Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Autophagy and Autophagy-related...
Genome stability
Autophagy-related proteins...
Autophagy-related proteins...
Autophagy-related proteins...
Conclusions and future...
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(14):5329-5344. doi:10.7150/ijbs.76134 This issue Cite
Review
Autophagy-related Proteins in Genome Stability: Autophagy-Dependent and Independent Actions
Ye Zhang1#, Ran Guo2#, Shan-Shan Wang1, Xiao-You Jiang1, Hong-Yan Cui1, Yang Guo1, Xiao-Yu Song1 ![]() , Qi-Qiang Guo1
, Qi-Qiang Guo1 ![]() , Liu Cao1
, Liu Cao1 ![]()
1. College of Basic Medical Science, Health Sciences Institute, Key Laboratory of Medical Cell Biology, Ministry of Education, China Medical University, Shenyang, Liaoning Province, P. R. China.
2. Department of Orthopedics, Shengjing Hospital of China Medical University, Shenyang, Liaoning Province, P.R. China.
#These authors contributed equally to this work.
Received 2022-6-14; Accepted 2022-8-13; Published 2022-8-21
Abstract

It is emerging that autophagy-related proteins regulate the adaptive response to DNA damage in maintaining genome stability at multiple pathways. Here, we discuss recent insights into how autophagy-related proteins participate in DNA damage repair processes, influence chromosomal instability, and regulate the cell cycle through autophagy-dependent and independent actions. There is increasing awareness of the importance of these pathways mediated by autophagy-related proteins to DNA damage response (DDR), and disturbances in these regulatory connections may be linked to genomic instability participated in various human diseases, such as cancer and aging.
Keywords: Autophagy-related proteins, Genome Stability, Autophagy, Non-autophagic functions
Introduction
Actively maintaining the integrity of the genome is essential for the healthy survival of an organism. DNA is the most basic unit of human genetics, and its damage and mutation are usually affected by many extrinsic factors (such as ionizing radiation, ultraviolet radiation, and chemical compounds) and intrinsic factors (such as free radicals produced as by-products of the organism's metabolism and replication errors). Therefore, cells need to evolve efficient mechanisms to sense and repair this damage to ensure their survival.
Autophagy is an essential biological behavior that plays a significant role in recycling cellular components and damaged organelles in response to various stressful conditions, such as metabolic stress and genomic instability. Although autophagy occurs in the cytoplasm, the deletion of autophagy related proteins can also lead to cellular DNA damage and the accumulation of genomic instability. Autophagy-related proteins are significant autophagy executors. Accumulating evidence suggests that autophagy-related proteins can not only regulate various stages of the autophagy pathway, but also participate in cellular biological behaviors in an autophagy-independent manner. Herein, we discuss recent insights into how autophagy-related proteins maintain genome stability through participation in DNA damage repair processes, influencing chromosomal instability, and regulating the cell cycle in an autophagy-dependent and independent manner.
Autophagy and Autophagy-related Proteins
Organisms are in a dynamic homeostasis of constant renewal. Autophagy is the primary scavenging mechanism in cells by which cytoplasmic components are delivered to lysosomes, where they are degraded. However, the function of autophagy is not just to remove substances from cells; it is a system to maintain homeostasis, help cells complete renewal, and provide energy [1].
Functionally, various autophagy-related proteins regulate different phases of the autophagy pathway, including origin of autophagy, formation of pre-autophagy structures (PAS), the extension of the membrane and maturity of autophagosomes, transport, recognition of autophagosomes, fusion with lysosomal membrane, and final degradation of autophagic lysosomal contents. In mammalian cells, autophagy is regulated by about 20 core ATG proteins and activated by the mTOR signaling pathway. The proteins involved in the process can be summarized in the following five units: (1) ULK1 kinase complex, including ULK1/2, ATG13, RB1CC1/FIP200, and ATG101, mainly responsible for initiating autophagy, receiving abnormal cell signals, recruiting ATG protein into PAS, and controlling the formation of autophagosomes [2]. Signals of intracellular energy depletion activate AMPK to inhibit mTORC1 and initiate autophagy to provide cells with energy and nutrients. This is the initial step of autophagy [3]. (2) Autophagy-specific class III phosphatidylinositol 3-kinase (PI3K) complex, including VPS34, VPS15, Beclin1, and ATG14L. Beclin1 interacts with VPS34, which activates VPS34 kinase activity to regulate autophagosome size and quantity [4, 5]. (3) ATG9A transportation system, including ATG9A, WIPI1/2, and ATG2A. ATG9A/Atg9 can be phosphorylated by ULK/Atg1, and the recruitment of LC3/Atg8 and WIPI1/2/Atg18 requires phosphorylated ATG9A/Atg9 [6]. (4) ATG12 ubiquitin coupling system, including ATG12, ATG7, ATG10, ATG5, and ATG16L1. ATG7 activates ATG12, and ATG12 is conjugated to ATG5 via Atg10. The conjugate is then able to be stabilized by the ATG16L protein and form the ATG12-ATG5-ATG16L complex, which is essential for the formation of the LC3-conjugated system [7-9]. (5) LC3 conjugate system, including LC3A/B/C, ATG7, ATG3, and ATG4A/B/C/D, is located downstream of the ATG12 ubiquitin coupling system. LC3 localizes to autophagosome membranes following post-translational modifications and is a classical autophagy marker in mammalian cells [10]. Both ubiquitin-like systems have a hand in subsequent autophagy processes (Figure 1).
Schematic overview of autophagy-related proteins involved in regulation of the autophagic pathway.
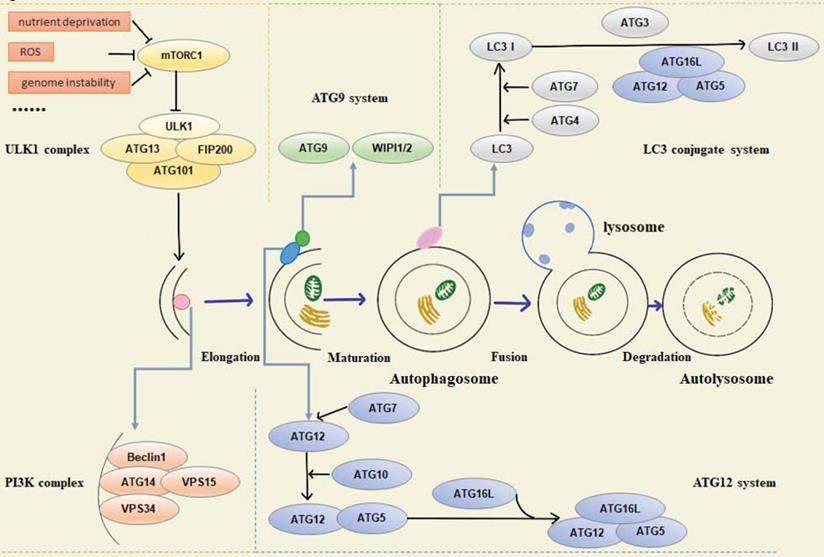
Genome stability
DNA, the basic unit of human genetics, and its damage and mutation are usually sensitive to environmental factors, including physical factors such as ionizing radiation, ultraviolet radiation, and chemical substances. Meanwhile, there are also many potential dangers in DNA replication and DNA damage repair (DDR); when something goes wrong, unfavorable mutations are likely to occur within the cells. To maintain the integrality and stabilization of the genome, DNA must be protected from damage caused by various environmental factors or spontaneous damage in the process of DNA metabolism. In response to these damages, eukaryotes have evolved the DDR pathway. DDR is a complicated signal transduction pathway that can quickly and accurately sense DNA damage and deliver these signals to cells to activate the response of cells to DNA damage. DDR involves a large number of proteins, and eukaryotic cells have evolved precise strategies to recruit and activate the correct factors at the right place and time [11].
In addition, cells are often exposed to certain stimulatory conditions that impede DNA replication, cause replication fork suspension, and increase fork instability. The resulting DNA replication stress prevents cells from completing DNA synthesis in a timely manner during interphase, leading to chromosomal breakage and genomic instability in mitosis [12, 13]. Regulating the operation of replication forks during replication errors is also an essential part of maintaining genome stability.
Cell cycle arrest is one of the most typical cellular response functions activated after DNA damage. It prevents DNA-damaged cells from entering mitosis and provides time for DDR. Cells use cell cycle checkpoints throughout the interphase to protect genomic integrity prior to mitosis and to block the cell cycle in time when unstable states occur. The cell cycle is mainly regulated by cyclin-dependent kinases (CDKs), which drive the G1/S transition and then regulate the DNA replication process in the S phase, which completes most of the DNA synthesis [14, 15]. When the cell cycle reaches S phase and G2 phase, mitotic cyclins are expressed, and CDKs activity continues to increase [16].
Improving chromosome instability has become a vital focus of the mitotic process to protect genomic integrity. Telomeres are susceptible to mitotic pressure and are thought to be the region where DDR is initiated. The maintaining telomere stability is essential to protect genome integrity and mitochondrial integrity [17].
It has been proven that autophagy plays an essential role in maintaining genomic stability [18]. When cells are under starvation or stress, abnormal mitochondria can produce high levels of ROS, which can lead to DNA damage. Autophagy contributes to the removal of all irretrievably oxidized biomolecules in cells, and the redox homeostasis maintained by autophagy significantly contributes to maintaining genomic stability. This is a crucial reason why autophagy is included in the system for maintaining genomic homeostasis [19]. It has also been shown that NDP52 and P62 regulate retrotransposon insertion in the genome through selective degradation of retrotransposon RNA by autophagy, suggesting that autophagy can buffer genetic variation in the physiological sense by degrading retrotransposon RNA [20]. However, another study showed that methyltransferase-like 14 (METTL14) positively regulated genome-wide repair by regulating N6-methyladenosine (m6A) methylation modification and DDB2 translation. Under UVB irradiation, METTL14 was downregulated by NBR1-mediated selective autophagy [21]. This suggested that autophagy played different roles in regulating genome homeostasis under different circumstances.
However, reports continue to indicate that autophagic proteins also have many non-autophagic functions, including regulating neuronal growth and axon growth, participating in DNA damage responses, and participating in type I IFN signaling [22-26]. The mechanism of autophagy proteins regulating genomic homeostasis in the nucleus has attracted a certain degree of attention. Interestingly, autophagy proteins can maintain genome homeostasis not only through autophagy-dependent pathways, but also through autophagy-independent pathways, exerting their non-autophagic functions in the nucleus. This article reviewed the current research on autophagy-related proteins in maintaining genome stability, to figure out the effects of autophagy-related proteins in the nucleus.
Autophagy-related proteins maintain genome stability through autophagy-dependent pathways
The autophagy pathway plays a vital role in maintaining all aspects of nuclear genome homeostasis, and autophagy-deficient cells are likely to experience genomic instability. DNA damage, chromosomal instability, and cell cycle arrest are common events [27, 28]. Activation of autophagy has been shown to reduce genomic instability [29, 30]. Recent studies have indicated that autophagy is directly participated in DDR in yeast and mammalian cells, regulating levels of a critical repair protein, RBBP8/CtIP [31]. When autophagy is activated, a variety of autophagy-related proteins can directly or indirectly affect genome homeostasis. For example, in mammals, autophagy regulates the protein level of active RHOA by regulating SQSTM1/p62. When autophagy is interrupted, active RHOA accumulates in the region surrounding the intermediate and leads to cytokinesis failure, multinucleation, and aneuploidy [32]. We discussed recent insights into how autophagy-related proteins participate in DNA damage repair processes, influence chromosomal instability, and regulate the cell cycle in an autophagy-dependent manner.
Autophagy function of autophagy-related proteins and DNA damage
Genome stability is essential throughout the life of a cell. Maintaining genomic homeostasis requires constant counteracting of accumulated DNA damage. Unrepaired DNA damage can cause a series of serious consequences, including cell cycle arrest and aging, apoptosis, cellular dysfunction, and mutation accumulation. Therefore, DNA damage repair is crucial in maintaining genomic homeostasis. DNA damage often occurs in the nucleus and is caused by environmental factors (UV light, ionizing radiation) and internal factors (ROS) [33]. In addition, when autophagy is impaired, oxidative stress will also lead to the accumulation of DNA damage. Therefore, maintaining intact autophagy is crucial in preventing DNA damage [34].
The mechanism of autophagy involved in DDR has been extensively studied and discussed. What is clear is that DNA damage can activate autophagy pathways and mitigate the state of genomic instability [33]. DNA double-strand break (DSB) is a very common incident in DNA damage. Taking DSB as an example of a representative DNA damage event, DSB can activate ATM, which leads to the LKB1-mediated phosphorylation of AMPKα subunit at Thr172. AMPK then activates TSC2 and ULK1, leading to the inhibition of mTOR, ATG1 activation, and autophagosome formation [3].
There is also evidence that autophagy mediated by Unc-51-like kinase 1 (Ulk1) and LC3 also plays an important role in combating genomic instability. Ulk1 and LC3 are two core components of autophagy. Ulk1 is involved in the induction of autophagy, while the phosphatidylethanolamine-conjugated form of LC3 is currently the most accurate autophagosome marker protein [35]. It has been reported that both LC3 and pUlk1 have nuclear localization. When LC3 and Ulk1 are silenced, autophagy is inhibited, and micronuclei, a diagnostic marker of genomic instability, is accumulated; silencing either one leads to an increase in the generation frequency of micronuclei and promote genomic instability [36]. Gao et al. found that in response to DNA damage, p53 increased the transcriptional level of Ulk1/Ulk2 and initiated sustained autophagy. By upregulating Ulk1/2, p53 promotes Ulk1/2 and other autophagy-initiating components such as ATG13, FIP200, and ATG101 to produce Ulk1 kinase complex, preparing for the occurrence of autophagy. In this process, AMPK and sesn1/2 levels are also upregulated, resulting in inhibition of mTOR kinase activity, thereby eliminating the inhibition of mTOR on autophagy. Disinhibition of mTOR further increases autophagic flux, resulting in sustained high levels of autophagy. If DNA damage cannot be repaired and cells are in a sub-death state of high-intensity autophagy for a long time, continuous autophagy will lead to non-apoptotic cell death [37].
FIP200 is an adhesion plaque kinase that is involved in autophagy. Studies have shown that FIP200KO MEFs show nuclear γ-H2AX staining after exposure to ionizing radiation (IR), indicating that DDR is defective at this time, and FIP200 is involved in DNA damage repair. Similar results showed that autophagy was inhibited in the presence of other DNA damage inducers such as camptothecin and etoposide, as well as in the use of 3-methyladenine. These suggest that FIP200 is likely to be involved in DDR through the autophagy pathway [38].
Beclin 1 is a key member of the autophagy protein family, and functions as a core component of the class III PI3K/Vps34 complex required for autophagosome formation and maturation [39]. UVRAG products can be associated with the Beclin 1/Bcl-2/PI(3)KC3 complex, in which UVRAG and Beclin 1 interact and codependently induce autophagy through their coiled-coil domain (CCD) [40]. UVRAG interacts with Beclin 1 to modulate the DNA damage signal, and Beclin 1 siRNA can significantly reduce the expression level of UVRAG. Experiments have shown that UVRAG ΔCCD mutant shows substantially damaged binding to Beclin 1, resulting in a more severe extent of radiation-induced DSBs. In mammalian cells, DSB is mainly repaired by two biological mechanisms, non-homologous end joining (NHEJ) and homologous recombination (HR). The core of HR repair DSB is Rad51, which is to promote homologous pairing between DNA double strands; 53BP1 is a sensor of DNA damage and a promoter of non-homologous end joining (NHEJ). Following irradiation treatment, 53BP1 was significantly enhanced in Beclin 1, UVRAG or ATG5 knockdown cells compared to control cells. This suggests that the involvement of Beclin 1 in DDR may be related to autophagy [41]. In addition, when the allele of Beclin1 is lost, autophagy deficiency will activate the DNA damage response in breast tumor cells, promote gene amplification, and cooperate with apoptosis defects to promote the occurrence of breast tumors. The research results also showed that Beclin 1 could play a role in resisting DNA damage or DNA damage repair [27].
Autophagy has been shown to occur in the cytoplasm. How does it participate in the function of the nucleus? Research shows that p62 plays a vital role in cytoplasmic communication during autophagy in regulating nuclear genome homeostasis [42, 43]. P62 is amomentous autophagy adaptor protein that binds to ubiquitinated protein polymersand delivers them to autophagosomes. In the cytoplasm, p62 acts as a receptor on autophagosomes to direct autophagy and autophagosome maturation. When p62 is reduced, cellular polyploidy increases, and autophagic maturation is impaired. Mitochondrial coding protein LRPPRC is combined with one of the microtubule-associated protein family MAP1S to promote autophagy initiation and development. Liver-specific deletion of LRPPRC results in a decrease in p62, which causes autophagy damage that negatively affects cells [44]. P62 has nuclear localization signals and nuclear export signals that enable it to shuttle between the cytoplasm and nucleus. In the nucleus, the repair process of DNA damage is affected by p62, which is specifically located in the nucleus. The inhibition of autophagy will lead to an increase in the level of nuclear p62, which in turn leads to a decrease in chromatin ubiquitination. This ubiquitination is essential for the recruitment of downstream factors of the DSB repair pathway, such as the 53BP1 and BRCA1/BARD1 complex [45]. In terms of mechanism, the ubiquitination reaction induced by DSB is triggered through the RNF8-dependent adjoint of ubiquitin on histone H1 [46]. RNF8 is recruited to the injury site to initiate H2A/H2AX ubiquitination via K63. Subsequently, RNF8 amplifies this cascade by recruiting an E3 ligase, RNF168, which promotes the monoubiquitination of histone H2A/H2AX to transmit DNA damage signals and activate the DDR program [47]. However, the interaction of the LIM-binding (LB) domain of p62 and the MU1 domain of RNF168 inhibits the E3 ligase activity of RNF168, resulting in damaged chromatin ubiquitination and decreased recruitment of DNA repair proteins after DNA damage [48]. Furthermore, when autophagy is inhibited or defective, there is another regulatory mechanism through which p62 accumulation affects DNA repair which has been reported in the literature through proteasomal degradation of filamin A (FLNA) and RAD51 in the nucleus to inhibit HR-directed DSB repair. The role of FLNA in DDR is to recruit RAD51 through interaction with BRCA1/2. The interaction of p62 and FLNA promotes proteasomal degradation of FLNA and RAD51 in the nucleus, reduces nuclear RAD51 levels, and slows DNA repair, especially through HR [49]. These results suggested that the accumulation of p62 caused by autophagy defects and nuclear import of p62 could disturb DSB repair in both NHEJ and HR pathways. In addition, when autophagy is inhibited, the accumulation of p62 leads to the production of ROS and ER stress. The increase of ROS levels under oxidative stress can also lead to DNA damage. Preventing the accumulation of p62 or ROS can reduce the damage of autophagy-deficient cells [50]. Clinically, Christoph Burdelski et al. found that this accumulation of p62 caused genomic instability and promoted rapid proliferation of prostate cancer cells, leading to tumor recurrence [51]. The phenomenon of p62 accumulation caused by inhibition of autophagy has been confirmed in many cancer cells, such as oral cancer, colon cancer, breast cancer, and so on [52, 53].
Studies have shown that both Ulk1 and LC3 participate in DSB. γ-H2AX (phosphorylated histone H2AX Ser139) is a classic marker of DSB. Both LC3-II and pUlk1 show nuclear localization, and can combine with γ-H2AX, Rad51, or PARP-1 to maintain genomic stability. Sunitinib can increase the level of γ-H2AX and enhance autophagic flux. When Sunitinib is administered, the binding of LC3, pUlk1 and Rad51 is weakened, indicating that autophagy may limit the role of LC3 and pUlk1 in DSB repair [36].
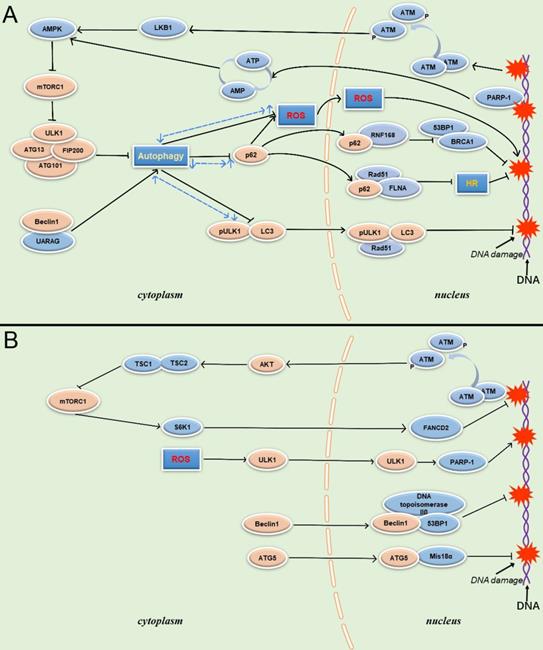
PARP-1 is another protein involved in autophagy regulation during DDR, and plays a pivotal role in the interaction between autophagy and DDR. PARP-1 is an important sensor of DNA breaks, and its catalytic activity is activated up to 500-fold on the site of DNA breaks. It is also an important recruitment molecule, and can concentrate the key molecules required for DNA fracture repair at the damaged site in a short time [54]. Meanwhile, overactivation of PARP-1 leads to ATP depletion, and AMPK can activate TSC1/2 to inhibit mTOR after sensing energy consumption to activate autophagy, enabling cells to obtain nutrients and energy. However, in response to DNA damage, the energy collapse caused by the over-activation of PARP-1 and induced high intensity continuous autophagy is a death signal for the cell. Although autophagy is a protective factor in the response to DNA damage, PARP-1deficient cells are better rescued than cells undergoing PARP-1induced persistent autophagy, resulting in an overall reduction in cell death [55] (Figure 2).
Schematic overview of autophagy-related proteins involved in DNA damage through the autophagy pathway and non-autophagic pathway. (A) Autophagy related proteins involved in DNA damage through the autophagy dependent pathway. (B) Autophagy related proteins involved in DNA damage through the non- autophagic pathway.
Autophagy function of autophagy-related proteins and chromosome instability
In the nucleus, chromosomal instability is inextricably linked to DNA damage. Chromosome instability (CIN) refers to the abnormal condition of chromosome integrity or chromosome number in the nucleus. CIN cells produce higher levels of ROS, leading to DNA damage and cell death. In CIN cells, autophagy activity also becomes more sensitive. Knockout of autophagy-related proteins Atg1 or Atg18 resulted in a remarkable increase in oxidative stress and DNA damage levels in CIN cells [56], indicating that autophagy plays a rescued role in CIN cells, and the improvement of autophagy level promotes the survival of CIN cells.
Telomeres are essential structures that chromosomes have evolved to maintain their stability. Telomeres are tandem TTAGGG DNA repeats that preserve genomic stability by recruiting protein complexes called shelterins, which protect the ends of chromosomes from being identified as DSBs [57]. Due to the loss of telomere sequence caused by replication shortening, the dysfunctional telomeres will activate the typical DNA damage response. Cells with telomere dysfunction have increased chromosomal fusion, resulting in genomic instability, which macroscopically contributes to the pathogenesis and progression of cancer. Autophagy suppresses this chromosomal instability by maintaining cellular metabolic homeostasis and inhibiting tumor progression [28, 58].
Autophagy can limit chromosomal instability, which could also explain its tumor suppressor function from a genetic perspective. Beclin1+/- cells are deficient in autophagy and susceptible to stimulation by metabolic stress. This innate defect in autophagy-deficient cells limits their survival; however, the cells increase the rate of gene mutation and chromosomal instability in order to survive, and even develop aneuploidy, a sign of tumorigenesis [28]. Some studies treated Beclin 1 as a dose-dependent tumor suppressor gene and found that Beclin 1-/- mice had early embryonic death. Meanwhile, Beclin 1-/- mutant ES cells had serious defects in autophagy, and Beclin 1+/- mutant mice had high incidence of spontaneous tumors [59]. The monoallele deletion of BECN1 also accelerated tumorigenesis in mice in the ovarian cancer model, suggesting that autophagy is a suppressor of ovarian cancer [60].
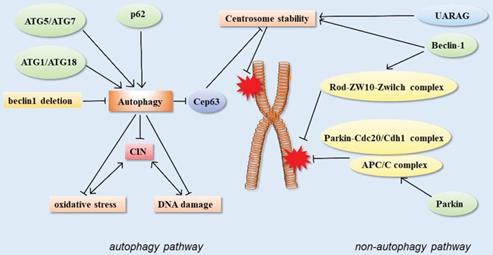
In addition, the centrosome number is associated with correct chromosome separation and genomic stability during mitosis. The ubiquitin-proteasome system is considered to be the main way to regulate the number of centrosomes, but recent studies have shown that autophagy also regulates the number of centrosomes. Autophagy-deficient cells contain multiple Cep63 dots, which increase the number of centrosomes. In wild-type cells, Cep63 is eliminated by p62-mediated autophagy. In mouse models, p62 -/- MEFs were also found to carry more centrosomes than normal. In vivo, p62-/- mouse hematopoietic cells had autophagy defects, and also contained multiple centrosomes. These results suggested that p62-mediated autophagy could regulate chromosome stability by regulating centrosome number [61] (Figure 3).
Schematic of autophagy-related proteins involved in regulating chromosome stability through the autophagy pathway and non-autophagic pathway.
Autophagy function of autophagy-related proteins involved in cell cycle regulation
If DNA damage is not repaired for a while, cells escape from the growth-inhibitory phase, activating cell cycle checkpoints, resulting in a cell cycle arrest state that allows time for DNA damage repair, known as cell cycle arrest [62]. After genome integrity is repaired, cells are released from cell cycle arrest. However, if DNA damage cannot be efficiently restored, cells will either continuously stagnate until death, or begin to replicate with unstable genomes. Autophagy-related proteins act on cell cycle regulation through the autophagy pathway, maintain genome stability, and regulate cell proliferation.
In C.elegans, bec-1/Beclin1, atg-18/wipi1/2 all promote the cell cycle process. Autophagy mediated by Bec-1/Beclin1 may be effective as an exocrine system. Since autophagy genes are observed from C. elegans to human, the function of bec-1/Beclin1 in C. elegans is a good reference for the proliferation of human tumor cells [63]. Genomic instability can directly or indirectly cause tumorigenesis. In recent years, studies have found that Beclin-1 induced autophagy can inhibit tumorigenesis through various mechanisms. Mechanically, this protective behavior may be related to three pathways. First, Beclin-1 induce autophagy and cell cycle arrest [64-66]. Second, as above, Beclin-1 expression places restrictions on chromosomal instability and reduces the occurrence of genetic mutations and DNA damage [28]. The third mechanism is related to autophagy-induced immune responses [67]. The Autophagy inhibitor 3-MA, acting on the Beclin 1/PIK3C III complex significantly, delays IR-induced G2/M phase arrest when cells are exposed to IR, suggesting that Beclin 1 may be associated with mitosis and the cell cycle [63]. A possible mechanism is that IR enhances the combination of Beclin 1/PLK1 and Beclin 1/CDC25C, suggesting that Beclin 1 may be involved in IR-induced G2/M arrest by combining with PLK1 and CDC25C [68].
ULK1-ATG13, the most upstream of the autophagy initiation complex, is phosphorylated by mTORC1 and AMPK to induce autophagy. Moreover, ULK1 and ATG13, the substrates of cdK1-CCNB/Cyclin B, are essential for cell cycle. Cdk1-induced phosphorylation of ULK1-ATG13 promotes autophagy and cell cycle progression. Studies have shown that double knockout of ULK1 and ATG13 significantly inhibits cell cycle and tumor cell proliferation in vitro and in vivo [69]. Ulk1-ATG13 not only provides a molecular mechanism to maintain autophagy, but also link autophagy to cell cycle regulation [70].
An interesting study showed that overexpression of Atg7 enhanced neural crest cell production in unilateral developmental neural tubes of chicken embryos. The Atg7 gene is located upstream of autophagy, which is responsible for inducing autophagy and removing damaged macromolecules and organelles when cell homeostasis is under various pressures [71]. In neural tube cells, the upregulation of the Atg7 gene can activate autophagy. At the same time, the upregulation of Atg7 can significantly accelerate the cell entering the S phase, which means that Atg7 regulates cell cycle progression [72]. However, the specific mechanisms and roles of Atg7 in cell cycle regulation seem to depend on the situation. Atg7 is a decisive factor in the arrest of the unstable cell cycle in serum-deficient and amino acid-deficient mouse embryonic fibroblasts [73]. However, other studies have shown that Atg7 can inhibit the effect of the CDK inhibitor p27 and control cell proliferation [74]. Wang et al. believed that, in neural stem cells (NSC), Atg7 and P62 might inhibit the G1 to S phase of the cell cycle through the autophagy pathway, thus inhibiting the differentiation of NSCs and promoting cell survival [75]. Meanwhile, the inhibition of autophagy by Atg7 deficiency may accelerate apoptosis and arrest the cell cycle in G0/G1 phase under glucose starvation [76].
The autophagy protein system is extensive. In addition to the above-mentioned vital proteins located upstream, other autophagy-related proteins participate in the regulation of the cell cycle. ATG10 is highly expressed in gastric cancer (GC) and may act as an oncogene to regulate the cell cycle and promote abnormal cell proliferation [77]. ATG4 is a kind of cysteine protease that is necessary for the formation of autophagosomes. ATG4B is the mammalian ortholog of yeast ATG4. In various cancer cells, the knockout of ATG4B can inhibit autophagy and induce cell cycle arrest, the main mechanism of which is to trigger the LKB1-AMPK energy response pathway to stop the G1/S period transition [78].
MTORC1 is a pivotal regulator of the G1 phase and autophagy. Regulation of the G1 phase is usually achieved by regulating cyclin D1 mRNA and protein ubiquitin-proteasome degradation. Everolimus specifically inhibits the mTORC1 signaling pathway and activates the autophagy pathway, blocking cell cycle progression at the late stage of G1 phase. There are two mechanisms: first, mTORC1 inhibitors trigger cyclin D1 nuclear output, ubiquitination, and 26S proteasome degradation by activating GSK-3β kinase [79]. Second, in addition to the ubiquitin-proteasome pathway, Everolimus can also promote cyclin D1 degradation through the autophagy pathway [80].
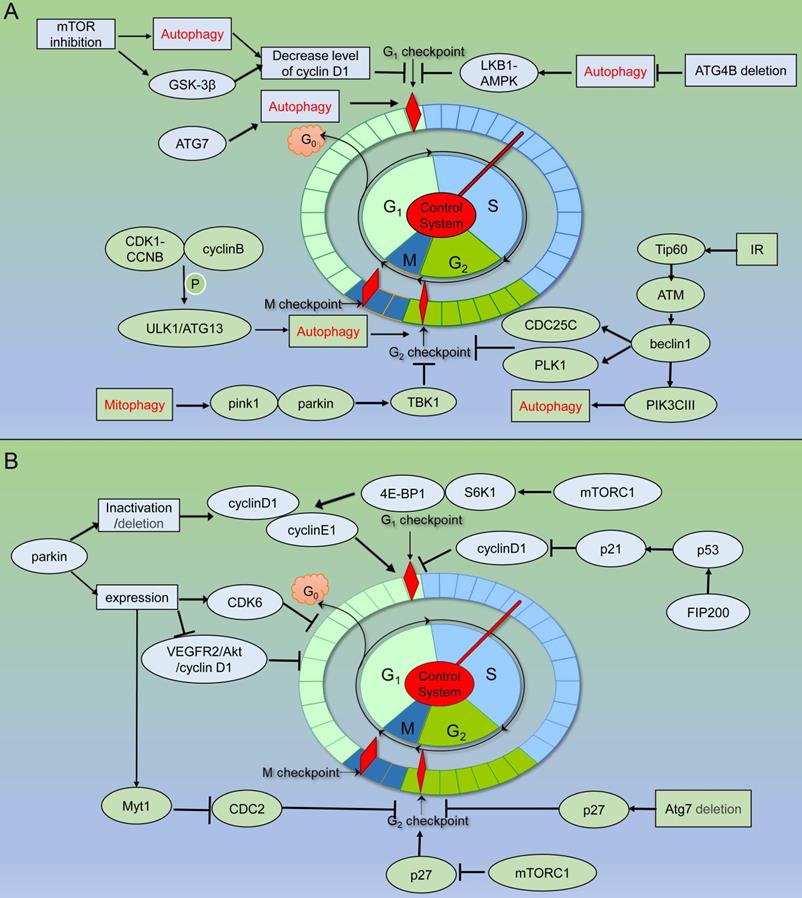
PINK1 and Parkin play essential roles as upstream factors in mitophagy targeting the clearance of damaged mitochondria, but their roles in maintaining genome homeostasis have not been explored sufficiently. PINK1/Parkin pathway can activate TBK1, and the phosphorylated TBK1 is isolated from the centrosome to the damaged mitochondria, resulting in G2/M phase arrest. In addition, ATM is an important regulatory protein in DDR. PINK1 and Parkin also interact with ATM genetically, but there seems to be no direct protein communication [81] (Figure 4) (Table 1).
Schematic of autophagy related proteins involved in cell cycle regulation. (A) Autophagy related proteins involved in cell cycle regulation through the autophagy dependent pathway. (B) Autophagy related proteins involved in cell cycle regulation through the non- autophagic pathway.
Autophagy-related proteins involved in genome stability through autophagy-dependent pathway
| Autophagy-related proteins | Target molecule | Functions | References |
|---|---|---|---|
| p62 | RNF168 | reduces the recruitment of DNA repair protein | [45] |
| FLNA and RAD51 | inhibits HR-directed DSB repair | [49] | |
| ULK1/LC3 | P53, γ -H2AX, Rad51, PARP-1 | promote DSB repair | [36] |
| Beclin1 | UVRAG | is involved in DDR and protect chromosome stability | [40, 41] |
| PLK1 and CDC25C | is involved in IR-induced G2/M arrest | [68] | |
| ULK1/ATG13 | CDK1-CCNB/cyclin B | are involved in cell cycle progression and cell proliferation | [69, 70] |
| ATG1/ATG18 | protect chromosome stability | [56] | |
| ATG7 | accelerates the progression of cells entering S phase | [72] | |
| deficiency of ATG7 blocks G1/S, G0/G1 phase | [75, 76] | ||
| ATG10 | promotes cell proliferation | [77] | |
| ATG4 | LKB1-AMPK | inhibits the progression of cells entering the G1/S transition | [78] |
| mTORC1 | cyclin D1 | mTORC1 inhibition activates autophagy and induces G1 cell cycle arrest | [79, 80] |
| PINK1/parkin | TBK1 | inhibits the progression of cells entering the G2/M transition | [81] |
| FIP200 | is involved in DNA repair | [38] |
Autophagy-related proteins maintain genome stability through autophagy-independent pathways
Autophagic proteins are interlinked to form a complete autophagy pathway maintaining cell homeostasis and genome stability. However, more and more studies have offered eloquent proof that the non-autophagy function of autophagy proteins also plays an essential role in cells. These proteins are involved in a wide range of cellular functions [22-26]. In the nucleus, autophagy proteins can also participate in teams that control genome homeostasis through non-autophagic pathways. While summarizing that autophagy proteins protect genomic homeostasis through the autophagy pathway, we also determined the mechanisms by which autophagy proteins function through non-autophagy pathways when several of the above-mentioned genomic abnormalities occur in cells.
Non-autophagic function of autophagy-related proteins involved in DNA damage
Cells complete DNA replication during mitosis, but under the attack of replication inhibitors, DNA replication forks are stalled, genome stability is disrupted, and the mitotic process is interrupted. mTOR is crucial for the resurrection of stagnant replication forks. Protein kinase B (PKB/AKT) has been shown to activate mTORC1 by phosphorylation of the TSC1/TSC2 complex, followed by phosphorylation of S6K and 4E-BP1. Phosphorylation of 4E-BP1 promotes DNA repair and replication fork survival, and the activated S6K1 performs DNA repair through FANCD2 [82, 83]. ATR-dependent activation of mTORC1 can also regulate nuclear F-actin to promote replication fork repair under replication stress [84]. There is an interaction between mTOR and the FA protein pathway, which is the classical repair pathway that regulates replication fork restart. Both can regulate the reboot of stalled replication forks via aphidicolin, an inhibitor of DNA polymerase alpha that causes fork stall and through the same pathway protects newly synthesized DNA strands from degradation by exonuclease [85]. However, in another case, the mTOR signaling pathway played a role in accelerating DNA damage caused by deletion of the breast cancer-related gene BRCA1. In BRCA1-deficient cells, mTORC2 is overactivated, so BRCA1-deficient breast cancer cells may be dependent on mTORC2 signaling and more sensitive to its inhibition, further suggesting that mTOR plays a role in driving DNA damage events in tumor cells [86]. So how does the mTOR signal mediate DNA damage? Studies have shown that this link also appears to be related to the phosphorylation of AKT. First, AKT can inhibit the activation of mTORC1 by TSC 1/2; at the same time, AKT can dissociate PRAS40, which inhibits mTORC1 autophosphorylation, from mTORC1 components [87, 88]. Second, AKT is a direct phosphorylation target of ATM, an important sensor protein in DDR [89]. It has been reported that direct DNA damage after rapamycin treatment cannot be detected by pulsed-field electrophoresis, indicating that, when mTOR is inhibited, DDR activation is more likely to occur than significant DNA damage [90].
Promoter hypermethylation leads to the epigenetic inactivation of MLH1 (mutL homolog 1) in the context of CpG island methylation phenotype (CIMP), resulting in DNA mismatch repair deficiency (MMR-D) [91]. Autophagy-related gene 5 (ATG5) locates in the cytoplasm and participates in autophagy through interaction with ATG12, promoting the formation of autophagosomes [92]. It has been reported that ATG5 in the cytoplasm can translocate to the nucleus. In the nucleus, ectopic ATG5 loses its activity of recruiting ATG12 to participate in autophagy, and exerts a DNA damage function independent of its autophagy activity. After ATG5 enters the nucleus, it interacts with Mis18α and promotes the hypermethylation of the hMLH1 promoter CpG island by promoting the hypermethylation of hMLH1, thereby causing microsatellite instability and DNA mismatch repair deficiency [93].
P62 can also participate in DNA damage through non-autophagy pathways, and the mechanism is associated with high ROS levels. It has been reported liver cells transfected with P62 have increased levels of stem cell marker DLK1, which enhances ROS levels, an important inducer of DNA damage and chromosomal instability [94]. P62 induces ROS production by activating NADPH oxidase in a DLK1-dependent manner, thereby compromising genome homeostasis, inducing inflammation, and promoting tumorigenesis [95]. Is DNA damage caused by ROS related to autophagy-related proteins? It has been found that after H2O2 treatment, most of the cytoplasmic ULK1 can localize to the nucleus and regulate the activity of DDR protein PARP1 in a kinase-dependent manner. PARP1 is a core protein in DDR and cell death mechanisms under oxidative stress. By enhancing PARP1 activity, ULK1 promotes the energy consumption and cell death under oxidative stress. This pathway proves that ULK1 can act as a DNA damage factor in the Atg7-independent autophagy pathway, but this view does not deny the possibility that ULK1 promotes autophagy-mediated cell death [96].
Interestingly, some studies have demonstrated that Beclin 1 can be involved in DDR independent of the autophagy pathway [97]. Beclin 1 can transfer from cytoplasm to nucleus after cells are exposed to IR. DNA topoisomerase IIβ is one of the most significant proteins interacting with nuclear Beclin 1, and the knockdown of DNA topoisomerase IIβ can inhibit the activity of HR and NHEJ pathways. DNA topoisomerase IIβ and Beclin 1 are able to co-localize with the DSB repair protein p53-binding protein 1 (53bP1) after exposure to IR. It has been speculated that Beclin 1 protects genome stability by cooperating with DNA topoisomerase IIβ. Interestingly, Beclin 1 loses its ability to localize to DNA damage sites when DNA topoisomerase IIβ is completely silenced, suggesting that DNA topoisomerase IIβ might directly recruit Beclin 1 to the DNA damage site, recruit DNA damage repair proteins, and promote DNA damage repair (Figure 2).
Non-autophagic function of autophagy-related proteins involved in regulating chromosome stability
Monoallelic loss of Beclin 1 has been reported to cause chromosomal disorders, resulting in chromosomal instability [28]. However, it remains to be determined whether all of these phenomena are caused by autophagy defects and whether they reflect the non-autophagy-dependent functions of Beclin 1. Increasing evidences suggests that autophagy proteins participate in genome homeostasis regulation through non-autophagy pathways. Studies have demonstrated that Beclin 1 depletion leads to severe chromosomal aggregation defects. The mechanism is related to the decrease in the core components of several kinetochore components, including ZW10, CENP-E, and CENP-F. Among them, Beclin 1 can directly interact with the external kinetochore component Zwint-1. Beclin-1 acts upstream of the Rod-ZW10-Zwilch (RZZ) complex to facilitate the accurate localization of motilin to the spindle during mitosis due to its direct binding to kinetochore constituent proteins [98]. In addition, Beclin 1 and/or UVRAG also modulate centrosome stability. Centrosome amplification can lead to spindle malformation and chromosome segregation errors, resulting in chromosomal instability. There was a significant increase in the number of multicentrosomes when Beclin 1 or UVRAG was knocked down, whereas the number of multicentrosomes increased to a lesser extent when ATG5 is knocked down. These results suggested that Beclin 1 and/or UVRAG may regulate centrosome stability independently of autophagy, prevent abnormal centrosome amplification, and maintain genomic stability and normal mitosis [41].
In addition, the E3 ubiquitin ligase parkin can interact with anaphase promoter complex/ cyclosome (APC/C) co-activators Cdc20 and CDH1, and further through the Parkin-Cdc20/Cdh1 complex and the APC/C complex co-regulate mitosis. The deletion of Parkin leads to abnormal levels of mitotic regulators, which may lead to chromosomal instability, the disruption of genomic homeostasis, chromosomal dislocation, and events such as hysteresis, aneuploidy, and cytokinesis defects. These could be closely related to tumorigenesis [99] (Figure 3).
Non-autophagic function of autophagy-related proteins involved in the cell cycle regulation
Atg7 combines with the tumor suppressor p53 to regulate the transcription of cell cycle inhibitor p21CDKN1A, and p21CDKN1A expression cannot be properly induced in Atg7-deficient cells. The binding of Atg7 and p53 is increased under metabolic stress and starvation. Under starvation, Atg7-deficient cells inhibit p53-mediated cell cycle arrest, whereas under sustained metabolic stress, p53-mediated cell death is increased in Atg7-deficient cells. Therefore, when Atg7 is absent, p53-dependent cell cycle arrest and cell death become more sensitive. The cell cycle defect is independent of the E1-like enzymatic activity of ATG7 and also independent of regulation of autophagy [73]. Furthermore, in human bladder cancer (BC), ATG7 knockdown induces cell cycle stagnation in the G2/M phase by promoting P27 expression, which can inhibit tumor development. One possible mechanism is that inhibition of ATG7 expression can stabilize ETS2 mRNA, thus reducing the transcription of mir-196b and the expression of mir-196b. miR-196b can bind to and degrade the 3'UTR of FOXO1 mRNA. Reduced miR-196b expression levels stabilize FOXO1 mRNA and ultimately promote p27 transcription and weaken BC tumorigenic growth [74].
CDC2 regulates the transition from G2 phase to mitosis, and dephosphorylation of CDC2 at Tyr15 is a key step in CDC2 activation [100]. Parkin brings about increasing expression of Myt1, followed by phosphorylation of CDC2 at Tyr15, which induces G2/M cell cycle arrest [101]. Parkin can not only regulate G2/M cell cycle arrest, but is also the main regulator of G1/S arrest. Knockdown or inactivation of parkin results in massive accumulation of cyclin D1 and cyclin E1, accelerating cell cycle progression [102]. Interestingly, parkin mRNA and protein expression are significantly decreased in breast cancer, but no significant changes in cyclin D1 and cyclin E levels are observed, while cyclin-dependent kinase 6 (CDK6) expression level is significantly increased. This suggests an inhibitory role of parkin in the development of breast cancer, and the mechanism may involve a novel association between parkin and CDK6 [103]. Furthermore, the AKT pathway accelerates G1/S cell cycle progression by increasing cyclin D1 levels, and AKT activation requires T308 phosphorylation of PDK1 and S473 phosphorylation of the TORC2 complex [104]. Regulation of VEGF-VEGFR2 on cell cycle progression and cell viability also relate to activation of the PI3K/AKT pathway [105]. The study found that parkin could act on TORC2 and significantly affect the expression of VEGFR-2, which suggested that the mechanism of parkin-mediated cell proliferation inhibition could involve the VEGFR2/AKT/cyclin D1 pathway [106].
mTOR also regulates the cell cycle through non-autophagy pathways in response to intracellular and extracellular signals. The regulation of G1 phase progression by mTOR also depends on the activity of the 4E-BP1 and S6K1 pathways, which can regulate the transcription of cyclin D and cyclin E [107]. In addition, mTORC1 blocks the nuclear function of p27 KIP1 as a CDK inhibitor by affecting its localization [108]. Some studies have found that the phosphorylation of 4E-BP1 and S6K1 still exists during G2/M, and the mTOR pathway also seems to be the guardian of genome stability by regulating the cell cycle during G2/M [109, 110].
As a focal adhesion kinase (FAK) protein inhibitor, FIP200 inhibits FAK kinase activity by binding to FAK, thereby inhibiting its biological function [111]. Studies have found that FIP200 can inhibit the G1/S phase progression, cell proliferation, and clone formation of human breast cancer cells. The mechanism is to enhance the activity of the p21 promoter by stabilizing the half-life of upstream p53 and reducing the level of cyclin D1 in breast cancer cells [112].
In addition, it was also reported that the predicted Atg10 homolog (SpAtg10) of the schizosaccharides was essential to maintaining the normal cell cycle process, but it did not combine with Atg12 through autophagic Ubl conjugation pathways [113] (Figure 4) (Table 2).
Autophagy-related proteins involved in genome stability through non-autophagic pathway
| Autophagy-related proteins | Target molecule | Functions | References |
|---|---|---|---|
| mTORC1 | S6K/FANCD2, 4E-BP1 | promotes DNA repair and replication fork survival | [81,82] |
| 4E-BP /S6K1/cyclin D and cyclin E | regulates G1 phase progression | [107] | |
| S6K1 | stabilizes the genome in G2/M phase | [109] | |
| ATG5 | Mis18α | microsatellite instability and DNA mismatch repair deficiency | [93] |
| p62 | DLK1/NADPH oxidase/ROS | induces genomic instability | [95] |
| ULK1 | PARP-1 | is as a DNA damage factor | [96] |
| Beclin1 | DNA topoisomerase IIβ/53bP1 | is involved in DNA repair | [97] |
| Zwint-1 | Protects chromosomestability | [98] | |
| ATG7 | p53 | Regulates cell cycle during metabolic stress; | [73] |
| ETS2/miRNA196b/FOXO1/p27 Axis | Knockdown of ATG7 induces cell cycle arrest in G2/M phase. | [74] | |
| parkin | Cdc20/Cdh1/APC/C | Protects chromosomestability | [99] |
| Myt1/CDC2 | Induces cell cycle arrest in G2/M phase; | [101] | |
| cyclin D1 and cyclin E1 | Knockdown or inactivation of PARK2 accelerates cell cycle progression; | [102] | |
| VEGFR2/Akt/cyclin D1 | Promoted G1 phase cell-cycle arrest and mitigated the proliferation. | [106] | |
| FIP200 | p21/cyclin D1 | Inhibits G1-S phase progression and cell proliferation | [112] |
| SpATG10 | Is essential for normal cell cycle progression in S. pombe. | [113] |
Autophagy-related proteins affect human disease by regulating genome stability
Autophagy-related proteins affect tumor development by regulating genome stability
Autophagy inhibits cell carcinogenesis by maintaining genomic stability in normal cells. However, once the tumor environment is established, autophagy is conducive to the survival and development of cancer cells in the tumor microenvironment [114, 115]. During the stage of tumorigenesis, cancer cells lose cell cycle control and exhibit immortal proliferation. Arresting the cell cycle, especially at the G2/M checkpoint, may be an effective method to inhibit tumor progression. The study found that Beclin 1 mutant mice had a relatively high incidence of cancer, including breast tumors, lymphoma, and hepatocellular carcinoma [59, 116]. Based on these findings, it can be inferred that that Beclin-1 can block G2/M phase to delay cell cycle progression and induce autophagy and Beclin 1 may be a tumor suppressor factor. The same results have been demonstrated in human cancers, where Beclin 1 expression was reduced in a number of human cancers, including glioblastoma, ovarian cancer, and esophageal cancer [117-119]. However, it has been reported that Beclin1 expression is increased in colorectal cancer cells and gastric cancer cells compared with normal cells [120]. No biological macromolecule is absolutely good or bad, and these results may suggest that Beclin1 plays distinct roles in different tissues.
UVB and UVA radiation can damage DNA, which is an important cause of skin cancer [121]. The tumor suppressor p53 can mediate the apoptosis and clearance of radiation-injured skin cells, but excessive clearance of skin cells can damage the skin barrier function. AKT/mTOR can negatively regulate cell apoptosis, inhibit autophagy and prevent cell cycle arrest, and cells prevent excessive apoptosis of skin cells through the AKT/mTOR anti-apoptotic signaling pathway [122, 123]. However, the overactivation of AKT/mTOR will promote the development of epidermal tumors. Therefore, the inhibition of mTOR complex and AKT may be a promising strategy for preventing photocarcinogenesis [124].
Myelodysplastic syndrome (MDS) can transform into acute myeloid leukemia (AML) and is caused by genomic instability and somatic mutations within hematopoietic stem cells (HSPC). Because MicroRNA-146a (miR-146a) is located adjacent to the distal deletion region and the deletion of miR-146a is an initiating event in del (5q) myeloid malignancies (which is dependent on the nuclear factor kappa B (NF-κB) control system), it is related to the pathogenesis of human MDS [125]. P62 can recruit TRAF6 and initiate NF-κB signaling. Knockdown of p62 or disruption of p62-TRAF6 binding can lead to cell cycle arrest and apoptosis in MDS/AML cell lines and clinical samples [126]. Importantly, in the study, the lack of p62 had little effect on the lifespan and function of normal HSPCs. This suggested that disturbing p62/TRAF6 binding may hold promise for treating miR-146a-deficient leukemia [127].
Autophagy-related proteins affect cellular senescence by regulating genome stability
Mitotic slippage refers to the cellular process in which mitosis is forced to stall and slip into interphase using antimitotic drugs, when chromosomes do not segregate properly and the cytoplasm does not divide perfectly. Sliding cells may continue to proliferate in a genomically unstable form, or they may arrest in the next post-interphase slip and eventually develop into cellular senescence, with senescence-associated secretory phenotypes (SASP) [128]. SASP factors cause paracrine and promote tumor formation. Following cell slippage, autophagy will be induced by ER stress and the AMPK/mTOR/ULK1 axis. Both pharmacological inhibition of autophagy and silencing of ATG5 can prevent the generation of SASP and reduce the paracrine tumorigenicity caused by SASP. Then, cells enter the S phase while inducing DNA damage and replication stress, reducing cell viability, bypassing cellular senescence, accelerating cell death, and preventing tumorigenesis [129].
Furthermore, following DNA damage, cascades signaling of genomic instability are integrated into PGC-1β-dependent mitochondrial biogenesis, which is conducive to ROS-mediated DDR and cell cycle arrest as well as cellular senescence [130]. Additionally, there is evidence that reducing mitochondrial content in vivo through mTORC 1 inhibition or PGC-1β deletion could delay aging in mouse livers.
Conclusions and future perspective
The non-autophagic functions of autophagy proteins are receiving increasing attention, among which many functions related to the maintenance of genome stability in the nucleus of autophagy proteins are essential for cell survival. These functions involve DNA damage repair, protection of chromosome stability, and cell cycle regulation. We summarized the roles of autophagy proteins in maintaining genome stability in the nucleus through both autophagy-dependent and autophagy-independent pathway. Some autophagy proteins can act through both autophagy-dependent and autophagy-independent pathways, while others can only act through a certain pathway. These autophagy proteins are closely related to tumorigenesis and development of various tumors and the process of cell senescence, which could open door to new ideas for the prevention and treatment of diseases. This enriches the background of these autophagic proteins, indicating a non-autophagic function in the nucleus. Research on the non-autophagy function of autophagy proteins is currently a hot issue among scientists. Therefore, fully revealing their functions, especially their non-autophagic functions, could lead to new theories and in-depth understanding of the biological significance of autophagy-related proteins.
Abbreviations
AML: acute myeloid leukemia; APC/C: anaphase promoter complex/ cyclosome; BC: bladder cancer; CIN: Chromosome instability; CCD: coiled-coil domain; CIMP: CpG island methylation phenotype; CDK6: cyclin-dependent kinase 6; CDKs: cyclin-dependent kinases; DDR: DNA damage response; DSB: DNA double-strand break; FLNA: filamin A; FAK: focal adhesion kinase; GC: gastric cancer; HSPC: hematopoietic stem cells; HR: homologous recombination; IR: ionizing radiation; LB: LIM-binding; METTL14: methyltransferase-like 14; miR-146a: microRNA-146a; MMR-D: mismatch repair deficiency; MLH1: mutL homolog 1; MDS: Myelodysplastic syndrome; m6A: N6-methyladenosine; NSC: neural stem cells; NHEJ: non-homologous end joining; NF-κB: nuclear factor kappa B; 53bP1: p53-binding protein 1; PAS: pre-autophagy structures; PKB/AKT: Protein kinase B; ROS: reactive oxygen species; RZZ: Rod-ZW10-Zwilch; SASP: senescence-associated secretory phenotypes; Ulk1: Unc-51-like kinase 1.
Acknowledgements
This work was supported by National Key R&D Program of China (2016YFC1302400); Key projects of National Natural Fund of China (82030091); Natural Science Foundation of China (82073089, 81770001); Ministry of Education Innovation Team Development Plan (IRT13101/17R107); Science Foundation from Liaoning Province (2021JH2/10300023, 2019JH2/10300, Z18-4-021), Natural Science Foundation of Liaoning Province of China (2018225083, JC2019039).
Author Contributions
Q.G., L.C., and X.S. designed the review; Y.Z. and R.G. searched for literatures and wrote the manuscript; Y.Z., R.G., S.W., X.J., H.C., and Y.G. organized literaturs; Y.Z. drew the pattern charts. All authors have read and approved the manuscript and agree with publication in this journal.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728-41
2. Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132-9
3. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132-41
4. Fogel AI, Dlouhy BJ, Wang C, Ryu SW, Neutzner A, Hasson SA. et al. Role of membrane association and Atg14-dependent phosphorylation in beclin-1-mediated autophagy. Mol Cell Biol. 2013;33:3675-88
5. Furuya N, Yu J, Byfield M, Pattingre S, Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy. 2005;1:46-52
6. Papinski D, Schuschnig M, Reiter W, Wilhelm L, Barnes CA, Maiolica A. et al. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell. 2014;53:471-83
7. Metlagel Z, Otomo C, Takaesu G, Otomo T. Structural basis of ATG3 recognition by the autophagic ubiquitin-like protein ATG12. Proc Natl Acad Sci U S A. 2013;110:18844-9
8. Otomo C, Metlagel Z, Takaesu G, Otomo T. Structure of the human ATG12~ATG5 conjugate required for LC3 lipidation in autophagy. Nat Struct Mol Biol. 2013;20:59-66
9. Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T. et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116:1679-88
10. Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805-12
11. Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 2017;58:235-63
12. Yoder FE, Vincent RA Jr, Morgan SK, Grush OC. Chromosome fragile sites. Cancer Genet Cytogenet. 1985;14:369
13. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
14. Widrow RJ, Hansen RS, Kawame H, Gartler SM, Laird CD. Very late DNA replication in the human cell cycle. Proc Natl Acad Sci U S A. 1998;95:11246-50
15. Diffley JF. Regulation of early events in chromosome replication. Curr Biol. 2004;14:R778-86
16. Akopyan K, Silva Cascales H, Hukasova E, Saurin AT, Mullers E, Jaiswal H. et al. Assessing kinetics from fixed cells reveals activation of the mitotic entry network at the S/G2 transition. Mol Cell. 2014;53:843-53
17. Hayashi MT, Cesare AJ, Fitzpatrick JA, Lazzerini-Denchi E, Karlseder J. A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat Struct Mol Biol. 2012;19:387-94
18. Zhang D, Tang B, Xie X, Xiao YF, Yang SM, Zhang JW. The interplay between DNA repair and autophagy in cancer therapy. Cancer Biol Ther. 2015;16:1005-13
19. Bu W, Hao X, Yang T, Wang J, Liu Q, Zhang X. et al. Autophagy Contributes to the Maintenance of Genomic Integrity by Reducing Oxidative Stress. Oxid Med Cell Longev. 2020: 2015920.
20. Guo H, Chitiprolu M, Gagnon D, Meng L, Perez-Iratxeta C, Lagace D. et al. Autophagy supports genomic stability by degrading retrotransposon RNA. Nat Commun. 2014;5:5276
21. Yang Z, Yang S, Cui YH, Wei J, Shah P, Park G. et al. METTL14 facilitates global genome repair and suppresses skin tumorigenesis. Proc Natl Acad Sci U S A. 2021;118:e2025948118
22. Wang L, Xu XB, You WW, Lin XX, Li CT, Qian HR. et al. The cytoplasmic nuclear shuttling of Beclin 1 in neurons with Alzheimer's disease-like injury. Neurosci Lett. 2017;661:63-70
23. Saleiro D, Mehrotra S, Kroczynska B, Beauchamp EM, Lisowski P, Majchrzak-Kita B. et al. Central role of ULK1 in type I interferon signaling. Cell Rep. 2015;11:605-17
24. Ilha J, do Espirito-Santo CC, de Freitas GR. mTOR Signaling Pathway and Protein Synthesis: From Training to Aging and Muscle Autophagy. Adv Exp Med Biol. 2018;1088:139-51
25. Wang B, Iyengar R, Li-Harms X, Joo JH, Wright C, Lavado A. et al. The autophagy-inducing kinases, ULK1 and ULK2, regulate axon guidance in the developing mouse forebrain via a noncanonical pathway. Autophagy. 2018;14:796-811
26. Zhou X, Babu JR, da Silva S, Shu Q, Graef IA, Oliver T. et al. Unc-51-like kinase 1/2-mediated endocytic processes regulate filopodia extension and branching of sensory axons. Proc Natl Acad Sci U S A. 2007;104:5842-7
27. Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S. et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21:1621-35
28. Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K. et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367-81
29. Xie R, Wang F, McKeehan WL, Liu L. Autophagy enhanced by microtubule- and mitochondrion-associated MAP1S suppresses genome instability and hepatocarcinogenesis. Cancer Res. 2011;71:7537-46
30. Vessoni AT, Filippi-Chiela EC, Menck CF, Lenz G. Autophagy and genomic integrity. Cell Death Differ. 2013;20:1444-54
31. Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R. et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471:74-9
32. Belaid A, Cerezo M, Chargui A, Corcelle-Termeau E, Pedeutour F, Giuliano S. et al. Autophagy plays a critical role in the degradation of active RHOA, the control of cell cytokinesis, and genomic stability. Cancer Res. 2013;73:4311-22
33. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071-8
34. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY. et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062-75
35. Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy. 2021;17:1-382
36. Yan S, Liu L, Ren F, Gao Q, Xu S, Hou B. et al. Sunitinib induces genomic instability of renal carcinoma cells through affecting the interaction of LC3-II and PARP-1. Cell Death Dis. 2017;8:e2988
37. Gao W, Shen Z, Shang L, Wang X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ. 2011;18:1598-607
38. Bae H, Guan JL. Suppression of autophagy by FIP200 deletion impairs DNA damage repair and increases cell death upon treatments with anticancer agents. Mol Cancer Res. 2011;9:1232-41
39. Sinha S, Levine B. The autophagy effector Beclin 1: a novel BH3-only protein. Oncogene. 2008;27(Suppl 1):S137-48
40. Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH. et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688-99
41. Park JM, Tougeron D, Huang S, Okamoto K, Sinicrope FA. Beclin 1 and UVRAG confer protection from radiation-induced DNA damage and maintain centrosome stability in colorectal cancer cells. PLoS One. 2014;9:e100819
42. Pankiv S, Lamark T, Bruun JA, Overvatn A, Bjorkoy G, Johansen T. Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. J Biol Chem. 2010;285:5941-53
43. Hewitt G, Korolchuk VI. Repair, Reuse, Recycle: The Expanding Role of Autophagy in Genome Maintenance. Trends Cell Biol. 2017;27:340-51
44. Li W, Dai Y, Shi B, Yue F, Zou J, Xu G. et al. LRPPRC sustains Yap-P27-mediated cell ploidy and P62-HDAC6-mediated autophagy maturation and suppresses genome instability and hepatocellular carcinomas. Oncogene. 2020;39:3879-92
45. Wang Y, Zhang N, Zhang L, Li R, Fu W, Ma K. et al. Autophagy Regulates Chromatin Ubiquitination in DNA Damage Response through Elimination of SQSTM1/p62. Mol Cell. 2016;63:34-48
46. Thorslund T, Ripplinger A, Hoffmann S, Wild T, Uckelmann M, Villumsen B. et al. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature. 2015;527:389-93
47. Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R. et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435-46
48. Feng Y, Klionsky DJ. Autophagy regulates DNA repair through SQSTM1/p62. Autophagy. 2017;13:995-6
49. Hewitt G, Carroll B, Sarallah R, Correia-Melo C, Ogrodnik M, Nelson G. et al. SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy. 2016;12:1917-30
50. Eskelinen EL. The dual role of autophagy in cancer. Curr Opin Pharmacol. 2011;11:294-300
51. Burdelski C, Reiswich V, Hube-Magg C, Kluth M, Minner S, Koop C. et al. Cytoplasmic Accumulation of Sequestosome 1 (p62) Is a Predictor of Biochemical Recurrence, Rapid Tumor Cell Proliferation, and Genomic Instability in Prostate Cancer. Clin Cancer Res. 2015;21:3471-9
52. Inui T, Chano T, Takikita-Suzuki M, Nishikawa M, Yamamoto G, Okabe H. Association of p62/SQSTM1 excess and oral carcinogenesis. PLoS One. 2013;8:e74398
53. Ren SX, Shen J, Cheng AS, Lu L, Chan RL, Li ZJ. et al. FK-16 derived from the anticancer peptide LL-37 induces caspase-independent apoptosis and autophagic cell death in colon cancer cells. PLoS One. 2013;8:e63641
54. Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517-28
55. Munoz-Gamez JA, Rodriguez-Vargas JM, Quiles-Perez R, Aguilar-Quesada R, Martin-Oliva D, de Murcia G. et al. PARP-1 is involved in autophagy induced by DNA damage. Autophagy. 2009;5:61-74
56. Liu D, Shaukat Z, Xu T, Denton D, Saint R, Gregory S. Autophagy regulates the survival of cells with chromosomal instability. Oncotarget. 2016;7:63913-23
57. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585-621
58. Mar FA, Debnath J, Stohr BA. Autophagy-independent senescence and genome instability driven by targeted telomere dysfunction. Autophagy. 2015;11:527-37
59. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077-82
60. Delaney JR, Patel CB, Bapat J, Jones CM, Ramos-Zapatero M, Ortell KK. et al. Autophagy gene haploinsufficiency drives chromosome instability, increases migration, and promotes early ovarian tumors. PLoS Genet. 2020;16:e1008558
61. Watanabe Y, Honda S, Konishi A, Arakawa S, Murohashi M, Yamaguchi H. et al. Autophagy controls centrosome number by degrading Cep63. Nat Commun. 2016;7:13508
62. Bento CF, Renna M, Ghislat G, Puri C, Ashkenazi A, Vicinanza M. et al. Mammalian Autophagy: How Does It Work? Annu Rev Biochem. 2016;85:685-713
63. Ames K, Da Cunha DS, Gonzalez B, Konta M, Lin F, Shechter G. et al. A Non-Cell-Autonomous Role of BEC-1/BECN1/Beclin1 in Coordinating Cell-Cycle Progression and Stem Cell Proliferation during Germline Development. Curr Biol. 2017;27:905-13
64. Koneri K, Goi T, Hirono Y, Katayama K, Yamaguchi A. Beclin 1 gene inhibits tumor growth in colon cancer cell lines. Anticancer Res. 2007;27:1453-7
65. Huerta S. Recent advances in the molecular diagnosis and prognosis of colorectal cancer. Expert Rev Mol Diagn. 2008;8:277-88
66. Won KY, Kim GY, Lim SJ, Kim YW. Decreased Beclin-1 expression is correlated with the growth of the primary tumor in patients with squamous cell carcinoma and adenocarcinoma of the lung. Hum Pathol. 2012;43:62-8
67. Xu Y, Liu XD, Gong X, Eissa NT. Signaling pathway of autophagy associated with innate immunity. Autophagy. 2008;4:110-2
68. Liang N, Liu X, Zhang S, Sun H. The role of Beclin 1 in IR-induced crosstalk between autophagy and G2/M cell cycle arrest. Cell Signal. 2019;62:109353
69. Li Z, Zhang X. Phospho-regulation and function of ULK1-ATG13 during the cell cycle. Autophagy. 2021;17:1054-6
70. Li Z, Tian X, Ji X, Wang J, Chen H, Wang D. et al. ULK1-ATG13 and their mitotic phospho-regulation by CDK1 connect autophagy to cell cycle. PLoS Biol. 2020;18:e3000288
71. Juhasz G, Erdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007;21:3061-6
72. Wang G, Chen EN, Liang C, Liang J, Gao LR, Chuai M. et al. Atg7-Mediated Autophagy Is Involved in the Neural Crest Cell Generation in Chick Embryo. Mol Neurobiol. 2018;55:3523-36
73. Lee IH, Kawai Y, Fergusson MM, Rovira, II, Bishop AJ, Motoyama N. et al. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science. 2012;336:225-8
74. Zhu J, Li Y, Tian Z, Hua X, Gu J, Li J. et al. ATG7 Overexpression Is Crucial for Tumorigenic Growth of Bladder Cancer In vitro and In vivo by Targeting the ETS2/miRNA196b/FOXO1/p27 Axis. Mol Ther Nucleic Acids. 2017;7:299-313
75. Wang JL, Wang JJ, Cai ZN, Xu CJ. The effect of curcumin on the differentiation, apoptosis and cell cycle of neural stem cells is mediated through inhibiting autophagy by the modulation of Atg7 and p62. Int J Mol Med. 2018;42:2481-8
76. Liu Q, Shi X, Zhou X, Wang D, Wang L, Li C. Effect of autophagy inhibition on cell viability and cell cycle progression in MDAMB231 human breast cancer cells. Mol Med Rep. 2014;10:625-30
77. An J, Liu Y, Duo S, Ma X, An L, Yan Y. et al. Podofilox suppresses gastric cancer cell proliferation by regulating cell cycle arrest and the c-Myc/ATG10 axis. Exp Ther Med. 2021;22:1203
78. Liu PF, Hsu CJ, Tsai WL, Cheng JS, Chen JJ, Huang IF. et al. Ablation of ATG4B Suppressed Autophagy and Activated AMPK for Cell Cycle Arrest in Cancer Cells. Cell Physiol Biochem. 2017;44:728-40
79. Hashemolhosseini S, Nagamine Y, Morley SJ, Desrivieres S, Mercep L, Ferrari S. Rapamycin inhibition of the G1 to S transition is mediated by effects on cyclin D1 mRNA and protein stability. J Biol Chem. 1998;273:14424-9
80. Pirtoli L, Belmonte G, Toscano M, Tini P, Miracco C. Comment on "Everolimus induces G1 cell arrest through autophagy-mediated protein degradation of cyclin D1 in breast cancer cells". Am J Physiol Cell Physiol. 2020;318:C448-C9
81. Sarraf SA, Sideris DP, Giagtzoglou N, Ni L, Kankel MW, Sen A. et al. PINK1/Parkin Influences Cell Cycle by Sequestering TBK1 at Damaged Mitochondria, Inhibiting Mitosis. Cell Rep. 2019;29:225-35 e5
82. Guo F, Li J, Du W, Zhang S, O'Connor M, Thomas G. et al. mTOR regulates DNA damage response through NF-kappaB-mediated FANCD2 pathway in hematopoietic cells. Leukemia. 2013;27:2040-6
83. Ma Y, Vassetzky Y, Dokudovskaya S. mTORC1 pathway in DNA damage response. Biochim Biophys Acta Mol Cell Res. 2018;1865:1293-311
84. Lamm N, Read MN, Nobis M, Van Ly D, Page SG, Masamsetti VP. et al. Nuclear F-actin counteracts nuclear deformation and promotes fork repair during replication stress. Nat Cell Biol. 2020;22:1460-70
85. Nolan M, Knudson K, Holz MK, Chaudhury I. Fanconi anemia and mTOR pathways functionally interact during stalled replication fork recovery. FEBS Lett. 2021;595:595-603
86. Krieger KL, Hu WF, Ripperger T, Woods NT. Functional Impacts of the BRCA1-mTORC2 Interaction in Breast Cancer. Int J Mol Sci. 2019;20:5876
87. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648-57
88. Thedieck K, Polak P, Kim ML, Molle KD, Cohen A, Jeno P. et al. PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS One. 2007;2:e1217
89. Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J. et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160-6
90. Bandhakavi S, Kim YM, Ro SH, Xie H, Onsongo G, Jun CB. et al. Quantitative nuclear proteomics identifies mTOR regulation of DNA damage response. Mol Cell Proteomics. 2010;9:403-14
91. Sinicrope FA, Rego RL, Halling KC, Foster N, Sargent DJ, La Plant B. et al. Prognostic impact of microsatellite instability and DNA ploidy in human colon carcinoma patients. Gastroenterology. 2006;131:729-37
92. Devis-Jauregui L, Eritja N, Davis ML, Matias-Guiu X, Llobet-Navas D. Autophagy in the physiological endometrium and cancer. Autophagy. 2021;17:1077-95
93. Sun SY, Hu XT, Yu XF, Zhang YY, Liu XH, Liu YH. et al. Nuclear translocation of ATG5 induces DNA mismatch repair deficiency (MMR-D)/microsatellite instability (MSI) via interacting with Mis18alpha in colorectal cancer. Br J Pharmacol. 2021;178:2351-69
94. Block K, Gorin Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat Rev Cancer. 2012;12:627-37
95. Kessler SM, Laggai S, Barghash A, Schultheiss CS, Lederer E, Artl M. et al. IMP2/p62 induces genomic instability and an aggressive hepatocellular carcinoma phenotype. Cell Death Dis. 2015;6:e1894
96. Joshi A, Iyengar R, Joo JH, Li-Harms XJ, Wright C, Marino R. et al. Nuclear ULK1 promotes cell death in response to oxidative stress through PARP1. Cell Death Differ. 2016;23:216-30
97. Xu F, Fang Y, Yan L, Xu L, Zhang S, Cao Y. et al. Nuclear localization of Beclin 1 promotes radiation-induced DNA damage repair independent of autophagy. Sci Rep. 2017;7:45385
98. Fremont S, Gerard A, Galloux M, Janvier K, Karess RE, Berlioz-Torrent C. Beclin-1 is required for chromosome congression and proper outer kinetochore assembly. EMBO Rep. 2013;14:364-72
99. Lee SB, Kim JJ, Nam HJ, Gao B, Yin P, Qin B. et al. Parkin Regulates Mitosis and Genomic Stability through Cdc20/Cdh1. Mol Cell. 2015;60:21-34
100. Norbury C, Blow J, Nurse P. Regulatory phosphorylation of the p34cdc2 protein kinase in vertebrates. EMBO J. 1991;10:3321-9
101. Lee MH, Cho Y, Jung BC, Kim SH, Kang YW, Pan CH. et al. Parkin induces G2/M cell cycle arrest in TNF-alpha-treated HeLa cells. Biochem Biophys Res Commun. 2015;464:63-9
102. Gong Y, Zack TI, Morris LG, Lin K, Hukkelhoven E, Raheja R. et al. Pan-cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat Genet. 2014;46:588-94
103. Tay SP, Yeo CW, Chai C, Chua PJ, Tan HM, Ang AX. et al. Parkin enhances the expression of cyclin-dependent kinase 6 and negatively regulates the proliferation of breast cancer cells. J Biol Chem. 2010;285:29231-8
104. Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem. 2005;280:40406-16
105. Knizetova P, Ehrmann J, Hlobilkova A, Vancova I, Kalita O, Kolar Z. et al. Autocrine regulation of glioblastoma cell cycle progression, viability and radioresistance through the VEGF-VEGFR2 (KDR) interplay. Cell Cycle. 2008;7:2553-61
106. Yeo CW, Ng FS, Chai C, Tan JM, Koh GR, Chong YK. et al. Parkin pathway activation mitigates glioma cell proliferation and predicts patient survival. Cancer Res. 2012;72:2543-53
107. Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol Cell Biol. 2004;24:200-16
108. Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782-7
109. Cuyas E, Corominas-Faja B, Joven J, Menendez JA. Cell cycle regulation by the nutrient-sensing mammalian target of rapamycin (mTOR) pathway. Methods Mol Biol. 2014;1170:113-44
110. Xu XY, Zhang Z, Su WH, Zhang Y, Yu YQ, Li YX. et al. Characterization of p70 S6 kinase 1 in early development of mouse embryos. Dev Dyn. 2009;238:3025-34
111. Abbi S, Ueda H, Zheng C, Cooper LA, Zhao J, Christopher R. et al. Regulation of focal adhesion kinase by a novel protein inhibitor FIP200. Mol Biol Cell. 2002;13:3178-91
112. Melkoumian ZK, Peng X, Gan B, Wu X, Guan JL. Mechanism of cell cycle regulation by FIP200 in human breast cancer cells. Cancer Res. 2005;65:6676-84
113. Flanagan MD, Whitehall SK, Morgan BA. An Atg10-like E2 enzyme is essential for cell cycle progression but not autophagy in Schizosaccharomyces pombe. Cell Cycle. 2013;12:271-7
114. Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G. et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51-64
115. Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H. et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717-29
116. Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A. et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809-20
117. Miracco C, Cosci E, Oliveri G, Luzi P, Pacenti L, Monciatti I. et al. Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumours. Int J Oncol. 2007;30:429-36
118. Shen Y, Li DD, Wang LL, Deng R, Zhu XF. Decreased expression of autophagy-related proteins in malignant epithelial ovarian cancer. Autophagy. 2008;4:1067-8
119. Chen Y, Lu Y, Lu C, Zhang L. Beclin-1 expression is a predictor of clinical outcome in patients with esophageal squamous cell carcinoma and correlated to hypoxia-inducible factor (HIF)-1alpha expression. Pathol Oncol Res. 2009;15:487-93
120. Ahn CH, Jeong EG, Lee JW, Kim MS, Kim SH, Kim SS. et al. Expression of beclin-1, an autophagy-related protein, in gastric and colorectal cancers. APMIS. 2007;115:1344-9
121. Besaratinia A, Synold TW, Chen HH, Chang C, Xi B, Riggs AD. et al. DNA lesions induced by UV A1 and B radiation in human cells: comparative analyses in the overall genome and in the p53 tumor suppressor gene. Proc Natl Acad Sci U S A. 2005;102:10058-63
122. Gottlieb TM, Leal JF, Seger R, Taya Y, Oren M. Cross-talk between Akt, p53 and Mdm2: possible implications for the regulation of apoptosis. Oncogene. 2002;21:1299-303
123. Strozyk E, Kulms D. The role of AKT/mTOR pathway in stress response to UV-irradiation: implication in skin carcinogenesis by regulation of apoptosis, autophagy and senescence. Int J Mol Sci. 2013;14:15260-85
124. Carr TD, DiGiovanni J, Lynch CJ, Shantz LM. Inhibition of mTOR suppresses UVB-induced keratinocyte proliferation and survival. Cancer Prev Res (Phila). 2012;5:1394-404
125. Starczynowski DT, Kuchenbauer F, Argiropoulos B, Sung S, Morin R, Muranyi A. et al. Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat Med. 2010;16:49-58
126. Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT. et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343-54
127. Fang J, Starczynowski DT. Genomic instability establishes dependencies on acquired gene regulatory networks: A novel role of p62 in myeloid malignancies with del(5q). Mol Cell Oncol. 2015;2:e1014219
128. Cheng B, Crasta K. Consequences of mitotic slippage for antimicrotubule drug therapy. Endocr Relat Cancer. 2017;24:T97-T106
129. Jakhar R, Luijten MNH, Wong AXF, Cheng B, Guo K, Neo SP. et al. Autophagy Governs Protumorigenic Effects of Mitotic Slippage-induced Senescence. Mol Cancer Res. 2018;16:1625-40
130. Correia-Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J. et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016;35:724-42
Author contact
![]() Corresponding authors: Liu Cao, College of Basic Medical Science, Health Sciences Institute, Key Laboratory of Medical Cell Biology, Ministry of Education, China Medical University, Shenyang, Liaoning Province, P. R. China, 110122. E-mail: lcaoedu.cn. Tel: +86-18900911888; Qi-Qiang Guo, College of Basic Medical Science, Health Sciences Institute, Key Laboratory of Medical Cell Biology, Ministry of Education, China Medical University, Shenyang, Liaoning Province, P. R. China, 110122. E-mail: qqguoedu.cn; Tel: +86-18900910432; Xiao-Yu Song, College of Basic Medical Science, Health Sciences Institute, Key Laboratory of Medical Cell Biology, Ministry of Education, China Medical University, Shenyang, Liaoning Province, P. R. China, 110122. Email: xysongedu.cn. Tel: +86-18900910183.
Corresponding authors: Liu Cao, College of Basic Medical Science, Health Sciences Institute, Key Laboratory of Medical Cell Biology, Ministry of Education, China Medical University, Shenyang, Liaoning Province, P. R. China, 110122. E-mail: lcaoedu.cn. Tel: +86-18900911888; Qi-Qiang Guo, College of Basic Medical Science, Health Sciences Institute, Key Laboratory of Medical Cell Biology, Ministry of Education, China Medical University, Shenyang, Liaoning Province, P. R. China, 110122. E-mail: qqguoedu.cn; Tel: +86-18900910432; Xiao-Yu Song, College of Basic Medical Science, Health Sciences Institute, Key Laboratory of Medical Cell Biology, Ministry of Education, China Medical University, Shenyang, Liaoning Province, P. R. China, 110122. Email: xysongedu.cn. Tel: +86-18900910183.