ganesan AK, Ho H, Bodemann B, Petersen S, Aruri J, Koshy S. et al. Genome-wide siRNA-based functional genomics of pigmentation identifies novel genes and pathways that impact melanogenesis in human cells. PLoS Genet. 2008;4:e1000298
26. Bowman SL, Bi-Karchin J, Le L, Marks MS. The road to lysosome-related organelles: Insights from Hermansky-Pudlak syndrome and other rare diseases. Traffic. 2019;20:404-35
27. Ahuja S, Knudsen L, Chillappagari S, Henneke I, Ruppert C, Korfei M. et al. MAP1LC3B overexpression protects against Hermansky-Pudlak syndrome type-1-induced defective autophagy in vitro. Am J Physiol Lung Cell Mol Physiol. 2016;310:L519-31
28. Nakatani Y, Nakamura N, Sano J, Inayama Y, Kawano N, Yamanaka S. et al. Interstitial pneumonia in Hermansky-Pudlak syndrome: significance of florid foamy swelling/degeneration (giant lamellar body degeneration) of type-2 pneumocytes. Virchows Arch. 2000;437:304-13
29. Lajoie P, Guay G, Dennis JW, Nabi IR. The lipid composition of autophagic vacuoles regulates expression of multilamellar bodies. J Cell Sci. 2005;118:1991-2003
30. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132-41
31. Vizza D, Perri A, Toteda G, Lupinacci S, Perrotta I, Lofaro D. et al. Rapamycin-induced autophagy protects proximal tubular renal cells against proteinuric damage through the transcriptional activation of the nerve growth factor receptor NGFR. Autophagy. 2018;14:1028-42
32. Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, Dean M. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med. 2004;350:1296-303
33. Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I. et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425-34
34. Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T. et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032-6
35. Andreeva AV, Kutuzov MA, Voyno-Yasenetskaya TA. Regulation of surfactant secretion in alveolar type II cells. Am J Physiol Lung Cell Mol Physiol. 2007;293:L259-71
36. Osanai K, Mizuno S, Toga H, Takahashi K. Trafficking of newly synthesized surfactant protein B to the lamellar body in alveolar type II cells. Cell Tissue Res. 2020;381:427-38
37. Osanai K, Mason RJ, Voelker DR. Trafficking of newly synthesized surfactant protein A in isolated rat alveolar type II cells. Am J Respir Cell Mol Biol. 1998;19:929-35
38. Zhou L, Lim L, Costa RH, Whitsett JA. Thyroid transcription factor-1, hepatocyte nuclear factor-3beta, surfactant protein B, C, and Clara cell secretory protein in developing mouse lung. J Histochem Cytochem. 1996;44:1183-93
39. Schmitz G, Muller G. Structure and function of lamellar bodies, lipid-protein complexes involved in storage and secretion of cellular lipids. J Lipid Res. 1991;32:1539-70
40. Cheong H, Wu J, Gonzales LK, Guttentag SH, Thompson CB, Lindsten T. Analysis of a lung defect in autophagy-deficient mouse strains. Autophagy. 2014;10:45-56
41. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1-222
42. Maniscalco WM, Wilson CM, Gross I, Gobran L, Rooney SA, Warshaw JB. Development of glycogen and phospholipid metabolism in fetal and newborn rat lung. Biochim Biophys Acta. 1978;530:333-46
43. Yeganeh B, Lee J, Ermini L, Lok I, Ackerley C, Post M. Autophagy is required for lung development and morphogenesis. J Clin Invest. 2019;129:2904-19
44. Morimoto M, Kopan R. rtTA toxicity limits the usefulness of the SP-C-rtTA transgenic mouse. Dev Biol. 2009;325:171-8
45. Kondomerkos DJ, Kalamidas SA, Kotoulas OB, Hann AC. Glycogen autophagy in the liver and heart of newborn rats. The effects of glucagon, adrenalin or rapamycin. Histol Histopathol. 2005;20:689-96
46. Fader CM, Sanchez D, Furlan M, Colombo MI. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic. 2008;9:230-50
47. Ikeda H, Shiojima I, Oka T, Yoshida M, Maemura K, Walsh K. et al. Increased Akt-mTOR signaling in lung epithelium is associated with respiratory distress syndrome in mice. Mol Cell Biol. 2011;31:1054-65
48. Yanagi S, Kishimoto H, Kawahara K, Sasaki T, Sasaki M, Nishio M. et al. Pten controls lung morphogenesis, bronchioalveolar stem cells, and onset of lung adenocarcinomas in mice. J Clin Invest. 2007;117:2929-40
49. Hahn DR, Na CL, Weaver TE. Reserve autophagic capacity in alveolar epithelia provides a replicative niche for influenza A virus. Am J Respir Cell Mol Biol. 2014;51:400-12
50. Morishita H, Kanda Y, Kaizuka T, Chino H, Nakao K, Miki Y. et al. Autophagy Is Required for Maturation of Surfactant-Containing Lamellar Bodies in the Lung and Swim Bladder. Cell Rep. 2020;33:108477
51. Perl AK, Wert SE, Nagy A, Lobe CG, Whitsett JA. Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc Natl Acad Sci U S A. 2002;99:10482-7
52. Tichelaar JW, Lu W, Whitsett JA. Conditional expression of fibroblast growth factor-7 in the developing and mature lung. J Biol Chem. 2000;275:11858-64
53. Wert SE, Glasser SW, Korfhagen TR, Whitsett JA. Transcriptional elements from the human SP-C gene direct expression in the primordial respiratory epithelium of transgenic mice. Dev Biol. 1993;156:426-43
54. Sauer B. Inducible gene targeting in mice using the Cre/lox system. Methods. 1998;14:381-92
55. Eskelinen EL. Maturation of autophagic vacuoles in Mammalian cells. Autophagy. 2005;1:1-10
56. Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A. et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113-22
57. Sun F, Xiao G, Qu Z. Isolation of Murine Alveolar Type II Epithelial Cells. Bio Protoc. 2017 7
58. Mao P, Wu S, Li J, Fu W, He W, Liu X. et al. Human alveolar epithelial type II cells in primary culture. Physiol Rep. 2015 3
Author contact
![]() Corresponding authors: Liu Cao, College of Basic Medical Sciences, China Medical University, Shenyang, Liaoning Province, China. E-mail: lcaoedu.cn, Tel: +86(24)31939636; Xiaoyu Song, Key Laboratory of Medical Cell Biology, Ministry of Education, China Medical University, Shenyang, China. E-mail: xysongedu.cn.
Corresponding authors: Liu Cao, College of Basic Medical Sciences, China Medical University, Shenyang, Liaoning Province, China. E-mail: lcaoedu.cn, Tel: +86(24)31939636; Xiaoyu Song, Key Laboratory of Medical Cell Biology, Ministry of Education, China Medical University, Shenyang, China. E-mail: xysongedu.cn.