Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(1):311-330. doi:10.7150/ijbs.78525 This issue Cite
Review
Insights into the roles and pathomechanisms of ceramide and sphigosine-1-phosphate in nonalcoholic fatty liver disease
Cheng Zhu1*, Qian Huai1*, Xu Zhang1, Hanren Dai1, Xiaolei Li1 ![]() , Hua Wang1,2
, Hua Wang1,2 ![]()
1. Department of Oncology, the First Affiliated Hospital of Anhui Medical University, Hefei, Anhui, China.
2. Inflammation and Immune Mediated Diseases Laboratory of Anhui Province, Anhui Medical University, Hefei, Anhui, China.
*These authors have contributed equally to this work and share first authorship.
Received 2022-8-31; Accepted 2022-11-12; Published 2023-1-1
Abstract

Non-alcoholic fatty liver disease (NAFLD), as one of the main causes of chronic liver disease worldwide, encompasses a spectrum of liver conditions that are not caused by other etiology, such as overt alcohol consumption, from simple steatosis to more aggressive non-alcoholic steatohepatitis (NASH) that involves liver inflammation and fibrosis, and to the lethal cirrhosis that may result in liver cancer and liver failure. The molecular mechanisms governing the transition from steatosis to NASH remain not fully understood, but the hepatic lipidome is extensively altered in the setting of steatosis and steatohepatitis, which also correlate with disease progression. With the tremendous advancement in the field of lipidomics in last two decades, a better understanding of the specific role of sphingolipids in fatty liver disease has taken shape. Among the numerous lipid subtypes that accumulate, ceramides are particularly impactful. On the one hand, excessive ceramides deposition in the liver cause hepatic steatosis. On the other hand, ceramides as lipotoxic lipid have significant effects on hepatic inflammation, apoptosis and insulin resistance that contribute to NAFLD. In this review, we summarize and evaluate current understanding of the multiple roles of ceramides in the onset of fatty liver disease and the pathogenic mechanisms underlying their effects, and we also discuss recent advances and challenges in pharmacological interventions targeting ceramide metabolism for the treatment of NAFLD.
Keywords: Ceramides, NAFLD, NASH, lipid metabolism, therapeutics.
Introduction
NAFLD is the most common chronic liver disease, paralleling the worldwide increase of obesity [1, 2]. NAFLD is a term that encompasses a group of pathologies ranging simple steatosis, NASH and cirrhosis. Fatty liver, or steatosis, is a disorder of deregulated lipid metabolism and is manifested by the excessive accumulation of lipid droplets in the cytosol of hepatocytes leading to toxicity. Hepatic damage is thought to be the main driving event behind the progression of NAFLD, causing injured or dying hepatocytes to release proinflammatory factors. Subsequent activation and infiltration of immune cells lead to inflammation and activation of hepatic stellate cells leads to fibrosis, furthering NAFLD progression [3-5].
NAFLD is also a lipotoxic disease characterized by insulin resistance, oxidative stress, adipokine secretion by adipocytes, endotoxins released by gut microbiota, and endoplasmic reticulum stress [6, 7]. For many years, the most accepted explanation for NAFLD progression is the two-hit hypothesis. According to this hypothesis, the first hit is represented by lipid accumulation in the liver in the presence of metabolic alterations, such as high-fat diet, obesity and insulin resistance. Excess of lipids would then increase liver vulnerability to different factors (second-hits), which might be responsible for the progression from NAFLD to NASH [8, 9]. The two-hit hypothesis is now obsolete, as it is inadequate to explain the several molecular and metabolic changes that take place in NAFLD. The multiple hit hypothesis considers multiple insults acting together on genetically predisposed subjects to induce NAFLD and provides a more accurate explanation of NAFLD pathogenesis. The multiple hit hypothesis refers to a perfect storm of multiple cells working simultaneously and interacting with each other during the development of NAFLD, leading to increased liver inflammation and liver fibrosis [8, 10]. Although the deposition of large amounts of lipids in the liver and elevated circulating lipid levels are considered early events in NAFLD, it is less associated with subsequent progression of NAFLD. While simple steatosis follows a relatively benign clinical course, in some cases it may progress to NASH characterized with cell death and chronic inflammation, the level of ceramides and complex sphingolipids with ceramides as precursor are changed evidently in the liver, which may be one of the important factors for the progression of NAFLD [11, 12]. Although a large number of studies have made much progress for understanding of the mechanisms involved in the complex pathophysiology of NAFLD, there are no current FDA-approved medications for NAFLD. Lifestyle modifications of weight loss (5%-10%) through methods, such as hypocaloric diets, aerobic exercise, and resistance training have been strongly advocated [13-15]. Thus, there is a crucial need to identify multiple targets in NAFLD pathogenesis for the development of diagnostic markers and targeted therapeutics.
Sphingolipids comprise a highly dynamic, diverse, and complex class of bioactive molecules that act as both structural components of cellular membranes and signaling molecules in mammalian cells. Ceramides are a class of bioactive sphingolipids consisting of a fatty acid chain and are also considered as the central molecules in sphingolipid metabolism. Besides being indispensable components of cell membranes, ceramides act as signaling molecules to coordinate various cellular functions, such as cell proliferation, apoptosis, senescence as well as stress response in health and disease [16-18]. Ceramides can be mainly synthesized through three pathways, including de novo pathway, sphingomyelinase pathway and salvage pathway [19-21]. The bioactive sphingolipid metabolites ceramide and sphingosine-1-phosphate (S1P) are a recent addition to the lipids accumulated in obesity and have emerged as important molecular players in various metabolic syndromes, such as fatty liver disease, and insulin resistance [22, 23]. Ceramide has been shown to accumulate in parallel to neutral lipids in livers of obese people and animals. Evidence has shown convincingly that sphingolipids, ceramide in particular, contribute to key steps in NAFLD onset, including the disruption of insulin sensitivity and mitochondrial metabolism, the metabolic derangement, and the stimulation of cell death [24, 25]. In this review, we provide an overview of the pathogenic mechanisms by which ceramides mediate the pathogenesis and progression of NAFLD and we also discuss potential therapeutic modalities targeting ceramide metabolism for the treatment of NAFLD.
Lipids accumulation in NAFLD
Lipid metabolism disorders are the primary causes for the occurrence and progression of fatty liver diseases. NAFLD is a condition of fat accumulation in the liver in combination with metabolic dysfunction in the form of overweight or obesity and insulin resistance. Hepatic steatosis is classified into various degrees based on lipid percentage in hepatocytes, which can further induce organelle dysfunction, cell injury, hepatocyte ballooning, apoptosis, and other pathophysiological changes that promote disease progression in NAFLD [26]. The liver controls the metabolism of fatty acids through several molecular mechanisms, including the uptake and export of fatty acids, de novo fatty acid generation, and fatty acid oxidation. When the balance between these pathways is altered, hepatic lipid accumulation commences, and long-term activation of inflammatory and fibrotic pathways can progress to worsen the liver disease [27].
Accumulation of lipids in hepatocytes is closely related to the interaction between liver and adipose tissue. The main lipid uptake for the liver is through lipolysis in adipose tissue, which releases fatty acids into the blood that are then taken up by the liver by membrane proteins called fatty acid transporters. The main features of adipose tissue dysfunction include enlarged or compressed adipocytes and chronic low-grade inflammation, which can lead to increased levels of lipolysis in adipocytes and increased free fatty acids production. Circulating free fatty acid can also be generated from the absorption of dietary. Several studies identified that the development of NAFLD is often associated with Western dietary patterns, particularly excessive dietary fat intake [28, 29]. It is well known that circulating free fatty acid pool in an obese state is held accountable for the majority of liver lipids in NAFLD. Uptake of circulating free fatty acid by hepatocytes is largely dependent on both the concentration of plasma fatty acids and the capacities of membrane-bound fatty acid transport proteins (FATPs), as well as cluster of differentiation 36 (CD36) [30, 31]. The significance of CD36 in the pathogenesis of NAFLD onset has been studied because modulation of its expression in the liver directly effects hepatic steatosis. A study exploring the role of CD36 in lipid metabolism revealed that localization of CD36 on the plasma membranes of hepatocytes and CD36 palmitoylation are markedly increased in the liver of mice with NASH, which promotes free fatty acid uptake by hepatocytes and inhibits fatty acid β-oxidation, leading to intracellular lipid accumulation and increased inflammatory response. Blockade of CD36 palmitoylation in mice with NAFLD decreased its localization on the hepatocellular plasma membrane and impaired its function as free fatty acid transporter, suggesting that CD36 or some of its functional regulators may be a promising therapeutic approach for the prevention and treatment of NAFLD [32]. CD36 can also interact with insulin-induced gene-2 (INSIG2) to increase the level of SREBP1 synthesis, promote de novo synthesis of fatty acid and induce hepatic steatosis [33]. Of the six mammalian FATP isoforms, FATP isoforms 2 and 5 are the major isoforms present in the liver. In a high-fat fed mouse model, knockout or knockdown of FATP2 and FATP5 reduces hepatocyte fatty acid uptake, the level of triglyceride deposition in the liver and resisted diet-induced obesity in mice, indicating a role of FATP-mediated lipid uptake as a facilitator of hepatic steatosis [34]. Following uptake, hydrophobic fatty acids do not diffuse freely in the cytosol and must instead be shuttled between different organelles by specific fatty acid binding proteins (FABP). FABP1, originally referred to as liver-FABP or L-FABP, is almost exclusively expressed in the liver. L-FABP promotes the intracellular transport of long-chain fatty acids and regulates cellular fatty acid uptake and lipid metabolism processes. It has been demonstrated that L-FABP levels are significantly increased in patients with NAFLD and can promote disease progression and insulin resistance processes [35]. Therefore, enhanced intracellular trafficking of fatty acids in the liver of NAFLD patients may be shunting harmful fatty acids to storage, thereby promoting steatosis.
One of the main sources of fatty acids is de novo lipogenesis, a pathway in which fatty acids are synthesized from non-lipid precursors. Aside from triglyceride synthesis from externally derived fatty acids, the liver can also synthesize fatty acids de novo from a range of carbon sourced such as glucose, amino acid, which are then diverted to lipid droplets for storage as triglyceride. Three essential enzymes, acetyl-CoA carboxylase (ACC), Fatty acid synthase (FAS), and stearoyl-CoA desaturase-1 (SCD1), regulate de novo lipogenesis in liver. Individuals with NAFLD have increased rates of de novo lipogenesis and this is a major factor contributing to increase lipid deposition. Consistent with these observations, the expression of ACC and FAS are increased in the liver of patients with NAFLD or NASH. In ob/ob mice fed a high-carbohydrate diet, transient genetic inhibition of ATP-citrate lyase (ACLY) reduces liver lipid content. By contrast, using in vivo isotope tracing, it was shown that liver-specific inhibition of ACLY in mice cannot suppress fructose-induced lipogenesis. Dietary fructose is converted to acetate by the gut microbiota, and this supplies lipogenic acetyl-CoA independently of ACLY. Depletion of the microbiota or silencing of hepatic acetate-CoA synthetase 2, which generates acetyl-CoA from acetate, potently suppresses the conversion of bolus fructose into hepatic acetyl-CoA and fatty acids [36]. The exact mechanism that drives de novo lipogenesis in NAFLD remains unknown, but activation of SREBP-1c and ChREBP due to increased circulating insulin and glucose in the context of whole-body insulin resistance has been suggested as a central driver [37]. While limited, the available data collectively indicate that failure to regulate de novo lipogenesis is a central feature of liver lipid accumulation in NAFLD patients. Increased de novo lipogenesis represents an important mechanism driving hepatic triglyceride accumulation in fatty liver disease.
Fatty acids can be oxidized by multiple pathways. In fact, hepatic lipid accumulation leads to a compensatory increased oxidation, which mainly involves mitochondria. Activation of PPARα that is expressed at high levels in liver induces the transcription of a range of genes involved in mitochondrial and extramitochondrial fatty acid oxidation, thereby reducing hepatic lipid levels, and both whole-body and hepatocyte-specific ablation of PPARα in mice lead to reduced transcription of hepatic genes related to mitochondrial β-oxidation. Notwithstanding the relationship between PPARα activity and hepatic lipid metabolism, studies of PPARα expression levels in patients with steatosis or NASH are conflicting, reporting unchanged, increased and decreased PPARα expression [38, 39]. The current data on fatty acid oxidation in NAFLD are conflicting, but even in studies suggesting augmented oxidation of fatty acids appear inadequate in clearing the liver of lipids. fatty acid oxidation in dysfunctional mitochondria—a characteristic of NAFLD—produces excessive ROS may ultimately facilitate disease progression by inducing oxidative stress and inflammation [40].
Toxic lipids deposited in the liver can lead to endoplasmic reticulum stress, mitochondrial damage to mediate hepatocyte injury and inflammatory responses [28, 41]. Several studies have reported that the ratio of unsaturated fatty acids to saturated fatty acids (SFA) in free fatty acids is closely related to the progression of NAFLD. The accumulation of unsaturated fatty acids has no significant effect on liver cell viability, but the hepatic accumulation of saturated fatty acids promotes apoptosis and induces liver damage [42]. Triglyceride molecules represent the major form of storage and transport of fatty acids within cells and in the plasma. NAFLD is characterized by excess triglyceride accumulation within the liver. Whereas previously it was considered that excess triglyceride stores in the setting of NAFLD contributed to lipotoxicity, emerging concepts suggest that increased triglyceride storage and is instead protective against fatty acids-mediated hepatotoxicity. It has been shown that inhibition of triglyceride synthesis reduces hepatic steatosis, but increases oxidative stress, inflammation levels and liver injury [43]. During NAFLD, specific lipids, such as palmitic acid, cholesterol and ceramides have a destructive effect on hepatocytes, and these lipotoxic substances can affect cellular function through various mechanisms, including endoplasmic reticulum stress, activation of death receptors and mitochondrial dysfunction [44-46]. Cholesterol homeostasis is mainly controlled by the liver. A balance between the availability of free and esterified cholesterol is critically important for hepatocellular function. When the synthesis and uptake of cholesterol exceed its removal, free cholesterol accumulates in hepatocytes. The role of cholesterol accumulation in NAFLD is an emerging topic, because it can further contribute to the alteration of the cellular function status. Free cholesterol overload in hepatocytes leads to ER stress, mitochondrial dysfunction, toxic oxysterols, and cholesterol crystallization in lipid droplets, thereby inducing hepatic cell death and inflammation [47, 48]. Statins inhibit the synthesis of lipid cholesterol and prevent NAFLD and its progression [49]. Although storage of triglycerides in the lipid droplets is a classical feature of NAFLD throughout the disease progression, triglycerides are presumed inert lipids that do not directly cause liver damage. Like cholesterol, ceramides play pivotal roles in all stages of NAFLD development, and in turn, lowering ceramides ameliorates NAFLD pathologies at multiple level [50-52].
Ceramides biosynthesis pathway
Ceramides are a class of bioactive sphingolipids consisting of a fatty acid chain that form the backbone of more complex molecules, such as glucosylceramides and sphingomyelins. Ceramides with specific chain lengths have differing effects on both physiological and pathophysiological [53, 54]. In addition to their structural role in the cell membrane, ceramides act as second messengers to coordinate various cellular processes, such as proliferation and apoptosis as well as stress response in health and disease [55, 56]. Ceramide production takes place by three different pathways: the de novo synthesis pathway, the sphingomyelin pathway, and the salvage pathway (Figure 1).
Three main pathways of ceramides synthesis in mammals. Ceramide is synthesized in three main ways, with de novo pathway occurring primarily in the endoplasmic reticulum, sphingomyelinase pathway occurring primarily in the lysosome, and salvage pathway occurring primarily in the Golgi apparatus. The three modes of synthesis work together to maintain the balance of ceramide metabolism in vivo.
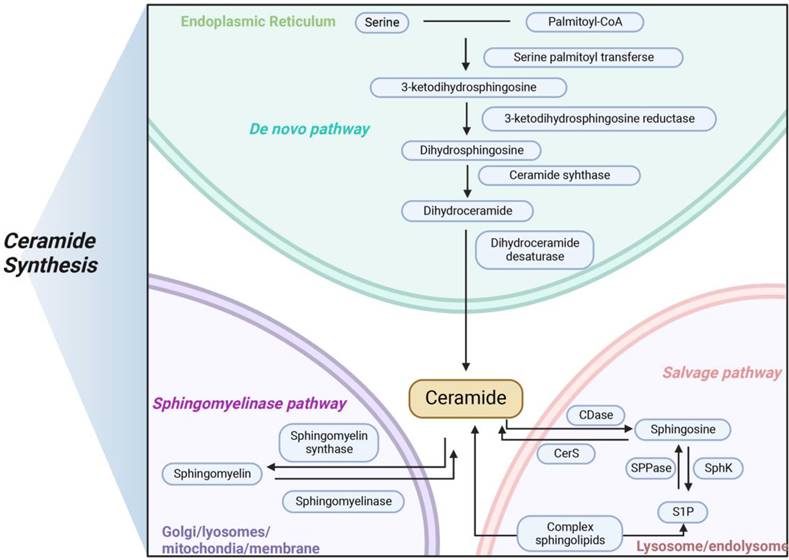
Ceramide de novo synthesis begins in the endoplasmic reticulum with the condensation of L-serine and palmitoyl-CoA by serine palmitoyltransferase (SPT) to form 3-ketosphinganine which is then converted to sphinganine by 3-ketosphinganine reductase in an NADPH-dependent manner. Ceramide synthases (CerS) attach acyl-CoAs with different chain lengths to sphinganine to form dihydroceramides [57]. Then, dihydroceramides are further dehydrogenated by dihydroceramide desaturase (DES) to form ceramide [58]. After de novo synthesis in endoplasmic reticulum, ceramides are either taken to the Golgi by vesicular transport to be glycosylated into glucosylceramides or delivered by ceramide transport protein. When ceramides reach the Golgi, they are modified with a polar head to form sphingomyelin. Several of the enzymes required for de novo ceramide synthesis, including SPT and CerS. In mammals, there are large subunits of SPT, SPTLC1, 2 and 3, with SPTLC1 pairing with either SPTLC2 or SPTLC3 to form an active, heterodimeric complex. Two essential subunits, SPTLC1 and SPTLC2, are required for enzyme function. Other subunits, such as SPTLC3, alter substrate specificity, allowing alternative fatty or amino acids to be incorporated to produce less abundant sphingolipid species. In mammals, six CerS isoforms, called CerS1-CerS6, are expressed, with each generating dihydroceramides with specific acyl chain lengths, although redundancies exist between the different isoforms [59]. These six CerS enzymes with a tissue-specific expression pattern, exhibit strict specificity in terms of the length of the fatty acid added to the sphingoid base and determine the fatty acid composition of sphingolipids in the cells [60, 61]. Excepting CerS3, five of the six CerS enzymes are expressed in the intestinal mucosal cells, leading to the generation of a diverse ceramide pool with variable acyl chain lengths ranging from C14 to C34. CerS2 is the most abundant CerS in hepatocytes which explains the observations that its product C24:1 ceramide is the most abundant ceramide species in the mouse liver [62, 63].
The second important metabolic pathway for the acute production of ceramides is the sphingomyelinase pathway. Sphingolipids and ceramides can be interconverted by the action of sphingomyelinase and sphingomyelin synthase. The phosphodiester bond in the middle of sphingolipids can be hydrolyzed by sphingomyelinase to produce phosphorylcholine and ceramides. Sphingomyelinases can be divided into three types according to the optimal pH differences and subcellular localization: neutral sphingomyelinase, alkaline sphingomyelinase and acid sphingomyelinase [64, 65]. While alkaline sphingomyelinase is mainly localized in the gastrointestinal tract and to some extent in the liver, neutral sphingomyelinase and acid sphingomyelinase are ubiquitous and account for the generation of ceramide in specific intracellular compartments, predominantly in the plasma membrane and lysosomes, respectively [66-68].
The salvage pathway of long chain sphingoid bases, leading to the re-generation of sphingolipids, has been estimated to contribute from 50% to 90% of sphingolipid biosynthesis. This pathway involves the degradation of glucosylceramide, sphingomyelin, and other complex sphingolipids, in acidic organelles such as the lysosome or late endosome, to form ceramides [69]. A growing body of evidence is starting to point toward roles for ceramide generated through the salvage pathway in many biological responses, such as cell growth arrest, cell signaling and transport [21]. Ceramides can be catabolized by acid ceramidases to form a sphingosine base and a free fatty acid chain, both of which are able to leave the lysosome, in contrast to ceramide which does not appear to leave the lysosome. The long chain sphingoid bases released from the lysosome may then re-enter pathways for synthesis of ceramide and/or S1P. In humans, five different ceramidases are known, which are differentially localized and are categorized according to their catalytic pH optimum: acid ceramidase (ASAH1), neutral ceramidase (ASAH2) and alkaline ceramidase (ACER1, ACER2, and ACER3). ASAH2 is highly expressed in the intestinal brush border membrane, ACER1 is often localized in the endoplasmic reticulum, ACER2 is mainly located in the Golgi complex, and ACER3 is widely expressed in the organism [70]. In summary, these ceramidases have different cellular and substrate preferences, working together to regulate the body's ceramide levels.
Liver ceramides contribution to NAFLD
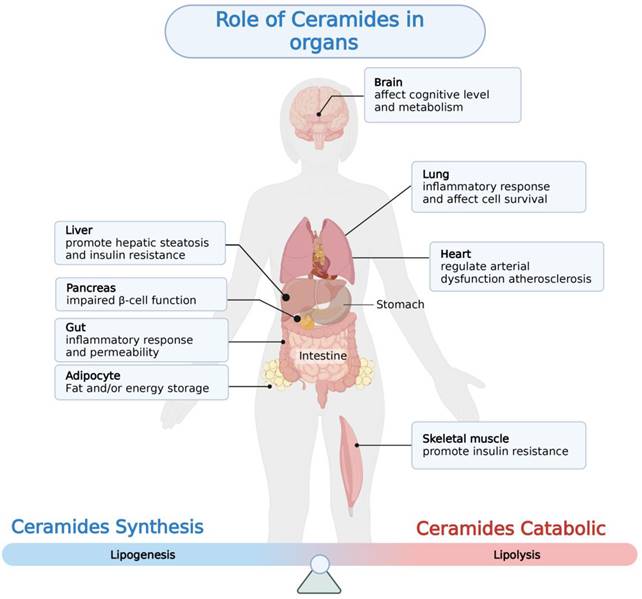
With the rapid development of lipidomics approaches using mass spectrometry, cohort studies have revealed strong correlations between serum and liver ceramides levels and NAFLD pathologies in humans. Aberrant accumulation of ceramide in tissues is thought to be a negative regulator of glucose tolerance and lipid metabolism, which can modulate pathophysiological processes in a variety of diseases including insulin resistance, cardiovascular disease, coronary atherosclerosis, and diabetes [71-74] (Figure 2, Table 1). Evidence suggests that ceramide contributes to key steps in the onset and progression of NAFLD (Figure 3).
Summary of studies on ceramide modulation in different disease.
| Diseases | Ceramide modulation | Reference Number |
|---|---|---|
| Alzheimer's disease | C18:1/18:0, C18:1/20:0↑ | [166] |
| Cer-C16:0, C18:0, C20:0, C24:0 | [167] | |
| Cer-C16:0, C18:0, C20:0, C24:0 | [168] | |
| Multiple sclerosis | plasma: C16-Cer, C24:1-Cer, C16-GlcCer, C24:1-GlcCer ↑ | [169] |
| cerebrospinal fluid: C16:0, C24:0-Cer ↑ | [170] | |
| Cardiovascular disease | Ceramide ratio: Cer18:1\/C16:0、Cer18/C18:0 and Cer18.1\/C24:1↑ | [171] |
| Cer16:0\/C24:0 ↑or Cer24:0\/C16:0 ↓ | [172] | |
| Type 2 diabetic subjects | C16:0-Cer, C18:0-Cer, C22:0-Cer and C24:0-Cer ↑ | [173] |
| Cer-C18:0, C20:0, C22:0, DHCer-C22:0 | [174] | |
| NASH | dihydroceramides (16:0, 22:0, and 24:1) and lactosylceramides↑ | [109] |
| Colorectal cancer | Cer-C16:0, C24:0, C24:1↑ | [175] |
| Rheumatoid arthritis | Cer-C16:0, C22:0, C23:0, C24:1 | [176] |
Aberrant ceramides accumulation in a variety of tissues can lead to the development of a variety of metabolism-related diseases. This figure depicts the pathological changes resulting from abnormal ceramide accumulation in different tissues.
Ceramides on hepatocytes, hepatic stellate cells, macrophages and hepatocellular carcinoma cells in the progression of NAFLD.
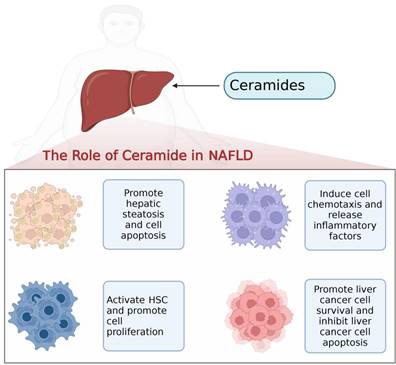
Roles of ceramides in NAFLD
NAFLD represents a major chronic liver disease worldwide, characterized as progressive and developing in different stages. While simple steatosis follows a relatively benign clinical course, in some cases it may progress to NASH; a state characterized with cell death and chronic inflammation [75, 76]. The onset and progression of NAFLD is associated with chronic elevation of ceramide content in hepatocytes. The emerging pathogenic roles of abnormal accumulation of ceramides in NAFLD, including inducing insulin resistance, promoting apoptosis, increasing endoplasmic reticulum stress and mitochondrial oxidative stress, and ultimately leading to steatosis, inflammation and fibrosis, have been demonstrated [25, 77, 78]. One recent clinical study has reported that ceramides and other sphingolipid levels are significantly elevated in the liver of patients with NAFLD and NASH, and that ceramide levels are positively correlated with the degree of oxidative stress and inflammation in the liver [79]. Similarly, Theodore et al. also found that ceramides were highly enriched in inflammatory steatosis hepatocytes by using in single-cell metabolomic analysis of the metabolic state of fatty acid-stimulated human hepatocyte lines, with the results validated in vivo and in line with previous reports [80]. Based on these findings, ceramides have been shown to serve as biomarkers of NAFLD. The function of ceramides in NAFLD is also closely related to the length of the contained acyl chains. Wang et al. found that C16-ceramide synthesized by CerS5 and CerS6 promotes endoplasmic reticulum stress, leading to decreased liver function and metabolic imbalance in patients [81]. However, CerS2-mediated synthesis of ultra-long chain ceramides protects against palmitic acid-induced lipotoxic endoplasmic reticulum stress in hepatocytes and attenuates insulin resistance levels and hepatic steatosis [82]. In the presence of excess energy, excessive intake of saturated fatty acids increases plasma and intrahepatic ceramide levels more than polyunsaturated fatty acids [83-85]. In the high-fat-fed mice model, the level of intrahepatic ceramides deposition in mice was significantly increased compared to the control group [86], implying that using different dietary patterns could regulate the level of ceramides. In addition, Dong et al. found that compared with control mice, hyperhomocysteinemia (HHcy) mice exhibited hepatic steatosis with a notable increase in ceramide-related metabolites and subsequent upregulation of ceramide synthesis genes. Furthermore, downregulation of ceramide levels through omega-3 polyunsaturated fatty acid supplementation ameliorates hepatic lipid accumulation, thus suggesting that ceramide may be a potential therapeutic target for the treatment of patients with hepatic steatosis [87]. In addition, induction of liver-specific overexpression of acid ceramidase or deletion of CerS6 expression can also reduce ceramide levels, normalize lipid uptake and sphingolipid metabolism, and improve diet-induced hepatic steatosis [88].
Ceramides induced hepatic insulin resistance
The pathophysiological mechanism of hepatic insulin resistance is very complex. On the one hand, accumulation of various lipids in the liver are key drivers of systemic and tissue-specific insulin resistance. On the other hand, tissue inflammation can induce insulin resistance [89, 90]. Early investigations of ceramide-induced insulin resistance were performed primarily in skeletal muscle, and more recently, studies have confirmed that ceramides have been explored as an important mediators of insulin resistance in adipose and liver [20, 91-93] (Figure 4). Nevertheless, there are some contradictory findings on the role of ceramides in the induction of insulin resistance. In a study of obese non-diabetic patients, hepatic ceramide levels were not significantly correlated with insulin resistance [94]. However, Hady et al. found a significant accumulation of intrahepatic ceramide in obese patients with diabetes, which was positively correlated with blood glucose levels, suggesting that ceramide may contribute to the induction of hepatic insulin resistance [95]. In animal experiments, Xia et al. using overexpressing acid ceramidase mice found that the overexpressed acid ceramidase to consume excess ceramide in the liver, reduces intrahepatic ceramides deposition and improves hepatic insulin resistance [88]. A recent study by Chaurasia et al. using ablation of the enzyme dihydroceramides desaturase 1 (DES1) from whole animals or tissue-specific deletion in the liver and/or adipose tissue found that inhibition of DES1 resolved hepatic steatosis and insulin resistance in mice caused by leptin deficiency or obesogenic diets, enhanced activation of AKT/PKB in vivo, and elicited a broad spectrum of metabolic benefits characterized by improvements in glucose and lipid handling. Meanwhile, ceramides also slow fatty acid turnover by increasing fatty acid storage in hepatocytes and reducing fat burning in adipose tissue. These findings highlight the role of ceramides in hepatic insulin resistance and also indicate that DES1 as a target that could be used to develop new treatments for various metabolic disorders [96].
Ceramides can be involved in the inhibition of insulin signaling. Ceramide is able to inhibit AKT signaling pathway through both PP2A and PKCζ mechanisms and Inhibition of IRS-1 phosphorylation and the function GLUT4. Ceramide can also be involved in processes such as endoplasmic reticulum stress and mitochondrial dysfunction. Adiponectin released from adipose tissue can also be involved in the regulation of ceramide synthesis.
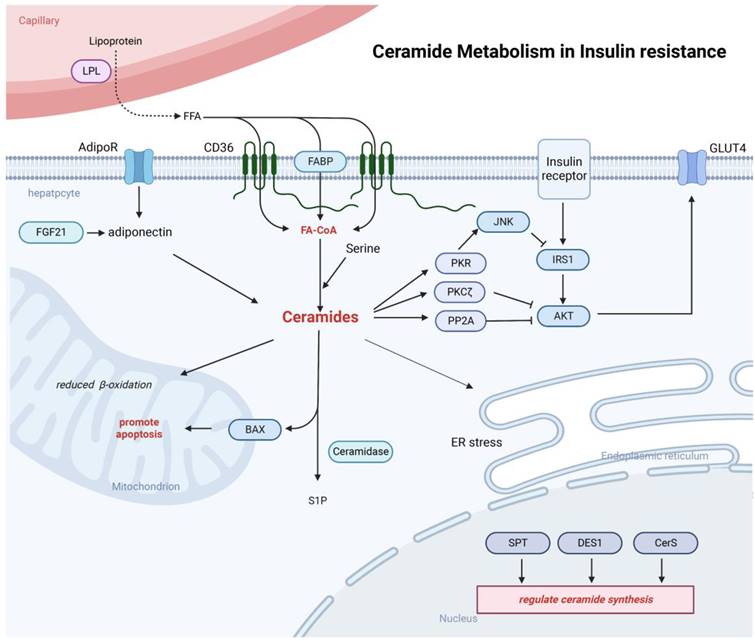
In the liver, the ceramide mechanism of action on insulin resistance has not been fully elucidated. Several mechanisms have been proposed to explain how ceramides might induce insulin resistance, including: 1) ceramides impair insulin activation of AKT through two mechanisms: impaired AKT translocation via activation of PKCζ or activation of PP2A. Activation of PKCζ by ceramide can interact with AKT to induce AKT chelation into specific regions of the plasma membrane called caveolae and prevent AKT recruitment to the plasma membrane [97]. Ceramides can also induce insulin resistance by activating PP2A, consequently hampering translocation of AKT from the cytoplasm to the plasma membrane. It has been shown that inhibition of PP2A in rats during AKT activation promotes hepatic insulin resistance, suggesting that insufficient activation of PP2A can also induce the development of insulin resistance [98]; 2) In hepatocytes and muscle, ceramide uses an alternative pathway in modulation of insulin signaling that requires the phosphorylation and activation of PKR. Usually, insulin inhibits PKR activity by suppressing its phosphorylation. In liver cells, ceramide-activated PKR interferes with the insulin signaling pathway by phosphorylating IRS1 at Ser312 in an IKK/JNK-dependent manner, thus preventing the binding of IRS1 to PI3K [99]; 3) a third pathway relates to the link between ceramides and GLUT4. Ceramide may inhibit glucose signaling by forming microstructural domains in phosphatidylcholine membranes, causing changes in plasma membrane fluidity, and avoiding GLUT4 fusion into the plasma membrane, thereby reducing glucose uptake and promoting insulin resistance. In addition, ceramide can also regulate the insulin resistance process by affecting free fatty acids, inflammatory cytokines, and glucocorticoids in various ways [100, 101]. Adiponectin, a common adipocyte secretory factor, can enhance insulin sensitivity, increase ceramidase activity, and promote ceramide catabolism [102]. In rodents, adiponectin could be activated by the metabolically active hormone fibroblast growth factor 21 (FGF-21), while diminishing accumulation of ceramides in obese animals. Moreover, FGF-21 lowers blood glucose levels and enhances insulin sensitivity in diabetic mice and diet-induced obese mice, only when adiponectin is functionally present [103].
Altering the acyl chain composition of ceramides may be a novel way of modulating insulin resistance and glucose metabolism. CerS1 and CerS6 induced ceramides of specific acyl chain lengths can lead to skeletal muscle insulin resistance, while ultra-long chain ceramides can improve the body's glucose metabolism levels [82, 104]. One previous study has demonstrated that the level of C18:0 ceramide production in skeletal muscle was reduced in mice lacking CerS1, which effectively prevented obesity-induced insulin resistance [104]. CerS2 null mice, which is unable to synthesize very long acyl chain (C22-C24) ceramides, exhibited glucose intolerance despite normal insulin secretion from the pancreas [19, 105].
Role of ceramides in NASH
Pathological NASH develops when fat accumulation in hepatocytes is characterized by lobular inflammation and hepatocellular ballooning. The final stages of NASH are advanced fibrosis/cirrhosis, hepatocellular carcinoma, and liver failure. NAFLD has a strong association with obesity, type 2 diabetes, and hypertension, and is thus considered the hepatic manifestation of the metabolic syndrome. Inflammation, the central feature in the progression from simple steatosis to NASH, is subsequently triggered by metabolic overload and ensuing hepatocyte injury [106-108]. Increasing intrahepatic ceramide levels in patients with NASH remains somewhat controversial. One study has indicated that hepatic ceramides are not only increased in insulin-resistant humans with NASH but also correlate with hepatic oxidative stress and inflammation, suggesting that intrahepatic ceramide levels may play a role during progression of simple steatosis to NASH in humans [109]. However, another recent study has indicated that NAFLD is characterized by distinct changes in the liver lipidome with steatosis and the development of NASH does not result in further changes in the lipidome [79]. Although ceramides accumulation in the liver and the progression of NASH are closely related, the underlying mechanisms by which ceramide involved in the different steps of NAFLD progression remain unclear. During the progression of NASH, saturated free fatty acids induce sublethal and lethal hepatocyte toxicity, in which endoplasmic reticulum stress is an important manifestation of sublethal hepatocyte toxicity. To cope with ER stress, the unfold protein response (UPR) is activated. UPR is a feedback regulatory system in which the endoplasmic reticulum detects the pressure of unfolded proteins in the lumen of the endoplasmic reticulum through intracellular signaling and is able to control the elimination of misfolded proteins in the endoplasmic reticulum to restore endoplasmic reticulum homeostasis. The unfolded protein mainly consists of three signaling branches containing inositol requiring enzyme-1 (IRE1), activating transcription factor 6 (ATF6), and protein kinase RNA-like endoplasmic reticulum kinase (PERK). In vitro model of palmitate-induced NASH, lipotoxic hepatocytes release extracellular vesicles (EV) containing ceramides mainly in an endoplasmic reticulum sensor-dependent manner to promote macrophage chemotaxis, and the use of IRE1 inhibitors or knockdown of its expression level reduces NASH inflammation as well as release of EVs. Activated IRE1A promotes the transcription of SPT through X-box binding protein 1 (XBP1), induces ceramides biosynthesis and EV release, thereby exacerbating the level of inflammation and the degree of liver injury in vivo. Meanwhile, the levels of XBP1, SPT and EV in the liver in NASH patients are significantly increased [52]. Besides, innate immune abnormalities in the liver also play an important role in the pathogenic mechanisms of ceramides in NAFLD. Palmitic acid stimulates hepatocytes to produce S1P-rich EVs, and during this process, sphingosine kinases 1 and 2 are involved in regulating S1P enrichment levels in EVs, and then S1P exacerbates the level of inflammation in the liver by binding to S1P receptors on the surface of macrophages [110]. Neutrophil aggregation is an early event in the NASH mouse model. In contrast to the beneficial effects during infection, neutrophils usually have deleterious effects on NASH through the production of ROS, cytokines, proteases and neutrophil extracellular traps (NETs). Neutrophil elastase (NE), another active protease secreted by neutrophils, has been shown to improve diets induced obesity and insulin resistance in mice. NE knockout improved western diet induced obesity, serum lipid disorders, steatosis and liver inflammation in NASH mice, which was possibly attributed to the potential of NE in regulating hepatic ceramides metabolism [111].
Ceramides have been reported as effectors in autophagy regulation, and are known to mediate apoptotic pathways. Thus, ceramides may play an important regulatory role in the autophagy function in the pathogenesis of NASH. However, the role of ceramides in autophagy is controversial. Ceramides were reported to either promote early autophagy and apoptosis or attenuate autophagy [112, 113]. Noticeably, blockade of ceramide synthesis by myriocin could alleviate NASH severity, and improve the serum transaminases, triglycerides, cholesterol, liver pathology and fatty acid metabolism. Additionally, myriocin could also reverse the impaired autophagy function, indicating an important regulatory role of ceramides in autophagy function in the pathogenesis of NASH [114]. Acid sphingomyelinase, a specific mechanism of ceramide generation, is required for the activation of key pathways that regulate hepatic steatosis and fibrosis, including endoplasmic reticulum stress and autophagy, and targeting acid sphingomyelinase can increase methionine cycling and phosphatidylcholine metabolism to slow the progression of NASH [115]. Unlike the short-chain ceramides described above that exacerbate NASH, the available evidence suggests that long-chain ceramides promote hepatocyte survival. Wang et al. found that ACER3 is upregulated in livers of patients with NASH and in mouse livers with NASH induced by a palmitate-enriched western diet (PEWD). Knocking out Acer3 was found to augment PEWD-induced elevation of C18:1-ceramide and alleviate early inflammation and fibrosis but not steatosis in mouse livers with NASH, suggesting that targeting ACER3 represents a novel approach to prevention and intervention of NASH [116]. Another study has shown that treatment with exogenous C24-ceramide also inhibited lipid synthesis and reduced intrahepatic lipid accumulation in a high-fat mouse model [117]. Overall, most experiments have confirmed that excessive deposition of ceramides in the liver can promote NASH progression, exacerbate the level of liver inflammation, and promote oxidative stress and liver injury. However, due to the complexity of ceramide function, the exact mechanisms of its role in the different steps of NAFLD progression require further investigation.
Role of ceramides in liver fibrosis
Liver fibrosis, which is manifested as excessive accumulation of extracellular matrix (ECM) proteins, is a common sequela to diverse chronic liver insults. Liver fibrosis is characterized by activation and proliferation of hepatic stellate cell (HSC), which are the major source of matrix-producing myofibroblasts [118]. Compared to the roles in steatosis and liver inflammation, the role of ceramides in liver fibrosis is less clear. Liver fibrosis is accompanied by the hepatic accumulation of ceramide and sphingolipids, and excess ceramide regulates HSC activation and promotes hepatocyte apoptosis. However, the specific role of ceramides in liver fibrosis remains controversial. On the one hand, abnormal hepatic accumulation of ceramides has been observed in several mouse models of liver fibrosis, and as ceramide accumulates, cells undergo apoptosis and trigger the fibrotic events. For example, in mice on which fibrosis was induced with carbon tetrachloride treatment, the degree of fibrosis and inflammation in the liver is positively correlated with circulatory and intrahepatic ceramide levels [55]. Activated HSCs produce ceramides via upregulation of key enzymes of ceramides synthesis pathway, and in turn, ceramides can activate HSCs and promote those cells to differentiate into profibrotic myofibroblasts, resulting in liver fibrosis [119]. On the other hand, excess ceramides in HSCs attenuated liver fibrosis in mice. Pharmacologic inhibition or genetic knockout of acid ceramidase in HSC could inhibit YAP/TAZ signaling pathway activity, increase ceramides accumulation in HSCs and prevent liver fibrosis, implicating ceramide as a critical regulator of YAP/TAZ signaling and HSC activation and highlighting acid ceramidase as a therapeutic target for the treatment of fibrosis [120]. Thus, ceramide is believed to be important in regulating apoptosis of hepatocytes. It is also a potential, major target for the treatment of NASH and fibrosis.
Dihydroceramide and S1P play also important roles in the progression of liver fibrosis. Sphingosine kinase is a key regulator of the dynamic ceramide/S1P rheostat balance. Ceramide usually inhibits proliferation and promotes apoptosis, while the further metabolite S1P stimulates growth and suppresses apoptosis. Repeated injections of purified hematopoietic stem cells markedly reduced liver fibrosis and improved liver function, which was increased by the co-administration of FTY720, a partial S1P receptor agonist, which enhanced the retention of hematopoietic stem cells inside the liver [121]. In addition, increased dihydroceramide led to impairment of autophagic flux, and aggravated the progression of liver fibrosis. Due to the complex role of ceramide in liver fibrosis, targeting ceramide and its precursors could be one of the therapeutic approaches for liver fibrosis. Treatment with rapamycin recovered autophagic flux by regulating dihydroceramide could be a potential strategic approach for providing therapy for liver fibrosis [58]. Chronic treatment with myriocin inhibited ceramide and lipid accumulation and improved liver fibrosis in the HFD-fed rat model, suggesting that ceramide synthesis could potentially be a target for the treatment of NAFLD [55].
Role of ceramides in liver cancer
Metabolic alterations are a well-established hallmark of cancer. Deregulated lipid metabolism has been strongly correlated with the onset and progression of liver cancer. Ceramide not only regulates the differentiation, proliferation and apoptosis of tumor cells, but also mediates the inflammatory response in tumors [122, 123]. Li et al. found that ceramide levels decreased but sphingomyelin level increased in hepatocellular carcinoma tissues by examining lipid expression in patients [124]. But Bao M et al. found that ceramides, S1P, SM levels and species were significantly increased in hepatocellular carcinoma [125]. These two paradoxical results may be due to changes in the signaling cascade of multiple cells during the development of liver cancer, resulting in dysregulation of enzyme expression related to ceramide metabolism. Ceramides are closely related to cell apoptosis and autophagy in tumors. In liver cancer, melatonin induced cellular autophagy by regulating the level of JNK phosphorylation, while specific acid sphingomyelinase-induced ceramide production participates in melatonin-mediated liver cancer cell death, suggesting that ceramide can modulate melatonin-induced autophagy and apoptosis in liver cancer cells [126]. Systemic administration of nano-liposomal C6-ceramide to mice engrafted with liver cancer reduced tumor vascularization, induced tumor cell apoptosis, and ultimately blocked tumor growth [127]. Ceramide can also regulate hepatocellular carcinoma cell migration. ACER2, one of the key enzymes that regulates ceramide hydrolysis, is overexpressed in hepatocellular carcinoma tissues and cell lines, and ACER2 knockdown resulted in decreased tumor cell growth and migration. Mechanically, ACER2 positively regulated the protein level of sphingomyelin phosphodiesterase-like 3B (SMPDL3B). Of note, ACER2/SMPDL3B promoted ceramide hydrolysis as well as S1P production that induced tumor cell growth and migration [128]. As mentioned above, acid sphingomyelinase regulates the homeostasis of sphingolipids, including ceramides and S1P. Acid sphingomyelinase in the liver inhibits tumor growth through cytotoxic macrophage accumulation and tissue inhibitor of metalloproteinase 1 production by hepatic myofibroblasts in response to S1P [129].
The functional sphingolipid rheostat between ceramide and S1P has been well characterized in cancer therapy. Notably, the exogenous addition of acid sphingomyelinase or ceramide has been shown to augment the anti-tumor efficacy of sorafenib [130]. Further data showed an upregulation of ceramide-induced cell death upon the stimulation of tumor cells with sorafenib and vorinostat [131]. In addition, nano-liposome C6-ceramide (LipC6) in combination with anti-CTLA4 antibody represents a novel therapeutic approach with significant potential in activating anti-tumor immune response. In mice with liver cancer, injection of LipC6 alone can reduce the amount of TAM in the tumor, enhance the anti-tumor immune response and inhibit the growth of hepatocellular carcinoma, suggesting that LipC6 might increase the efficacy of immunotherapy [132]. Tellingly, LipC6 in combination with anti‐CTLA4 antibody, but not anti‐PD‐1 antibody, significantly slowed tumor growth, enhanced tumor‐infiltrating CD8+ T cells, and suppressed tumor‐resident Tregs, representing a novel therapeutic approach with significant potential in activating anti-tumor immune response [133].
Role of ceramide in organ-crosstalk
Ceramides in adipose-liver crosstalk
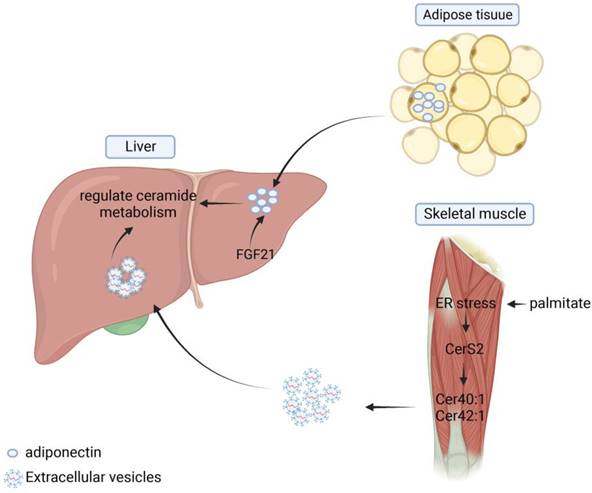
Metabolic syndromes, including NAFLD, can induce impaired adipose tissue function and release lipids to other organs, further leading to altered metabolic function, such as hepatic steatosis. Several studies have shown that there is organ-organ crosstalk in hepatic steatosis and that various lipids such as ceramides play a mediating role between different tissues and help maintain the homeostasis of the body's internal environment. In adipose tissue, inhibition of ceramide synthesis or promotion of ceramide hydrolysis can reduce the level of inflammation and hepatic steatosis within adipose tissue and improve systemic metabolic disturbances [134]. Xia et al. showed that induction of acid ceramidase overexpression in adipose tissue reduced the level of hepatic steatosis and alleviated insulin resistance more rapidly and significantly than direct overexpression of the enzyme in the liver [88]. Turpin's research group analyzed the differences in sphingolipid enrichment in different organs and tissues. The result showed that C16:0 was predominantly distributed in human adipose tissue, but that both C16:0 and CerS6 were significantly elevated in adipose and liver tissues in the obese population and in HFD-induced mouse models. In liver-specific CerS6 knockout mice, researchers found that mitochondrial β-oxidation and hepatic lipid metabolism levels were significantly increased in BAT, which not only improved energy supply but also reduced hepatic steatosis and insulin resistance [82]. In addition, adiponectin secreted and produced in adipose tissue regulates glucose and lipid homeostasis by exerting pleiotropic effects on liver, pancreatic beta cells, kidney, heart and other tissues [102]. Via adiponectin receptors, AdipoR1 and AdipoR2, adiponectin stimulates the deacylation of ceramide to produce S1P, thereby reducing ceramide accumulation and improving hepatic steatosis [101]. FGF21 is a member of the fibroblast growth factor (FGF) superfamily, which increases the level of glucose and lipid utilization and regulates energy metabolism. William et al. suggested the existence of adiponectin-FGF21-ceramidase axis, the specific process of which is that FGF21 regulates the process of sphingolipid metabolism by inducing the level of lipocalin expression up and decreases ceramide levels, regulating sphingolipid metabolic processes (Figure 5) [103].
The role of ceramides in organ crosstalk. Adipose tissue can secrete adiponectin and thus regulate ceramide metabolism in the liver. Skeletal muscle can release extracellular vesicles containing Cer40:1 and Cer42:1 under endoplasmic reticulum stress conditions. These EVs can be absorbed by liver and regulate ceramide metabolism.
Ceramides and gut-liver axis
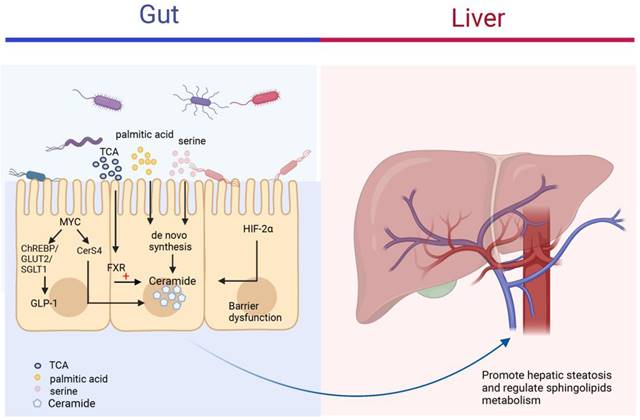
The intestine plays an important role in the body's metabolism and physiology by transporting derived products to the liver with the help of the portal vein, which affects the pathophysiological functions of the liver [135, 136]. When the microenvironment in the intestine is altered, the intestinal flora is involved in liver disease progression by activating various signaling pathways, such as FXR, hypoxia-inducible factor 2α, which directly or indirectly regulate ceramide synthesis and affect blood circulation and intrahepatic levels of fatty acids, ceramide and other sphingolipids (Figure 6) [137, 138]. The level of ceramide synthesized within the intestinal flora varies somewhat under different dietary conditions. Di et al. gave germ-free mice, normal mice, and low germ mice fed linoleic or α-linolenic acid and analyzed the differences in circulating and intrahepatic fatty acid and sphingolipid deposition levels. The results showed that mouse intestinal microflora modulated circulating sphingolipid content and influenced the enrichment of Cer(d18:1/20:0), Cer(d18:1/24:1), and SM(d18:1/24:1) in the liver [139]. In Asah2-/- mice using a combination of dietary models of NAFLD/NASH, IgA secretion levels were altered in the intestine of Asah2-/- mice compared to control mice, leading to increased synthesis of IgA-associated bacteria and their derivatives, decreased inhibition of sphingolipid synthesis, and improved hepatic inflammatory response [140]. Different gut microbiota also differs in their effects on ceramide, lipids produced by Bacteroidetes can be transferred to the hepatic portal vein; Bacteroides thetaiotaomicron can increase ceramide levels in the liver and regulate sphingolipid metabolism [141]. FXR is a nuclear receptor for bile acid specific response that regulates metabolic homeostasis in the intestine. Studies have shown that taurine-conjugated bile acids (TCA) can inhibit the process of small intestinal ceramide de novo synthesis and slow down the NAFLD process by inhibiting FXR [142]. In intestine-specific FXR knockout mice, the expression of ceramide synthesis genes SMPD3/4, SPTLC2 and CERS4 were suppressed in the intestine, and ceramide synthesis levels were reduced, which contributed to the improvement of HFD-induced obesity, insulin resistance and hepatic steatosis [143], MYC has an important role as a proto-oncogene in cell proliferation, apoptosis and metabolic processes. The increased expression of intestinal MYC was observed in both obese people and mouse models, suggesting that intestinal MYC may be involved in the development of obesity-related metabolic syndrome. Luo et al. found that intestinal MYC not only regulates glucagon-like peptide-1 (GLP-1) secretion through ChREBP and GLUT2/SGLT1, but also regulates CerS4 expression and thus promotes ceramide synthesis. Intestinal-derived GLP-1 and ceramide enter the circulation as metabolic signals to the liver and various organs throughout the body, affecting the development of metabolic diseases such as obesity, insulin resistance, and fatty liver [144].
The roles of ceramides in the gut-liver axis. The figure mainly depicts the different mechanisms by which the different contents of the intestine can regulate ceramide synthesis. Gut microbiota can increase the risk of NAFLD through the synthesis of ceramide by uptake of palmitic acid and serine in the intestine and transfer to the liver via the portal vein to regulate sphingolipid metabolism in the liver. FXR and HIF-2α in the gut also regulate ceramide levels in the intestine and circulation.
Ceramides in other organ-liver crosstalk
Ceramides are mostly synthesized in the liver and affect sphingolipid metabolism in several organs throughout the body via the blood circulation. In cardiovascular disease, ceramides could cause endothelial dysfunction, affect the release of inflammatory factors and metalloproteinases, and promote atherosclerosis formation. Ceramides of different lengths have similar effects in cardiac insufficiency and liver diseases. High levels of C16:0 ceramide is detrimental to cardiac function, while very long chain ceramide C24:0 reduces the risk of cardiac events. Treatment with myriocin reduced high levels of ceramide-induced atherosclerosis and decreased hepatic lipid aggregation in mice [53]. CYP7A1 in the liver is the rate-limiting enzyme in the bile acid synthesis pathway that can influence the progression of atherosclerosis. FGF15/19 binding to FGF receptor 4 could inhibit CYP7A1 expression in the liver and reduce circulating ceramide levels, which plays an important role in the prevention of obesity, insulin resistance and fatty liver disease [145]. Except for cardiovascular, ceramides also play an important role in the metabolic interaction between skeletal muscle and the liver. Palmitate-induced cellular stress activates the de novo synthesis pathway of CerS2, inducing secretion of the long-chain ceramides Cer40:1 and Cer42:1. Ceramides are packaged into EV, secreted through the accumulation of dihydroceramide and induce UPR activation in naïve myotubes. Palmitate-induced endoplasmic reticulum stress in skeletal muscle can regulate systemic sphingolipid metabolism and mediate lipotoxic endoplasmic reticulum stress in adipose tissue and liver by activating CerS2 expression, promoting the accumulation of long-chain ceramides, and the release of EV containing ceramides [73]. During obesity, skeletal muscle exhibits excess storage of various bioactive lipids, including diacylglycerol and ceramides, and by measuring changes in ceramide species in the skeletal muscle of dietary obese mice, researchers found increased levels of C18:0, which enhances skeletal muscle insulin resistance and accelerates the development of metabolic diseases [104].
Ceramide and Sphingolipid metabolism as therapeutic target for NAFLD
Given the role of ceramides in the onset and pathogenesis of NAFLD, therapeutic approaches that target ceramide production could thus be of interest, including inhibition of ceramide synthesis or reduction of ceramide deposition levels. Next, we will further outline the drugs involved in ceramide as a therapeutic target for NAFLD (Table 2).
Drugs targeting ceramide and Sphingosine-1 Phosphate for NAFLD.
| Types | Targets | Reference Number |
|---|---|---|
| P053 | A selective inhibitor of ceramide synthase 1 | [104] |
| Myriocin | The inhibitor of SPT | [150, 177] |
| CB1R | Inhibit the activator of SPT and expression of CerS1 and CerS6 | [155] |
| Fenofibrates | lowering plasma triglycerides (TG) and ceramide | [156] |
| exenatide | The mimics of GLP-1 | [160] |
| Liraglutide | The activator of GLP-1 | [159] |
| K145 | The inhibitor of SPHK2 | [153, 154] |
| NCT | The activator of HNF-4α | [178] |
| lipofermata | The inhibitor of FATP2 | [179] |
| hepano | ayurvedic polyherbal formulation | [157] |
| saroglitazar | a dual PPAR α/γ agonist | [157] |
| Fenretinide | Target DES and reduce the de novo synthesis of ceramide | [180, 181] |
| Fumonisin B1 | The inhibitor of CerS | [146, 147] |
| Safingol | The inhibitor of SPHK1 | [181] |
| LipC6 | containing short chain C6-Ceramide | [132] |
Direct inhibition of ceramide synthesis process
Because of the closely correlation between ceramide deposition levels and NAFLD progression, inhibition of ceramide synthesis and reduction of ceramide key enzyme expression can improve NAFLD progression and stop the onset of a range of disease endpoints in NAFLD. A variety of ceramide synthase inhibitors have been identified that are able to target different CerS isoforms separately. Fumonisin B1, which was isolated from a fusariotoxin family associated with human esophageal cancer pathogenesis, was one of the first CerS inhibitors to be discovered and has a significant inhibitory effect on ceramide synthesis. However, fumonisin B1 is hepatotoxic, which leads to some limitations in its clinical application [146, 147]. Turner et al. found that the selective CerS inhibitor P053 specifically inhibited CerS1 expression, thereby reducing C18:0 ceramide levels in cells and mouse skeletal muscle and improving insulin resistance [104]. Another potential mechanism for reducing ceramide synthesis is the inhibition of the expression of the ceramide precursor which is dihydroceramide. Veeriah et al. found that administration of the potent HNF-4α agonist N-trans-caffeoyltyramine led to increased dihydroceramide levels by inhibiting dihydroceramide conversion to ceramides and promoted reversal of hepatic steatosis through a mechanism involving the stimulation of lipophagy by dihydroceramides [148]. LipC6 as a non-toxic hydrophilic liposome carrier has been found to be clinically useful in the treatment of hepatocellular carcinoma. Recent studies have shown that LipC6 can reverse the imbalance of liver lipid metabolism induced by MCD feeding and reduce liver lipid deposition and inflammation levels by regulating AMPK/Nrf2 pathway and protecting antioxidant signaling pathway in a model of NAFLD/NASH [149].
Indirect inhibition of ceramide production by inhibiting sphingolipid metabolism
SPT is a key enzyme in ceramide synthesis, and reduction of ceramide synthesis can be achieved by inhibiting SPT. Myriocin is a widely studied SPT inhibitor that irreversibly inhibits SPT expression and reduces ceramide levels, playing an important role in the treatment of diabetes, hepatic steatosis and atherosclerosis [150, 151]. Interestingly, the novel immunomodulatory molecule FTY720, an analogue of myriocin, does not inhibit SPT, but is able to bind to S1P receptors, inhibit S1P lyase expression and participate in the regulation of sphingolipid metabolic homeostasis [152]. In addition to the widely known myriocin, researchers have identified various other SPT inhibitors such as 3-(2-amino-ethyl)-5-[3-(4-butoxyl-phenyl)-propylidene] -thiazolidine-2, 4-dione (K145), cannabinoid-1- receptor (CB1R), etc. In NAFLD, K145 significantly improved fat accumulation in the liver of mice, and more importantly, it had no effect on lipid accumulation in normal liver [153, 154]. CB1R inhibitors inhibit SPT activity while also suppressing CerS1 and CerS6 expression and reducing intrahepatic ceramide content [155].
Suppression of free fatty acid (FFA) levels
Excessive deposition of FFA in vivo can promote ceramide synthesis in cells and tissues. Inhibition of FFA release may become one of the ways to regulate ceramide and mitigate the progression of metabolism-related diseases. Fenofibrate is a common lipid-regulating drug, and a recent study has shown that fenofibrate can slow the development of chronic inflammation by inhibiting hepatic ceramide synthase and reducing the accumulation of ceramides and lipids in the plasma and liver [156]. In clinical studies, both saroglitazar and hepano were used for the treatment of hyperlipidemia and showed good inhibitory effects on ceramide. In addition, hepano has a superior modulating effect on sphingomyelins than saroglitazar, which has a significant hepatoprotective effect [157].
Target to glucagon-like peptide-1 (GLP-1)
Liraglutide is a type of glucagon-like peptide-1 receptor agonist (GLP-1Ra) that acts directly on hepatocytes to alleviate hepatic steatosis [158]. GLP-1Ra limit food intake and weight gain, additional beneficial properties in the context of obesity/insulin-resistance. Liraglutide could prevent accumulation of C16 and C24-ceramides/sphingomyelins species, inflammation and initiation of fibrosis in MCD-diet-fed mice liver, suggesting beneficial hepatic actions independent of weight loss and global hepatic steatosis [159]. Other GLP-1Ra can also reduce ceramide synthesis, for example, exenatide can regulate lipid levels in type 2 diabetic patients based on improving glucose metabolism and insulin resistance, and significantly reduce circulating levels of sphingomyelin, ceramide, phosphatidylcholine and other lipids [160].
Modification of dietary interventions and exercise
Both dietary interventions and changes in exercise patterns can reverse the elevation of ceramide in obesity, NAFLD, and other metabolic syndromes. Previous studies have confirmed that long-term adoption of Western diet, especially with high lipid intake, can promote the evolution of NAFLD, leading to elevated circulating and intrahepatic ceramide levels [85]. In a cohort study of dietary interventions, Wang et al. found that plasma ceramide concentrations in patients were positively associated with cardiovascular disease risk, and that adoption of the Mediterranean diet attenuated elevated plasma levels of ceramide and delayed disease progression [161]. Exercise or lifestyle changes can also reduce the accumulation of ceramides [162]. After NASH patients received 1 year of lifestyle intervention to achieve weight loss, transcript levels of SPTLC2 and CERS1 in the liver were significantly reduced compared to controls without lifestyle intervention, and ceramide synthesis levels were reduced, reversing the NASH process [163]. High resistance exercise and aerobic exercise can significantly improve liver and skeletal muscle insulin resistance, hyperlipidemia, obesity and other diseases related to the metabolic syndrome [164]. Shepherd et al. also found a reduction in total ceramide levels in the muscles of obese men after continuous exercise training, with the most significant reduction in C18:0 ceramide levels in particular [165]. In conclusion, the use of the right dietary interventions and exercise modalities can reduce ceramide accumulation in the body and thus improve the NAFLD process.
Conclusion
Ceramide is considered the central molecule in sphingolipids metabolism and its modulation plays a key role in the development of several pathologies. Many in vivo and in vitro experiments have confirmed the closely relationship between abnormal ceramide metabolism and the pathogenesis of NAFLD. Due to the lack of effective drugs for the treatment of NAFLD, it is imperative to explore therapeutic modalities that may delay and reverse the disease. Given the important role of ceramides in orchestrating the progression of NAFLD, targeting ceramides offers a new therapy for the disease. Several therapeutic strategies focused on ceramide or other sphingolipid metabolizing enzymes that act as direct targets, including CerS, SPT, however, SPT is not a viable therapeutic target, owing to safety issues that result from the extreme diminution of all sphingolipids. In the future, treatments that focus on a more limited collection of harmful ceramide subseeds may represent a new strategy for therapeutic interventions. Currently, the vast majority of targeted drugs against ceramide are still in the preclinical stage and clinical trials are needed to confirm the effectiveness of the relevant drugs in humans. Due to the complexity of ceramide function, further study of the exact mechanisms of ceramide in disease development will facilitate the improvement of therapeutic strategies. Many questions remain to be explored at the present time, such as the specific functions of different chain-length ceramides remain unclear; whether the overall role of ceramides in liver fibrosis is pro- or anti-fibrotic; and whether the functions of ceramides are consistent in different organ tissues. Answering the above questions can help promote the possibility of ceramide-related drugs as a treatment for NAFLD. With the rapid development of single-cell metabolomics and transcriptomics, it has also increased our understanding of ceramide function in NAFLD and facilitated the search for potential therapeutic approaches targeting ceramide metabolism.
Abbreviations
ACER1-3: alkaline ceramidase; ACLY: ATP-citrate lyase; ASAH1: acid ceramidase; ASAH2: neutral ceramidase; ASMase/ASM: acid sphingomyelinase; ATF6: activating transcription factor 6; CB1R: cannabinoid-1-receptor; CD36: fatty acid translocase; CerS: ceramide synthase; DES: dihydroceramide desaturase; EV: extracellular vesicles; FFA: free fatty acids; FABP: fatty acid binding protein; FATP: fatty acid transport proteins; GlcCer: glucosylceramide; GLP-1: glucagon-like peptide-1; HFD: High-fat diet; HHcy: Hyperhomocysteinemia; HSC: hepatic stellate cell; INSIG2: insulin-induced gene-2; IKK: IκB kinase; IRE1: inositol requiring enzyme-1; K145: 3-(2-amino-ethyl)-5-[3-(4-butoxyl-phenyl)-propylidene] -thiazolidine-2, 4-dione; MCD: methionine and choline-deficient; NAFLD: Non-alcoholic fatty liver disease; NASH: Non-alcoholic steatohepatitis; NET: neutrophil extracellular traps; NE: neutrophil elastase; PERK: protein kinase RNA-like endoplasmic reticulum kinase; PEWD: palmitate-enriched Western diet; PKA: protein kinase A; PKR: RNA-dependent protein kinase; sAC: soluble adenylyl cyclase SFA saturated fatty acids; S1P: Sphingosine 1-Phosphate; SM: sphingomyelin; SMPDL3B: sphingomyelin phosphodiesterase-like 3B; SPT: serine palmitoyltransferase; TCA: taurine-conjugated bile acids; UPR: unfold protein response; XBP1: X-box binding protein 1.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant/Award Numbers: 82100628, 82270622, and 82270641), the Postdoctoral Science Foundation of China (Grant/Award Numbers: 2021M690184, 2021M690186 and 2021M700183), and the Natural Science Foundation of Anhui Province (Grant Number. 2108085QH313).
Author Contributions
C.Z. and Q.H. contributed equally to this article. C.Z. and Q.H. drafted the original article and prepared the figures. X.Z. and H.D. searched for relevant literature and wrote this paper. X.L. proposed research design, searched for relevant literature and wrote this paper. H.W. offered professional advice and revision. All authors critically reviewed the content and approved the final version of this paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lazarus JV, Mark HE, Anstee QM, Arab JP, Batterham RL, Castera L. et al. Advancing the global public health agenda for NAFLD: a consensus statement. Nat Rev Gastroenterol Hepatol. 2022;19:60-78
2. Zhou J, Zhou F, Wang W, Zhang XJ, Ji YX, Zhang P. et al. Epidemiological Features of NAFLD From 1999 to 2018 in China. Hepatology (Baltimore, Md). 2020;71:1851-64
3. Scorletti E, Carr RM. A new perspective on NAFLD: Focusing on lipid droplets. Journal of hepatology. 2022;76:934-45
4. Yki-Järvinen H, Luukkonen PK, Hodson L, Moore JB. Dietary carbohydrates and fats in nonalcoholic fatty liver disease. Nature reviews Gastroenterology & hepatology. 2021;18:770-86
5. Younossi ZM. Non-alcoholic fatty liver disease - A global public health perspective. Journal of hepatology. 2019;70:531-44
6. Stefan N, Cusi K. A global view of the interplay between non-alcoholic fatty liver disease and diabetes. The Lancet Diabetes & Endocrinology. 2022;10:284-96
7. Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F. et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell metabolism. 2015;21:739-46
8. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836-46
9. Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184:2537-64
10. Kotlyarov S, Bulgakov A. Lipid Metabolism Disorders in the Comorbid Course of Nonalcoholic Fatty Liver Disease and Chronic Obstructive Pulmonary Disease. Cells. 2021 10
11. Mashek DG. Hepatic lipid droplets: A balancing act between energy storage and metabolic dysfunction in NAFLD. Mol Metab. 2021;50:101115
12. Dorairaj V, Sulaiman SA, Abu N, Abdul Murad NA. Nonalcoholic Fatty Liver Disease (NAFLD): Pathogenesis and Noninvasive Diagnosis. Biomedicines. 2021 10
13. Ziolkowska S, Binienda A, Jablkowski M, Szemraj J, Czarny P. The Interplay between Insulin Resistance, Inflammation, Oxidative Stress, Base Excision Repair and Metabolic Syndrome in Nonalcoholic Fatty Liver Disease. Int J Mol Sci. 2021 22
14. El-Agroudy NN, Kurzbach A, Rodionov RN, O'Sullivan J, Roden M, Birkenfeld AL. et al. Are Lifestyle Therapies Effective for NAFLD Treatment? Trends Endocrinol Metab. 2019
15. Loeffelholz CV, Roth J, Coldewey SM, Birkenfeld AL. The Role of Physical Activity in Nonalcoholic and Metabolic Dysfunction Associated Fatty Liver Disease. Biomedicines. 2021 9
16. Kalinichenko LS, Gulbins E, Kornhuber J, Muller CP. Sphingolipid control of cognitive functions in health and disease. Prog Lipid Res. 2022;86:101162
17. Choi RH, Tatum SM, Symons JD, Summers SA, Holland WL. Ceramides and other sphingolipids as drivers of cardiovascular disease. Nature reviews Cardiology. 2021;18:701-11
18. Mah M, Febbraio M, Turpin-Nolan S. Circulating Ceramides- Are Origins Important for Sphingolipid Biomarkers and Treatments? Frontiers in endocrinology. 2021;12:684448
19. Park WJ, Song JH, Kim GT, Park TS. Ceramide and Sphingosine 1-Phosphate in Liver Diseases. Molecules and cells. 2020;43:419-30
20. Aburasayn H, Al Batran R, Ussher JR. Targeting ceramide metabolism in obesity. Am J Physiol Endocrinol Metab. 2016;311:E423-35
21. Castro BM, Prieto M, Silva LC. Ceramide: a simple sphingolipid with unique biophysical properties. Prog Lipid Res. 2014;54:53-67
22. Alexaki A, Gupta SD, Majumder S, Kono M, Tuymetova G, Harmon JM. et al. Autophagy regulates sphingolipid levels in the liver. J Lipid Res. 2014;55:2521-31
23. Iqbal J, Walsh MT, Hammad SM, Hussain MM. Sphingolipids and Lipoproteins in Health and Metabolic Disorders. Trends in endocrinology and metabolism: TEM. 2017;28:506-18
24. Chaurasia B, Summers SA. Ceramides - Lipotoxic Inducers of Metabolic Disorders. Trends in endocrinology and metabolism: TEM. 2015;26:538-50
25. Hajduch E, Lachkar F, Ferré P, Foufelle F. Roles of Ceramides in Non-Alcoholic Fatty Liver Disease. Journal of clinical medicine. 2021 10
26. Hydes T, Alam U, Cuthbertson DJ. The Impact of Macronutrient Intake on Non-alcoholic Fatty Liver Disease (NAFLD): Too Much Fat, Too Much Carbohydrate, or Just Too Many Calories? Frontiers in nutrition. 2021;8:640557
27. Sanyal AJ. Mechanisms of Disease: pathogenesis of nonalcoholic fatty liver disease. Nature clinical practice Gastroenterology & hepatology. 2005;2:46-53
28. Esler WP, Bence KK. Metabolic Targets in Nonalcoholic Fatty Liver Disease. Cell Mol Gastroenterol Hepatol. 2019
29. Duwaerts CC, Maher JJ. Macronutrients and the Adipose-Liver Axis in Obesity and Fatty Liver. Cell Mol Gastroenterol Hepatol. 2019;7:749-61
30. Pei K, Gui T, Kan DF, Feng HC, Jin YQ, Yang Y. et al. An Overview of Lipid Metabolism and Nonalcoholic Fatty Liver Disease. Biomed Research International. 2020. 2020
31. Charlton M, Viker K, Krishnan A, Sanderson S, Veldt B, Kaalsbeek AJ. et al. Differential expression of lumican and fatty acid binding protein-1: new insights into the histologic spectrum of nonalcoholic fatty liver disease. Hepatology. 2009;49:1375-84
32. Zhao L, Zhang C, Luo X, Wang P, Zhou W, Zhong S. et al. CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non-alcoholic steatohepatitis. Journal of hepatology. 2018;69:705-17
33. Zeng H, Qin H, Liao M, Zheng E, Luo X, Xiao A. et al. CD36 promotes de novo lipogenesis in hepatocytes through INSIG2-dependent SREBP1 processing. Mol Metab. 2022;57:101428
34. Geng Y, Faber KN, de Meijer VE, Blokzijl H, Moshage H. How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease? Hepatol Int. 2021;15:21-35
35. Higuchi N, Kato M, Tanaka M, Miyazaki M, Takao S, Kohjima M. et al. Effects of insulin resistance and hepatic lipid accumulation on hepatic mRNA expression levels of apoB, MTP and L-FABP in non-alcoholic fatty liver disease. Experimental and therapeutic medicine. 2011;2:1077-81
36. Zhao S, Jang C, Liu J, Uehara K, Gilbert M, Izzo L. et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature. 2020;579:586-91
37. Smith GI, Shankaran M, Yoshino M, Schweitzer GG, Chondronikola M, Beals JW. et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. The Journal of clinical investigation. 2020;130:1453-60
38. Sinha RA, Rajak S, Singh BK, Yen PM. Hepatic Lipid Catabolism via PPARα-Lysosomal Crosstalk. International journal of molecular sciences. 2020 21
39. Montagner A, Polizzi A, Fouché E, Ducheix S, Lippi Y, Lasserre F. et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut. 2016;65:1202-14
40. Simões ICM, Amorim R, Teixeira J, Karkucinska-Wieckowska A, Carvalho A, Pereira SP. et al. The Alterations of Mitochondrial Function during NAFLD Progression-An Independent Effect of Mitochondrial ROS Production. International journal of molecular sciences. 2021 22
41. Willis SA, Bawden SJ, Malaikah S, Sargeant JA, Stensel DJ, Aithal GP. et al. The role of hepatic lipid composition in obesity-related metabolic disease. Liver international: official journal of the International Association for the Study of the Liver. 2021;41:2819-35
42. Alkhouri N, Dixon LJ, Feldstein AE. Lipotoxicity in nonalcoholic fatty liver disease: not all lipids are created equal. Expert Rev Gastroenterol Hepatol. 2009;3:445-51
43. Seebacher F, Zeigerer A, Kory N, Krahmer N. Hepatic lipid droplet homeostasis and fatty liver disease. Semin Cell Dev Biol. 2020
44. Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. Journal of hepatology. 2018;68:280-95
45. Horn CL, Morales AL, Savard C, Farrell GC, Ioannou GN. Role of Cholesterol-Associated Steatohepatitis in the Development of NASH. Hepatology communications. 2022;6:12-35
46. Tilg H, Adolph TE, Dudek M, Knolle P. Non-alcoholic fatty liver disease: the interplay between metabolism, microbes and immunity. Nature metabolism. 2021;3:1596-607
47. Min HK, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J. et al. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012;15:665-74
48. Wang X, Cai B, Yang X, Sonubi OO, Zheng Z, Ramakrishnan R. et al. Cholesterol Stabilizes TAZ in Hepatocytes to Promote Experimental Non-alcoholic Steatohepatitis. Cell Metab. 2020
49. Hlušička J, Arora M, Brůha R, Žák A. Statins and liver. Casopis lekaru ceskych. 2022;161:80-3
50. Lair B, Laurens C, Van Den Bosch B, Moro C. Novel Insights and Mechanisms of Lipotoxicity-Driven Insulin Resistance. International journal of molecular sciences. 2020 21
51. Green CD, Maceyka M, Cowart LA, Spiegel S. Sphingolipids in metabolic disease: The good, the bad, and the unknown. Cell metabolism. 2021;33:1293-306
52. Dasgupta D, Nakao Y, Mauer AS, Thompson JM, Sehrawat TS, Liao CY. et al. IRE1A Stimulates Hepatocyte-Derived Extracellular Vesicles That Promote Inflammation in Mice With Steatohepatitis. Gastroenterology. 2020;159:1487-503.e17
53. Cirillo F, Piccoli M, Ghiroldi A, Monasky MM, Rota P, La Rocca P. et al. The antithetic role of ceramide and sphingosine-1-phosphate in cardiac dysfunction. Journal of cellular physiology. 2021;236:4857-73
54. Kucuk S, Niven J, Caamano J, Jones SW, Camacho-Muñoz D, Nicolaou A. et al. Unwrapping the mechanisms of ceramide and fatty acid-initiated signals leading to immune-inflammatory responses in obesity. The international journal of biochemistry & cell biology. 2021;135:105972
55. Jiang M, Li C, Liu Q, Wang A, Lei M. Inhibiting Ceramide Synthesis Attenuates Hepatic Steatosis and Fibrosis in Rats With Non-alcoholic Fatty Liver Disease. Front Endocrinol (Lausanne). 2019;10:665
56. Li Y, Talbot CL, Chaurasia B. Ceramides in Adipose Tissue. Front Endocrinol (Lausanne). 2020;11:407
57. Gault CR, Obeid LM, Hannun YA. An overview of sphingolipid metabolism: from synthesis to breakdown. Advances in experimental medicine and biology. 2010;688:1-23
58. Lee AY, Lee JW, Kim JE, Mock HJ, Park S, Kim S. et al. Dihydroceramide is a key metabolite that regulates autophagy and promotes fibrosis in hepatic steatosis model. Biochemical and biophysical research communications. 2017;494:460-9
59. Raichur S. Ceramide Synthases Are Attractive Drug Targets for Treating Metabolic Diseases. Frontiers in endocrinology. 2020;11:483
60. Montefusco DJ, Allegood JC, Spiegel S, Cowart LA. Non-alcoholic fatty liver disease: Insights from sphingolipidomics. Biochemical and biophysical research communications. 2018;504:608-16
61. Skácel J, Slusher BS, Tsukamoto T. Small Molecule Inhibitors Targeting Biosynthesis of Ceramide, the Central Hub of the Sphingolipid Network. Journal of medicinal chemistry. 2021;64:279-97
62. Park WJ, Park JW. The effect of altered sphingolipid acyl chain length on various disease models. Biological chemistry. 2015;396:693-705
63. Grösch S, Schiffmann S, Geisslinger G. Chain length-specific properties of ceramides. Prog Lipid Res. 2012;51:50-62
64. Insausti-Urkia N, Solsona-Vilarrasa E, Garcia-Ruiz C, Fernandez-Checa JC. Sphingomyelinases and Liver Diseases. Biomolecules. 2020 10
65. Parveen F, Bender D, Law SH, Mishra VK, Chen CC, Ke LY. Role of Ceramidases in Sphingolipid Metabolism and Human Diseases. Cells. 2019 8
66. Xiang H, Jin S, Tan F, Xu Y, Lu Y, Wu T. Physiological functions and therapeutic applications of neutral sphingomyelinase and acid sphingomyelinase. Biomed Pharmacother. 2021;139:111610
67. Norris GH, Blesso CN. Dietary sphingolipids: potential for management of dyslipidemia and nonalcoholic fatty liver disease. Nutr Rev. 2017;75:274-85
68. Kono M, Dreier JL, Ellis JM, Allende ML, Kalkofen DN, Sanders KM. et al. Neutral ceramidase encoded by the Asah2 gene is essential for the intestinal degradation of sphingolipids. J Biol Chem. 2006;281:7324-31
69. Kitatani K, Idkowiak-Baldys J, Hannun YA. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal. 2008;20:1010-8
70. Coant N, Sakamoto W, Mao C, Hannun YA. Ceramidases, roles in sphingolipid metabolism and in health and disease. Adv Biol Regul. 2017;63:122-31
71. Borodzicz S, Rudnicka L, Mirowska-Guzel D, Cudnoch-Jedrzejewska A. The role of epidermal sphingolipids in dermatologic diseases. Lipids Health Dis. 2016;15:13
72. Kim D, Lee NR, Park SY, Jun M, Lee K, Kim S. et al. As in Atopic Dermatitis, Nonlesional Skin in Allergic Contact Dermatitis Displays Abnormalities in Barrier Function and Ceramide Content. J Invest Dermatol. 2017;137:748-50
73. McNally BD, Ashley DF, Hanschke L, Daou HN, Watt NT, Murfitt SA. et al. Long-chain ceramides are cell non-autonomous signals linking lipotoxicity to endoplasmic reticulum stress in skeletal muscle. Nat Commun. 2022;13:1748
74. Regnier M, Polizzi A, Guillou H, Loiseau N. Sphingolipid metabolism in non-alcoholic fatty liver diseases. Biochimie. 2019;159:9-22
75. Svegliati-Baroni G, Pierantonelli I, Torquato P, Marinelli R, Ferreri C, Chatgilialoglu C. et al. Lipidomic biomarkers and mechanisms of lipotoxicity in non-alcoholic fatty liver disease. Free Radic Biol Med. 2019;144:293-309
76. Legaki AI, Moustakas, II, Sikorska M, Papadopoulos G, Velliou RI, Chatzigeorgiou A. Hepatocyte Mitochondrial Dynamics and Bioenergetics in Obesity-Related Non-Alcoholic Fatty Liver Disease. Curr Obes Rep. 2022
77. Poss AM, Summers SA. Too Much of a Good Thing? An Evolutionary Theory to Explain the Role of Ceramides in NAFLD. Front Endocrinol (Lausanne). 2020;11:505
78. Nikolova-Karakashian M. Sphingolipids at the Crossroads of NAFLD and Senescence. Adv Cancer Res. 2018;140:155-90
79. Ooi GJ, Meikle PJ, Huynh K, Earnest A, Roberts SK, Kemp W. et al. Hepatic lipidomic remodeling in severe obesity manifests with steatosis and does not evolve with non-alcoholic steatohepatitis. J Hepatol. 2021;75:524-35
80. Rappez L, Stadler M, Triana S, Gathungu RM, Ovchinnikova K, Phapale P. et al. SpaceM reveals metabolic states of single cells. Nat Methods. 2021;18:799-805
81. Gosejacob D, Jager PS, Vom Dorp K, Frejno M, Carstensen AC, Kohnke M. et al. Ceramide Synthase 5 Is Essential to Maintain C16:0-Ceramide Pools and Contributes to the Development of Diet-induced Obesity. J Biol Chem. 2016;291:6989-7003
82. Turpin SM, Nicholls HT, Willmes DM, Mourier A, Brodesser S, Wunderlich CM. et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014;20:678-86
83. Gadgil MD, Sarkar M, Sands C, Lewis MR, Herrington DM, Kanaya AM. Associations of NAFLD with circulating ceramides and impaired glycemia. Diabetes Res Clin Pract. 2022;186:109829
84. Luukkonen PK, Zhou Y, Sadevirta S, Leivonen M, Arola J, Oresic M. et al. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J Hepatol. 2016;64:1167-75
85. Rosqvist F, Kullberg J, Ståhlman M, Cedernaes J, Heurling K, Johansson HE. et al. Overeating Saturated Fat Promotes Fatty Liver and Ceramides Compared With Polyunsaturated Fat: A Randomized Trial. The Journal of clinical endocrinology and metabolism. 2019;104:6207-19
86. Sztolsztener K, Konstantynowicz-Nowicka K, Harasim-Symbor E, Chabowski A. Time-Dependent Changes in Hepatic Sphingolipid Accumulation and PI3K/Akt/mTOR Signaling Pathway in a Rat Model of NAFLD. Int J Mol Sci. 2021 22
87. Dong YQ, Zhang XZ, Sun LL, Zhang SY, Liu B, Liu HY. et al. Omega-3 PUFA ameliorates hyperhomocysteinemia-induced hepatic steatosis in mice by inhibiting hepatic ceramide synthesis. Acta Pharmacol Sin. 2017;38:1601-10
88. Xia JY, Holland WL, Kusminski CM, Sun K, Sharma AX, Pearson MJ. et al. Targeted Induction of Ceramide Degradation Leads to Improved Systemic Metabolism and Reduced Hepatic Steatosis. Cell Metab. 2015;22:266-78
89. Petersen MC, Shulman GI. Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol Sci. 2017;38:649-65
90. Cuomo P, Capparelli R, Iannelli A, Iannelli D. Role of Branched-Chain Amino Acid Metabolism in Type 2 Diabetes, Obesity, Cardiovascular Disease and Non-Alcoholic Fatty Liver Disease. Int J Mol Sci. 2022 23
91. Kolb R, Sutterwala FS, Zhang W. Obesity and cancer: inflammation bridges the two. Curr Opin Pharmacol. 2016;29:77-89
92. Preuss C, Jelenik T, Bodis K, Mussig K, Burkart V, Szendroedi J. et al. A New Targeted Lipidomics Approach Reveals Lipid Droplets in Liver, Muscle and Heart as a Repository for Diacylglycerol and Ceramide Species in Non-Alcoholic Fatty Liver. Cells. 2019 8
93. Juchnicka I, Kuzmicki M, Szamatowicz J. Ceramides and Sphingosino-1-Phosphate in Obesity. Front Endocrinol (Lausanne). 2021;12:635995
94. Kumashiro N, Erion DM, Zhang D, Kahn M, Beddow SA, Chu X. et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A. 2011;108:16381-5
95. Razak Hady H, Blachnio-Zabielska AU, Szczerbinski L, Zabielski P, Imierska M, Dadan J. et al. Ceramide Content in Liver Increases Along with Insulin Resistance in Obese Patients. J Clin Med. 2019 8
96. Chaurasia B, Tippetts TS, Mayoral Monibas R, Liu J, Li Y, Wang L. et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science. 2019;365:386-92
97. Turban S, Hajduch E. Protein kinase C isoforms: mediators of reactive lipid metabolites in the development of insulin resistance. FEBS letters. 2011;585:269-74
98. Galbo T, Perry RJ, Nishimura E, Samuel VT, Quistorff B, Shulman GI. PP2A inhibition results in hepatic insulin resistance despite Akt2 activation. Aging. 2013;5:770-81
99. Yang X, Nath A, Opperman MJ, Chan C. The double-stranded RNA-dependent protein kinase differentially regulates insulin receptor substrates 1 and 2 in HepG2 cells. Molecular biology of the cell. 2010;21:3449-58
100. Roszczyc-Owsiejczuk K, Zabielski P. Sphingolipids as a Culprit of Mitochondrial Dysfunction in Insulin Resistance and Type 2 Diabetes. Front Endocrinol (Lausanne). 2021;12:635175
101. Blachnio-Zabielska AU, Hady HR, Markowski AR, Kurianiuk A, Karwowska A, Gorski J. et al. Inhibition of Ceramide De Novo Synthesis Affects Adipocytokine Secretion and Improves Systemic and Adipose Tissue Insulin Sensitivity. Int J Mol Sci. 2018 19
102. Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH. et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med. 2011;17:55-63
103. Holland WL, Adams AC, Brozinick JT, Bui HH, Miyauchi Y, Kusminski CM. et al. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab. 2013;17:790-7
104. Turner N, Lim XY, Toop HD, Osborne B, Brandon AE, Taylor EN. et al. A selective inhibitor of ceramide synthase 1 reveals a novel role in fat metabolism. Nature communications. 2018;9:3165
105. Apostolopoulou M, Gordillo R, Gancheva S, Strassburger K, Herder C, Esposito I. et al. Role of ceramide-to-dihydroceramide ratios for insulin resistance and non-alcoholic fatty liver disease in humans. BMJ Open Diabetes Res Care. 2020 8
106. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol. 2018;15:349-64
107. Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. 2017;66:1300-12
108. Schwabe RF, Tabas I, Pajvani UB. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology. 2020;158:1913-28
109. Apostolopoulou M, Gordillo R, Koliaki C, Gancheva S, Jelenik T, De Filippo E. et al. Specific Hepatic Sphingolipids Relate to Insulin Resistance, Oxidative Stress, and Inflammation in Nonalcoholic Steatohepatitis. Diabetes care. 2018;41:1235-43
110. Liao CY, Song MJ, Gao Y, Mauer AS, Revzin A, Malhi H. Hepatocyte-Derived Lipotoxic Extracellular Vesicle Sphingosine 1-Phosphate Induces Macrophage Chemotaxis. Front Immunol. 2018;9:2980
111. Chen J, Liang B, Bian D, Luo Y, Yang J, Li Z. et al. Knockout of neutrophil elastase protects against western diet induced nonalcoholic steatohepatitis in mice by regulating hepatic ceramides metabolism. Biochem Biophys Res Commun. 2019;518:691-7
112. Byrnes K, Blessinger S, Bailey NT, Scaife R, Liu G, Khambu B. Therapeutic regulation of autophagy in hepatic metabolism. Acta Pharm Sin B. 2022;12:33-49
113. Jiang W, Ogretmen B. Ceramide stress in survival versus lethal autophagy paradox: ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Autophagy. 2013;9:258-9
114. Yang RX, Pan Q, Liu XL, Zhou D, Xin FZ, Zhao ZH. et al. Therapeutic effect and autophagy regulation of myriocin in nonalcoholic steatohepatitis. Lipids Health Dis. 2019;18:179
115. Garcia-Ruiz C, Mato JM, Vance D, Kaplowitz N, Fernandez-Checa JC. Acid sphingomyelinase-ceramide system in steatohepatitis: a novel target regulating multiple pathways. J Hepatol. 2015;62:219-33
116. Wang K, Li C, Lin X, Sun H, Xu R, Li Q. et al. Targeting alkaline ceramidase 3 alleviates the severity of nonalcoholic steatohepatitis by reducing oxidative stress. Cell Death Dis. 2020;11:28
117. Kim YR, Lee EJ, Shin KO, Kim MH, Pewzner-Jung Y, Lee YM. et al. Hepatic triglyceride accumulation via endoplasmic reticulum stress-induced SREBP-1 activation is regulated by ceramide synthases. Exp Mol Med. 2019;51:1-16
118. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiological reviews. 2008;88:125-72
119. Moles A, Tarrats N, Morales A, Dominguez M, Bataller R, Caballeria J. et al. Acidic sphingomyelinase controls hepatic stellate cell activation and in vivo liver fibrogenesis. Am J Pathol. 2010;177:1214-24
120. Alsamman S, Christenson SA, Yu A, Ayad NME, Mooring MS, Segal JM. et al. Targeting acid ceramidase inhibits YAP/TAZ signaling to reduce fibrosis in mice. Sci Transl Med. 2020 12
121. King A, Houlihan DD, Kavanagh D, Haldar D, Luu N, Owen A. et al. Sphingosine-1-Phosphate Prevents Egress of Hematopoietic Stem Cells From Liver to Reduce Fibrosis. Gastroenterology. 2017;153:233-48.e16
122. Simon J, Ouro A, Ala-Ibanibo L, Presa N, Delgado TC, Martinez-Chantar ML. Sphingolipids in Non-Alcoholic Fatty Liver Disease and Hepatocellular Carcinoma: Ceramide Turnover. Int J Mol Sci. 2019 21
123. Grbcic P, Car EPM, Sedic M. Targeting Ceramide Metabolism in Hepatocellular Carcinoma: New Points for Therapeutic Intervention. Curr Med Chem. 2020;27:6611-27
124. Li Z, Guan M, Lin Y, Cui X, Zhang Y, Zhao Z. et al. Aberrant Lipid Metabolism in Hepatocellular Carcinoma Revealed by Liver Lipidomics. Int J Mol Sci. 2017 18
125. Bao M, Chen Z, Xu Y, Zhao Y, Zha R, Huang S. et al. Sphingosine kinase 1 promotes tumour cell migration and invasion via the S1P/EDG1 axis in hepatocellular carcinoma. Liver international: official journal of the International Association for the Study of the Liver. 2012;32:331-8
126. Ordonez R, Fernandez A, Prieto-Dominguez N, Martinez L, Garcia-Ruiz C, Fernandez-Checa JC. et al. Ceramide metabolism regulates autophagy and apoptotic cell death induced by melatonin in liver cancer cells. J Pineal Res. 2015;59:178-89
127. Tagaram HR, Divittore NA, Barth BM, Kaiser JM, Avella D, Kimchi ET. et al. Nanoliposomal ceramide prevents in vivo growth of hepatocellular carcinoma. Gut. 2011;60:695-701
128. Liu B, Xiao J, Dong M, Qiu Z, Jin J. Human alkaline ceramidase 2 promotes the growth, invasion, and migration of hepatocellular carcinoma cells via sphingomyelin phosphodiesterase acid-like 3B. Cancer Sci. 2020;111:2259-74
129. Osawa Y, Suetsugu A, Matsushima-Nishiwaki R, Yasuda I, Saibara T, Moriwaki H. et al. Liver acid sphingomyelinase inhibits growth of metastatic colon cancer. J Clin Invest. 2013;123:834-43
130. Jakobi K, Beyer S, Koch A, Thomas D, Schwalm S, Zeuzem S. et al. Sorafenib Treatment and Modulation of the Sphingolipid Pathway Affect Proliferation and Viability of Hepatocellular Carcinoma In Vitro. Int J Mol Sci. 2020 21
131. Zhang G, Park MA, Mitchell C, Hamed H, Rahmani M, Martin AP. et al. Vorinostat and sorafenib synergistically kill tumor cells via FLIP suppression and CD95 activation. Clin Cancer Res. 2008;14:5385-99
132. Li G, Liu D, Kimchi ET, Kaifi JT, Qi X, Manjunath Y. et al. Nanoliposome C6-Ceramide Increases the Anti-tumor Immune Response and Slows Growth of Liver Tumors in Mice. Gastroenterology. 2018;154:1024-36.e9
133. Qi X, Wu F, Kim SH, Kaifi JT, Kimchi ET, Snyder H. et al. Nanoliposome C6-Ceramide in combination with anti-CTLA4 antibody improves anti-tumor immunity in hepatocellular cancer. FASEB J. 2022;36:e22250
134. Morigny P, Boucher J, Arner P, Langin D. Lipid and glucose metabolism in white adipocytes: pathways, dysfunction and therapeutics. Nat Rev Endocrinol. 2021;17:276-95
135. Albillos A, Gottardi A, Rescigno M. The gut-liver axis in liver disease: pathophysiological basis for therapy. J Hepatol. 2019
136. Zhang X, Ji X, Wang Q, Li JZ. New insight into inter-organ crosstalk contributing to the pathogenesis of non-alcoholic fatty liver disease (NAFLD). Protein Cell. 2018;9:164-77
137. Bauer KC, Littlejohn PT, Ayala V, Creus-Cuadros A, Finlay BB. Nonalcoholic Fatty Liver Disease and the Gut-Liver Axis: Exploring an Undernutrition Perspective. Gastroenterology. 2022
138. Rohrhofer J, Zwirzitz B, Selberherr E, Untersmayr E. The Impact of Dietary Sphingolipids on Intestinal Microbiota and Gastrointestinal Immune Homeostasis. Front Immunol. 2021;12:635704
139. Di Rienzi SC, Johnson EL, Waters JL, Kennedy EA, Jacobson J, Lawrence P. et al. The microbiome affects liver sphingolipids and plasma fatty acids in a murine model of the Western diet based on soybean oil. J Nutr Biochem. 2021;97:108808
140. Gu X, Sun R, Chen L, Chu S, Doll MA, Li X. et al. Neutral Ceramidase Mediates Nonalcoholic Steatohepatitis by Regulating Monounsaturated Fatty Acids and Gut IgA(+) B Cells. Hepatology. 2021;73:901-19
141. Johnson EL, Heaver SL, Waters JL, Kim BI, Bretin A, Goodman AL. et al. Sphingolipids produced by gut bacteria enter host metabolic pathways impacting ceramide levels. Nature Communications. 2020 11
142. Jiang CT, Xie C, Li F, Zhang LM, Nichols RG, Krausz KW. et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. Journal of Clinical Investigation. 2015;125:386-402
143. Jiang CT, Xie C, Lv Y, Li J, Krausz KW, Shi JM. et al. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nature Communications. 2015 6
144. Luo Y, Yang S, Wu X, Takahashi S, Sun L, Cai J. et al. Intestinal MYC modulates obesity-related metabolic dysfunction. Nat Metab. 2021;3:923-39
145. Shang Q, Guo GL, Honda A, Saumoy M, Salen G, Xu G. FGF15/19 protein levels in the portal blood do not reflect changes in the ileal FGF15/19 or hepatic CYP7A1 mRNA levels. J Lipid Res. 2013;54:2606-14
146. Régnier M, Polizzi A, Lukowicz C, Smati S, Lasserre F, Lippi Y. et al. The protective role of liver X receptor (LXR) during fumonisin B1-induced hepatotoxicity. Archives of toxicology. 2019;93:505-17
147. Dellafiora L, Galaverna G, Dall'Asta C. Mechanisms of Fumonisin B1 Toxicity: A Computational Perspective beyond the Ceramide Synthases Inhibition. Chemical research in toxicology. 2018;31:1203-12
148. Veeriah V, Lee SH, Levine F. Long-term oral administration of an HNF4alpha agonist prevents weight gain and hepatic steatosis by promoting increased mitochondrial mass and function. Cell Death Dis. 2022;13:89
149. Zanieri F, Levi A, Montefusco D, Longato L, De Chiara F, Frenguelli L. et al. Exogenous Liposomal Ceramide-C6 Ameliorates Lipidomic Profile, Energy Homeostasis, and Anti-Oxidant Systems in NASH. Cells. 2020 9
150. Zabielski P, Daniluk J, Hady HR, Markowski AR, Imierska M, Górski J. et al. The effect of high-fat diet and inhibition of ceramide production on insulin action in liver. Journal of cellular physiology. 2019;234:1851-61
151. Kurek K, Piotrowska DM, Wiesiolek-Kurek P, Lukaszuk B, Chabowski A, Gorski J. et al. Inhibition of ceramide de novo synthesis reduces liver lipid accumulation in rats with nonalcoholic fatty liver disease. Liver Int. 2014;34:1074-83
152. Berdyshev EV, Gorshkova I, Skobeleva A, Bittman R, Lu X, Dudek SM. et al. FTY720 inhibits ceramide synthases and up-regulates dihydrosphingosine 1-phosphate formation in human lung endothelial cells. J Biol Chem. 2009;284:5467-77
153. Shi Y, Wei Q, Liu Y, Yuan J. The alleviating effect of sphingosine kinases 2 inhibitor K145 on nonalcoholic fatty liver. Biochemical and biophysical research communications. 2021;580:1-6
154. Lee SY, Hong IK, Kim BR, Shim SM, Sung Lee J, Lee HY. et al. Activation of sphingosine kinase 2 by endoplasmic reticulum stress ameliorates hepatic steatosis and insulin resistance in mice. Hepatology (Baltimore, Md). 2015;62:135-46
155. Cinar R, Godlewski G, Liu J, Tam J, Jourdan T, Mukhopadhyay B. et al. Hepatic cannabinoid-1 receptors mediate diet-induced insulin resistance by increasing de novo synthesis of long-chain ceramides. Hepatology (Baltimore, Md). 2014;59:143-53
156. Camacho-Muñoz D, Kiezel-Tsugunova M, Kiss O, Uddin M, Sundén M, Ryaboshapkina M. et al. Omega-3 carboxylic acids and fenofibrate differentially alter plasma lipid mediators in patients with non-alcoholic fatty liver disease. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2021;35:e21976
157. Sarkar S, Kumari D, Gupta SK, Sharma V, Mukhi S, Kamboj P. et al. Saroglitazar and Hepano treatment offers protection against high fat high fructose diet induced obesity, insulin resistance and steatosis by modulating various class of hepatic and circulating lipids. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2021;144:112357
158. Ji J, Feng M, Huang Y, Niu X. Liraglutide inhibits receptor for advanced glycation end products (RAGE)/reduced form of nicotinamide-adenine dinucleotide phosphate (NAPDH) signaling to ameliorate non-alcoholic fatty liver disease (NAFLD) in vivo and vitro. Bioengineered. 2022;13:5091-102
159. Somm E, Montandon SA, Loizides-Mangold U, Gaïa N, Lazarevic V, De Vito C. et al. The GLP-1R agonist liraglutide limits hepatic lipotoxicity and inflammatory response in mice fed a methionine-choline deficient diet. Translational research: the journal of laboratory and clinical medicine. 2021;227:75-88
160. Zhang L, Hu Y, An Y, Wang Q, Liu J, Wang G. The Changes of Lipidomic Profiles Reveal Therapeutic Effects of Exenatide in Patients With Type 2 Diabetes. Frontiers in endocrinology. 2022;13:677202
161. Wang DD, Toledo E, Hruby A, Rosner BA, Willett WC, Sun Q. et al. Plasma Ceramides, Mediterranean Diet, and Incident Cardiovascular Disease in the PREDIMED Trial (Prevención con Dieta Mediterránea). Circulation. 2017;135:2028-40
162. Gaggini M, Pingitore A, Vassalle C. Plasma Ceramides Pathophysiology, Measurements, Challenges, and Opportunities. Metabolites. 2021 11
163. Promrat K, Longato L, Wands JR, de la Monte SM. Weight loss amelioration of non-alcoholic steatohepatitis linked to shifts in hepatic ceramide expression and serum ceramide levels. Hepatol Res. 2011;41:754-62
164. Mendham AE, Goedecke JH, Zeng Y, Larsen S, George C, Hauksson J. et al. Exercise training improves mitochondrial respiration and is associated with an altered intramuscular phospholipid signature in women with obesity. Diabetologia. 2021;64:1642-59
165. Shepherd SO, Cocks M, Meikle PJ, Mellett NA, Ranasinghe AM, Barker TA. et al. Lipid droplet remodelling and reduced muscle ceramides following sprint interval and moderate-intensity continuous exercise training in obese males. International journal of obesity (2005). 2017;41:1745-54
166. Panchal M, Gaudin M, Lazar AN, Salvati E, Rivals I, Ayciriex S. et al. Ceramides and sphingomyelinases in senile plaques. 2014; 65: 193-201.
167. Filippov V, Song MA, Zhang K, Vinters HV, Tung S, Kirsch WM. et al. Increased ceramide in brains with Alzheimer's and other neurodegenerative diseases. Journal of Alzheimer's disease: JAD. 2012;29:537-47
168. Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K. et al. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2070-5
169. Kurz J, Brunkhorst R, Foerch C, Blum L, Henke M, Gabriel L. et al. The relevance of ceramides and their synthesizing enzymes for multiple sclerosis. Clinical science (London, England: 1979). 2018;132:1963-76
170. Vidaurre OG, Haines JD, Katz Sand I, Adula KP, Huynh JL, McGraw CA. et al. Cerebrospinal fluid ceramides from patients with multiple sclerosis impair neuronal bioenergetics. Brain: a journal of neurology. 2014;137:2271-86
171. Havulinna AS, Sysi-Aho M, Hilvo M, Kauhanen D, Hurme R, Ekroos K. et al. Circulating Ceramides Predict Cardiovascular Outcomes in the Population-Based FINRISK 2002 Cohort. 2016; 36: 2424-30.
172. Ji R, Akashi H, Drosatos K, Liao X, Jiang H, Kennel PJ. et al. Increased de novo ceramide synthesis and accumulation in failing myocardium. JCI insight. 2017 2
173. Razak Hady H, Błachnio-Zabielska AU, Szczerbiński Ł, Zabielski P, Imierska M, Dadan J. et al. Ceramide Content in Liver Increases Along with Insulin Resistance in Obese Patients. Journal of clinical medicine. 2019 8
174. Wigger L, Cruciani-Guglielmacci C, Nicolas A, Denom J, Fernandez N, Fumeron F. et al. Plasma Dihydroceramides Are Diabetes Susceptibility Biomarker Candidates in Mice and Humans. Cell Reports. 2017;18:2269-79
175. Chen L, Chen H, Li Y, Li L, Qiu Y, Ren J. Endocannabinoid and ceramide levels are altered in patients with colorectal cancer. Oncology reports. 2015;34:447-54
176. Kosinska MK, Liebisch G, Lochnit G, Wilhelm J, Klein H, Kaesser U. et al. Sphingolipids in human synovial fluid-a lipidomic study. PloS one. 2014 p. e91769
177. Yu Z, Peng Q, Li S, Hao H, Deng J, Meng L. et al. Myriocin and d-PDMP ameliorate atherosclerosis in ApoE-/- mice via reducing lipid uptake and vascular inflammation. Clinical science (London, England: 1979). 2020;134:439-58
178. Veeriah V, Lee SH, Levine F. Long-term oral administration of an HNF4α agonist prevents weight gain and hepatic steatosis by promoting increased mitochondrial mass and function. Cell death & disease. 2022;13:89
179. Kim JL, Mestre B, Malitsky S, Itkin M, Kupervaser M, Futerman AH. Fatty acid transport protein 2 interacts with ceramide synthase 2 to promote ceramide synthesis. The Journal of biological chemistry. 2022;298:101735
180. Rahmaniyan M, Curley RW Jr, Obeid LM, Hannun YA, Kraveka JM. Identification of dihydroceramide desaturase as a direct in vitro target for fenretinide. The Journal of biological chemistry. 2011;286:24754-64
181. Mody N, McIlroy GD. The mechanisms of Fenretinide-mediated anti-cancer activity and prevention of obesity and type-2 diabetes. Biochemical pharmacology. 2014;91:277-86
Author contact
![]() Corresponding author: Hua Wang, Department of Oncology, the First Affiliated Hospital of Anhui Medical University, Hefei, Anhui, China and Inflammation and Immune Mediated Diseases Laboratory of Anhui Province, Anhui Medical University, Hefei, Anhui, China. E-mail: wanghuaedu.cn; Xiaolei Li, Department of Oncology, the First Affiliated Hospital of Anhui Medical University, Hefei, Anhui, China. E-mail: lixiaoleiedu.cn.
Corresponding author: Hua Wang, Department of Oncology, the First Affiliated Hospital of Anhui Medical University, Hefei, Anhui, China and Inflammation and Immune Mediated Diseases Laboratory of Anhui Province, Anhui Medical University, Hefei, Anhui, China. E-mail: wanghuaedu.cn; Xiaolei Li, Department of Oncology, the First Affiliated Hospital of Anhui Medical University, Hefei, Anhui, China. E-mail: lixiaoleiedu.cn.