Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(7):2994-3005. doi:10.7150/ijbs.71381 This issue Cite
Research Paper
The sphingosine kinase inhibitor SKI-V suppresses cervical cancer cell growth
Yan Zhang1*, Long Cheng2*, Xin Shi3*, Yu Song4*, Xiao-yu Chen5, Min-bin Chen1, Jin Yao6 ![]() , Zhi-qing Zhang3
, Zhi-qing Zhang3 ![]() , Shang Cai7
, Shang Cai7 ![]()
1. Department of Radiotherapy and Oncology, Affiliated Kunshan Hospital of Jiangsu University, Kunshan, China.
2. Department of Interventional Radiology, Dushu Lake Hospital Affiliated to Soochow University, Medical Center of Soochow University, Suzhou Dushu Lake Hospital, Suzhou, China.
3. Department of Neurology and Clinical Research Center of Neurological Disease, The Second Affiliated Hospital of Soochow University, Suzhou, China.
4. Department of Oncology, The Affiliated Zhangjiagang Hospital of Soochow University, Suzhou, China.
5. Changshu Hospital Affiliated to Nanjing University of Chinese Medicine, Changshu, China.
6. The Affiliated Eye Hospital, Nanjing Medical University, Nanjing, China.
7. Department of Radiotherapy & Oncology, The Second Affiliated Hospital of Soochow University, Institute of Radiation Oncology, Soochow University, Suzhou, China.
*Co-first authors.
Received 2022-1-24; Accepted 2022-3-31; Published 2022-4-18
Abstract

Overexpression and/or overactivation of sphingosine kinase 1/2 (SphK1/2) is important for tumorigenesis and progression of cervical cancer. The current study examined the potential activity and signaling mechanisms of SKI-V, a non-lipid small molecule SphK inhibitor, against cervical cancer cells. In different primary and immortalized cervical cancer cells, SKI-V exerted significant anti-cancer activity by inhibiting cell viability, colony formation, proliferation, cell cycle progression and cell migration. Significant apoptosis activation was detected in SKI-V-treated cervical cancer cells. Significantly, SKI-V also provoked programmed necrosis cascade in cervical cancer cells, as it induced mitochondrial p53-cyclophilin-D-adenine nucleotide translocator-1 (ANT1) complexation, mitochondrial membrane potential collapse, reactive oxygen species production and the release of lactate dehydrogenase into the medium. Further, SKI-V blocked SphK activation and induced ceramide accumulation in primary cervical cancer cells, without affecting SphK1/2 expression. SKI-V-induced cytotoxicity in cervical cancer cells was largely inhibited by sphingosine-1-phosphate or the SphK1 activator K6PC-5, but was sensitized by adding the short-chain ceramide C6. Moreover, SKI-V inhibited Akt-mTOR (mammalian target of rapamycin) activation in primary cervical cancer cells, and its cytotoxicity was mitigated by a constitutively-active Akt. In vivo, daily intraperitoneal injection of SKI-V significantly inhibited subcutaneous primary cervical cancer xenograft growth in nude mice. Together, the SphK inhibitor SKI-V suppresses cervical cancer growth in vitro and in vivo.
Keywords: Cervical cancer, sphingosine kinase (SphK), SKI-V, Akt-mTOR, Cancer growth.
Introduction
Cervical cancer is one of the most common types of cancer and the third-most common cause of death from cancer among women [1, 2]. It is responsible for about 8-10% of all cancer deaths of women every year [3, 4]. Human papillomavirus (HPV) infection is associated with over 90% of all cervical cancer cases [5]. The widespread use of cervical screening [6], along with other measurers, has significantly reduced the mortality, and the overall five-year survival rate is close to 72% [3, 4]. The current treatments for cervical cancer include surgery, radiation therapy (used in all stages), cisplatin-based chemotherapy, and molecularly-targeted therapies [7, 8]. The prognosis of metastatic, recurrent and other advanced cervical cancers remains extremely poor. Indeed, the five year survival rate decrease to 30-40% for women with stage III cervical cancers and only 15% or fewer for those with stage IV cancers [4].
Due to the limited success with current therapies for the management of advanced cervical cancer, there is increased interest in the development of novel targeted therapeutics [9, 10]. Sphingosine kinases (SphKs), including SphK1 and SphK2, are conserved lipid kinases that catalyze formation sphingosine-1-phosphate (S1P) from the precursor sphingolipid [11-15]. Sphingolipid metabolites, ceramide, sphingosine and S1P [11-15], are vital lipid second messengers involved in diverse cellular processes, including apoptosis regulation, cell proliferation and cell survival as well as cell migration, invasion, metastasis, and tumor neovascularization [11-15].
Kim et al., have shown that SphK1 expression is dramatically increased in cervical cancer tissues and cell lines [16]. Moreover, SphK1 upregulation is associated with tumor size, invasion depth, lymph node metastasis, stage, and lymphovascular invasion [16]. Importantly, elevated SphK1 expression also correlated with poor prognosis and is an independent prognostic factor for predicting poor recurrence-free survival [16]. Xu et al., demonstrated that SphK2 expression is elevated in cervical cancer cells [17]. ABC294640, a specific SphK2 inhibitor [18, 19], potently inhibited cervical cancer cell growth in vitro and in vivo [17]. These evidences supported that targeting SphK1/2 could achieve significant activity against cervical cancer cells.
SKI-V (CAS No. 24418-86-8) is a non-competitive and non-lipid small molecule SphK inhibitor, and the IC50 is close to 2 μM for SphK [20, 21]. SKI-V potently suppressed SphK activity and depleted S1P, inducing apoptosis in bladder cancer cells [21]. In immunocompetent BALB/c mice, SKI-V intraperitoneal injection arrested growth of the mammary adenocarcinoma xenograft [21]. The SphK inhibitor was relative safe and no significant toxicity was detected at doses up to 75 mg/kg in Swiss-Webster mice and BALB/c nude mice [21]. The potential activity and the underlying mechanisms of SKI-V against cervical cancer cells were examined in the present study.
Materials and methods
Chemicals and reagents. SKI-V was obtained from MedChemExpress (Beijing, China). JC-1, EdU (5-Ethynyl-2'-deoxyuridine), DAPI (4',6-diamidino-2-phenylindole), TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) and CellROX dyes, as well as Annexin V and propidium iodide (PI) were purchased from Thermo-Fisher Invitrogen Co. (Shanghai, China). Antibodies were all purchased from Cell Signaling Technologies (Beverly, MA). Cell culture reagents were obtained from Hyclone (Logan, UT). Puromycin, polybrene, N-acetyl-L-cysteine (NAC), z-DEVD-fmk, z-VAD-fmk, sphingosine 1-phosphate (S1P), cyclosporin A, LY294002, PD98059 and U0126 were from Sigma-Aldrich (St. Louis, Mo). K6PC-5, SKI-II, FTY720, and ABC294640 were obtained from Selleck (Beijing, China). C6 ceramide and S1P were described in our previous studies [19, 22].
Cell culture. A total of three female patients (pCCa-1, pCCa-2 and pCCa-3) with gynecology and obstetrics (FIGO) stages IIA-IIB cervical cancers (squamous cell carcinoma), at age 49/55/67, were enrolled in authors institutions. The written-informed consent was provided from each patient. All patients underwent standardized treatments, including radical hysterectomy, concurrent chemo-radiation and chemotherapy. The surgery-isolated fresh tumor tissue specimens were washed with PBS and minced into small pieces. The tissues were then incubated in phenol-red free DMEM/F12 medium plus type I collagenase and DNase I for 3h. The obtained primary cells were filtrated through the nylon cell strainer (BD) [23]. Cells were then centrifuged, collected, washed and resuspended in phenol-red free DMEM/F12 plus 10% FBS. Fibroblasts, blood vessel cells, immune cells and other non-cancerous cells in the medium supernatant were immediately removed after cancer cell attachment. The exact same procedure was applied to adjacent normal cervical tissues to obtain the primary human cervical epithelial cells (HCerEpC). HeLa cervical cancer cell line and Ect1/E6E7 cervical epithelial cell line were purchased from the Cell Bank of Shanghai Institute of Biological Science of CAS (Shanghai, China). The immortalized cells were cultured in high glucose (17.51 mM) DMEM/F-12 medium plus 10% FBS (Gibco, Suzhou, China). The protocols were approved by the Ethics Committee of Soochow University, in accordance to the Declaration of Helsinki.
Quantitative real-time reverse transcriptase polymerase chain reaction (qRT-PCR) assay. As described [24, 25], TRIzol reagents were added to cells or fresh tissues. Total RNA was reversely transcripted to cDNA. qRT-PCR assays were carried out by the ABI7600 Prism system using the SYBR Green PCR kit, and the product melting temperature was calculated. Quantification of targeted mRNA was through the 2-∆∆Ct method[24, 26]. GAPDH mRNA was tested as the internal control. All verified primers were purchased from Genechem (Shanghai, China).
Western blotting and co-immunoprecipitation. As described [24, 25], aliquots of 30-40 μg proteins per treatment were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA). Western blotting protocols were reported previously [25, 27-29], and the ImageJ software was utilized for data quantification. The mitochondria fraction lysates were achieved using the described protocol [30, 31] and lysates (500 μg per treatment) were pre-cleared and incubated with anti-Cyclophilin-D (CyPD) antibody. The proteins that were immunoprecipitated with CyPD were captured and tested by Western blotting analyses.
The SphK activity assay. Following treatment, cells and tissues were homogenized and centrifuged, and the supernatant was obtained. The SphK activity was measured by the described protocol [32].
Ceramide assay. Cells were plated into six-well plates and treated with SKI-V. The detailed protocols of analyzing total cellular ceramides were described previously [28, 33, 34]. Ceramides were expressed as fmol by nmol of phospholipid.
Cyclophilin-D (CyPD) shRNA. CyPD shRNA lentiviral particles were purchased from Santa Cruz Biotech and were added to cultured cervical cancer cells for 24h. Thereafter, puromycin (2.0 μg/mL) was added to select stable cells for 4-5 passages. CyPD silencing in stable cells was verified by Western blotting.
Constitutively-active mutant Akt1. The recombinant adenoviral construct encoding the constitutively-active Akt1 (caAkt1, S473D) was provided by Dr. Xu [35], which was stably transduced to pCCa-1 cells. Cells were then distributed into 192-well plates, and single stable cells with caAkt1 were verified by Western blotting.
Cellular function assays, including Cell Counting Kit-8 (CCK-8) assay of cell viability, [H3] DNA incorporation ELISA (enzyme-linked immunosorbent assay), nuclear EdU staining, colony formation, Trypan blue staining of cell death, propidium iodide (PI)-FACS (fluorescence-activated cell sorting) detection of cell cycle progression, “Transwell” assays were described in detail in our previous studies [27, 36, 37]. Cell apoptosis-related assays, including the caspae-3 activity assay, the nuclear TUNEL staining, Annexin V-PI FACS, were described in detail in our previous studies [27, 36, 37]. Cell necrosis detection by measuring medium LDH contents was described elsewhere [38].
Mouse xenograft studies. Animal protocols in the present study were approved by IACUC and Ethics Board of Soochow University. Five to six week-old BALB/c nude mice (half male half female, 18.3-19.5g) were purchased from the Animal Center of Soochow University and were maintained indoors at standard conditions. pCCa-1 cells (5 × 106 cells per mouse, in 200 μL DMEM/Matrigel solution, no serum) were subcutaneously (s.c.) injected into the flanks of the nude mice. pCCa-1 xenograft tumors were then established with 20 days of cell inoculation, and each xenograft tumor was close to 100 mm3, and it was the time when treatment was initiated. The mice body weights and tumor volumes were measured every six days using the digital calipers [28].
Statistical analysis. Data were normally distributed and were presented as mean ± standard deviation (SD). Statistical analyses were carried out by SPSS 23.0 (SPSS Co., Chicago, IL). Unpaired student's T-test was employed to compare two groups. One-way ANOVA with the Scheffe' and Tukey Test was employed for comparison of multiple groups. P values of <0.05 were considered as statistically significant.
Results
SKI-V exerts significant anti-cancer activity in cultured cervical cancer cells
The primary human cervical cancer pCCa-1 cells were cultivated in complete medium and treated with SKI-V at gradually-increased concentrations (from 1 to 30 μM). CCK-8 assays were carried out to examine cell viability and results demonstrated that SKI-V inhibited pCCa-1 cell viability in a concentration-dependent manner (Figure 1A). The viability (CCK-8 OD) reduction by SKI-V was significant at 3-30 μM concentrations, but not at 1 μM (Figure 1A). In addition, the SphK inhibitor showed a time-dependent response in decreasing pCCa-1 cell viability (Figure 1A), as it required at least 48h to cause a significant effect (Figure 1A) and lasted for at least 96h (Figure 1A). The colony formation assay results in Figure 1B further supported the anti-survival activity of SKI-V in pCCa-1 cells, and the significantly decreased number of pCCa-1 cell colonies was observed following SKI-V (3-30 μM) treatment (Figure 1B). The positive trypan blue staining is a characteristic marker of cell death. SKI-V, in a concentration-dependent response, increased the number of trypan blue-positive pCCa-1 cells, supporting its cytotoxic effect to cervical cancer cells (Figure 1C).
Further experimental results showed that SKI-V (3-30 μM) treatment significantly decreased [H3] DNA incorporation (ELISA OD) in pCCa-1 cells (Figure 1D), suggesting that the SphK inhibitor suppressed cervical cancer cell proliferation. Moreover, the EdU-positive nuclei ratio in pCCa-1 cells was robustly decreased following SKI-V (3-30 μM) treatment (Figure 1E), further supporting the anti-proliferative activity of SKI-V. The titration experiments in Figure 1A-E showed that SKI-V at 10 µM resulted in significant anti-cervical cancer activity and this concentration was selected for the following experiments.
The PI-FACS assays were then carried out to study the potential effect of SKI-V on cell cycle progression. Results implied that SKI-V (10 μM) resulted in G1-S cell cycle arrest in pCCa-1 cells (Figure 1F), causing increased G1-phase cell ratio but decreased S-phase cell ratio (Figure 1F). The in vitro cell mobility assay results showed that SKI-V (10 μM) potently inhibited pCCa-1 cell migration, tested by the “Transwell” assay (Figure 1G).
SKI-V exerts significant anti-cancer activity in cultured cervical cancer cells. Patient-derived primary human cervical cancer cells (pCCa-1, pCCa-2, and pCCa-3, from three patients) (A-J), the established HeLa cell line (H-J), the primary human cervical epithelial cells (HCerEpC) (K and L) or Ect1/E6E7 cervical epithelial cell line(K and L) were cultivated in FBS-containing complete medium and treated with SKI-V at the applied concentrations. Control cells were treated with the vehicle control (0.1% DMSO, “Veh”). Cells were further cultured in the conditional medium for the applied time periods, cell viability (CCK-8 OD, A , H and K), colony formation (B), cell death (by testing Trypan blue ratio, C) and cell proliferation (by measuring [H3] DNA incorporation and the EdU-positive nuclei ratio D, E, I and L) as well as cell cycle progression (PI-FACS assays, F) and cell migration (“Transwell” assays, G and J) were tested. For EdU staining assays, five random views of total 1, 500 cell nuclei per treatment were included to calculate the average EdU ratio (% vs. DAPI). For all “Transwell” assays, five random microscopy views of each condition were included to calculate the average number of migrated cells. Data were presented as mean ± standard deviation (SD, n=5). * P < 0.05 vs. “Veh” treatment. “N.S.” stands for the non-statistical difference (P > 0.05, K and L). The experiments were repeated five times with similar results obtained. Scale bar = 100 μm (E, G, I and L).
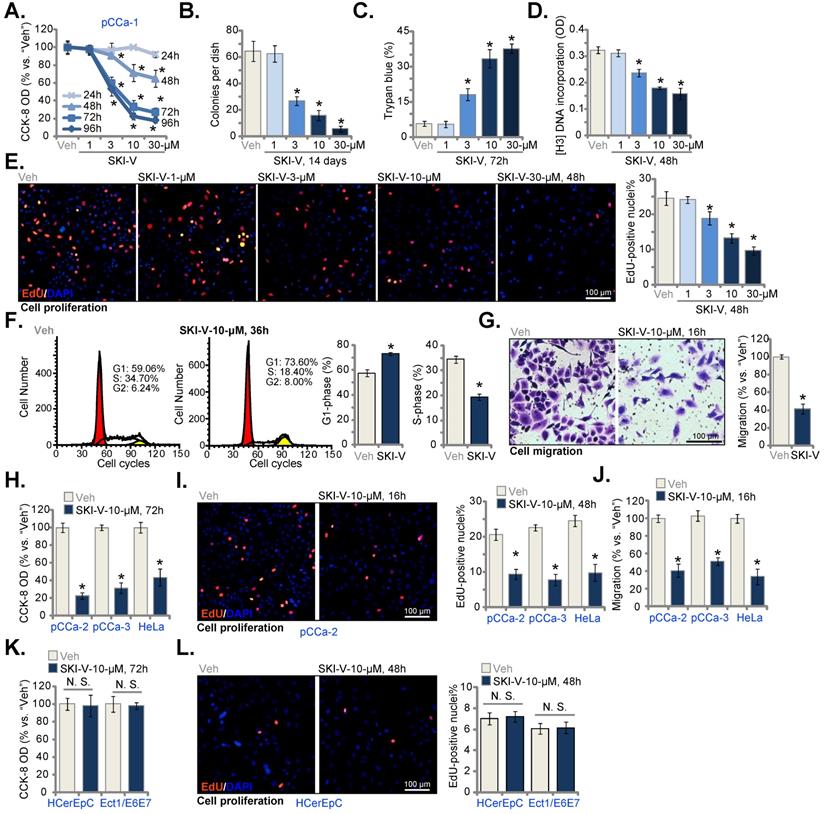
SKI-V provokes apoptosis in cervical cancer cells. Patient-derived primary human cervical cancer cells (pCCa-1, pCCa-2, and pCCa-3) (A-E), the established HeLa cell line (D and E), the primary human cervical epithelial cells (HCerEpC) (F and G) or Ect1/E6E7 cervical epithelial cell line (F and G) were cultivated in FBS-containing complete medium and treated with SKI-V (10 μM). Control cells were treated with the vehicle control (0.1% DMSO, “Veh”). Cells were further cultured in the conditional medium for the applied time periods, caspase-3 activation (A, D and F) was tested. Cell apoptosis was tested by nuclear TUNEL staining (B, E and G, with results quantified) and Annexin V FACS (C) assays. Data were presented as mean ± standard deviation (SD, n=5). * P < 0.05 vs. “Veh” treatment. “N.S.” stands for the non-statistical difference (P > 0.05, F and G). Scale bar = 100 μm (B).
Whether SKI-V could exert significant anti-cancer activity in other cervical cancer cells was studied. The primary cervical cancer cells that were derived from two other patients, pCCa-2 and pCCa-3, as well as the immortalized HeLa cells were cultivated and treated with SKI-V (10 μM). The SphK inhibitor resulted in significant cytotoxicity in the cervical cancer cells, leading to robust viability (CCK-8 OD) reduction (Figure 1H). Moreover, in the cervical cancer cells, SKI-V largely inhibited cell proliferation (tested by EdU-positive nuclei ratio, Figure 1I) and migration (Figure 1J). In the primary human cervical epithelial cells (HCerEpC) and immortalized Ect1/E6E7 epithelial cells, the very same SKI-V treatment failed to significantly inhibit cell viability (CCK-8 OD, Figure 1K) and proliferation (tested by EdU staining assays, Figure 1L), indicating a cancer cell specific effect of the SphK inhibitor.
SKI-V provokes apoptosis in cervical cancer cells
SphK inhibition will cause S1P depletion and ceramide accumulation, leading to cell apoptosis [11-14]. We therefore tested whether SKI-V could provoke apoptosis in pCCa-1 cervical cancer cells. As shown in Figure 2A, following SKI-V (10 μM) treatment, the caspase-3 activity was dramatically enhanced. Significant apoptosis was detected and SKI-V robustly increased the TUNEL-positive nuclei ratio (Figure 2B) and Annexin V-positive staining (Figure 2C) in pCCa-1 cells. Significantly, SKI-V provoked apoptosis activation in other cervical cancer cells as well. The SphK1 inhibitor significantly increased the caspase-3 activity (Figure 2D) and the TUNEL-positive nuclei ratio (Figure 2E) in primary pCCa-2 and pCCa-3 cells as well as in the established HeLa cells. These results demonstrated that SKI-V induced apoptosis in cervical cancer cells. In HCerEpC and Ect1/E6E7 epithelial cells, treatment with SKI-V however failed to significantly increase the caspase-3 activity (Figure 2F) and TUNEL-positive nuclei ratio (Figure 2G), again showing the cancer cell specific effect.
SKI-V provokes programmed necrosis in cervical cancer cells
To examine whether apoptosis was the only mechanism of SKI-V-induced cytotoxicity in cervical cancer cells, the apoptosis inhibitors were applied, including the caspase-3 inhibitor z-DEVD-fmk and the pan caspase inhibitor z-VAD-fmk. The nuclear TUNEL staining assay results demonstrated that the two caspase inhibitors blocked apoptosis activation (evidenced by the TUNEL staining assays) in the pCCa-1 primary cervical cancer cells (Figure 3A). However, the caspase inhibitors only partially attenuated SKI-V-induced viability (CCK-8 OD) reduction (Figure 3B) and cell death (Figure 3C). These results implied that besides apoptosis, the SphK inhibitor could also induce other forms of cell death in cervical cancer cells.
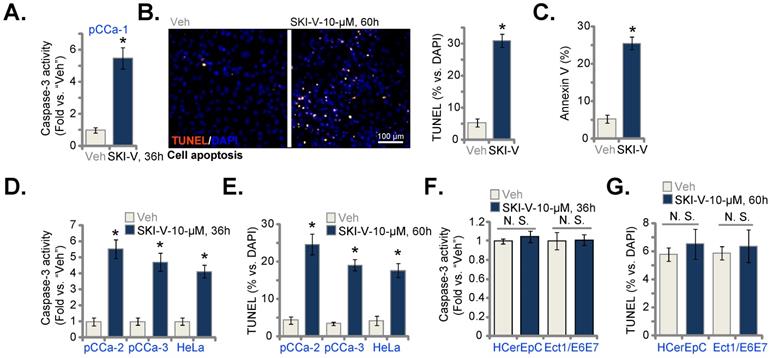
Recent studies have identified a mitochondria-dependent pathway of programmed necrosis, [39-43]. A number of anti-cancer agents could provoke the programmed necrosis cascade, contributing to cancer cell death [44-48]. The mitochondrial immunoprecipitation (mito-IP) assay results in Figure 3D showed that SKI-V treatment induced p53-CyPD-adenine nucleotide translocator-1 (ANT1) complexation in the mitochondria of pCCa-1 cells, known as the initial step of programmed necrosis cascade induction [39, 49, 50]. The SphK inhibitor also induced mitochondrial membrane potential (MMP) collapse in pCCa-1 cells, evidenced by JC-1 green monomer accumulation (Figure 3E). Reactive oxygen species (ROS) levels were tested by CellROX intensity assays, and results found that ROS were significantly increased in SKI-V-treated pCCa-1 cells (Figure 3F). The increased lactate dehydrogenase (LDH) releasing in the medium confirmed necrosis-induced by SKI-V in pCCa-1 cells (Figure 3G). Importantly, cyclosporin A (CsA), the CyPD inhibitor [48, 51], or shRNA-induced silencing of CyPD (“shCyPD”) inhibited SKI-V-induced viability (CCK-8 OD) reduction (Figure 3H) and cell death (Figure 3I). CyPD silencing was verified by Western blotting assays (Figure 3J). The antioxidant N-acetyl-L-cysteine (NAC) attenuated SKI-V-induced pCCa-1 cell death and apoptosis in pCCa-1 primary cells (Figure 3K).
SKI-V provokes programmed necrosis in cervical cancer cells. The pCCa-1 primary human cervical cancer cells were pretreated for 1h with the caspase-3 inhibitor z-DEVD-fmk (40 μM), the pan caspase inhibitor z-VAD-fmk (40 μM) or the vehicle control, followed by SKI-V (10 μM) treatment; Cells were further cultured in the conditional medium for the applied time periods, cell apoptosis, viability and death were tested by nuclear TUNEL staining (A), CCK-8 (B) and Trypan blue staining (C) assays, respectively. Patient-derived primary human cervical cancer cells (pCCa-1, pCCa-2, and pCCa-3) (D-G, L-N), the established HeLa cell line (L-N), the primary human cervical epithelial cells (HCerEpC) (O) or Ect1/E6E7 cervical epithelial cell line (O) were treated with SKI-V (10 μM) or the vehicle control (0.1% DMSO, “Veh”) for applied time periods, mitochondrial p53-CyPD-ANT1 association was tested by mitochondrial immunoprecipitation (mito-IP) assays (D), their expression was examined as well (D, “Inputs”); Mitochondrial membrane potential (MMP) reduction, ROS production and cell necrosis were tested by JC-1 staining (E and L), CellROX staining (F and M) and medium LDH release (G, N and O) assays, respectively. The pCCa-1 cells expressing the CyPD shRNA (“shCyPD”), with cyclosporin A (“CsA”, 10 μM) pretreatment (for 1h) or with scramble control shRNA plus 0.1% DMSO treatment (“DMSO+shC”) were treated with SKI-V (10 μM) or the vehicle control; Cells were further cultured for the applied time periods, cell viability and death were tested by CCK-8 (H) and Trypan blue staining (I) assays, respectively. Expression of listed proteins was shown (J). The pCCa-1 primary cells were pretreated with N-acetyl-L-cysteine (NAC, 500 μM) for 30 min, followed by SKI-V (10 μM) stimulation, and cells were cultured for applied time periods, and cell death (by measuring Trypan blue-positive cell ratio) and apoptosis (by measuring TUNEL-positive nuclei ratio) were tested (K). Data were presented as mean ± standard deviation (SD, n=5). * P < 0.05 vs. “Veh” treatment. # P < 0.05 vs. “DMSO” pretreatment (A-C). # P < 0.05 (K). # P < 0.05 vs. “DMSO+shC” cells (H and I). “N.S.” stands for the non-statistical difference (P > 0.05, O). Scale bar = 100 μm (E and F).
In pCCa-2 and pCCa-3 primary cells as well as in the established HeLa cells, treatment with SKI-V similarly induced MMP reduction (tested by JC-1 green monomer accumulation, Figure 3L), ROS production (tested by CellROX intensity increase, Figure 3M) and cell necrosis (LDH releasing to the medium, Figure 3N). In HCerEpC and immortalized Ect1/E6E7 epithelial cells, the very same SKI-V treatment however failed to induce cell necrosis and medium LDH contents were not significantly changed (Figure 3O). These results indicated that programmed necrosis cascade activation contributed to SKI-V-induced cytotoxicity in cervical cancer cells.
SKI-V inhibits SphK in cervical cancer cells
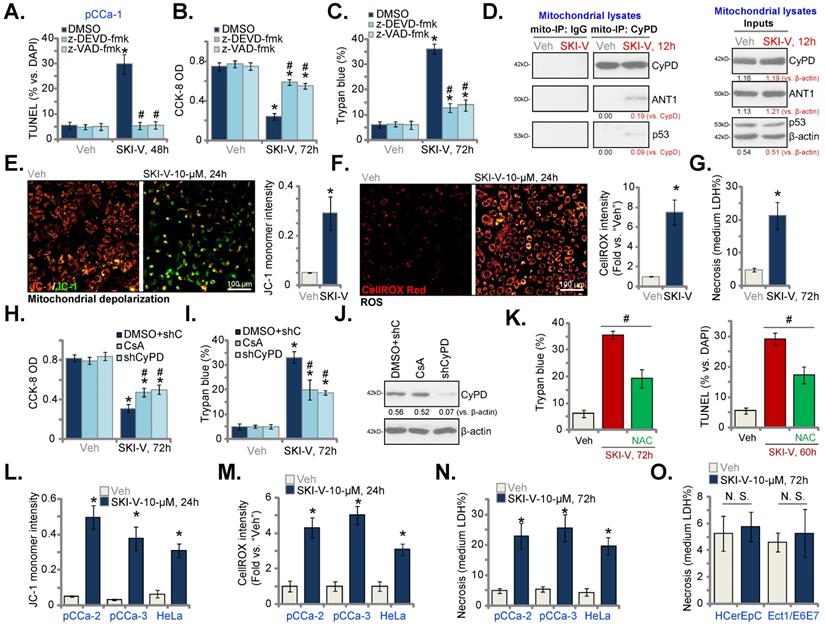
Next, we tested whether SKI-V indeed blocked SphK activation. In pCCa-1 and pCCa-2 primary cervical cancer cells, treatment with SKI-V (10 μM, 6h) robustly decreased the SphK activity (Figure 4A), and induced ceramide accumulation (Figure 4B). The mRNA (Figure 4C) and protein (Figure 4D) expression of SphK1 and SphK2 were however unchanged after SKI-V treatment. Importantly, exogenously adding S1P or treatment with the SphK1 activator K6PC-5 [52-54] largely inhibited SKI-V-induced viability (CCK-8 OD) reduction (Figure 4E), cell death (Trypan blue assays, Figure 4F) and apoptosis (tested by nuclear TUNEL staining, Figure 4G) in pCCa-1 cells. On the contrast, a short-chain ceramide C6 [22, 55, 56] augmented SKI-V-induced cytotoxicity and apoptosis in pCCa-1 cells (Figure 4H-J). These results implied that SKI-V-induced cytotoxicity in cervical cancer cells was associated with SphK blockage. We also compared the anti-cervical cancer cell activity of SKI-V with other known SphK inhibitors, including SKI-II [57-59], FTY720 [60, 61] and the SphK2 specific inhibitor ABC294640 [18, 19, 62]. As shown in pCCa-1 and pCCa-2 primary cells, SKI-V-induced cytotoxicity (viability reduction, Figure 4K) and death (tested by LDH releasing to the medium, Figure 4L) were more significant than other SphK inhibitors (at the same concentration).
SKI-V inhibits SphK in cervical cancer cells. Patient-derived primary human cervical cancer cells (pCCa-1 and pCCa-2) were treated with SKI-V (10 μM) or the vehicle control (0.1% DMSO, “Veh”). Cells were further cultured in the conditional medium for the applied time periods, the SphK activity (A), cellular ceramide contents (B) and expression of SphK1/2 (both mRNA and protein, C and D) were shown. pCCa-1 cervical cancer cells were pretreated for 1h with S1P (10 μM), K6PC-5 (10 μM) or 0.1% DMSO, followed by SKI-V (10 μM) treatment and cultured for applied time periods, cell viability, death and apoptosis were tested by CCK-8 (E), Trypan blue staining (F) and nuclear TUNEL staining (G),assays, respectively. pCCa-1 cells were treated with SKI-V (3/10 μM), or together with C6 ceramide (10 μg/mL), cells were further cultured for the applied time periods, and cell viability (H), death (I) and apoptosis (J) were tested similarly. pCCa-1 or pCCa-2 cells were treated with 10 μM of SKI-V, SKI-II, FTY720 or ABC294640 for the applied time periods, cell viability and death were tested by CCK-8 (K) and Trypan blue staining (L) assays, respectively. Data were presented as mean ± standard deviation (SD, n=5). * P < 0.05 vs. “Veh” treatment. # P < 0.05 vs. “DMSO” pretreatment group (E-G). # P < 0.05 vs. no C6 ceramide co-treatment (H-J). # P < 0.05 vs. SKI-V treatment (K and L). “N.S.” stands for the non-statistical difference (P > 0.05, C).
SKI-V inhibits Akt-mTOR activation in cervical cancer cells
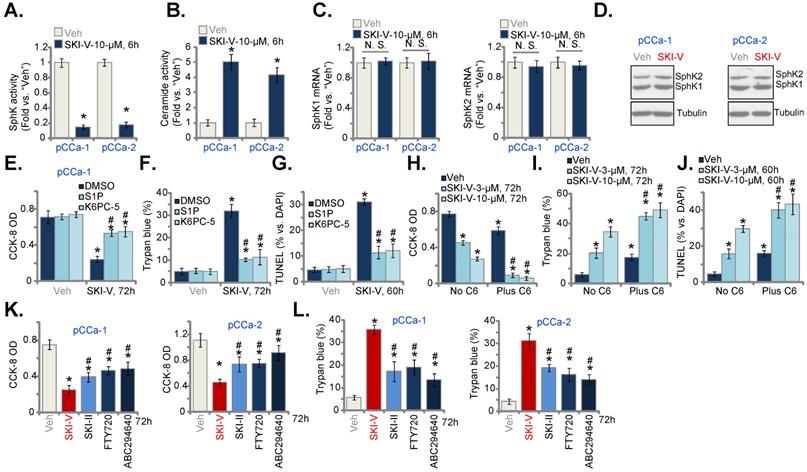
The results above suggested that there could be SphK inhibition-independent mechanism participating in SKI-V-induced anti-cervical cancer cell activity. Akt-mammalian target of rapamycin (mTOR) overactivation is essential for cervical cancer progression [63-65]. In pCCa-1 and pCCa-2 cervical cancer cells, SKI-V (10 μM, 3h) significantly suppressed phosphorylation of Akt (Ser-473) and S6K (Thr-389) (Figure 5A), indicating that SKI-V inhibited Akt-mTOR cascade activation in cervical cancer cells. To study the association between SKI-V-induced Akt-mTOR inhibition and cytotoxicity, an adenovirus-encoded constrictively-active Akt1 (caAkt1, S473D) construct [35] was stably transduced to pCCa-1 cells, which restored Akt-S6K1 phosphorylation in SKI-V (10 μM, 3h)-treated cells (Figure 5B). Significantly, in pCCa-1 cells SKI-V-induced viability (CCK-8 OD) reduction (Figure 5C) and cell death (tested by Trypan blue staining increase, Figure 5D) were mitigated by caAkt1. Moreover, apoptosis induction in SKI-V-treated pCCa-1 cells, evidenced by increased TUNEL-positive nuclei ratio, was ameliorated by caAkt1 as well (Figure 5E).
SKI-V inhibits Akt-mTOR activation in cervical cancer cells. Patient-derived primary human cervical cancer cells (pCCa-1 and pCCa-2) were treated with SKI-V (10 μM) or the vehicle control (0.1% DMSO, “Veh”), and cells were cultured in the conditional medium for 3h, expression of listed proteins was shown (A and H). pCCa-1 cells expressing the adenovirus-encoded constrictively-active Akt1 (“caAkt1”, S473D) construct or the empty vector (“Vec”) were treated with SKI-V (10 μM), the parental control cells were treated with the vehicle control (0.1% DMSO, “Veh”). Cells were further cultured in the conditional medium for the applied time periods, expression of listed proteins was shown (B); Cell viability, death and apoptosis were tested by CCK-8 (C), Trypan blue staining (D) and nuclear TUNEL staining (E) assays, respectively. The pCCa-1 primary cells were pretreated with LY294002 (1 μM) for 30 min, followed by SKI-V (10 μM) stimulation, and cells were cultured for applied time periods; Cell death (by measuring Trypan blue-positive cell ratio) and apoptosis (by measuring TUNEL-positive nuclei ratio) were tested (F). pCCa-1 cells were treated with 10 μM of SKI-V or FTY720 for 3h, p-p85 and total p85 expression was shown (G).pCCa-1 primary cells were pretreated with PD98059 (10 μM) and U0126 (10 μM) for 1h, followed by SKI-V (10 μM) stimulation, and cells were cultured for applied time periods; Cell viability (by measuring CCK-8 OD) and death (by measuring Trypan blue-positive cell ratio) were tested (I). Data were presented as mean ± standard deviation (SD, n=5). * P < 0.05 vs. “Veh” treatment (A). # P < 0.05 (B-F and I). “N.S.” stands for the non-statistical difference (P > 0.05, H).
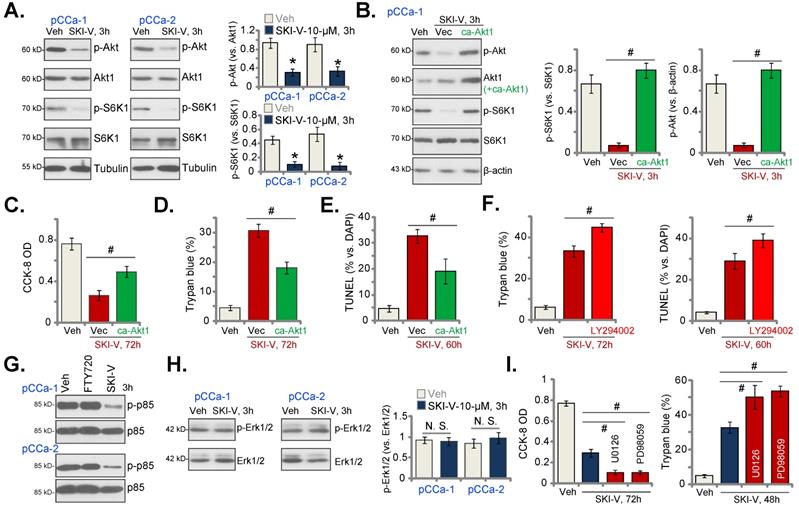
The PI3K-Akt-mTOR pan inhibitor, LY294002 [66], intensified SKI-V-induced cytotoxicity and apoptosis in pCCa-1 primary cervical cancer cells (Figure 5F). These results implied that Akt-mTOR inactivation participated in SKI-V-induced cytotoxicity in cervical cancer cells. SKI-V potently inhibited PI3K activation (p85 phosphorylation) in pCCa-1 and pCCa-2 cervical cancer cells (Figure 5G). Yet, FTY720 failed to inhibit p85 phosphorylation (revised Figure 5G). Therefore, PI3K-Akt-mTOR inactivation could be the unique action by SKI-V, independent of SphK inhibition.
Interestingly, treatment with the SphK inhibitor failed to inhibit Erk-MAPK activation (Erk1/2 phosphorylation, Figure 5H). Yet, the Erk-MAPK inhibitors, including PD98059 and U0126, augmented SKI-V-induced viability reduction (Figure 5I) and cell death (Figure 5H). These results implied that Erk-MAPK activation serves as an endogenous resistant mechanism for SKI-V-induced activity against cervical cancer cells, and Erk-MAPK inhibition could sensitize SKI-V-induced activity.
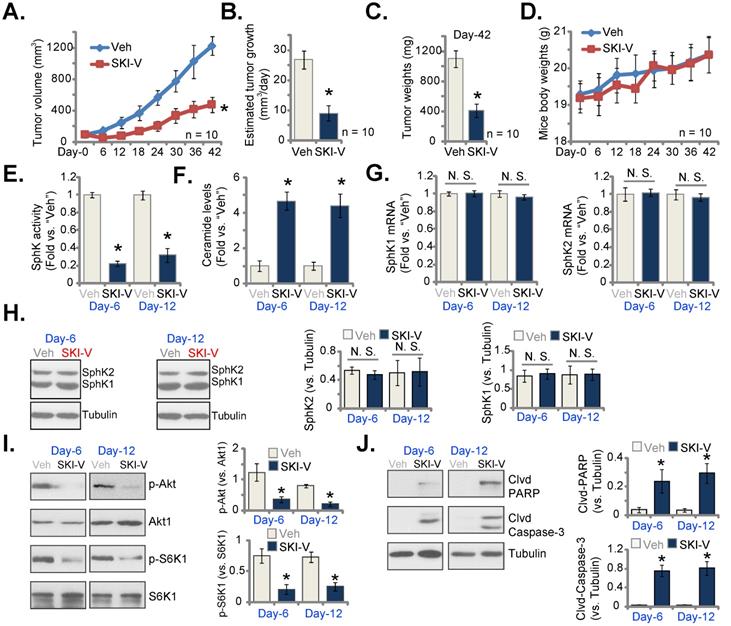
SKI-V administration inhibits cervical cancer cell growth in nude mice. pCCa-1 xenograft-bearing nude mice were subject to intraperitoneal (i.p.) injection of SKI-V (at 25 mg/kg body weight, daily for 15 days) or the vehicle control (“Veh”), the tumor volumes (A) and the mice body weights (D) were recorded every six days (“Day-0” to “Day-42”). The estimated daily tumor growth was calculated using the formula described (B). At “Day-42” all tumors were separated carefully and tumor weights recorded (C). In the described tumor tissue lysates, SphK activity (E), ceramide contents (F), expression of listed mRNAs (G) and proteins (H-J) were tested, with results quantified. Data were presented as mean ± standard deviation (SD). n=10 stands for 10 mice per group (A-D). For E-J expression of listed genes and proteins in five small pieces of each tumor xenograft was tested (n=5), and results were combined and quantified. * P < 0.05 vs. “Veh” treatment. “N.S.” stands for the non-statistical difference (P > 0.05, H).
SKI-V administration inhibits cervical cancer cell growth in nude mice
At last, we tested the potential activity of SKI-V against cervical cancer cells in vivo. A significant number of pCCa-1 cells (five million cells per mouse) were inoculated and subcutaneously (s.c.) injected to the flanks of nude mice. pCCa-1 xenograft tumors were then established with 20 days of cell inoculation, and each xenograft tumor was close to 100 mm3 in volume (labeled as “Day-0”). The xenograft-bearing mice were then randomly assigned into two groups, receiving intraperitoneal (i.p.) injection of SKI-V (at 25 mg/kg body weight, daily for 15 days) or the vehicle control (“Veh”). Figure 6A demonstrated that SKI-V injection significantly inhibited subcutaneous pCCa-1 xenograft tumor growth in nude mice. We also calculated the estimated daily tumor growth using the described formula [67]. The growth of SKI-V-treated tumors was significantly slower than vehicle administration (Figure 6B). Xenograft tumors of the two groups were isolated at “Day-42” and weighted individually. As shown, SKI-V-treated tumors were significantly lighter than vehicle controls (Figure 6C). There was no significant difference in the mice body weights (Figure 6D).
At “Day-6” and “Day-12”, six hours after SKI-V/Veh administration, we carefully isolated one tumor of each group and total four xenograft tumors were obtained. The tumors were cut into small pieces and tumor tissue lysates were obtained. As shown the SphK activity in SKI-V-treated tumor tissues was dramatically decreased (Figure 6E), while ceramide contents were significantly increased (Figure 6F). The mRNA (Figure 6G) and protein (Figure 6H) expression of SphK1 and SphK2 in pCCa-1 tumor tissues were unchanged after SKI-V treatment. Importantly, Akt-mTOR activation was significantly inhibited in SKI-V-treated pCCa-1 tumor tissues, as the levels of phosphorylated Akt1 and S6K were dramatically decreased (Figure 6I). In addition, increased cleavages of PARP and caspase-3 were detected in pCCa-1 tumor tissues with SKI-V administration, indicating apoptosis activation (Figure 6J).
Discussion
Cervical cancer is the third most common malignancy among women, causing over 275, 000 deaths globally each year [1, 2]. The effective molecularly-targeted therapies for cervical cancer are urgently needed, particularly in developing countries [9, 10]. SphKs, including SphK1 and SphK2, are important therapeutic targets of cervical cancer [16, 17]. SphK inhibitors, SKI-II [57-59] and FTY720 [60, 61], potently inhibited survival and induced apoptosis in established cervical cancer cells [16]. Moreover, FTY720 robustly inhibited in vivo tumor growth in a patient-derived xenograft (PDX) model of cervical cancer [16].
SKI-V is a non-lipid small molecule SphK inhibitor. We found that SKI-V treatment in cultured cervical cancer cells potently inhibited SphK activity and induced ceramide accumulation. In different primary cervical cancer cells (pCCa-1/-2/-3) and established HeLa cell line, SKI-V exerted significant anti-cancer activity, as it inhibited cell viability, colony formation, proliferation, cell cycle progression (causing G1-S arrest) and cell migration. Significant apoptosis activation was detected as well in the SKI-V-treated cells. In vivo, daily intraperitoneal injection of a single dose of SKI-V (25 mg/kg body weight) robustly suppressed subcutaneous pCCa-1 xenograft growth in nude mice. SphK inhibition, ceramide accumulation and apoptosis induction were detected in SKI-V-treated xenograft tissues. Importantly, treatment with the SphK inhibitor failed to induce significant cytotoxicity and apoptosis in non-cancerous cervical epithelial cells. Moreover, the nude mice were well-tolerated to the SKI-V treatment regimen, showing no apparent toxicities. Therefore, SKI-V could be a promising therapeutic option with important translational value for cervical cancer.
Existing studies have shown that a number of different anti-cancer agents can induce programmed necrosis cascade in cancer cells. Zhang et al., have shown that berberine induced both apoptosis and programmed necrosis in prostate cancer cells, and necrosis contributed more than apoptosis in contributing berberine-induced cytotoxicity in prostate cancer cells [48]. Conversely, inhibition of CyPD-p53 cascade potently attenuated berberine-induced cytotoxicity in prostate cancer cells [48]. Qin et al. have shown that salinomycin induced programmed necrosis cascade activation in glioma cells [47]. Conversely, CyPD silencing or inhibition significantly attenuated salinomycin-induced glioma cell necrosis and cytotoxicity [47]. Guo et al., showed that AICAR (5-Aminoimidazole-4-carboxamide riboside or acadesine) induced ROS production and AMP-activated protein kinase (AMPK)-independent programmed necrosis cascade in prostate cancer cells [46].
In colorectal cancer (CRC) cells the SphK inhibitor PF-543 provoked programmed necrosis cascade by inducing mitochondrial p53-CyPD association, mitochondrial membrane potential reduction and the release of LDH to the medium. Conversely, CyPD silencing or inhibition largely attenuated CRC cell necrotic death by PF-543. The Cyp-D inhibitor cyclosporin A largely inhibited the in vivo anti-tumor activity by PF-543 in xenograft mice [45]. In the present study we showed that SKI-V induced programmed necrosis cascade in cervical cancer cells as well. The SphK inhibitor induced mitochondrial p53-CyPD-ANT1 complexation, mitochondrial membrane potential collapse, ROS production and the release of LDH into the medium. CyPD inhibition (by CsA) or silencing (by targeted shRNA) mitigated SKI-V-induced cytotoxicity in primary cervical cancer cells. Therefore, besides apoptosis, simultaneous activation of the programmed necrosis cascade should be one of important reasons for the superior anti-cervical cancer cell activity by SKI-V. Indeed, we found that SKI-V was significantly more potent than other known SphK inhibitors (SKI-II, FTY720 and ABC294640) in killing cervical cancer cells.
PI3K-Akt-mTOR is often dysregulated and overactivated in cervical cancer due to various genetic mutations, including PTEN depletion, PI3KCA mutation and sustained activation or mutation of multiple receptor tyrosine kinases (RTKs) [63-65]. The PI3K-Akt-mTOR cascade could be a valuable and promising therapeutic target and the biomarker predicting the prognosis of cervical cancer [63-65]. Moreover, PI3K-Akt-mTOR cascade is essential for the virus/host cell crosstalk in HPV-positive cervical cancer [64]. A number of different agents or genetic methods targeting this cascade can efficiently inhibit cervical cancer cell growth and induce cell death [63, 68-71]. Here we found that SKI-V potently inhibited Akt-mTOR activation in primary cervical cancer cells, and restoring Akt activation by caAkt1 ameliorated SKI-V-induced cytotoxicity in cervical cancer cells. Akt-mTOR inactivation was also detected SKI-V-treated xenograft tissues. Thus, Akt-mTOR inhibition is another important mechanism of SKI-V-induced anti-cervical cancer activity.
Conclusion
The SphK inhibitor SKI-V suppresses cervical cancer growth in vitro and in vivo. It could have important translational value for cervical cancer.
Acknowledgements
Funding Statement and Acknowledgements. This work is supported by the National Natural Science Foundation of China (81970823 and 81974388), Science Foundation of Jiangsu Health Commission (M2021081), a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, Research Project of Medical Talent of Suzhou (GSWS2021025), and by Suzhou Science and Technology Development Program (SKJY2021028 and SYS2019056), Suzhou Municipal Health and Family Planning Commission (KJXW2019016), Research Project of Jiangsu Province Health Committee (Z2019054).
Ethics Statement. This study was approved by Ethics Committee of Affiliated Kunshan Hospital of Soochow University.
Author Contribution Statement. All the listed authors designed the study, performed the experiments and the statistical analysis, and wrote the manuscript and revise it. Authors have read the manuscript and approved the final submission.
Data Availability Statement. All data are available upon request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71:7-33
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7-30
3. Canfell K, Kim JJ, Brisson M, Keane A, Simms KT, Caruana M. et al. Mortality impact of achieving WHO cervical cancer elimination targets: a comparative modelling analysis in 78 low-income and lower-middle-income countries. Lancet. 2020;395:591-603
4. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209-49
5. Bosch FX, de Sanjose S. The epidemiology of human papillomavirus infection and cervical cancer. Dis Markers. 2007;23:213-27
6. Nygard M. Screening for cervical cancer: when theory meets reality. BMC Cancer. 2011;11:240
7. Einhorn N, Trope C, Ridderheim M, Boman K, Sorbe B, Cavallin-Stahl E. A systematic overview of radiation therapy effects in cervical cancer (cervix uteri). Acta Oncol. 2003;42:546-56
8. Chemoradiotherapy for Cervical Cancer Meta-analysis C. Reducing uncertainties about the effects of chemoradiotherapy for cervical cancer: individual patient data meta-analysis. Cochrane Database Syst Rev. 2010: CD008285.
9. Wang Q, Peng H, Qi X, Wu M, Zhao X. Targeted therapies in gynecological cancers: a comprehensive review of clinical evidence. Signal Transduct Target Ther. 2020;5:137
10. Zagouri F, Sergentanis TN, Chrysikos D, Filipits M, Bartsch R. Molecularly targeted therapies in cervical cancer. A systematic review. Gynecol Oncol. 2012;126:291-303
11. Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012;22:50-60
12. Vadas M, Xia P, McCaughan G, Gamble J. The role of sphingosine kinase 1 in cancer: oncogene or non-oncogene addiction? Biochim Biophys Acta. 2008;1781:442-7
13. Shida D, Takabe K, Kapitonov D, Milstien S, Spiegel S. Targeting SphK1 as a new strategy against cancer. Curr Drug Targets. 2008;9:662-73
14. Alemany R, van Koppen CJ, Danneberg K, Ter Braak M, Meyer Zu Heringdorf D. Regulation and functional roles of sphingosine kinases. Naunyn Schmiedebergs Arch Pharmacol. 2007;374:413-28
15. Maceyka M, Payne SG, Milstien S, Spiegel S. Sphingosine kinase, sphingosine-1-phosphate, and apoptosis. Biochim Biophys Acta. 2002;1585:193-201
16. Kim HS, Yoon G, Ryu JY, Cho YJ, Choi JJ, Lee YY. et al. Sphingosine kinase 1 is a reliable prognostic factor and a novel therapeutic target for uterine cervical cancer. Oncotarget. 2015;6:26746-56
17. Xu L, Jin L, Yang B, Wang L, Xia Z, Zhang Q. et al. The sphingosine kinase 2 inhibitor ABC294640 inhibits cervical carcinoma cell growth. Oncotarget. 2018;9:2384-94
18. Yang J, Yang C, Zhang S, Mei Z, Shi M, Sun S. et al. ABC294640, a sphingosine kinase 2 inhibitor, enhances the antitumor effects of TRAIL in non-small cell lung cancer. Cancer Biol Ther. 2015;16:1194-204
19. Xun C, Chen MB, Qi L, Tie-Ning Z, Peng X, Ning L. et al. Targeting sphingosine kinase 2 (SphK2) by ABC294640 inhibits colorectal cancer cell growth in vitro and in vivo. J Exp Clin Cancer Res. 2015;34:94
20. French KJ, Upson JJ, Keller SN, Zhuang Y, Yun JK, Smith CD. Antitumor activity of sphingosine kinase inhibitors. J Pharmacol Exp Ther. 2006;318:596-603
21. French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL. et al. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003;63:5962-9
22. Chen MB, Jiang Q, Liu YY, Zhang Y, He BS, Wei MX. et al. C6 ceramide dramatically increases vincristine sensitivity both in vivo and in vitro, involving AMP-activated protein kinase-p53 signaling. Carcinogenesis. 2015;36:1061-70
23. Lee HL, Park SH, Kim TM, Jung YY, Park MH, Oh SH. et al. Bee venom inhibits growth of human cervical tumors in mice. Oncotarget. 2015;6:7280-92
24. Bai JY, Li Y, Xue GH, Li KR, Zheng YF, Zhang ZQ. et al. Requirement of Galphai1 and Galphai3 in interleukin-4-induced signaling, macrophage M2 polarization and allergic asthma response. Theranostics. 2021;11:4894-909
25. Liu YY, Chen MB, Cheng L, Zhang ZQ, Yu ZQ, Jiang Q. et al. microRNA-200a downregulation in human glioma leads to Galphai1 over-expression, Akt activation, and cell proliferation. Oncogene. 2018;37:2890-902
26. Wang Y, Liu YY, Chen MB, Cheng KW, Qi LN, Zhang ZQ. et al. Neuronal-driven glioma growth requires Galphai1 and Galphai3. Theranostics. 2021;11:8535-49
27. Zheng J, Zhang Y, Cai S, Dong L, Hu X, Chen MB. et al. MicroRNA-4651 targets bromodomain-containing protein 4 to inhibit non-small cell lung cancer cell progression. Cancer Lett. 2020;476:129-39
28. Xu M, Wang Y, Zhou LN, Xu LJ, Jin ZC, Yang DR. et al. The therapeutic value of SC66 in human renal cell carcinoma cells. Cell Death Dis. 2020;11:353
29. Wang SS, Lv Y, Xu XC, Zuo Y, Song Y, Wu GP. et al. Triptonide inhibits human nasopharyngeal carcinoma cell growth via disrupting Lnc-RNA THOR-IGF2BP1 signaling. Cancer Lett. 2019;443:13-24
30. Zhou LN, Li P, Cai S, Li G, Liu F. Ninjurin2 overexpression promotes glioma cell growth. Aging (Albany NY). 2019;11:11136-47
31. Li G, Zhou LN, Yang H, He X, Duan Y, Wu F. Ninjurin 2 overexpression promotes human colorectal cancer cell growth in vitro and in vivo. Aging (Albany NY). 2019;11:8526-41
32. Tauseef M, Kini V, Knezevic N, Brannan M, Ramchandaran R, Fyrst H. et al. Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circ Res. 2008;103:1164-72
33. Gong L, Yang B, Xu M, Cheng B, Tang X, Zheng P. et al. Bortezomib-induced apoptosis in cultured pancreatic cancer cells is associated with ceramide production. Cancer Chemother Pharmacol. 2014;73:69-77
34. Chen MB, Yang L, Lu PH, Fu XL, Zhang Y, Zhu YQ. et al. MicroRNA-101 down-regulates sphingosine kinase 1 in colorectal cancer cells. Biochem Biophys Res Commun. 2015;463:954-60
35. Zha JH, Xia YC, Ye CL, Hu Z, Zhang Q, Xiao H. et al. The Anti-Non-Small Cell Lung Cancer Cell Activity by a mTOR Kinase Inhibitor PQR620. Front Oncol. 2021;11:669518
36. Zhu XR, Peng SQ, Wang L, Chen XY, Feng CX, Liu YY. et al. Identification of phosphoenolpyruvate carboxykinase 1 as a potential therapeutic target for pancreatic cancer. Cell Death Dis. 2021;12:918
37. Chen MB, Liu YY, Xing ZY, Zhang ZQ, Jiang Q, Lu PH. et al. Itraconazole-Induced Inhibition on Human Esophageal Cancer Cell Growth Requires AMPK Activation. Mol Cancer Ther. 2018;17:1229-39
38. Wang TB, Geng M, Jin H, Tang AG, Sun H, Zhou LZ. et al. SREBP1 site 1 protease inhibitor PF-429242 suppresses renal cell carcinoma cell growth. Cell Death Dis. 2021;12:717
39. Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149:1536-48
40. Tsujimoto Y, Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis. 2007;12:835-40
41. Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans. 2006;34:232-7
42. Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H. et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652-8
43. Halestrap A. Biochemistry: a pore way to die. Nature. 2005;434:578-9
44. Xia YC, Zha JH, Sang YH, Yin H, Xu GQ, Zhen J. et al. AMPK activation by ASP4132 inhibits non-small cell lung cancer cell growth. Cell Death Dis. 2021;12:365
45. Ju T, Gao D, Fang ZY. Targeting colorectal cancer cells by a novel sphingosine kinase 1 inhibitor PF-543. Biochem Biophys Res Commun. 2016;470:728-34
46. Guo F, Liu SQ, Gao XH, Zhang LY. AICAR induces AMPK-independent programmed necrosis in prostate cancer cells. Biochem Biophys Res Commun. 2016;474:277-83
47. Qin LS, Jia PF, Zhang ZQ, Zhang SM. ROS-p53-cyclophilin-D signaling mediates salinomycin-induced glioma cell necrosis. J Exp Clin Cancer Res. 2015;34:57
48. Zhang LY, Wu YL, Gao XH, Guo F. Mitochondrial protein cyclophilin-D-mediated programmed necrosis attributes to berberine-induced cytotoxicity in cultured prostate cancer cells. Biochem Biophys Res Commun. 2014;450:697-703
49. Montero J, Dutta C, van Bodegom D, Weinstock D, Letai A. p53 regulates a non-apoptotic death induced by ROS. Cell Death Differ. 2013;20:1465-74
50. Baumann K. Cell death: multitasking p53 promotes necrosis. Nat Rev Mol Cell Biol. 2012;13:480-1
51. Zhou C, Chen Z, Lu X, Wu H, Yang Q, Xu D. Icaritin activates JNK-dependent mPTP necrosis pathway in colorectal cancer cells. Tumour Biol. 2016;37:3135-44
52. Shao JJ, Peng Y, Wang LM, Wang JK, Chen X. Activation of SphK1 by K6PC-5 Inhibits Oxygen-Glucose Deprivation/Reoxygenation-Induced Myocardial Cell Death. DNA Cell Biol. 2015;34:669-76
53. Ji F, Mao L, Liu Y, Cao X, Xie Y, Wang S. et al. K6PC-5, a novel sphingosine kinase 1 (SphK1) activator, alleviates dexamethasone-induced damages to osteoblasts through activating SphK1-Akt signaling. Biochem Biophys Res Commun. 2015;458:568-75
54. Hong JH, Youm JK, Kwon MJ, Park BD, Lee YM, Lee SI. et al. K6PC-5, a direct activator of sphingosine kinase 1, promotes epidermal differentiation through intracellular Ca2+ signaling. J Invest Dermatol. 2008;128:2166-78
55. Yang L, Zheng LY, Tian Y, Zhang ZQ, Dong WL, Wang XF. et al. C6 ceramide dramatically enhances docetaxel-induced growth inhibition and apoptosis in cultured breast cancer cells: a mechanism study. Exp Cell Res. 2015;332:47-59
56. Yu T, Li J, Sun H. C6 ceramide potentiates curcumin-induced cell death and apoptosis in melanoma cell lines in vitro. Cancer Chemother Pharmacol. 2010;66:999-1003
57. Yang L, Weng W, Sun ZX, Fu XJ, Ma J, Zhuang WF. SphK1 inhibitor II (SKI-II) inhibits acute myelogenous leukemia cell growth in vitro and in vivo. Biochem Biophys Res Commun. 2015;460:903-8
58. Li PH, Wu JX, Zheng JN, Pei DS. A sphingosine kinase-1 inhibitor, SKI-II, induces growth inhibition and apoptosis in human gastric cancer cells. Asian Pac J Cancer Prev. 2014;15:10381-5
59. Chiba Y, Takeuchi H, Sakai H, Misawa M. SKI-II, an inhibitor of sphingosine kinase, ameliorates antigen-induced bronchial smooth muscle hyperresponsiveness, but not airway inflammation, in mice. J Pharmacol Sci. 2010;114:304-10
60. Pchejetski D, Bohler T, Brizuela L, Sauer L, Doumerc N, Golzio M. et al. FTY720 (fingolimod) sensitizes prostate cancer cells to radiotherapy by inhibition of sphingosine kinase-1. Cancer Res. 2010;70:8651-61
61. LaMontagne K, Littlewood-Evans A, Schnell C, O'Reilly T, Wyder L, Sanchez T. et al. Antagonism of sphingosine-1-phosphate receptors by FTY720 inhibits angiogenesis and tumor vascularization. Cancer Res. 2006;66:221-31
62. Antoon JW, White MD, Meacham WD, Slaughter EM, Muir SE, Elliott S. et al. Antiestrogenic effects of the novel sphingosine kinase-2 inhibitor ABC294640. Endocrinology. 2010;151:5124-35
63. Bahrami A, Hasanzadeh M, Hassanian SM, ShahidSales S, Ghayour-Mobarhan M, Ferns GA. et al. The Potential Value of the PI3K/Akt/mTOR Signaling Pathway for Assessing Prognosis in Cervical Cancer and as a Target for Therapy. J Cell Biochem. 2017;118:4163-9
64. Bossler F, Hoppe-Seyler K, Hoppe-Seyler F. PI3K/AKT/mTOR Signaling Regulates the Virus/Host Cell Crosstalk in HPV-Positive Cervical Cancer Cells. Int J Mol Sci. 2019 20
65. Husseinzadeh N, Husseinzadeh HD. mTOR inhibitors and their clinical application in cervical, endometrial and ovarian cancers: a critical review. Gynecol Oncol. 2014;133:375-81
66. Brunn GJ, Williams J, Sabers C, Wiederrecht G, Lawrence JC Jr, Abraham RT. Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. EMBO J. 1996;15:5256-67
67. Gao YY, Ling ZY, Zhu YR, Shi C, Wang Y, Zhang XY. et al. The histone acetyltransferase HBO1 functions as a novel oncogenic gene in osteosarcoma. Theranostics. 2021;11:4599-615
68. Zhang W, Zhou Q, Wei Y, Da M, Zhang C, Zhong J. et al. The exosome-mediated PI3k/Akt/mTOR signaling pathway in cervical cancer. Int J Clin Exp Pathol. 2019;12:2474-84
69. Song L, Liu S, Zhang L, Yao H, Gao F, Xu D. et al. MiR-21 modulates radiosensitivity of cervical cancer through inhibiting autophagy via the PTEN/Akt/HIF-1alpha feedback loop and the Akt-mTOR signaling pathway. Tumour Biol. 2016;37:12161-8
70. Bai X, Ma Y, Zhang G. Butein suppresses cervical cancer growth through the PI3K/AKT/mTOR pathway. Oncol Rep. 2015;33:3085-92
71. Rashmi R, DeSelm C, Helms C, Bowcock A, Rogers BE, Rader JL. et al. AKT inhibitors promote cell death in cervical cancer through disruption of mTOR signaling and glucose uptake. PLoS One. 2014;9:e92948
Author contact
![]() Corresponding author: dryaojincom (J. Y.); zhiqing630com (Z. Z.); drshangcaiedu.cn (S. C.)
Corresponding author: dryaojincom (J. Y.); zhiqing630com (Z. Z.); drshangcaiedu.cn (S. C.)