Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
CD38 in MM
CD38 mAb in MM
Combined therapy based on NK...
Discussion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(5):1974-1988. doi:10.7150/ijbs.68148 This issue Cite
Review
Regulation of CD38 on Multiple Myeloma and NK Cells by Monoclonal Antibodies
Hao-Tian Wu1, Xiang-Yu Zhao1,2 ![]()
1. Peking University People's Hospital, Peking University Institute of Hematology, Beijing Key Laboratory of Hematopoietic Stem Cell Transplantation, National Clinical Research Center for Hematologic Disease, No. 11 South Street of Xizhimen, Xicheng District, Beijing 100044, China.
2. Collaborative Innovation Center of Hematology, China.
Received 2021-10-17; Accepted 2022-1-19; Published 2022-2-21
Abstract

CD38 is highly expressed on multiple myeloma (MM) cells and plays a role in regulating tumor generation and development. CD38 monoclonal antibodies (mAbs) have been used as an effective therapy for MM treatment by various mechanisms, including complement-dependent cytotoxic effects, antibody-dependent cell-mediated cytotoxicity, antibody-dependent cellular phagocytosis, programmed cell death, enzymatic modulation, and immunomodulation. Although CD38 mAbs inhibit the proliferation and survival of MM cells, there are substantial side effects on antitumoral NK cells. The NK-mediated immune response needs to be further evaluated to minimize the adverse effects of NK cell loss. The killing effect of CD38 mAbs on CD38high NK cells should be minimized and the potential combination of CD38low/- NK cells and CD38 mAbs should be maximized to better benefit from their therapeutic efficacy against MM. CD38 mAb effects against MM can be maximized by combination therapies with immunomodulatory imide drugs (IMiDs), proteasome inhibitors (PIs), anti-programmed death 1 (PD-1)/programmed death ligand 1 (PD-L1) antibodies, or cellular therapies for the treatment of MM, especially in patients with relapsed or refractory MM (R/R MM) and drug-resistant MM.
Keywords: multiple myeloma, CD38, monoclonal antibody, NK cells, combination therapy
Introduction
Multiple Myeloma (MM) is hematological cancer with B cell lineage disorder, characterized by the expansion of malignant plasma cells in the bone marrow [1]. Despite the use of high-dose chemotherapy in combination with autologous stem cell transplantation and other emerging new weapons, the prognosis of MM is disappointing. Novel therapeutic approaches have been tested in the last few years including new immunomodulatory drugs (IMiDs), proteasome inhibitors (PIs), and monoclonal antibodies (mAbs) [2, 3].
In 1980, during the pioneering analysis of the human lymphocyte surface, Reinherz and Schlossman discovered CD38, a 45-kDa peptide chain consisting of intracellular, transmembrane, and extracellular domains with two to four N-linked oligosaccharide chains of sialic acid residues [4]. As a transmembrane glycoprotein with receptor-mediated adhesion function and ectoenzymatic activity, CD38 is ubiquitously expressed on MM cells, as well as some regulatory T cells (Tregs) and natural killer (NK) cells [5, 6]. The characteristic high CD38 surface density in MM cells leads to the development of anti-CD38 mAbs [7]. After decades of efforts, the aspiration to accurately target MM cells has begun to bear fruit in 2015, with Food and Drug Administration (FDA) approval of the CD38 mAb, Daratumumab [8]. CD38 mAbs are transforming MM treatment in virtue of their distinct activity as a single agent or in combinations and the manageable toxicity [9].
At the meantime, a relatively high level of CD38 is expressed on NK cells, inducing antibody-dependent cellular cytotoxicity (ADCC) to kill tumor cells, raising a question of whether the CD38 mAbs have a positive or negative impact on NK cells and what strategy should be used to maximize immune effects against MM cells [9].
This review will focus on the tissue distribution and function of CD38, its structure, function, and treatment with CD38 mAbs. We will examine the role of NK cells in MM development and the effects of CD38 mAbs on NK cells. Finally, we will discuss the efficacy of CD38 mAbs in combination with other treatments to maximize their immune response.
CD38 in MM
CD38 distribution
CD38 is present on MM and NK cells, monocytes, and B and T cells, in descending order of CD38 expression level. It can serve as a membrane receptor, with its ligand CD31, to participant in endothelial adhesion. It also triggers signaling as a coreceptor with other membrane molecules including TCR and BCR complexes on lymphocytes and CD16 on NK cells [10]. Besides these peripheral blood mononuclear cells (PBMCs), CD38 is expressed in nonhematopoietic cells as well, including prostatic epithelial cells, airway smooth muscle cells, and corneal suprabasal limbal epithelial cells [11-13]. As a single chain glycoprotein with single transmembrane segment, the CD38 topological membrane orientation differs, in various tissues. In its most common type II orientation, CD38's catalytic domain faces the extracellular environment [14, 15], while in its type III orientation, the catalytic domain faces the cytoplasm [16, 17]. The different orientations in different tissues have functional implications as enzymatic substrates can be consumed and products can be produced in extracellular or intracellular compartments [18].
Multiple myeloma patients have the highest level of CD38 expression (~105) in plasma cells, followed by NK cells (~104). In relapsed or refractory myeloma (R/R MM) patients, the CD38 expression levels are relatively high in NK cells, regulatory B cells (Bregs), Tregs and myeloid-derived suppressor cells (MDSCs) compared with CD8+ and CD4+ T cells, indicating that CD38 is involved in providing the immunosuppressive environment for tumor growth [19].
CD38 as an ectoenzyme promotes MM proliferation
The ectoenzyme activities of CD38 vary considerably, three of which, nicotinamide adenine dinucleotide (NAD)+ hydrolase, adenosine diphosphate ribose cyclase (ADPRC), and cyclic adenosine diphosphate ribose (cADPR) hydrolase, were verified via anti-CD38 monoclonal antibody [20]. The NAD+ hydrolase mediates the conversion of NAD+ to adenosine diphosphate ribose (ADPR). Although CD38 shares a common evolutionary ancestor with ADPRC, their enzymatic functions for the homeostasis of NAD+ and cADPR have evolved divergently. The ADPRC function is limited to generating cADPR from NAD+ (cyclase) while CD38 has multiple roles in the production and degradation of cADPR [21]. CD38 also catalyzes the production of nicotinic acid adenine dinucleotide phosphate (NAADP) from nicotinamide adenine dinucleotide phosphate (NADP)+. Both ADPR and NAADP act as intracellular secondary messengers and play an essential role in Ca2+ influx by activating transient receptor potential melastatin-2 (TRPM2) in immunocytes to regulate many physiological processes [22]. Moreover, the soluble extracellular domain of CD38 has been shown to mediate adenosine diphosphate (ADP) ribosylation of several proteins, including CD38 itself [23].
The NAD+-CD38-cADPR-Ca2+ axis, based on the ectoenzyme activity of CD38, regulates the tumor microenvironment by a series of reactions following Ca2+ flux. The high level of Ca2+ in cytoplasm supports cytoskeleton remodeling, plasma membrane invagination and Warburg effect (Figure 1) [24, 25]. Targeting CD38 in vivo blocks mitochondrial transfer and improves animal survival in mice, demonstrating a consistently reduced tumor burden in bone marrows [26]. It has been shown that the inhibition of CD38 expression could lead to tumor response, vital to achieving tumor control [27].
Schematic representation of CD38 in promoting MM proliferation and preventing MM death. CD38 directs the cyclization of extracellular NAD+ to intracellular cADPR (or ADPR) and triggers Ca2+ release to cytoplasm in MM cells. Intracellular CD38 contributes to a low level of NAD+ in cytoplasm. The high level of Ca2+ supports membrane invagination, cytoskeleton remodeling, and metabolic alteration. The high Ca2+ level in cytoplasm with mitochondria dysfunction and the low intracellular NAD+ level both leads to the Warburg effect. Besides, CD38 can protect MM cells from apoptosis caused by a miRNA, miR-26a. CD38 is also one of the three ectoenzymes in the CD38/CD203a/CD73 pathway that produces ADO, which not only regulates immune response in hypoxic conditions but also suppresses the antitumor immune response by recruiting MDSCs and Treg cells, inhibiting the activity of T effector cells and thus favoring tumor progression.
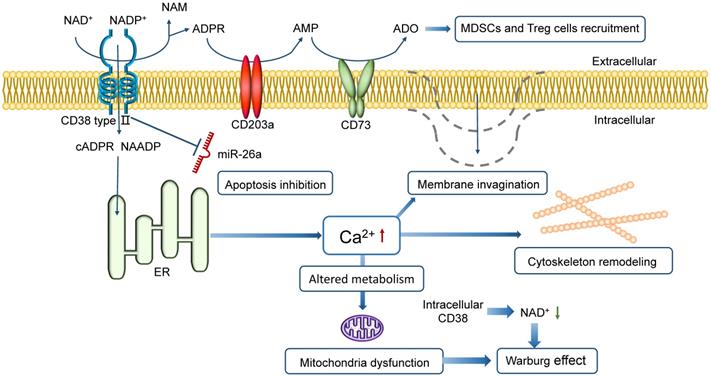
CD38 prevents MM death
CD38 can protect tumor cells from all forms of cell death, including apoptosis, necrosis, and autophagy [28]. It is an inhibitor of apoptosis in MM cells that blocks suppression of cell growth and apoptosis caused by miR-26a, a miRNA that induces apoptosis in MM [29]. Besides, CD38 is one of the three ectoenzymes in the CD38/CD203a/CD73 pathway that produces adenosine (ADO), a nucleoside regulating immune response upon oxygen deficit environment. ADO also restrains the antitumor effect by MDSCs and Treg recruitment, inhibition of the activity of T effector cells and thus tumor progression. A higher expression level of CD38 in the tumor microenvironment is likely to confer a poorer prognosis [30, 31].
CD38 functions in NK cells
NK cells are large granular effector lymphocytes of the innate immune system that can target tumor cells without prior sensitization. The primary mechanism of tumor killing via NK cells involves releasing cytotoxic molecules through a degranulation process. During the degranulation and release, perforin polymerizes and forms pores in the target cell membrane to facilitate granzymes delivery into target cells, inducing apoptosis through different pathways, including caspases-3/7 activation, cytochrome C release from mitochondria driven by Bak/Bax, and IL-1β regulation by caspase-1 [32]. Another mechanism involves activating death receptors of the tumor necrosis factor receptor (TNFR) superfamily which have high expression on target cells. Examples include CD95 (Fas) and TNF-related apoptosis-inducing ligand (TRAIL)-receptor (TRAIL-R) that are activated by their ligands, Fas ligand (FasL) and TRAIL, present on NK cell membranes or secreted by NK-derived exosomes. The oligomerized death receptors recruit Fas-associated death domain (FADD), which binds procaspase-8, allowing the tumor killing activation and spurring apoptotic pathways [33]. Also, NK cells are armed with functional Fcγ receptor (FcγR) III/CD16. Thus ADCC is another pathway to kill target cells (Figure 2a) [34].
Schematic mechanism representation of cytolytic functions in NK cells. a) NK cells kill MM cells through perforin and granzymes release, death receptors activation and ADCC. The perforin and granzymes induce apoptotic cell death by different pathways, such as caspases-3/7 activation, cytochrome C release from mitochondria and IL-1β regulation by caspase-1. Another mechanism involves activating death receptors of TNFR superfamily expressed on target cells, which binds procaspase-8 after activation of their ligands on NK cells, allowing its activation and triggering apoptotic pathways. ADCC is another pathway to kill the target cells, especially when patients receive specific mAbs treatment. b) The cytotoxicity is dependent on the expression of activating and inhibitory receptors on NK cell membrane. They are engaged by specific ligands on target cells. The inhibitory ligands HLA-I are widely expressed on most healthy cells and the he inhibitory signal predominate the response in a healthy cell with a low quantity of activating ligands, blocking cell lysis. However, MM cells often downregulate HLA-I and allow effector NK cells to be activated by ITAMs in their cytoplasmic domains, resulting in the production of cytokines, granzyme B, and perforins and leading to tumor cell killing. Also, a strong capacity of ADCC is able to overcome these inhibitory signals.
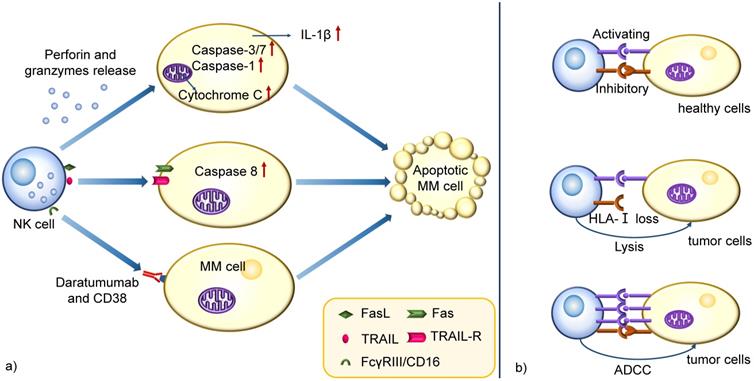
When an NK cell encounters a cell, it does not necessarily induce cell killing. Instead, the cytotoxicity relies on the expression of activating and inhibitory receptors on NK cell membrane, engaged by specific ligands expressed on target cells. Activating receptors include KIR2DS1, KIR3DS1, and natural cytotoxicity receptors (NCRs) and inhibitory receptors include KIR2DL1, KIR3DL1, and NKG2A [35].
It is known that almost all of the healthy cells in human body express the inhibitory ligands human leukocyte antigen class I (HLA-I). The inhibitory signal will predominate the response in a healthy cell with a low quantity of activating ligands, blocking cell lysis [36]. However, tumor cells often downregulate HLA-I and allow effector NK cells to be activated by immunoreceptor tyrosine-based activation motifs (ITAMs) in intracellular domains, resulting in the release of cytokines, perforins and granzyme B and leading to tumor cell death. A strong capacity of ADCC is also able to overcome these inhibitory signals. (Figure 2b) [37, 38].
The resistance of MM cells to NK-mediated lysis is mainly associated with HLA-I-dependent mechanism. A high level of HLA-I on MM cell membranes may be involved in protecting these cells from NK-mediated attack and contribute to the immune evasion [39].
It is the loss of NK functional abilities rather than decreased numbers that results in monoclonal gammopathy of undetermined significance (MGUS) for MM progression [40]. The malignant progression is accompanied with cytotoxicity decrease in NK cells and conversion to exhaustion due to changes in NK receptors, for example, the downregulation of NKG2D, NKp30, and DNAX accessory molecule-1 (DNAM-1) receptors. Additionally, a higher expression of programmed death 1 (PD-1) on NK membranes and the binding to its ligand on MM cells contribute to the impaired immune response and MM progression [41].
The early phases of hematopoietic differentiation and commitment towards the common lymphoid progenitor stage require CD38 expression together with other specific markers [42]. In later stages of immuno-hematopoietic cells, some differences exist in the CD38 expression pattern. While some of mature cells start to decrease the expression of CD38, the majority of NK cells constitutively express CD38 [43]. It is physically or functionally clustered on these so-called conventional NK (cNK) cells with high level of CD38 as well as low-affinity receptor Fc RIII/CD16 [44-46]. The interaction between CD38 and some particular molecules is needed for initiation of specific transcription, secretion of cytokines, and activation of lymphocytic effector functions [45, 46]. For conventional CD16+ NK cells, the association between CD38 and CD16 promotes the CD38-mediated activation of intracellular signaling pathways, resulting in the initiation of Ca2+ flux, zeta-chain-associated protein kinase 70 (ZAP70) tyrosine phosphorylation, mitogen-activated protein kinase (MAPK) activation inducing IFN-γ secretion and cytotoxicity crucial for the immune response [47]. The interleukin-2 (IL-2)-dependent protein expression and CD38 association with CD16 on CD16+ NK cells are also central for the effector cytotoxic phenotype of lytic machinery [43]. Furthermore, CD38 in CD16- NK cells mediate the production of adenosine, inducing the inhibition of CD4+ T cells that can be reverted by blockade with CD38 antibodies (Figure 3) [48]. It is believed that the knockdown of CD38 mediates a significant change in aerobic metabolism, including mitochondrial respiratory capacity enhancement, NAD+ boosting and Ca2+ transport. Besides, recent studies also suggest that it induces a long-term persistence of memory-like cells [49, 50].
Mechanism of action of CD38 in CD38high NK cell. In CD16+ NK cells, the association with CD16 is instrumental for the CD38-mediated intracellular signaling events. The high level of Ca2+ induces ZAP70 tyrosine phosphorylation, MAPK activation, and IFN-γ secretion. While in CD16- NK cells, the main function of CD38 is on the membrane. It mediates the production of adenosine, which induces the inhibition of CD4+ T cells, bringing the negative effect in immune response.
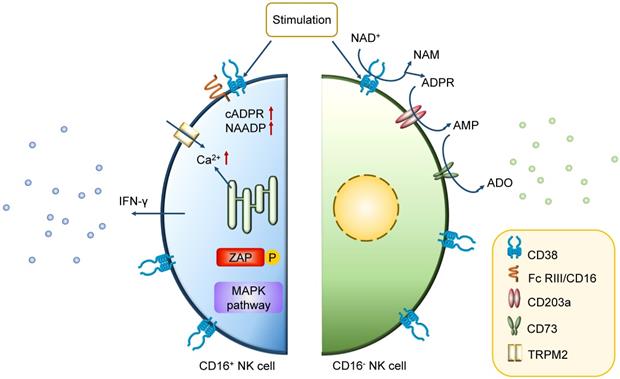
Recently, an outstanding subset of NK cells (g-NK cells) have entered our vision. They account for 3~10% of NK cells in healthy samples and vary from the conventional NK cells by a lack of FcεRIγ expression [51]. While encountering Cytomegalovirus (CMV) infections, the proportion of g-NK cells will increase to 25~30% [52]. Further studies have indicated that g-NK cells are persistent memory-like cells (4~9 months). These g-NK cells have a minimal level of CD38 on surface [51]. Although CD38 is vital in cNK cells, it is not a necessary property during immune response such as ADCC against MM. Because of the defect in expression of activation markers including CD38, NKp46 and NKp30, g-NK cells act as memory-like cells, with less IFN-γ secretion than activated cNK cells, but have stronger antitumor potential than cNK cells after CD16 crosslinking [53].
CD38 mAb in MM
Structure of CD38 mAbs
Daratumumab and Isatuximab are conventional CD38-specific mAbs, both derived from human CD38-immunized mice. Daratumumab was generated from gene-altered mice carrying genomic loci encoding human IgH and IgL, while Isatuximab was obtained from wild-type mice [54]. Daratumumab has been approved for monotherapy use [55]. The chimeric antibody was produced by genetic fusion of the VH and VL domains of the mouse monoclonal antibody to human IgG1 and kappa domains, respectively [56].
A constrained peptide approach showed that the epitope of Daratumumab was targeted to two β-strands containing amino acids 233~246 and 267~280 of CD38, and the specific amino acid residues were identified, in particular serine at position 274. The Daratumumab epitope on CD38 was speculated to offer a favorable structural arrangement for hexamer formation, allowing efficient C1q docking and initiation of the classical complement pathway [57].
CD38 mAbs induce complement dependent cytotoxicity
As the most effective inducer of complement dependent cytotoxicity (CDC), Daratumumab is currently believed as one of the most important mechanisms of MM cell killing in the clinic, efficiently inducing high levels of CDC at a low concentration in bone marrow microenvironment [58, 59]. Therefore, Daratumumab emerged as the preferred CD38 mAb in the clinical treatment of MM [59, 60].
The classical CDC pathway starts with a binding of CD38-CD38 mAb complex and the classic complement-activating protein C1q. It leads to the generation of anaphylatoxins (C3a, C4a, and C5a), chemo-attractants, and opsonins. Additionally, terminal complement pathway activation generates membrane attack complexes (MAC), which build pores in the MM cell membrane, resulting in direct cell lysis [61, 62]. The Daratumumab-mediated CDC depends on CD38 expression level, and a high CD38 expression on MM cell membranes strongly improves CDC activity of Daratumumab [63].
CD38 mAbs induce ADCC
In the presence of PBMCs enriched for many NK cells, Ab-dependent killing via FcR+ effector cell is also considered a vital mechanism for immune therapy [64, 65]. The 51Cr-release assay evaluates the capacity of mAb-induced effector cell-dependent lysis of MM cells [66] and provides evidence that the lysis is dose-dependent and effective in the presence of bone marrow-derived mesenchymal stem/stromal cells (BMSCs) and in patient-derived PBMCs [59].
It has been reported that CD32c and CD16 are the key FcγRs on NK cells and CD64 is mainly present on macrophages, DC cells, neutrophils, and eosinophils [67]. MM cell death occurs via NK cell degranulation and release of perforins and granzymes and via activation of receptors inducing MM cell apoptosis [68, 69]. The secretion of pro-inflammatory cytokines, such as interferon-γ (IFN-γ), TNFα, and granulocyte-macrophage colony-stimulating factor (GM-CSF), is triggered by CD16 engagement, contributing to the recruitment of adaptive immune cells [70]. However, the ADCC lysis is reduced following Daratumumab treatment, probably due to decreased NK cell numbers. In most patients, PBMCs isolated during CD38 mAb treatment can still induce some level of ADCC, indicating that the remaining group of NK cells maintain cytotoxic functionality [71]. ADCC is widely observed in CD38 mAb treatment and is the main mechanism of Isatuximab-induced tumor cell death in several lines of experiments. Its efficiency depends on CD38 expression [63, 72].
CD38 mAbs induce macrophage-mediated phagocytosis
Human macrophage-mediated antibody-dependent cellular phagocytosis (ADCP) is another efficient killing mechanism of Daratumumab by rapid and sequential engulfing of multiple MM cells [54]. The percentage of macrophages and the number of remaining MM cells are the main indicators for estimating the ADCP level of MM cells [54]. Although ADCP-induced phagocytosis is related to CD38 level on targeted cells, differences in CD38 expression levels of different target cell lines do not appear to be a convincible explanation for the differences in phagocytosis-mediated elimination [73].
Phagocytosis is suppressed via CD47/SIRPα activation. CD47 on MM cells can bind to SIRPα on macrophages, resulting in an inhibition of phagocytosis [74]. Recently, the neutralization of CD47 by its antibody has been shown to enhance ADCP of MM cells mediated by CD38 mAb [75-77]. Also, low-dose cyclophosphamide is used to improve Daratumumab-mediated ADCP against tumor cells, probably mediated by increased expression of FcγR on macrophages and reduced CD47 levels on tumor cells [78]. Moreover, it is also partly dependent on the ratio of monocytes to MM cells [63].
CD38 mAbs induce programmed cell death
Programmed cell death (PCD) mediated by Daratumumab has been observed in MM cells [79]. In CD38+ MM cell lines, PCD is induced by activating FcγRI-expressing cells cross-linking Daratumumab [79]. During cell death, phosphatidylserine translocation is a reversible process and initiation marker, whereas damage of the mitochondrial membrane potential and cell membrane integrity are irreversible [79]. Another anti-CD38 mAb, Isatuximab, has also shown significant antitumor activity by a more potent action of direct cell killing different from crosslinking, mediated by activation of caspases-7 and 8, lysosome permeabilization, upregulation of reactive oxygen species (ROS), as well as ADCC, CDC, and ADCP mentioned above [80, 81].
CD38 mAbs modulate enzymatic functions
Daratumumab mainly inhibits the cyclase activity and hydrolyzes cADPR in vitro, increasing NAD and ADPR levels, decreasing Ca2+ mobilization, and reducing signaling in MM cells, all of which induce cell death [57]. However, CD38 on tumor cells displays more NAD+ hydrolase than ADPRC activity. Thus, besides Daratumumab, a better CD38 mAb is required to inhibit the NAD+ hydrolase activity of CD38+ MM cells [82].
Another CD38 mAb, Isatuximab, almost completely inhibits the enzymatic activity of recombinant CD38 (5 nmol/L) at concentrations of 20~200 nmol/L, including its hydrolase activity [83]. CD38 is also suggested as the dominant enzyme for ADO generation in MM cells, alleviating an immunosuppressive microenvironment in MM patients. However, the certain evidence whether the inhibition of ADO-mediated signaling pathway has a direct impact on anti-MM activities of CD38 mAbs is still confusing because of the technical challenges of direct measurement of ADO level in patients [84, 85].
CD38 mAbs induce immunomodulation
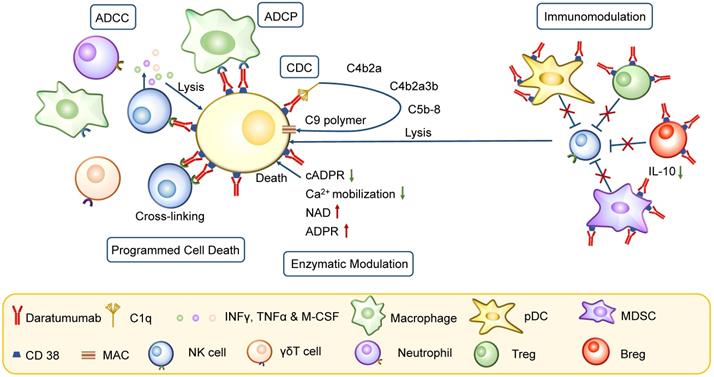
CD38 is widely expressed on immune cells and tumor cells. The CD38+ plasmacytoid dendritic cells (pDCs) confer a growth and survival advantage to MM cells by decreasing the pDC antitumor function [86]. Daratumumab induces progressive depletion of pDCs and prevents upregulated programmed death ligand 1 (PD-L1) expression on APCs to improve the disease response [87]. Besides pDCs, immune dysfunction in MM is also mediated by MDSCs, Tregs, and Bregs. The Tregs can be divided into 2 subpopulations, CD38+ Tregs that make up approximately 90% of the total Treg population and are more potent in suppressing T cell proliferation, and CD38- Tregs. Daratumumab eliminates the subset of CD38+ Tregs, while Tregs without CD38 expression are not affected [5]. MDSCs are also effectively killed by Daratumumab, as observed in CDC and ADCC assays [5]. Bregs also contribute to an immunosuppressive microenvironment by producing and releasing IL-10 and abrogating NK cell-mediated ADCC [88]. Since CD38 is present on these immunosuppressive cells, Daratumumab significantly induces immunomodulation during response against MM [88]. Daratumumab causes depletion of CD38+ Tregs, Bregs, and MDSCs and an increase in clonality and effector functions of T helper and T cytotoxic cells [89]. Isatuximab causes preferential depletion of Tregs by causing apoptosis, inhibiting cell proliferation, and relieving conventional T cells from suppression, thereby inducing immunomodulation in MM patients (Figure 4) [80, 81].
Mechanism of action of CD38 mAbs. CD38 mAb Daratumumab has pleiotropic mechanisms of action, including Fc-dependent immune-effector mechanisms, programmed cell death, enzymatic modulation and immunomodulation. The Fc-dependent immune-effector mechanisms consist of CDC, ADCC and ADCP. CDC is initiated by binding of C1q, leading to the activation of downstream complement proteins and resulting in assembly of the membrane attack complex (MAC); ADCC is achieved through the activation of FcRs on NK cells and other cells including neutrophils, macrophages and γδT cells; ADCP is mediated by macrophages and other immune cells, following interaction of the Fc frament of Daratumumab with FcRs on effector cells. Besides, PCD is activated by activating FcγRI-expressing cells cross-linking Daratumumab. In enzyme modulation, the reduction in cADPR and Ca2+ mobilization and the increase of NAD induce cell death. The immune dysfunction in MM is mediated by pDC, Tregs, MDSCs and Bregs. CD38 mAbs cause a depletion of these cells, which is crucial for effective immune response.
Effects of CD38 mAbs on NK cells
Although the CD38 mAbs have positive effects on MM cells, they can also interfere with immune cell populations, and their effect on NK cells is unclear and controversial [90].
CD38 mAbs are believed to cause an impaired immune response, especially the response mediated by NK cells. Depletion of CD38high NK cells has been observed in patients treated with Daratumumab [91]. Studies have shown that Daratumumab triggers a dose-dependent NK cell apoptosis, resulting in a significant reduction in primary NK cells. A strong increase in NK cell degranulation has been observed without target MM cells, indicating that Daratumumab may induce NK cell apoptosis by NK cell fratricide or NK-mediated cytotoxicity by ADCC [5, 92]. The 51Cr release assay and flow cytometry-based cytotoxicity assay indicated the existence of NK cells self-lyse, and Daratumumab integrity is necessary for the cytotoxicity [91]. However, other observations suggest that the residual CD38low/- NK population displays a high proliferative potential and functional activity. Therefore, CD38low/- NK cells play a significant role in the presence of CD38 mAb by inducing ADCC in the stressed tumor microenvironmental condition [93]. One of the uncertainties to predict the outcome of CD38 mAbs treatment is the g-NK cell. This subset of CD38low/- NK cells markedly enhances ADCC against MM cells during the CD38 mAb treatment and have longer persistence than CD38high cNK cells [94]. These two observations are difficult to reconcile, since both processes occur in a single MM patient. Whether their heterogeneity and different response levels to CD38 mAb in patients contribute to different outcomes following Daratumumab therapy requires more evidence (Figure 5) [55].
Effects of CD38 mAbs on CD38high and CD38low/- NK cells. CD38 mAbs are believed to cause an impaired immune response mediated by NK cells. Since CD38 is highly expressed on the majority of NK cells, Daratumumab induces NK cell apoptosis by fratricide or NK-mediated cytotoxicity by ADCC. A large quantity (85~90%) of CD38high NK cells are killed. However, the residual CD38low/- NK population displays a high proliferative potential and functional activity. The memory-like g-NK cells are activated after CD16 crosslinking. The effector CD38low/- NK cells show a high proliferative potential and functional activity in the presence of CD38 mAb by inducing ADCC in the stressed tumor microenvironmental condition.
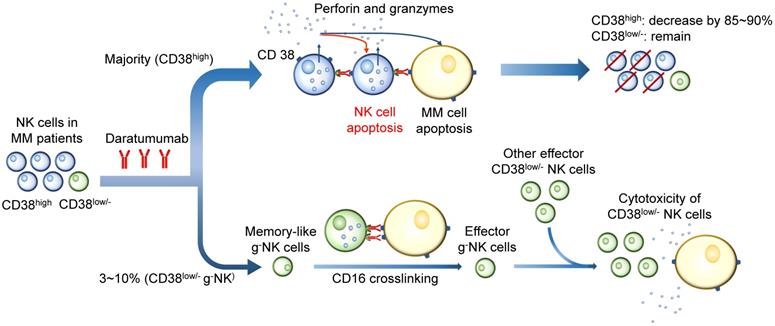
As discussed above, NK cells play a significant role in antitumor response in MM patients. There are consequently several combined therapies based on NK cells, trying to increase antitumor efficacy via variant ways, including regulating NK cell ligands on MM cells, increasing the number and activity of effector NK cells and better utilizing the antitumor effect of CD38low/- NK cells.
Combined therapy based on NK cells
CD38 mAb and immunomodulatory imide drugs
The combined therapy of CD38 mAbs with immunomodulatory imide drugs shows excellent performance in relapsed or refractory MM patients. The clinical use of IMiDs in MM patients has significantly improved their long-term survival and life quality. Thalidomide is widely used in newly diagnosed, relapsed, or refractory MM, especially as maintenance therapy after autologous stem cell transplantation (ASCT). Lenalidomide is a second-generation IMiD with fewer side effects in older patients than the first generation of IMiDs. Pomalidomide is a third-generation IMiD that is 10 times more potent than Lenalidomide and has shown impressive results in relapsed or refractory patients after Lenalidomide and Bortezomib therapy [95].
In the combined therapy, IMiDs increase the direct toxic effects of CD38 mAbs, including lysosomal-mediated apoptotic pathways, and increase CD38 expression on Treg cells, resulting in enhanced elimination of Treg cells [96, 97]. Also, preclinical data show that IMiDs synergize with Daratumumab in patients with IMiD-resistant tumors, indicating that their immune system still responds to the immunomodulatory effects of these agents [98].
During IMiD treatment, the transcription factors Ikaros (IKZF1) and Aiolos (IKZF3) are degraded. A recent study has found that CD38 mRNA expression on MM cells is increased on Ikaros and Aiolos degradation after Lenalidomide treatment. Thus, an alternate cell-intrinsic hypothesis might explain the immunoenhancing effect of CD38 mAbs combined with IMiDs. It is inferred that the transcriptional activation of interferon-stimulated genes (ISGs), including CD38, results from signal transduction after IFN-α/β binding to the type 1 interferon (IFN) receptor, activation of janus kinase 1 (JAK1), signal transducers and activators of transcription 1/2 (STAT1/2) and interferon regulatory factor 9 (IRF9), assembly of the IFN-stimulated gene factor 3 (ISGF3) complexes that translocate to the nucleus and bind to IFN-stimulated response elements [99]. Despite this, the CD38 expression on activated NK cells is not altered by Lenalidomide treatment despite a similar reduction in Ikaros, indicating that this process is specific to MM cells compared to immune cells [100].
Although CD38high NK cell population is reduced by CD38 mAb, it brings few destructive effects to the efficacy and safety during combined therapy, because IMiDs, such as Lenalidomide, markedly enhance NK cell-mediated ADCC by increasing the number and activity of activated CD16+ NK cells [71, 98, 101]. In addition, IMiDs also enhance CD38 mAb-mediated ADCP by promoting the tumoricidal activity of macrophages [102]. The IMiD-mediated recovery of CD16+ NK cells makes the combined therapy possibly a good choice to conquer the negative affect of reduction of CD38high NK cells in CD38 mAbs (Figure 6a) treatment [103].
Combined therapy against MM based on NK cells. a) CD38 mAbs Daratumumab with IMiDs Lenalidomide: CD38 expression in MM cells is inhibited by transcription factors Ikaros (IKZF1) and Aiolos (IKZF3). IMiD reduces the IKZF1/3 and increases the CD38, which is benefit for Daratumumab treatment. The combined therapy markedly enhances NK cell-mediated ADCC by increasing the number and activity of NK cells and eliminating Tregs. It can also promote macrophage-mediated ADCP. b) Daratumumab with PIs Bortezomib: An increasing level of DR5 and decreasing level of HLA-E enhance NK-mediated ADCC and reduce immune escape. However, the combination is associated with infusion-related reactions and accounts for risks of thrombocytopenia and neutropenia. c) Daratumumab with PD-1/PD-L1 mAb: The downregulation of PD-L1/PD-1 axis in MM cells and the activation of ADCC in NK cells show potential on durable control in drug-resistant MM. d) Daratumumab with NK cellular therapy: The combination aims to enhance the tumoricidal effect of CD38low/- NK cells. KHYG1 and g- NK cells are CD38low/- NK cells which prove to be effective to kill MM cells by ADCC. G-NK cells are also able to reduce the immune escape because of its low level of NKG2A expression. Besides, the CD38low/- NK cells engineered by CRISPR/Cas9 system is another method to maintain NK cell function during Daratumumab therapy.
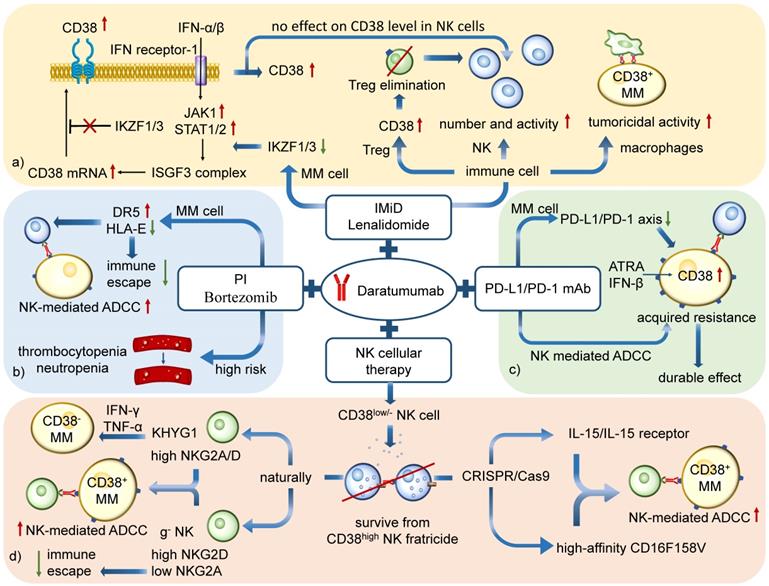
CD38 mAb and proteasome inhibitors
The combination of Daratumumab with proteasome inhibitors is beneficial in patients, including those over 65 years old and those with prior PI Bortezomib exposure. Bortezomib sensitizes MM cells to NK cells via upregulation of death receptor 5 (DR5) and selective reduction of HLA-E on MM cells, reducing the HLA-E-mediated immune escape in MM patients [104]. It is believed that PIs enhance the therapeutic efficacy of Daratumumab by sensitizing tumor cells for NK-mediated ADCC and improving MM cell lysis. The rate of overall response, minimal residual disease-free survival and progression-free survival rate are significantly higher in relapsed and refractory MM patients receiving combined therapy than those treated with PIs only [105, 106]. While the clinical evidence strongly suggests that the outcome of combined therapy is better than single drug therapy, there are few researches paying attention to the clinical efficacy in patients with different proportion of CD38high and CD38low/- NK cells.
However, it is of note that the combined therapy is associated with infusion-related reactions and accounts for higher rates of thrombocytopenia and neutropenia than treatment with bortezomib or dexamethasone alone, which may cause harmful effects to patients (Figure 6b) [107].
CD38 mAb and PD-L1/PD-1 mAb
PD-1 is a 288 aa type I transmembrane protein and belongs to the CD28 receptor family. PD-L1, also known as B7-H1 and CD274, is a 40kDa type I transmembrane glycoprotein. The engagement of the PD-1 receptor with PD-L1 activates PD-1 downstream from Src homology region 2 domain-containing protein tyrosine phosphatase (SHP-2) and dephosphorylates ZAP70, leading to the inhibition of T cell survival, proliferation and cytokine production, inducing T-cell exhaustion; it also promotes Tregs development and decreases NK cell cytotoxicity, granule exocytosis, and IFN-γ secretion through the pathway by protein kinase C (PKC), phosphoinositide 3-kinase (PI3K) , extracellular-signal-regulated kinase (ERK), and protein kinase B (AKT) activation [108, 109]. PD-L1 expression plays a dominant role in MM cells in immune inhibition and immune escape [110-112].
The anti-PD-1/PD-L1 antibody inhibits the negative impact of the PD-L1/PD-1 axis on treatment. However, previous data demonstrated that the tumor growth suppression and metastases by anti-PD-L1 antibody treatment lacked a complete durable response [110, 113], suggesting the existence of resistance mechanisms. Studies have demonstrated that the resistance indeed developed progressively during 5~7 weeks of PD-L1/PD-1 antibody treatment. Among the top 200 differentially-expressed genes, CD38 was identified as the only prominently upregulated gene/protein [114]. The level of CD38 mRNA and protein was significantly increased on anti-PD-L1 resistant tumor cells. The hypothesis for PD-1/PD-L1 blockade focuses on the infiltration of activated T cells and inflammatory changes that lead to all-trans-Retinoic acid (ATRA)- and IFN-β-mediated CD38 upregulation, which mediates immunosuppression via adenosine production and its effect on CD8+ cytotoxic T cells [114].
Interestingly, it has also been shown that increased expression of the PD-1/PD-L1 axis suppresses the activity of NK cells, the CD38 mAbs-mediated ADCC [115]. CD38 mAb Isatuximab, but not Daratumumab, blocks CD38 enzymatic activity of adenosine production [72]. Thus, the combined therapy of two kinds of antibodies is beneficial to MM patients, especially those who received anti-PD-1/PD-L1 antibodies and developed acquired resistance [114, 116]. Also, pre-treatment with clinically relevant agents such as ATRA and Panobinostat could be used to enhance CD38 expression. Preclinical studies to evaluate this treatment are in Phase Ⅰ trials [63, 117, 118]. Recently researches have confirmed a better Daratumumab-mediated cytotoxicity when a PD-L1/PD-1 mAb is added to single Daratumumab treatment, showing another advantage for the combined therapy [119]. The future is promising for this therapy (Figure 6c).
CD38 mAb and NK cellular therapy
CD38 plays a significant role in NK cell function by facilitating ADP-ribose pyrophosphate (ADRP) production and mobilization of intracellular Ca2+ necessary for cytolytic degranulation. However, the treatment with Daratumumab leads to a rapid elimination (85~90%) of CD38+ NK cells, lasting up to 6 months after the treatment [94, 120]. Since CD38low NK cells display cytotoxic superiority in NK-mediated antitumor response, their efficacy should be maximized [91].
It is reported that different NK cell subsets differ in the level of Killer-cell immunoglobulin-like receptors (KIR) surface expression that has a negative impact on Daratumumab-mediated ADCC [93]. Compared with primary expanded NK and NK-92 cells with high expression of CD38, KHYG1 cells have significantly lower CD38 surface expression. It is believed that low CD38 expression is enough to initiate the intracellular signaling pathways since other receptors, such as NKG2D on KHYG1 cells, can compensate for the lack of CD38-mediated effector functions [63]. A study on three different NK cell lines suggests that compared to primary expanded cells with high level of KIR2DL1, KIR2DL2/3, and KIR3DL1, the NK-92 and KHYG1 NK cell subsets have low cell surface expression of the inhibitory receptors but a high proportion of NKG2A, important for NK cell education. Besides, KHYG1 expresses a natural variant of low-affinity CD16, which is called CD16F158V. It is a high-affinity receptor which markedly enhances the tumoricidal immune response in the CD38low/- NK cells [121]. It is also shown that KHYG1 NK cells can be used in combination with CD38 mAb to specifically target and kill CD38high and even CD38low MM cells in vitro, possibly by releasing IFN-γ and TNF-α [122]. Therefore, KHYG1 NK cells could be considered as an NK cell pool with low CD38 expression that can survive from Daratumumab-mediated NK cell decrease while still capable of mounting a cytotoxic response against MM target cells. Furthermore, given that the patients with high pre-treatment CD38 levels on MM cells have a better response to Daratumumab monotherapy, high CD38 expression on MM cells seems satisfactory to maximize the clinical efficacy such as the combination treatment of CD16F158V CD38low NK cells and Daratumumab [120].
The natural CD38low NK cells account for a small fraction of blood NK cells and their generation in high numbers is clinically challenging. However, the CRISPR/Cas9 technology to delete CD38 on NK cells (CD38KO NK) can be used to enhance the capacity of NK-mediated ADCC during CD38 mAb therapy. These CD38KO NK cells are resistant to fratricide, showing superior persistence in immune-deficient mice pretreated with CD38 antibodies and enhancing ADCC activity against CD38+ MM cell lines and primary MM cells. To maximize the CD38 mAb-mediated ADCC, the combination of CD38 mAb and CD38KO NK maintains NK cell function during Daratumumab therapy. This combined treatment has other benefits, including changes in aerobic metabolism with higher mitochondrial respiratory capacity of CD38KO NK cells. These findings have demonstrated that adoptive immunotherapy using ex vivo expanded CD38KO NK cells can boost Daratumumab activity in MM [90].
FT538 is the first multiplexed engineered NK cell therapy generated by CRISPR/Cas9. Its high-affinity CD16F158V and IL-15/IL-15 receptor fusion promote a high level of the immune response, and the CD38KO property makes it possible to mitigate NK cell fratricide by CD38 mAb. FT538 therapy combined with Daratumumab against MM cells demonstrates a more effective MM control than Daratumumab therapy alone [123]. Recently a novel off-the-shelf product FT576 has come into our sight. FT576 comprises an anti-BCMA CAR, a high-affinity non-cleavable CD16, CD38 knockdown and IL-15/IL-15 receptor and has proved competent for ADCC against MM cells.
Selected ongoing and planned clinical trials on CD38 MoAb monotherapies and combined therapies in multiple myeloma
| Study | Phase | Patient | Treatment |
|---|---|---|---|
| NCT04151667 | 2 | NDMM in older adults (>65 years old) | IberDd vs DRd vs DVd |
| NCT03901963 | 3 | NDMM with minimal residual disease positive after frontline ASCT | Daratumumab and Lenalidomide vs Lenalidomide alone |
| NCT02316106 | 2 | SMM | Single agent 3 Daratumumab dose schedules (long, intermediate and short treatment duration) |
| NCT04975997 | 3 | RRMM | IberDd vs DVd |
| NCT02807454 | 2 | RRMM | Daratumumab and Durvalumab (PD-L1 MoAb) vs Pomalidomide, Daratumumab, Durvalumab and Dexamethasone |
| NCT02076009 | 3 | RRMM | DRd vs Lenalidomide and Dexamethasone |
| NCT04614636 | 1 | RRMM | FT538 monotherapy vs FT538 and Daratumumab vs FT538 and Elotuzumab (SLAMF7 MoAb) |
| NCT04700176 | 2 | MM Previously exposed to Daratumumab-Based Regimens | Progressed on DRd to be treated with DPd and ATRA vs progressed on DPd to be treated with DPd and ATRA |
IberDd, Daratumumab and Dexamethasone; DVd, Daratumumab, Bortezomib, and Dexamethasone; DRd, Daratumumab, Lenalidomide and Dexamethasone; DPd, Daratumumab, Pomalidomide, and Dexamethasone; RRMM, relapsed or refractory multiple myeloma; ATRA, all-trans retinoic acid; NDMM, newly diagnosed multiple myeloma; SMM, smoldering multiple myeloma; ASCT, autologous stem cell transplant.
It shows great capacity of immune response of CD38-deficient g-NK cells in MM. In addition to Fate's work with the FT538 and FT576 products, Indapta Therapeutics has shown that CD38-deficient g-NK cells can be preferentially expanded which could produce high efficacy in combination with daratumumab as an anti-myeloma product without resorting to genetic engineering [94]. The g-NK cell expresses infrequent NKG2A, upregulated NKG2C and elevated level of inhibitor of apoptosis proteins such as Bcl-2 [53, 94]. Compared with the performance of cNK cells in Daratumumab therapy, g-NK shows a greater CD38 mAb-induced cytotoxicity and improved persistence. The increased expression of perforin, granzyme, CD107a, IFN-γ and TNF-α in g-NK cells with the presence of Daratumumab are responsible for such phenomena [53]. The great level of ADCC induced by Daratumumab with accumulation of activating signals could overcome the inhibitory effects even when the NK cells are not HLA-mismatched. Besides, the cytolytic enzymes in g-NK cells are activated, inducing MM cell apoptosis after CD16 crosslinking. Furthermore, a great increase in the number of g-NK cells is observed in the mice receiving Daratumumab treatment, which is 10 times higher than that of cNK cells in peripheral blood [94].
G-NK cells also prevent immune evasion of MM cells. Some MM cell lines escape from immune system by upregulation of surficial HLA-E. HLA-E could inhibit the antitumor activity of IL2-activated NK cells by binding with NKG2A. Thus, the lack of NKG2A on g-NK cells could cut off the HLA-E evasion [124, 125]. G-NK cells share similar benefits in aerobic metabolism reprograming with CD38KO cells as well. Meanwhile, it shows great capacity of immune response in MM compared to KHYG1 cells for its potential against HLA-E mediated immune evasion. Since its potential efficacy and stable persistence, g-NK cells could be an excellent off-the-shelf product against R/R MM (Figure 6d) [94].
Discussion
CD38 mAb, with its impressive single-agent activity to kill MM cells, has attracted increasing attention. However, the outcome of MM patients with Daratumumab is uncertain, partly due to its confounding effects on NK cell lines, such as the clearance of CD38high NK cells and ADCC mainly mediated by NK cells with low or no CD38 membrane expression. The heterogeneity of NK cells with respect to quantity and functional changes might be related to the different outcomes in MM patients. Therefore, therapies combined with other treatments are considered helpful to maximize CD38 mAb effects against MM.
The combination of the CD38 mAb with an IMiD remarkably enhances NK cell-mediated ADCC by increasing the quantity and activity of NK cells, promoting the tumoricidal activity of macrophages, and enhancing CD38 mAb-mediated ADCP. Besides, another therapy combined with PIs improves overall response and progression-free survival among relapsed and refractory MM patients. But we have to pay attention to its infusion-related reactions and accounts for higher risk of thrombocytopenia and neutropenia. Combining the two antibodies, anti CD38 and anti PD-L1/PD-1, is beneficial for MM patients resistant to monotherapy. Furthermore, the cellular therapy with KHYG1, CD38KO NK cells or g-NK cells protects the immune activity of NK cells by upregulating the quantity or activity of CD38low/- NK cells and preventing the HLA-E-mediated immune evasion to maximize the effects of CD38 mAb against MM.
CD38 mAb is a milestone in MM treatment and the future of combined therapy will be more promising if more in-depth studies are conducted. For instance, there is no effective method to select patients who will have a successful outcome in the long term after CD38 mAb therapy and exclude those who will not benefit from the treatment. The management guidance on precision medicine for different patients is still lacking. Guidelines for various treatments for different groups of patients with relapsed or refractory MM and drug-resistant MM, including single-agent and combined therapies, are needed to set clinical standards and reduce the economic burden on patients.
Abbreviations
MM: multiple myeloma; mAbs: monoclonal antibodies; IMiDs: immunomodulatory imide drugs; PIs: proteasome inhibitors; PD-1: programmed death 1; PD-L1: programmed death ligand 1; R/R MM: relapsed or refractory multiple myeloma; Tregs: regulatory T cells; NK: natural killer; FDA: Food and Drug Administration; ADCC: antibody-dependent cellular cytotoxicity; PBMCs: peripheral blood mononuclear cells; Bregs: regulatory B cells; MDSCs: myeloid-derived suppressor cells; NAD: nicotinamide adenine dinucleotide; ADPRC: adenosine diphosphate ribose cyclase; cADPR: cyclic adenosine diphosphate ribose; ADPR: adenosine diphosphate ribose; NAADP: Nicotinic acid adenine dinucleotide phosphate; NADP: nicotinamide adenine dinucleotide phosphate; TRPM2: transient receptor potential melastatin-2; ADP: adenosine diphosphate; ADO: adenosine; TCA: tricarboxylic acid; TNFR: tumor necrosis factor receptor; TRAIL: TNF-related apoptosis-inducing ligand; TRAIL-R: TNF-related apoptosis-inducing ligand-receptor; FasL: Fas ligand; FADD: Fas-associated death domain; NCRs: natural cytotoxicity receptors; FcγR: Fcγ receptor; HLA-I: human leukocyte antigen class I; ITAMs: immunoreceptor tyrosine-based activation motifs; MGUS: monoclonal gammopathy of undetermined significance; DNAM-1: DNAX accessory molecule-1; cNK: conventional NK; ZAP70: zeta-chain-associated protein kinase 70; MAPK: mitogen-activated protein kinase; IL-2: interleukin-2; CMV: Cytomegalovirus; CDC: complement dependent cytotoxicity; MAC: membrane attack complexes; BMSCs: bone marrow-derived mesenchymal stem/stromal cells; IFN-γ: interferon-γ; GM-CSF: granulocyte-macrophage colony-stimulating factor; ADCP: antibody-dependent cellular phagocytosis; PCD: programmed cell death; ROS: reactive oxygen species; pDC: plasmacytoid dendritic cells; ASCT: autologous stem cell transplantation; JAK1: janus kinase 1; STAT1/2: signal transducers and activators of transcription 1/2; IRF9: interferon regulatory factor 9; IFNs: interferons; ISGF3: IFN-stimulated gene factor 3; ISGs: interferon-stimulated genes; DR5: death receptor 5; SHP-2: Src homology region 2 domain-containing protein tyrosine phosphatase; PKC: protein kinase C; PI3K: phosphoinositide 3-kinase; ERK: extracellular-signal-regulated kinase; AKT: protein kinase B; ATRA: all-trans-Retinoic acid; ADRP: ADP-ribose pyrophosphate; KIR: Killer-cell immunoglobulin-like receptors.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (Grant No. 81670166, 81870140, 82070184), the Beijing Municipal Science & Technology Commission (Z171100001017098), and Peking University People's Hospital Research and Development Funds (No. RDL2021-01).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kazandjian D. Multiple myeloma epidemiology and survival: A unique malignancy. Seminars in oncology. 2016;43:676-81
2. Kyle RA. Five decades of therapy for multiple myeloma: a paradigm for therapeutic models. Leukemia. 2005;19:910-2
3. Landgren O, Rajkumar SV. New Developments in Diagnosis, Prognosis, and Assessment of Response in Multiple Myeloma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016;22:5428-33
4. Laubach JP, Richardson PG. CD38-Targeted Immunochemotherapy in Refractory Multiple Myeloma: A New Horizon. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:2660-2
5. Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T. et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood. 2016;128:384-94
6. Crowell PD, Goldstein AS. Functional evidence that progenitor cells near sites of inflammation are precursors for aggressive prostate cancer. Mol Cell Oncol. 2017;4:e1279723
7. van de Donk NWCJ, Usmani SZ. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. 2018; 9.
8. Abramson HN. Monoclonal Antibodies for the Treatment of Multiple Myeloma: An Update. Int J Mol Sci. 2018;19:3924
9. van de Donk N, Richardson PG, Malavasi F. CD38 antibodies in multiple myeloma: back to the future. Blood. 2018;131:13-29
10. Liu Q, Kriksunov IA, Graeff R, Munshi C, Lee HC, Hao Q. Crystal Structure of Human CD38 Extracellular Domain. Structure. 2005;13:1331-9
11. Crowell PD, Goldstein AS. Functional evidence that progenitor cells near sites of inflammation are precursors for aggressive prostate cancer. Mol Cell Oncol. 2017;4:e1279723-e
12. Kotlikoff MI, Kannan MS, Solway J, Deng K-Y, Deshpande DA, Dowell M. et al. Methodologic advancements in the study of airway smooth muscle. Journal of Allergy and Clinical Immunology. 2004;114:S18-S31
13. Horenstein AL, Sizzano F, Lusso R, Besso FG, Ferrero E, Deaglio S. et al. CD38 and CD157 Ectoenzymes Mark Cell Subsets in the Human Corneal Limbus. Molecular Medicine. 2009;15:76-84
14. Zhao YJ, Lam CM, Lee HC. The membrane-bound enzyme CD38 exists in two opposing orientations. Science signaling. 2012;5:ra67
15. Franco L, Guida L, Bruzzone S, Zocchi E, Usai C, De Flora A. The transmembrane glycoprotein CD38 is a catalytically active transporter responsible for generation and influx of the second messenger cyclic ADP-ribose across membranes. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1998;12:1507-20
16. Liu J, Zhao YJ, Li WH, Hou YN, Li T, Zhao ZY. et al. Cytosolic interaction of type III human CD38 with CIB1 modulates cellular cyclic ADP-ribose levels. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:8283-8
17. Lee HC, Zhao YJ. Resolving the topological enigma in Ca(2+) signaling by cyclic ADP-ribose and NAADP. The Journal of biological chemistry. 2019;294:19831-43
18. Piedra-Quintero ZL, Wilson Z, Nava P, Guerau-de-Arellano M. CD38: An Immunomodulatory Molecule in Inflammation and Autoimmunity. Frontiers in immunology. 2020;11:597959
19. Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T. et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood. 2016;128:384-94
20. Zocchi E, Franco L, Guida L, Benatti U, Bargellesi A, Malavasi F. et al. A single protein immunologically identified as CD38 displays NAD+ glycohydrolase, ADP-ribosyl cyclase and cyclic ADP-ribose hydrolase activities at the outer surface of human erythrocytes. Biochemical and biophysical research communications. 1993;196:1459-65
21. Liu Q, Graeff R, Kriksunov IA, Jiang H, Zhang B, Oppenheimer N. et al. Structural Basis for Enzymatic Evolution from a Dedicated ADP-ribosyl Cyclase to a Multifunctional NAD Hydrolase*. Journal of Biological Chemistry. 2009;284:27637-45
22. Churchill GC, Okada Y, Thomas JM, Genazzani AA, Patel S, Galione A. NAADP mobilizes Ca(2+) from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell. 2002;111:703-8
23. Grimaldi JC, Balasubramanian S, Kabra NH, Shanafelt A, Bazan JF, Zurawski G. et al. CD38-mediated ribosylation of proteins. Journal of immunology (Baltimore, Md: 1950). 1995;155:811-7
24. Sun C, Liu X, Wang B, Wang Z, Liu Y, Di C. et al. Endocytosis-mediated mitochondrial transplantation: Transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity. Theranostics. 2019;9:3595-607
25. Lu L, Wang J, Yang Q, Xie X, Huang Y. The role of CD38 in HIV infection. AIDS Research and Therapy. 2021 18
26. Marlein CR, Piddock RE, Mistry JJ, Zaitseva L, Hellmich C, Horton RH. et al. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Research. 2019;79:2285
27. Chatterjee S, Daenthanasanmak A, Chakraborty P, Wyatt MW, Dhar P, Selvam SP. et al. CD38-NAD+Axis Regulates Immunotherapeutic Anti-Tumor T Cell Response. Cell Metabolism. 2018;27:85-100.e8
28. Morandi F, Airoldi I, Marimpietri D, Bracci C, Faini AC, Gramignoli R. CD38, a Receptor with Multifunctional Activities: From Modulatory Functions on Regulatory Cell Subsets and Extracellular Vesicles, to a Target for Therapeutic Strategies. Cells. 2019;8:1527
29. Hu Y, Liu H, Fang C, Li C, Xhyliu F, Dysert H. et al. Targeting of CD38 by the Tumor Suppressor miR-26a Serves as a Novel Potential Therapeutic Agent in Multiple Myeloma. Cancer research. 2020;80:2031-44
30. Goh S, Ng HHM, Chew V, Sim XN, Li H, Lim S. et al. CD38 is a good predictor of anti-PD-1 immunotherapy responsiveness in hepatocellular carcinoma. bioRxiv. 2019: 638981.
31. Wo YJ, Gan ASP, Lim X, Tay ISY, Lim S, Lim JCT. et al. The Roles of CD38 and CD157 in the Solid Tumor Microenvironment and Cancer Immunotherapy. 2020; 9: 26.
32. Martinez-Lostao L, Anel A, Pardo J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clinical Cancer Research. 2015;21:5047-56
33. Sun JC, Lanier LL. NK cell development, homeostasis and function: parallels with CD8⁺ T cells. Nature reviews Immunology. 2011;11:645-57
34. Zambello R, Barilà G, Manni S, Piazza F, Semenzato G. NK cells and CD38: Implication for (Immuno)Therapy in Plasma Cell Dyscrasias. Cells. 2020;9:768
35. Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC. et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annual review of immunology. 2001;19:197-223
36. Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nature immunology. 2008;9:495-502
37. Ljunggren HG, Kärre K. In search of the 'missing self': MHC molecules and NK cell recognition. Immunology today. 1990;11:237-44
38. Paul S, Lal G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Frontiers in immunology. 2017;8:1124 -
39. Gao M, Gao L, Yang G, Tao Y, Hou J, Xu H. et al. Myeloma cells resistance to NK cell lysis mainly involves an HLA class I-dependent mechanism. Acta Biochimica et Biophysica Sinica. 2014;46:597-604
40. Dosani T, Carlsten M, Maric I, Landgren O. The cellular immune system in myelomagenesis: NK cells and T cells in the development of MM and their uses in immunotherapies. Blood cancer journal. 2015;5:e306-e
41. Dosani T, Carlsten M, Maric I, Landgren O. The cellular immune system in myelomagenesis: NK cells and T cells in the development of myeloma [corrected] and their uses in immunotherapies. Blood cancer journal. 2015;5:e306
42. Higuchi Y, Zeng H, Ogawa M. CD38 expression by hematopoietic stem cells of newborn and juvenile mice. Leukemia. 2003;17:171-4
43. Nagler A, Lanier LL, Cwirla S, Phillips JH. Comparative studies of human FcRIII-positive and negative natural killer cells. Journal of immunology (Baltimore, Md: 1950). 1989;143:3183-91
44. Snoeck HW, Lardon F, Lenjou M, Nys G, Van Bockstaele DR, Peetermans ME. Differential regulation of the expression of CD38 and human leukocyte antigen-DR on CD34+ hematopoietic progenitor cells by interleukin-4 and interferon-gamma. Experimental hematology. 1993;21:1480-6
45. Funaro A, De Monte LB, Dianzani U, Forni M, Malavasi F. Human CD38 is associated to distinct molecules which mediate transmembrane signaling in different lineages. European journal of immunology. 1993;23:2407-11
46. Kitanaka A, Ito C, Coustan-Smith E, Campana D. CD38 ligation in human B cell progenitors triggers tyrosine phosphorylation of CD19 and association of CD19 with lyn and phosphatidylinositol 3-kinase. Journal of immunology (Baltimore, Md: 1950). 1997;159:184-92
47. Deaglio S, Zubiaur M, Gregorini A, Bottarel F, Ausiello CM, Dianzani U. et al. Human CD38 and CD16 are functionally dependent and physically associated in natural killer cells. Blood. 2002;99:2490-8
48. Morandi F, Horenstein AL, Chillemi A, Quarona V, Chiesa S, Imperatori A. et al. CD56brightCD16- NK Cells Produce Adenosine through a CD38-Mediated Pathway and Act as Regulatory Cells Inhibiting Autologous CD4+ T Cell Proliferation. Journal of immunology (Baltimore, Md: 1950). 2015;195:965-72
49. Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E. et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiological reviews. 2008;88:841-86
50. Hogan KA, Chini CCS, Chini EN. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Frontiers in immunology. 2019;10:1187
51. Hwang I, Zhang T, Scott JM, Kim AR, Lee T, Kakarla T. et al. Identification of human NK cells that are deficient for signaling adaptor FcRγ and specialized for antibody-dependent immune functions. Int Immunol. 2012;24:793-802
52. Lee J, Zhang T, Hwang I, Kim A, Nitschke L, Kim M. et al. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity. 2015;42:431-42
53. Zhang T, Scott JM, Hwang I, Kim S. Cutting edge: antibody-dependent memory-like NK cells distinguished by FcRγ deficiency. Journal of immunology (Baltimore, Md: 1950). 2013;190:1402-6
54. Overdijk MB, Verploegen S, Bögels M, van Egmond M, Lammerts van Bueren JJ, Mutis T. et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs. 2015;7:311-21
55. Nooka AK, Kaufman JL, Hofmeister CC, Joseph NS, Heffner TL, Gupta VA. et al. Daratumumab in multiple myeloma. 2019; 125: 2364-82.
56. Bannas P, Koch-Nolte F. Perspectives for the Development of CD38-Specific Heavy Chain Antibodies as Therapeutics for Multiple Myeloma. Frontiers in immunology. 2018;9:2559
57. van de Donk NW, Janmaat ML, Mutis T, Lammerts van Bueren JJ, Ahmadi T, Sasser AK. et al. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunological reviews. 2016;270:95-112
58. Teeling JL, French RR, Cragg MS, van den Brakel J, Pluyter M, Huang H. et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood. 2004;104:1793-800
59. de Weers M, Tai Y-T, van der Veer MS, Bakker JM, Vink T, Jacobs DCH. et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. The Journal of Immunology. 2011;186:1840
60. Lammerts van Bueren J, Jakobs D, Kaldenhoven N, Roza M, Hiddingh S, Meesters J. et al. Direct in vitro Comparison of Daratumumab with Surrogate Analogs of CD38 Antibodies MOR03087, SAR650984 and Ab79. Blood. 2014;124:3474 -
61. Janmaat ML, van de Donk NWCJ, van Bueren JL, Ahmadi T, Kate Sasser A, Jansson RK. et al. Discovery, Development, and Mechanisms of Action of the Human CD38 Antibody Daratumumab. Successful Drug Discovery. 2018 p. 153-95
62. Melis JP, Strumane K, Ruuls SR, Beurskens FJ, Schuurman J, Parren PW. Complement in therapy and disease: Regulating the complement system with antibody-based therapeutics. Molecular immunology. 2015;67:117-30
63. Nijhof IS, Groen RW, Lokhorst HM, van Kessel B, Bloem AC, van Velzen J. et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia. 2015;29:2039-49
64. Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nature medicine. 2000;6:443-6
65. Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P. et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754-8
66. Bleeker WK, Lammerts van Bueren JJ, van Ojik HH, Gerritsen AF, Pluyter M, Houtkamp M. et al. Dual Mode of Action of a Human Anti-Epidermal Growth Factor Receptor Monoclonal Antibody for Cancer Therapy. The Journal of Immunology. 2004;173:4699
67. Nimmerjahn F, Ravetch JV. Fcgamma receptors: old friends and new family members. Immunity. 2006;24:19-28
68. Krzewski K, Coligan JE. Human NK cell lytic granules and regulation of their exocytosis. Frontiers in immunology. 2012;3:335
69. Smyth MJ, Cretney E, Kelly JM, Westwood JA, Street SE, Yagita H. et al. Activation of NK cell cytotoxicity. Molecular immunology. 2005;42:501-10
70. Paul S, Lal G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Frontiers in immunology. 2017;8:1124
71. Casneuf T, Xu XS, Adams HC 3rd, Axel AE, Chiu C, Khan I. et al. Effects of daratumumab on natural killer cells and impact on clinical outcomes in relapsed or refractory multiple myeloma. Blood Adv. 2017;1:2105-14
72. Martin TG, Corzo K, Chiron M, Velde HV, Abbadessa G, Campana F. et al. Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab. Cells. 2019 8
73. Fumey W, Koenigsdorf J, Kunick V, Menzel S, Schütze K, Unger M. et al. Nanobodies effectively modulate the enzymatic activity of CD38 and allow specific imaging of CD38(+) tumors in mouse models in vivo. Scientific reports. 2017;7:14289
74. Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R. et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271-85
75. van Bommel PE, He Y, Schepel I, Hendriks MAJM, Wiersma VR, van Ginkel RJ. et al. CD20-selective inhibition of CD47-SIRPα "don't eat me" signaling with a bispecific antibody-derivative enhances the anticancer activity of daratumumab, alemtuzumab and obinutuzumab. Oncoimmunology. 2017;7:e1386361-e
76. Kauder SE, Kuo TC, Chen A, Harrabi O, Rocha SS, Doyle L. et al. ALX148 Is a High Affinity Sirpα Fusion Protein That Blocks CD47, Enhances the Activity of Anti-Cancer Antibodies and Checkpoint Inhibitors, and Has a Favorable Safety Profile in Preclinical Models. Blood. 2017;130:112 -
77. Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S. et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell. 2010;142:699-713
78. Rigalou A, Ryan A, Natoni A, Chiu C, Sasser K, O'Dwyer ME. Potentiation of Anti-Myeloma Activity of Daratumumab with Combination of Cyclophosphamide, Lenalidomide or Bortezomib Via a Tumor Secretory Response That Greatly Augments Macrophage-Induced ADCP. Blood. 2016;128:2101 -
79. Overdijk MB, Jansen JHM, Nederend M, Lammerts van Bueren JJ, Groen RWJ, Parren PWHI. et al. The Therapeutic CD38 Monoclonal Antibody Daratumumab Induces Programmed Cell Death via Fcγ Receptor-Mediated Cross-Linking. The Journal of Immunology. 2016;197:807
80. Moreno L, Perez C, Zabaleta A, Manrique I, Alignani D, Ajona D. et al. The Mechanism of Action of the Anti-CD38 Monoclonal Antibody Isatuximab in Multiple Myeloma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2019;25:3176-87
81. Moreno L, Zabaleta A, Alignani D, Ajona D, Lasa M, Maiso P. et al. Critical Analysis on the Mechanism of Action (MoA) of the Anti-CD38 Monoclonal Antibody Isatuximab in Multiple Myeloma (MM). Blood. 2016;128:2105 -
82. Baum N, Fliegert R, Bauche A, Hambach J, Menzel S, Haag F. et al. Daratumumab and Nanobody-Based Heavy Chain Antibodies Inhibit the ADPR Cyclase but not the NAD(+) Hydrolase Activity of CD38-Expressing Multiple Myeloma Cells. Cancers (Basel). 2020;13:76
83. Deckert J, Wetzel MC, Bartle LM, Skaletskaya A, Goldmacher VS, Vallée F. et al. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:4574-83
84. Cai T, Wetzel M, Nicolazzi C, Vallee F, Deckert J, Dumontet C. et al. Preclinical Characterization of SAR650984, a Humanized Anti-CD38 Antibody for the Treatment of Multiple Myeloma: P-288. 2013; 13.
85. Martin TG, Corzo K, Chiron M, Velde Hvd, Abbadessa G, Campana F. et al. Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab. Cells. 2019;8:1522
86. Chauhan D, Singh AV, Brahmandam M, Carrasco R, Bandi M, Hideshima T. et al. Functional interaction of plasmacytoid dendritic cells with multiple myeloma cells: a therapeutic target. Cancer cell. 2009;16:309-23
87. Stocker N, Gaugler B, Ricard L, de Vassoigne F, Marjanovic Z, Mohty M. et al. Daratumumab prevents programmed death ligand-1 expression on antigen-presenting cells in de novo multiple myeloma. Cancer Med. 2020;9:2077-84
88. Zhang L, Tai YT, Ho M, Xing L, Chauhan D, Gang A. et al. Regulatory B cell-myeloma cell interaction confers immunosuppression and promotes their survival in the bone marrow milieu. Blood cancer journal. 2017;7:e547
89. Jansen JHM, Boross P, Overdijk MB, van Bueren JJL, Parren PWHI, Leusen JHW. Daratumumab, a Human CD38 Antibody Induces Apoptosis of Myeloma Tumor Cells Via Fc Receptor-Mediated Crosslinking. Blood. 2012;120:2974 -
90. Naeimi Kararoudi M, Nagai Y, Elmas E, de Souza Fernandes Pereira M, Ali SA, Imus PH. et al. CD38 deletion of human primary NK cells eliminates daratumumab-induced fratricide and boosts their effector activity. Blood. 2020;136:2416-27
91. Wang Y, Zhang Y, Hughes T, Zhang J, Caligiuri MA, Benson DM. et al. Fratricide of NK Cells in Daratumumab Therapy for Multiple Myeloma Overcome by Ex vivo-Expanded Autologous NK Cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2018;24:4006-17
92. Sconocchia G, Titus JA, Mazzoni A, Visintin A, Pericle F, Hicks SW. et al. CD38 triggers cytotoxic responses in activated human natural killer cells. Blood. 1999;94:3864-71
93. Mahaweni NM, Bos GMJ, Mitsiades CS, Tilanus MGJ, Wieten L. Daratumumab augments alloreactive natural killer cell cytotoxicity towards CD38+ multiple myeloma cell lines in a biochemical context mimicking tumour microenvironment conditions. Cancer immunology, immunotherapy: CII. 2018;67:861-72
94. Bigley AB, Spade S, Agha NH, Biswas S, Tang S, Malik MH. et al. FcεRIγ-negative NK cells persist in vivo and enhance efficacy of therapeutic monoclonal antibodies in multiple myeloma. Blood Adv. 2021;5:3021-31
95. Shahzad R, Rachael AS, Suzanne L. Immunomodulatory Drugs (IMiDs) in Multiple Myeloma. Current Cancer Drug Targets. 2017;17:846-57
96. Jiang H, Acharya C, An G, Zhong M, Feng X, Wang L. et al. SAR650984 directly induces multiple myeloma cell death via lysosomal-associated and apoptotic pathways, which is further enhanced by pomalidomide. Leukemia. 2016;30:399-408
97. Feng X, Zhang L, Acharya C, An G, Wen K, Qiu L. et al. Targeting CD38 Suppresses Induction and Function of T Regulatory Cells to Mitigate Immunosuppression in Multiple Myeloma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2017;23:4290-300
98. Nijhof IS, Groen RW, Noort WA, van Kessel B, de Jong-Korlaar R, Bakker J. et al. Preclinical Evidence for the Therapeutic Potential of CD38-Targeted Immuno-Chemotherapy in Multiple Myeloma Patients Refractory to Lenalidomide and Bortezomib. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:2802-10
99. Fedele PL, Willis SN, Liao Y, Low MS, Rautela J, Segal DH. et al. IMiDs prime myeloma cells for daratumumab-mediated cytotoxicity through loss of Ikaros and Aiolos. Blood. 2018;132:2166-78
100. Fedele PL, Willis SN, Liao Y, Low MS, Rautela J, Segal DH. et al. IMiDs prime myeloma cells for daratumumab-mediated cytotoxicity through loss of Ikaros and Aiolos. Blood. 2018;132:2166-78
101. van der Veer MS, de Weers M, van Kessel B, Bakker JM, Wittebol S, Parren PW. et al. Towards effective immunotherapy of myeloma: enhanced elimination of myeloma cells by combination of lenalidomide with the human CD38 monoclonal antibody daratumumab. Haematologica. 2011;96:284-90
102. Busch L, Mougiakakos D, Büttner-Herold M, Müller MJ, Volmer DA, Bach C. et al. Lenalidomide enhances MOR202-dependent macrophage-mediated effector functions via the vitamin D pathway. Leukemia. 2018;32:2445-58
103. Casneuf T, Xu XS, Adams HC 3rd, Axel AE, Chiu C, Khan I. et al. Effects of daratumumab on natural killer cells and impact on clinical outcomes in relapsed or refractory multiple myeloma. Blood Adv. 2017;1:2105-14
104. Carlsten M, Namazi A, Reger R, Levy E, Berg M, St Hilaire C. et al. Bortezomib sensitizes multiple myeloma to NK cells via ER-stress-induced suppression of HLA-E and upregulation of DR5. Oncoimmunology. 2019;8:e1534664
105. van der Veer MS, de Weers M, van Kessel B, Bakker JM, Wittebol S, Parren PWHI. et al. The therapeutic human CD38 antibody daratumumab improves the anti-myeloma effect of newly emerging multi-drug therapies. Blood cancer journal. 2011;1:e41-e
106. Mateos MV, Dimopoulos MA, Cavo M, Suzuki K, Jakubowiak A, Knop S. et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. The New England journal of medicine. 2018;378:518-28
107. Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M. et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. The New England journal of medicine. 2016;375:754-66
108. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV. et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Molecular and cellular biology. 2005;25:9543-53
109. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H. et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. The Journal of experimental medicine. 2000;192:1027-34
110. Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA. et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nature communications. 2014;5:5241
111. Kang BN, Tirumurugaan KG, Deshpande DA, Amrani Y, Panettieri RA, Walseth TF. et al. Transcriptional regulation of CD38 expression by tumor necrosis factor-alpha in human airway smooth muscle cells: role of NF-kappaB and sensitivity to glucocorticoids. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2006;20:1000-2
112. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E. et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373-7
113. Chen L, Yi X, Goswami S, Ahn YH, Roybal JD, Yang Y. et al. Growth and metastasis of lung adenocarcinoma is potentiated by BMP4-mediated immunosuppression. Oncoimmunology. 2016;5:e1234570
114. Chen L, Diao L, Yang Y, Yi X, Rodriguez BL, Li Y. et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer discovery. 2018;8:1156-75
115. Zhu C, Song Z, Wang A, Srinivasan S, Yang G, Greco R. et al. Isatuximab Acts Through Fc-Dependent, Independent, and Direct Pathways to Kill Multiple Myeloma Cells. Frontiers in immunology. 2020;11:1771
116. Costa F, Marchica V, Storti P, Malavasi F, Giuliani N. PD-L1/PD-1 Axis in Multiple Myeloma Microenvironment and a Possible Link with CD38-Mediated Immune-Suppression. Cancers (Basel). 2021;13:164
117. Horenstein AL, Faini AC, Morandi F, Bracci C, Lanza F, Giuliani N. et al. The Circular Life of Human CD38: From Basic Science to Clinics and Back. Molecules (Basel, Switzerland). 2020 25
118. García-Guerrero E, Gogishvili T, Danhof S, Schreder M, Pallaud C, Pérez-Simón JA. et al. Panobinostat induces CD38 upregulation and augments the antimyeloma efficacy of daratumumab. Blood. 2017;129:3386-8
119. Verkleij CPM, Jhatakia A, Broekmans MEC, Frerichs KA, Zweegman S, Mutis T. et al. Preclinical Rationale for Targeting the PD-1/PD-L1 Axis in Combination with a CD38 Antibody in Multiple Myeloma and Other CD38-Positive Malignancies. 2020; 12: 3713.
120. Nijhof IS, Casneuf T, van Velzen J, van Kessel B, Axel AE, Syed K. et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood. 2016;128:959-70
121. Sarkar S, Chauhan S, Daly J, Natoni A, Fairfield H, Henderson R. et al. The CD38low natural killer cell line KHYG1 transiently expressing CD16F158V in combination with daratumumab targets multiple myeloma cells with minimal effector NK cell fratricide. Cancer Immunology, Immunotherapy. 2020 69
122. Sarkar S, Chauhan SKS, Daly J, Natoni A, Fairfield H, Henderson R. et al. The CD38(low) natural killer cell line KHYG1 transiently expressing CD16(F158V) in combination with daratumumab targets multiple myeloma cells with minimal effector NK cell fratricide. Cancer immunology, immunotherapy: CII. 2020;69:421-34
123. Janakiram M, Vij R, Siegel DS, Shih T, Weymer S, Valamehr B. et al. A Phase I Study of FT538, a First-of-Kind, Off-the-Shelf, Multiplexed Engineered, iPSC-Derived NK Cell Therapy As Monotherapy in Relapsed/Refractory Acute Myelogenous Leukemia and in Combination with Daratumumab or Elotuzumab in Relapsed/Refractory Multiple Myeloma. Blood. 2020;136:3 -
124. Sarkar S, van Gelder M, Noort W, Xu Y, Rouschop KM, Groen R. et al. Optimal selection of natural killer cells to kill myeloma: the role of HLA-E and NKG2A. Cancer immunology, immunotherapy: CII. 2015;64:951-63
125. Tognarelli S, Wirsching S, von Metzler I, Rais B, Jacobs B, Serve H. et al. Enhancing the Activation and Releasing the Brakes: A Double Hit Strategy to Improve NK Cell Cytotoxicity Against Multiple Myeloma. Frontiers in immunology. 2018;9:2743
Author contact
![]() Corresponding author: Xiang-Yu Zhao, Peking University People's Hospital, Peking University Institute of Hematology, Beijing Key Laboratory of Hematopoietic Stem Cell Transplantation, National Clinical Research Center for Hematologic Disease, No. 11 South Street of Xizhimen, Xicheng District, Beijing 100044, China; Collaborative Innovation Center of Hematology, China. Tel: 86-10-88325949; Fax: 86-10-88324577; E-mail: zhao_xyedu.cn.
Corresponding author: Xiang-Yu Zhao, Peking University People's Hospital, Peking University Institute of Hematology, Beijing Key Laboratory of Hematopoietic Stem Cell Transplantation, National Clinical Research Center for Hematologic Disease, No. 11 South Street of Xizhimen, Xicheng District, Beijing 100044, China; Collaborative Innovation Center of Hematology, China. Tel: 86-10-88325949; Fax: 86-10-88324577; E-mail: zhao_xyedu.cn.