Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(4):1594-1611. doi:10.7150/ijbs.68873 This issue Cite
Research Paper
Atractylenolide III ameliorates Non-Alcoholic Fatty Liver Disease by activating Hepatic Adiponectin Receptor 1-Mediated AMPK Pathway
Qian Li1#, Jia-Xin Tan1#, Yong He2, Fang Bai3, Shi-Wei Li3, Yi-Wen Hou1, Long-Shan Ji1, Ya-Ting Gao1, Xin Zhang1, Zhen-Hua Zhou4, Zhuo Yu4, Miao Fang1, Yue-Qiu Gao1 ![]() , Man Li1
, Man Li1 ![]()
1. Laboratory of Cellular Immunity, Shuguang Hospital, Affiliated to Shanghai University of Traditional Chinese Medicine, Shanghai, China.
2. Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China.
3. School of Life Science and Technology and Shanghai Institute for Advanced Immunochemical Studies, Shanghai Tech University, Shanghai, China.
4. Department of Hepatopathy, Shuguang Hospital, Affiliated to Shanghai University of Traditional Chinese Medicine, Shanghai, China.
#These authors made equal contributions to this work.
Received 2021-11-9; Accepted 2021-12-29; Published 2022-1-31
Abstract

Background: Nonalcoholic fatty liver disease (NAFLD) is the most frequent cause of chronic liver diseases worldwide. At present, there are no effective pharmacological therapies for NAFLD except lifestyle intervention-mediated weight loss. Atractylenolide III (ATL III), the major bioactive component found in Atractylode smacrocephala Koidz, has been shown to exert anti-oxidant, anti-tumor, anti-allergic response, anti-bacterial effects and cognitive protection. Here we investigate the therapeutic potential and underlying mechanisms of ATL III for the treatment of NAFLD.
Methods: Male C57BL/6J mice were fed a high-fat diet (HFD) and treated with ATL III. Lipid accumulation was analyzed by Oil Red O staining in liver tissues and free fatty acids (FFAs)-treated hepatocytes. AMP-activated protein (AMPK) and sirtuin 1(SIRT1) signaling pathways were inhibited by Compound C and EX527 in vitro, respectively. Small-interfering RNA (siRNA) was used to knockdown adiponectin receptor 1 (AdipoR1) expression in HepG2 cells.
Results: ATL III treatment ameliorated liver injury and hepatic lipid accumulation in the HFD-induced NAFLD mouse model as demonstrated by that ATL III administration significantly reduced serum levels of alanine aminotransferase, glutamic oxaloacetic transaminase, triglycerides, total cholesterol and low-density lipoprotein. Furthermore, treatment with ATL III alleviated hepatic oxidative stress, inflammation and fibrosis in the HFD feeding model. To study the underlying mechanisms, we performed Computer Aided Design assay and found that open-formed AdipoR1 and adiponectin receptor 2 were the potential receptors targeted by ATL III. Interestingly, HFD feeding or FFAs treatment only reduced hepatic AdipoR1 expression, while such reduction was abolished by ATL III administration. In addition, in vitro treatment with ATL III activated the AdipoR1 downstream AMPK /SIRT1 signaling pathway and reduced lipid deposition in HepG2 cells, which was diminished by silencing AdipoR1. Finally, inhibition of AMPK or SIRT1, the AdipoR1 downstream signaling, abolished the protective effects of ATL III on lipid deposition and oxidative stress in FFAs-treated HepG2 cells.
Conclusion: Our findings suggest that ATL III is a therapeutic drug for the treatment of NAFLD and such protective effect is mediated by activating hepatic AdipoR1-mediated AMPK/SIRT1 signaling pathway.
Keywords: ATL III, AdipoR1, oxidative stress, inflammation, AMPK, SIRT1
Introduction
Liver disease is one of the most serious health problems [1], and nonalcoholic fatty liver disease (NAFLD) is the most common type of liver diseases. NAFLD is characterized with lipid accumulation without significant alcohol consumption, followed by liver inflammation (non-alcoholic steatohepatitis, NASH) and occurs mostly in patients who are obese and have the metabolic syndrome [2-4]. Recently, the prevalence of NAFLD has increased dramatically over the last three decades to nearly 15%-30% in western countries [5]. An updated meta-analysis focusing on Asian epidemiological studies reported an NAFLD prevalence of 30% and an incidence of 50.9 cases per 1,000 person-years [6]. It has been projected that China will have the largest number of liver-related deaths from NAFLD by 2030 [7]. The criterion for diagnosis of NAFLD is the presence of more than 5% triglycerides (TG) stored in hepatocytes [8]. Many potential risk factors of NAFLD have been identified, such as overweight or obesity, diabetes and insulin resistance. However, the mechanisms underlying the progression of NAFLD have not been completely elucidated and there are currently no effective pharmacological therapies for NAFLD.
AMP-activated protein (AMPK) is a key sensor of cellular energy homeostasis and is activated by increased cellular adenosine mono-phosphate (AMP) and adenosine di-phosphate (ADP) [9]. Activation of AMPK results in a decrease in lipogenesis and an increase in fatty acid oxidation and lipolysis in adipocytes in vitro and in vivo [10]. The ability of AMPK to respond to altered AMP levels is dependent upon liver kinase B1 (LKB1), which phosphorylates AMPK [11]. AMPK exerts many functions via the regulation of sirtuin 1 (SIRT1), a protein that plays an important role in regulating cell energy metabolism, cell stress, and cell fate [12]. AMPK enhances SIRT1 activity by increasing cellular nicotinamide adenine dinucleotide (NAD+) levels, further leading to the deacetylation and regulation of downstream SIRT1 targets such as peroxisome proliferator-activated receptor gamma coactivator-1α (PGC1α) [13]. Adiponectin is the most abundant adipose-specific hormone and exerts its functions by binding to its specific receptors, adiponectin receptor protein 1 (AdipoR1) and adiponectin receptor protein 2 (AdipoR2). AdipoR1 is abundantly expressed in skeletal muscle and liver, whereas AdipoR2 is predominantly expressed in the liver. Accumulating evidence suggests that activation of AdipoR1 and AdipoR2 plays important roles in regulating glucose and lipid metabolism, inflammation and oxidative stress. In the liver, activation of AdipoR1 and AdipoR2 induces the activation of AMPK pathways and PPARα pathways, respectively, which makes the AdipoRs as a potential drug target for NAFLD therapy [14].
Up to now, there were no FDA proved pharmacological therapies for NAFLD except lifestyle intervention-mediated weight loss. Therefore, it is urgent to identify the new therapeutic targets and therapies for the treatment of NAFLD. In recent years, many herbs and phytochemicals have been used as complementary and alternative therapies for liver diseases [15, 16]. For example, Atractylenolide III (ATL III) is a sesquiterpene lactone and is the major bioactive component found in Atractylode smacrocephala Koidz. ATL III is also found in other medicinal herbs, such as Codonopsis pilosula, Atractylodes lancea, and Chloranthus henryi Hemsl. ATL III has been demonstrated to exhibit a series of benefits, including anti-oxidant, anti-tumor, anti-allergic response, anti-bacterial effects, and cognitive protection [17-19]. However, the effects of ATL III on NAFLD have not been explored.
In the current study, we examined the effects of ATL III on NAFLD in high-fat diet (HFD)-fed mice and free fatty acids (FFAs)-treated HepG2 cells. We found that AdipoR1 is the potential combination receptor with ATL III by using computer Aided Design assay. Then the impacts of ATL III on lipid accumulation and oxidation and the relevant mechanisms are investigated. Our study revealed that ATL III treatment effectively ameliorated NAFLD by regulating AdipoR1 mediated AMPK-SIRT1 signaling pathway, suggesting that ATL III has therapeutic potential for the treatment of NAFLD.
Materials and methods
Chemical reagent
Stock solutions of 20 mM oleic acid (OA, Sigma-Aldrich, USA) and 40 mM palmitic acid (PA, Sigma-Aldrich) prepared in culture medium containing 1% fatty-acid-free bovine serum albumin (BSA, Sigma-Aldrich) were conveniently diluted in culture medium to obtain the desired final concentrations. A FFAs mixture of 3 mM (OA and PA at the proportion of 2:1) was prepared as stock solution [20]. The 200 μg/ml stock solution of ATL III (purity ≥ 99%, Tauto Biotech, Shanghai) was prepared in dimethyl sulfoxide (DMSO) and the final concentration of DMSO was less 0.1% in all in vitro experiments.
Animals and administration of ATL III
Six-week old male C57BL/6J mice were used throughout our experiments. HFD (No. D12492) was purchased from Research Diets, Inc. (Middlesex County, NJ, USA). Mice were randomly divided into the following three experimental groups (each group n=12) (1) control diet group (CD): mice were fed normal chow diet and administrated normal saline by tail intravenous injection; (2) the HFD-fed induced NAFLD mice group (HFD+DMSO): mice were fed an HFD (contained 60% fat) for 16 weeks and administrated DMSO by tail intravenous injection; (3) the HFD-fed induced NAFLD mice group administrated with ATL III (HFD+ATLIII): mice maintained an HFD and were administrated 10 mg/kg ATLIII (purity ≥ 98%, St. Louis, MO, USA). The 40 mg/ml stock solution of ATL III (purity ≥ 99%, Tauto Biotech, Shanghai) was prepared in DMSO, and then the stock solution was diluted with 0.9% NaCl. The final concentration of ATL III was 1 mg/ml, and it was injected by tail vein. Mice in both HFD+DMSO and HFD+ATL III groups maintained an HFD for 16 weeks and were then treated with DMSO or ATL III every other day in the last 4 weeks.
All animal experiments were approved by the Committee of Animal Experimentation (No. PZSHUTCM210320005) and conducted at Shanghai university of Traditional Chinese Medicine in accordance with its Animal Experiment Guidelines.
Histological Analysis
Liver tissues were fixed with formalin and then embedded in paraffin. All fixed tissues were sliced into 4-μm slices and stained by hematoxylin and eosin (HE). All slices were examined using a light microscope (Olympus, Tokyo, Japan). The Ballooning Degeneration Score and NAFLD Activity Score were evaluated [21].
Biochemical Analysis
Serum, liver tissues and cells lysates were individually collected. The levels of ALT (alanine aminotransferase), AST (Glutamic oxaloacetic transaminase), TG, total cholesterol (TC), high-density lipoprotein (HDL), low-density lipoprotein (LDL), malondiadehyde (MDA), glutathione peroxidase (GSH-Px) and superoxide dismutase (SOD) were tested with commercial kits according to the manufacturer's instructions (Nanjing Jian Cheng Bioengineering Institute, China).
Prediction of the potential targets of ATL III
To identify the potential targets of ATL III, a molecular docking based computational target fishing simulation was designed and performed in this study. The genes (proteins) which were involved into the AMPK signaling pathway released from KEGG database (https://www.genome.jp/kegg/) were collected, and the structures of these proteins were found from RCSB (https://www.rcsb.org/). All structures of selected proteins were prepared by Protein Preparation Wizard of Schrödinger2020-3 [22], in which we added missing hydrogens and side chain, generated proper ionization state of residues in pH 7.0 ± 2.0, and performed structure minimization with Optimized Potentials for Liquid Simulations 3e (OPLS3e) force field [23]. The native ligands and the water of these proteins were removed for further reverse docking. The compound structure of ATL III was downloaded from PubChem [24], after which it was prepared by LigPrep of Schrödinger2020-3. During the ligand preparation, the force field was set as OPLS3e [23], and the ionization state was generated by Epik at a pH of 7.0 ± 2.0 [25]. Then we performed docking simulations to test the binding between ATL III and these proteins with the Glide v8.8 of Schrödinger [26, 27]. The standard precision mode and the OPLS2005 force field were used and other parameters were kept as default. Then, all the binding complexes of the candidate proteins with ATL III were collected and ranked together, and the top ranked complexes by docking score were selected for further analysis. Residues around ATL III in a range of 5Å were treated as flexible, and the optimization was performed in variable-dielectric generalized Born solvation model and OPLS4 force field.
Western Blot Analysis
Protein extracts were extracted from liver tissues or HepG2 cells using the Protein Lysis Kit (Beyotime, China) and then separated on 12% SDS-PAGE gels. Then, gels were transferred to polyvinylidene difluoride (PVDF) membranes and PVDF membranes were incubated with primary antibodies against AdipoR1, AdipoR2 (Affinity Biosciences, China), AMPK, phosphorylated-AMPK (p-AMPK), LKB1, phospho-LKB1 (p-LKB1) (Cell Signaling Technology, MA, USA), SIRT1, nuclear factor erythroid-2-related factor 2 (Nrf2), carnitine palmitoyltransferase-1A (CPT1A), silent information regulator transcription 3 (SIRT3), PGC1α and GAPDH (Proteintech Group, USA) overnight at 4 °C. The PVDF membranes were washed and incubated with secondary antibodies at room temperature for 1h. Finally, Omni-ECLTM Femto Light Chemiluminescence Kit (EpiZyme, China) was used to detect specific protein expression. The images were obtained using Chemi-Lumin One Ultra (Tanon, China).
Cell Culture and FFAs-induced steatosis of HepG2 cells
The hepatocellular carcinoma cell line HepG2 was purchased from Cell bank of the representative culture preservation committee of the Chinese Academy of Sciences, China. HepG2 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Gibco), 100 units/ml penicillin and 100 μg/ml streptomycin under a humidified atmosphere of 95% air and 5% CO2 at 37 °C. HepG2 cells were incubated with 1 mM FFAs to stimulate lipid accumulation for 24 h [28], and then were treated with ATL III (0-50 μg/ml) for 24h to evaluate its effects on lipid metabolism. The control group was treated with an equivalent volume of 1% BSA solution. The control and FFAs-treated groups were treated with the same volume of DMSO [29]. In in vivo exeriments, 25μg/ml ATL III was used to explore the protective mechanism.
Cell viability Assay
The cells (5×103 cells/well) were seeded in 96-well plates and cultivated overnight, and then cells were incubated with different concentrations of FFAs (0-1.5 mM) or ATL III (0-200 μg/ml) for 24 h or 48 h. Next, the cell counting kit-8 (CCK-8, AbMole Bioscience, USA) solution was added into the culture medium (10 μl/well), and the cultures were incubated for 2 h at 37 °C. The absorbance was measured at 450 nm by Microplate Reader (Bio-Rad; Hercules, CA, USA).
Oil Red O staining
Oil Red O staining was used to evaluate the intracellular lipid droplet levels. Frozen liver blocks were sectioned into 6-μm slices. The liver slices and fixed HepG2 cells were washed with PBS and stained by Oil Red O working solution (Oil Red O dye and diluent at a ratio of 5:2) for 30 min at 4 °C. Then the liver slices and cells were washed with 60% isopropyl alcohol to remove the excess dye, and washed with PBS. Finally, the stained liver slices and HepG2 cells were observed by the Olympus microscope (Olympus, Tokyo, Japan). To further quantify the intracellular lipid content, the images were analyzed using Image J software.
Measurement of intracellular reactive oxygen species (ROS)
The level of ROS was detected using fluorescent probe 2',7'-dichloro-dihydro-fluorescein diacetate (DCFH-DA, Nanjing Jian Cheng Bioengineering Institute, China). Cells were collected and incubated with DCFH-DA for 30 min at 37 °C in the dark. The fluorescence intensity was measured using flow cytometry (BD FACSCalibur, America).
Inhibitor experiments
Compound C is the selective and reversible AMPK inhibitor [30]. EX-527 is the selective SIRT1 inhibitor. The HepG2 cells were seeded into 6-well plates (5×105 cells/well) for 12 h, and then were pre-incubated with presence or absence Compound C (2 mM, AbMole Bioscience, USA) or EX 527 (10 μM, AbMole Bioscience, USA) for 1 h. Next, cells were treated with FFAs for 24 h, and were incubated with FFAs and ATL III for another 24 h [31, 32].
Interruption of AdipoR1
SiRNAs were chemically synthesized, annealed and transfected into 60-70% confluent HepG2 cells using Lipofectamine 3000 (Thermo Fisher Scientific, American) [33]. The sequence of the AdipoR1-targeting siRNAs was as follows: CAGCTTTCGTCCACTTCTA. Unrelated siRNA sequence was used as negative control. AdipoR1-targeting siRNAs and unrelated siRNA sequence were synthesized by Fubio Biomedical Technology Co., Ltd, China.
Enzyme-linked immunosorbent assay (ELISA)
Hyaluronic acid (HA), laminin (LN), Collage type VI (IV-C) and procollagen type III (PC III) were fibrosis-relevant proteins. The serum levels of HA, LN, IV-C and PC III were tested by ELISA according to the manufacture's recommended protocols (Vazyme Biotech Co, Ltd, China).
Statistical Analysis
Statistical analyses were performed using the SPSS software (version 17.0). Test of Homogeneity of variance was performed. When the variance was uniform, one-way ANOVA followed by Fisher's Least Significant Difference (LSD) test was performed to compare data between two groups from multiple groups. When the variance was not uniform, Kruskal-Wallis test was performed to compare multiple groups. If there were significant differences among multiple groups, Mann-Whiteney U was performed to compare median between two groups. Then the test standard (α=0.05) was corrected by using Bonferroni method. Results were represented as mean±SD. The tests were all two-tailed, and statistical significance was set as P <0.05.
Results
ATL III administration ameliorates liver steatosis in HFD-fed mice
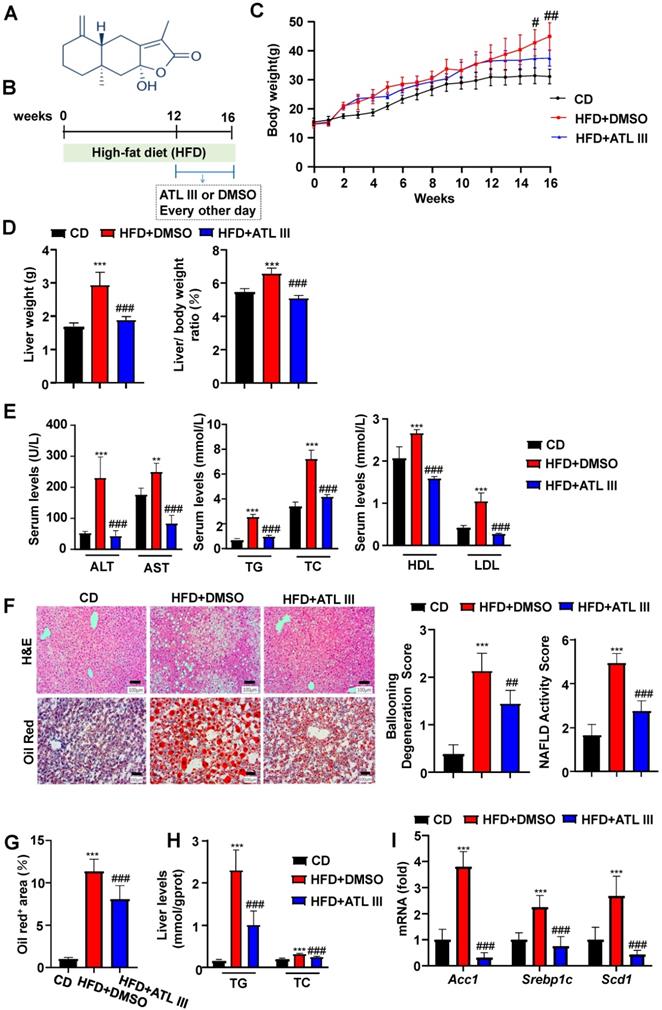
ATL III is the main active constituent of Atractylodes rhizome, and its structure is shown in Figure 1A. To determine the protective effects of ATL III on NAFLD in vivo, male C57BL/6J mice were fed a HFD for 12 weeks followed by ATL III administration for 4 weeks, or DMSO as a control (Figure 1B). ATL III administration reduced mouse body weight (Figure 1C), but didn't affect food intake after HFD feeding (Supplementary Figure S1). As expected, ATL III-treated mice had less liver weight as compared with DMSO-treated mice after HFD feeding (Figure 1D). Furthermore, ATL III administration significantly ameliorated HFD-induced liver injury and lipid accumulation as evidenced by lower serum levels of ALT, AST, TG, TC and LDL in ATL III-treated mice (Figure 1E). Moreover, H&E and Oil red O staining showed that lipid accumulation, Ballooning Degeneration Score and NAFLD Activity Score in the liver were significantly decreased in ATL III-treated mice after HFD feeding compared with DMSO-treated mice (Figure 1F and 1G). Similarly, Liver TG and TC levels were also remarkably reduced in ATL III-treated mice after HFD feeding (Figure 1H). RT-qPCR analyses showed that hepatic up-regulation of some lipogenesis-related key genes including Acc1, Srebp1c and Scd1 in HFD-fed mice was inhibited by ATL III treatment (Figure 1I). All the data strongly suggest that ATL III administration ameliorates HFD-induced liver injury and lipid accumulation.
ATL III administration ameliorates liver steatosis in HFD-fed mice. Mice were categorized into three groups including control diet group (CD), the HFD-fed induced NAFLD group (HFD+DMSO) and the HFD-fed induced NAFLD group administrated with ATL III (HFD+ATL III). A. The structure of ATL III is shown. B. Mice were fed with an HFD for 16 weeks to induce NAFLD and administered DMSO or ATL III by tail intravenous injection in the last 4 weeks. C. Dynamic changes of weight during the experiment stage were recorded. D. Liver weight and liver/body weight ratio (%) were analyzed. E. Alanine aminotransferase (ALT), Aspartate aminotransferase (AST), Triglycerides (TG), Total Cholesterol (TC), High-density lipoprotein (HDL) and Low-density lipoprotein (LDL). Serum ALT, AST, TG, TC, HDL and LDL levels were tested. F. H&E and Oil Red O-stained slices of liver tissues were shown. Ballooning Degeneration Score and NAFLD Activity Score were calculated. G. The quantitation analyses of Oil Red O-stained slices of liver tissues were shown. H. TG and TC levels in liver tissues were examined. The 'mmol/gprot' means that the protein content of tissue extracts was calculated as mmols per gram protein of the tissue. I. The levels of some lipogenesis-related key genes including Acc1, Srebp1c and Scd1 in the livers of HFD-fed mice were tested by RT-qPCR. Values represent means±SD. (n=12 mice in each group). **, compared with CD P<0.01, ***, compared with CD P<0.001; #, compared with HFD+DMSO P<0.05; ##, compared with HFD+DMSO P<0.01, ###, compared with HFD+DMSO P<0.001.
ATL III administration mitigates liver inflammation, fibrosis and oxidative stress in HFD-fed mice
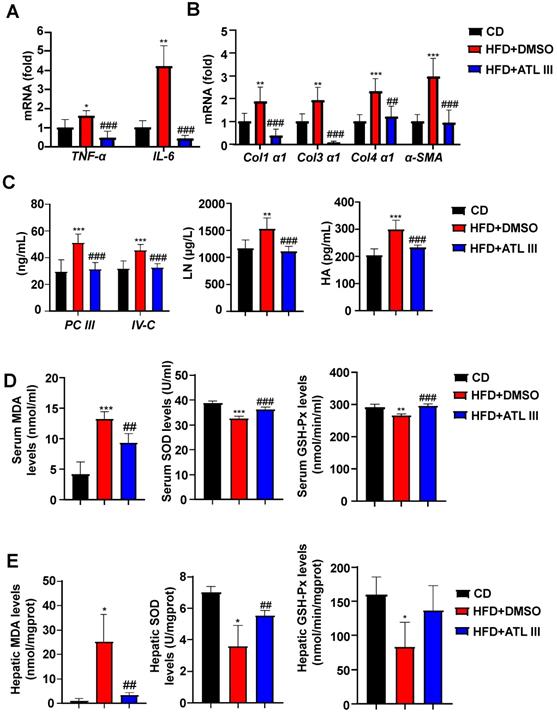
Next, we examined whether ATL III ameliorated other NAFLD phenotypes. As illustrated in Figure 2A-C, ATL III injection decreased liver inflammation and fibrosis as demonstrated by that the levels of several inflammatory and fibrogenic genes in liver and fibrosis-relevant proteins in serum were significantly decreased in ATL III-treated HFD mice compared with DMSO-treated mice. In addition, we found that serum and hepatic MDA levels were dramatically elevated in HFD-fed mice compared with CD-fed mice, and such elevation was markedly reduced by ATL III treatment (Figure 2D and 2E). Importantly, serum levels of SOD and GSH-Px were significantly decreased after HFD feeding, and ATL III administration restored levels of SOD and GSH-Px (Figure 2D and 2E). Hepatic SOD and GSH-Px levels were markedly decreased after HFD feeding, but only the up-regulated levels of SOD induced by ATL III administration were significant.
ATL III administration mitigates liver inflammation, fibrosis, and oxidative stress after HFD feeding. A. The levels of inflammatory factors including TNF-α and IL-6 in the livers of HFD-fed mice were tested by RT-qPCR. B. The levels of fibrogenic genes including Col1α1, Col3α1, Col4α1 and α-SMA were examined by RT-qPCR. C. Procollagen type III (PC III), Collage type VI (IV-C), Laminin (LN), Hyaluronic acid (HA). The expression levels of serum fibrosis-related proteins including PC III, IV-C, LN and HA were examined by ELISA. D. Malondialdehyde (MDA), Superoxide Dismutase (SOD), Glutathione peroxidase (GSH-Px). Serum MDA, SOD and GSH-Px levels were measured by chemocolorimetry. E. The expression levels of MDA, SOD and GSH-Px in liver tissues were examined by chemocolorimetry. The 'mgprot' mean that the protein content of tissue extracts and enzymatic activities were calculated as nmols and units per milligram protein of the tissue. Values represent means ± SD (n=12 mice in each group). *, Compared with CD P<0.05, **, compared with CD P<0.01, ***, compared with CD P<0.001; ##, compared with HFD+DMSO P<0.01, ###, compared with HFD+DMSO P<0.001.
Incubation with ATL III reduces FFAs-induced lipid accumulation and ROS production in HepG2 cells
To investigate the molecular mechanisms by which ATL III inhibits lipid accumulation in hepatocytes, we treated HepG2 cells with different doses of FFAs and/or ATL III for 24h and 48h. Cell viability assay revealed that ATL III administration didn't affect hepatocyte viability (Supplementary Figure S2). FFAs treatment markedly increased fat content in hepatocytes, which was significantly reduced by ATL III administration in a dose-dependent manner (Figure 3A). ATL III treatment notably limited FFAs-induced accumulation of intracellular TG and TC in HepG2 cells (Figure 3B). In the murine hepatocyte cell line AML12, the fat content and intracellular TG and TC levels were increased by FFAs treatment, and the up-regulated levels induced by FFAs were all inhibited after ATL III treatment (Supplementary Figure S3).
ATL III administration reduces FFAs-induced lipid accumulation in HepG2 cells. HepG2 cells were categorized into five groups including the control group, FFAs group, 12.5 µg/ml ATL III group, 25 µg/ml ATL III group and 50 µg/ml ATL III group. A. Lipids in HepG2 cells were stained by Oil Red O. Representative Oil Red O staining images were shown. The quantification of intracellular lipid content by Oil Red O staining was analyzed by calculating the area of intracellular lipid droplets. B. Intracellular TG and TC levels were measured by biochemical test kits. C. ROS expression levels in HepG2 cells were tested by flow cytometry. Representative flow cytometry analysis of ROS levels in HepG2 cells were shown. The analyses of ROS levels expressed by HepG2 cells were shown. D. MDA, SOD and GSH-Px levels expressed by HepG2 cells were measured. The above experiments were performed three times independently, and the results were displayed as mean±SD. **, Compared with Control group P<0.01 ***, compared with Control group P<0.001; #, compared with FFAs group P<0.05; ##, compared with FFAs group P<0.01; ###, compared with FFAs group P<0.001.
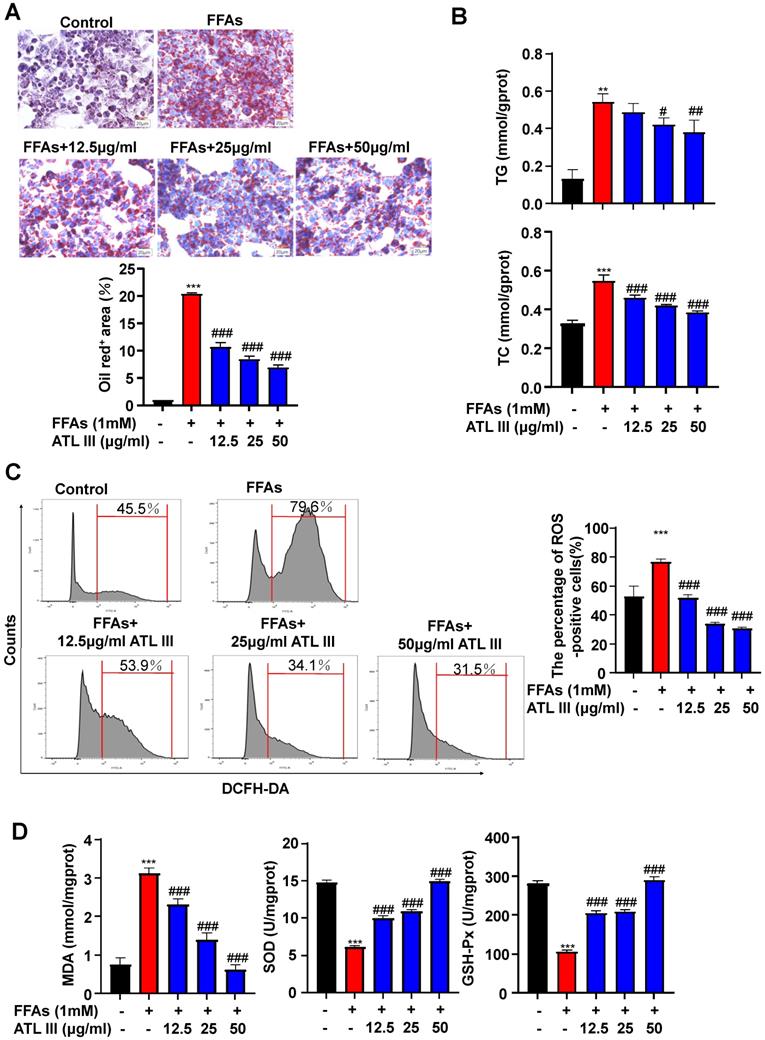
To determine whether ATL III inhibited the oxidative stress in hepatocytes, we measured ROS, MDA, SOD and GSH-Px levels in HepG2 cells after FFAs treatment. As illustrated in Figure 3C, ROS levels were increased by treatment with FFAs while ATL III treatment inhibited FFAs-induced ROS production in HepG2 cells. In addition, the up-regulation of MDA levels induced by FFAs was blocked by ATL III administration. Next, we examined the anti-oxidant factors SOD and GSH-Px, and we found that FFAs treatment reduced SOD and GSH-Px levels in HepG2 cells, which was restored by ATL III incubation (Figure 3D).
Computer Aided Design assay identifies AdipoR1 is a potential binding target of ATL III
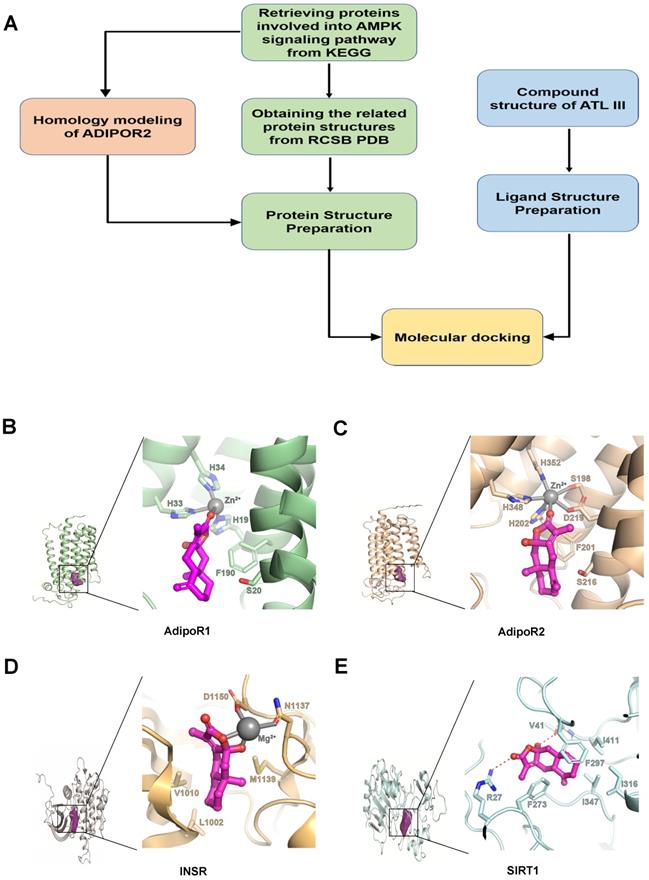
To understand the mechanisms by which ATL III ameliorates fat accumulation in hepatocytes, we searched ATL III targets by using Computer Aided Design assay, as shown in Figure 4A. The AMPK signaling pathway is well known to play an essential role in controlling the progression of NAFLD [34]. Therefore, we focused on the genes being involved in this pathway to see if there is a candidate binding target of ATL III. As a result, 79 AMPK-related protein structures were obtained. All these protein structures were taken as our target candidates and their binding abilities with ATL III were computationally evaluated with molecular docking simulations. Through the calculations, four potential targets were identified, including open-formed AdipoR1, open-formed AdipoR2, Insulin Receptor (INSR) and SIRT1 (Figure 4B-E).
The potential binding target of ATL III was identified by Computer Aided Design assay. A. The designed workflow for identifying potential drug targets of ATL III from the genes being involved in the AMPK pathway was shown. B. AdipoR1: Zoomed potential binding pattern of AdipoR1 and ATL III. ATL III formed hydrophobic interaction with F190 and coordinate bond with zinc ion of AdipoR1 (GlideScore:-3.266). C. AdipoR2: Zoomed potential binding pattern of AdipoR2 and ATL III. ATL III formed hydrophobic interaction with F201 and coordinate bond with zinc ion of AdipoR2 as a part of hexa-coordinated complex (Glidescore:-3.478). D. INSR: Zoomed potential binding pattern of INSR and ATL III. ATL III formed two coordinate bond with magnesium ion and hydrophobic interaction with L1002, V1010 and M1139 of INSR (GlideScore; -3.478). E. SIRT1: Zoomed potential binding pattern of SIRT1 and ATL III. ATL III formed hydrogen bond with V41, R27 and stable hydrophobic interaction with five residues: F273, F297, I316, I347, I411 of SIRT1 (GlideScore:-5.969). GlideScore was the docking score for estimating the binding affinity between a ligand and a protein. In this work, the contribution of the metal ions to the binding affinity was not considered.
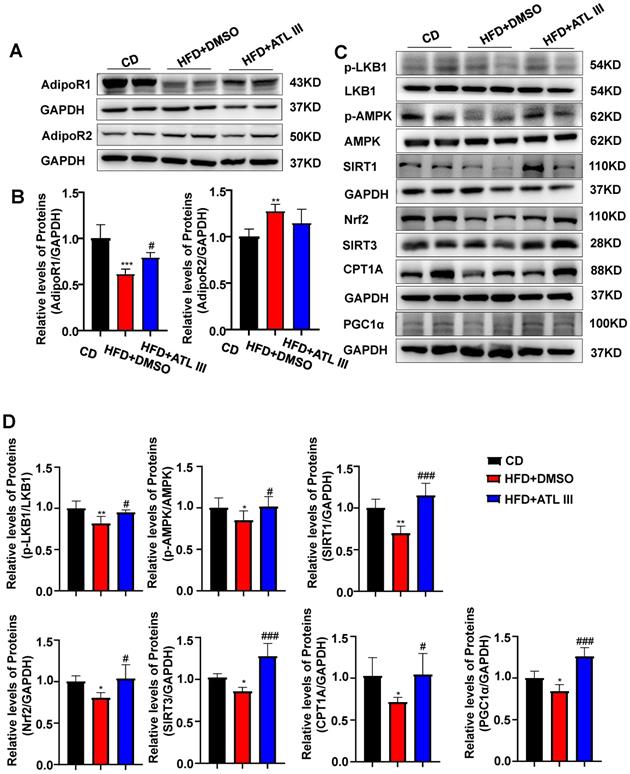
HFD feeding down-regulates hepatic expression of AdipoR1 and its downstream signaling in mice, which is partially restored by ATL III treatment
Among the four potential ATL III targets, AdipoR1 and AdipoR2 are well known to regulate lipid metabolism in the pathogenesis of NAFLD [35]. Then we examined hepatic expression of AdipoR1 and AdipoR2 in the HFD-induced NAFLD mouse model. As illustrated in Figure 5A, 5B and Supplementary Figure S4A, hepatic AdipoR1 levels were markedly decreased after HFD feeding while ATL III significantly restored hepatic AdipoR1 levels. However, hepatic AdipoR2 levels were increased in HFD-fed mice and showed a decreasing trend in ATL III-treated mice but didn't reach statistical significance. Most importantly, HFD feeding caused the down-regulation of AdipoR1-mediated downstream signaling including p-LKB1, p-AMPK, SIRT1, SIRT3, Nrf2, CPT1A and PGC1α protein levels, which were involved in lipid accumulation (LKB1 and AMPK), oxidative stress (SIRT3 and Nrf2) and fatty acid oxidation (CPT1A and PGC1α) [14]. Such down-regulation was partially reversed by ATL III administration (Figure 5C, 5D and Supplementary Figure S4B).
ATL III treatment restores the down-regulation of hepatic AdipoR1 expression and AdipoR1 signaling induced by HFD feeding in mice. A. Expression levels of hepatic AdipoR1 and AdipoR2 were examined in HFD mice. B. Relative levels of hepatic AdipoR1 and AdipoR2 were analyzed. C. AdipoR1 downstream signaling molecules including p-LKB1, p-AMPK, SIRT1, Nrf2, SIRT3, CPT1A and PGC1α were tested in HFD mice. D. Relative levels of p-LKB1, p-AMPK, SIRT1, Nrf2, SIRT3, CPT1A and PGC1α were analyzed. The experiments were performed three times independently, and values represent means ± SD.*, compared with CD P<0.05; **, compared with CD P<0.01; ***, compared with CD P<0.001; #, compared with HFD+DMSO P<0.05; ##, compared with HFD+DMSO P<0.01; ###, compared with HFD+DMSO P<0.001.
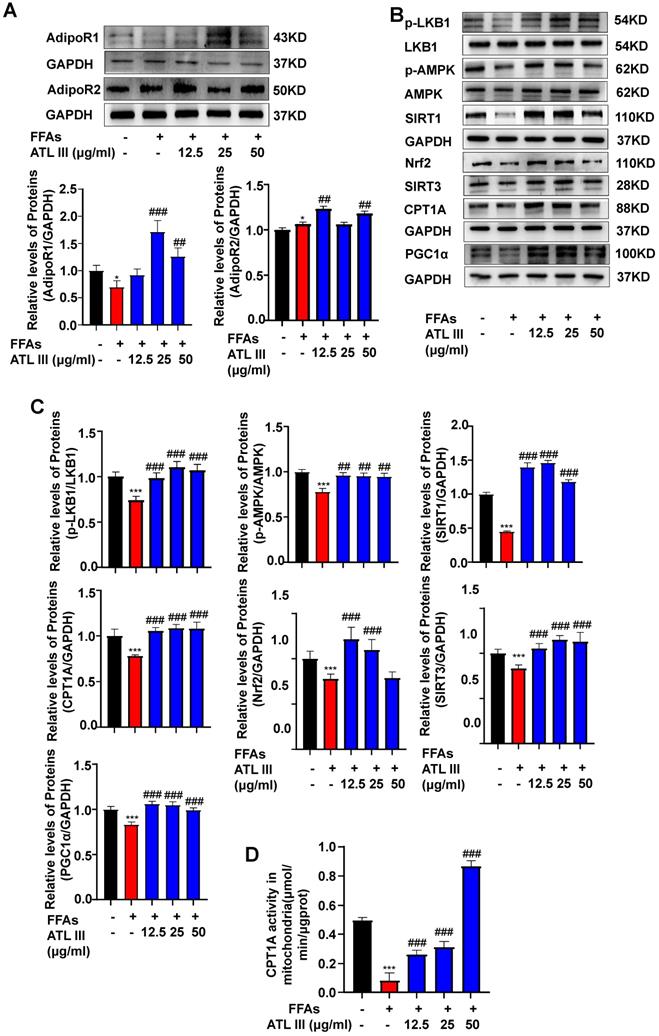
FFAs treatment down-regulates the expression of AdipoR1 and its downstream signaling in HepG2 cells, which is partially restored by ATL III treatment
To examine whether ATL III reverses FFAs-induced AdipoR1 down-regulation in vitro, HepG2 cells were directly treated with FFAs and/or ATL III. As illustrated in Figure 6A and Supplementary Figure S5A, FFAs treatment only reduced AdipoR1 protein levels in hepatocytes and such reduction was restored by ATL III treatment. FFAs treatment increased AdipoR2 levels, and such increase was strengthened by ATL III treatment, which suggested that AdioR2 maybe not involved in FFAs-induced steatosis of HepG2 cells. Furthermore, ATL III treatment partially abolished FFAs-induced down-regulation of AdipoR1-mediated signaling pathways (Figure 6B, 6C and Supplementary Figure S5B). In addition, we found that CPT1A activity was decreased by FFAs treatment, and such decrease was abolished by ATL III treatment (Figure 6D).
ATL III restores the down-regulated expression of AdipoR1-mediated AMPK-SIRT1 signaling molecules in HepG2 cells. A. HepG2 cells were categorized into five groups including the control group, FFAs group, 12.5 µg/ml ATL III group, 25 µg/ml ATL III group and 50 µg/ml ATL III group. AdipoR1 and AdipoR2 protein levels were examined by western-blot. Relative levels of hepatic AdipoR1 and AdipoR2 were analyzed. B. The expression levels of a number of key components of AdipoR1-mediated downstream signaling molecules were assessed by western blot. C. Relative levels of p-LKB1, p-AMPK, SIRT1, Nrf2, SIRT3, CPT1A and PGC1α were analyzed. D. The CPT1A activity was examined by using a CPT1A activity detection kit. The experiments were performed three times independently, and values represent means±SD. *, Compared with Control group P<0.05; ***, compared with Control group P<0.001; ##, compared with FFAs group P<0.01; ###, compared with FFAs group P<0.001.
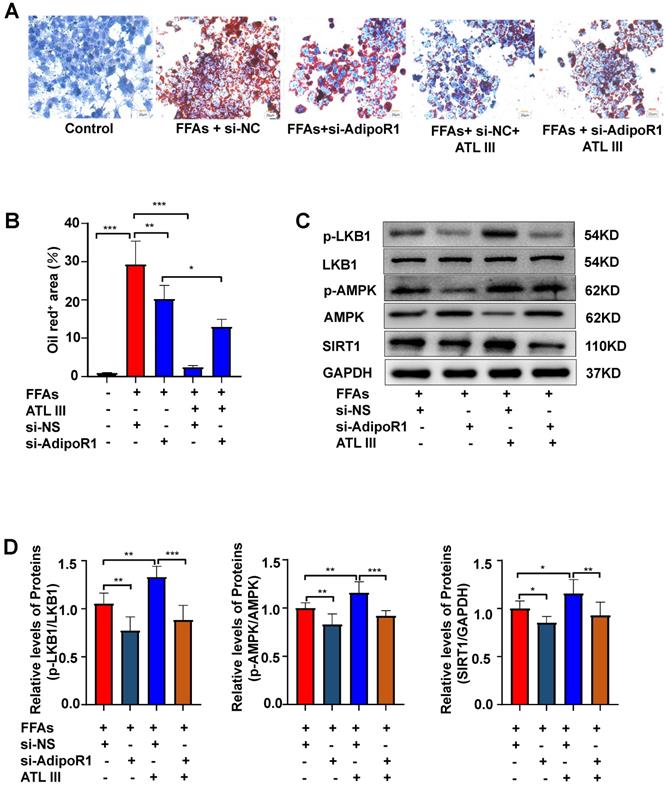
Silencing AdipoR1 abolishes ATL III-mediated amelioration of lipid accumulation and AdipoR1 downstream signaling in HepG2 cells
To further confirm whether ATL III inhibits lipid deposition through AdipoR1 in hepatocytes, the expression of AdipoR1 was silenced by transfecting AdipoR1 siRNA into HepG2 cells. It was noted that the inhibition of lipid deposition induced by ATL III was blunted by the interruption of AdipoR1, suggesting that AdipoR1 plays a crucial role in ATL III mediated-inhibition of lipid accumulation (Figure 7A and 7B). In addition, the interruption of AdipoR1 suppressed ATL III-mediated up-regulation of p-LKB1, p-AMPK and SIRT1 in HepG2 cells (Figure 7C, 7D and Supplementary Figure S6).
Silencing AdipoR1 abolishes ATL III-mediated amelioration of lipid accumulation and AdipoR1 downstream signaling in HepG2 cells. A. The expression of AdipoR1 was silenced by transfecting AdipoR1 siRNA into HepG2 cells. Lipids in HepG2 cells were stained by Oil Red O. Representative Oil Red O staining images were shown. B. The quantification of intracellular lipid content by Oil Red O staining was analyzed by calculating the area of intracellular lipid droplets. C. The expression levels of p-LKB1, p-AMPK and SIRT1 were examined by western blot. D. Relative levels of p-LKB1, p-AMPK and SIRT1 were analyzed. The experiments were performed four times independently, and values represent means±SD.*, P<0.05; **, P<0.01; ***, P<0.001.
Inhibition of AMPK or SIRT1, the AdipoR1 downstream signaling, abolishes the protective effects of ATL III on lipid deposition and oxidative stress in FFAs-treated HepG2 cells
The above results suggest that ATL III activates the AMPK and SIRT1 signaling pathways via the activation of AdipoR1. To investigate whether the AdipoR1 downstream AMPK and SIRT1 pathways contribute to the protective effects of ATL III, HepG2 cells were pre-treated with AMPK inhibitor (Compound C) or SIRT1 inhibitor (Ex 527) and then were treated with FFAs. As illustrated in Figure 8A and 8B, inhibition of AMPK or SIRT1 markedly enhanced FFAs-induced lipid accumulation. Importantly, the down-regulation of lipid accumulation induced by ATL III treatment was abolished by inhibiting AMPK or SIRT1.
Inhibition of AMPK or SIRT1 represses the inhibitory effects of ATL III on hepatic lipid deposition in FFAs-induced HepG2 cells. HepG2 cells were categorized into seven groups including the control group, FFAs group, ATL III group, Compound C group, EX527 group, Compound C combined with ATL III group and EX527 combined with ATL III group. HepG2 cells were pre-incubated with Compound C (AMPK inhibitor) or EX527 (SITR1 inhibitor) for 1h, and then were incubated with FFAs for 24h, and were incubated with ATL III for another 24h. A. Lipids in HepG2 cells were stained by Oil Red O. Representative Oil Red O staining images were shown. B. The quantification of intracellular lipid content by Oil Red O staining was shown in the panel. C. ROS expression levels in HepG2 cells were tested by flow cytometry. Representative flow cytometry analysis of ROS levels in HepG2 cells were shown. D. The analysis of ROS levels expressed by HepG2 cells was shown. E-G. MDA, SOD and GSH-Px levels were measured. The experiments were performed three times independently, and the results were displayed as mean ± SD. *, P<0.05; **, P<0.01; ***, P<0.001.
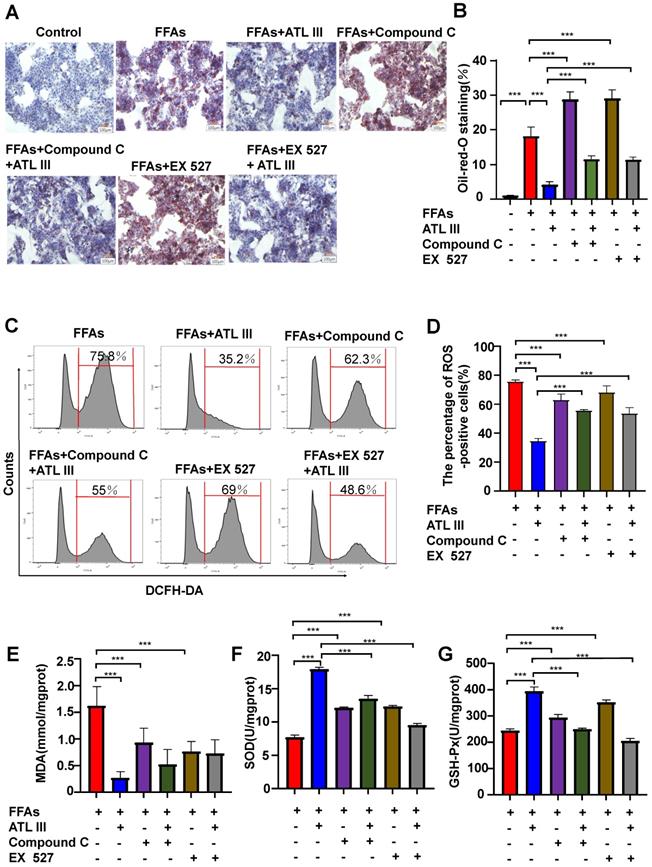
Next, the effects of inhibitors of AMPK/ SIRT1 pathway on oxidative stress in FFAs-treated HepG2 cells were explored. It was not expected that the ROS levels in FFAs-treated HepG2 cells treated were higher than that in HepG2 cells treated by AMPK or SIRT1 inhibitors (Figure 8C and 8D). Crucially, the down-regulation of ROS levels induced by ATL III in FFAs-treated HepG2 cells was markedly blocked by inhibiting AMPK or SIRT1.
In addition, ATL III exposure markedly reduced FFAs-induced MDA levels in HepG2 cells, but the reduction was not obviously inhibited by co-treatment with AMPK or SIRT inhibitors (Figure 8E). Interestingly, the decreased GSH-Px and SOD levels in HepG2 cells induced by FFAs was partly abolished by co-treatment with AMPK or SIRT1 inhibitors. The up-regulation of GSH-Px and SOD levels induced by ATL III in FFAs-treated HepG2 cells were remarkably blocked after inhibition of AMPK or SIRT1 (Figure 8F and Figure 8G), which suggested that AMPK/SIRT1 pathway was involved in the elevation of GSH-Px and SOD levels regulated by ATL III.
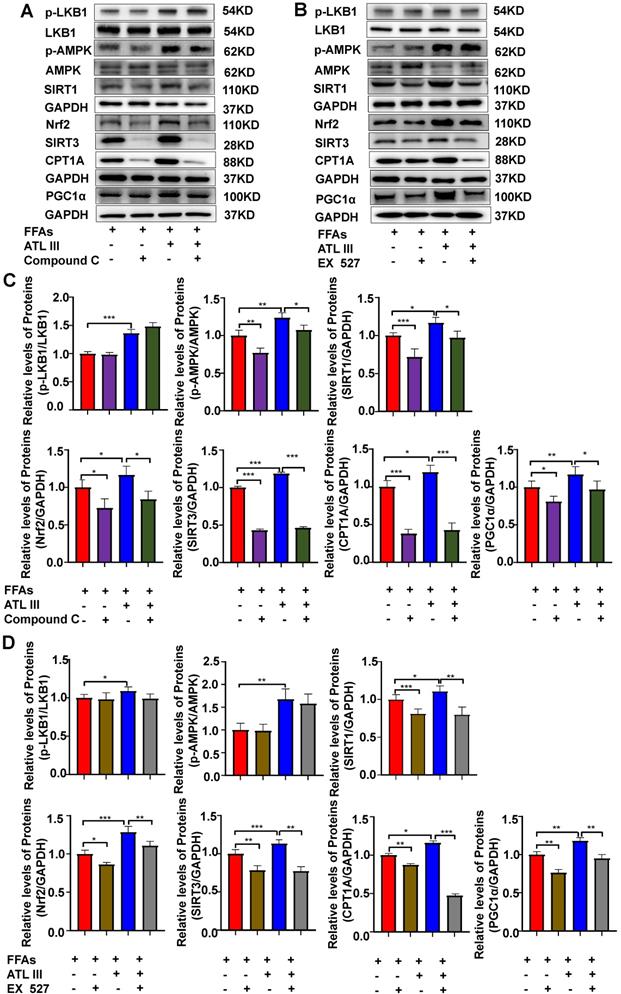
Inhibition of AMPK or SIRT1 abolishes the activated effects of ATL III on the SIRT1 downstream signaling in FFAs-treated HepG2 cells
To confirm the link between LKB1, AMPK and SIRT1, the protein levels of relevant signaling molecules in HepG2 cells were examined after inhibition of AMPK or SIRT1. As shown in Figure 9 and Supplementary Figure S7, the inhibition of AMPK or SIRT1 didn't affect the expression level of p-LKB1. The inhibition of AMPK reduced the up-regulation of SIRT1 induced by FFAs, but the inhibition of SIRT1 can't affect the up-regulation of p-AMPK. Further, the inhibition of AMPK or SIRT1 significantly reduced the protein levels of the SIRT1 downstream signaling pathways including Nrf2, SITR3, CPT1A and PGC1α. The above results reveal that AMPK acts as the primordial trigger for activating SIRT1 and its downstream signaling in FFAs-induced steatosis of HepG2 cells. Interestingly, the up-regulated expression of SIRT1 and its downstream signaling induced by ATL III in FFAs-treated HepG2 cells was partly blunted, which suggests that AMPK/SIRT1 signaling pathway is the crucial mechanism of ATL III ameliorating NAFLD.
Inhibition of AMPK or SIRT1 abolishes the ATL III-mediated activation of the SIRT1 downstream signaling in FFAs-treated HepG2 cells. A-B. Levels of p-LKB1, p-AMPK, SIRT1, Nrf2, SIRT3, CPT1A and PGC1α protein were examined by western-blot. C-D. Relative levels of p-LKB1, p-AMPK, SIRT1, Nrf2, SIRT3, CPT1A and PGC1α were analyzed. The experiments were performed three times independently, and the results were displayed as mean±SD. *, P<0.05; **, P<0.01; ***, P<0.001.
Discussion
NAFLD is a major health problem, which may occur in up to 20% of the general population worldwide [36]. NAFLD is characterized by the accumulation of lipids in the liver arising from multiple factors: augmented fatty acid uptake, increased de novo lipogenesis, decreased fatty acid oxidation and very low density lipoproteins secretion. Therefore, a therapeutic option that targets multiple aspects in the pathogenesis of NAFLD is considered to be an effective strategy to treat this disease. The beneficial effects of traditional Chinese medicine on the treatment of NAFLD have been observed and widely accepted in recent years [37], however, the underlying mechanisms remain largely unknown. ATL III, the major bioactive component found in Atractylode smacrocephala Koidz, has been demonstrated to exhibit a series of benefits, including anti-oxidant, anti-tumor, anti-allergic response, anti-bacterial effects, and cognitive protection [17-19], but the effects of ATL III in NAFLD have not been explored. Our current study clearly revealed that ATL III treatment improved NAFLD in HFD-fed mice and FFAs-treated HepG2 cells. Especially, ATL III decreased liver inflammation and fibrosis induced by HFD to the levels equal or inferior to the ones observed in CD group, suggesting that ATL III has the therapeutical potential for the treatment of NAFLD.
One of the important findings in the current study is the discovery of AdipoR1 as a potential binding target for ATL III. AMPK signaling pathway is well known to play an essential role in controlling the development of NAFLD [9]. Therefore, we focused on the genes being involved in this pathway to see if there is a candidate binding target of ATL III. The molecular docking based computational target fishing simulation was designed and performed in this study, and AdipoR1 and AdipoR2 were identified as the potential targets of ATL III. In addition, our data revealed that ATL III treatment restored hepatic expression of AdipoR1 that was markedly down-regulated in the HFD-induced NAFLD mouse model and FFAs-treated HepG2 cell model. Adiponectin is an adipocytokine that regulates glucose and lipid metabolism via the binding to its receptors AdipoR1 and AdipoR2, and the important roles of adiponectin and its receptors in regulating insulin resistance and lipid metabolism have been well documented [38, 39]. The AdipoR1/AdipoR2 dual agonist has been shown to improve both NASH and fibrosis in mouse models [35]. Therefore, ATL III may provide the pharmacological foundation for developing AdipoR1-based therapeutic agents on NAFLD.
Oxidative stress induced by free radicals plays an important role in the initiation and progression of NAFLD [40, 41]. Accumulating studies suggest that hepatic MDA and ROS levels are significantly increased, while some anti-oxidative stress proteins including GSH-Px and SOD levels are significantly declined in NAFLD patients [42-45]. Oxidative stress reflects an imbalance between bioavailability of ROS and the cellular antioxidant system that leads to a critical failure of cellular functions and eventually cell death [46]. On the one hand, the levels of ROS and MDA were meaningfully increased in the HFD mouse model and FFAs-treated cell model, which were reduced by ATL III treatment. On the other hand, the levels of GSH-Px and SOD were decreased in the HFD mouse model and FFAs-treated cell model, which were up-regulated by ATL III. All these results suggest that ATL III treatment ameliorates hepatic steatosis by decreasing intracellular lipid droplets and oxidative stress.
To clarify the inhibitory effects of ATL III on lipid accumulation and oxidative stress, we investigated the mechanism of ATL III regulating lipid metabolism-related signaling pathways in HepG2 cells. The AMPK/SIRT1 signaling pathway is an important pathway involved in regulating hepatic lipogenesis, which is regarded as a therapeutic target for the treatment of NAFLD [47, 48]. Our results showed that ATL III activated AMPK and SIRT1 signaling molecules, and further stimulated the downstream molecules including Nrf2, SIRT3, CPT1A, and PGC1α. When FFAs-treated HepG2 cells were treated with inhibitors of AMPK or SIRT1, the inhibitory effects of ATL III on lipid droplets in FFAs-treated HepG2 cells were blocked. In addition, inhibition of AMPK partly canceled out the increased protein levels of SIRT1 induced by FFAs, suggesting that AMPK was the downstream signaling molecule of SIRT1 activation induced by FFAs. Further, our data indicated that Nrf2, SIRT3, CPT1A and PGC1α were the downstream signaling molecules of SIRT1 because the activation of these pathways was suppressed by SIRT1 inhibitor. More importantly, the activation of AMPK /SITR1 signaling pathways induced by ATL III was partly blunted by AMPK inhibitor or SIRT1 inhibitor. Taken together, our results suggest that ATL III ameliorates NAFLD by activating multiple signaling pathways involved in lipid accumulation (LKB1 and AMPK), oxidative stress (SIRT3 and Nrf2) and fatty acid oxidation (CPT1A and PGC1α).
A schematic overview depicting ATL III ameliorates NAFLD by activating the AdipoR1-mediated AMPK/SIRT1 pathway. In both HFD-induced NAFLD mouse model and FFAs-induced HepG2 steatosis model, AdipoR1-mediated signaling pathway takes part in the protection roles of ATL III ameliorating NAFLD. Fatty liver induced by 3-month HFD feeding has prominent lipid accumulation with high levels of oxidative stress and fibrosis, which is inhibited by ATL III administration. In FFAs-induced HepG2 steatosis model, ATL III treatment activates AdipoR1-mediated AMPK/SIRT1 pathway. In one hand, ATL III increases SIRT3 and Nrf2 levels, which decreases oxidative stress. In another hand, ATL III increases PGC1 α and CPT1A levels, which contributes to fatty acid oxidation.
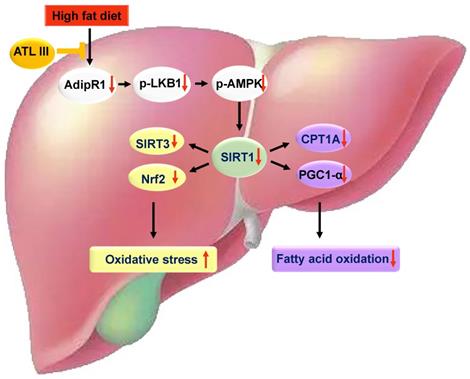
In summary, our results revealed that ATL III alleviates lipid accumulation and oxidative stress by promoting AdipoR1-mediated AMPK/SIRT1 pathways. Further investigation illustrates that this effect is mediated by regulating SIRT3, Nrf2, CPT1A and PGC1α expression. ATL III is a potential therapeutic candidate for the treatment of NAFLD.
Abbreviations
AdipoR1: adiponectin receptor protein 1; AdipoR2: adiponectin receptor protein 2; ADP: adenosine di-phosphate; ALT: alanine aminotransferase; AMP: adenosine mono-phosphate; AMPK: AMP-activated protein; AST: Glutamic oxaloacetic transaminase; ATL III: Atractylenolide III; BSA: bovine serum albumin; CCK-8: cell counting kit-8; CD: control diet group; IV-C: Collage type IV; CPT1A: carnitine palmitoyltransferase-1A; DCFH-DA: 2',7'-dichloro-dihydro-fluorescein diacetate; DMEM: Dulbecco's modified Eagle's medium; DMSO: dimethyl sulfoxide; FBS: fetal bovine serum; FFAs: free fatty acids; GSH-Px: glutathione peroxidase; HA: hyaluronic acid; HDL: high-density lipoprotein; HE: hematoxylin and eosin; HFD: high-fat diet; LDL: low-density lipoprotein; LKB1: liver kinase B1; LN: laminin; MDA: malondiadehyde; NAD+: nicotinamide adenine dinucleotide; NAFLD: nonalcoholic fatty liver disease; NASH: non-alcoholic steatohepatitis; Nrf2: nuclear factor erythroid-2-related factor 2; OA: oleic acid; PA: palmitic acid; PC III: procollagen type III; PGC1α: peroxisome proliferator-activated receptor gamma coactivator-1α; SIRT1: sirtuin 1; SIRT3: silent information regulator transcription 3; SOD: superoxide dismutase; TC: total cholesterol; TG: triglycerides.
Supplementary Material
Supplementary figures.
Supplementary methods.
Acknowledgements
This work was partly supported by National Natural Science Foundation of China (No. 82074155, 81874436, 81973773, China); “Shuguang Program” supported by Shanghai Education Development Foundation and Shanghai Municipal Education Commission (No. 18SG39, China); Program of Shanghai Academic/Technology Research Leader (No. 20XD1423500, China); Clinical Research Plan of SHDC (No. SHDC2020CR3089B, China); Shanghai Key Clinical Specialty Construction Project (No. shslczdzk01201, China); Shanghai Frontier Research Base of Disease and Syndrome Biology of inflammatory cancer transformation (No. 2021KJ03-12, China); Shanghai Sailing Program (No. 20YF1450200, China); Si Ming Foundation of Shuguang Hospital Affiliated to Shanghai University of Traditional Chinese Medicine (No. SGKJ-201912, China). In addition, this work was supported by Shanghai Key Laboratory of Traditional Chinese Medicine and Key Laboratory of Liver and Kidney Diseases (Shanghai University of Traditional Chinese Medicine), Ministry of Education.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hong M, Li S, Tan HY, Wang N, Tsao SW, Feng Y. Current Status of Herbal Medicines in Chronic Liver Disease Therapy: The Biological Effects, Molecular Targets and Future Prospects. International journal of molecular sciences. 2015;16:28705-45
2. Jin X, Yang YD, Chen K, Lv ZY, Zheng L, Liu YP. et al. HDMCP uncouples yeast mitochondrial respiration and alleviates steatosis in L02 and hepG2 cells by decreasing ATP and H2O2 levels: a novel mechanism for NAFLD. Journal of hepatology. 2009;50:1019-28
3. Zhu X, Xiong T, Liu P, Guo X, Xiao L, Zhou F. et al. Quercetin ameliorates HFD-induced NAFLD by promoting hepatic VLDL assembly and lipophagy via the IRE1a/XBP1s pathway. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association. 2018;114:52-60
4. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M. et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nature reviews Gastroenterology & hepatology. 2018;15:11-20
5. Ratziu V, Bellentani S, Cortez-Pinto H, Day C, Marchesini G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. Journal of hepatology. 2010;53:372-84
6. Li J, Zou B, Yeo YH, Feng Y, Xie X, Lee DH. et al. Prevalence, incidence, and outcome of non-alcoholic fatty liver disease in Asia, 1999-2019: a systematic review and meta-analysis. The lancet Gastroenterology & hepatology. 2019;4:389-98
7. Lee HW, Wong VW. Changing NAFLD Epidemiology in China. Hepatology (Baltimore, Md). 2019;70:1095-8
8. Watkins PA, Ellis JM. Peroxisomal acyl-CoA synthetases. Biochimica et biophysica acta. 2012;1822:1411-20
9. Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes & development. 2011;25:1895-908
10. Gaidhu MP, Fediuc S, Anthony NM, So M, Mirpourian M, Perry RL. et al. Prolonged AICAR-induced AMP-kinase activation promotes energy dissipation in white adipocytes: novel mechanisms integrating HSL and ATGL. Journal of lipid research. 2009;50:704-15
11. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nature reviews Cancer. 2009;9:563-75
12. Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587-91
13. Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC. et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056-60
14. Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M. et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nature medicine. 2007;13:332-9
15. Liao JC, Chang WT, Lee MS, Chiu YJ, Chao WK, Lin YC. et al. Antinociceptive and anti-inflammatory activities of Cuscuta chinensis seeds in mice. The American journal of Chinese medicine. 2014;42:223-42
16. Wong VW, Wong GL, Chan AW, Chu WC, Choi PC, Chim AM. et al. Treatment of non-alcoholic steatohepatitis with Phyllanthus urinaria: a randomized trial. Journal of gastroenterology and hepatology. 2013;28:57-62
17. Ji Y, Kang Z, Kang N, Zhao Y, Guo Q, Chen Y. Atractylenolide III Enhances the Anti-Neoplastic Efficacy of Docetaxel in Gastric Cancer Cell by Inhibiting Fibroblast Growth Factor Receptors 1, -2, and -4 Expression. Journal of environmental pathology, toxicology and oncology: official organ of the International Society for Environmental Toxicology and Cancer. 2019;38:217-27
18. Kang TH, Han NR, Kim HM, Jeong HJ. Blockade of IL-6 secretion pathway by the sesquiterpenoid atractylenolide III. Journal of natural products. 2011;74:223-7
19. Zhao H, Ji ZH, Liu C, Yu XY. Neuroprotection and mechanisms of atractylenolide III in preventing learning and memory impairment induced by chronic high-dose homocysteine administration in rats. Neuroscience. 2015;290:485-91
20. Gómez-Lechón MJ, Donato MT, Martínez-Romero A, Jiménez N, Castell JV, O'Connor JE. A human hepatocellular in vitro model to investigate steatosis. Chemico-biological interactions. 2007;165:106-16
21. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW. et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology (Baltimore, Md). 2005;41:1313-21
22. Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. Journal of computer-aided molecular design. 2013;27:221-34
23. Roos K, Wu C, Damm W, Reboul M, Stevenson JM, Lu C. et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. Journal of chemical theory and computation. 2019;15:1863-74
24. Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A. et al. PubChem Substance and Compound databases. Nucleic acids research. 2016;44:D1202-13
25. Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M. Epik: a software program for pK( a ) prediction and protonation state generation for drug-like molecules. Journal of computer-aided molecular design. 2007;21:681-91
26. Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT. et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. Journal of medicinal chemistry. 2004;47:1739-49
27. Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT. et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. Journal of medicinal chemistry. 2004;47:1750-9
28. Kim MH, Seong JB, Huh JW, Bae YC, Lee HS, Lee DS. Peroxiredoxin 5 ameliorates obesity-induced non-alcoholic fatty liver disease through the regulation of oxidative stress and AMP-activated protein kinase signaling. Redox biology. 2020;28:101315
29. Huang Y, Lang H, Chen K, Zhang Y, Gao Y, Ran L. et al. Resveratrol protects against nonalcoholic fatty liver disease by improving lipid metabolism and redox homeostasis via the PPARα pathway. Applied physiology, nutrition, and metabolism = Physiologie appliquee, nutrition et metabolisme. 2020;45:227-39
30. Latorre J, Ortega FJ, Liñares-Pose L, Moreno-Navarrete JM, Lluch A, Comas F. et al. Compounds that modulate AMPK activity and hepatic steatosis impact the biosynthesis of microRNAs required to maintain lipid homeostasis in hepatocytes. EBioMedicine. 2020;53:102697
31. Zhang J, Zhang SD, Wang P, Guo N, Wang W, Yao LP. et al. Pinolenic acid ameliorates oleic acid-induced lipogenesis and oxidative stress via AMPK/SIRT1 signaling pathway in HepG2 cells. European journal of pharmacology. 2019;861:172618
32. Prola A, Pires Da Silva J, Guilbert A, Lecru L, Piquereau J, Ribeiro M. et al. SIRT1 protects the heart from ER stress-induced cell death through eIF2α deacetylation. Cell death and differentiation. 2017;24:343-56
33. El-Andaloussi S, Lee Y, Lakhal-Littleton S, Li J, Seow Y, Gardiner C. et al. Exosome-mediated delivery of siRNA in vitro and in vivo. Nature protocols. 2012;7:2112-26
34. Smith BK, Marcinko K, Desjardins EM, Lally JS, Ford RJ, Steinberg GR. Treatment of nonalcoholic fatty liver disease: role of AMPK. American journal of physiology Endocrinology and metabolism. 2016;311:E730-e40
35. Xu H, Zhao Q, Song N, Yan Z, Lin R, Wu S. et al. AdipoR1/AdipoR2 dual agonist recovers nonalcoholic steatohepatitis and related fibrosis via endoplasmic reticulum-mitochondria axis. Nature communications. 2020;11:5807
36. Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K. et al. The diagnosis and management of non-alcoholic fatty liver disease: practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology (Baltimore, Md). 2012;55:2005-23
37. Liang Z, Chen X, Shi J, Hu H, Xue Y, Ung COL. Efficacy and safety of traditional Chinese medicines for non-alcoholic fatty liver disease: a systematic literature review of randomized controlled trials. Chinese medicine. 2021;16:9
38. Imamura M, Ogawa T, Sasaguri Y, Chayama K, Ueno H. Suppression of macrophage infiltration inhibits activation of hepatic stellate cells and liver fibrogenesis in rats. Gastroenterology. 2005;128:138-46
39. Tomita K, Oike Y, Teratani T, Taguchi T, Noguchi M, Suzuki T. et al. Hepatic AdipoR2 signaling plays a protective role against progression of nonalcoholic steatohepatitis in mice. Hepatology (Baltimore, Md). 2008;48:458-73
40. Farzanegi P, Dana A, Ebrahimpoor Z, Asadi M, Azarbayjani MA. Mechanisms of beneficial effects of exercise training on non-alcoholic fatty liver disease (NAFLD): Roles of oxidative stress and inflammation. European journal of sport science. 2019;19:994-1003
41. Hwang S, He Y, Xiang X, Seo W, Kim SJ, Ma J. et al. Interleukin-22 Ameliorates Neutrophil-Driven Nonalcoholic Steatohepatitis Through Multiple Targets. Hepatology (Baltimore, Md). 2020;72:412-29
42. Polimeni L, Del Ben M, Baratta F, Perri L, Albanese F, Pastori D. et al. Oxidative stress: New insights on the association of non-alcoholic fatty liver disease and atherosclerosis. World journal of hepatology. 2015;7:1325-36
43. Hajighasem A, Farzanegi P, Mazaheri Z. Effects of combined therapy with resveratrol, continuous and interval exercises on apoptosis, oxidative stress, and inflammatory biomarkers in the liver of old rats with non-alcoholic fatty liver disease. Archives of physiology and biochemistry. 2019;125:142-9
44. Irie M, Sohda T, Anan A, Fukunaga A, Takata K, Tanaka T. et al. Reduced Glutathione suppresses Oxidative Stress in Nonalcoholic Fatty Liver Disease. Euroasian journal of hepato-gastroenterology. 2016;6:13-8
45. Ding C, Zhao Y, Shi X, Zhang N, Zu G, Li Z. et al. New insights into salvianolic acid A action: Regulation of the TXNIP/NLRP3 and TXNIP/ChREBP pathways ameliorates HFD-induced NAFLD in rats. Scientific reports. 2016;6:28734
46. Colagar AH, Marzony ET. Ascorbic Acid in human seminal plasma: determination and its relationship to sperm quality. Journal of clinical biochemistry and nutrition. 2009;45:144-9
47. Schimmack G, Defronzo RA, Musi N. AMP-activated protein kinase: Role in metabolism and therapeutic implications. Diabetes, obesity & metabolism. 2006;8:591-602
48. Chyau CC, Wang HF, Zhang WJ, Chen CC, Huang SH, Chang CC. et al. Antrodan Alleviates High-Fat and High-Fructose Diet-Induced Fatty Liver Disease in C57BL/6 Mice Model via AMPK/Sirt1/SREBP-1c/PPARγ Pathway. International journal of molecular sciences. 2020 21
Author contact
![]() Corresponding authors: E-mail address: liman121000edu.cn (Man Li), gaoyueqiucom (Yue-Qiu Gao).
Corresponding authors: E-mail address: liman121000edu.cn (Man Li), gaoyueqiucom (Yue-Qiu Gao).