Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Molecular mechanisms of...
Pathophysiology of mitochondrial...
Conclusions and perspectives
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(14):5276-5290. doi:10.7150/ijbs.75402 This issue Cite
Review
Mitochondrial quality control in diabetic cardiomyopathy: from molecular mechanisms to therapeutic strategies
Chen Cai1,2*, Feng Wu1,2*, Jing He1,2*, Yaoyuan Zhang1,2, Nengxian Shi1,2, Xiaojie Peng1,2, Qing Ou1,2, Ziying Li1,2, Xiaoqing Jiang1,2, Jiankai Zhong3, Ying Tan1,2 ![]()
1. Department of Critical Care Medicine, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China.
2. Department of Critical Care Medicine, The First School of Clinical Medicine, Southern Medical University, Guangzhou 510515, China.
3. Department of Cardiology, Shunde Hospital, Southern Medical University (The First People's Hospital of Shunde), Foshan 528308, Guangdong, China.
*These authors contributed equally to this article.
Received 2022-5-22; Accepted 2022-7-18; Published 2022-8-15
Abstract

In diabetic cardiomyopathy (DCM), a major diabetic complication, the myocardium is structurally and functionally altered without evidence of coronary artery disease, hypertension or valvular disease. Although numerous anti-diabetic drugs have been applied clinically, specific medicines to prevent DCM progression are unavailable, so the prognosis of DCM remains poor. Mitochondrial ATP production maintains the energetic requirements of cardiomyocytes, whereas mitochondrial dysfunction can induce or aggravate DCM by promoting oxidative stress, dysregulated calcium homeostasis, metabolic reprogramming, abnormal intracellular signaling and mitochondrial apoptosis in cardiomyocytes. In response to mitochondrial dysfunction, the mitochondrial quality control (MQC) system (including mitochondrial fission, fusion, and mitophagy) is activated to repair damaged mitochondria. Physiological mitochondrial fission fragments the network to isolate damaged mitochondria. Mitophagy then allows dysfunctional mitochondria to be engulfed by autophagosomes and degraded in lysosomes. However, abnormal MQC results in excessive mitochondrial fission, impaired mitochondrial fusion and delayed mitophagy, causing fragmented mitochondria to accumulate in cardiomyocytes. In this review, we summarize the molecular mechanisms of MQC and discuss how pathological MQC contributes to DCM development. We then present promising therapeutic approaches to improve MQC and prevent DCM progression.
Keywords: Diabetic cardiomyopathy, mitochondrial quality control, mitochondrial fission, mitochondrial fusion, mitophagy
Introduction
Epidemiology and predisposing factors of diabetic cardiomyopathy (DCM)
From a pathophysiological viewpoint, DCM is a heart failure state primarily attributable to chronic myocardial metabolic disorder (Table 1). Clinically, myocardial damage etiology is always labeled as coronary artery disease (such as angina and myocardial infarction) in light of the irreplaceable role served by coronary arteries in managing heart blood flow [1]. However, some patients (especially middle-aged women) with no evidence of coronary artery disease detectable with imaging tools or functional tests may suffer from angina and myocardial infarction as a result of cardiac microvascular spasm or endothelial dysfunction [2]. In addition, several diseases of the heart muscle, including hypertrophic cardiomyopathy and arrhythmia-induced cardiomyopathy, clinically present with ischemia-related symptoms such as angina or ventricular wall motion abnormalities due to increased myocardial oxygen demand [3]. Consequently, the etiology of DCM in an individual patient is rarely unambiguous in practice and is frequently complicated by overlap between a classical ischemic component and discernable non-ischemic causes such as viral myocarditis, hypertension, diabetes, valvular disease, dyslipidemia, obesity, and unhealthy lifestyles [4] (Figure 1). Although research into the pathological mechanism of DCM is ongoing, we have not yet precisely defined its epidemiology and predisposing factors. Our aim, therefore, is to provide reference for the prevention of such diseases by reviewing the risk factors that can induce DCM or the pathological mechanism underlying DCM.
Proposed classification of DCM
| Stage of DCMa | Clinical phenotype |
|---|---|
| Stage I | Diastolic dysfunction with normal ejection fraction |
| Stage II | Combined systolic and diastolic dysfunction |
| Stage III | Systolic and diastolic dysfunction with microvascular disease/coronary atherosclerosis without obstructive coronary heart disease |
| Stage IV | Clinically overt ischemia/infarct causing HF |
aExcluding coronary heart disease, valvular disease and uncontrolled hypertension.
The contribution of altered metabolism to cardiovascular risk.
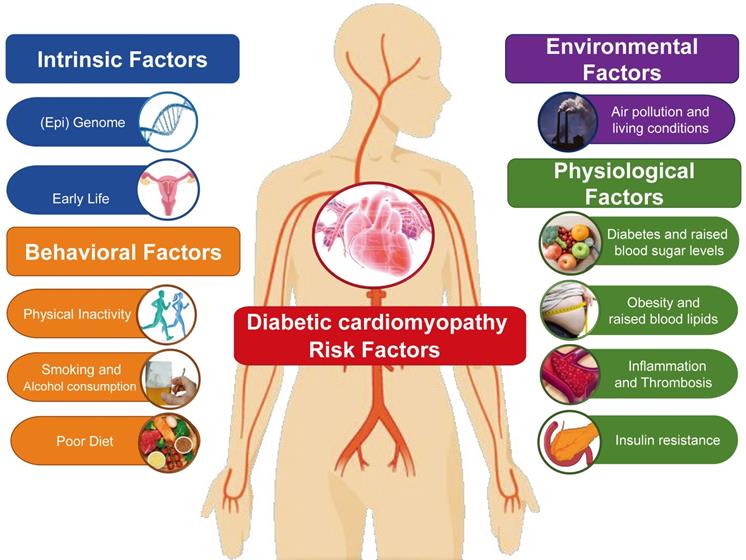
DCM affects more than 23 million people worldwide, including more than 15.5 million people in the United States [5]. Its prevalence in women (5%) is lower than in men (7.5%), and older women with DCM are more likely to have atypical ischemic heart disease symptoms such as a coronary microcirculation disorder, coronary microvascular injury, coronary artery dissection, or Takotsubo cardiomyopathy [6, 7]. In the United States, 6.6 million women are diagnosed with DCM each year, including 2.7 million patients with a history of myocardial infarction [8]. In the Framingham study, men and women with coronary heart disease were followed for up to 44 years [9].The results show that 40-year-old men have a higher lifetime risk of coronary heart disease than women. However, the incidence among women aged 65-94 is higher than among those aged 35-64; that is, that rates of morbidity and mortality due to coronary artery disease are higher among postmenopausal women [10]. Among the numerous other risk factors for DCM are obesity, hypertension, hyperglycemia, fat metabolism disorders, atherosclerosis, sedentary lifestyle, lack of exercise, smoking, and pressure [11]. These factors may interact with each other and directly or indirectly lead to the onset of DCM or affect the process of disease [12].
Age
Age is a key risk factor positively correlated with the prevalence of DCM [13]. This in large part reflects the increased incidence disorders in lipid metabolism and hyperlipidemia seen with increasing age. With declines in organ function, blood lipid metabolism is slowed, leading to hyperlipidemia and diabetes, which may be accompanied by other ailments such as hypertension [14, 15]. Such metabolic disorders can exacerbate coronary deposition of lipid plaque, which can progress to coronary atherosclerosis or microvascular damage, indirectly leading to the occurrence of DCM [16]. Although age may be related to the occurrence of DCM, it is not an inevitable factor. Men over 45 years of age and women over 55 years of age have similar increased risk of DCM. Moreover, the DCM mortality rate among women aged 35 to 44 is higher than among men of the same age [17].The reason may be that there is a lack of knowledge about DCM-inducing factors, including the roles of blood sugar, blood fat and body weight. Obesity, work stress, smoking, sedentary lifestyle, lack of exercise and metabolic syndrome are considered to be the arch criminals leading to DCM [18, 19]. Age is only one factor contributing to the pathogenesis of DCM. Indeed, DCM can even occur in some younger patients, including patients under 20 years old. In those cases, genetic factors and family genetic history play key roles. For example, in cases of early-onset cardiomyopathy, comorbidities involving abnormal blood lipid metabolism are often present.
Genetic factors
Cardiovascular disease is one of more than 3000 diseases in which genetic factors play a role [20]. Studies have shown that patients with coronary heart disease, hypertension, and other cardiovascular diseases, are more than four times more likely to die of similar diseases than the general population. If the parents of monozygotic twins suffer from cardiovascular disease, the probability of cardiovascular disease in both twins is more than six times higher than in the general population. DCM is a complex and multifactorial cardiovascular disease and it, too, is affected by genetic as well as environmental factors [21, 22].
Coronary atherosclerosis is the main cause of coronary artery stenosis and DCM. Low density lipoprotein (LDL) cholesterol molecules contribute to the development and progression of atherosclerosis because it is the main type of lipid absorbed by cell within atherosclerotic lesion. Therefore, genetic changes affecting plasma LDL levels are an important factor in the development of DCM [23]. Familial hypercholesterolemia caused by genetic factors will be accompanied by high levels of LDL and cholesterol, which accelerates development of atherosclerosis and coronary heart disease [24-26]. Nonetheless, the development of DCM still mainly depends on an individual's health behavior. Without the influence of genetic factors, a high salt diet, high-fat diet, long periods sitting and other stressful lifestyle or working environment factors will also lead to the onset of DCM [27].
Smoking
Smoking is considered to be an independent cause of DCM. It is obviously related to the onset of DCM and coronary heart disease [28, 29]. Studies have found that smokers are 70% more likely to develop DCM than non-smokers. Carbon monoxide, thromboxane, nicotine and other harmful components in cigarette smoke adversely affect the patient's vasodilator function and then cause vascular dysfunction and damage to the vascular endothelium [30, 31]. Nicotine in cigarettes can also cause thickening and abnormal contraction of blood vessel walls. In addition, increased levels of cadmium and lead in the blood of smokers can induce oxidative damage to the vascular endothelial structure, reducing the oxygen supply of myocardial cells while increasing oxygen consumption by myocardial cells, leading to myocardial hypoxia [32].
Smoking also promotes abnormal lipid metabolism, aggravating atherosclerosis. Nicotine promotes oxidation of LDL, after which the oxidized LDL bind to receptors on endothelial cell surfaces and form lipid plaques over time [33]. Vascular endothelial dysfunction caused by smoking can also promote the release of inflammatory factors and increase the secretion of prostaglandin F2α metabolites into the circulation, promoting the development of atherosclerosis [34, 35]. In patients with acute myocardial infarction, smoking can increase levels of thrombolysis and expression of inflammatory factors, which will aggravate myocardial ischemia directly related to smoking [36, 37]. Smoking cessation reduces the number of serum inflammatory markers, and the longer the patient goes without smoking, the more obvious the effect will be.
Diet
Both epidemiological and experimental results show that high intake of animal fat and cholesterol is positively correlated with the incidence of DCM [38]. High cholesterol and triglyceride caused by diets high in fat, calories, salt and sugar can contribute to the development hyperlipidemia and DCM as well as hypertension [39, 40]. A healthy diet is a primary factor contributing to DCM prevention. The benefits of the Mediterranean diet are related to the reduction various cardiovascular risk factors, including inflammation, vascular endothelial injury, insulin resistance and especially the formation of atherosclerosis and arterial stenosis [41]. Importantly, the benefits of a healthy diet are not limited to DCM, and a good diet should come first in any effort to reduce cardiovascular risk.
Mental stress
Mental stress is a major risk factor for DCM, though the mechanism by which stress contributes to the pathogenesis of coronary atherosclerosis or DCM has not yet been confirmed [42, 43]. Nonetheless, clinical studies have shown that mental stress can be the main factor underlying increases in the prevalence of DCM and the triggering factor for acute myocardial ischemia (as indicated by sudden death, myocardial infarction, ST segment depression) in patients with coronary atherosclerosis [44, 45].
In a laboratory environment, mental stress can cause myocardial ischemia in some patients. The prevalence of myocardial ischemia caused by mental stress depends on the stressor used, the patient group, and especially the diagnostic tool. In fact, ischemic attacks caused by mental stress are usually “silent,” and their severity and scope are smaller than those caused by exercise stress tests. Patients with myocardial ischemia caused by mental stress tend to have higher scores in terms of aggression, anger, and hostility. These psychological characteristics are related to higher cardiovascular responsiveness. After being under tremendous pressure, the heart rate and blood pressure will increase rapidly, and the increase is greater. The mechanism of the myocardial ischemia induced by mental stress may be an increase in myocardial oxygen demand due to the increased heart rate and blood pressure [46, 47]. It is noteworthy that psychological ailments such as depression and anxiety can lead to or affect the condition of patients with DCM [48]. Depression can directly affect the mortality rate among patients with DCM [49, 50]. Attention should therefore be paid to the evaluation and treatment of psychological factors in patients with DCM.
Obesity
Obesity is generally considered to be a risk factor for DCM, in part because a high-calorie/fat diet contributes to the progression of atherosclerosis. Epidemiological studies now tend to support this argument. However, angiographic studies have shown little or no correlation between total fat mass and coronary atherosclerosis [51]. Obesity, especially centripetal obesity, is closely related to the traditional risk factors of DCM and dyslipidemia. This may be due to the high homo-cysteinemia, high lipoprotein levels and increased thrombosis in obese people. Blood lipids are often beyond the standard range in obese individuals, and high calorie diets lead to elevations in LDL, triglycerides and blood pressure, thereby promoting the formation and/or progression of coronary atherosclerosis [52, 53]. Fat in obese people often accumulates within important organs and blood vessels, which leads to abnormal glucose and lipid metabolism, increases in blood pressure, myocardial load, and myocardial oxygen consumption while increasing body weight. Metabolic processes are greatly altered within cardiomyocytes due elevated sugar or calorie sources and low self-consumption rate. The resultant dysregulation of fat and glucose metabolism can lead to an imbalance in mitochondrial oxidative phosphorylation and energy metabolism and serious myocardial injury.
In addition to increasing myocardial load and dyslipidemia, obesity has adverse effects on coronary circulation. These include coronary vasomotor dysfunction and coronary artery occlusion [54].Obesity associated with dyslipidemia easily leads to increases in coronary and microvascular resistance, which is the primary factor contributing to the pathogenesis of coronary microvascular disease. Moreover, adipose tissue with infiltrating macrophages in obese patients are a key source of pro-inflammatory mediators, which can induce microvascular inflammation and myocardial hypoperfusion and further release of pro-inflammatory mediators into the coronary circulation [55, 56]. These mediators impair coronary microcirculation and are a main cause of DCM and heart failure in obese patients. More importantly, in addition to their underlying diseases, obese people are often sedentary, eat an unhealthy diet, and engage in other bad habits. The interaction among these habits hinders the formation of coronary collateral circulation [57]. In addition, obese people often exhibit insulin resistance or type 2 diabetes as well as hyperlipidemia and hyper-fibrinogen [58, 59], all of which are predisposing factors for atherosclerosis and arterial stenosis and, ultimately, DCM.
Sedentary lifestyle
Many studies suggest that a sedentary lifestyle can increase the risk of coronary atherosclerosis or DCM [60]. The rapid development of modern science and technology has changed people's daily live and commuting, resulting in a significant decline in people's activity level. Many office workers and the elderly now have a sedentary lifestyle characterized by long periods of inactivity. This lack of activity/exercise can lead to obesity, hypertension, arrhythmia, atherosclerosis, slowed blood flow, and thrombosis [61, 62]. The coronary atherosclerosis or coronary artery stenosis that often develops as a result of this lifestyle becomes the main cause of DCM. However, interventions that reduce sedentary behavior may improve cardiovascular health and well-being.
Molecular mechanisms of mitochondrial dysfunction in DCM
Mechanisms of DCM
DCM is a special cardiac complication characterized by chronic cardiomyopathy caused by diabetes [10]. The pathological process includes oxidative stress, energy metabolism, inflammatory reaction and increased myocardial cell apoptosis in cardiac myocytes [10, 63] (Table 2). Cardiac manifestations include early diastolic dysfunction, cardiac hypertrophy, ventricular dilatation and systolic dysfunction, which eventually lead to heart failure [64]. In diabetic mice, studies have showed the evidence of impaired mitochondrial function in heart tissue, which is related to mitochondrial ultrastructural defects [65]. Insulin resistance leads to ROS overproduction in cardiomyocytes [66]. Studies have also shown that hyperglycemia can lead to the fragmentation of mitochondria in cardiomyocytes, induce mitochondrial division, and produce mitochondrial ROS [67]. Additionally, high glucose can induce mitochondrial division in cardiomyocytes due to an increase in the o-glcnacylation of Drp1 and the decrease of Drp1 in Ser637 phosphorylation. In addition, high glucose decreased the content of OPA1 and augmented its glycosylation. Increasing OPA1 protein level or decreasing OPA1 glycosylation could block hyperglycemia-related mitochondrial division. Clinical trials show that antagonizing ROS by antioxidants alone is not enough to attenuate DCM [68, 69]. A more effective strategy is to improve the overall ability of mitochondrial quality control to maintain healthy mitochondrial pools needed to support cardiac contractility.
Summary of metabolic processes involved in the pathophysiology of DCM
| Pathological mechanism | Pathophysiological pathway | Structural change | Functional alteration |
|---|---|---|---|
| Deranged Ca2+ homeostasis | Calcium leak from ryanodine receptor; Reduced sarcolemmal elimination of Ca2+; Prolonged Ca2+ transients | Mitochondrial leakage of toxic proteins; Myocardial cytotoxicity | Prolonged diastolic relaxation time; Myocardial stiffness; Impaired relaxation |
| Abnormal fatty acid metabolism | Increased systemic lipolysis; Loss of metabolic flexibility; Increased utilization of free fatty acids | Cardiac steatosis; Lipotoxicity; Myocyte apoptosis | Increased O2 consumption; Pathologic cardiac remodeling; Systolic dysfunction |
| Hyperglycemia | Activation of protein kinase C pathways; Production of free radicals | Myocardial necrosis; Dystrophic calcification | Myocardial fibrosis; LV hypertrophy; Diastolic dysfunction |
| Myocardial fibrosis | Transforming growth factor-β; Matrix metalloproteinase-2; Smooth muscle actin | Interstitial fibrosis; LV hypertrophy; Intimal thickening of microvasculature | Diastolic dysfunction; Systolic dysfunction |
| AGE/RAGE | Janus kinase pathway; MAPK activation | Cross-linking of extracellular matrix; Reduction of myocardial compliance Myocardial fibrosis | Prolonged isovolumetric relaxation time; Elevated LV end-diastolic diameter |
| ROS | Diacylglycerol Protein kinase C; NADPH-oxidase pathway | Oxidative myocardial injury; Mitochondrial damage Cardiac fibrosis | Myocardial stiffness; Diastolic dysfunction |
| Inflammation | NF-κB; Tumor necrosis factor-a; Interleukin-6 | Inflammatory myocardial injury | Systolic dysfunction |
| Cardiac autonomic neuropathy | Hyperadrenergic state; Increased activation of b-receptors and RAAS | Interstitial fibrosis | Diastolic dysfunction |
| Altered protein homeostasis | Impaired ubiquitin proteasome system | Proteotoxicity; Myocardial cell damage | Pathological remodeling in diabetic hearts of animals |
| Microvascular dysfunction | Upregulation of vascular endothelial growth factor pathway | Fibrosis of capillaries | Impaired myocardial functional reserve |
RAGE: AGE-specific receptor, NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells, RAAS: Renin-angiotensin-aldosterone system.
Mechanisms of mitochondrial dysfunction
In recent years, the prevalence and mortality of cardiovascular diseases, such as atherosclerosis, hypertension, myocardial hypertrophy and diabetic cardiomyopathy, are increasing all over the world, which are the main causes of death and disability. Mitochondria are organelles with double membrane structure, which can meet the high energy requirement of heart metabolism through oxidative phosphorylation [70-72]. Mitochondria are extremely sensitive to the changes of their environment. When the external environment, such as impaired nutrition provision and decreased oxygen supply, mitochondria are able to make corresponding metabolic adaptation [73]. However, these protective mitochondrial adaptations, including mitochondrial dynamics regulation, mitophagy activation, mitochondrial biogenesis augmentation, mitochondrial bioenergetics improvement and mitochondrial anti-oxidative capacity intensification are usually impaired in many cardiovascular diseases [74], which is therefore accompanied by mitochondrial respiratory chain dysfunction, ATP synthesis disorder, oxidative stress and mitochondrial integrity loss [75, 76]. In dysfunctional mitochondria, the decoupling of electron transport chain leads to ROS production and ATP depletion, causing extensive damage to cardiomyocytes and activating cell apoptosis or necrosis [77]. At present, the relationship between mitochondrial dysfunction and cardiovascular diseases has been confirmed [78]. Among them, abnormal mitochondrial dynamics and impaired mitochondrial function have been found to be closely associated with decreased mitochondrial biosynthesis, increased mitochondria lipid oxidative damage, and excessive mitochondrial DNA breakage [79-81]; these alterations have been identified as the pathophysiological basis of a variety of cardiovascular diseases.
Mitochondrion is the power source of cells. ATP is synthesized by the metabolites of fatty acids, glucose and amino acids [82, 83]; these alterations are primarily occurred in mitochondria [84]. In addition, mitochondria play an important role in calcium homeostasis [85], hemoglobin synthesis, fatty acid oxidation, ROS production and clearance, and cell growth and apoptosis regulation [86, 87]. Therefore, the normal structure and function of mitochondria have a role in meeting the energy requirements of vital activities, maintaining the homeostasis of cellular environment and regulating cell growth.
Mitochondrial dysfunction refers to the structural and functional abnormalities of mitochondria caused by the accumulation of ROS in cells under the stimulation of various damage factors, such as ischemia and hypoxia [88]. The features of mitochondrial dysfunction include a decrease of membrane potential, a reduction in mitochondrial biosynthesis, a drop in ATP synthesis and a damage to mitochondrial respiratory chain. Besides, the well-known outcome of mitochondrial dysfunction is ROS overproduction at the mitochondrial respiratory chain, causing oxidative damage to cell and mitochondrial protein, lipid and DNA [89]. Small ROS production will further augment mitochondria damage and thus promote mitochondrial ROS production, which forms a vicious circle of mitochondrial damage. Once the ROS production exceeds the scavenging capacity of antioxidant system, cellular oxidative stress and tissue damage occur. It is necessary to point out that most of the ROS in the heart are produced by the decoupling of mitochondrial respiratory chain.
Pathophysiology of mitochondrial quality control (MQC) in DCM
Mitochondrial fusion and fission dynamics
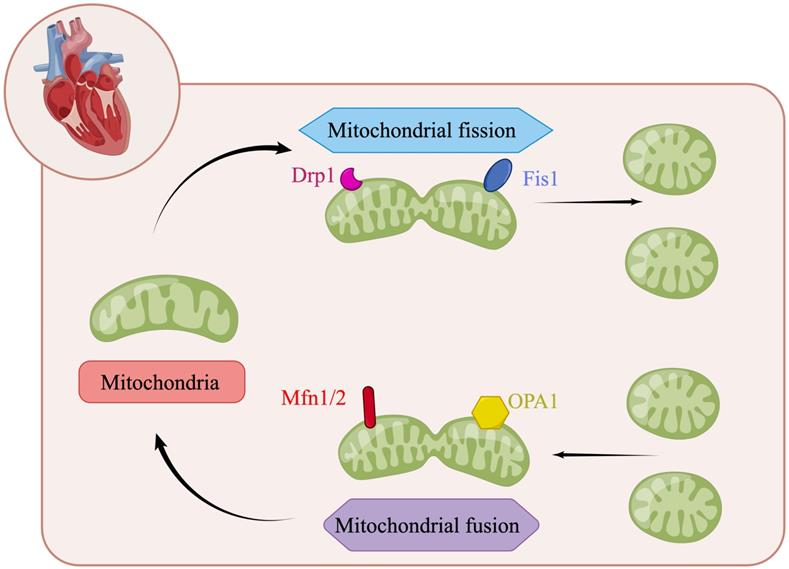
Mitochondria are highly dynamic organelles in most mammalian cells. Through continuous fusion and division, the shape, size and quantity of mitochondria can be changed to meet the metabolic needs of cells (Figure 2) [66, 90]. In mitochondrial dynamics, the regulation of mitochondrial division and fusion is a group of dynamic-related GTP enzymes [91], including fusion of mitochondrial endocardium and mitochondrial outer membrane. Mitochondrial fusion protein Mfn1/2 promotes mitochondrial outer membrane fusion whereas OPA1 promotes fusion of inter membrane of mitochondria [92]. Mammalian Mfn1/2 is a highly similar protein (human similarity is about 80%), which is composed of 737 and 757 amino acid sequences, respectively [93]. They have homology, about 80% similar structure related sequences, and have homogeneity and heterogeneity in physical function. They will form Mfn1 homopolymer, Mfn2 homopolymer and Mfn1/2 heteropolymer. The second component regulating mitochondrial fusion is OPA1, a transmembrane protein closely related to mitochondrial inner membrane, which is expressed in a variety of variants through alternative splicing and post-translational proteolysis, resulting in short (s) and long (L) subtypes. Deletion or mutation of any of these genes will lead to embryo death and mitochondrial dysfunction. The depletion of Mfn1 and/or Mfn2 in cells leads to poor cell growth, and the decrease of cell respiration due to decreased mitochondrial membrane potential. Knockout of Mfn1 or Mfn2 is also associated with embryo death due to placental defects [94]. OPA1 deficiency was characterized by mitochondrial fragmentation, decreased cristae and oxidative phosphorylation [95]. The regulatory factors of mitochondrial division are Drp1, Mff, Fis1, Mid49 and Mid51. Drp1-dependent mitosis can be divided into four steps [96-99]: translocation of Drp1 to the outer membrane of mitochondria, subsequent high-level assembly, hydrolysis of GTP, and final disassembly. Drp1 is an 80 kDa dynein GTPase superfamily protein [100, 101]. It mainly exists in the cytoplasm in the form of dimer/tetramer, shuttling between the cytoplasm and mitochondria. The recruitment of Drp1 from the cytoplasm to the outer membrane of mitochondria is an important step in mitochondrial division. Recruitment from cytoplasm to mitochondria is mediated by several outer mitochondrial membrane proteins, including Mff, Fis1, Mid49 and Mid51 [96, 102]. Drp1 is recruited into mitochondria through a receptor anchored on the outer membrane of mitochondria [103]. Once recruited, Drp1 further assembles around the mitochondrial tubules to form an oligomeric ring, which contracts and splits mitochondria in a GTP dependent process [104]. Mff is an important factor in the recruitment of Drp1 during mitosis, and its overexpression leads to increased mitosis [105]. In contrast, other drp1 receptors, Mid49 and Mid51, seem to recruit inactive forms of drp1 because their overexpression inhibits mitochondrial division [106].
The regulation of mitochondrial dynamics. Mitochondrial fusion protein Mfn1/2 promotes mitochondrial outer membrane fusion whereas OPA1 promotes fusion of inter membrane of mitochondria. The regulatory factors of mitochondrial division are Drp1, Mff, Fis1, Mid49 and Mid51. (By Figdraw (www.figdraw.com)).

Mitochondrial fusion and fission dynamics in DCM
Mitochondrial morphological changes in cardiomyocytes were observed in ob/ob mice [107]. In these mice, mitochondria appeared as abnormally large 'mega-mitochondria' [107]. Cardiomyocytes from neonatal rats exposed prenatally to diabetes or a high-fat diet exhibited impaired mitochondrial dynamics, which resulted in shorter, wider mitochondria than those from control rats [108]. These hyperglycemia-induced changes in mitochondrial dynamics seemed to be gender-specific; in fact, the male hearts contained post-translational modifications known to impair mitochondrial dynamics [108].
Another study demonstrated that Drp1 was significantly upregulated in cardiomyocytes during the progression of DCM, whereas Mfn1 and 2 were markedly downregulated [109]. Genetic ablation of Drp1 was found to protect the heart against hyperglycemic damage by sustaining cardiomyocyte viability and function [110]. In diabetic hearts, Drp1 phosphorylation at Ser616 was induced, and this alteration was followed by cardiomyocyte hypertrophy and mitochondrial dysfunction, suggesting that post-translational modifications of mitochondrial dynamics proteins may contribute to mitochondrial dysfunction during DCM [111]. In the setting of DCM, other phosphorylated forms of Drp1 have also been detected, such as p-Drp1Ser637 [112], p-Drp1Ser579 [113] and p-Drp1Ser600 [113, 114]. Moreover, increased O-GlcNAcylation of Drp1 at threonine 585 and 586 was observed in diabetic heart mitochondria, correlating with elevated mitochondrial fragmentation in cardiomyocytes [115]. Interestingly, lipid overload during diabetes was associated with Drp1 acetylation at lysine 642, which then promoted mitochondrial fission and cardiomyocyte death [116]. In contrast, transfection of a nonacetylated Drp1 mutant (K642R) prevented hyperglycemia-induced cardiomyocyte hypertrophy and dysfunction [116, 117].
Regarding mitochondrial fusion, an in vitro model of DCM (primary cultured neonatal rat cardiomyocytes treated with high glucose) exhibited elevated Mfn1/2 ubiquitination, a reduced mitochondrial membrane potential, increased mPTP opening and diminished ATP production [118]. Hyperglycemia also induced O-GlcNAcylation of OPA1 in neonatal cardiac myocytes, thus reducing the mitochondrial length and suppressing complex IV activity [119]. Of note, a fructose-rich diet stimulated mitochondrial fragmentation in the heart by suppressing Mfn2 and inducing Drp1 without influencing OPA1 [120], suggesting that different kinds of sugar may distinctly impact the regulators of mitochondrial dynamics in diabetic hearts.
Several upstream signals of abnormal mitochondrial dynamics have been described in the setting of DCM, including translocase of outer mitochondrial membrane 70 (Tom70) [121], cyclin C [122], sirtuin 1 (Sirt1) [123], estrogen [124], Gp78 [125], insulin [126] and norepinephrine [127]. The molecular effects of these upstream signals on mitochondrial dynamics during DCM are detailed in Table 3. In turn, the activation of mitochondrial fission in hyperglycemia-treated cardiomyocytes has multiple downstream effects, including cardiomyocyte apoptosis, oxidative stress, myocardial fibrosis [128], mitochondrial membrane potential reduction, insulin pathway deactivation, insulin resistance [129], delayed mitochondrial respiration and mitochondrial calcium overload [130]. Together, these alterations may eventually induce mitochondrial dysfunction and reduce cardiomyocyte viability, accelerating the development of DCM.
Upstream signals of mitochondrial dynamics in the setting of DCM
| Upstream regulator | Mechanism | Reference |
|---|---|---|
| Tom70 | Tom70 enhances high-glucose and high-fat treatment-induced mitochondrial superoxide production, resulting in Drp1-induced mitochondrial fission. | [121] |
| Cyclin C | Cyclin C translocates to the cytoplasm and binds to cyclin-dependent kinase 1 to promote Drp1 phosphorylation at Ser616. | [122] |
| Sirt1 | Sirt1 deficiency promotes Akt activation, thus increasing Drp1 activity, culminating in excessive mitochondrial fission and ROS production. | [123] |
| Estrogen | Estrogen upregulates Drp1 and downregulates Mfn2 in diabetic rats. | [124] |
| Gp78 | The Gp78-ubiquitin proteasome system promotes the ubiquitination of Mfn1/2. | [125] |
| Insulin | Insulin treatment increases OPA1 protein levels, promotes mitochondrial fusion, increases the mitochondrial membrane potential and elevates both intracellular ATP production and oxygen consumption in cardiomyocytes. | [126] |
| Norepinephrine | Norepinephrine acts through α1-adrenergic receptors to increase cytoplasmic Ca2+ levels, thus activating calcineurin and promoting Drp1 migration to mitochondria. | [127] |
Mitophagy
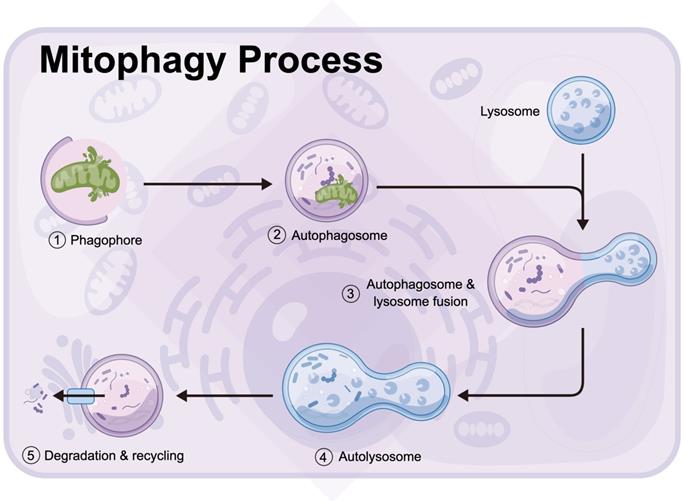
Mitophagy is a process in which autophagosomes selectively target to phagocytize dysfunctional or damaged mitochondria and transfer them to lysosomes for cell recycling (Figure 3) [131, 132]. The division of mitochondria is considered by biologists to be mainly to separate the mitochondria of daughter cells with damaged membrane potential [133, 134]. The daughter mitochondria with normal membrane potential can fuse with other mitochondria [135]. The stability of mitochondrial internal environment requires a perfect balance between mitochondrial phagocytosis and mitochondrial biogenesis [136, 137]. Mitophagy recognizes target mitochondria through LC3 adaptor through the ubiquitin-dependent and -independent pathways [138]. The first step of ubiquitin-dependent mitophagy is the ubiquitination of mitochondrial substrate, which is the recognized by LC3 adapter [139, 140]. At present, the most studied mitophagy pathway in mammals is regulated by the putative kinases such as PINK1 and Parkin, which is under the control of phosphatase and tensin homolog PTEN [141]. PINK1 is a sensor of mitochondrial polarization. In healthy polarized mitochondria, PINK1 is introduced into mitochondria by transporter outer membrane complex and into mitochondria by transporter inner membrane complex. Under normal conditions, the basal PINK1 level was maintained at a low level [142]. In the process of mitochondrial depolarization, the decrease of mitochondrial membrane potential is related to the accumulation of PINK1 on the outer membrane of mitochondria, which, together with ubiquitin ligase Parkin, controls the elimination of defective mitochondria [143]. In addition, PINK1 can phosphorylate the Ser65 site of ubiquitin like (UBL) domain, activate the E3 ligase activity of Parkin and promote the recruitment of Parkin in the outer membrane of mitochondria [144, 145]. There are many Parkin substrates on the mitochondrial outer membrane, including mitochondrial fusion protein Mfn1/2, mitochondrial outer membrane translocator, mitochondrial transporter and voltage dependent anion channel VDAC [146]. By interacting with LC3 aptamer on the membrane, autophagosomes were recruited to selectively encapsulate the modified receptor mitochondria, and finally the damaged mitochondria were transferred to lysosomes for degradation. LC3 adaptor proteins include p62, NBR1, OPTN, NDP53 and TAX1BP1, but only OPTN and NDP52 are considered as their main substrates.
Mitophagy is a process in which autophagosomes selectively target to phagocytize dysfunctional or damaged mitochondria and transfer them to lysosomes for cell recycling (by Figdraw (www.figdraw.com)).
In addition to the above ubiquitin dependent pathway and LC3 binding mediated mitochondrial autophagy, there are some damaged mitochondria that can also be recognized by LC3 adapter in a ubiquitin independent manner. These LC3 receptors are located in mitochondria, which can directly bind to LC3 and transfer damaged mitochondria to lysosomes for degradation. These receptors include BNIP3, Nix, Fundc1, PHB2 [147-149].
Mitophagy in DCM
The changes in mitophagy during DCM were evaluated using the mito-Keima assay in cardiomyocytes from GFP-LC3 mice fed a high-fat diet [150]. Mitophagy was significantly induced after three weeks and further increased after two months of high-fat-diet treatment, accompanied by an increased end-diastolic pressure-volume relationship, lipid accumulation and cardiac hypertrophy [150]. Knocking out Parkin partially suppressed cardiomyocyte mitophagy, elevated lipid accumulation and worsened diastolic dysfunction in response to high-fat-diet feeding [150]. These data suggested that mitophagy is enhanced in the early stage of DCM. Although mitophagy is cardioprotective, endogenous mitophagy fails to prevent the progression of DCM. At the molecular level, mitophagy induction has been described as an adaptive response to increased fatty acid oxidation in the heart, because Parkin expression seems to correlate with the levels of acetyl CoA carboxylase 2, a regulator of long-chain fatty acid transport into mitochondria [151].
Using a novel dual-fluorescent mitophagy reporter (mt-Rosella), Kobayashi et al. traced dysfunctional mitochondria that were degraded in lysosomes, and found that chronic hyperglycemic stress impaired mitophagy in heart tissues from type-1 diabetic mice [152]. In another study, mice were fed a high-fat diet for 10 weeks, and changes in FUNDC1-dependent mitophagy were monitored in the early stage of DCM [153]. FUNDC1 expression was slightly elevated and therefore mitophagy was moderately enhanced in diabetic heart tissues [153]. However, ablation of FUNDC1 exacerbated myocardial inflammation, oxidative stress and cardiomyocyte apoptosis, suggesting that FUNDC1-dependent mitophagy is a defensive program, despite its failure to halt the progression of DCM [153].
Mitochondrial morphological changes in cardiomyocytes from diabetic mice were observed using electron microscopy [154]. Most of the cardiomyocytes contained dissociated mitochondria that lacked typical contacts, and some of the mitochondria exhibited complete destruction of the cristae, “watering” of the matrix, and remains that resembled large vacuoles [154, 155]. Expanded sarcoplasmic reticular profiles and glycogen accumulation were often recorded, and the contractile apparatus of the cardiomyocytes exhibited displaced myofibrils and tortuous Z-disks that were mismatched between adjacent myofibrils [154]. However, activation of Parkin-induced mitophagy partly reversed the ultrastructural changes in cardiomyocyte mitochondria and prevented myocardial tissue disorganization [154]. These findings confirmed that enhanced mitophagy, rather than basal mitophagy, is needed to sustain mitochondrial structure and myocardial homeostasis [156].
The protective mechanisms of increased mitophagy have been widely reported. Mitophagy was shown to attenuate mitochondria-induced cardiomyocyte apoptosis by reducing OMM hyperpermeability, cytochrome c release and oxidative stress [157]. In addition to inhibiting apoptosis, mitophagy was found to repress cardiac ferroptosis, necroptosis and lipid peroxidation [153]. Activation of Parkin-induced mitophagy maintained the mitochondrial membrane potential, normalized mitochondrial energy metabolism and balanced the redox response in the heart [158]. During the development of DCM, mitophagy enhanced cardiomyocyte mitochondrial regeneration and biogenesis [159] and normalized the cardiac mitochondrial morphology and bioenergetics [160]. By improving mitochondrial homeostasis [161], mitophagy was found to reduce lipid accumulation and increase both diastolic function (end-diastolic pressure-volume relationship) and systolic function (end-systolic pressure-volume relationship) in diabetic hearts [150]. Moreover, the activation of mitophagy reduced body weight, improved hyperglycemic control, inhibited dyslipidemia, attenuated myocardial fibrosis and suppressed the inflammatory response (as evidenced by reduced transforming growth factor β1, hydroxyproline and brain natriuretic peptide levels) in the hearts of diabetic rats [162].
Targeted pharmacological approaches to activate mitophagy may be useful clinical therapeutic strategies to retard the progression and/or improve the prognosis of DCM (Table 4). The US Food and Drug Association has approved the anti-diabetic drug empagliflozin for the treatment of HF, regardless of diabetes status [163]. Empagliflozin was reported to prevent HF by increasing the autophagic vacuole number and reducing myocardial fibrosis in diabetic hearts [164, 165]. Liraglutide, a glucagon‑like peptide‑1 receptor agonist that has been used to treat diabetes and obesity, was shown to activate Sirt1, a protein deacetylase that depends on both adenosine monophosphate-activated protein kinase (AMPK) and NAD [158]. Liraglutide thereby increased mitophagy activity in diabetic hearts; however, deletion of Parkin significantly abrogated these cardioprotective effects [158].
Targeted pharmacological or non-pharmacological therapeutic strategies to activate mitophagy for the treatment of DCM
| Therapeutic strategy | Mechanism | Reference |
|---|---|---|
| Empagliflozin | Empagliflozin prevents diabetic HF by increasing the autophagic vacuole number in the heart, thus reducing myocardial fibrosis. | [164] |
| Ginseng Dingzhi Decoction | Ginseng Dingzhi Decoction activates mitophagy and thus ameliorates myocardial hypertrophy, heart function and mitochondrial homeostasis following high-glucose stimulation. | [169] |
| Liraglutide | Liraglutide activates the AMPK- and NAD‑dependent protein deacetylase Sirt1, thus increasing Parkin-induced mitophagy in diabetic hearts. | [158] |
| Melatonin | Melatonin increases the number of typical autophagosomes engulfing mitochondria through Parkin-induced mitophagy in diabetic hearts, thus reducing cardiac remodeling. | [160] |
| Hydrogen sulfide | Hydrogen sulfide facilitates Parkin translocation onto mitochondria and thus promotes mitophagy in the heart, ultimately reducing mitochondrial fragmentation, enhancing mitochondrial respiratory chain activity, suppressing mitochondrial apoptosis and improving cardiac function in db/db mice. | [170] |
| Alisporivir | Alisporivir upregulates PINK1 and Parkin mRNA expression in the heart tissues of diabetic mice. | [154] |
| D-β-hydroxybutyrate-(R)-1,3 butanediol monoester (ketone ester) diet | A ketone ester diet improves cytosolic E3 ubiquitin ligase translocation onto mitochondria and reinforces LC3-induced autophagosome formation, thus enhancing cardiac systolic and diastolic function in animals with type-2 diabetes mellitus. | [168] |
Melatonin, a regulator of the biological clock, was reported to increase the number of typical autophagosomes engulfing mitochondria in the heart, thus preventing hyperglycemia-induced cardiac remodeling during DCM [166]; however, knocking out Parkin partly compromised these beneficial effects [160]. In the hearts of db/db mice, the gasotransmitter hydrogen sulfide facilitated Parkin translocation onto mitochondria, thereby promoting mitophagy, improving cardiac function, reducing mitochondrial fragmentation, enhancing mitochondrial respiratory chain activity and suppressing mitochondrial apoptosis [167]. Alisporivir, a non-immunosuppressive cyclosporin derivative and selective inhibitor of the mPTP, exerted cardioprotective effects by upregulating PINK1 and Parkin mRNA expression in the heart tissues of diabetic mice, thus inducing mitophagy [154].
The D-β-hydroxybutyrate-(R)-1,3 butanediol monoester (ketone ester) diet was reported as a non-pharmacological approach to enhancing cardiac mitophagy [168]. The ketone ester diet enhanced the resistance of mitochondria to oxidative stress, inhibited mPTP opening and increased mitochondrial succinyl-CoA:3-oxoacid-CoA transferase expression in cardiomyocytes [168]. In addition, the ketone ester diet improved cytosolic E3 ubiquitin ligase translocation onto mitochondria and reinforced LC3-induced autophagosome formation in cardiomyocytes, leading to better cardiac systolic and diastolic function in mice with type-2 diabetes mellitus [168].
Conclusions and perspectives
The quality of mitochondria is maintained through the synthesis of new mitochondria, fusion and division, and the elimination of damaged mitochondria by mitophagy (Figure 4). With the increase of age, the changes of mitochondrion division and fusion process and the inhibition of mitophagy will lead to the decrease of mitochondrial biogenesis and the damage to mitochondrial clearance, which contributes to a series of metabolic disorders and pathophysiological diseases. In this review, we briefly discuss the roles of mitochondrial dynamics and mitophagy in regulating DCM. Meanwhile, the potential targeted therapies against mitochondrial dysfunction are also introduced in this review. However, it requires more attention to describe the detailed action afforded by MQC in DCM. Besides, new therapeutic approaches with a focus on MQC in the setting of DCM are the next clinical issue in the world.
Physiological MQC is an endogenous defense program that restores the mitochondrial integrity and homeostasis in response to mitochondrial damage; however, hyperglycemia compromises this protective mechanism. Mitochondrial fission is overactivated in diabetic hearts, while fusion is markedly inhibited, resulting in extensive mitochondrial fragmentation. Under physiological conditions, mitophagy can engulf fragmented mitochondria; however, this process is inhibited under high-glucose conditions, so dysfunctional mitochondria accumulate within cardiomyocytes. Likewise, mitochondrial biogenesis can regenerate or replicate mitochondria, but hyperglycemia suppresses this process by inhibiting AMPK/PGC-1α. When MQC is blunted, mitochondrial dysfunction cannot be rectified, so the mitochondrial quality and quantity are further diminished.
Acknowledgements
Funding
This study is supported by the National Natural Science Foundation of China (NO. 82102262) and Guangdong Basic and Applied Basic Research Foundation (NO. 2021A1515010977 and NO. 2020A1515110174).
Author Contributions
Chen Cai, Feng Wu and Jing He collected and analyzed the article and/or data. Yaoyuan Zhang, Nengxian Shi, and Xiaojie Peng contributed to manuscript preparation. Qing Qu, Ziying Li and Xiaoqing Jiang made the pictures and tables. Jiankai Zhong and Ying Tan contributed to manuscript writing and editing.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mosterd A, Hoes AW. Clinical epidemiology of heart failure. Heart. 2007;93:1137-46
2. Snipelisky D, Chaudhry SP, Stewart GC. The Many Faces of Heart Failure. Card Electrophysiol Clin. 2019;11:11-20
3. Hoffman TM. Chronic Heart Failure. Pediatr Crit Care Med. 2016;17:S119-23
4. Dillmann WH. Diabetic Cardiomyopathy. Circ Res. 2019;124:1160-2
5. Jia G, Whaley-Connell A, Sowers JR. Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia. 2018;61:21-8
6. McSweeney JC, Rosenfeld AG, Abel WM, Braun LT, Burke LE, Daugherty SL. et al. Preventing and Experiencing Ischemic Heart Disease as a Woman: State of the Science: A Scientific Statement From the American Heart Association. Circulation. 2016;133:1302-31
7. Zhu H, Zhou H. Novel Insight into the Role of Endoplasmic Reticulum Stress in the Pathogenesis of Myocardial Ischemia-Reperfusion Injury. Oxid Med Cell Longev. 2021;2021:5529810
8. Mehta LS, Beckie TM, DeVon HA, Grines CL, Krumholz HM, Johnson MN. et al. Acute Myocardial Infarction in Women: A Scientific Statement From the American Heart Association. Circulation. 2016;133:916-47
9. Kannel WB. Prevalence and clinical aspects of unrecognized myocardial infarction and sudden unexpected death. Circulation. 1987;75:Ii4-5
10. Lorenzo-Almorós A, Cepeda-Rodrigo JM, Lorenzo Ó. Diabetic cardiomyopathy. Rev Clin Esp. 2020
11. Peer N, Baatiema L, Kengne AP. Ischaemic heart disease, stroke, and their cardiometabolic risk factors in Africa: current challenges and outlook for the future. Expert review of cardiovascular therapy. 2021;19:129-40
12. Shaw LJ, Shaw RE, Merz CN, Brindis RG, Klein LW, Nallamothu B. et al. Impact of ethnicity and gender differences on angiographic coronary artery disease prevalence and in-hospital mortality in the American College of Cardiology-National Cardiovascular Data Registry. Circulation. 2008;117:1787-801
13. Martín-Fernández B, Gredilla R. Mitochondria and oxidative stress in heart aging. Age (Dordrecht, Netherlands). 2016;38:225-38
14. Costantino S, Paneni F, Cosentino F. Ageing, metabolism and cardiovascular disease. The Journal of physiology. 2016;594:2061-73
15. Jakovljevic DG. Physical activity and cardiovascular aging: Physiological and molecular insights. Exp Gerontol. 2018;109:67-74
16. Niccoli G, Scalone G, Crea F. Acute myocardial infarction with no obstructive coronary atherosclerosis: mechanisms and management. European heart journal. 2015;36:475-81
17. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML. et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women's Health Initiative randomized controlled trial. Jama. 2002;288:321-33
18. Dal Canto E, Ceriello A, Rydén L, Ferrini M, Hansen TB, Schnell O. et al. Diabetes as a cardiovascular risk factor: An overview of global trends of macro and micro vascular complications. European journal of preventive cardiology. 2019;26:25-32
19. Al-Mallah MH, Sakr S, Al-Qunaibet A. Cardiorespiratory Fitness and Cardiovascular Disease Prevention: an Update. Current atherosclerosis reports. 2018;20:1
20. Ylä-Herttuala S, Baker AH. Cardiovascular Gene Therapy: Past, Present, and Future. Molecular therapy: the journal of the American Society of Gene Therapy. 2017;25:1095-106
21. Musunuru K, Hickey KT, Al-Khatib SM, Delles C, Fornage M, Fox CS. et al. Basic concepts and potential applications of genetics and genomics for cardiovascular and stroke clinicians: a scientific statement from the American Heart Association. Circulation Cardiovascular genetics. 2015;8:216-42
22. Fox CS, Hall JL, Arnett DK, Ashley EA, Delles C, Engler MB. et al. Future translational applications from the contemporary genomics era: a scientific statement from the American Heart Association. Circulation. 2015;131:1715-36
23. Sharma K, Baliga RR. Genetics of Dyslipidemia and Ischemic Heart Disease. Current cardiology reports. 2017;19:46
24. Khera AV, Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nature reviews Genetics. 2017;18:331-44
25. Musunuru K, Kathiresan S. Genetics of Common, Complex Coronary Artery Disease. Cell. 2019;177:132-45
26. Wang L, Fan C, Topol SE, Topol EJ, Wang Q. Mutation of MEF2A in an inherited disorder with features of coronary artery disease. Science (New York, NY). 2003;302:1578-81
27. Nordlie MA, Wold LE, Kloner RA. Genetic contributors toward increased risk for ischemic heart disease. Journal of molecular and cellular cardiology. 2005;39:667-79
28. Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol. 2004;43:1731-7
29. Huxley RR, Woodward M. Cigarette smoking as a risk factor for coronary heart disease in women compared with men: a systematic review and meta-analysis of prospective cohort studies. Lancet. 2011;378:1297-305
30. Rigotti NA, Pasternak RC. Cigarette smoking and coronary heart disease: risks and management. Cardiology clinics. 1996;14:51-68
31. Shen Y, Wang X, Liu Y, Singhal M, Gürkaşlar C, Valls AF. et al. STAT3-YAP/TAZ signaling in endothelial cells promotes tumor angiogenesis. Sci Signal. 2021;14:eabj8393
32. Leistikow BN. Smoking and ischemic heart disease disparities between studies, genders, times, and socioeconomic strata. Journal of cardiovascular translational research. 2009;2:267-73
33. Siasos G, Tsigkou V, Kokkou E, Oikonomou E, Vavuranakis M, Vlachopoulos C. et al. Smoking and atherosclerosis: mechanisms of disease and new therapeutic approaches. Current medicinal chemistry. 2014;21:3936-48
34. Messner B, Bernhard D. Smoking and cardiovascular disease: mechanisms of endothelial dysfunction and early atherogenesis. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:509-15
35. Golbidi S, Edvinsson L, Laher I. Smoking and Endothelial Dysfunction. Current vascular pharmacology. 2020;18:1-11
36. Wang Z, Wang D, Wang Y. Cigarette Smoking and Adipose Tissue: The Emerging Role in Progression of Atherosclerosis. Mediators of inflammation. 2017;2017:3102737
37. Sriram G, Milling LE, Chen JK, Kong YW, Joughin BA, Abraham W. et al. The injury response to DNA damage in live tumor cells promotes antitumor immunity. Sci Signal. 2021;14:eabc4764
38. DiNicolantonio JJ, Lucan SC, O'Keefe JH. The Evidence for Saturated Fat and for Sugar Related to Coronary Heart Disease. Progress in cardiovascular diseases. 2016;58:464-72
39. Ruiz-Canela M, Bes-Rastrollo M, Martínez-González MA. The Role of Dietary Inflammatory Index in Cardiovascular Disease, Metabolic Syndrome and Mortality. Int J Mol Sci. 2016 17
40. Ulbricht TL, Southgate DA. Coronary heart disease: seven dietary factors. Lancet. 1991;338:985-92
41. Whayne TF Jr. Ischemic heart disease and the Mediterranean diet. Current cardiology reports. 2014;16:491
42. Wirtz PH, von Känel R. Psychological Stress, Inflammation, and Coronary Heart Disease. Current cardiology reports. 2017;19:111
43. Richards SH, Anderson L, Jenkinson CE, Whalley B, Rees K, Davies P. et al. Psychological interventions for coronary heart disease. The Cochrane database of systematic reviews. 2017;4:Cd002902
44. Steptoe A, Kivimäki M. Stress and cardiovascular disease. Nature reviews Cardiology. 2012;9:360-70
45. Boland TA, Lee VH, Bleck TP. Stress-induced cardiomyopathy. Critical care medicine. 2015;43:686-93
46. Cas LD, Metra M, Nodari S, Nardi M, Giubbini R, Visioli O. [Stress and ischemic heart disease]. Cardiologia (Rome, Italy). 1993;38:415-25
47. Baffi TR, Lordén G, Wozniak JM, Feichtner A, Yeung W, Kornev AP. et al. mTORC2 controls the activity of PKC and Akt by phosphorylating a conserved TOR interaction motif. Sci Signal. 2021 14
48. Suárez-Mendoza A, Petersen-Aranguren F, Almeida-Velasco A, Robles-García R, Camacho Á, Fresán-Orellana A. Psychometric evaluation of the Hospital Anxiety and Depression Scale in Mexican adults with ischemic and hypertensive cardiomyopathy. Archivos de cardiologia de Mexico. 2019;89:221-6
49. Hare DL, Toukhsati SR, Johansson P, Jaarsma T. Depression and cardiovascular disease: a clinical review. European heart journal. 2014;35:1365-72
50. Ma H, Wang Y, Xue Y, Huang D, Kong Y, Zhao X. et al. The effect of Xinkeshu tablets on depression and anxiety symptoms in patients with coronary artery disease: Results from a double-blind, randomized, placebo-controlled study. Biomed Pharmacother. 2019;112:108639
51. Alexander JK. Obesity and coronary heart disease. The American journal of the medical sciences. 2001;321:215-24
52. Jokinen E. Obesity and cardiovascular disease. Minerva pediatrica. 2015;67:25-32
53. Oliveira MM, Lourenco MV, Longo F, Kasica NP, Yang W, Ureta G. et al. Correction of eIF2-dependent defects in brain protein synthesis, synaptic plasticity, and memory in mouse models of Alzheimer's disease. Sci Signal. 2021 14
54. Bagi Z, Broskova Z, Feher A. Obesity and coronary microvascular disease - implications for adipose tissue-mediated remote inflammatory response. Current vascular pharmacology. 2014;12:453-61
55. Owan T, Litwin SE. Is there a cardiomyopathy of obesity? Current heart failure reports. 2007;4:221-8
56. Tiwari S, Ndisang JF. The role of obesity in cardiomyopathy and nephropathy. Current pharmaceutical design. 2014;20:1409-17
57. Nakamura M, Sadoshima J. Cardiomyopathy in obesity, insulin resistance and diabetes. The Journal of physiology. 2020;598:2977-93
58. Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nature reviews Cardiology. 2011;8:30-41
59. Samson AL, Garnish SE, Hildebrand JM, Murphy JM. Location, location, location: A compartmentalized view of TNF-induced necroptotic signaling. Sci Signal. 2021 14
60. Wu NN, Tian H, Chen P, Wang D, Ren J, Zhang Y. Physical Exercise and Selective Autophagy: Benefit and Risk on Cardiovascular Health. Cells. 2019 8
61. Li S, Culver B, Ren J. Benefit and risk of exercise on myocardial function in diabetes. Pharmacological research. 2003;48:127-32
62. Winzer EB, Woitek F, Linke A. Physical Activity in the Prevention and Treatment of Coronary Artery Disease. J Am Heart Assoc. 2018 7
63. Chang X, Lochner A, Wang HH, Wang S, Zhu H, Ren J. et al. Coronary microvascular injury in myocardial infarction: perception and knowledge for mitochondrial quality control. Theranostics. 2021;11:6766-85
64. Murtaza G, Virk HUH, Khalid M, Lavie CJ, Ventura H, Mukherjee D. et al. Diabetic cardiomyopathy - A comprehensive updated review. Prog Cardiovasc Dis. 2019;62:315-26
65. Vásquez-Trincado C, García-Carvajal I, Pennanen C, Parra V, Hill JA, Rothermel BA. et al. Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol. 2016;594:509-25
66. Zhu H, Toan S, Mui D, Zhou H. Mitochondrial quality surveillance as a therapeutic target in myocardial infarction. Acta Physiol (Oxf). 2021;231:e13590
67. Bubb KJ, Drummond GR, Figtree GA. New opportunities for targeting redox dysregulation in cardiovascular disease. Cardiovasc Res. 2020;116:532-44
68. Saito T, Hamano K, Sadoshima J. Molecular mechanisms and clinical implications of multiple forms of mitophagy in the heart. Cardiovasc Res. 2020
69. Lone AM, Giansanti P, Jørgensen MJ, Gjerga E, Dugourd A, Scholten A. et al. Systems approach reveals distinct and shared signaling networks of the four PGE(2) receptors in T cells. Sci Signal. 2021;14:eabc8579
70. Huang S, Li Z, Wu Z, Liu C, Yu M, Wen M. et al. DDAH2 suppresses RLR-MAVS-mediated innate antiviral immunity by stimulating nitric oxide-activated, Drp1-induced mitochondrial fission. Sci Signal. 2021 14
71. Crooks DR, Maio N, Lang M, Ricketts CJ, Vocke CD, Gurram S. et al. Mitochondrial DNA alterations underlie an irreversible shift to aerobic glycolysis in fumarate hydratase-deficient renal cancer. Sci Signal. 2021 14
72. Zheng H, Zhu H, Liu X, Huang X, Huang A, Huang Y. Mitophagy in Diabetic Cardiomyopathy: Roles and Mechanisms. Front Cell Dev Biol. 2021;9:750382
73. Liao H, Qi Y, Ye Y, Yue P, Zhang D, Li Y. Mechanotranduction Pathways in the Regulation of Mitochondrial Homeostasis in Cardiomyocytes. Front Cell Dev Biol. 2020;8:625089
74. Poznyak AV, Ivanova EA, Sobenin IA, Yet SF, Orekhov AN. The Role of Mitochondria in Cardiovascular Diseases. Biology (Basel). 2020 9
75. Ledderose C, Bromberger S, Slubowski CJ, Sueyoshi K, Aytan D, Shen Y. et al. The purinergic receptor P2Y11 choreographs the polarization, mitochondrial metabolism, and migration of T lymphocytes. Sci Signal. 2020 13
76. Zhou H, Toan S. Pathological Roles of Mitochondrial Oxidative Stress and Mitochondrial Dynamics in Cardiac Microvascular Ischemia/Reperfusion Injury. Biomolecules. 2020 10
77. Hu Z, Ju F, Du L, Abbott GW. Empagliflozin protects the heart against ischemia/reperfusion-induced sudden cardiac death. Cardiovasc Diabetol. 2021;20:199
78. Diaz-Vegas A, Sanchez-Aguilera P, Krycer JR, Morales PE, Monsalves-Alvarez M, Cifuentes M. et al. Is Mitochondrial Dysfunction a Common Root of Noncommunicable Chronic Diseases? Endocr Rev. 2020;41:491-517
79. Larson-Casey JL, He C, Carter AB. Mitochondrial quality control in pulmonary fibrosis. Redox Biol. 2020;33:101426
80. Anzell AR, Maizy R, Przyklenk K, Sanderson TH. Mitochondrial Quality Control and Disease: Insights into Ischemia-Reperfusion Injury. Mol Neurobiol. 2018;55:2547-64
81. Tahrir FG, Langford D, Amini S, Mohseni Ahooyi T, Khalili K. Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J Cell Physiol. 2019;234:8122-33
82. Moehlman AT, Youle RJ. Mitochondrial Quality Control and Restraining Innate Immunity. Annu Rev Cell Dev Biol. 2020;36:265-89
83. Shoger KE, Cheemalavagu N, Cao YM, Michalides BA, Chaudhri VK, Cohen JA. et al. CISH attenuates homeostatic cytokine signaling to promote lung-specific macrophage programming and function. Sci Signal. 2021;14:eabe5137
84. Glanz VY, Sobenin IA, Grechko AV, Yet SF, Orekhov AN. The role of mitochondria in cardiovascular diseases related to atherosclerosis. Front Biosci (Elite Ed). 2020;12:102-12
85. Tan Y, Mui D, Toan S, Zhu P, Li R, Zhou H. SERCA Overexpression Improves Mitochondrial Quality Control and Attenuates Cardiac Microvascular Ischemia-Reperfusion Injury. Mol Ther Nucleic Acids. 2020;22:696-707
86. Roca-Portoles A, Tait SWG. Mitochondrial quality control: from molecule to organelle. Cell Mol Life Sci. 2021;78:3853-66
87. Huang J, Li R, Wang C. The Role of Mitochondrial Quality Control in Cardiac Ischemia/Reperfusion Injury. Oxid Med Cell Longev. 2021;2021:5543452
88. Bozi LHM, Campos JC, Zambelli VO, Ferreira ND, Ferreira JCB. Mitochondrially-targeted treatment strategies. Mol Aspects Med. 2020;71:100836
89. Dubois-Deruy E, Peugnet V, Turkieh A, Pinet F. Oxidative Stress in Cardiovascular Diseases. Antioxidants (Basel, Switzerland). 2020 9
90. Zhou H, Ren J, Toan S, Mui D. Role of mitochondrial quality surveillance in myocardial infarction: From bench to bedside. Ageing Res Rev. 2021;66:101250
91. Forte M, Schirone L, Ameri P, Basso C, Catalucci D, Modica J. et al. The role of mitochondrial dynamics in cardiovascular diseases. Br J Pharmacol. 2021;178:2060-76
92. Silva-Palacios A, Zazueta C, Pedraza-Chaverri J. ER membranes associated with mitochondria: Possible therapeutic targets in heart-associated diseases. Pharmacol Res. 2020;156:104758
93. Zhu H, Tan Y, Du W, Li Y, Toan S, Mui D. et al. Phosphoglycerate mutase 5 exacerbates cardiac ischemia-reperfusion injury through disrupting mitochondrial quality control. Redox Biol. 2021;38:101777
94. Rebelo AR, Garcez M, Homem CC. Tumor start-up: mitochondrial fusion makes it happen. Embo j. 2020;39:e106927
95. Hu C, Shu L, Huang X, Yu J, Li L, Gong L. et al. OPA1 and MICOS Regulate mitochondrial crista dynamics and formation. Cell Death Dis. 2020;11:940
96. Jin JY, Wei XX, Zhi XL, Wang XH, Meng D. Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol Sin. 2021;42:655-64
97. Wang J, Zhou H. Mitochondrial quality control mechanisms as molecular targets in cardiac ischemia-reperfusion injury. Acta Pharm Sin B. 2020;10:1866-79
98. Wang J, Toan S, Zhou H. Mitochondrial quality control in cardiac microvascular ischemia-reperfusion injury: New insights into the mechanisms and therapeutic potentials. Pharmacol Res. 2020;156:104771
99. Wang J, Toan S, Zhou H. New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury. Angiogenesis. 2020;23:299-314
100. Zinsmaier KE. Mitochondrial Miro GTPases coordinate mitochondrial and peroxisomal dynamics. Small GTPases. 2020:1-27
101. Zhou H, Wang J, Hu S, Zhu H, Toanc S, Ren J. BI1 alleviates cardiac microvascular ischemia-reperfusion injury via modifying mitochondrial fission and inhibiting XO/ROS/F-actin pathways. J Cell Physiol. 2019;234:5056-69
102. Wang J, Zhu P, Toan S, Li R, Ren J, Zhou H. Pum2-Mff axis fine-tunes mitochondrial quality control in acute ischemic kidney injury. Cell Biol Toxicol. 2020;36:365-78
103. Wang S, Zhu H, Li R, Mui D, Toan S, Chang X. et al. DNA-PKcs interacts with and phosphorylates Fis1 to induce mitochondrial fragmentation in tubular cells during acute kidney injury. Sci Signal. 2022;15:eabh1121
104. Xian H, Liou YC. Functions of outer mitochondrial membrane proteins: mediating the crosstalk between mitochondrial dynamics and mitophagy. Cell Death Differ. 2021;28:827-42
105. Fenton AR, Jongens TA, Holzbaur ELF. Mitochondrial dynamics: Shaping and remodeling an organelle network. Curr Opin Cell Biol. 2021;68:28-36
106. Wang L, Kriegstein A. Mitochondria Control Cortical Cell Fate after Mitosis. Dev Cell. 2020;55:120-2
107. Stacchiotti A, Favero G, Giugno L, Golic I, Korac A, Rezzani R. Melatonin Efficacy in Obese Leptin-Deficient Mice Heart. Nutrients. 2017 9
108. Larsen TD, Sabey KH, Knutson AJ, Gandy TCT, Louwagie EJ, Lauterboeck L. et al. Diabetic Pregnancy and Maternal High-Fat Diet Impair Mitochondrial Dynamism in the Developing Fetal Rat Heart by Sex-Specific Mechanisms. Int J Mol Sci. 2019 20
109. Ma T, Huang X, Zheng H, Huang G, Li W, Liu X. et al. SFRP2 Improves Mitochondrial Dynamics and Mitochondrial Biogenesis, Oxidative Stress, and Apoptosis in Diabetic Cardiomyopathy. Oxid Med Cell Longev. 2021;2021:9265016
110. Catanzaro MP, Weiner A, Kaminaris A, Li C, Cai F, Zhao F. et al. Doxorubicin-induced cardiomyocyte death is mediated by unchecked mitochondrial fission and mitophagy. Faseb j. 2019;33:11096-108
111. Wu QR, Zheng DL, Liu PM, Yang H, Li LA, Kuang SJ. et al. High glucose induces Drp1-mediated mitochondrial fission via the Orai1 calcium channel to participate in diabetic cardiomyocyte hypertrophy. Cell Death Dis. 2021;12:216
112. Feng X, Wang S, Yang X, Lin J, Man W, Dong Y. et al. Mst1 Knockout Alleviates Mitochondrial Fission and Mitigates Left Ventricular Remodeling in the Development of Diabetic Cardiomyopathy. Front Cell Dev Biol. 2020;8:628842
113. Valera-Alberni M, Joffraud M, Miro-Blanch J, Capellades J, Junza A, Dayon L. et al. Crosstalk between Drp1 phosphorylation sites during mitochondrial remodeling and their impact on metabolic adaptation. Cell Rep. 2021;36:109565
114. Galvan DL, Long J, Green N, Chang BH, Lin JS, Schumacker P. et al. Drp1S600 phosphorylation regulates mitochondrial fission and progression of nephropathy in diabetic mice. J Clin Invest. 2019;129:2807-23
115. Gawlowski T, Suarez J, Scott B, Torres-Gonzalez M, Wang H, Schwappacher R. et al. Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-β-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J Biol Chem. 2012;287:30024-34
116. Hu Q, Zhang H, Gutiérrez Cortés N, Wu D, Wang P, Zhang J. et al. Increased Drp1 Acetylation by Lipid Overload Induces Cardiomyocyte Death and Heart Dysfunction. Circ Res. 2020;126:456-70
117. Froggatt HM, Harding AT, Chaparian RR, Heaton NS. ETV7 limits antiviral gene expression and control of influenza viruses. Sci Signal. 2021 14
118. Wang Y, Wang Y, Li F, Zhang X, Li H, Yang G. et al. Spermine Protects Cardiomyocytes from High Glucose-Induced Energy Disturbance by Targeting the CaSR-gp78-Ubiquitin Proteasome System. Cardiovasc Drugs Ther. 2021;35:73-85
119. Pereira GC, Lee L, Rawlings N, Ouwendijk J, Parker JE, Andrienko TN. et al. Hexokinase II dissociation alone cannot account for changes in heart mitochondrial function, morphology and sensitivity to permeability transition pore opening following ischemia. PLoS One. 2020;15:e0234653
120. Federico M, Zavala M, Vico T, López S, Portiansky E, Alvarez S. et al. CaMKII activation in early diabetic hearts induces altered sarcoplasmic reticulum-mitochondria signaling. Sci Rep. 2021;11:20025
121. Wang P, Wang D, Yang Y, Hou J, Wan J, Ran F. et al. Tom70 protects against diabetic cardiomyopathy through its antioxidant and antiapoptotic properties. Hypertens Res. 2020;43:1047-56
122. Ponce JM, Coen G, Spitler KM, Dragisic N, Martins I, Hinton A Jr. et al. Stress-Induced Cyclin C Translocation Regulates Cardiac Mitochondrial Dynamics. J Am Heart Assoc. 2020;9:e014366
123. Tao A, Xu X, Kvietys P, Kao R, Martin C, Rui T. Experimental diabetes mellitus exacerbates ischemia/reperfusion-induced myocardial injury by promoting mitochondrial fission: Role of down-regulation of myocardial Sirt1 and subsequent Akt/Drp1 interaction. Int J Biochem Cell Biol. 2018;105:94-103
124. Apaijai N, Charoenphandhu N, Ittichaichareon J, Suntornsaratoon P, Krishnamra N, Aeimlapa R. et al. Estrogen deprivation aggravates cardiac hypertrophy in nonobese Type 2 diabetic Goto-Kakizaki (GK) rats. Biosci Rep. 2017 37
125. Wang Y, Gao P, Wei C, Li H, Zhang L, Zhao Y. et al. Calcium sensing receptor protects high glucose-induced energy metabolism disorder via blocking gp78-ubiquitin proteasome pathway. Cell Death Dis. 2017;8:e2799
126. Parra V, Verdejo HE, Iglewski M, Del Campo A, Troncoso R, Jones D. et al. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NFκB-Opa-1 signaling pathway. Diabetes. 2014;63:75-88
127. Pennanen C, Parra V, López-Crisosto C, Morales PE, Del Campo A, Gutierrez T. et al. Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin signaling pathway. J Cell Sci. 2014;127:2659-71
128. Ding M, Dong Q, Liu Z, Liu Z, Qu Y, Li X. et al. Inhibition of dynamin-related protein 1 protects against myocardial ischemia-reperfusion injury in diabetic mice. Cardiovasc Diabetol. 2017;16:19
129. Watanabe T, Saotome M, Nobuhara M, Sakamoto A, Urushida T, Katoh H. et al. Roles of mitochondrial fragmentation and reactive oxygen species in mitochondrial dysfunction and myocardial insulin resistance. Exp Cell Res. 2014;323:314-25
130. Wang J, Toan S, Li R, Zhou H. Melatonin fine-tunes intracellular calcium signals and eliminates myocardial damage through the IP3R/MCU pathways in cardiorenal syndrome type 3. Biochem Pharmacol. 2020;174:113832
131. Killackey SA, Philpott DJ, Girardin SE. Mitophagy pathways in health and disease. J Cell Biol. 2020 219
132. Saito T, Hamano K, Sadoshima J. Molecular mechanisms and clinical implications of multiple forms of mitophagy in the heart. Cardiovasc Res. 2021;117:2730-41
133. Li W, He P, Huang Y, Li YF, Lu J, Li M. et al. Selective autophagy of intracellular organelles: recent research advances. Theranostics. 2021;11:222-56
134. Bai T, Wang F, Zheng Y, Liang Q, Wang Y, Kong J. et al. Myocardial redox status, mitophagy and cardioprotection: a potential way to amend diabetic heart? Clin Sci (Lond). 2016;130:1511-21
135. Zhou H, He L, Xu G, Chen L. Mitophagy in cardiovascular disease. Clin Chim Acta. 2020;507:210-8
136. Jiang X, Cai S, Jin Y, Wu F, He J, Wu X. et al. Irisin Attenuates Oxidative Stress, Mitochondrial Dysfunction, and Apoptosis in the H9C2 Cellular Model of Septic Cardiomyopathy through Augmenting Fundc1-Dependent Mitophagy. Oxid Med Cell Longev. 2021;2021:2989974
137. Catheline SE, Bell RD, Oluoch LS, James MN, Escalera-Rivera K, Maynard RD. et al. IKKβ-NF-κB signaling in adult chondrocytes promotes the onset of age-related osteoarthritis in mice. Sci Signal. 2021;14:eabf3535
138. Moro L. Mitochondria at the Crossroads of Physiology and Pathology. J Clin Med. 2020 9
139. Eckl EM, Ziegemann O, Krumwiede L, Fessler E, Jae LT. Sensing, signaling and surviving mitochondrial stress. Cell Mol Life Sci. 2021;78:5925-51
140. Bartlett JJ, Trivedi PC, Pulinilkunnil T. Autophagic dysregulation in doxorubicin cardiomyopathy. J Mol Cell Cardiol. 2017;104:1-8
141. Andres AM, Tucker KC, Thomas A, Taylor DJ, Sengstock D, Jahania SM. et al. Mitophagy and mitochondrial biogenesis in atrial tissue of patients undergoing heart surgery with cardiopulmonary bypass. JCI Insight. 2017;2:e89303
142. Morales PE, Arias-Durán C, Ávalos-Guajardo Y, Aedo G, Verdejo HE, Parra V. et al. Emerging role of mitophagy in cardiovascular physiology and pathology. Mol Aspects Med. 2020;71:100822
143. Zhang R, Krigman J, Luo H, Ozgen S, Yang M, Sun N. Mitophagy in cardiovascular homeostasis. Mech Ageing Dev. 2020;188:111245
144. Zhang H, Yang N, He H, Chai J, Cheng X, Zhao H. et al. The zinc transporter ZIP7 (Slc39a7) controls myocardial reperfusion injury by regulating mitophagy. Basic Res Cardiol. 2021;116:54
145. Jimenez RE, Kubli DA, Gustafsson Å B. Autophagy and mitophagy in the myocardium: therapeutic potential and concerns. Br J Pharmacol. 2014;171:1907-16
146. Morciano G, Patergnani S, Bonora M, Pedriali G, Tarocco A, Bouhamida E. et al. Mitophagy in Cardiovascular Diseases. J Clin Med. 2020 9
147. Ajoolabady A, Aslkhodapasandhokmabad H, Aghanejad A, Zhang Y, Ren J. Mitophagy Receptors and Mediators: Therapeutic Targets in the Management of Cardiovascular Ageing. Ageing Res Rev. 2020;62:101129
148. Wang J, Zhu P, Li R, Ren J, Zhang Y, Zhou H. Bax inhibitor 1 preserves mitochondrial homeostasis in acute kidney injury through promoting mitochondrial retention of PHB2. Theranostics. 2020;10:384-97
149. Wang J, Zhu P, Li R, Ren J, Zhou H. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. 2020;30:101415
150. Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M. et al. Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ Res. 2019;124:1360-71
151. Shao D, Kolwicz SC Jr, Wang P, Roe ND, Villet O, Nishi K. et al. Increasing Fatty Acid Oxidation Prevents High-Fat Diet-Induced Cardiomyopathy Through Regulating Parkin-Mediated Mitophagy. Circulation. 2020;142:983-97
152. Kobayashi S, Patel J, Zhao F, Huang Y, Kobayashi T, Liang Q. Novel Dual-Fluorescent Mitophagy Reporter Reveals a Reduced Mitophagy Flux in Type 1 Diabetic Mouse Heart. J Am Osteopath Assoc. 2020;120:446-55
153. Pei Z, Liu Y, Liu S, Jin W, Luo Y, Sun M. et al. FUNDC1 insufficiency sensitizes high fat diet intake-induced cardiac remodeling and contractile anomaly through ACSL4-mediated ferroptosis. Metabolism. 2021;122:154840
154. Belosludtseva NV, Starinets VS, Mikheeva IB, Serov DA, Astashev ME, Belosludtsev MN. et al. Effect of the MPT Pore Inhibitor Alisporivir on the Development of Mitochondrial Dysfunction in the Heart Tissue of Diabetic Mice. Biology (Basel). 2021 10
155. Zhou H, Zhu P, Wang J, Zhu H, Ren J, Chen Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2alpha-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018;25:1080-93
156. Zhou H, Wang J, Zhu P, Zhu H, Toan S, Hu S. et al. NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting Mff-required mitochondrial fission by CK2alpha. Basic Res Cardiol. 2018;113:23
157. Law BA, Liao X, Moore KS, Southard A, Roddy P, Ji R. et al. Lipotoxic very-long-chain ceramides cause mitochondrial dysfunction, oxidative stress, and cell death in cardiomyocytes. Faseb j. 2018;32:1403-16
158. Qiao H, Ren H, Du H, Zhang M, Xiong X, Lv R. Liraglutide repairs the infarcted heart: The role of the SIRT1/Parkin/mitophagy pathway. Mol Med Rep. 2018;17:3722-34
159. Wang S, Zhao Z, Fan Y, Zhang M, Feng X, Lin J. et al. Mst1 inhibits Sirt3 expression and contributes to diabetic cardiomyopathy through inhibiting Parkin-dependent mitophagy. Biochim Biophys Acta Mol Basis Dis. 2019;1865:1905-14
160. Wang S, Zhao Z, Feng X, Cheng Z, Xiong Z, Wang T. et al. Melatonin activates Parkin translocation and rescues the impaired mitophagy activity of diabetic cardiomyopathy through Mst1 inhibition. J Cell Mol Med. 2018;22:5132-44
161. Zhou H, Zhu P, Wang J, Toan S, Ren J. DNA-PKcs promotes alcohol-related liver disease by activating Drp1-related mitochondrial fission and repressing FUNDC1-required mitophagy. Signal transduction and targeted therapy. 2019;4:56
162. Elrashidy RA, Ibrahim SE. Cinacalcet as a surrogate therapy for diabetic cardiomyopathy in rats through AMPK-mediated promotion of mitochondrial and autophagic function. Toxicol Appl Pharmacol. 2021;421:115533
163. Larkin HD. FDA Expands Empagliflozin Heart Failure Indication. Jama. 2022;327:1219
164. Mizuno M, Kuno A, Yano T, Miki T, Oshima H, Sato T. et al. Empagliflozin normalizes the size and number of mitochondria and prevents reduction in mitochondrial size after myocardial infarction in diabetic hearts. Physiol Rep. 2018;6:e13741
165. Zhou H, Wang S, Zhu P, Hu S, Chen Y, Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018;15:335-46
166. Zhou H, Yue Y, Wang J, Ma Q, Chen Y. Melatonin therapy for diabetic cardiomyopathy: A mechanism involving Syk-mitochondrial complex I-SERCA pathway. Cell Signal. 2018;47:88-100
167. Zhou H, Zhu P, Wang J, Toan S, Ren J. DNA-PKcs promotes alcohol-related liver disease by activating Drp1-related mitochondrial fission and repressing FUNDC1-required mitophagy. Signal transduction and targeted therapy. 2019;4:56
168. Thai PN, Miller CV, King MT, Schaefer S, Veech RL, Chiamvimonvat N. et al. Ketone Ester D-β-Hydroxybutyrate-(R)-1,3 Butanediol Prevents Decline in Cardiac Function in Type 2 Diabetic Mice. J Am Heart Assoc. 2021;10:e020729
169. Wang J, Chen P, Cao Q, Wang W, Chang X. Traditional Chinese Medicine Ginseng Dingzhi Decoction Ameliorates Myocardial Fibrosis and High Glucose-Induced Cardiomyocyte Injury by Regulating Intestinal Flora and Mitochondrial Dysfunction. Oxid Med Cell Longev. 2022;2022:9205908
170. Sun Y, Lu F, Yu X, Wang B, Chen J, Lu F. et al. Exogenous H(2)S Promoted USP8 Sulfhydration to Regulate Mitophagy in the Hearts of db/db Mice. Aging Dis. 2020;11:269-85
Author contact
![]() Corresponding author: Ying Tan, E-mail: tanying0924com.
Corresponding author: Ying Tan, E-mail: tanying0924com.