Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Generation and growth of lipid...
The role of lipid droplet...
Lipid droplet-mitochondrion...
Lipophagy regulates adipose...
The role of lipid droplets in...
Concluding Remarks
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(16):6176-6188. doi:10.7150/ijbs.77051 This issue Cite
Review
Lipid Droplets: A Cellular Organelle Vital for Thermogenesis
Lupeng Chen1, Yi Jin1, Jian Wu1, Zhuqing Ren1,2 ![]()
1. Key Laboratory of Agriculture Animal Genetics, Breeding and Reproduction of the Ministry of Education, College of Animal Science and Technology, Huazhong Agricultural University, Wuhan 430070, China.
2. Hubei Hongshan Laboratory, Wuhan, China.
Received 2022-7-12; Accepted 2022-10-9; Published 2022-10-24
Abstract

Mammals maintain a constant core body temperature through adaptive thermogenesis which includes shivering and non-shivering thermogenesis. Non-shivering thermogenesis relies primarily on mitochondrial uncoupling protein 1 (UCP1) in thermogenic fat (including brown and beige adipose tissue) to burn substrates, such as fatty acids (FAs), and convert chemical energy into heat. Lipid droplets (LDs), which are organelles that store lipids, are present in large numbers in thermogenic fat and are essential for adipose thermogenesis. Upon cold stimulation, LDs rapidly release FAs through autophagy or lipase-mediated lipolysis and rapidly translocate FAs into the mitochondria by interacting with mitochondria to burn and so promote thermogenesis. In addition, LD proteins promote the expression of UCP1 by activating the transcriptional activity of thermogenesis-related proteins. Here, the progress of research on the important role of LDs in thermogenesis is reviewed, mainly in terms of LD proteins, LD-organelle interactions, and LD autophagy (lipophagy). The emerging rationale for the involvement of LDs in each thermogenic pathway is described and the remaining unanswered questions in this field are highlighted.
Keywords: Lipid droplets, Thermogenesis, Brown/beige adipose tissue, Lipophagy
Introduction
Adiposes are often classified into thermogenic and non-thermogenic fats, which define their thermogenic capability [1, 2]. Non-thermogenic fat is represented by white adipose tissue [3] (WAT), which is characterized by large unilocular lipid droplets (LDs) and a low range of mitochondria with low levels of uncoupling protein 1 (UCP1). White adipose tissue is responsible for the body's energy storage by storing it in unilocular LD as triglycerides. In contrast, thermogenic adipose includes brown adipocytes and beige adipocytes [4], which are characterized by multilocular LDs and mitochondria with high-density cristae structures expressing UCP1 that can efficiently consume energy in the form of heat [5].
Mammals maintain a constant core body temperature through adaptive thermogenesis which includes shivering and non-shivering thermogenesis [6, 7] in response to cold stimuli. Thermogenic adipose tissue plays a key role in non-shivering thermogenesis in the form of an ineffective enzymatic cycle [8] or by relying on UCP1 to deregulate respiration during ATP synthesis and consuming chemical energy to form heat. The sources of energy for non-shivering thermogenesis are diverse and include glucose [6, 9, 10], fatty acids (FAs) [11], succinate [12] and branched-chain amino acids [13, 14]. Although there are multiple options for energy sources in thermogenesis, FAs are the primary molecules selected for adipose thermogenesis. In response to cold stimulation, the adipose tissue thermogenesis program is activated, and LDs in adipocytes release FAs by lipases, which is burned as fuel by the mitochondria to produce heat. This may explain why brown and beige adipocytes contain large amounts of LDs. To determine how adipocytes regulate lipid metabolism, such as FA metabolism, to generate heat, it is necessary to understand the important organelles that store, transfer, and metabolize lipids in LDs.
Generation and growth of lipid droplets in thermogenic fat
LDs are widespread dynamic organelles that consist of a monolayer of phospholipids encapsulated in neutral lipids, with a variety of specific LD-associated proteins [15-17]. LDs are found in a variety of tissues in animals, including the heart, liver, kidney, and adipose tissue, with thermogenic fat being the tissue containing a large number of LDs, demonstrating the importance of LDs in adipose tissue. The biogenesis of LDs occurs in the endoplasmic reticulum (ER), in a highly regulated biological process [15, 16, 18]. First, FAs and coenzyme A (CoA) are catalyzed by acyl CoA ligases to form acetyl CoA, whose acyl groups are transferred to diacylglycerol (DAG), cholesterol, and eventually triglyceride (TAG) and cholesteryl ester (CE) catalyzed by Acyl-CoA: diacylglycerol acyltransferases (DGATs) and acyl-CoA: cholesterol acyltransferase enzymes (ACATs) in the ER membrane [17]; following this, the neutral lipids undergo liquid-liquid phase separation and nucleation [19]. Second, the nucleated neutral lipid domains grow and bud towards the cytoplasmic side by the action of seipin [20-24], lipid droplet assembly factor 1 (LDAF1) [25, 26], fat storage-inducing transmembrane protein 2 (FIT2) [27, 28], and other factors, and are finally shed from the ER. Third, a variety of lipid synthases comprising the newly generated LDs, such as GPAT4 [29-31] and DGAT2 [32], catalyze the formation of TAG directly on the surface of LDs and thus promote the expansion of LDs (Figure 1).
Model of lipid droplet generation, growth, and fusion in adipose tissue. ER, endoplasmic reticulum; LLPS, liquid-liquid phase separation; TAG, Triglyceride; Plin1, perilipin1; AGPAT, acylglycerolphosphate acyltransferase; DAG, diacylglycerol; DGAT2, Acyl-CoA: diacylglycerol acyltransferase 2; GPAT4, glycerol-3 phosphate acyltransferase 4; CIDE, cell death-inducing DFF45-like effector; PAP, phosphatidic acid phosphatase; LDCS, LD contact sites; GTP, guanosine triphosphate; GDP, guanosine diphosphate; PUFA, polyunsaturated fatty acids.
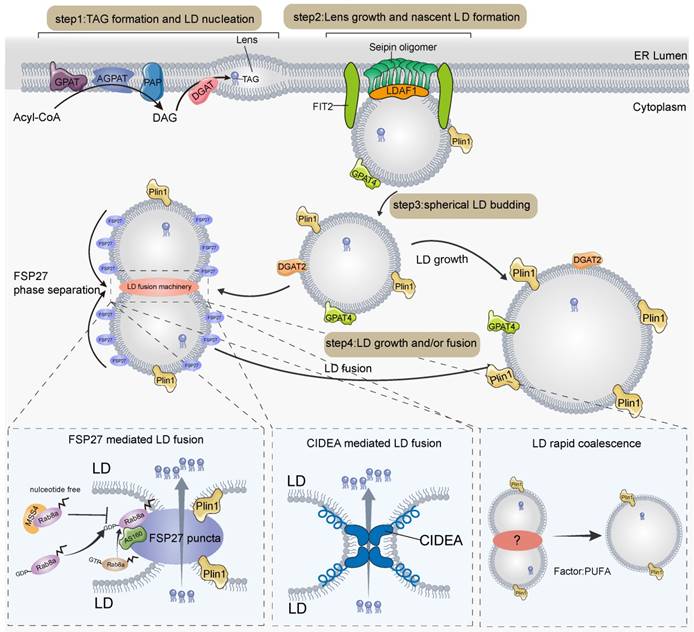
In addition, LDs can fuse to form larger droplets in response to cell death-inducing DFF45-like effector (CIDE) family proteins [33]. CIDE family proteins consist of CIDEA, CIDEB, and CIDEC (also known as fat-specific protein 27 (FSP27)) are highly expressed in adipose tissue, suggesting that LD fusion is essential for maintaining the morphology and stability of LDs in adipose tissue [34]. CIDE is homogeneously distributed on the LD surface, but upon fusion of LDs, liquid-liquid phase separation occurs and rapidly accumulates at LD contact sites (LDCS). This forms highly plastic and lipid-permeable fusion plates that contain unique lipid channels that allow neutral lipid flow from donor to recipient LDs, thereby mediating atypical LD fusion [35, 36]. CIDE proteins regulate LD fusion by recruiting other regulators to LD contact sites; for example, CIDEC recruits Rab8a and Plin1 to positively regulate LD fusion activity [37, 38] (Figure 1). Among them, plin1 can promote CIDEC-mediated LD fusion by expanding pore size or increasing LD surface tension [37]. Rab8a-GDP but not Rab8a-GTP binds to the C-terminal structural domain of CIDEC, which leads to the recruitment of Rab8a to the LDCS and the formation of a complex with Rab8a's GTPase-activating protein (GAP) AS160 and CIDEC [38], thereby promoting LD fusion and growth. Another family member that regulates LD fusion is CIDEA [33, 39-41]. CIDEA is specifically highly expressed in brown adipose tissue and promotes LD fusion during adipose differentiation [41]. The amphipathic helix in CIDEA is essential for LD fusion, which helps CIDEA to embed in the LD phospholipid monolayer and bind phosphatidic acid (PA). The N-terminal structural domain of CIDEA docks with the C-terminal dimerization region of the trans-complex and is enriched in the LDCS, interacting with the cone-shaped phospholipid PA, which may increase the permeability of the phospholipid barrier and facilitate LD fusion through lipid transfer [39]. In the liver, CIDEB localizes to LDCS and promotes lipid exchange and LD fusion in small LD-containing hepatocytes and large LD-containing hepatocytes [42]. In contrast, it is unknown whether CIDEB regulates LD fusion in adipocytes, as CIDEB is mainly expressed in the liver and kidney [43].
In white adipocytes, LDs eventually fuse to form large unilocular LDs, which occupy most of the cellular space [35, 36]. However, in brown or beige adipocytes, LDs are numerous and much smaller in diameter than the large unilocular LDs of white adipocytes, and the small multilocular LDs are essential for the efficient flow of hydrolyzed free fatty acids (FFA) into neighboring mitochondria for β-oxidation [44] and heat production. This implies that the LDs of thermogenic adipocytes cannot fuse indefinitely at room temperature or cold conditions; therefore, a mechanism is required to inhibit the indefinite fusion of LDs. MSS4, an atypical guanine nucleotide exchange factor (GEF) of Rab8a, maintains Rab8a in a nucleotide-free state by converting Rab8a-GDP to Rab8a-GTP, which allows separation of LD contact sites and thus inhibits CIDEC-mediated LD fusion [38]. In brown adipose tissue, there is an FSP27 isoform, FSP27β, whose deletion leads to a larger diameter and a smaller number of LDs with a white adipocyte phenotype [45]. Mechanistically, FSP27β can form a complex with CIDEA in brown adipose tissue and inhibit the formation of CIDEA dimers, thus inhibiting CIDEA from regulating the fusion of LDs in brown adipocytes, ensuring the stable presence of smaller LDs in brown adipocytes, and avoiding excessive fusion of LDs [45, 46]. In addition, in white fat, histone deacetylase 6 (HDAC6) deacetylates CIDEC, leading to CIDEC destabilization, thereby reducing LD fusion. In contrast, FAs, by promoting the dissociation of CIDEC from HDAC6, can prevent CIDEC deacetylation and thus promote LD fusion [47].
LDs are organelles generated from the ER. The generation of LDs is divided into processes including nucleation, nascent LD formation, budding and maturation. First, FAs are catalyzed by a series of enzymes to catalyze the synthesis of TAG, which forms lens structures in the ER by LLPS. Second, lens grows and forms nascent LD driven by seipin, LDAF1, FIT2, and other factors. Third, spherical LD that have grown to a particular stage buds out from the ER. Finally, further maturation of LDs by growth or fusion. DGAT2 and GPAT4 on the surface of LDs promote the growth and expansion of LDs through local lipid synthesis. LD fusion is mediated by CIDE proteins. CIDEC aggregates with LDCS by LLPS and regulates LD fusion via interactions with Plin1, AS160, and Rab8a. CIDEA promotes LD fusion by forming a trans-complex in LDCS. In addition, LDs can also become larger by transient fusion and PUFA facilitates this process, in which it has not been determined whether proteins are involved.
The role of lipid droplet proteins in adipose thermogenesis
LDs are coated with various proteins, including members of the PLIN family and lipolytic enzymes, which are essential for LD function. The most important role of LDs in thermogenic lipids is to maintain body temperature by rapidly producing FAs for mitochondrial burning and heat generation in response to cold stimulation. The rapid mobilization of lipids depends on the lipolytic enzymes adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL) on the LD surface. Under cold stimulation conditions, the skin transmits the perceived cold signal to the hypothalamus, which activates brown adipose tissue (BAT) via norepinephrine (NA) released from the sympathetic nerve endings. Norepinephrine (NA) binds to β-adrenoceptors (β-ARs) and then to Gα G-proteins that activate adenylate cyclase (AC), thereby increasing cellular cyclic adenosine monophosphate (cAMP) concentrations. cAMP further activates protein kinase A (PKA), leading to phosphorylation of Plin1, which in turn activates ATGL [48, 49]. ATGL hydrolyzes triglycerides stored in LDs to DAG and FFA, which is the rate-limiting step in triglyceride hydrolysis. HSL phosphorylated by PKA then hydrolyzes DAG to FFA and monoacylglycerol, which can be hydrolyzed to glycerol and FFA by HSL, MGL, and ABHD5. The FFA released by the above steps is transported to the mitochondria by FABP to activate UCP1 as well as to be burned as a substrate, eventually dissipating H+ potential energy through proton leakage and generating heat [48]. Prolonged cold stimulation induces a large uptake of extracellular circulating lipids by brown adipocytes, which are mainly stored in the TG-rich lipoprotein (TRL) [50]. However, brown adipocytes do not seem to be able to directly absorb, process and break down TRL particles. The main site of TRL breakdown is the endothelium adjacent to the adipocytes. The endothelial cells take up TRL via cluster of differentiation 36 (CD36) and then TRL is broken down by LAL [51]. The released FAs are transported intracellularly via fatty acid transport protein 1 (FATP1) on the surface of brown adipocytes [52]. Some of these FAs are transported to mitochondria for β-oxidation to promote thermogenesis through the synergistic action of carnitine palmitoyltransferase (CPT)1a and CPT2 transporter [53], while others are re-esterified to neutral lipids and stored in newly formed small LDs to alleviate lipotoxicity (Figure 2). The formation of small multilocular LDs in brown adipocytes increases the contact area between LD and lipase, which effectively promotes lipolysis and FAs transport to mitochondria close to LDs, thus accelerating mitochondrial β-oxidation and thermogenesis. Thus, thermogenesis is a precisely regulated process involving numerous factors. As the executioner of LD function, LD proteins play an important role in thermogenesis. The thermogenic effects of LD proteins can be divided into two categories: one regulating lipolysis and the other regulating the expression of UCP1, a key protein for thermogenesis.
Cold stimulation-mediated lipid droplet breakdown and thermogenesis. AC, adenylate cyclase; PKA, protein kinase A; CGI-58, comparative gene identification-58; Plin1, perilipin1; ATGL, adipose triglyceride lipase; FABP4: fatty acid binding protein 4; HSL, hormone-sensitive lipase; cAMP, cyclic adenosine monophosphate; MGL, monoacylglycerol lipase; FATP1: fatty acid transport protein 1; CPT1a, carnitine palmitoyltransferase 1A; CPT2, carnitine palmitoyltransferase 2.
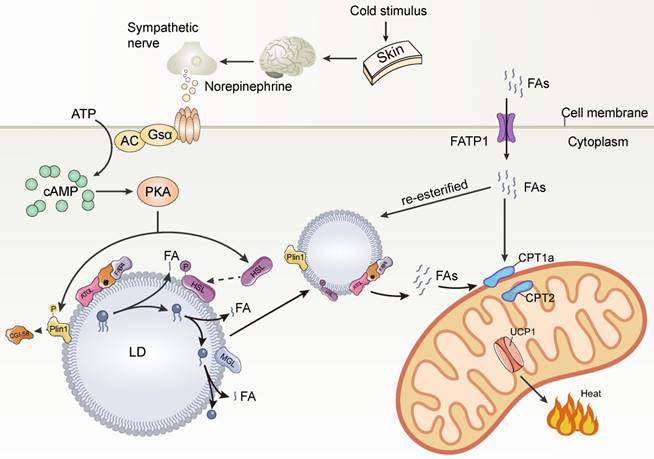
In response to cold stimulation, sympathetic nerves are activated to release norepinephrine which binds to β-AR on brown and beige cells and dissociates the receptor-coupled trimeric Gs protein and activates AC, leading to cAMP synthesis. cAMP further activates PKA. PKA phosphorylates Plin1 leading to the release of CGI-58 sequestered by Plin1. CGI-58 interacts with FABP4 to facilitate ATGL activity to enhance lipolysis. PKA also phosphorylates HSL and causes its translocation to LD to further promote lipolysis. Cold stimulation induces FATP1 to transport FAs from the extracellular into brown adipocytes. A portion of FAs were transported to mitochondria for β-oxidation to promote thermogenesis through the synergistic action of CPT1α and CPT2 transport proteins, while another portion of FAs were re-esterified to neutral lipids for storage in newly formed small LDs to alleviate lipotoxicity.
Lipid droplet proteins regulate lipolysis
Members of the PLIN family of intrinsic LD proteins, including Plin1-5, are important for regulating LD growth, maintaining LD dynamics [54] and thermogenesis. It has been shown that plin2 deficiency induces subcutaneous WAT browning in the groin at room temperature and activates the thermogenic program [55]. Notably, a 20% sucrose diet induced subcutaneous WAT in plin2-deficient mice exhibiting significant browning and a significant increase in thermogenic gene expression, suggesting that Plin2 is a mediator of diet-induced adipose browning [55]. Similarly, Plin3 deletion stimulates browning and thermogenic gene expression in inguinal white adipose tissue (iWAT) [56]. Plin3-knockout mice exhibit enhanced cold tolerance and enhanced basal and stimulated lipolysis in iWAT, inducing activation of peroxisome proliferator-activated receptor alpha (PPARα). Plin3 deficiency promotes PPARα target gene and uncoupling protein 1 expression, and the formation of multi-compartment LD under cold stimulation [56]. It is suggested that plin2 and plin3 serve as intrinsic protective factors that regulate adipocyte thermogenesis by limiting lipid metabolism and thermogenic gene expression. Plin5, a LD protein highly expressed in oxidative tissues [57], is involved in lipolysis regulation. Experimental data from mouse myoblasts suggest that Plin5 activates ATGL and promotes lipolysis by recruiting CGI-58 to the LD surface [58]. However, another study showed that Plin5 interacts with ATGL lipolysis and inhibits LD breakdown [59]. This suggests that Plin5 may have a two-sided role in the regulation of lipolysis. In addition, Plin5 is abundantly expressed in BAT and mediates LD-mitochondrial interactions, which are essential for BAT thermogenesis (we describe this in detail below).
Carboxylesterase 3 (Ces3) is a hydrolase with extensive activity in the liver and adipose tissue [60]. Normally, ces3 is localized to the ER [61-63], however, it is transferred from the ER to LDs after cold exposure or isoproterenol (ISO) treatment and becomes the major LD-targeting protein [64]. It has been shown that Ces3 translocation to LDs is dependent on PKA-activated pathways, but the precise mechanism by which PKA regulates Ces3 translocation between the two organelles remains to be further investigated. Under β-adrenergic or cold conditions, Ces3 acts as a non-classical lipid hydrolase for lipolysis and promotes BAT thermogenesis. Interfering with Ces3 expression or inhibiting Ces3 enzymatic activity decreased β-oxidation and mitochondrial biosynthesis-related genes, UCP1 expression, and oxygen consumption rate in differentiated 3T3-L1 cells. This suggests an important role of Ces3 in ISO or cold stimulation-induced enhancement of mitochondrial function and activation of the adipogenic thermogenic program [64]. Mechanistically, Ces3 inhibition downregulates the expression of Ucp1 and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) via peroxisome proliferator-activated receptor γ (PPARγ), which attenuates the thermogenic program of adipocytes and leads to marked impairment in the ability of mice to protect their body temperature in the cold [64]. However, how precisely Ces3 regulates the expression of Ucp1, e.g., the direct interaction factors downstream of Ces3, remains to be investigated in depth.
Vacuolar protein sorting 13C (VPS13C), a mammalian ortholog of yeast Vps13 [65, 66], is highly expressed in mouse BAT [67]. Biochemical experiments have shown that VPS13C is an LD-associated protein in adipocytes that is closely associated with the multilocular phenotype of adipocytes [67]. Research data demonstrated that VPS13C has a suppressive effect on the expression of genes related to the regulation of LD catabolism and thermogenesis [67]. Silencing of VPS13C decreased LD size and triglyceride content and increased the release of basal free FAs. Mechanistically, this is due to the deletion of vps13c, leading to an increase in ATGL, but not HSL-targeted LDs, which promotes LD hydrolysis. In addition, deletion of VPS13C upregulates thermogenesis-related genes, such as PPARα, PGC-1α, UCP1, and CIDEA, suggesting that VPS13C can inhibit thermogenesis [67]. Furthermore, how VPS13C localizes to LDs remains unknown, and analysis of the VPS13C sequence revealed the presence of a pair of amphiphilic slices in the plane with the membrane, suggesting that VPS13C may have an intramembrane-anchoring structural domain and directly bind LDs. Previous studies have shown that yeast VPS13C homologs can mediate organelle interactions and that human VPS13C can interact with vesicle-associated membrane protein-associated proteins A and B (VAPA and VAPB) and is an ER protein known to mediate ER-organelle interactions and lipid transfer. Whether VPS13C regulates the interactions between LDs and other organelles in adipocytes and whether such interactions are associated with thermogenesis remains to be further investigated.
Hypoxia-inducible gene 2 (Hig2) is a protein containing 63 amino acids and is expressed in both adipose and liver tissues and is localized to the surface of LDs [68, 69]. At 23°C, adipocyte-specific Hig2 deficiency altered the adipose tissue distribution in HFD-fed mice. Epididymal WAT (eWAT) body weight was significantly lower in Hig2AdKO animals fed a high-fat diet, indicating that Hig2-specific deficiency reduced fat storage deposition. However, at 30 °C, the phenomenon in which Hig2-specific deficiency reduced fat storage deposition disappeared. This indicates that Hig2-specific deficiency reduces fat storage and requires activation of non-shivering thermogenesis [69]. But how hig2 deficiency promotes fat release requires further investigation.
Lipid droplet proteins regulate the expression of thermogenic genes
CIDEA not only promotes LD fusion but also plays an important role in thermogenesis. CIDEA, which has a different expression profile in humans and mice, is only expressed in mouse BAT, whereas it is expressed in human WAT and BAT [69]. In human adipocytes, CIDEA knockout downregulates the expression of thermogenic genes, such as PGC1α and UCP1, whereas re-expression of CIDEA reverses the downregulation of thermogenic genes caused by CIDEA deletion [69]. In addition, CIDEA knockout reduced mitochondrial respiration, proton leakage, and maximal respiratory capacity of beige adipocytes. Mechanistically, during browning, CIDEA translocates from LDs to the nucleus and specifically binds to the Liver X Receptor alpha (LXRα), thereby attenuating the inhibition of UCP1 enhancer activity by LXRα and enhancing the binding of PPARγ to the UCP1 enhancer, which drives UCP1 transcription for thermogenesis [69]. In addition, Transcriptome data analysis revealed genes participating in global thermogenic processes, for example, adaptive thermogenesis, fatty acid oxidation, mitochondrial transport, electron transport chain, were downregulated in CIP-deficient human beige adipocytes, indicating the control of thermogenic and metabolic functions by CIDEA [70] (Figure 3).
Lipid droplet proteins regulate thermogenic gene expression. MUFA, monounsaturated fatty acids; PPARα, peroxisome proliferator-activated receptor alpha; PPARγ, peroxisome proliferator-activated receptor γ; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; LXRα, Liver X Receptor alpha; SIRT, silencing information regulator factor 2-related enzyme 1.
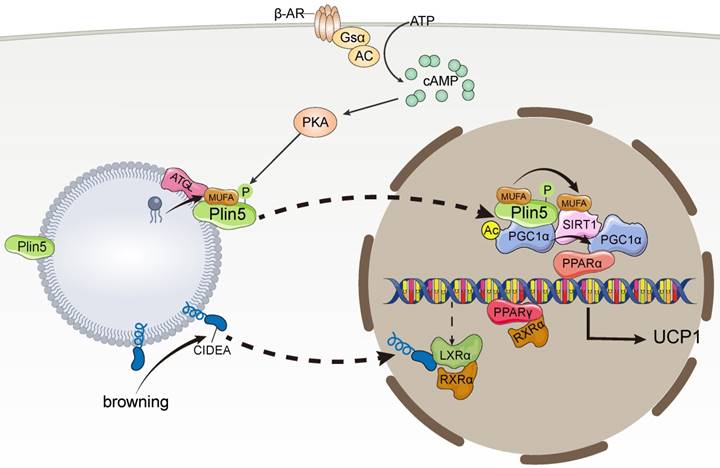
However, the function of CIDEA in the brown fat of mice differs considerably from that in humans. Under cold stimulation, brown fat in CIDEA-null mice exhibited a higher metabolic rate, body temperature, and lipolysis rate, suggesting greater thermogenic activity [71]. Zhou et al. found that CIDEA localizes to mitochondria and cooperates with and suppresses the uncoupling activity of UCP1 [71].
In another study using transgenic aP2-hCidea mice that overexpressed human CIDEA in all adipose tissues, UCP1 activity was significantly inhibited in BAT mitochondria isolated from aP2-hCidea mice [72]. Interestingly, Fischer et al. found that CIDEA itself did not localize to the mitochondria, contradicting the findings of Zhou et al. [71]. Fischer suggested that Zhou's findings may have resulted from contamination of membrane fractions during the experiment. Fischer suggested that CIDEA may indirectly inhibit UCP1 activity, while mice increased the total amount of UCP1 in the tissues to match the diminished thermogenic capacity of the UCP1 protein through a properly balanced compensatory mechanism [72].
Plin5 not only regulates lipolysis directly on the surface of LDs but also acts as a transcriptional coactivator in response to catecholamine stimulation and participates in lipolysis. Mechanistically, catecholamine facilitates PKA phosphorylation of Plin5, leading to the translocation of Plin5 from LDs to the nucleus and the formation of transcriptional complexes with PGC-1α and silencing information regulator factor 2-related enzyme 1 (SIRT1) [73], which deacetylates PPARγ in a ligand-dependent manner [74, 75]. Plin5 binds to SIRT1 and disinhibits its deacetylase activity, which in turn promotes PGC-1α promoter activity to activate PGC-1α target genes, thereby promoting the transcription of thermogenesis-related mitochondrial biogenesis, and oxidative function-related genes, ultimately leading to lipolysis [73]. Interestingly, catecholamine-activated PKA phosphorylation events promote Plin5 binding of LD-derived monounsaturated fatty acids (MUFAs) mediated by ATGL, which facilitates Plin5 nuclear translocation [76]. Plin5-carried MUFAs induce conformational changes in SIRT1 in the nucleus by directly binding to SIRT1, thereby activating SIRT1 [76]. Ultimately, MUFAs enhance the transcriptional activity of PGC-1α in a SIRT1-dependent manner, which in turn promotes the expression of thermogenesis-related genes [76] (Figure 3).
Aifm2, a FAD-dependent NADH/NAD oxidoreductase, was identified as an LD-associated protein that is highly enriched in BAT [77]. It was shown that Aifm2 in BAT and WAT promotes oxygen consumption, uncoupled respiration, and thermogenesis during cold and diet-induced thermogenesis and plays a key role in glycolysis and glucose oxidation [77]. In response to cold stimulation or β-adrenergic stimulation, Aifm2 translocates from LDs to the outer mitochondrial inner membrane, maintains high cytoplasmic NAD levels by oxidizing NADH, and maintains a stable rate of glycolysis and glucose oxidation [77]. Aifm2 increases overall mitochondrial activity in BAT cells and transfers electrons to the electron transport chain, providing fuel for thermogenesis and promoting uncoupled respiration and thermogenesis [77]. However, there are many questions regarding the role of Aifm2 in thermogenesis that require further investigation, e.g., the molecular mechanism of Aifm2 transfer from the LDs to mitochondria in response to cold stimulation.
Cold exposure stimulates ATGL-mediated lipolysis and production of MUFA. Plin5 phosphorylated by PKA preferentially binds MUFA and translocates to the nucleus. MUFA enhances PGC-1α/PPARα signaling by activating SIRT1 and promotes mitochondrial oxidative metabolism and UCP1 expression. During the induction of browning, CIDEA is transferred from lipid droplets to the nucleus in a concentration-dependent manner. CIDEA binds LXRα and inhibits its repression of UCP1 enhancer RXRα activity, thereby enhancing PPARγ activity and activating UCP1 transcription.
Lipid droplet-mitochondrion contacts to regulate thermogenesis
In addition to the presence of multilocular LDs, the most characteristic feature of thermogenic adipocytes is the high content of mitochondria and the consequent brown and beige coloration. Numerous studies have shown that there is an extensive network of interactions between LDs and mitochondria [78, 79] in a variety of oxidative tissues, including brown adipose tissue [80, 81], the heart [82], and type I skeletal muscle [83]. LD-mitochondrial interactions can be subdivided into two categories according to their strength: (I) dynamic interactions, which can be disrupted by high-speed centrifugal forces (9,000×g). Benador et al. [78] used 9,000×g centrifugal force to isolate mitochondria from mouse brown adipocytes that interact with LDs and named them peridroplet mitochondria (PDM). (II) Stable interactions that could not be disrupted by high-speed centrifugation (228,000×g). Cui et al. [81] used ultracentrifugation at 228,000×g, which also failed to separate LDs from mitochondria; these were named LD-anchored mitochondria (LDAM) (Figure 4). Organelle complexes formed by anchoring LDs to the mitochondria are considered to be permanently intercalated.
Mechanism of lipid droplet-mitochondrial interactions in thermogenesis. Mfn2, mitofusin 2.
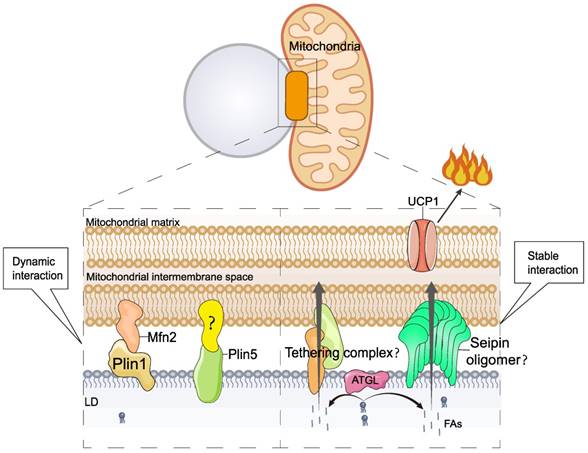
Under cold stimulation, LDs-mitochondrial contact is increased in BAT, which is thought to facilitate the rapid consumption of fat and heat production [80, 84]. The interaction between LDs and mitochondria is precisely regulated, and studies have shown that several proteins are involved in regulating LD-mitochondrion interactions, including mitofusin 2 (Mfn2) [85], Plin5, and others. In BAT, Mfn2 regulates LD-mitochondrial interactions by specifically interacting with Plin1, and this interaction requires Mfn2 GTPase activity [85]. Mfn2-regulated LD-mitochondrial interactions respond to cold or adrenergic stimulation. Adipose-specific Mfn2 knockout mice (Mfn2-adKO) of BAT exhibit higher lipid accumulation and larger LDs [85]. Lipid accumulation was not due to an increase in lipid synthesis genes but to a decrease in the rate of lipolysis. In addition, the BAT mitochondria of Mfn2-adKO animals showed significantly reduced levels of complexes I and III, which inhibited the respiratory capacity of BAT. Impaired lipolysis and oxidative capacity result in reduced thermogenesis in Mfn2-adKO mice, which are unable to maintain body temperature during cold exposure [85].
Plin5, a LD protein, regulates LD-mitochondrial interactions. Cold stimulation induces elevated BAT Plin5 expression, which promotes BAT FA uptake, mitochondrial biogenesis, cristae packaging, and oxidative function, thereby enhancing UCP1-dependent mitochondrial respiration and thermogenesis [86]. Plin5 overexpression in BAT also leads to increased insulin sensitivity and reduced inflammation in iWAT [86]. In addition, Plin5 knockdown reduces mitochondrial oxygen consumption rate and mitochondrial DNA content [69].
Interactions between LDs and mitochondria are widely believed to facilitate the rapid entry of FAs into the mitochondria. However, the mechanism by which FAs enter mitochondria from LDs is poorly understood. Seipin can form nanoscale tubular interfaces that facilitate the flow of lipids from the ER into nascent LDs [20, 21, 23, 24]. Interestingly, recent studies have found that seipin is localized at the contact site between LDs and mitochondria [22]. Therefore, seipin may regulate LD-mitochondrial interactions and mediate the transfer of FAs from LDs to mitochondria. However, induction of cAMP/PKA signaling in the absence of Bscl2 results in increased lipolysis and fatty acid oxidation in WAT and BAT, as well as browning of WAT [87], suggesting a potential inhibition of lipolysis and thermogenesis by seipin, which does not seem to support the above hypothesis. In conclusion, the mechanism of FA transfer between LDs and mitochondria requires further investigation.
Cold stimulation induces significant LD-mitochondrial interactions in thermogenic adipose tissue. Two mechanisms of LD-mitochondrial interactions have been identified. I) Dynamic Interaction. This process is regulated by the Plin1-Mfn2 complex and Plin5; however, the presence of the Plin5 interaction factor is unclear. II) Stable Interaction. This process is known as permanent LD-mitochondrial interaction or LD-mitochondrial anchoring. The molecular mechanism of LD-mitochondrial anchoring is poorly understood. Seipin, a protein with a lipid transport channel structure, is found to localize to the LD-mitochondrial contact site. Therefore, it is hypothesized that seipin may mediate LD-mitochondria interactions and direct FA transport between LDs and mitochondria.
Lipophagy regulates adipose thermogenesis
Autophagy, an important pathway for degrading cellular contents to maintain normal cellular function and dynamic homeostasis [88], has also been found to be involved in LD degradation [89-91]. The degradation of LDs by autophagy is mainly dependent on acidic hydrolases of lysosomes such as the LIPA/LAL (lipase A, lysosomal acid), and the process of autophagy-mediated LD degradation is called lipophagy which is divided into three main pathways: macrolipophagy, microlipophagy, and chaperone-mediated autophagy [89-93].
Macrolipophagy begins with the segregation of LDs by autophagosomes, which are then delivered to lysosomes for turnover [94]. Macrolipophagy consists of microtubule-associated protein 1A / 1B-light chain 3 (LC3-II)-dependent phagocytosis of some LDs to form lipoautophagosomes, which subsequently fuse with lysosomes to form autolysosomes [94]. During microlipophagy, LDs and lysosomes undergo lipid transfer via transient contact [95, 96]. Chaperone-mediated autophagy facilitates the transfer of LD proteins Plin2 and Plin3 from HSP70 to lysosomes for degradation, thereby increasing the accessibility of cytoplasmic lipases to LDs [97, 98].
In BAT, cold stimulation activates autophagy in hypothalamic pro-opiomelanocortin (POMC) neurons and lipophagy in BAT via neural activation, promoting LC3 localization to LDs and formation of lipoautophagosomes, leading to lipophagy and promoting FA utilization [99] (Figure 5). Inhibition of autophagy of POMC or BAT leads to impaired BAT lipophagy, reduced uncoupled respiration rate, and lipid accumulation in mice, making them unable to maintain normal body temperature under cold exposure [99]. This indicates that BAT lipophagy has an important role for thermogenic function in mice. In addition, the interface between LDs and autophagosomes provides a platform for LC3 to interact with lipolytic enzymes, and LC3 interacts with ATGL to promote lipolysis [99]. Under cold stimulation, lipophagy and lipolysis have complementary effects on FA mobilization.
Mechanism of lipophagy mediated thermogenesis. LAL, lysosomal acid lipase.
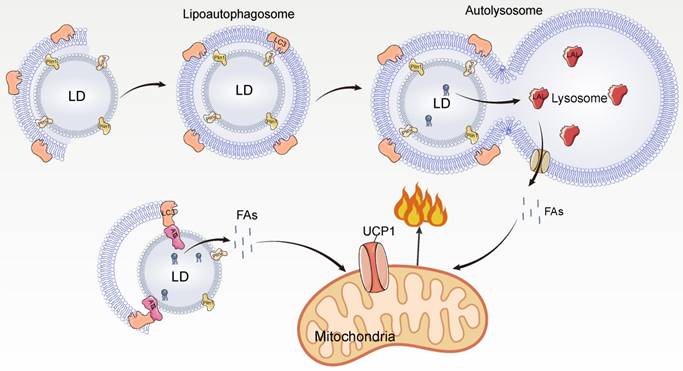
Whether microautophagy occurs in BAT and promotes lipolytic and thermogenic functions remains unclear. Notably, both macroautophagy and microautophagy are extremely important for the regulation of lipid metabolism in the liver [94, 96], suggesting that macroautophagy and microautophagy can function in the same tissue. Microautophagy-mediated transient interactions between lysosomes and LDs rapidly degrade neutral lipids, which is important for the rapid mobilization of lipolysis. In response to cold stimulation, the body can rapidly respond and mobilize lipids; therefore, microautophagy may occur in BAT to rapidly mobilize lipids.
Smaller LDs have higher surface resolution and are more conducive to lipolysis. In the liver, larger LDs are reduced by ATGL lipolysis, and smaller LDs are eliminated by lipophagy [90]. In BAT, LDs may need to be smaller for rapid lipolysis. Current studies suggest that there are two main ways in which LDs become smaller: one is a lipolytic process involving lipolytic enzymes [93, 100] and the other is the generation of smaller LDs by budding from larger LDs [101]. The process of LD budding has been demonstrated in vitro, but it remains to be confirmed whether it occurs abundantly and rapidly in vivo in response to lipolysis. WAT has unilocular LDs that become multilocular after browning, and how unilocular LDs are converted to multilocular LDs is not known. LD budding provides the possibility for browning. Thus, LD budding may play an important role in rapid lipid utilization and browning of WAT.
Cold stimulation strongly activates lipophagy in BAT. The currently identified lipophagy in BAT is macroautophagy. Microtubule-associated protein 1A/1B-light chain 3 (LC3-II)-mediated macroautophagy engulfs LD to form a lipoautophagosome, which later fuses with lysosomes leading to TAG hydrolysis by lysosomal acid lipase (LAL) to hydrolyze and release FAs. In addition, LC3-II can directly interact with ATGL on the surface of LDs and promote lipolysis.
The role of lipid droplets in the futile cycle of thermogenesis
Futile metabolic cycling mechanisms, in which one ATP-consuming reaction takes place concurrently with an inverse energetic reaction, are the basis of UCP1-independent thermogenesis. Other than ATP depletion and energy loss in the form of heat, these futile cycles have no overall effect. Previous studies have shown the existence of three futile cycling mechanisms, one being the Ca2+ cycle thermogenesis [10]. The second is the creatine cycling, which is related to phosphorylation and dephosphorylation of creatine [102]. There is no direct relationship between these two futile cycling mechanisms and LDs. The third one is the futile cycle between TAG and FAs [103, 104]. TAG is catalyzed by lipase and eventually forms glycerol and FAs, which in turn can be re-esterified to TAG. This futile cycle can be induced by thiazolidinediones (TZDs) [103], cold exposure [104], and leptin [105] by promoting lipolysis. Therefore, LD proteins involved in lipolysis such as ATGL, Plin1, and CGI-58, Plin5 may have an effect on this cycle. The LD-mitochondrial interface is considered to be a good site for metabolic reactions, and LD-mitochondrial interactions are not only involved in lipolysis but also promote LD expansion by facilitating triglyceride synthesis [78]. Therefore, LD-mitochondrial interactions may be involved in the ineffective cycle of TAG, but experimental data are lacking to support this hypothesis.
Concluding Remarks
Although the role of LDs in thermogenesis has been well studied, there are still many unanswered questions. For example, it is still unknown which protein mediates the anchoring of LDs to mitochondria. It has been shown that Plin5 mediates LD-mitochondrial interactions, but deletion of Plin5 does not affect LD-mitochondrial anchoring. In addition, the mechanism of FA transfer between LDs and the mitochondria remains unknown. Direct FA transfer between LDs and the mitochondria is thought to be beneficial in alleviating cellular lipotoxicity. This direct transfer of FAs in organelles may be directly regulated by proteins but may also be a direct result of membrane fusion. In conclusion, it would be of great interest to explore the mechanism of FA transfer between LD and mitochondria.
Previous studies have shown that LDs of different sizes in the liver have different protein compositions, indicating different functions and fates. Large LDs in the liver are subjected to ATGL-mediated lipolysis and become smaller, while small LDs are degraded via the autophagic pathway. Brown or beige fat contains multilocular LDs, and there are differences in size between LDs. The protein composition on the surface of LDs of different sizes isolated from BAT differs; however, it remains unknown whether differences between LD proteins affect lipolysis as well as the function of LDs. In summary, a detailed understanding of LD-organelle interactions and LD dynamics will help in understanding the role of LDs in thermogenesis.
Abbreviations
UCP1: uncoupling protein 1; LDs: lipid droplets; WAT: white adipose tissue; BAT: brown adipose tissue; TAG: triglycerides; FAs: fatty acids; LDAF1: lipid droplet assembly factor 1; FIT2: fat storage-inducing transmembrane protein 2; GPAT4: lycerol-3 phosphate acyltransferase 4; DGAT2: acyl-CoA:diacylglycerol acyltransferase; CIDEA: cell death-inducing DFF45-like effector family proteins A; CIDEB: cell death-inducing DFF45-like effector family proteins B; CIDEC: cell death-inducing DFF45-like effector family proteins C; LDCS: lipid droplets contact sites; PA: phosphatidic acid; FFA: free fatty acids; HDAC6: histone deacetylase 6; Plin1: perilipin 1; DAG: diacylglycerol; PAP: phosphatidic acid phosphatase; GTP: Guanosine triphosphate; GDP: guanosine diphosphate; PUFA: polyunsaturated fatty acids; LLPS: liquid-liquid phase separation; ATGL: adipose triglyceride lipase; HSL: hormone-sensitive lipase; NA: norepinephrine; BAT: brown adipose tissue; β-ARs: β-adrenoceptors; PKA: protein kinase A; AC: adenylate cyclase; iWAT: white adipose tissue; PPARα: peroxisome proliferator-activated receptor alpha; Ces3: Carboxylesterase 3; ISO: isoproterenol; PPARγ: peroxisome proliferator-activated receptor γ; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; Hig2: Hypoxia-inducible gene 2; LXRα: Liver X Receptor alpha; SIRT1: silencing information regulator factor 2-related enzyme 1; MUFAs: monounsaturated fatty acids; PDM: peridroplet mitochondria; LDAM: LD-anchored mitochondria; Mfn2: mitofusin 2; POMC: pro-opiomelanocortin; LAL: lysosomal acid lipase; FATP1: fatty acid transport protein 1; CPT1a: carnitine palmitoyltransferase 1A; CPT2: carnitine palmitoyltransferase 2.
Acknowledgements
Funding
This work was supported by the National Key Research and Development Program of China (No. 2021YFF1000601), the National Natural Science Foundation of China (No. 32172700), the Joint Funds of the National Natural Science Foundation of China (No. U20A2052), and the Fundamental Research Funds for the Central Universities (No. 2662022DKPY002).
Author contributions
Lupeng Chen provided the conception, wrote the manuscript. Yi Jin and Jian Wu contributed to the reference collection and revised the manuscript. Zhuqing Ren supervised the manuscript. All authors contributed to the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Shamsi F, Wang C-H, Tseng Y-H. The evolving view of thermogenic adipocytes - ontogeny, niche and function. Nat Rev Endocrinol. 2021;17:726-44
2. Tan CY, Ishikawa K, Virtue S, Vidal-Puig A. Brown adipose tissue in the treatment of obesity and diabetes: Are we hot enough? J Diabetes Investig. 2011;2:341-50
3. Montanari T, Pošćić N, Colitti M. Factors involved in white-to-brown adipose tissue conversion and in thermogenesis: a review. Obes Rev. 2017;18:495-513
4. Harms M, Seale P. Brown and beige fat: development, function and therapeutic potential. Nat Med. 2013;19:1252-63
5. Sun W, Modica S, Dong H, Wolfrum C. Plasticity and heterogeneity of thermogenic adipose tissue. Nat Metab. 2021;3:751-61
6. Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277-359
7. Lowell BB, Spiegelman BM. Towards a molecular understanding of adaptive thermogenesis. Nature. 2000;404:652-60
8. Chouchani ET, Kazak L, Spiegelman BM. New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell Metab. 2019;29:27-37
9. Keinan O, Valentine JM, Xiao H, Mahata SK, Reilly SM, Abu-Odeh M. et al. Glycogen metabolism links glucose homeostasis to thermogenesis in adipocytes. Nature. 2021;599:296-301
10. Ikeda K, Kang Q, Yoneshiro T, Camporez JP, Maki H, Homma M. et al. UCP1-independent signaling involving SERCA2b-mediated calcium cycling regulates beige fat thermogenesis and systemic glucose homeostasis. Nat Med. 2017;23:1454-65
11. Cohen P, Kajimura S. The cellular and functional complexity of thermogenic fat. Nat Rev Mol Cell Biol. 2021;22:393-409
12. Mills EL, Pierce KA, Jedrychowski MP, Garrity R, Winther S, Vidoni S. et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature. 2018;560:102-6
13. Yoneshiro T, Wang Q, Tajima K, Matsushita M, Maki H, Igarashi K. et al. BCAA catabolism in brown fat controls energy homeostasis through SLC25A44. Nature. 2019;572:614-9
14. Ma Q-X, Zhu W-Y, Lu X-C, Jiang D, Xu F, Li J-T. et al. BCAA-BCKA axis regulates WAT browning through acetylation of PRDM16. Nat Metab. 2022;4:106-22
15. Walther TC, Chung J, Farese RV. Lipid Droplet Biogenesis. Annu Rev Cell Dev Biol. 2017;33:491-510
16. Jackson CL. Lipid droplet biogenesis. Curr Opin Cell Biol. 2019;59:88-96
17. Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. 2019;20:137-55
18. Wilfling F, Haas JT, Walther TC, Farese RV. Lipid droplet biogenesis. Curr Opin Cell Biol. 2014;29:39-45
19. Thiam AR, Ikonen E. Lipid Droplet Nucleation. Trends Cell Biol. 2021;31:108-18
20. Zoni V, Khaddaj R, Lukmantara I, Shinoda W, Yang H, Schneiter R. et al. Seipin accumulates and traps diacylglycerols and triglycerides in its ring-like structure. Proc Natl Acad Sci U S A. 2021 118
21. Salo VT, Li S, Vihinen H, Hölttä-Vuori M, Szkalisity A, Horvath P. et al. Seipin Facilitates Triglyceride Flow to Lipid Droplet and Counteracts Droplet Ripening via Endoplasmic Reticulum Contact. Developmental Cell. 2019 50
22. Combot Y, Salo VT, Chadeuf G, Hölttä M, Ven K, Pulli I. et al. Seipin localizes at endoplasmic-reticulum-mitochondria contact sites to control mitochondrial calcium import and metabolism in adipocytes. Cell Reports. 2022;38:110213
23. Prasanna X, Salo VT, Li S, Ven K, Vihinen H, Jokitalo E. et al. Seipin traps triacylglycerols to facilitate their nanoscale clustering in the endoplasmic reticulum membrane. PLoS Biol. 2021;19:e3000998
24. Rao MJ, Goodman JM. Seipin: harvesting fat and keeping adipocytes healthy. Trends Cell Biol. 2021;31:912-23
25. Chung J, Wu X, Lambert TJ, Lai ZW, Walther TC, Farese RV Jr. LDAF1 and Seipin Form a Lipid Droplet Assembly Complex. Dev Cell. 2019;51:551-63.e7
26. Goodman JM. LDAF1 Holds the Key to Seipin Function. Dev Cell. 2019;51:544-5
27. Gross DA, Zhan C, Silver DL. Direct binding of triglyceride to fat storage-inducing transmembrane proteins 1 and 2 is important for lipid droplet formation. Proc Natl Acad Sci U S A. 2011;108:19581-6
28. Chen F, Yan B, Ren J, Lyu R, Wu Y, Guo Y. et al. FIT2 organizes lipid droplet biogenesis with ER tubule-forming proteins and septins. J Cell Biol. 2021 220
29. Olarte M-J, Kim S, Sharp ME, Swanson JMJ, Farese RV, Walther TC. Determinants of Endoplasmic Reticulum-to-Lipid Droplet Protein Targeting. Developmental Cell. 2020 54
30. Pagac M, Cooper DE, Qi Y, Lukmantara IE, Mak HY, Wu Z. et al. SEIPIN Regulates Lipid Droplet Expansion and Adipocyte Development by Modulating the Activity of Glycerol-3-phosphate Acyltransferase. Cell Reports. 2016;17:1546-59
31. Wilfling F, Wang H, Haas JT, Krahmer N, Gould TJ, Uchida A. et al. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Developmental Cell. 2013;24:384-99
32. Bhatt-Wessel B, Jordan TW, Miller JH, Peng L. Role of DGAT enzymes in triacylglycerol metabolism. Arch Biochem Biophys. 2018 655
33. Gao G, Chen F-J, Zhou L, Su L, Xu D, Xu L. et al. Control of lipid droplet fusion and growth by CIDE family proteins. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2017;1862:1197-204
34. Gao G, Chen F-J, Zhou L, Su L, Xu D, Xu L. et al. Control of lipid droplet fusion and growth by CIDE family proteins. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862:1197-204
35. Lyu X, Wang J, Wang J, Yin Y-S, Zhu Y, Li L-L. et al. A gel-like condensation of Cidec generates lipid-permeable plates for lipid droplet fusion. Developmental Cell. 2021;56:2592-606.e7
36. Gong J, Sun Z, Wu L, Xu W, Schieber N, Xu D. et al. Fsp27 promotes lipid droplet growth by lipid exchange and transfer at lipid droplet contact sites. Journal of Cell Biology. 2011;195:953-63
37. Sun Z, Gong J, Wu H, Xu W, Wu L, Xu D. et al. Perilipin1 promotes unilocular lipid droplet formation through the activation of Fsp27 in adipocytes. Nature Communications. 2013;4:1594
38. Wu L, Xu D, Zhou L, Xie B, Yu L, Yang H. et al. Rab8a-AS160-MSS4 regulatory circuit controls lipid droplet fusion and growth. Developmental Cell. 2014;30:378-93
39. Barneda D, Planas-Iglesias J, Gaspar ML, Mohammadyani D, Prasannan S, Dormann D. et al. The brown adipocyte protein CIDEA promotes lipid droplet fusion via a phosphatidic acid-binding amphipathic helix. Elife. 2015;4:e07485
40. Chen F-J, Yin Y, Chua BT, Li P. CIDE family proteins control lipid homeostasis and the development of metabolic diseases. Traffic. 2020 21
41. Wu L, Zhou L, Chen C, Gong J, Xu L, Ye J. et al. Cidea controls lipid droplet fusion and lipid storage in brown and white adipose tissue. Sci China Life Sci. 2014;57:107-16
42. Xu W, Wu L, Yu M, Chen F-J, Arshad M, Xia X. et al. Differential Roles of Cell Death-inducing DNA Fragmentation Factor-α-like Effector (CIDE) Proteins in Promoting Lipid Droplet Fusion and Growth in Subpopulations of Hepatocytes*♦. Journal of Biological Chemistry. 2016;291:4282-93
43. Li JZ, Ye J, Xue B, Qi J, Zhang J, Zhou Z. et al. Cideb Regulates Diet-Induced Obesity, Liver Steatosis, and Insulin Sensitivity by Controlling Lipogenesis and Fatty Acid Oxidation. Diabetes. 2007;56:2523-32
44. Rambold AS, Cohen S, Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Developmental Cell. 2015;32:678-92
45. Nishimoto Y, Nakajima S, Tateya S, Saito M, Ogawa W, Tamori Y. Cell death-inducing DNA fragmentation factor A-like effector A and fat-specific protein 27β coordinately control lipid droplet size in brown adipocytes. Journal of Biological Chemistry. 2017;292:10824-34
46. Nishimoto Y, Tamori Y. CIDE Family-Mediated Unique Lipid Droplet Morphology in White Adipose Tissue and Brown Adipose Tissue Determines the Adipocyte Energy Metabolism. Journal of Atherosclerosis and Thrombosis. 2017;24:989-98
47. Qian H, Chen Y, Nian Z, Su L, Yu H, Chen F-J. et al. HDAC6-mediated acetylation of lipid droplet-binding protein CIDEC regulates fat-induced lipid storage. J Clin Invest. 2017;127:1353-69
48. Peirce V, Carobbio S, Vidal-Puig A. The different shades of fat. Nature. 2014;510:76-83
49. Zhang Z, Yang D, Xiang J, Zhou J, Cao H, Che Q. et al. Non-shivering Thermogenesis Signalling Regulation and Potential Therapeutic Applications of Brown Adipose Tissue. Int J Biol Sci. 2021;17:2853-70
50. Sponton CH, de Lima-Junior JC, Leiria LO. What puts the heat on thermogenic fat: metabolism of fuel substrates. Trends Endocrinol Metab. 2022;33:587-99
51. Fischer AW, Jaeckstein MY, Gottschling K, Heine M, Sass F, Mangels N. et al. Lysosomal lipoprotein processing in endothelial cells stimulates adipose tissue thermogenic adaptation. Cell Metab. 2021;33:547-64.e7
52. Wu Q, Kazantzis M, Doege H, Ortegon AM, Tsang B, Falcon A. et al. Fatty acid transport protein 1 is required for nonshivering thermogenesis in brown adipose tissue. Diabetes. 2006;55:3229-37
53. Lee J, Ellis JM, Wolfgang MJ. Adipose fatty acid oxidation is required for thermogenesis and potentiates oxidative stress-induced inflammation. Cell Rep. 2015;10:266-79
54. Bickel PE, Tansey JT, Welte MA. PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim Biophys Acta. 2009;1791:419-40
55. Libby AE, Bales ES, Monks J, Orlicky DJ, McManaman JL. Perilipin-2 deletion promotes carbohydrate-mediated browning of white adipose tissue at ambient temperature. J Lipid Res. 2018;59:1482-500
56. Lee YK, Sohn JH, Han JS, Park YJ, Jeon YG, Ji Y. et al. Deficiency Stimulates Thermogenic Beige Adipocytes Through Activation. Diabetes. 2018;67:791-804
57. Wolins NE, Quaynor BK, Skinner JR, Tzekov A, Croce MA, Gropler MC. et al. OXPAT/PAT-1 is a PPAR-induced lipid droplet protein that promotes fatty acid utilization. Diabetes. 2006;55:3418-28
58. Granneman JG, Moore HH, Mottillo EP, Zhu Z. Functional interactions between Mldp (LSDP5) and Abhd5 in the control of intracellular lipid accumulation. J Biol Chem. 2009;284:3049-57
59. Wang H, Bell M, Sreenivasan U, Sreenevasan U, Hu H, Liu J. et al. Unique regulation of adipose triglyceride lipase (ATGL) by perilipin 5, a lipid droplet-associated protein. J Biol Chem. 2011;286:15707-15
60. Sanghani SP, Quinney SK, Fredenburg TB, Davis WI, Murry DJ, Bosron WF. Hydrolysis of irinotecan and its oxidative metabolites, 7-ethyl-10-[4-N-(5-aminopentanoic acid)-1-piperidino] carbonyloxycamptothecin and 7-ethyl-10-[4-(1-piperidino)-1-amino]-carbonyloxycamptothecin, by human carboxylesterases CES1A1, CES2, and a newly expressed carboxylesterase isoenzyme, CES3. Drug Metab Dispos. 2004;32:505-11
61. Lian J, Wei E, Wang SP, Quiroga AD, Li L, Di Pardo A. et al. Liver specific inactivation of carboxylesterase 3/triacylglycerol hydrolase decreases blood lipids without causing severe steatosis in mice. Hepatology. 2012;56:2154-62
62. Lian J, Quiroga AD, Li L, Lehner R. Ces3/TGH deficiency improves dyslipidemia and reduces atherosclerosis in Ldlr(-/-) mice. Circ Res. 2012;111:982-90
63. Lian J, Nelson R, Lehner R. Carboxylesterases in lipid metabolism: from mouse to human. Protein Cell. 2018;9:178-95
64. Yang L, Li X, Tang H, Gao Z, Zhang K, Sun K. A Unique Role of Carboxylesterase 3 (Ces3) in β-Adrenergic Signaling-Stimulated Thermogenesis. Diabetes. 2019;68:1178-96
65. Leonzino M, Reinisch KM, De Camilli P. Insights into VPS13 properties and function reveal a new mechanism of eukaryotic lipid transport. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2021;1866:159003
66. Velayos-Baeza A, Vettori A, Copley RR, Dobson-Stone C, Monaco AP. Analysis of the human VPS13 gene family. Genomics. 2004;84:536-49
67. Ramseyer VD, Kimler VA, Granneman JG. Vacuolar protein sorting 13C is a novel lipid droplet protein that inhibits lipolysis in brown adipocytes. Molecular Metabolism. 2018;7:57-70
68. DiStefano MT, Danai LV, Roth Flach RJ, Chawla A, Pedersen DJ, Guilherme A. et al. The Lipid Droplet Protein Hypoxia-inducible Gene 2 Promotes Hepatic Triglyceride Deposition by Inhibiting Lipolysis*. Journal of Biological Chemistry. 2015;290:15175-84
69. DiStefano MT, Roth Flach RJ, Senol-Cosar O, Danai LV, Virbasius JV, Nicoloro SM. et al. Adipocyte-specific Hypoxia-inducible gene 2 promotes fat deposition and diet-induced insulin resistance. Molecular Metabolism. 2016;5:1149-61
70. Laurencikiene J, Stenson BM, Arvidsson Nordström E, Agustsson T, Langin D, Isaksson B. et al. Evidence for an Important Role of CIDEA in Human Cancer Cachexia. Cancer Research. 2008;68:9247-54
71. Zhou Z, Yon Toh S, Chen Z, Guo K, Peng Ng C, Ponniah S. et al. Cidea-deficient mice have lean phenotype and are resistant to obesity. Nature Genetics. 2003;35:49-56
72. Fischer AW, Shabalina IG, Mattsson CL, Abreu-Vieira G, Cannon B, Nedergaard J. et al. UCP1 inhibition in Cidea-overexpressing mice is physiologically counteracted by brown adipose tissue hyperrecruitment. American Journal of Physiology-Endocrinology and Metabolism. 2016;312:E72-E87
73. Gallardo-Montejano VI, Saxena G, Kusminski CM, Yang C, McAfee JL, Hahner L. et al. Nuclear Perilipin 5 integrates lipid droplet lipolysis with PGC-1α/SIRT1-dependent transcriptional regulation of mitochondrial function. Nat Commun. 2016;7:12723
74. Qiang L, Wang L, Kon N, Zhao W, Lee S, Zhang Y. et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Pparγ. Cell. 2012;150:620-32
75. Xu Y, Yu T, Ma G, Zheng L, Jiang X, Yang F. et al. Berberine modulates deacetylation of PPARγ to promote adipose tissue remodeling and thermogenesis via AMPK/SIRT1 pathway. Int J Biol Sci. 2021;17:3173-87
76. Najt CP, Khan SA, Heden TD, Witthuhn BA, Perez M, Heier JL. et al. Lipid Droplet-Derived Monounsaturated Fatty Acids Traffic via PLIN5 to Allosterically Activate SIRT1. Mol Cell. 2020;77:810-24.e8
77. Nguyen HP, Yi D, Lin F, Viscarra JA, Tabuchi C, Ngo K. et al. Aifm2, a NADH Oxidase, Supports Robust Glycolysis and Is Required for Cold- and Diet-Induced Thermogenesis. Mol Cell. 2020 77
78. Benador IY, Veliova M, Mahdaviani K, Petcherski A, Wikstrom JD, Assali EA. et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics that Support Lipid Droplet Expansion. Cell Metabolism. 2018 27
79. Wang J, Fang N, Xiong J, Du Y, Cao Y, Ji W-K. An ESCRT-dependent step in fatty acid transfer from lipid droplets to mitochondria through VPS13D-TSG101 interactions. Nature Communications. 2021;12:1252
80. Yu J, Zhang S, Cui L, Wang W, Na H, Zhu X. et al. Lipid droplet remodeling and interaction with mitochondria in mouse brown adipose tissue during cold treatment. Biochim Biophys Acta. 2015;1853:918-28
81. Cui L, Mirza AH, Zhang S, Liang B, Liu P. Lipid droplets and mitochondria are anchored during brown adipocyte differentiation. Protein Cell. 2019;10:921-6
82. Li L, Zhang H, Wang W, Hong Y, Wang J, Zhang S. et al. Comparative proteomics reveals abnormal binding of ATGL and dysferlin on lipid droplets from pressure overload-induced dysfunctional rat hearts. Sci Rep. 2016;6:19782
83. Zhang H, Wang Y, Li J, Yu J, Pu J, Li L. et al. Proteome of skeletal muscle lipid droplet reveals association with mitochondria and apolipoprotein a-I. J Proteome Res. 2011;10:4757-68
84. Cui L, Liu P. Two Types of Contact Between Lipid Droplets and Mitochondria. Front Cell Dev Biol. 2020;8:618322
85. Boutant M, Kulkarni SS, Joffraud M, Ratajczak J, Valera-Alberni M, Combe R. et al. Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 2017;36:1543-58
86. Gallardo-Montejano VI, Yang C, Hahner L, McAfee JL, Johnson JA, Holland WL. et al. Perilipin 5 links mitochondrial uncoupled respiration in brown fat to healthy white fat remodeling and systemic glucose tolerance. Nature Communications. 2021;12:3320
87. Zhou H, Lei X, Benson T, Mintz J, Xu X, Harris RB. et al. Berardinelli-Seip congenital lipodystrophy 2 regulates adipocyte lipolysis, browning, and energy balance in adult animals. J Lipid Res. 2015;56:1912-25
88. Zhao YG, Codogno P, Zhang H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nature Reviews Molecular Cell Biology. 2021;22:733-50
89. Kaushik S, Cuervo AM. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy. 2016;12:432-8
90. Schott MB, Weller SG, Schulze RJ, Krueger EW, Drizyte-Miller K, Casey CA. et al. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J Cell Biol. 2019;218:3320-35
91. Schulze RJ, Sathyanarayan A, Mashek DG. Breaking fat: The regulation and mechanisms of lipophagy. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2017;1862:1178-87
92. Filali-Mouncef Y, Hunter C, Roccio F, Zagkou S, Dupont N, Primard C. et al. The ménage à trois of autophagy, lipid droplets and liver disease. Autophagy. 2022;18:50-72
93. Grabner GF, Xie H, Schweiger M, Zechner R. Lipolysis: cellular mechanisms for lipid mobilization from fat stores. Nat Metab. 2021;3:1445-65
94. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M. et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131-5
95. Mijaljica D, Prescott M, Devenish RJ. Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy. 2011;7:673-82
96. Schulze RJ, Krueger EW, Weller SG, Johnson KM, Casey CA, Schott MB. et al. Direct lysosome-based autophagy of lipid droplets in hepatocytes. Proc Natl Acad Sci U S A. 2020;117:32443-52
97. Kaushik S, Cuervo AM. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol. 2015;17:759-70
98. Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. 1990;15:305-9
99. Martinez-Lopez N, Garcia-Macia M, Sahu S, Athonvarangkul D, Liebling E, Merlo P. et al. Autophagy in the CNS and Periphery Coordinate Lipophagy and Lipolysis in the Brown Adipose Tissue and Liver. Cell Metab. 2016;23:113-27
100. Zechner R, Zimmermann R, Eichmann TO, Kohlwein SD, Haemmerle G, Lass A. et al. FAT SIGNALS-lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012;15:279-91
101. Thiam AR, Antonny B, Wang J, Delacotte J, Wilfling F, Walther TC. et al. COPI buds 60-nm lipid droplets from reconstituted water-phospholipid-triacylglyceride interfaces, suggesting a tension clamp function. Proc Natl Acad Sci U S A. 2013;110:13244-9
102. Kazak L, Chouchani ET, Jedrychowski MP, Erickson BK, Shinoda K, Cohen P. et al. A creatine-driven substrate cycle enhances energy expenditure and thermogenesis in beige fat. Cell. 2015;163:643-55
103. Guan HP, Li Y, Jensen MV, Newgard CB, Steppan CM, Lazar MA. A futile metabolic cycle activated in adipocytes by antidiabetic agents. Nat Med. 2002;8:1122-8
104. Flachs P, Adamcova K, Zouhar P, Marques C, Janovska P, Viegas I. et al. Induction of lipogenesis in white fat during cold exposure in mice: link to lean phenotype. Int J Obes (Lond). 2017;41:372-80
105. Reidy SP, Weber JM. Accelerated substrate cycling: a new energy-wasting role for leptin in vivo. Am J Physiol Endocrinol Metab. 2002;282:E312-7
Author contact
![]() Corresponding author: Zhuqing Ren, Key Laboratory of Agriculture Animal Genetics, Breeding and Reproduction of the Ministry of Education, College of Animal Science and Technology, Huazhong Agricultural University, Wuhan 430070, China. Tel: +8613297980341; E-mail: renzqhzau.edu.cn.
Corresponding author: Zhuqing Ren, Key Laboratory of Agriculture Animal Genetics, Breeding and Reproduction of the Ministry of Education, College of Animal Science and Technology, Huazhong Agricultural University, Wuhan 430070, China. Tel: +8613297980341; E-mail: renzqhzau.edu.cn.