Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Brief of DNA Replication Origin...
Replication Origin Licensing and...
Replication Origin Licensing and...
Replication Origin Licensing...
Conclusion and Future...
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(3):1211-1219. doi:10.7150/ijbs.67529 This issue Cite
Review
DNA Replication Licensing Factors: Novel Targets for Cancer Therapy via Inhibiting the Stemness of Cancer Cells
Shaoran Song1,2, Yaochun Wang1,2 ![]() , Peijun Liu1,2
, Peijun Liu1,2 ![]()
1. Center for Translational Medicine, the First Affiliated Hospital of Xi'an Jiaotong University, Xi'an, China
2. The Key Laboratory for Tumor Precision Medicine of Shaanxi Province, The First Affiliated Hospital, Xi'an Jiaotong University, Xi'an, Shaanxi 710061, China
Received 2021-9-27; Accepted 2021-12-17; Published 2022-1-1
Abstract

The replication licensing factors strictly regulate the DNA replication origin licensing process to guarantee the stability of the genome. Numerous experimental studies have recently demonstrated that the replication licensing factors as oncogenes are essential for the occurrence and development of cancers. Drug resistance, being one of the main characteristics of cancer stem cells, can cause a high recurrence rate and a low survival rate in patients with different cancers. However, the function of the replication licensing factors in cancer stemness remains unclear. The following article highlights the most recent research on DNA replication origin licensing factors in cancer and their function in anti-cancer drug resistance. Moreover, this article proposes a new perspective that replication licensing factors as chemotherapy shield affect anti-cancer drug resistance by promoting the stemness of cancer cells.
Keywords: replication initiation factors, cancer stem cell, stemness, drug resistance
Introduction
Cancer is anticipated to be the leading cause of mortality worldwide in the twenty-first century, posing the most significant impediment to extending life expectancy. According to estimates from the GLOBOCAN in 2020, there were 19.3 million new cancer cases and 9.9 million cancer deaths worldwide [1]. Although traditional treatment methods such as anti-cancer drugs, surgical resection, combined chemotherapy, and radiotherapy have achieved significant results, increasing numbers of patients inevitably face higher tumor recurrence rates and metastasis. The aggressive characteristics of cancer are partially due to cancer stem cells (CSCs) being resistant to conventional anti-cancer treatments.
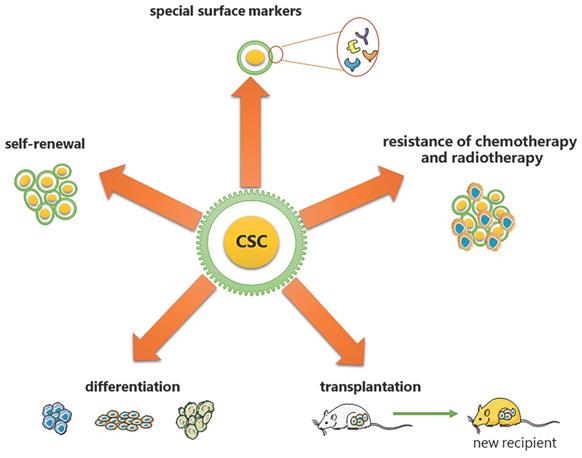
CSCs are considered to have the exclusive ability to grow malignant cell populations indefinitely, which is recognized as the primary cause of cancer recurrence and metastasis [2-5]. Pierce and colleagues described CSCs for the first time as malignant cells with a high proliferative ability and a limited capacity for differentiation under normal homeostatic conditions [6]. Nowadays, a substantial body of research has established that CSCs are critical in a variety of tumor types, including leukemia [7], prostate cancer [8], breast cancer [9], and colorectal cancer [10]. Based on the analysis from previous research, CSCs have been proven to have the following five most characteristic properties (as shown in Figure 1): (1) self-renewal: tumor-sphere formation in vitro and tumorigenic ability in vivo [11, 12]; (2) the ability of differentiation; (3) the ability of transplantation [13]; (4) resistance of conventional chemotherapy and radiotherapy [14]; and (5) unique surface markers [15]. Additionally, CSCs were verified to be dormant [15]. Moreover, identifying and isolating CSCs from solid tumors based on their characteristics has been widely employed to develop more effective strategies for cancer eradication. However, the hierarchical structure of CSCs is shown to be more malleable than previously believed, complicating their complete elimination [2].
Five most characteristic properties of CSCs. (1) self-renewal: tumor-sphere formation in vitro/ tumorigenic ability in vivo; (2) the ability of differentiation; (3) the ability of transplantation; (4) resistance of conventional chemotherapy and radiotherapy; (5) unique surface markers.
DNA replication in eukaryotic cells requires a precise synthesis process. However, with long-term amplification, DNA replication errors continue to accumulate, leading to genome instability, which is one of the hallmarks of cancer [16-18]. Furthermore, increased genomic instability accelerates clonal evolution, resulting in more aggressive clones and stubborn drug resistance [19]. As a result, controlling DNA replication's origin is a critical mechanism for cancer elimination [20]. Moreover, the determination of DNA replication origins in eukaryotes involves two subsequent steps: (1) “licensing”: identification of the pre-RC site; (2) “firing”: activation of DNA synthesis [21]. "Licensing" is a critical step in enforcing spatial and temporal constraints on DNA replication. Factors that participate in DNA replication origin licensing include the origin recognition complex (ORC, comprising the six subunits ORC1-6), cell division cycle 6 (CDC6), CDC10‑dependent transcript 1, (also known as DNA replication factor CDT1), and the mini-chromosome maintenance (MCM) helicase complex. These licensing factors have been identified as oncogenes, making them attractive potential therapeutic targets [22]. Furthermore, there is mounting evidence that dysregulation of DNA replication licensing factors may influence cancer occurrence and progression via regulating CSCs stemness maintenance [23, 24].
This article highlights the current DNA replication licensing factors and their role in drug resistance in normal stem cells and CSCs, significantly increasing the replication licensing factors' potential as novel therapeutic targets. However, more research is urgently needed to elucidate the regulatory mechanisms of DNA replication licensing factors and their related pathways for the differentiation, self-renewal, and differentiation of cancer stem cells, which may eventually strengthen our ability to promote cancer treatment and prevention technologies through the elimination of cancer stem cells.
Brief of DNA Replication Origin Licensing
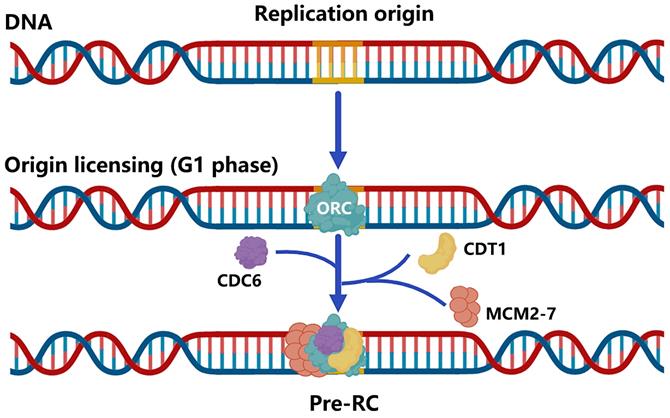
DNA replication origin licensing requires a series of different proteins to act in turn. To begin with, the evolutionarily conservative ORC features a gap that permits DNA to enter through the pentameric ORC ring's central channel and interact with it via ATP [25, 26]. These binding sites, known as replication origins, are essential for loading replicative helicases [27-29]. Using cryo-EM, Li et al. revealed that subunits of ORC could cooperate with the initiator specific motif (ISM) and β-hairpins to bend DNA, which is essential for the loading of MCM2-7 [30]. Second, CDC6, another AAA+ ATPase, loads to the replication origin and seals the previous gap on ORC, trapping DNA in the ORC⸱CDC6 toroid [25, 26]. However, the mechanism by which CDC6 regulates the loading of MCM2-7 through the change of the ATP hydrolysis cycle remains unclear. Simultaneously, MCM2-7 and CDT1 form a stable OCCM complex to ensure that the opening of the hexamer can smoothly pass through the DNA that has already been loaded with ORC and CDC6 [31]. The research of Yuan et al. discovered that both CDC6 and ORC2 interact directly with the MCM3 WH domain [32]. Thus, these factors collectively form the pre-initiation complex (pre-RC), which "licenses" the origins to replicate DNA and progress to the next phase of the cell cycle [21] (Figure 2).
Brief of DNA replication origin licensing. Sequential loading of replication licensing factors on all potential origins in the genome makes replication initiation restricted to the G1 phase. Firstly, the origin recognition complex (ORC, comprising the six subunits ORC1-6) with ATPase activity is recruited to replication origins, followed by cell division cycle 6 (CDC6) and CDC10-dependent transcript 1 (CDT1) binding to the ORC, and finally the mini-chromosome maintenance helicase complex (MCM2-7) loading into the complex to form Pre-RC.
Replication Origin Licensing and Cancers
To ensure the high accuracy of replication, strictly control only one replication origin licensing per cell cycle. Once inappropriate replication origin licensing occurs in the same cell cycle, it amplifies the nuclear genome, a process known as re-replication. Mutations such as overexpression of CDT1/ CDC6 or depletion of geminin have been reported to lead to re-replication rapidly [33, 34]. Besides that, Re-replication is usually accompanied by the appearance of DNA damage, genomic stress, or instability, which is related to cell cycle arrest, senescence, and apoptosis. Thus, replication licensing is highly associated with multiple clinical pathogenesis and tumorigenesis.
Replication origin licensing factors and cell cycle progression and proliferation
The rapid and uncontrolled proliferation caused by multiple genetic mutations is necessary for carcinogenesis [16, 17, 35]. Uncontrolled proliferation and/ or enhanced genomic instability make most tumor cells suffer from high replication stress. Consistent with this, it has been reported that replication licensing factors are overexpressed in various cancer cell lines and function as oncogenes [36-38]. Since only licensed DNA is allowed to enter the S phase from the G1 phase. The destruction of replication licensing can lead to G1-S phase arrest, inhibiting cell proliferation. Previous studies have demonstrated decreased ORC6 expression induced by the siRNA knock-down approach triggered cell cycle arrest in the G1 phase. This cell cycle regulation is associated with wild-type p53 [37]. Moreover, the normal loading of MCM proteins onto chromatin during the G1 phase was impeded after the deletion of either CDC6 or CDT1, thereby stalling the cell cycle progression [39]. In vitro studies on mouse embryonic fibroblasts and fetal and adult diploid tissues revealed that global or tissue-specific ORC1 deficiency impairs DNA replication, cell lineage expansion, and organ development [40].
Replication origin licensing factors and cell apoptosis and survival
Reduced expression of licensing factors leads to more apoptosis in different cancers. In cell lines of gastric cancer, osteosarcoma, and cervical cancer, down-regulation of CDC6 promotes cell apoptosis, which negatively impacts growth in vivo and in vitro [40-42]. Moreover, partial overexpression of MCMs can enhance cell proliferation significantly and suppress apoptosis, whereas knockdown of MCMs reverses these effects [43-45]. Notably, subcellular localization of replication licensing factors was found to correlate significantly with cell apoptosis in cancer cells. The perinuclear accumulation of ORC1 caused by the absence of normal ORC1 modifications such as monoubiquitylation and hyperphosphorylation was demonstrated to induce cell apoptosis.
Similarly, ORC2 could restore the uniform nuclear localization of ORC1 and prevent the induction of apoptosis [41]. Furthermore, since increased cytoplasmic MCM2 promotes cell apoptosis, tumor samples with cytoplasmic MCM2 demonstrated better prognoses [42]. Hence, targeting replication licensing factors through epigenetic modification or subcellular localization may be an appealing strategy for inducing cancer cell death and improving survival.
Replication origin licensing factors and metastasis
Metastasis consists of two major steps: dissemination and colonization. Numerous investigations have implicated replication licensing factors as the key molecules regulating almost all the steps of metastasis by targeting key genes. By suppressing the ERK/JNK signaling pathway, decreased ORC1 inhibited cancer cell invasion and migration [43]. Trans-well migration assays demonstrated that silencing CDC6 dramatically reduced the ability of UMUC3 and T24 cells to migrate [39]. In HCT-116 cells, CDC6 overexpression was associated with the reduction of E-cadherin, indicating CDC6 may have an important role in the metastasis of HPV-associated cancers [44]. A similar regulating effect was identified in Sideridou's research [45]. Consequently, replication licensing factors play an important role in cancer metastasis.
Replication Origin Licensing and Drug-Resistance
ORC and drug-resistance
The ORC complex serves as a platform for initial assembly during the DNA replication process. Various studies have been conducted to determine its role in the development and treatment of cancers and how its aberrant expression affects the susceptibility to traditional cancer therapy. For example, it has been observed that in colon cancer cells HCT-116 (wt-p53) with lower expression of ORC6 were more sensitive to 5-fluorouracil (5-FU) and cisplatin treatment than the control group, as a result of p53 phosphorylation regulation [37]. In response to gemcitabine treatment, Plk1-mediated phosphorylation of ORC1/2 at the start of replication was elevated, and replication initiation was increased, leading to resistance to chemotherapeutic agents [46]. The resistance of gemcitabine may also involve a p53-dependent manner, but the specific molecular mechanism needs further study. Another study suggested that siORC1 enhanced the sensitivity of U2OS cells to hydroxyurea (HU), although the removal of other origin licensing factors, such as ORC6 or CDC6, did not. Nevertheless, in non-tumor cells, the enhanced sensitivity to HU was not detected [47]. Compared with non-cancer cells, tumor cells may depend on the origin licensing capacity due to their higher oxidative and replication stress. Consequently, targeting origin licensing factors can make cancer cells more sensitive to chemotherapy drugs.
MCM2-7 and drug-resistance
The MCM2-7 complex, as the core of the replication initiation permission complex, is also involved in the formation of DNA helicase, which is responsible for the melting and unwinding of the double helix during DNA synthesis [21, 48, 49]. Recently, genomic analyses also identified MCMs as gene candidates for acquired drug resistance in several types of cancer [50, 51]. MCM2 has been shown to be strongly related to the Vemurafenib resistance induced by up-regulated expression of CDC7 [52]. Due to a p53-dependent apoptotic response, MCM2 deletion can also increase the sensitivity of ovarian cancer cells to carboplatin [53]. Additionally, down-regulation of MUS81 promotes apoptosis by inducing S-phase arrest and activation of MCM2, thereby increasing the sensitivity of EOC cells to Olaparib [54]. Microarray analysis revealed that the transcriptional expression of MCM2 in the cisplatin-resistant ovarian carcinoma cell was twice that of the non-drug-resistant group [55]. However, the specific mechanism remains to be explored. Research by Mitali Das and colleagues indicated that excessive amounts of MCMs could be used as a backup for replication stress in cervical cancer cells; moreover, its regulatory mechanism of sensitivity to cisplatin depends on the HPV status of the cells [56]. Furthermore, recent work by Wang et al. indicated that the EGFR pathway can regulate the interaction of MCM8 and other DNA replication licensing factors, hence preserving the clonogenic and tumorigenic potential of GSCs [57].
Furthermore, lentivirus-mediated MCM7 silencing could significantly sensitize chronic lymphocytic leukemia cells to fludarabine [58]. It has also been demonstrated that MCM7 plays an important role in cell cycle arrest, apoptosis, and cell death caused by BVP-induced DNA damage [59]. Furthermore, the underlying mechanism involves regulating retinoblastoma protein (Rb) and the expression of checkpoint control proteins [59]. Interestingly, statin drugs could reduce the expression of MCM7 and RB via activating ER and autophagy signaling pathways, which induced the growth-inhibitory effects in TamR cells [60, 61]. Therefore, these findings suggest that MCMs might be a potential target for drug-resistant cancer cells.
CDC6 and drug-resistance
Several types of research have revealed that despite its function during DNA replication licensing, CDC6 also regulates mitosis exit from yeast cells to human cells by interacting with Cdk1 [62-64]. In addition, CDC6 was found to be highly expressed in a variety of cancer cells, including drug-resistant cancer cells [39, 65]. Recent studies have elucidated that CDC6 could promote mitotic slippage by inhibiting CDK1; thus, cancer cells avoid apoptosis and exhibit PTX resistance [66]. CDC6 was predicted to play a pivotal role in ovarian cancer treated with decitabine [67]. Interestingly, both bladder cancer cells and CDDP-resistant bladder cancer cells became more sensitive to CDDP when CDC6 was down-regulated [63]. Because the CDDP-induced S phase arrest was abolished under the deletion of CDC6, which led to aberrant mitosis by inactivating the ATR-Chk1-Cdc25C pathway [39]. CDC6 and Human antigen R (HuR) were found to be positively correlated with malignant behaviors and oxaliplatin (L-OHP) resistance [68]. Mechanistically, it has been proved that HuR can bind to CDC6 3'-UTR, thus regulating the sensitivity of CRC cells to L-OHP [68]. Therefore, CDC6 may be a novel molecular target to overcome drug resistance.
CDT1 and drug-resistance
According to Stathopoulou and colleagues, anticancer chemotherapeutic agents can degrade CDT1 in various ways [69]. However, only a few researchers have mentioned a relationship between CDT1 and anti-cancer drug resistance [70, 71]. Intriguingly, stability or activation of CDT1 or acute depletion of CDT2 could result in re-replication, which radio-sensitizes head and neck squamous cell carcinoma (HNSCC) cells [72]. Numerous studies published in the last few years have clarified CDT1's prognostic function in various types of cancer. However, further investigation requires further investigation to determine whether it has the same effect on anti-cancer treatment resistance as other replication licensing factors (Table 1).
Replication licensing factors and drug-resistance.
| DNA replication licensing proteins | Anti-cancer agent | Cancer cell line | reference |
|---|---|---|---|
| ORC1-6 | 5-Fluorouracil, cisplatin | HCT-116 | [37] |
| gemcitabine | Panc-1, BxPC-3 | [46] | |
| Hydroxyurea, Hydrogen peroxide | U20S, HeLa, MDA-MB-231 | [47] | |
| CDC6 | decitabine | ovarian cancer cell | [67] |
| paclitaxel | MDA-MB-231, HepG2, BGC823, SGC7901 | [65, 66, 82] | |
| cisplatin | UMUC3 | [39] | |
| oxaliplatin | HT-29, HCT116 | [68] | |
| MCM2 | Vemurafenib | A375, WM983B | [52] |
| carboplatin | A2780 | [53] | |
| Olaparib | A2780, SKOV3 | [54] | |
| cisplatin | PE01, PE01CDDP | [55] | |
| Trichostatin A | HCT116 | [71] | |
| Geinstein | LNCaP, PC3 | [70] | |
| MCM4 | cisplatin | SiHa, ME180, CaSki, and C-33A, | [56] |
| MCM5 | camptothecins | MMRU | [50] |
| doxorubicin | AML-2 | [51] | |
| MCM6 | pemetrexed | NSCLC cell | [83] |
| MCM7 | fludarabine | MEC-1, EHEB | [58] |
| cisplatin | T24, T24R2 | [84] | |
| vinblastine (VBL) | KB-3-1, KB-v1 | [85] | |
| Oxaliplatin, Etoposide | SW480 | [86] | |
| Gemcitabine, 5-Fluorouracil | Panc1, Colo-357 | [86] | |
| Breviscapine | LNCap, PC3, C4-2B | [59] | |
| tamoxifen | MCF7 TamR, T47D TamR | [61] | |
| CDT1 | Trichostatin A | HCT116 | [71] |
| Geinstein | LNCaP, PC3 | [70] |
Replication Origin Licensing Factors and Stemness
Replication origin licensing factors in normal stem cells
Stemness is the ability of a cell to remain undifferentiated (self-renewal) while having the ability to differentiate into any other cell type (potential) [73]. Stemness exits in both normal stem cells and CSCs. According to retrospective investigations, replication licensing factors are required to maintain stem cell pluripotency. MCMs loading is restricted to occurring in the G1 phase to prevent re-replication. Initially, partial depletion of MCMs proteins was confirmed to result in cancer and stem cell deficiencies in vitro and in vivo [74, 75]. Although less MCMs loading was sufficient for normal proliferation, cells with excess MCMs loading were more resistant to DNA damage and replication stress [69, 76]. Further research has reported that rapid MCMs loading as an inherent feature of stem cells helps maintain pluripotency and slows differentiation [23]. Moreover, CDT1 and CDC6 were expressed extensively in pluripotent stem cells, similar to findings from mouse embryonic stem cell research [77].
Replication origin licensing factors in CSCs
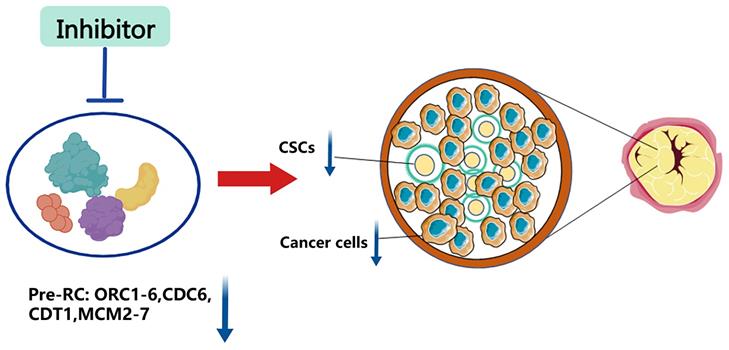
Since normal stem cells and CSCs share many characteristics, under certain conditions or stimulation, normal stem cells can be transformed into CSCs. Additionally, cancers will arise when the pluripotent stem cells of epigenetic organisms are inoculated into heterozygous sites in vivo [78-80]. Thus, we hypothesize that replication licensing factors could maintain the aggressive characteristics of CSCs, such as rapid proliferation, metastasis, and drug resistance, which were observed in normal stem cells. Numerous experimental studies have also reinforced this view. Mechanically, genes that contribute to stemness regulation, such as p53, c-MYC, and RB, are also proven to exist in the replication licensing pathway regulating cancer stemness [47, 60, 61, 75, 81]. As a consequence, replication licensing factors could positively shield cancer cells with stemness against chemotherapeutic treatment. The expression level of replication initiation factors in cancer cells is significantly higher than that of normal cells, and tumor stem cells are even higher than the former. To this end, we propose using inhibitors that specifically target replication initiation factors to destroy tumors. It can extirpate not only normal tumor cells but also CSCs (Figure 3). As a result, further research on selective inhibitors of replication initiation factors is urgently needed in the future.
Inhibition of replication origin licensing factors targeting both cancer cells and CSCs. Since replication licensing factor expression is higher in cancer cells, especially in cancer stem cells, than in normal cells, inhibitors that selectively target replication initiation factors could eradicate both cancer cells and cancer stem cells.
Possible mechanisms of tumor recurrence due to replication licensing factors
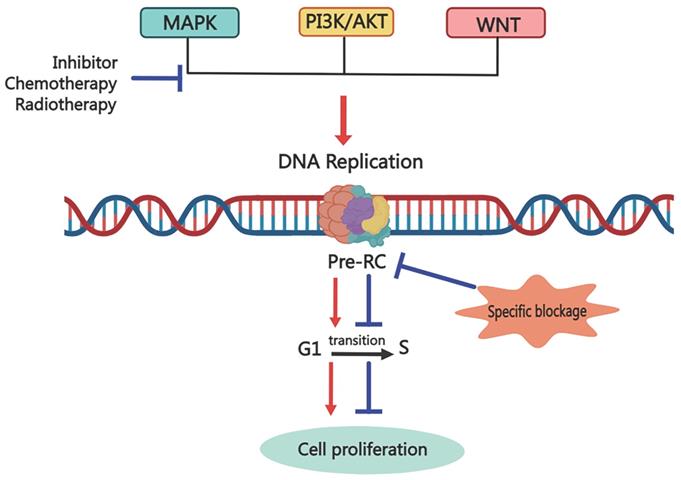
Although there is a relatively complete system for cancer treatment, traditional treatments such as surgery, adjuvant radiotherapy, and chemotherapy can no longer cope with the rapidly increasing rate of recurrence and metastasis. And molecular targeted therapy emerges in time. It is a revolutionary way to selectively target specific molecules with drugs or other substances to prevent cancer cells from proliferation and spread. Although these selective molecular therapy drugs have achieved good results, we have found that patients will still relapse and metastasize after a period of targeted drug treatment. So, what is the root of cancer recurrence and metastasis? Here, we give a possible mechanism for it, as shown in Figure 4. When molecular target therapy agents work on one signal transduction pathway, they block the downstream cell events, including cell proliferation, invasion and metastasis, cell cycle regulation, angiogenesis, and so on. However, after a while, the cunning tumor cells choose to increase the signal transduction of other pathways to promote the downstream cell events, leading to cancer relapse and metastasis. But all the cell events are based on the DNA replication process, and replication licensing is the key step of the DNA replication process. Thus, the destruction of replication licensing factors, the cancer shield, may be a fundamental treatment to eliminate tumors.
Possible mechanism for targeting Pre-RC to eliminate cancer cells via inhibiting the cancer stemness. Common anti-cancer treatments block one signal transduction pathway to inhibit downstream biological events, including cell proliferation, invasion, and migration. However, cunning tumor cells can trigger cancer recurrence and metastasis by enhancing the transduction of other signaling pathways. Targeting the replication licensing process specifically, which is the foundation for all biological functions, could eradicate cancer thoroughly.
Conclusion and Future Perspectives
This review briefly reviewed the licensing process during replication initiation and the function of replication licensing factors in tumorigenesis and development. The current information on replication licensing factors on the regulation of drug resistance in various cancer cells was given in a summary. By retrospecting the role of the replication licensing factors in stem cells, cancer cells, and cancer stem cells, we found that highly expressed replication licensing factors maintain the aggressive characteristics of CSCs, thus making them stronger than non-stem cancer cells in response to DNA damage and replication stress caused by chemotherapeutic agents. Therefore, we postulated that the replication licensing factors, as a shield for cancer, could maintain the stemness of CSCs and lead to cancer recurrence and metastasis. With the continuous deepening of the understanding and research of cancer stemness, new anti-cancer methods targeting cancer stem cells are needed to reduce the possibility of tumor recurrence. Therefore, understanding the mechanism of the replication licensing factors maintaining stemness and regulating drug resistance can significantly benefit us in cancer research and treatment.
Abbreviations
CSCs: cancer stem cells; ORC: origin recognition complex; CDC6: cell division cycle 6; CDT1: CDC10‑dependent transcript 1; MCM: mini-chromosome maintenance helicase complex; pre-RC: pre-initiation complex; HU: hydroxyurea; 5-FU: 5-fluorouracil; Rb: retinoblastoma protein; HNSCC: head and neck squamous cell carcinoma.
Acknowledgements
The project was funded by the National Natural Science Foundation of China (No. 82173365), the Natural Science Basic Research Project of Shaanxi Province (No. 2020JM-362), the Key projects of Natural Science Foundation of Shaanxi Province (No. 2021JZ-36), the Institutional foundation of The First Affiliated Hospital of Xi'an Jiaotong University (No. 2019ZYTS-19 and No. 2020QN-05), and the Clinical Research Award of the First Affiliated Hospital of Xi'an Jiaotong University, China (No. XJTU1AF-CRF-2017-007).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I. et al. Erratum: Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians. 2020;70:313
2. Batlle E, Clevers H. Cancer stem cells revisited. Nature medicine. 2017;23:1124-34
3. Reya T, Morrison S, Clarke M. et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-11
4. Nguyen L, Vanner R, Dirks P. et al. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12:133-43
5. Schulenburg A, Bramswig K, Herrmann H. et al. Neoplastic stem cells: current concepts and clinical perspectives. Crit Rev Oncol Hematol. 2010;76:79-98
6. Pierce G, Speers W. Tumors as caricatures of the process of tissue renewal: prospects for therapy by directing differentiation. Cancer research. 1988;48:1996-2004
7. Lapidot T, Sirard C, Vormoor J. et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645-8
8. Qin J, Liu X, Laffin B. et al. The PSA(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell. 2012;10:556-69
9. Al-Hajj M, Wicha M, Benito-Hernandez A. et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983-8
10. O'Brien C, Pollett A, Gallinger A. et al. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106-10
11. Zhu J, Wang S, Chen Y. et al. miR-19 targeting of GSK3β mediates sulforaphane suppression of lung cancer stem cells. The Journal of nutritional biochemistry. 2017;44:80-91
12. Chen L, Wang A, Dong B. et al. A new prospect in cancer therapy: targeting cancer stem cells to eradicate cancer. Chin J Cancer. 2012;31:564-72
13. Tang C, Ang B, Pervaiz S. Cancer stem cell: target for anti-cancer therapy. FASEB J. 2007;21:3777-85
14. Deonarain M, Kousparou C, Epenetos A. Antibodies targeting cancer stem cells: a new paradigm in immunotherapy? MAbs. 2009;1:12-25
15. Yadav A, Desai N. Cancer Stem Cells: Acquisition, Characteristics, Therapeutic Implications, Targeting Strategies and Future Prospects. Stem Cell Rev Rep. 2019;15:331-55
16. Abbas T, Keaton M, Dutta A. Genomic instability in cancer. Cold Spring Harbor perspectives in biology. 2013;5:a012914
17. Hanahan D, Weinberg R. The hallmarks of cancer. Cell. 2000;100:57-70
18. Petropoulos M, Champeris Tsaniras S, Taraviras S. et al. Replication Licensing Aberrations, Replication Stress, and Genomic Instability. Trends in biochemical sciences. 2019;44:752-64
19. Vassilev A, DePamphilis M. Links between DNA Replication, Stem Cells and Cancer. Genes (Basel). 2017;8:45
20. Wittig K, Sansam C, Noble T. et al. The CRL4DTL E3 ligase induces degradation of the DNA replication initiation factor TICRR/TRESLIN specifically during S phase. Nucleic Acids Res. 2021;8:10507-23
21. Fragkos M, Ganier O, Coulombe P. et al. DNA replication origin activation in space and time. Nat Rev Mol Cell Biol. 2015;16:360-74
22. Mahadevappa R, Neves H, Yuen S. et al. DNA Replication Licensing Protein MCM10 Promotes Tumor Progression and Is a Novel Prognostic Biomarker and Potential Therapeutic Target in Breast Cancer. Cancers (Basel). 2018;10:282
23. Matson J, Dumitru R, Coryell P. et al. Rapid DNA replication origin licensing protects stem cell pluripotency. Elife. 2017;6:e30473
24. Alvarez S, Diaz M, Flach J. et al. Replication stress caused by low MCM expression limits fetal erythropoiesis and hematopoietic stem cell functionality. Nat Commun. 2015;6:8548
25. Sun J, Evrin C, Samel S. et al. Cryo-EM structure of a helicase loading intermediate containing ORC-Cdc6-Cdt1-MCM2-7 bound to DNA. Nat Struct Mol Biol. 2013;20:944-51
26. Sun J, Kawakami H, Zech J. et al. Cdc6-induced conformational changes in ORC bound to origin DNA revealed by cryo-electron microscopy. Structure (London, England: 1993). 2012;20:534-44
27. Remus D, Beuron F, Tolun G. et al. Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell. 2009;139:719-30
28. Bleichert F. Mechanisms of replication origin licensing: a structural perspective. Curr Opin Struct Biol. 2019;59:195-204
29. Evrin C, Clarke P, Zech J. et al. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20240-5
30. Li N, Lam W, Zhai Y. et al. Structure of the origin recognition complex bound to DNA replication origin. Nature. 2018;559:217-22
31. Zhai Y, Cheng E, Wu H. et al. Open-ringed structure of the Cdt1-Mcm2-7 complex as a precursor of the MCM double hexamer. Nature structural & molecular biology. 2017;24:300-8
32. Yuan Z, Riera A, Bai L. et al. Structural basis of Mcm2-7 replicative helicase loading by ORC-Cdc6 and Cdt1. Nature structural & molecular biology. 2017;24:316-24
33. Vaziri C, Saxena S, Jeon Y. et al. A p53-dependent checkpoint pathway prevents rereplication. Mol Cell. 2003;11:997-1008
34. Melixetian M, Ballabeni A, Masiero L. et al. Loss of Geminin induces rereplication in the presence of functional p53. J Cell Biol. 2004;165:473-82
35. Hanahan D, Weinberg R. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
36. McNairn A, Gilbert D. Overexpression of ORC subunits and increased ORC-chromatin association in transformed mammalian cells. J Cell Biochem. 2005;96:879-87
37. Gavin E, Song B, Wang Y. et al. Reduction of Orc6 expression sensitizes human colon cancer cells to 5-fluorouracil and cisplatin. PLoS One. 2008;3:e4054
38. Lau E, Tsuji T, Guo L. et al. The role of pre-replicative complex (pre-RC) components in oncogenesis. FASEB J. 2007;21:3786-94
39. Chen S, Chen X, Xie G. et al. Cdc6 contributes to cisplatin-resistance by activation of ATR-Chk1 pathway in bladder cancer cells. Oncotarget. 2016;7:40362-76
40. Okano-Uchida T, Kent L, Ouseph M. et al. Endoreduplication of the mouse genome in the absence of ORC1. Genes & development. 2018;32:978-90
41. Saha T, Ghosh S, Vassilev A. et al. Ubiquitylation, phosphorylation and Orc2 modulate the subcellular location of Orc1 and prevent it from inducing apoptosis. Journal of cell science. 2006;119:1371-82
42. Aihemaiti G, Kurata M, Nogawa D. et al. Subcellular localization of MCM2 correlates with the prognosis of ovarian clear cell carcinoma. Oncotarget. 2018;9:28213-25
43. Xiong W, Xie C, Qiu Y. et al. Origin recognition complex subunit 1 regulates cell growth and metastasis in glioma by altering activation of ERK and JNK signaling pathway. Molecular and cellular probes. 2020;49:101496
44. Faghihloo E, Sadeghizadeh M, Shahmahmoodi S. et al. Cdc6 expression is induced by HPV16 E6 and E7 oncogenes and represses E-cadherin expression. Cancer gene therapy. 2016;8:45
45. Sideridou M, Zakopoulou R, Evangelou K. et al. Cdc6 expression represses E-cadherin transcription and activates adjacent replication origins. The Journal of cell biology. 2011;195:1123-40
46. Song B, Liu X, Rice S. et al. Plk1 phosphorylation of orc2 and hbo1 contributes to gemcitabine resistance in pancreatic cancer. Molecular cancer therapeutics. 2013;12:58-68
47. Zimmerman K, Jones R, Petermann E. et al. Diminished origin-licensing capacity specifically sensitizes tumor cells to replication stress. Molecular cancer research: MCR. 2013;11:370-80
48. Woodward AM, Gohler T, Luciani MG. et al. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J Cell Biol. 2006;173:673-83
49. Takahashi TS, Wigley DB, Walter JC. Pumps, paradoxes and ploughshares: mechanism of the MCM2-7 DNA helicase. Trends Biochem Sci. 2005;30:437-44
50. Gao K, Lockwood W, Li J. et al. Genomic analyses identify gene candidates for acquired irinotecan resistance in melanoma cells. International journal of oncology. 2008;32:1343-9
51. Song J, Choi C, Yeom H. et al. Monitoring the gene expression profiles of doxorubicin-resistant acute myelocytic leukemia cells by DNA microarray analysis. Life sciences. 2006;79:193-202
52. Maiorano D, Moreau J, Méchali M. XCDT1 is required for the assembly of pre-replicative complexes in Xenopus laevis. Nature. 2000;404:622-5
53. Deng M, Sun J, Xie S. et al. Inhibition of MCM2 enhances the sensitivity of ovarian cancer cell to carboplatin. Molecular medicine reports. 2019;20:2258-66
54. Zhong A, Zhang H, Xie S. et al. Inhibition of MUS81 improves the chemical sensitivity of olaparib by regulating MCM2 in epithelial ovarian cancer. Oncology reports. 2018;39:1747-56
55. Macleod K, Mullen P, Sewell J. et al. Altered ErbB receptor signaling and gene expression in cisplatin-resistant ovarian cancer. Cancer research. 2005;65:6789-800
56. Das M, Prasad S, Yadav S. et al. HPV-type-specific response of cervical cancer cells to cisplatin after silencing replication licensing factor MCM4. Tumour biology: the journal of the International Society for Oncodevelopmental Biology Medicine. 2015;36:9987-94
57. Wang X, Zhang L, Song Y. et al. MCM8 is regulated by EGFR signaling and promotes the growth of glioma stem cells through its interaction with DNA-replication-initiating factors. Oncogene. 2021;40:4615-24
58. Xu Y, Zhou X, Li Y. et al. Suppression of minichromosome maintenance 7 expression sensitizes chronic lymphocytic leukemia cells to fludarabine. Leukemia lymphoma. 2019;60:1266-74
59. Guan Y, Yang D, Nong S. et al. Breviscapine (BVP) inhibits prostate cancer progression through damaging DNA by minichromosome maintenance protein-7 (MCM-7) modulation. Biomedicine pharmacotherapy. 2017;93:103-16
60. Li J, Liu J, Liang Z. et al. Simvastatin and Atorvastatin inhibit DNA replication licensing factor MCM7 and effectively suppress RB-deficient tumors growth. Cell death disease markers. 2017;8:e2673
61. Liang Z, Li W, Liu J. et al. Simvastatin suppresses the DNA replication licensing factor MCM7 and inhibits the growth of tamoxifen-resistant breast cancer cells. Scientific reports. 2017;7:41776
62. El Dika M, Laskowska-Kaszub K, Koryto M. et al. CDC6 controls dynamics of the first embryonic M-phase entry and progression via CDK1 inhibition. Dev Biol. 2014;396:67-80
63. Calzada A, Sacristan M, Sanchez E. et al. Cdc6 cooperates with Sic1 and Hct1 to inactivate mitotic cyclin-dependent kinases. Nature. 2001;412:355-8
64. Yim H, Erikson R. Cell division cycle 6, a mitotic substrate of polo-like kinase 1, regulates chromosomal segregation mediated by cyclin-dependent kinase 1 and separase. Proc Natl Acad Sci U S A. 2010;107:19742-7
65. Liu T, Sun H, Liu S. et al. The suppression of DUSP5 expression correlates with paclitaxel resistance and poor prognosis in basal-like breast cancer. Int J Med Sci. 2018;15:738-47
66. He Y, Yan D, Zheng D. et al. Cell Division Cycle 6 Promotes Mitotic Slippage and Contributes to Drug Resistance in Paclitaxel-Treated Cancer Cells. PloS one. 2016;11:e0162633
67. Wang Y, Qiu S. Prediction of key genes in ovarian cancer treated with decitabine based on network strategy. Oncol Rep. 2016;35:3548-58
68. Cai J, Wang H, Jiao X. et al. The RNA-Binding Protein HuR Confers Oxaliplatin Resistance of Colorectal Cancer By Upregulating CDC6. Mol Cancer Ther. 2019;18:1243-54
69. McIntosh D, Blow J. Dormant origins, the licensing checkpoint, and the response to replicative stresses. Cold Spring Harbor perspectives in biology. 2012;4:a012955
70. Majid S, Dar A, Saini S. et al. Regulation of minichromosome maintenance gene family by microRNA-1296 and genistein in prostate cancer. Cancer research. 2010;70:2809-18
71. Liu Y, He G, Wang Y. et al. MCM-2 is a therapeutic target of Trichostatin A in colon cancer cells. Toxicology letters. 2013;221:23-30
72. Vanderdys V, Allak A, Guessous F. et al. The Neddylation Inhibitor Pevonedistat (MLN4924) Suppresses and Radiosensitizes Head and Neck Squamous Carcinoma Cells and Tumors. Molecular cancer therapeutics. 2018;17:368-80
73. Weissman I. Stem cells: units of development, units of regeneration, and units in evolution. Cell. 2000;100:157-68
74. Pruitt S, Bailey K, Freeland A. Reduced Mcm2 expression results in severe stem/progenitor cell deficiency and cancer. Stem cells. 2007;25:3121-32
75. Kunnev D, Rusiniak M, Kudla A. et al. DNA damage response and tumorigenesis in Mcm2-deficient mice. Oncogene. 2010;29:3630-8
76. Blow J, Ge X, Jackson D. How dormant origins promote complete genome replication. Trends in biochemical sciences. 2011;36:405-14
77. Ballabeni A, Park I, Zhao R. et al. Cell cycle adaptations of embryonic stem cells. Proc Natl Acad Sci U S A. 2011;108:19252-7
78. Boroviak T, Loos R, Bertone P. et al. The ability of inner-cell-mass cells to self-renew as embryonic stem cells is acquired following epiblast specification. Nat Cell Biol. 2014;16:516-28
79. Saitou M, Yamaji M. Primordial germ cells in mice. Cold Spring Harbor perspectives in biology. 2012;4:a008375
80. Brons I, Smithers L, Trotter M. et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature. 2007;448:191-5
81. Shen L, Nishioka T, Guo J. et al. Geminin functions downstream of p53 in K-ras-induced gene amplification of dihydrofolate reductase. Cancer research. 2012;72:6153-62
82. Zhao B, Zhang J, Chen X. et al. Mir-26b inhibits growth and resistance to paclitaxel chemotherapy by silencing the CDC6 gene in gastric cancer. Archives of medical science: AMS. 2019;15:498-503
83. Hou J, Lambers M, den Hamer B. et al. Expression profiling-based subtyping identifies novel non-small cell lung cancer subgroups and implicates putative resistance to pemetrexed therapy. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2012;7:105-14
84. Kim S, Ho J, Jin H. et al. Upregulated expression of BCL2, MCM7, and CCNE1 indicate cisplatin-resistance in the set of two human bladder cancer cell lines: T24 cisplatin sensitive and T24R2 cisplatin resistant bladder cancer cell lines. Investigative clinical urology. 2016;57:63-72
85. Wang J, Tai L, Tzang C. et al. 1p31, 7q21 and 18q21 chromosomal aberrations and candidate genes in acquired vinblastine resistance of human cervical carcinoma KB cells. Oncology reports. 2008;19:1155-64
86. Bryant V, Elias R, McCarthy S. et al. Suppression of Reserve MCM Complexes Chemosensitizes to Gemcitabine and 5-Fluorouracil. Mol Cancer Res. 2015;13:1296-305
Author contact
![]() Corresponding authors: Peijun Liu and Yaochun Wang. Center for Translational Medicine, the First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, Shaanxi, China. Email: liupeijunxjtu.edu.cn (P. J. L.) and wangycedu.cn (Y.C.W.)
Corresponding authors: Peijun Liu and Yaochun Wang. Center for Translational Medicine, the First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, Shaanxi, China. Email: liupeijunxjtu.edu.cn (P. J. L.) and wangycedu.cn (Y.C.W.)