Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(1):294-310. doi:10.7150/ijbs.78097 This issue Cite
Research Paper
Direct targeting of sEH with alisol B alleviated the apoptosis, inflammation, and oxidative stress in cisplatin-induced acute kidney injury
Juan Zhang1,2*, Zhi-Lin Luan1*, Xiao-Kui Huo1*, Min Zhang1, Christophe Morisseau3, Cheng-Peng Sun1 ![]() , Bruce D. Hammock3
, Bruce D. Hammock3 ![]() , Xiao-Chi Ma1
, Xiao-Chi Ma1 ![]()
1. College of Pharmacy, Second Affiliated Hospital, Dalian Medical University, Dalian 116044, China.
2. School of Pharmaceutical Sciences, Health Science Center, Shenzhen University, Shenzhen 518061, China.
3. Department of Entomology and Nematology, UC Davis Comprehensive Cancer Center, University of California, Davis, CA 95616, United States.
*These authors contributed equally to this work.
Received 2022-8-17; Accepted 2022-11-8; Published 2023-1-1
Abstract

Acute kidney injury (AKI) is a pathological condition characterized by a rapid decrease in glomerular filtration rate and nitrogenous waste accumulation during hemodynamic regulation. Alisol B, from Alisma orientale, displays anti-tumor, anti-complement, and anti-inflammatory effects. However, its effect and action mechanism on AKI is still unclear. Herein, alisol B significantly attenuated cisplatin (Cis)-induced renal tubular apoptosis through decreasing expressions levels of cleaved-caspase 3 and cleaved-PARP and the ratio of Bax/Bcl-2 depended on the p53 pathway. Alisol B also alleviated Cis-induced inflammatory response (e.g. the increase of ICAM-1, MCP-1, COX-2, iNOS, IL-6, and TNF-α) and oxidative stress (e.g. the decrease of SOD and GSH, the decrease of HO-1, GCLC, GCLM, and NQO-1) through the NF-κB and Nrf2 pathways. In a target fishing experiment, alisol B bound to soluble epoxide hydrolase (sEH) as a direct cellular target through the hydrogen bond with Gln384, which was further supported by inhibition kinetics and surface plasmon resonance (equilibrium dissociation constant, KD = 1.32 μM). Notably, alisol B enhanced levels of epoxyeicosatrienoic acids and decreased levels of dihydroxyeicosatrienoic acids, indicating that alisol B reduced the sEH activity in vivo. In addition, sEH genetic deletion alleviated Cis-induced AKI and abolished the protective effect of alisol B in Cis-induced AKI as well. These findings indicated that alisol B targeted sEH to alleviate Cis-induced AKI via GSK3β-mediated p53, NF-κB, and Nrf2 signaling pathways and could be used as a potential therapeutic agent in the treatment of AKI.
Keywords: Alisol B, acute kidney injury, cisplatin, soluble epoxide hydrolase, nephrotoxicity
Introduction
cis-Diamminedichloroplatinum (II) (cisplatin, Cis) is a chemotherapeutic drug to be used for the treatment of solid tumors due to its efficacy in slowing cancer growth [1-5]. Its clinical application is frequently complicated as significant nephrotoxicity, ototoxicity, and neurotoxicity [1, 2]. 50-100 mg/m2 of Cis dose induces nephrotoxicity in approximately one-third of the patients, resulting in the occurrence of acute kidney injury (AKI) [6, 7]. As the most common reason for renal consultation, AKI is a pathological condition with the characteristics of the nitrogenous waste accumulation and the rapid decrease of glomerular filtration rate during hemodynamic regulation [8]. The continuously growing incidence of AKI is a global health concern for the scientific community [9]. Although its mechanism is still limited, some factors, such as cell apoptosis, inflammation, and oxidative stress, are associated with Cis-induced AKI [10]. Cis usually accumulates in renal proximal tubule segment of nephron, and cross-links genomic DNA to distort the duplex structure and stall the replication fork, triggering the activation of the p53 signaling pathway to induce cell apoptosis [1, 2, 11-14]. Furthermore, Cis exposure results in the inflammatory response to release a mass of proinflammatory cytokines, contributing to the development of renal tissue damage and renal failure [1, 2, 15]. Additionally, accumulating Cis in renal tubule induces oxidative stress to produce reactive oxygen species (ROS), such as superoxide anion and the hydroxyl radical, resulting in the exacerbation of Cis-stimulated apoptosis and inflammation [1, 2, 16]. Therefore, regulating apoptosis, inflammation, and oxidative stress serves as a preferred strategy for attenuating Cis-induced AKI.
Polyunsaturated fatty acids (PUFAs) belong to the number of the ω-3 and ω-6 families, and function as a vital role for maintaining the physiological health [17-19]. The best studied of PUFAs arachidonic acid (AA), locates in membranes as the form of phospholipids, is metabolized by cytochrome P450s (CYPs) into bioactive derivatives [20]. As the most representative metabolite, epoxyeicosatrienoic acids (EETs), from AA produced by CYP450 oxidases, have received a great attention from scientists due to their physiological effects [20-22]. However, soluble epoxide hydrolase (sEH) is able to rapidly metabolize EETs, causing the loss of their multiple effects [18, 23-28]. sEH (genetic name Ephx2) is widely distributed in tissues, particularly kidney and liver [29, 30]. Its C-terminal mediates the hydrolysis of bioactive epoxy fatty acids (EpFAs), such as EETs [20, 31], while the role of the N-terminal phosphatase is poorly understood. Recently, the expression and activity of sEH have been reported in connection with kidney diseases [32], and Ephx2 genetic deletion and inhibition of sEH both attenuate renal diseases [32-34]. For example, sEH inhibitors, AUDA and AR9273, significantly attenuated the renal functions (e.g. creatinine (Cr) and blood urea nitrogen (BUN)) in Cis-induced AKI [29, 34, 35]. Therefore, sEH inhibition to enhance the level of EETs has become an attractive strategy to treat diseases related to inflammation, such as AKI and lung injury [36, 37].
The effects of natural medicines in the prevention and treatment of kidney diseases, such as AKI and CKD, are receiving increasing attention in recent years [38]. Alismatis Rhizoma has been widely and historically used in clinical practice for the treatment of dysuria, edema, diarrhea, hypertension, hyperlipidemia, and nephropathy [39, 40]. Alisol B possesses a four-ring protostane skeleton with a side chain at C-17, and is the main constituent of Alismatis Rhizoma which only is known to exist in the genus Alisma and be served as characteristic markers of this genus [41-43]. Pharmacological studies focused on alisol B have revealed its multiple biological effects, including anti-complement, anti-allergy, anti-inflammation, and others [44]. A study by Zhao et al. demonstrated that alisol B could alleviate non-alcoholic steatohepatitis via reducing the accumulation of hepatocyte lipid, inflammation, and lipotoxicity in the animal model [45]. Furthermore, its analogue alisol B 23-acetate alleviated the course of AKI and chronic kidney disease (CKD) [43, 46], and the network pharmacology study focused on diabetic nephropathy indicated the reno-protective potential of alisol B [47]. Herein, we investigated beneficial effects of alisol B toward renal tubular apoptosis, inflammatory response, and oxidative stress in the Cis-induced AKI mouse model, and revealed its drug target and underlying mechanism.
Materials and Methods
Chemicals and reagents
Alisol B was isolated from A. orientale by authors, and identified through 1H and 13C NMR spectra. Recombinant human sEH was purchased from Cayman Chemical and used to detect the KD value of alisol B with sEH. Hematoxylin and eosin (H&E) staining kit was obtained from Beyotime (Shanghai, China). The primary antibodies were purchased from Proteintech (Wuhan, China), Cell Signaling Technology (CST, Danvers, MA, USA), Abcam (Cambridge, United Kingdom), Boster (Wuhan, China) and Abclonal (Wuhan, China). Detailed information for the primary antibodies were as following: p53 (10442-1-AP, Proteintech), PARP (9542S, CST), Bcl-2 (3498S, CST), Bax (ab182734, Abcam), Caspase3 (ab49822, Abcam), Cleave-Caspase3 (9664S, CST), β-actin (BM0627, Boster), MCP-1 (66272-1-lg, Proteintech), ICAM-1 (60299-1-lg, Proteintech), iNOS (13120S, CST), IL-6 (21865-1-AP, Proteintech), p65 (8242T, CST), Grp78 (ab21685, abcam), 4-HNE (ab48506, Abcam), TNF-α (60291-1-lg, Proteintech), 8-OXO (ab206461, Abcam), HO-1 (A1346, ABclonal), Nrf2 (ab31163, Abcam), Keap1 (10503-2-AP, Proteintech), sEH (10833-1-AP, Proteintech), Kim-1 (14971S, CST), GSK3β (A2081, ABclonal), and p-GSK3β (5558P, CST).
Animal models
Wild-type (WT) C57BL/6 mice were obtained from the Experimental Animal Center of Dalian Medical University (Dalian, China), sEH (gene name Ephx2, responsible for encoding the sEH protein) gene knockout (KO) mice (21-24 g) were obtained from Cyagen Biosciences Inc. (Suzhou, China), and kept under a light/dark cycle of 12 h per day at a controlled temperature. The genotyping and confirmation of the sEH KO mice are shown in Figure S5. All the animal experiments were in line with the Institutional Animal Care and Use Committee of Dalian Medical University (NO. AEE19044).
Male C57BL/6 WT mice (23-25 g) were randomly classified into 6 groups (12/group): the control group, alisol B (60 mg/kg) group, Cis group, Cis + alisol B (15 mg/kg) group, Cis + alisol B (30 mg/kg) group, and Cis + alisol B (60 mg/kg) group. Mice were pretreated with alisol B (15, 30, or 60 mg/kg, dissolved in 10% hydroxypropyl β-cyclodextrin) via oral gavage for seven days. On Day 5, mice in Cis and Cis + alisol B groups were subjected (i.p.) to Cis (20 mg/kg). Mice were sacrificed on Day 8, and the blood and kidneys were collected. The mice in control group were administrated with the vehicle (10% hydroxypropyl β-cyclodextrin and saline).
Measurement of plasma and kidney
The plasma and kidney samples were prepared and stored at -80 °C after the mice were sacrificed. Cr, BUN, malonyldiadehyde (MDA), glutathione (GSH) and superoxide dismutase (SOD) were measured using the corresponding assay kits (Jiancheng Bioengineering Institute, Nanjing, China).
Renal pathological assessments and immunohistochemical staining
Formalin-fixed and paraffin-embedded renal sections were stained using H&E or periodic acid-Schiff's (PAS) to identify injured tubules according to standard protocols. The acute tubular necrosis (ATN) score was determined on the basis of the previous criteria [48]. Immunohistochemical staining were performed in a routine procedure. In brief, renal sections were incubated with 3% H2O2, blocked with 5% BSA at 37 °C, and incubated with primary antibodies at 4 °C overnight. The sections were treated with a horseradish peroxidase-conjugated secondary antibody at 37 °C. After 30 min, the sections were visualized with diaminobenzidine. Immunohistochemical signals were quantified by Image J software.
Quantitative real-time PCR
Total RNA was extracted from C57BL/6 mice and sEH KO mice by using TRIzol reagent (Accurate Biotechnology, Changsha, China), and the reverse transcription experiment was performed by using its corresponding kit to afford the cDNA. The PCR experiments were performed on an CFX96 Real-time System (Bio-Rad, California, USA) with MonAmp SYBR Green qPCR SuperMix (Monad, Shanghai, China). All measured values were normalized with β-actin according to the ΔΔCT method. These primers were listed in Table S1.
Western blot
Kidney samples were extracted with RIPA Lysis buffer with the cocktail, and then centrifugated at 4 °C for 15 min to afford the supernatants used for the determination of the protein concentrations and Western blot analysis. The proteins were subjected to 10%-12% SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membranes, blocked with 5% skim milk, and incubated with the primary antibodies overnight at 4 °C. The membranes were subsequently incubated with a horseradish peroxidase-conjugated secondary antibody for 1 h, and detected using the Tanon 5200- ECL detection system. Image J software was used for the quantitative analysis.
LC-MS analysis for Epoxyeicosatrienoic acids (EETs) and dihydroxyeicosatrienoic acid (DHETs)
The levels of EETs and DHETs were measured as previously described [49-51]. Kidney samples were homogenized with water to afford the supernatant after the centrifugation at 20,000 g for the analysis using LC-MS/MS.
Flow cytometry
Mice were anesthetized and perfused with normal saline to collect the kidneys in the ice plate. The kidney samples were cut into pieces (about 1-2 mm), and washed by PBS for tree times. The samples were digested by the collagenase at 37 °C for 30 min to afford renal cells after the centrifugation. Renal cells were stained by Annexin V and propidium iodide (PI) at 4 °C for 30 min, and then analyzed using the flow cytometer.
Cell culture and treatment
HK-2 cells were cultured in Dulbecco's modified eagle medium (DMEM) with 10% fetal bovine serum (FBS) at 37 °C in humidified air containing 5% CO2 at 37 °C. Cells were seeded into the 6-well plate overnight, and then pretreated with or without alisol B (20 μM) and LiCl (5 mM, a GSK3β inhibitor) for 1 h before the challenge with Cis (20 μM). After 24 h, cells were harvested for PCR and Western blot analyses.
Target protein identification for alisol B
The protein target of alisol B was identified using a pulled-down assay [52]. Epoxy-activated Sepharose 6B beads (GE Healthcare, Chicago, USA) was used for the coupling of alisol B. HEK293 lysates were treated with alisol B-coupled beads or alisol B overnight at 4 °C, the bead-captured proteins were washed with PBS, then the beads were boiled with loading buffer for 10 min. The supernatant was separated by 10% SDS-PAGE, followed by the silver staining, Western blot, and LC-MS/MS analysis.
Cellular thermal shift assay (CETSA)
HEK293 lysates were incubated with alisol B or the vehicle for 30 min, and then heated at different temperatures for 3 min. After the centrifugation of the heated lysates at 20,000 g for 20 min at 4 °C, the supernatants were analyzed by Western blot.
Solvent-induced protein precipitation (SIP) assay
HEK293 lysates were incubated with alisol B or the vehicle as our previous method [52, 53]. The lysates were incubated with alisol B or the vehicle for 30 min, and then treated with acetone: ethanol: acetic acid (AEA, 50: 50: 0.1) for 20 min at 37 °C. After the centrifugation at 20,000 g for 20 min at 4 °C, the supernatants were analyzed by Western blot.
Drug affinity responsive target stability (DARTS) assay
Before the treatment with pronase (15 min at room temperature), HEK293 lysates were incubated with alisol B (5, 10, and 20 μM) for 1 h, and then isolated by SDS-PAGE for Western blot detection [52].
Surface plasmon resonance (SPR) assay
Biacore T200 instruments (GE Healthcare) were used to analyze the binding affinity of alisol B with sEH as previously described [51].
Soluble epoxide hydrolase activity assessment
The inhibitory activity of alisol B towards sEH was also assayed as previously described [54, 55]. The sEH (0.1 ng/mL) was pre-incubated with alisol B for 3 min, and then added the probe PHOME (10 μM). After 20 min incubation at 37 °C, the fluorescence signal was recorded on a microplate reader.
Molecular dynamics simulation
The interaction of alisol B with sEH (PDB: 4OCZ) was analyzed using GROMACS package as previous methods [56, 57].
Statistical analysis
Statistical analysis was carried out using one-way or two-way ANOVA with the Prism software package. Data were presented as means ± standard error of the mean (SEM). Results were considered significant at p<0.05.
Results
Alisol B ameliorated Cis-induced AKI in mice
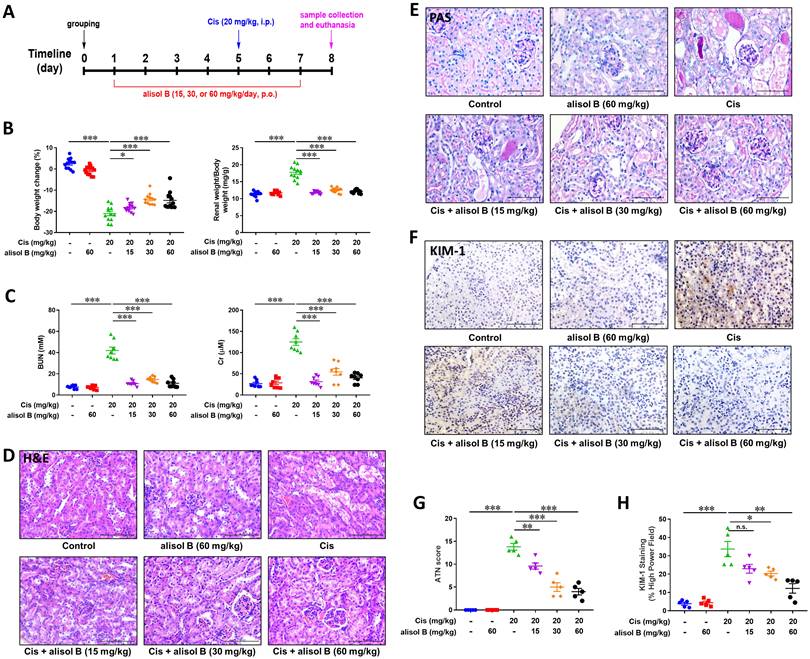
In order to investigate the effect of alisol B in Cis-induced AKI, mice were treated with alisol B (15, 30, and 60 mg/kg) and then treated with Cis (20 mg/kg) as described in Figure 1A. Cis (20 mg/kg) significantly decreased mouse body weight and increased the mouse renal weight/body weight, whereas these changes were reversed by alisol B (15, 30, and 60 mg/kg) in a dose-dependent manner (Figure 1B&S1). Furthermore, Cis (20 mg/kg) treatment resulted in renal dysfunction, such as elevated levels of BUN and Cr and obvious renal histological damage (Figure 1C-E). Alisol B administration markedly improved renal function impacted by Cis. The increase of BUN and creatinine induced by Cis was dose-dependently attenuated by alisol B (15, 30, and 60 mg/kg) (Figure 1C). The results of H&E and PAS staining revealed that Cis (20 mg/kg) treatment led to the extensive necrosis of the proximal tubules compared with the control group (Figure 1D, E, & G). However, the mice in control and alisol B-treated groups exhibited relatively normal morphology with alleviated renal tubule damage (15, 30, and 60 mg/kg) (Figure 1D, E, & G), which was further supported by the result of of the kidney injury molecule 1 (KIM-1) staining (Figure 1F&H). These results suggested that alisol B protected against Cis-induced AKI.
Alisol B ameliorated Cis-induced AKI in mice. (A) Schematic diagram of Cis-induced AKI with alisol B treatment. Mice were intragastrically administrated with alisol B (15, 30, or 60 mg/kg/day) daily for 7 days and intravenously treated with Cis (20 mg/kg) on day 4. The animals were sacrificed on day 8; (B) Body weight and kidney weight of mice were measured daily; data were presented as mean ± SEM, n=12, *p<0.05, ***p<0.001; (C) Measurement of blood urea nitrogen (BUN) and serum creatinine (Cr); data were presented as mean ± SEM, n=8, *** p<0.001; (D) Representative images showing H&E staining on renal sections from mice in different groups; (E) Representative images showing PAS staining on renal sections from mice in different groups; (F) Immunostaining of kidney injury molecule 1 (KIM-1), a specific renal injury marker, on renal sections from mice in different groups. (G) ATN scoring of histopathological features in different groups of mice; data were presented as mean ± SEM, n=5, **p<0.01, ***p<0.001; (H) Quantitative analysis of KIM-1 positive staining in different groups; data were presented as mean ± SEM, n=5, *p<0.05, ** p<0.01, ***p<0.001.
Alisol B attenuated Cis-induced renal cell apoptosis through p53 signaling pathway
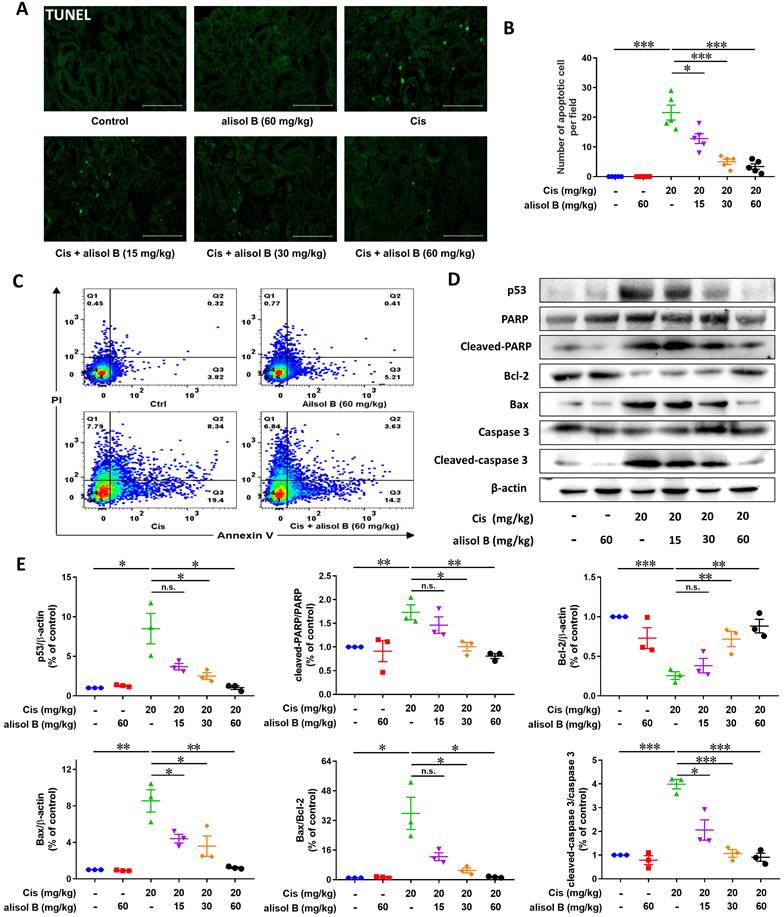
Cis treatment resulted in significant cell apoptosis in the kidneys, while alisol B (15, 30, and 60 mg/kg) reduced the number of TUNEL-positive nuclei (Figure 2A&B), which was further supported by the flow cytometry experiment through the Annexin-V/PI staining showing that alisol B remarkably attenuated Cis-induced renal cell apoptosis (Figure 2C&S2). In parallel, Western blot analyses showed that alisol B (15, 30, and 60 mg/kg) reversed the increase of expression levels of cleaved-caspase 3, cleaved-PARP, p53, and the ratio of Bax/Bcl-2 (Figure 2D&E). Therefore, alisol B exerted anti-apoptotic effect through p53 pathway in Cis-induced AKI mice.
Alisol B inhibited renal cell apoptosis induced by Cis in mice. (A) Representative images of TUNEL staining on the renal sections from mice in different groups; (B) Quantitative analysis of TUNEL staining; data were presented as mean ± SEM, n=5, *p<0.05, ***p<0.001; (C) Representative images of Annexin-V/PI staining on the renal cells from mice in different groups analyzed by flow cytometry; (D) Western blot showing the effect of alisol B on protein levels of genes important in cell apoptosis after Cis challenge; (E) Quantitative analysis for the protein levels in (D); data were presented as mean ± SEM, n=3, *p<0.05, ** p<0.01, ***p<0.001, n.s.= no significance.
Alisol B alleviated Cis-induced renal inflammation through NF-κB signaling pathway
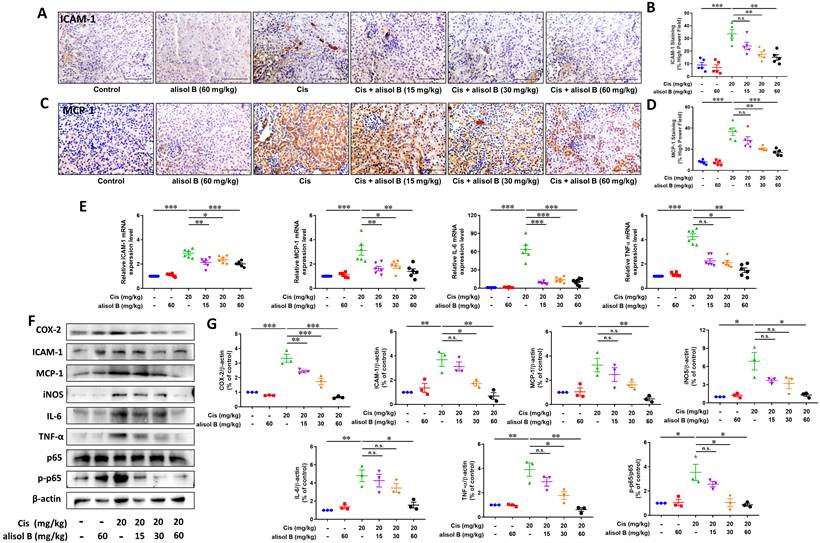
Cis treatment results in the upregulation of various proinflammatory cytokines and chemokines in the kidneys. To examine the effect of alisol B against renal inflammation on the basis of nuclear factor-kappa B (NF-κB) signaling pathway, we determined expression levels of NF-κB p-p65 and its downstream target proteins and genes, such as inducible nitric oxide synthase (iNOS), intracellular adhesion molecule-1 (ICAM-1), interleukin-6 (IL-6), monocyte chemoattractant protein-1 (MCP-1), tumor necrosis factor-alpha (TNF-α), and cyclooxygenase-2 (COX-2). As shown in Figure 3A-D, renal sections from Cis-treated mice displayed significantly increased staining of ICAM-1 and MCP-1, while alisol B (15, 30, and 60 mg/kg) markedly reduced staining of ICAM-1 and MCP-1 induced by Cis (Figure 3A-D). Consistent with the immunostaining results, alisol B (15, 30, and 60 mg/kg) dose-dependently regulated protein and mRNA expression levels of MCP-1 and ICAM-1 in Cis-induced mice (Figure 3E-G). Cis treatment resulted in the increase of the phosphorylated p65 expression level and its downstream target proteins and genes IL-6, iNOS, TNF-α, and COX-2, whereas their expression levels were dose-dependently decreased in Cis-induced mice via alisol B (15, 30, and 60 mg/kg, Figure 3E-G). Collectively, alisol B protected Cis-induced inflammation via NF-κB signaling pathway.
Alisol B reduced inflammation in the kidneys of Cis-treated mice. (A) Immunostaining of intracellular adhesion molecule-1 (ICAM-1) on renal sections from mice in different groups; (B) Quantitative analysis of ICAM-1 positive staining in different groups; data were presented as mean ± SEM, n=5, ** p<0.01, ***p<0.001, n.s.= no significance; (C) Immunostaining of monocyte chemoattractant protein-1 (MCP-1) on renal sections from mice in different groups; (D) Quantitative analysis of MCP-1 positive staining in different groups; data were presented as mean ± SEM, n=5 per group, * p<0.05, ***p<0.001; (E) qPCR analysis showing the mRNA levels of inflammation-related genes, ICAM-1, MCP-1, IL-6 and TNF-α, in mouse kidneys from different groups; data were presented as mean ± SEM, n=6, *p<0.05, ** p<0.01, ***p<0.001, n.s.= no significance; (F) Western blot demonstrating the effect of alisol B on expression levels of COX-2, ICAM-1, MCP-1, iNOS, IL-6, TNF-α, p-p65, and p65 after Cis challenge; (G) Quantitative analysis for the protein levels in (F); data were presented as mean ± SEM, n=3, *p<0.05, ** p<0.01, n.s.= no significance.
Alisol B alleviated Cis-induced renal oxidative and endoplasmic reticulum stresses through Nrf2 signaling pathway
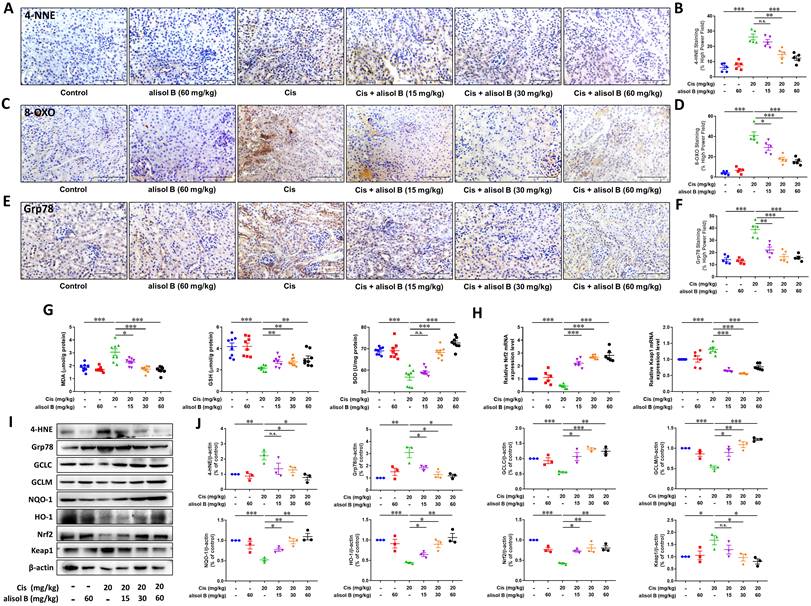
Oxidative and endoplasmic reticulum (ER) stresses are the important pathological course of AKI, therefore, we investigated whether alisol B protected against Cis-induced AKI by attenuating oxidative and ER stresses. We firstly examined the immunostaining of markers for reactive oxygen species (ROS) production and ER stress, 8-oxo-2′-deoxyguanosine (8-OXO), glucose-regulated protein 78 (Grp78), and 4-hydroxynonenal (4-HNE). The immunostaining results showed that alisol B (15, 30, and 60 mg/kg) markedly reduced Cis-induced expressions of 4-HNE, 8-OXO, and Grp78 (Figure 4A-F). Furthermore, alisol B (15, 30, and 60 mg/kg) reduced the MDA level, and enhanced GSH and SOD levels in Cis-induced AKI mice (Figure 4G). As a key transcription factor, nuclear factor erythroid 2-related factor 2 (Nrf2) is in charge of oxidative and ER stresses, we found that alisol B (15, 30, and 60 mg/kg) could activate Nrf2 signaling pathway via upregulating expression levels of HO-1, GCLC, GCLM, NQO-1, and Nrf2, and downregulating Keap1 mRNA and expression levels (Figure 4H-J) in Cis-induced AKI mice. These findings indicated that alisol B alleviated Cis-induced oxidative and ER stresses via Nrf2 signaling pathway.
Alisol B decreased oxidative stress and ER stress in Cis-induced AKI. (A-F) Representative images and quantitative analysis of immunostaining for 4-HNE (A&B), 8-OXO (C&D), and Grp78 (E&F) on renal sections from mice in different groups; data were presented as mean ± SEM, n=5 per group, * p<0.05, ** p<0.01, ***p<0.001, n.s.= no significance; (G) Measurement of renal MDA, GSH, and SOD from mice in different groups; data were presented as mean ± SEM, n=8, *p<0.05, ** p<0.01, ***p<0.001, n.s.= no significance; (H) qPCR analysis showing the mRNA levels of oxidative stress-related genes, Nrf2 and Keap1, in mouse kidneys from different groups; data were presented as mean ± SEM, n=6 per group, ***p<0.001; (I) Western blot demonstrating the effect of alisol B on expression levels of 4-HNE, Grp78, GCLC, GCLM, NQO-1, HO-1, Nrf2, and Keap1 after Cis challenge; (J) Quantitative analysis for the protein levels in (I); data were presented as mean ± SEM, n=3, *p<0.05, ** p<0.01, *** p<0.001, n.s.= no significance.
sEH as a direct cellular target of alisol B
In order to identify the direct cellular targets of alisol B, its epoxyactivated conjugated Sepharose beads were performed as previously described [52], and used as a small-molecule affinity reagent to capture its binding proteins. As described in Figure 5A, a protein band at ~63 kDa was identified as soluble epoxide hydrolase (sEH) by LC-MS/MS analysis, which was further supported by Western blot (Figure 5B&S3). CETSA, SIP, and DARTS were also used to monitor drug target engagement. The results of CETSA, SIP, and DARTS showed that alisol B alleviated effects of temperature, organic solvent, and pronase on the stability of sEH (Figure 5D&E), revealing the direct interaction between sEH and alisol B. The SPR technique was used to analyze the affinity of alisol B and sEH, which demonstrated their equilibrium dissociation constant (KD = 1.32 μM, Figure 5F). In addition, the inhibition kinetic result revealed its inhibition constant (Ki) value of 5.97 μM (Figure 5G&H). The aforementioned results suggested the direct binding of alisol B to sEH.
sEH is a direct target of alisol B. (A) Identification of cellular target of alisol B using pull-down technology coupled with LC-MS/MS. HEK293 lysate was incubated with control beads or alisol B beads. The binding proteins were detected by silver staining and LC-MS/MS analysis; (B) The binding proteins were detected by Western blot; (C) HEK293 lysate was incubated with alisol B in the presence or absence of pronase (5 µg/mL); (D) HEK293 lysate were exposed to alisol B (50 µM) or vehicle followed by a cellular thermal shift assay; (E) HEK293 lysate were exposed to alisol B (50 µM) or vehicle followed by a SIP assay; (F) SPR plot of alisol B with sEH; (G) Michealis-Menten plot of alisol B against sEH; (H) Lineweaver-Burk plot of alisol B against sEH; (I) The interaction of alisol B with sEH analyzed by molecular dynamics; (J-M) Gln384Ala mutation abolished the binding of alisol B with sEH.
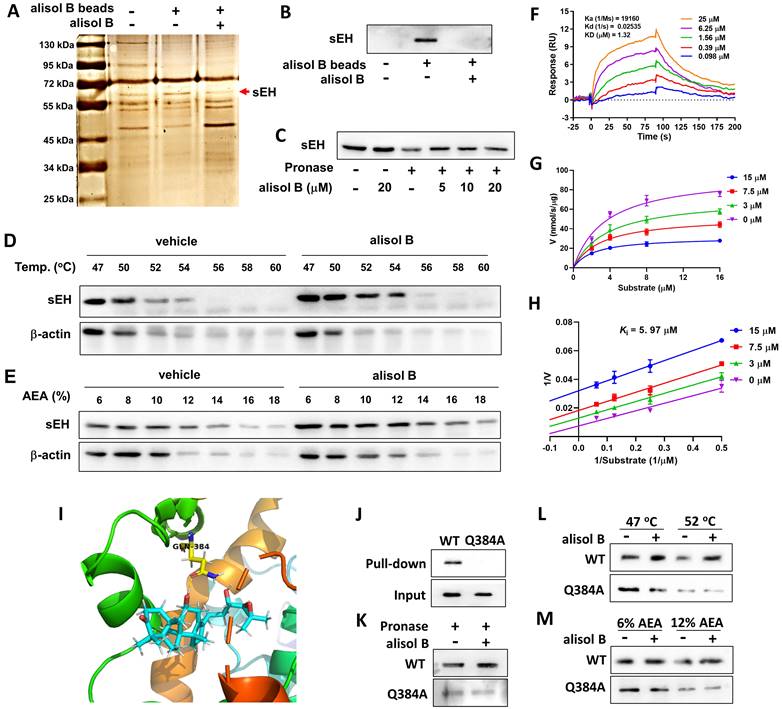
In order to understand the interaction of alisol B and sEH, molecular dynamics stimulation was used. As described in Figure 5I&S4, alisol B could bind to sEH via the hydrogen bond interaction with Gln384 in the catalytic cavity of sEH, therefore Gln384 was mutated into Gln384Ala in order to confirm the role of Gln384. It is worth noting that Gln384Ala abolished the binding of alisol B with sEH (Figure 5J), demonstrating that alisol B interacted with amino acid residue Gln384 of sEH. Similarly, CETSA, SIP, and DARTS assays with Gln384Ala mutated sEH further supported our findings (Figure 5K-M).
Alisol B suppressed the sEH activity to stabilize the level of EETs in Cis-induced AKI, further the inhibition of GSK3β
The result of Sepharose beads demonstrated that the sEH served as a direct cellular target of alisol B, and it is involved in the hydrolytic metabolism of natural epoxides, such as EpFAs (e.g. EETs), to form the corresponding vicinal diol, such as DHETs [58], therefore, LC-MS/MS was applied for analyzing the effect of alisol B on the sEH activity and its metabolites in Cis-induced mice. As shown in Figure 6A-F, Cis exposure reduced levels of 8,9-EET, 11,12-EET, and 14,15-EET, and increased levels of 8,9-DHET, 11,12-DHET, and 14,15-DHET, while the reverse results were observed after administration of alisol B (15, 30, and 60 mg/kg). The ratio of EETs and DHETs suggested that alisol B suppressed the sEH activity in vivo (Figure 6G-I).
Alisol B inhibited sEH activity in the kidneys from mice with Cis-induced AKI. (A-C) Levels of EETs regulated by alisol B in the mice with Cis-induced AKI, 8,9-EET (A), 11,12-EET (B), and 14,15-EET (C); data were presented as mean ± SEM, n=8, *p<0.05, ** p<0.01, ***p<0.001; (D-F) Levels of DHETs regulated by alisol B in the mice with Cis-induced AKI, 8,9-DHET (D), 11,12-DHET (E), and 14,15-DHET (F); data were presented as mean ± SEM, n=8, *p<0.05, ** p<0.01, ***p<0.001; (G-I) The ratio of EETs and DHETs regulated by alisol B in the mice with Cis-induced AKI, 8,9-EET/8,9-DHET (G), 11,12-EET/11,12-DHET (H), and 14,15-EET/14,15-DHET (I); data were presented as mean ± SEM, n=8, *p<0.05, **p<0.01, ***p<0.001; (J) Western blot demonstrating the effect of alisol B on protein levels of GSK-3β after Cis challenge; (K) Quantitative analysis for the protein levels in (J); data were presented as mean ± SEM, n=3, *p<0.05, ** p<0.01, ***p<0.001, n.s.= no significance.
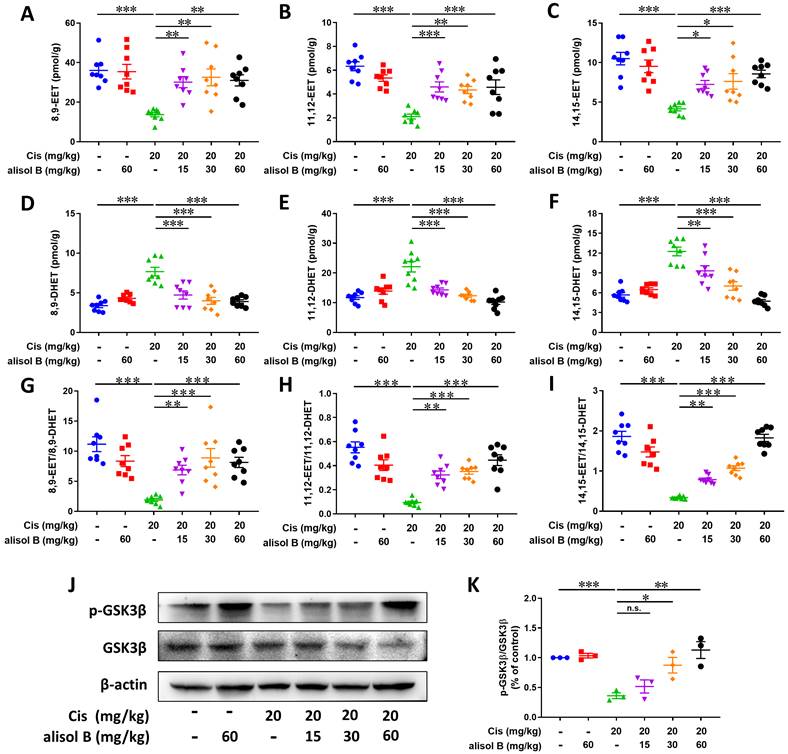
Endogenous EETs can regulate the activity of glycogen synthase kinase 3beta (GSK3β) that is a serine/threonine kinase in connection with apoptosis, inflammation, and oxidative stress via regulating p53, NF-κB, and Nrf2 signaling pathways [59-61]. Therefore, we also determined the effect of sEH inhibition by alisol B on GSK3β. As described in Figure 6J&K, the decreased protein level of p-GSK3β by Cis-induced AKI was significantly improved by alisol B administration, which suggested that GSK3β served as a downstream key pathway of sEH in Cis-induced AKI.
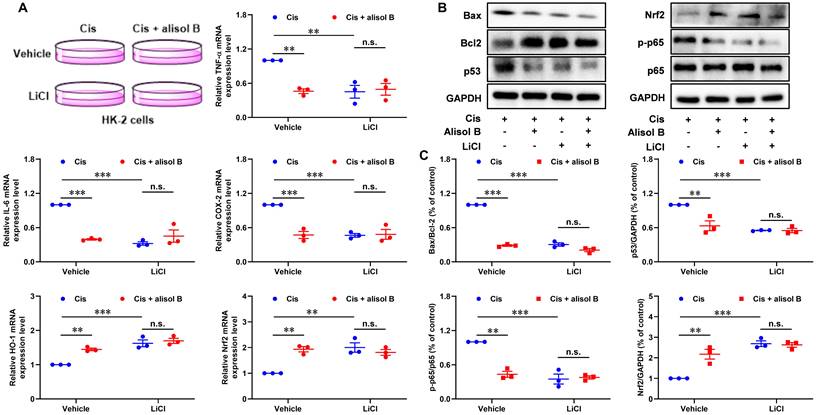
In order to investigate the role of GSK3β in the treatment of alisol B for Cis-mediated AKI, we performed the GSK3β inhibition experiment in Cis-stimulated HK-2 cells (Figure 7). LiCl, a GSK3β inhibitor, decreased the ratio of Bax/Bcl-2 and expression levels of p53 and phosphorylated p65, and enhanced the Nrf2 expression level in Cis-exposed HK-2 cells (Figure 7B&C). It is worth noting that alisol B did not display further effects toward Bax/Bcl-2, p53, p-p65, and Nrf2 in Cis-stimulated HK-2 cells after the inhibition of GSK3β by LiCl. Similarly, the inhibition of GSK3β by LiCl abolished effects of alisol B toward mRNA expressions of TNF-α, IL-6, COX-2, HO-1, and Nrf2 in Cis-stimulated HK-2 cells (Figure 7A). These results demonstrated that alisol B suppressed the sEH activity to enhance the EETs level, triggering GSK3β-mediated p53, NF-κB, and Nrf2 pathways to attenuate Cis-induced AKI.
Inhibition of GSK3β by LiCl abolished the reno-protective effect of alisol B in vitro. (A) qPCR analysis showing the mRNA levels of inflammation-related genes, TNF-α, IL-6, COX-2, NQO-1, HO-1, and Nrf2, in Cis-stimulated HK-2 cells with or without alisol B or LiCl; data were presented as mean ± SEM, n=3, ** p<0.01, ***p<0.001, n.s.= no significance. (B) Effects of inhibition of GSK3β by LiCl abolished anti-apoptosis, anti-inflammatory, and anti-oxidation effects of alisol B in Cis-stimulated HK-2 cells. (C) Quantitative analysis for the protein levels in (B); data were presented as mean ± SEM, n=3, ** p<0.01, ***p<0.001, n.s.= no significance.
sEH genetic deletion abolished the renal protective effect of alisol B in Cis-induced AKI
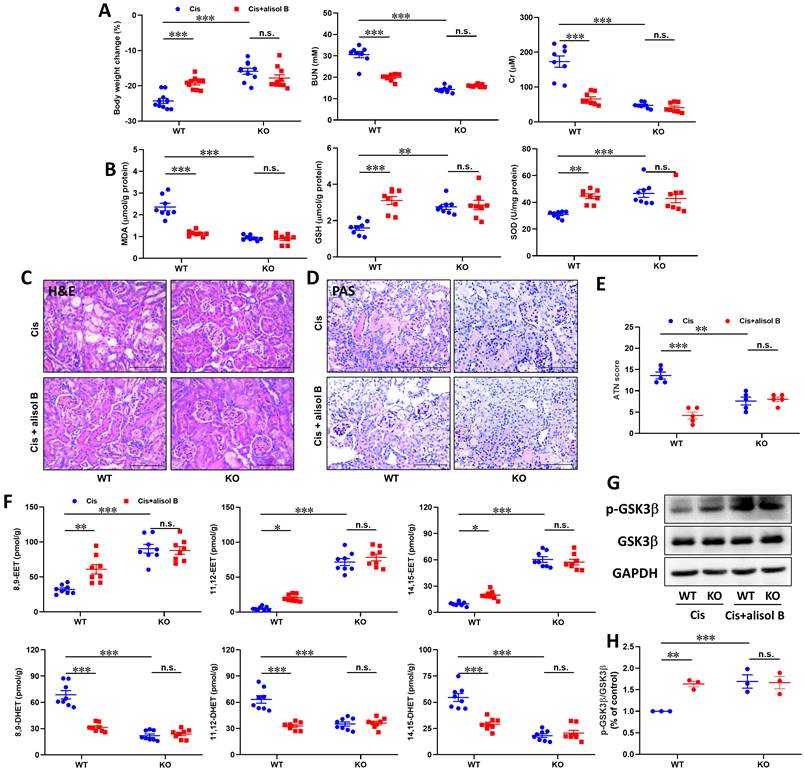
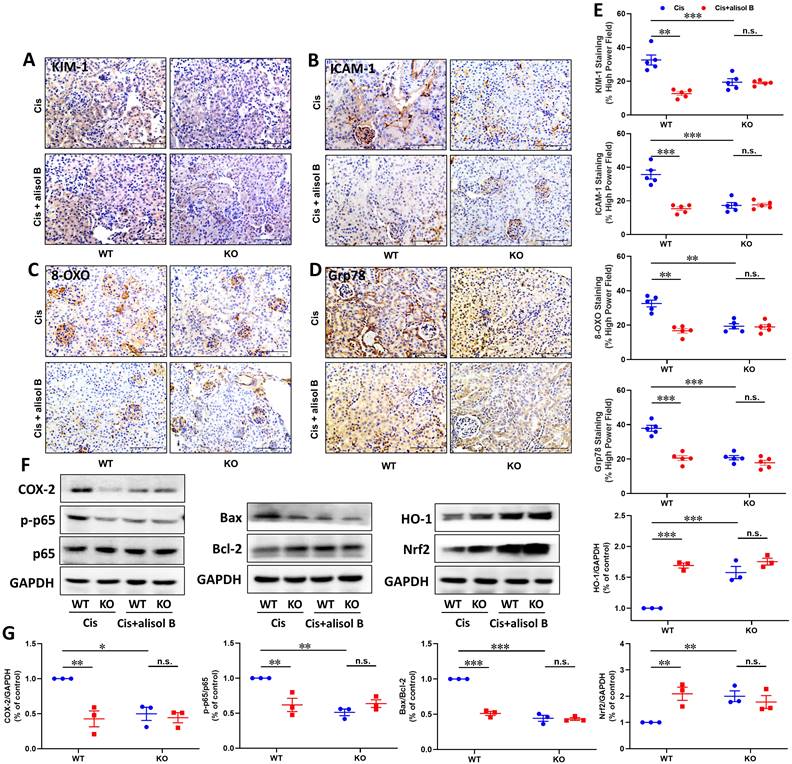
To further determine the effect of sEH in the treatment of AKI by alisol B, WT and sEH KO (sEH-/-, Figure S5) mice were administrated with Cis and alisol B (60 mg/kg). In contrast to WT mice, sEH KO declined renal function and the change of renal morphology induced by Cis (Figure 8A-E, S6, & S7). Meanwhile, Ephx2 genetic deletion also over shadowed the protective effect of alisol B (Figure 8A-E, S6, & S7). The results of EETs and DHETs analyzed by LC-MS/MS illustrated that sEH genetic deletion reversed the reduction of EETs and the increase in DHETs induced by Cis, and promoted the phosphorylation of GSK3β (Figure 8F-H). The effect of alisol B was abolished by sEH genetic deletion (Figure 8F-H). Furthermore, Ephx2 genetic deletion suppressed Cis-induced oxidative and ER stresses and inflammation (Figure 9A-E), downregulated expression levels of COX-2 and p-p65 and the ratio of Bax/Bcl-2, promoted HO-1 and Nrf2 expressions in Cis-induced AKI mice (Figure 9F&G), and abolished the effect of alisol B as well (Figure 9A-G). These results demonstrated that sEH genetic deletion abolished the renal protective effect of alisol B, which further supported sEH being a target of alisol B in the AKI.
Ephx2 KO mice abolished the renal protective effect of alisol B. (A) Measurement of Body weight change, BUN and Cr in WT and sEH-/- mice with Cis-induced AKI treated with alisol B; data were presented as mean ± SEM, n=8-10, *** p<0.001, n.s.= no significance; (B) Measurement of renal MDA, GSH, and SOD from WT and sEH-/- mice with Cis-induced AKI treated with alisol B; data were presented as mean ± SEM, n=8, ** p<0.01, ***p<0.001, n.s.= no significance; (C-D) Representative images of H&E (C) and PAS (D) staining in WT and sEH-/- mice with Cis-induced AKI treated with alisol B; (E) ATN scoring of histopathological features in different groups of mice; data were presented as mean ± SEM, n=5, ** p<0.01, ***p<0.001; n.s.= no significance; (F) Levels of EETs and DHETs regulated by alisol B in WT and sEH-/- mice with Cis-induced AKI treated with alisol B; data were presented as mean ± SEM, n=8, *p<0.05, ** p<0.01, *** p<0.001, n.s.= no significance; (G) Ephx2 KO abolished effects of alisol B on expression levels of p-GSK3β and GSK3β in Cis-induced AKI; (H) Quantitative analysis for the protein level in (G); data were presented as mean ± SEM, n=3, ** p<0.01, ***p<0.001, n.s.= no significance.
Ephx2 KO mice abolished the protective effect of alisol B in the apoptosis, inflammation, and oxidative stress. (A-D) Representative images of immunostaining for KIM-1 (A), ICAM-1 (B), 8-OXO (C), and Grp78 (D) in WT and sEH-/- mice with Cis-induced AKI treated with alisol B; (E) Quantitative analysis of immunostaining for KIM-1, ICAM-1, 8-OXO, and Grp78; data were presented as mean ± SEM, n=5, ** p<0.01, ***p<0.001; n.s.= no significance; (F) Ephx2 KO abolished effects of alisol B on expression levels of proteins related to inflammation, apoptosis, and oxidative stress in Cis-induced AKI; (G) Quantitative analysis for the protein levels in (F); data were presented as mean ± SEM, n=3, *p<0.05, ** p<0.01, ***p<0.001, n.s.= no significance.
Discussion
Since 1978, Cis, a platinum-based alkylating compound, has become an antineoplastic agent to treat solid cancers, including cervical, bladder, and small and non-small cell lung cancers [62]. The accumulation of Cis in renal proximal epithelial cells leads to DNA damage, mitochondrial destruction, renal cell apoptosis, inflammatory response, and excessive generation of ROS [63], therefore, its clinical application is greatly limited [64]. It is commonly felt in the renal cancer field that lives could be saved if one could increase the dose of Cis at a chemotherapeutic agent if renal damage could be avoided. Although various approaches have been introduced to overcome the side effect of Cis-mediated nephrotoxicity, to date no effective medical treatment strategy has been established for Cis-induced AKI apart from renal replacement therapy. Thus, novel effective therapeutic agents are urgently required to protect patients under Cis-based chemotherapy from renal damage.
As a critical health problem worldwide, AKI causes high mortality and morbidity in the clinic, and increases the risk of CKD and ESRD [65-67]. In this study, alisol B, a triterpene from A. orientale, was shown to preserve renal function after Cis challenge in mice, and served as a potential protector against Cis-induced AKI. The result of the target fishing experiment demonstrated that alisol B bound to sEH as a direct cellular target through the hydrogen bond with Gln384, which was supported by the results of the inhibition kinetics (Ki = 5.97 μM) and surface plasmon resonance (KD = 1.32 μM). Inhibition of sEH by alisol B stabilized the level of EETs to alleviate renal tubular apoptosis, inflammatory response, and oxidative stress via GSK3β-mediated p53, NF-κB, and Nrf2 signaling pathways. Additionally, sEH genetic deletion alleviated the course of Cis-induced AKI and abolished the reno-protective effect of alisol B in Cis-induced AKI mice, revealing that the inhibition of sEH by alisol B to enhance the level of EETs resulted in its renal protective effect through the regulation of apoptosis, oxidative stress, and inflammation. These findings suggested that alisol B could served as a potential candidate for the treatment of AKI.
A. orientale, namely “Ze Xie” in Chinese is widely used as a traditional Chinese medicine (TCM) in the treatment of various diseases, such as hyperlipidemia and hypertension [68, 69]. Most beneficial effects of A. orientale are attributed to its characteristic protostane-type triterpenoids [42, 70-73]. Alisol B, one of major protostane-type triterpenes from A. orientale [41, 42], has multiple biological effects, including anti-complement, anti-allergy, and anti-inflammation [44], and its 23-acetate displays the protective effect in ischemia-reperfusion-induced AKI [43]. Similar to alisol B 23-acetate, alisol B alleviated Cis-induced renal tubule damage, and improved the renal function in Cis-induced AKI mice as well.
The sEH exists in almost all the living organisms. In mammals it is widely distributed in tissues but in particularly high in liver and kidney [29, 30], and mediates the metabolism of epoxides, such as EETs [29]. Recently, sEH inhibitors exert anti-inflammation, analgesia, anti-fibrosis, cardioprotection, and renoprotection, because of stabilization of the level of EETs, the epoxide metabolites of arachidonic acid (AA) [7, 30, 37, 74, 75]. The expression and activity of sEH have been reported to be associated with kidney diseases [32], and Ephx2 genetic deletion and chemical inhibition of sEH both attenuate renal diseases [32-34]. For example, sEH inhibitors, AUDA and AR9273, significantly attenuated Cis-induced AKI through the decrease of BUN and Cr levels [29, 34, 35]. Moreover, Ephx2 genetic deletion ameliorates the course of Cis-mediated AKI as well [34]. Therefore, natural and synthetic sEH inhibitors with favorable pharmacokinetics and pharmacodynamics may act as promising therapeutics for the treatment of Cis-induced AKI. In this study, alisol B was found to bind to sEH via inactivating with Gln384 (KD = 1.32 μM) and displayed an inhibitory effect with a Ki value of 5.97 μM, resulting in its reno-protective effect in vivo. Furthermore, sEH genetic deletion attenuated the course of Cis-induced AKI, and abolished the protective effect of alisol B.
EETs are bioactive lipid mediators converted from AA by cytochrome P450 2J (CYP2J) or 2C (CYP2C) and the substrates of sEH as well [20]. A great body of studies demonstrated that the level of EETs was significantly decreased, and the level of its corresponding diols was remarkable increased in the renal injury [76]. Meanwhile, administration of 14,15-EET or improving the level of EETs via suppressing sEH reduced cell apoptosis, inflammatory response, and oxidative stress by GSK3β-mediated p53, NF-κB, and Nrf2 signaling pathways in the kidney and central nervous system diseases [51, 59, 61], such as AKI, Parkinson's and Alzheimer's diseases. In addition, administration of EET analogs EET-F01 and EET-A could alleviate Cis-induced nephrotoxicity via suppressing apoptosis, inflammation, and oxidative stress [77, 78]. Therefore, inhibition of sEH by alisol B to ehance the EETs level is connected with apoptosis, oxidative and ER stresses, and inflammation. The p53 overexpression was observed in Cis-induced AKI mice, and suppressing the p53 expression in Cis-induced AKI ameliorated the apoptosis, suggested the role of p53 in Cis-induced nephrotoxicity [11-14]. In this study, alisol B reduced Cis-mediated cell apoptosis by blocking the p53 pathway involved in caspase 3, Bax, Bcl-2, and PARP. Alisol B renoprotection may be also related to decreased inflammation in Cis-treated kidney [64]. This speculation is supported by the findings that sEH inhibition by alisol B markedly reduced proinflammatory cytokine expression, including IL-6, TNF-α, ICAM-1, iNOS, COX-2, and MCP-1, which are associated with and probably causative of Cis-induced AKI.
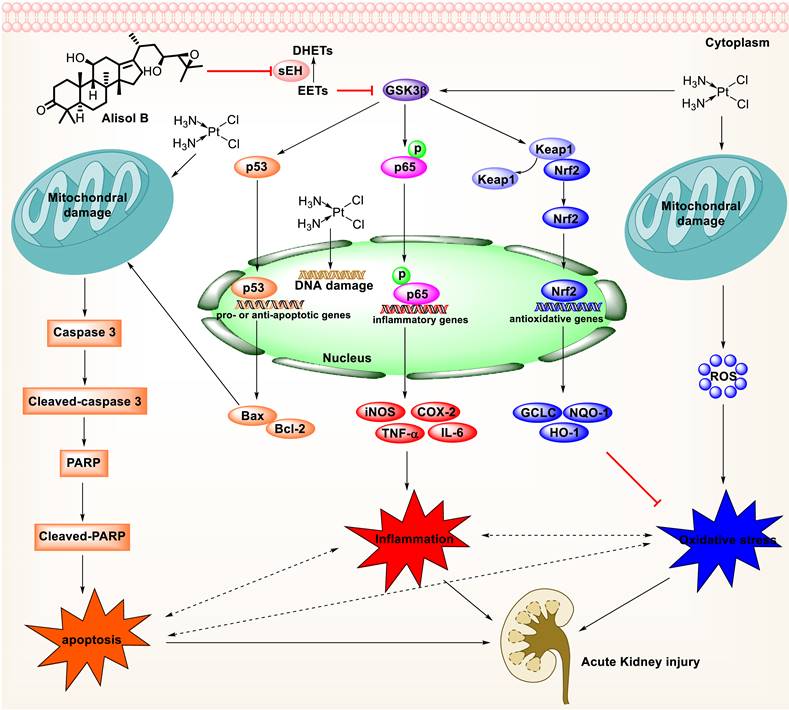
Furthermore, oxidative and ER stresses have long been considered as important contributors to Cis-induced AKI [2, 79]. Oxidative stress promotes histopathological changes through the formation of MDA and the reduction of SOD and GSH levels [80, 81]. Inhibition of sEH by alisol B significantly alleviated Cis-induced oxidative stress by reducing MDA level and increasing SOD and GSH levels. Consistently, alisol B treatment significantly reduced oxidative and ER stresses through the Nrf2 activation after Cis treatment. Moreover, the up-regulated level of Grp78, an ER chaperone [82], is inhibited by alisol B. In addition, sEH deficiency attenuated Cis-induced apoptosis, oxidative and ER stresses, and inflammation, and the effect of alisol B in these fields was abolished in Cis-induced sEH KO mice as well (Figure 10).
Mechanism of targeting sEH with alisol B in the treatment of Cis-induced AKI.
Conclusion
In summary, alisol B significantly attenuated renal tubular apoptosis, inflammatory response, and oxidative stress in Cis-induced AKI. Furthermore, alisol B targeted sEH to alleviate Cis-induced AKI via GSK3β-mediated p53, NF-κB, and Nrf2 signaling pathways, which was supported by the sEH genetic deletion experiment in vivo, suggesting that alisol B was a potential candidate to treat drug-induced AKI.
Abbreviations
AKI: acute kidney injury; BUN: blood urea nitrogen; CETSA: cellular thermal shift assay; CKD: chronic kidney disease; Cis: cisplatin; COX-2: cyclooxygenase-2; Cr: creatinine; DARTS: drug affinity responsive target stability; DHETs: dihydroxyeicosatrienoic acid; EETs: epoxyeicosatrienoic acids; ESRD: end stage renal disease; GSH: glutathione; ICAM-1: intracellular adhesion molecule-1; IL-6: interleukin-6; iNOS: inducible nitric oxide synthase; KO: knockout; MCP-1: monocyte chemoattractant protein-1; MDA: malonyldiadehyde; NF-κB: nuclear factor-kappa B; PAS: periodic acid-Schiff's; PVDF: polyvinylidene difluoride; sEH: soluble epoxide hydrolase; SIP: solvent-induced protein precipitation; SOD: superoxide dismutase; SPR: surface plasmon resonance; TNF-α: tumor necrosis factor-alpha.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Funding
This work is supported by National Natural Science Foundation of China (No. 82274069), Distinguished professor of Liaoning Province (No. XLYC2002008), Revolutionizing Innovative, Visionary Environmental Health Research Program of the National Institute of Environmental Health Sciences (No. R35 ES030443), Superfund Basic Research Program of the National Institutes of Environmental Health Sciences (No. P42 ES04699), China Postdoctoral Science Foundation (No. 2022M720095), and Dalian Young Star of Science and Technology (No. 2019RQ123).
Author contributions
C.P.S., X.C.M., and B.D.H. designed this experiment and revised this manuscript. J.Z., C.P.S., Z.L.L., X.K.H., M.Z., and C.M. performed experiments, analyzed data, wrote, and revised the manuscript. J.Z. and Z.L.L. analyzed data. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All the animal experiments were in line with the Institutional Animal Care and Use Committee of Dalian Medical University (NO. AEE19044).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573-584
2. Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int. 2008;73:994-1007
3. Fan CC, Tsai ST, Lin CY. et al. EFHD2 contributes to non-small cell lung cancer cisplatin resistance by the activation of NOX4-ROS-ABCC1 axis. Redox Biol. 2020;34:101571
4. Sun Y, Qiao Y, Liu Y. et al. ent-Kaurane diterpenoids induce apoptosis and ferroptosis through targeting redox resetting to overcome cisplatin resistance. Redox Biol. 2021;43:101977
5. Wang Y, Zhang M, Bi R. et al. ACSL4 deficiency confers protection against ferroptosis-mediated acute kidney injury. Redox Biol. 2022;51:102262
6. Lebwohl D, Canetta R. Clinical development of platinum complexes in cancer therapy: an historical perspective and an update. Eur J Cancer. 1998;34:1522-1534
7. Shiraishi F, Curtis LM, Truong L. et al. Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am J Physiol Renal Physiol. 2000;278:F726-F736
8. Volarevic V, Djokovic B, Jankovic MG. et al. Molecular mechanisms of cisplatin-induced nephrotoxicity: a balance on the knife edge between renoprotection and tumor toxicity. J Biomed Sci. 2019;26:25
9. Hu J, Chen R, Liu S. et al. Global Incidence and Outcomes of Adult Patients with Acute Kidney Injury After Cardiac Surgery: A Systematic Review and Meta-Analysis. J Cardiothorac Vasc Anesth. 2016;30:82-89
10. Lopes JA, Jorge S. The RIFLE and AKIN classifications for acute kidney injury: a critical and comprehensive review. Clin Kidney J. 2013;6:8-14
11. Jiang M, Yi X, Hsu S. et al. Role of p53 in cisplatin-induced tubular cell apoptosis: dependence on p53 transcriptional activity. Am J Physiol Renal Physiol. 2004;287:F1140-F1147
12. Wei Q, Dong G, Yang T. et al. Activation and involvement of p53 in cisplatin-induced nephrotoxicity. Am J Physiol Renal Physiol. 2007;293:F1282-F1291
13. Luo J, Li M, Tang Y. et al. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc Natl Acad Sci U S A. 2004;101:2259-2264
14. Ibanez-Cabellos JS, Seco-Cervera M, Picher-Latorre C. et al. Acute depletion of telomerase components DKC1 and NOP10 induces oxidative stress and disrupts ribosomal biogenesis via NPM1 and activation of the P53 pathway. Biochim Biophys Acta Mol Cell Res. 2020;1867:118845
15. Ramesh G, Reeves WB. Inflammatory cytokines in acute renal failure. Kidney Int Suppl. 2004: S56-S61.
16. Baliga R, Ueda N, Walker PD. et al. Oxidant mechanisms in toxic acute renal failure. Drug Metab Rev. 1999;31:971-997
17. Laye S, Nadjar A, Joffre C. et al. Anti-inflammatory effects of omega-3 fatty acids in the brain: Physiological mechanisms and relevance to pharmacology. Pharmacol Rev. 2018;70:12-38
18. Pallas M, Vazquez S, Sanfeliu C. et al. Soluble epoxide hydrolase inhibition to face neuroinflammation in Parkinson's disease: A new therapeutic strategy. Biomolecules. 2020;10:701
19. Feuerer N, Marzi J, Brauchle EM. et al. Lipidome profiling with Raman microspectroscopy identifies macrophage response to surface topographies of implant materials. Proc Natl Acad Sci U S A. 2021;118:e2113694118
20. Sun CP, Zhang XY, Morisseau C. et al. Discovery of soluble epoxide hydrolase inhibitors from chemical synthesis and natural products. J Med Chem. 2021;64:184-215
21. Wang Y, Wagner KM, Morisseau C. et al. Inhibition of the soluble epoxide hydrolase as an analgesic strategy: A review of preclinical evidence. J Pain Res. 2021;14:61-72
22. An G, Lee KSS, Yang J. et al. Target-mediated drug disposition-a class effect of soluble epoxide hydrolase inhibitors. J Clin Pharmacol. 2021;61:531-537
23. Fuller-Pace FV, Southern PJ. Temporal analysis of transcription and replication during acute infection with lymphocytic choriomeningitis virus. Virology. 1988;162:260-263
24. Deng J, Yang H, Haak VM. et al. Eicosanoid regulation of debris-stimulated metastasis. Proc Natl Acad Sci U S A. 2021;118:e2107771118
25. Pu Y, Yang J, Chang L. et al. Maternal glyphosate exposure causes autism-like behaviors in offspring through increased expression of soluble epoxide hydrolase. Proc Natl Acad Sci U S A. 2020;117:11753-11759
26. Fishbein A, Wang W, Yang H. et al. Resolution of eicosanoid/cytokine storm prevents carcinogen and inflammation-initiated hepatocellular cancer progression. Proc Natl Acad Sci U S A. 2020;117:21576-21587
27. Bergmann CB, McReynolds CB, Wan D. et al. sEH-derived metabolites of linoleic acid drive pathologic inflammation while impairing key innate immune cell function in burn injury. Proc Natl Acad Sci U S A. 2022;119:e2120691119
28. Wang Y, Yang J, Wang W. et al. Soluble epoxide hydrolase is an endogenous regulator of obesity-induced intestinal barrier dysfunction and bacterial translocation. Proc Natl Acad Sci U S A. 2020;117:8431-8436
29. Newman JW, Morisseau C, Hammock BD. Epoxide hydrolases: their roles and interactions with lipid metabolism. Prog Lipid Res. 2005;44:1-51
30. Morisseau C, Hammock BD. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol. 2013;53:37-58
31. Gomez GA, Morisseau C, Hammock BD. et al. Structure of human epoxide hydrolase reveals mechanistic inferences on bifunctional catalysis in epoxide and phosphate ester hydrolysis. Biochemistry. 2004;43:4716-4723
32. Liu JY. Inhibition of Soluble Epoxide Hydrolase for Renal Health. Front Pharmacol. 2018;9:1551
33. Parrish AR, Chen G, Burghardt RC. et al. Attenuation of cisplatin nephrotoxicity by inhibition of soluble epoxide hydrolase. Cell Biol Toxicol. 2009;25:217-225
34. Liu Y, Webb HK, Fukushima H. et al. Attenuation of cisplatin-induced renal injury by inhibition of soluble epoxide hydrolase involves nuclear factor kappaB signaling. J Pharmacol Exp Ther. 2012;341:725-734
35. Liu Y, Lu X, Nguyen S. et al. Epoxyeicosatrienoic acids prevent cisplatin-induced renal apoptosis through a p38 mitogen-activated protein kinase-regulated mitochondrial pathway. Mol Pharmacol. 2013;84:925-934
36. Zhao WY, Yan JJ, Zhang M. et al. Natural soluble epoxide hydrolase inhibitors from Inula britanica and their potential interactions with soluble epoxide hydrolase: Insight from inhibition kinetics and molecular dynamics. Chem Biol Interact. 2021;345:109571
37. Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov. 2009;8:794-805
38. Chen DQ, Hu HH, Wang YN. et al. Natural products for the prevention and treatment of kidney disease. Phytomedicine. 2018;50:50-60
39. Tian T, Chen H, Zhao YY. Traditional uses, phytochemistry, pharmacology, toxicology and quality control of Alisma orientale (Sam.) Juzep: a review. J Ethnopharmacol. 2014; 158 Pt A: 373-387
40. Shu Z, Pu J, Chen L. et al. Alisma orientale: Ethnopharmacology, Phytochemistry and Pharmacology of an Important Traditional Chinese Medicine. Am J Chin Med. 2016;44:227-251
41. Li HM, Chen XJ, Luo D. et al. Protostane-type triterpenoids from Alisma orientale. Chem Biodivers. 2017 14
42. Liu X, Li SL, Zhou Y. et al. Characterization of protostane triterpenoids in Alisma orientalis by ultra-performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2010;24:1514-1522
43. Luan ZL, Ming WH, Sun XW. et al. A naturally occurring FXR agonist, alisol B 23-acetate, protects against renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2021;321:F617-F628
44. Lee JW, Kobayashi Y, Nakamichi Y. et al. Alisol-B, a novel phyto-steroid, suppresses the RANKL-induced osteoclast formation and prevents bone loss in mice. Biochem Pharmacol. 2010;80:352-361
45. Zhao Z, Deng ZT, Huang S. et al. Alisol B alleviates hepatocyte lipid accumulation and lipotoxicity via regulating RARalpha-PPARgamma-CD36 cascade and attenuates non-alcoholic steatohepatitis in mice. Nutrients. 2022;14:2411
46. Chen H, Wang MC, Chen YY. et al. Alisol B 23-acetate attenuates CKD progression by regulating the renin-angiotensin system and gut-kidney axis. Ther Adv Chronic Dis. 2020;11:2040622320920025
47. Fang J, Wang C, Zheng J. et al. Network pharmacology study of Yishen capsules in the treatment of diabetic nephropathy. PLoS One. 2022;17:e0273498
48. Tavares MB, Chagas de Almeida Mda C, Martins RT. et al. Acute tubular necrosis and renal failure in patients with glomerular disease. Ren Fail. 2012;34:1252-1257
49. Wang W, Yang J, Zhang J. et al. Lipidomic profiling reveals soluble epoxide hydrolase as a therapeutic target of obesity-induced colonic inflammation. Proc Natl Acad Sci U S A. 2018;115:5283-5288
50. Yang J, Schmelzer K, Georgi K. et al. Quantitative profiling method for oxylipin metabolome by liquid chromatography electrospray ionization tandem mass spectrometry. Anal Chem. 2009;81:8085-8093
51. Sun CP, Zhou JJ, Yu ZL. et al. Kurarinone alleviated Parkinson's disease via stabilization of epoxyeicosatrienoic acids in animal model. Proc Natl Acad Sci U S A. 2022;119:e2118818119
52. Wang LC, Liao LX, Lv HN. et al. Highly selective activation of heat shock protein 70 by allosteric regulation provides an insight into efficient neuroinflammation inhibition. EBioMedicine. 2017;23:160-172
53. Zhang X, Wang Q, Li Y. et al. Solvent-Induced Protein Precipitation for Drug Target Discovery on the Proteomic Scale. Anal Chem. 2020;92:1363-1371
54. Sun CP, Zhang J, Zhao WY. et al. Protostane-type triterpenoids as natural soluble epoxide hydrolase inhibitors: Inhibition potentials and molecular dynamics. Bioorg Chem. 2020;96:103637
55. Liu ZB, Sun CP, Xu JX. et al. Phytochemical constituents from Scutellaria baicalensis in soluble epoxide hydrolase inhibition: Kinetics and interaction mechanism merged with simulations. Int J Biol Macromol. 2019;133:1187-1193
56. Lv X, Bai R, Yan JK. et al. Investigation of the inhibitory effect of protostanes on human carboxylesterase 2 and their interaction: Inhibition kinetics and molecular stimulations. Int J Biol Macromol. 2021;167:1262-1272
57. Sun CP, Yan JK, Yi J. et al. The study of inhibitory effect of natural flavonoids toward beta-glucuronidase and interaction of flavonoids with beta-glucuronidase. Int J Biol Macromol. 2020;143:349-358
58. Morisseau C. Role of epoxide hydrolases in lipid metabolism. Biochimie. 2013;95:91-95
59. Deng BQ, Luo Y, Kang X. et al. Epoxide metabolites of arachidonate and docosahexaenoate function conversely in acute kidney injury involved in GSK3beta signaling. Proc Natl Acad Sci U S A. 2017;114:12608-12613
60. Ghosh A, Comerota MM, Wan DB. et al. An epoxide hydrolase inhibitor reduces neuroinflammation in a mouse model of Alzheimer's disease. Science Translational Medicine. 2020;12:eabb1206
61. Sun CP, Zhang XY, Zhou JJ. et al. Inhibition of sEH via stabilizing the level of EETs alleviated Alzheimer's disease through GSK3beta signaling pathway. Food Chem Toxicol. 2021;156:112516
62. Ghosh S. Cisplatin: The first metal based anticancer drug. Bioorg Chem. 2019;88:102925
63. McSweeney KR, Gadanec LK, Qaradakhi T. et al. Mechanisms of cisplatin-induced acute kidney injury: Pathological mechanisms, pharmacological interventions, and genetic mitigations. Cancers (Basel). 2021;13:1572
64. Manohar S, Leung N. Cisplatin nephrotoxicity: a review of the literature. J Nephrol. 2018;31:15-25
65. Chertow GM, Burdick E, Honour M. et al. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol. 2005;16:3365-3370
66. He L, Wei Q, Liu J. et al. AKI on CKD: heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017;92:1071-1083
67. Kurzhagen JT, Dellepiane S, Cantaluppi V. et al. AKI: an increasingly recognized risk factor for CKD development and progression. J Nephrol. 2020;33:1171-1187
68. Fang CY, Lou DY, Zhou LQ. et al. Natural products: potential treatments for cisplatin-induced nephrotoxicity. Acta Pharmacol Sin. 2021;42:1951-1969
69. Wu Z, Raven PH, Hong D. et al. Flora of China Illustrations. Beijing, St. Louis: Science Press; Missouri Botanical Garden. 1998
70. Hur JM, Choi JW, Park JC. Effects of methanol extract of Alisma orientale rhizome and its major component, alisol B 23-acetate, on hepatic drug metabolizing enzymes in rats treated with bromobenzene. Arch Pharm Res. 2007;30:1543-1549
71. Peng GP, Tian G, Huang XF. et al. Guaiane-type sesquiterpenoids from Alisma orientalis. Phytochemistry. 2003;63:877-881
72. Yoshikawa M, Yamaguchi S, Matsuda H. et al. Crude drugs from aquatic plants. V. On the constituents of alismatis rhizoma. (3). Stereostructures of water-soluble bioactive sesquiterpenes, sulfoorientalols a, b, c, and d, from Chinese alismatis rhizoma. Chem Pharm Bull (Tokyo). 1994;42:2430-2435
73. Fukuyama Y, Pei-Wu G, Rei W. et al. 11-deoxyalisol C and alisol D: new protostane-type triterpenoids from Alisma plantago-aquatica. Planta Med. 1988;54:445-447
74. Li L, Li N, Pang W. et al. Opposite effects of gene deficiency and pharmacological inhibition of soluble epoxide hydrolase on cardiac fibrosis. PLoS One. 2014;9:e94092
75. Hu DY, Luo Y, Li CB. et al. Oxylipin profiling of human plasma reflects the renal dysfunction in uremic patients. Metabolomics. 2018;14:104
76. Luo Y, Wu MY, Deng BQ. et al. Inhibition of soluble epoxide hydrolase attenuates a high-fat diet-mediated renal injury by activating PAX2 and AMPK. Proc Natl Acad Sci U S A. 2019;116:5154-5159
77. Imig JD, Hye Khan MA, Burkhan A. et al. Kidney-targeted epoxyeicosatrienoic acid analog, EET-F01, reduces inflammation, oxidative stress, and cisplatin-induced nephrotoxicity. Int J Mol Sci. 2021;22:2793
78. Khan MA, Liu J, Kumar G. et al. Novel orally active epoxyeicosatrienoic acid (EET) analogs attenuate cisplatin nephrotoxicity. FASEB J. 2013;27:2946-2956
79. Perse M, Veceric-Haler Z. Cisplatin-induced rodent model of kidney injury: Characteristics and challenges. Biomed Res Int. 2018;2018:1462802
80. Galgamuwa R, Hardy K, Dahlstrom JE. et al. Dichloroacetate prevents cisplatin-induced nephrotoxicity without compromising cisplatin anticancer properties. J Am Soc Nephrol. 2016;27:3331-3344
81. Garcia-Gimenez JL, Garces C, Roma-Mateo C. et al. Oxidative stress-mediated alterations in histone post-translational modifications. Free Radic Biol Med. 2021;170:6-18
82. Bertolotti A, Zhang Y, Hendershot LM. et al. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326-332
Author contact
![]() Corresponding authors: College of Pharmacy, Second Affiliated Hospital, Dalian Medical University, Dalian, China. E-mail: suncp146com (C.P. Sun); maxc1978com (X.C. Ma). Department of Entomology and Nematology, UC Davis Comprehensive Cancer Center, University of California, Davis, CA 95616, United States. E-mail: bdhammockedu (B.D. Hammock).
Corresponding authors: College of Pharmacy, Second Affiliated Hospital, Dalian Medical University, Dalian, China. E-mail: suncp146com (C.P. Sun); maxc1978com (X.C. Ma). Department of Entomology and Nematology, UC Davis Comprehensive Cancer Center, University of California, Davis, CA 95616, United States. E-mail: bdhammockedu (B.D. Hammock).