Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Molecular Pathophysiology of ICM
Mitochondrial Quality Control...
Regulation of MQC for Management...
Future Perspectives and...
Summary and Conclusion
Funding
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(2):426-448. doi:10.7150/ijbs.76223 This issue Cite
Review
Molecular Mechanisms of Mitochondrial Quality Control in Ischemic Cardiomyopathy
Xing Chang1, Ruxiu Liu1 ![]() , Ruibing Li2, Youyou Peng5, Pingjun Zhu4
, Ruibing Li2, Youyou Peng5, Pingjun Zhu4 ![]() , Hao Zhou3
, Hao Zhou3 ![]()
1. Guang'anmen Hospital of China Academy of Chinese Medical Sciences, Beijing, China.
2. Department of Clinical Laboratory Medicine, The First Medical Centre, Medical School of Chinese People's Liberation Army, Beijing, China.
3. Senior Department of Cardiology, The Sixth Medical Centre of People's Liberation Army General Hospital, Beijing, China.
4. Department of Respiratory and Critical Care Medicine, The Second Medical Center & National Clinical Research Center for Geriatric Diseases, Chinese PLA General Hospital, Beijing, China.
5. Montverde Future Academy Shanghai, 88 Jianhao Road, Pudong New District, Shanghai, China.
Received 2022-6-16; Accepted 2022-10-20; Published 2023-1-1
Abstract

Ischemic cardiomyopathy (ICM) is a special type of coronary heart disease or an advanced stage of the disease, which is related to the pathological mechanism of primary dilated cardiomyopathy. Ischemic cardiomyopathy mainly occurs in the long-term myocardial ischemia, resulting in diffuse myocardial fibrosis. This in turn affects the cardiac ejection function, resulting in a significant impact on myocardial systolic and diastolic function, resulting in a decrease in the cardiac ejection fraction. The pathogenesis of ICM is closely related to coronary heart disease. Mainly due to coronary atherosclerosis caused by coronary stenosis or vascular occlusion, causing vascular inflammatory lesions and thrombosis. As the disease progresses, it leads to long-term myocardial ischemia and eventually ICM. The pathological mechanism is mainly related to the mechanisms of inflammation, myocardial hypertrophy, fibrosis and vascular remodeling. Mitochondria are organelles with a double-membrane structure, so the composition of the mitochondrial outer compartment is basically similar to that of the cytoplasm. When ischemia-reperfusion induces a large influx of calcium into the cell, the concentration of calcium ions in the mitochondrial outer compartment also increases. The subsequent opening of the membrane permeability transition pore in the inner mitochondrial membrane and the resulting calcium overload induces the homeostasis of cardiomyocytes and activates the mitochondrial pathway of apoptosis. Mitochondrial Quality Control (MQC), as an important mechanism for regulating mitochondrial function in cardiomyocytes, affects the morphological structure/function and lifespan of mitochondria. In this review, we discuss the role of MQC (including mitophagy, mitochondrial dynamics, and mitochondrial biosynthesis) in the pathogenesis of ICM and provide important evidence for targeting MQC for ICM.
Keywords: Ischemic cardiomyopathy, mitochondrial quality control, mitophagy, mitochondrial dynamics, mitochondrial biogenesis, calcium signal
Introduction
ICM is a special type or late stage of coronary heart disease. It is mainly due to the long-term ischemia/hypoxia caused most commonly by coronary atherosclerosis, which leads to myocardial hypertrophy or stiffness, congestive heart failure and/or arrhythmia [1-3]. Other conditions that can lead to ICM include acute myocardial infarction accompanied by coronary microvascular damage or endothelial damage, coronary artery spasm, embolism, congenital coronary artery malformation and coronary arteritis [4]. The heart is different from other organs. When oxygen consumption by the heart increases, it can only meet that demand through coronary blood flow. Consequently, coronary stenosis that causes decreases regional blood flow will often lead to myocardial ischemia [5-7]. The main causes of coronary stenosis and myocardial ischemia are coronary atherosclerosis, arterial thrombosis, vascular inflammation/injury and coronary microvascular injury/abnormal coronary artery structure [8, 9]. The clinical symptoms of ICM are angina pectoris, heart failure, arrhythmia, thrombosis/embolism, dyspnea and so on (Fig. 1). Some patients also exhibit pulmonary edema. In late stage of ICM, heart failure, serious arrhythmia, and even death will occur [10]. Numerous studies have shown that the development of the pathological mechanism of ICM is closely related to mitochondrial quality control (MQC). The MQC includes mechanisms such as mitophagy, mitochondrial biosynthesis, mitochondrial fusion and fission. MQC can maintain a certain level of mitochondrial homeostasis and mitochondrial energy metabolism, thereby maintaining the homeostasis of the intracellular environment and the physiological function of cardiomyocytes. In this review, we discuss the role of MQC in different pathological mechanisms of ICM, and provide references for targeted therapy of the complex mechanisms of ICM.
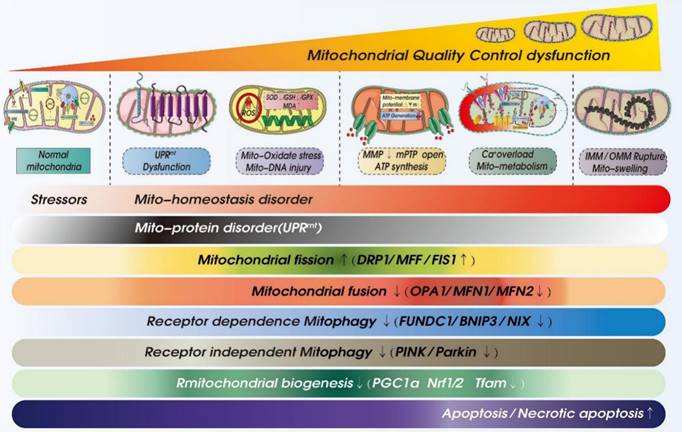
ROS-mediated mitochondrial dysfunction in ICM. Legend: Oxidative stress damage caused by excessive accumulation of ROS can seriously damage mitochondrial function. When mitochondria are dysfunctional, mitochondrial inner and outer membrane permeability (OMM) functions are also dysfunctional, which leads to abnormal opening of mitochondrial permeability transition pore (mPTP). It is also accompanied by calcium homeostasis disorder and tricarboxylic acid circulation disorder. Calcium signal conduction dysfunction and mitochondrial energy metabolism (mitochondrial respiration) dysfunction also occur.
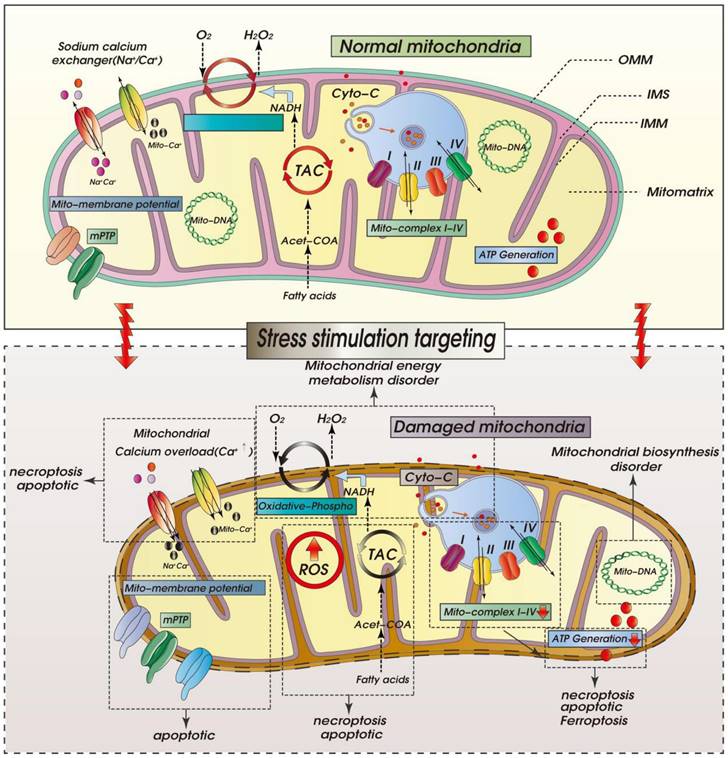
Molecular Pathophysiology of ICM
Inflammation
The pathogenesis of ICM involves coronary artery stenosis caused by atherosclerosis, which affects the supply of blood oxygen, but its essence is inflammatory [11]. This inflammatory process underlying ICM involves the participation of a variety of pathological mechanisms and is a stress response to myocardial tissue injury or coronary microvascular endothelial injury. It is also an inevitable mechanism contributing to the healing process after myocardial injury [12]. In the late stage of acute myocardial infarction, there will be a long-lasting inflammatory reaction within the myocardium or vascular endothelium, which can lead to the rupture of coronary atherosclerotic plaque and cause more serious damage. Neutrophils, lymphocytes, and monocyte macrophages in the blood circulation will be activated and release interleukin-6 (IL-6). This rapid increase of inflammatory markers and inflammatory cells in the circulation after myocardial infarction is consistent with the increase of the recurrence rate of cardiovascular events and is a predictor of recurrent cardiovascular events [13].
The explosive inflammatory response can cause great injury to myocardium [14]. Common cardiovascular risk factors such as high sugar, high fat, high salt diet, smoking, hypertension and insulin resistance can lead to chronic inflammation, redox imbalance and mitochondrial homeostasis disorder [15]. In addition, oxidized or modified LDL will attract white blood cells into the coronary intima, which are phagocytosed by macrophages, resulting in the formation of foam cells further development of atherosclerotic plaques. The growth of foam cells will gradually increase the coronary arterial occlusion, leading to a mismatch between myocardial oxygen demand and oxygen supply, which can result in myocardial infarction or myocardial ischemic injury [16]. Moreover, the inflammatory reaction can be spread by modified LDL, leading to coronary endothelial or microvascular injury, fiber proliferation reaction and thrombosis in subintimal arteries, which can exacerbate acute coronary artery stenosis and myocardial ischemic injury [17]. Notably, within tissues under the stress of coronary ischemia, the inflammatory response also plays a transient protective role. The inflammatory response indirectly maintains the integrity of cardiac structure and cardiac function. However, if the inflammation persists and is not effectively controlled, it will lead to ICM.
Cardiomyocyte necrosis and apoptosis is accompanied by inflammatory reaction. This pro-inflammatory stage aims to remove damaged cardiomyocytes or vascular endothelial cells. However, in the process of removing damaged cells and promoting wound healing may lead to excessive activity of fibroblasts and replacement of cardiomyocytes with active fibroblasts, resulting in scar formation or myocardial fibrosis, which is also one of the reasons for serious myocardial fibrosis and myocardial hypertrophy are seen in later stages of ICM [18, 19]. In coronary artery endothelial cells, pro-inflammatory cytokines also induce the expression of adhesion molecules and promote immune cell adhesion to endothelium and transendothelial migration. It can also induce explosive production of reactive oxygen species (ROS), accelerate the process of endothelial cell apoptosis, stimulate the formation of collagen in microvascular endothelial cells and dysfunction of the fibrinolytic system [20]. It is therefore an important pathological basis for microvascular injury, the no reflow phenomenon and ICM in the later stage of myocardial ischemia and in acute myocardial infarction [18].
The inflammatory mechanism regulating NLRP3 is closely related to mitochondria, which play a key role in the initiation and regulation of NLRP3 inflammatory bodies or inflammasomes. NLRP3 activation can further induce mitochondrial homeostasis imbalance, induce NLRP3 de‑ubiquitination and the release of mitochondrially derived molecules and destroy mitochondrial DNA [21]. When mitophagy is blocked, mitochondrial ROS will have a cumulative effect that acts indirectly to activate NLRP3. Static NLRP3 is located within the endoplasmic reticulum. However, when inflammatory bodies are activated, NLRP3 is redistributed to the space around the nucleus and colocalizes with mitochondria, resulting in the destruction of mitochondrial structure and respiratory chain function, which further enhances production of ROS [22, 23].
It appears that mitochondria not only regulate NLRP3 activation, but are also the docking site for inflammatory body assembly. In stress stimulated cells, NLRP3 is activated and associated with mitochondria and mitochondrial associated membranes (MAMs) [24]. Mitochondria may also act as an “adapter” for NLRP3 activation, and the ROS production and redox imbalance associated with mitochondrial damage are important drivers of NLRP3 inflammatory body activation.
Current evidence suggests that mechanism regulating inflammatory is an important pathological factor contributing to ICM, and ROS bursts and NLRP3 activation mediated by stress-induced mitochondrial dysfunction may be key factors inducing ICM inflammation. Mitochondria may act as a “Trojan horse” to induce inflammatory storms, but they have dual regulatory roles, as can maintain the basic function of cardiomyocytes in the context of oxidative stress and inflammation [23].
Oxidative stress
Oxidative stress is defined as tissue destruction caused by an imbalance between ROS production and cell redox, which will have a variety of negative effects on the metabolic function and physiological activities of cardiomyocytes [25]. It is also an important regulatory mechanism that induces apoptosis and other forms of programmed cell death and is a key factor contributing to myocardial and vascular endothelial injury in ICM.
ROS are short-lived, low-molecular weight compounds, mainly derived from various redox reactions. Increases in ROS can alter cell signaling proteins and produce corresponding functional consequences that mediate the pathophysiological process of ICM [26]. ROS are mainly produced in the mitochondria. In healthy myocardium, ROS are an accidental by‑product of mitochondrial respiration, and their levels are controlled by antioxidant enzymes. Under many pathological conditions, it is difficult to maintain redox balance, and when the antioxidant cell defense system is inhibited by oxidative stress, mitochondrial dysfunction, apoptosis or other forms of programmed cell death can occur. Within the myocardium, excessive accumulation of ROS lead disruption of calcium homeostasis, cardiac remodeling induced by transforming growth factor signal transduction, apoptosis and necrosis [27]. In the pathogenesis of ICM, ROS production also interacts with inflammatory processes, fibrosis and dysregulation of mitochondrial homeostasis and has negative effects on angiogenesis, vascular tone and endothelial cell genome stability.
Under normal physiological conditions, ROS are rarely produced in the heart. Superoxide leaking from the mitochondrial electron transport chain is quickly reduced to water and molecular oxygen by the antioxidant enzyme system. The heart can cope with redox changes and shows a significant adaptive response to maintain normal contractility. Mitochondria are the “hub” of cell redox process. Many mitochondrial stress signals participate in intercellular signal transduction and interaction mechanisms to regulate adaptation of cardiomyocytes to exogenous and endogenous stimuli. Oxidative stress is sensed by mitochondria, which initiate the release of stress signals that regulate membrane polarization, adenine nucleotide levels, ROS production, excitation/contraction coupling and opening/closing of membrane permeability transition pores [28]. These mechanisms are key determinants of the recovery response of cardiomyocytes after ischemic injury.
As shown in Figure 1, within ischemic cardiomyocytes, a rapid increase of ROS induces electron leakage from mitochondrial respiratory complexes I and III, which disrupts release of cardiolipin. Resultant changes in the phospholipid composition of the inner mitochondrial membrane will lead to the instability of the electron transport chain complex and super-complex, ultimately leading to mitochondrial structural disruption and dysfunction [29]. As a result, cell energy metabolism (ATP production) will be diminished, leading to additional ROS production and completion of a vicious cycle. When the compensatory response of cardiomyocytes is continuously stimulated, there is excessive proliferation and hypertrophy of fibroblasts, which leads to development of myocardial fibrosis and cardiac dysfunction [30]. In hearts affected by ICM, chronic aseptic inflammation caused by myocardial ischemia, excessive accumulation of ROS will mediate the release of inflammatory cytokines and activate matrix metalloproteinases, resulting in collagen degradation. This may lead to myofibril disorder and ventricular remodeling and dilation. The ROS production stimulated by inflammatory cells also disrupts cellular ion exchange by inhibiting the activities of sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2a) and sarcolemmal (Na+ K+) Ca2+-ATPase, which results in decreased Ca2+ efflux and increased Ca2+ influx, ultimately leading to intracellular Ca2+ overload and cell death [31].
ROS mediated mitochondrial dysfunction is particularly damaging to the coronary vascular system, as the amount of oxygen delivered to the myocardium is greater than to other tissues or organs. The development of ICM increases the duration of myocardial ischemia and can lead to necrosis. Although early reperfusion can restore blood flow to the affected area, the myocardial injury caused by reperfusion injury may be much higher than that caused by ischemia itself. This is in large part because reperfusion may induce the explosive ROS production, and the resultant oxidative stress injury to the myocardium may be more serious than the ischemic injury [32]. Targeting the regulation of mitochondrial function or energy metabolism may be an important target for relief of oxidant stress in ICM, and this intervention should run through all stages of ICM.
Cell death
The cell death seen in patients with ICM has significant prognostic value with respect to cardiac function, quality of life and survival after coronary revascularization. Although numerous animal studies and clinical investigations have been performed to explore the appropriate detection modalities for cardiomyocyte death (e.g., dobutamine stress echocardiography [33], late contrast enhancement of magnetic resonance imaging [34], and technetium-99m single positron emission computed tomography [35]), it remains a mystery as to where cardiomyocyte death occurs exactly and how cardiomyocyte death is induced in different stages of ICM.
Apoptosis is a form of programmed cell death that contributes significantly to cardiomyocyte loss during ICM. It is characterized by sequential activation of caspase family enzymes, which is reportedly the downstream signaling cascade induced by mitochondria damage, endoplasmic reticulum (ER) stress or extracellular signals. Mitochondrial dysfunction promotes the leakage of mitochondria-localized pro-apoptotic factors (i.e., Cyt-C, Smac and HtrA2/Omi) into cytosol stimulating activation of caspase-3/-9. ER stress is an adaptive response to attenuate at the transcription level the buildup of abnormal proteins within the ER. However, uncontrolled ER stress augments the expression of caspase-12, which contributes to caspase-3 activation. In addition to organelle-mediated apoptosis, extracellularly stimulated apoptosis is initiated via the death receptor [36]. In brief, the binding of an apoptotic ligand (e.g., FasL or TNF-α) to its death receptor promotes the formation of death-inducing signaling complex (DISC).
Necrosis accounts for only about 90,000 cardiomyocyte deaths 2 h after coronary artery occlusion and has no effect on the non-ischemic myocardium. Interestingly, necroptosis, a regulated form of necrosis, reportedly accounts for more than 50% of cardiomyocyte death during acute myocardial ischemia. Necroptosis is primarily induced by the activation of intracellular serine/threonine kinases receptor interacting protein kinase 1 (RIPK1) and RIPK3 as a result of inflammatory responses or oxidative stress [37]. This signal transduction pathway is accepted as the classical necroptotic cell death pathway involved in cardiomyocyte death during myocardial ischemia, myocardial infarction, and post-infarction cardiac injury. On the other hand, RIPK3 also phosphorylates PGAM5, a mitochondrial protein phosphatase participating in mitochondrial fission and mitophagy [38]. Cyclophilin D (CypD), is phosphorylated by PGAM5 at Ser31, which enhances the mPTP opening rate. As a complex channel bridging the inner and outer mitochondrial membranes, the long-term opening of mPTPs leads to mitochondrial swelling and ATP exhaust. Ca2+-calmodulin-dependent protein kinase (CaMKII) is another phosphorylation substrate of RIPK3 [37]. Like CypD, phosphorylated CaMKII is capable enhancing mPTP opening through a mechanism involving ROS overload.
It appears that levels of both RIPK3 and phospho-MLKL are significantly upregulated in ischemic cardiomyocytes. Genetic or pharmacological blockade of necroptosis pathway molecules increases resistance to ischemic challenge and reduces infarct size, with a prominent improvement in cardiac function [39]. In addition to evidence from animal experiments, the necroptotic ratio, as revealed by RIPK1/3 staining and TUNEL, is significantly increased in myocardial samples from patients with ICM undergoing cardiac transplantation. By contrast, control samples (from the hearts of patients who died from non-cardiac causes) showed only faint and very scarce staining of RIPK1/3 staining and TUNEL [40]. Interestingly, a subsequent study reported that RIPK3 expression is primarily upregulated in the left ventricle of failing hearts 6 weeks after 60-min coronary occlusion, whereas RIPK3 expression is downregulated in right ventricle. On the other hand, MLKL expression is only elevated in the right ventricle and is unhanged in the left ventricle of failing heart 6 weeks after 60-min coronary occlusion [41]. The basis of these discrepancies is still unclear but indicate different responses and/or different signals regulating necroptotic cell death in left/right ventricles during myocardial ischemia.
Fibrosis
At later stages, ICM is accompanied by myocardial hypertrophy and heart failure, which are closely related to myocardial fibrosis. Myocardial fibrosis is an important part of tissue repair after myocardial ischemic injury. It is the result of persistent and repeatedly aggravated myocardial ischemia and hypoxia caused by coronary stenosis [42]. Myocardial fibrosis is the product of multiple pathological processes, including reactive interstitial fibrosis and perivascular fibrosis. Myocardial fibrosis can disrupt intracellular energy metabolism and the redox balance, adversely affecting the nutrition supply to myocardium [43] and inducing chronic inflammation, vascular remodeling and cell death. It can also lead to the development of ICM and, eventually, heart failure. With progressive myocardial fibrosis, the resultant cardiac cavity expansion can lead to increased ventricular wall thickness with multifocal white fiber cords or patches, and even transmural scarring [44]. The endocardium thickens and organic mural thrombosis is seen. Fibrotic injury of the myocardium is also accompanied by local atrophy with hypertrophy in areas adjacent to the fibrosis. Progression of the condition induces increases in heart volume and weight, which becomes the pathological basis of heart failure at later stages of ICM.
The interaction between fibroblasts and cardiomyocytes plays an important role in the course of myocardial fibrosis. Myofibroblasts can induce and regulate cardiomyocyte hypertrophy under stress or myocardial injury. Cardiac fibroblasts and cardiomyocytes interact through biochemical mediators such as TGF-β, angiotensin II (Ang-II) and various interleukins [45]. Signaling between fibroblasts and cardiomyocytes also occurs through crosstalk via gap junctional proteins and biomechanical interactions. With progression of myocardial fibrosis in later stage ICM, as the thickness of the ventricular muscle and the stress on the ventricular wall increase, many cells are experience increased biomechanical strain. Mechanically sensitive adhesion proteins such as integrin and cadherin mediate mechanical signals between fibroblasts and cardiomyocytes and their microenvironment and indirectly stimulate the production of extracellular matrix (ECM) [46].
Excessive deposition of ECM within heart tissue leads to hardening of the fibrotic tissue and reduces the compliance of the myocardium. The purpose of fibrosis is to promote or accelerate deposition of connective tissue to maintain the integrity and structure of injured myocardial tissue. However, This adaptive response gradually leads to scarring of the damaged tissue [47]. As cardiomyocytes are replaced by fibroblasts, myocardial tissue is disrupted and cardiac function is diminished.
Mitochondrial-derived ROS (mtROS)-induced TGF-β binds to cell surface receptors, leading to phosphorylation of the Smad transcription factor family to initiate gene expression, which may be a key factor in the proliferation and/or differentiation of fibroblasts. TGF-β-induced gene expression in fibroblasts requires mtROS produced by complex-III, and excessive accumulation of ROS can be detected in the mitochondrial matrix and cytoplasm. Application of antioxidants that target mtROS produced by complex-III reduces TGF-β-induced expression of pro-fibrotic genes [48], suggesting mtROS is an important regulator of normal TGF-β-mediated gene expression and fibrosis formation. Oxidative stress and inflammatory responses arising from mitochondrial dysfunction are the key factors contributing to myocardial fibrosis. Therapies targeting mitochondrial quality surveillance may be important targets for the treatment of myocardial fibrosis.
Calcium signaling disorder
Dysregulation of Ca2+ homeostasis is closely related to the pathogenesis of ICM. Ca2+ overload within cardiomyocytes can be induced by inflammatory responses, oxidative stress injury and mitochondrial dysfunction, and can stimulate progression of both myocardial fibrosis and myocardial hypertrophy [49]. The Ca2+ distribution is uneven within the myocardium. In many cell types cytosolic free Ca2+ acts as a second messenger transducing signals from extracellular stimuli. In cardiomyocytes at rest, the cytosolic Ca2+ concentration in cardiomyocytes is tightly regulated to around 10-7-10-8 mol/L through the action of SERCA2a and sarcolemmal (Na+ K+) Ca2+-ATPase. Transient changes in the level of cytosolic Ca2+ within cardiomyocytes and vascular smooth muscle mediate excitation-contraction coupling in these cells. Therefore, levels of cytosolic Ca2+ are strictly regulated [50]. Within the ischemic myocardium, extracellular signals can increase the cytosolic Ca2+concentration by increasing extracellular Ca2+ influx through opening of Ca2+channels in cell membrane and Na+/Ca2+ exchange and release of Ca2+ from intracellular stores in the sarcoplasmic reticulum. Within the ischemic myocardium excessive influx of extracellular Ca2+ and release of store Ca2+ can lead to Ca2+ overload [51]. Within vascular smooth muscle cells, the increased contractility caused by Ca2+ overload can lead to increased total peripheral blood flow resistance and hypertension, and gradually destroy the structural integrity of arteries and arteriolar walls. Vascular Ca2+ overload exacerbates arteriosclerosis in various animal models and coronary plaques in human, thereby contributing to the pathogenesis of ICM [52].
Mitochondria are the energy suppliers for cells. They also monitor signals from the extracellular and intracellular environment, including growth factors, ROS and DNA damage signals. In the animal models of ICM, mitochondrial dysfunction is a key factor contributing to myocardial Ca2+ dysregulation and injury [53]. Mitochondria are also important nodes for regulation of the cytosolic free Ca2+ concentration by interacting with the endo/sarcoplasmic reticulum (ER/SR) through “mitochondria ER contacts.” The molecular assembly that forms this connection provides a local environment that enhances the exchange of Ca2+ and other signals between the two organelles. Consequently, ischemia will not only cause mitochondrial oxidative stress damage, but also induces ER/SR stress, which leads to release of Ca2+ from the ER/SR into the mitochondria causing mitochondrial Ca2+ overload [54]. The resultant mitochondrial dysfunction is considered to be a “transit-station” between ER stress and cardiomyocyte Ca2+ overload [150].
Mitochondria are formed by a double membrane. The inner membrane contains the components involved in oxidative phosphorylation (ATP production), while the outer membrane controls mitochondrial dynamics, including their fission and fusion. In addition to helping to control levels of cytosolic Ca2+ and intracellular Ca2+ signaling, mitochondrial Ca2+ uptake also plays key role in regulating ATP production, autophagy, and cell death. Ca2+ is taken up into mitochondria primarily via mitochondrial calcium transporter. Dysregulation of mitochondrial Ca2+ homeostasis may trigger the opening of mPTPs in the inner membrane, which can lead to uncoupling of oxidative phosphorylation, mitochondrial swelling and mitochondrial inner or outer membrane damage [55]. Under normal conditions, mPTPs allow positive ions to enter the mitochondrial matrix. When affected by ICM, however, mitochondrial permeability is changed, enabling the soluble protein apoptosis inducing factor (AIF) to be released into the cytosol, where it induces apoptosis [56].
In the pathogenesis of ICM, intracellular Ca2+ homeostasis is closely related to ROS-mediated oxidative stress. The redox balance within cardiac and vascular myocytes is governed by the balance between ROS production and the activities of antioxidant enzymes. Although the relationship between oxidative stress and Ca2+ overload associated with ICM remains to be further clarified, ROS formation under various experimental and clinical conditions may directly or indirectly disrupt intracellular calcium homeostasis [57]. For instance, excessive accumulation of ROS can interfere with Ca2+ channels in both the SR and plasma membrane, resulting in extracellular Ca2+ influx and SR Ca2+ release. As mentioned earlier, upon reperfusion of the coronary artery and myocardium, the large influx of oxygen molecules into the ischemic tissue can also lead to the explosive production of ROS and myocardial mitochondrial damage. This will adversely affect mitochondrial oxidative phosphorylation, resulting in a lack of cell energy, which will, in turn, lead to SERCA dysfunction and release of excessive levels of Ca2+ from the SR, mitochondrial Ca2+ overload, and activation of apoptosis-related signaling [56].
In summary, the pathological mechanism underlying ICM is multifactorial and involves inflammation, oxidative stress, cell death, fibrosis and dysregulation of Ca2+ homeostasis (Table 1). Moreover, these mechanisms interact with each other, exacerbating the myocardial injury. Mitochondria can directly regulate intracellular Ca2+ homeostasis through the MCU and provide energy for cardiomyocytes through oxidative phosphorylation to promote cardiomyocyte survival. Mitochondrial can directly affect the regulatory mechanisms governing apoptosis through release of apoptotic signaling molecules that trigger the caspase apoptosis pathway. It is therefore necessary to further clarify the regulatory roles of mitochondria in the multiple mechanisms contributing to the pathogenesis of ICM.
Molecular Pathophysiology of ICM
| Mitochondrial quality control | Regulators | Model |
|---|---|---|
| Inflammation | IL-6/ICAM-1/MMP-9/TNF- α/IL-13/TLR/NLRP3 | Cardiomyocytes/endothelial cells |
| Oxidative stress | ROS/SOD/CAT/GSH/ GPX/TreX | Cardiomyocytes/endothelial cells |
| Cell death | Cyt-C/Smac/HtrA2/Omi/Caspase-3/Caspase-9/RIPK3/MLKL/CypD/CaMKII/ | Cardiomyocytes/endothelial cells |
| Fibrosis | α-SMA/cyto-C/TNF- α/ERK1-2/Smad/TGF-β | Cardiomyocytes |
| Calcium signal disorder | Cyto-C/SMAC/Omi/HTR2A /AIF/MCU1-MCU2/SERCA/RyR/CaMKII | Cardiomyocytes/endothelial cells |
Mitochondrial Quality Control (MQC) in ICM
MQC in cardiomyocyte bioenergetics and metabolism (aerobic respiration, anaerobic glycolysis)
There is currently no specific treatment for ICM. The main treatment methods involve adjusting living habits and control of blood pressure, blood lipid and blood glucose. As shown in Figure 2 and Table 2, recent studies have shown MQC (including mitochondrial dynamics, mitochondrial autophagy, and mitochondrial biosynthesis) to be centrally involved in the pathological mechanism underlying ICM and have put forward new research directions and targeted treatment strategies.
Regulatory mechanisms involved in MQC in cell homeostasis and ICM. MQC directly or indirectly regulates cellular internal and external stress signaling pathways, affects the activity of cardiomyocytes and microvascular endothelial cells, and contributes to multiple pathological mechanisms, including oxidative stress, cardiomyocyte hypertrophy, fibrosis, and Ca2+ homeostasis disorders, as well as apoptotic/necroptotic processes.
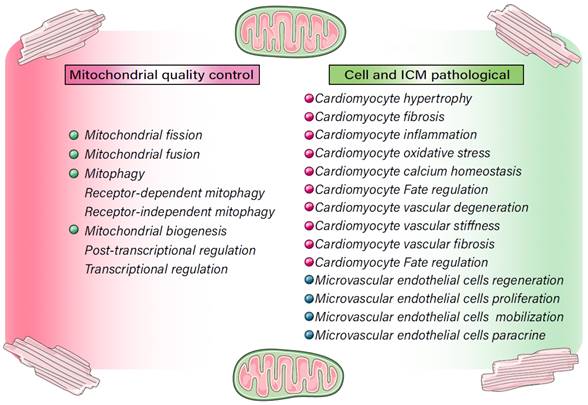
Mechanism of MQC in ICM
| Mitochondrial Quality Control | Protein substrate | Full name | Regulators | Target cell |
|---|---|---|---|---|
| Mitochondrial fission | Drp1/DLP1 | Dynamin-related protein 1 | ERK (extracellular regulated protein kinases) | Cardiomyocytes/Cardiac microcirculation endothelial cells (CMECs) |
| Fis1 | Fission 1 | TBC1D15 (TBC domain family member 15) | Cardiomyocytes | |
| Mff | Mitochondrial fission factor | 1.Voltage-dependent anion channel 1/hexokinase 2 (HK2/VDAC1) 2. DUSP1 (Dual-specificity protein phosphatase1) | Cardiac microcirculation endothelial cells (CMECs) | |
| Mitochondrial fusion | OPA1 | Optic Atrophy 1 | 1. TNFα receptor 2 (TNFR2) 2. AMPKα2 (AMP-activated protein kinase) | Cardiomyocytes |
| Mfn1 | Mitofusin1 | DNA-dependent protein kinase catalytic subunits (DNA-Pkcs) | Cardiomyocytes | |
| Mfn2 | Mitofusin2 | DNA-dependent protein kinase catalytic subunits (DNA-Pkcs) | Cardiomyocytes | |
| Mitophagy (Receptor-dependent) | FUNDC1 | FUN14 domain-containing 1 | 1. Serine/threonine kinase casein kinase2α (CK2α) 2. Nuclear receptor subfamily 4 group A member 1 (NR4A1) | Cardiomyocytes/Cardiac microcirculation endothelial cells (CMECs) |
| BNIP3 | BCL2/adenovirus E1B 19-kDa interacting protein 3 | 1. JNK(c-Jun N-terminal kinase) 2. Dual-specificity protein phosphatase1 (DUSP1) | Cardiomyocytes | |
| NIX | Nip3-like protein X | |||
| Mitophagy (Receptor-independent) | PINK1/Parkin | PTEN induced putative kinase 1 | AMP-activated protein kinase-α2 (AMPKα2) | Cardiomyocytes/Cardiac microcirculation endothelial cells (CMECs) |
| Mitochondrial biogenesis (Post-transcriptional regulation) | PGC-1 α/PPARγ | Peroxisome proliferator activated receptor γ /Peroxisomeproliferator-activated receptor-γ coactivator-1α | Cardiomyocytes | |
| NRF1/2 | Nuclear respiratory factor 1/2 | Cardiomyocytes | ||
| Mitochondrial biogenesis (Transcriptional- regulation) | TFAM | Transcription Factor A, Mitochondrial | Cardiomyocytes |
Levels of active energy metabolism in the heart are the highest in the body, as the heart requires large amounts of energy to maintain systolic activity. As an adaptation to the high energy demand needed to drive the contraction cycle and maintaining ionic homeostasis, mitochondrial density within cardiomyocytes is high. Within mature cardiomyocytes, mitochondria account for one-third of the total cell volume. Moreover, they are relatively stable structures and closely related to sarcoplasmic reticulum and myofibrils, enabling them to provide energy in the form of ATP and creatine and contribute to Ca2+ regulation [58]. About 90% of cellular ATP produced within the myocardium is used to support the systolic/diastolic cycle. Even slight changes in mitochondrial function can lead to significant changes in cardiomyocyte energy production and damage to cardiovascular health. Mitochondria are thus deeply involved in the processes ongoing with cardiomyocytes when the heart is ischemic, or cardiomyocytes are in a state of stress.
The mitochondrial energy metabolism system is composed of metabolic fuel transport and degradation pathways as well as fatty acid β-oxidation, electron transport chain, molecular motors (ATPase) and feedback signals all acting in concert in a non-equilibrium steady state. The role of mitochondrial oxidative phosphorylation in the heart is to maintain the high phosphorylation potential to replace ATP catabolized during systolic/diastolic cycling [59]. Oxidative phosphorylation is an aerobic metabolic process in which electrons pass through the inner membrane of mitochondria via the electron transfer chain, ultimately reducing oxygen to water molecules. Through this reaction series, protons are pumped against their concentration gradient (ΔPHm) into the mitochondrial membrane gap [60].
The mitochondrial energy metabolism system perfectly adapts ATP synthesis to ATP hydrolysis, which is closely related to the integrity of mitochondrial morphology and structure and the normal maintenance of energy metabolism. Because the structure, function and energy metabolism level of mitochondria directly affect the metabolic processes of cardiomyocytes, MQC is an essential regulatory factor governing the survival cycle of cardiomyocytes. Myocardial energy consumption mainly depends on oxidized fatty acids, glucose, lactic acid, and ketones, which are used to produce ATP through oxidative phosphorylation. However, this is not absolute and related to the metabolic, nutritional, and stress state of the body [61]. During movement, lactic acid accounts for a large proportion of the myocardial energy supply. The myocardium contains high levels of lactate dehydrogenase isozyme (LDH) 1, which catalyzes the dehydrogenation of lactic acid to, putting the reduced form of the coenzyme nicotinamide adenine dinucleotide (NADH) and pyruvate into the tricarboxylic acid (TCA) cycle to provide energy.
Mitochondria are where sugars, fatty acids and amino acids are oxidized to release the energy used for oxidative phosphorylation during ATP production. More than 95% of ATP in the mitochondria is produced through oxidation. Under normal physiological conditions, 60-90% of myocardial ATP is produced by the oxidation of fatty acids. The common pathway for final oxidation and oxidative phosphorylation is the tricarboxylic acid cycle [62]. These molecules are then used to reduce oxygen and release the energy used to synthesize ATP. When the heart is in an “energy starvation state,” aerobic oxidative metabolism of fatty acids and pyruvate is significantly inhibited. The heart then depends largely on increased sugar intake and glycolysis to provide energy. When the heart in an ischemic state, glycolysis is accelerated with the increase of glucose intake, and the activity of important enzymes is maintained by providing limited amounts of ATP [63]. However, when ischemia is severe, there is a decrease of glucose intake, depletion of glycogen, translocation and inactivation of phosphofructokinase, and inhibition of glyceraldehyde-3-phosphate dehydrogenase, all of which inhibit glycolysis and reduce ATP production. After ischemia/reperfusion, the recovery of fatty acid energy metabolism is faster than glucose oxidation, and the imbalance between glycolysis and glucose oxidation can further cause accumulation of metabolites (e.g., lactic acid and H+) exacerbating cardiomyocyte damage [64]. At this time, MQC plays a key regulatory role in cell metabolism.
As shown in Figure 3, mitochondrial energy metabolism plays important regulatory roles at different stages of ICM and reperfusion injury. In acute myocardial ischemia, the oxygen supply and nutrition/energy supply are lacking, which disrupts intracellular Ca2+ and other ion homeostasis. At the same time, there is a loss mitochondrial energy metabolism and mitochondrial membrane potential. As a result of the decrease in oxidative phosphorylation, ATP synthesis is insufficient, and the cell death program is initiated. During reperfusion, the oxygen and nutrient supplies are recovered, but the supply exceeds the metabolic ability of cardiomyocytes, which the accumulation of mitochondrial ROS and an increase in mitochondrial membrane potential, which stimulates abnormal opening of mPTPs. In addition, the overactivation of oxidative phosphorylation and the tricarboxylic acid cycle leads to excessive ATP and ROS production and, ultimately, mitochondrial dysregulation and oxidative stress damage [61, 65].
Mechanisms underlying mitochondrial homeostasis in cellular energy metabolism. Myocardial ischemia/reperfusion injuries contribute to the pathogenesis of ICM. During myocardial hypoxia/ischemia, and nutrients decline while Ca2+, Na+ and K+ channels open, lead to intracellular Ca2+ overload and disruption of intracellular ion homeostasis leading to a loss of mitochondrial membrane potential and decline of oxidative phosphorylation. This in turn will lead to a decline in ATP synthesis and cell energy metabolism disorders. After reperfusion, intracellular oxygen levels rise sharply, as do nutrient levels. In addition, cardiomyocytes exhibit Ca2+ overload and mitochondrial MCU dysfunction, exacerbation the exacerbating the Ca2+ overload. The transient increase in in oxygen supply leads to accumulation of ROS and increases in the mitochondrial membrane potential, which stimulates the abnormal opening of mPTPs. Oxidative phosphorylation and the tricarboxylic acid cycle are over-activated due the oversupply of blood oxygen and nutrients, leading to the overproduction of ATP and ROS, ultimately causing oxidative stress and disruption of mitochondrial homeostasis.
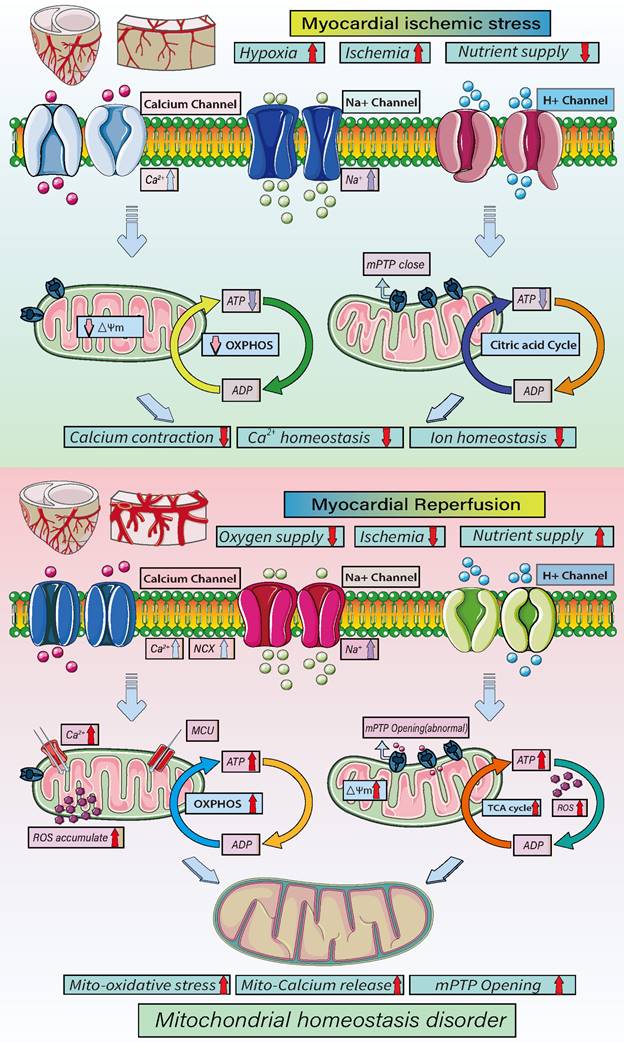
In addition, the dysregulation of mitochondrial dynamics, mitophagy and biosynthesis caused by acute ischemia and reperfusion directly affects the cell metabolism and death processes. It has been observed, for example, that feeding mice a high-fat diet (HFD, 60 kcal% fat) activates mitophagy, which alters cardiac mitochondrial energy metabolism and fatty acid oxidation and indirectly affects the synthesis of mitochondrial DNA. However, blocking parkin-mediated mitophagy leads to mitochondrial dysfunction and lipid accumulation, exacerbating myocardial injury and cardiac systolic/diastolic dysfunction [66], whereas activation of mitophagy using Tat Beclin1 reduces mitochondrial dysfunction, improves regulation of mitochondrial energy metabolism and fatty acid oxidation, reduces lipid accumulation, prevents cardiac diastolic dysfunction and reduces metabolic myocardial injury [67]. In addition, PINK1-Parkin-mediated mitophagy can shift mitochondrial substrate preference from carbohydrate to fatty acid [68].
Hexokinase II (HK-II) catalyzes the first step in glucose metabolism in cardiomyocytes and is deeply involved in mitochondrial energy metabolism. Moreover, HK-II also acts as a signaling molecule that suppresses cell death mediated via the mitochondrial pathway. During myocardial ischemia, HK-II serves as a sensor of metabolic disorder that enhances mitophagy to maintain basic energy metabolism and resist stress-induced mitochondrial damage [69]. Dissociation of HK-II from mitochondria under ischemic conditions induces mitochondrial recruitment and activation of parkin and enhances ubiquitination of mitochondrial proteins, thereby increasing levels of mitophagy and protecting against cell death [70]. However, the loss of mitochondrial HK-II under ischemia disrupts the stability of mitochondrial contact sites and leads to the degradation of BcL‑xL, increased permeability of the outer mitochondrial membrane, and release of cytochrome C. In addition, during reperfusion, the loss of HK-II stimulate ROS production and the opening of mPTPs, resulting in myocardial injury [71].
MQC in cardiomyocyte hypertrophy and fibrosis
Cardiomyocyte hypertrophy refers to increases in cardiomyocyte volume, diameter, width or length and the number of sarcomeres. Cardiomyocyte proliferation occurs when myocardial hypertrophy is excessive, which a key reason for myocardial hypertrophy and heart failure seen in the late stages of ICM [72]. Cardiomyocytes are terminal cells with high contraction and expansion characteristics. They generally do not independently proliferate, but able to increase in size a hypertrophic state.
The contractile proteins within cardiomyocytes include myosin, actin and tropomyosin. Experiments show that when the heart is stimulated by increased load, or the heart is hypertrophic, long-term excessive pressure and/or load can lead to contractile protein dysfunction and increased ventricular wall stress, resulting in myocardial hypertrophy [73]. Increased energy consumption induced by stress or myocardial dysfunction alters cell metabolism to increase mitochondrial work, which can trigger secondary diseases and worsen ICM, eventually leading to end-stage cardiomyocyte hypertrophy. Evidence shows that patients with myocardial hypertrophy and animal models of myocardial hypertrophy show excessive accumulation of mitochondrial ROS and decreased endogenous antioxidant enzyme activity. This is accompanied by mitochondrial dysfunction involving disruption of mitochondrial oxidative phosphorylation and mitochondrial protein homeostasis and [74].
In 3-week- and 6-month-old cardiac hypertrophy model mice, mitochondria exhibit an abnormal structure with increased numbers of smaller than normal mitochondria. Expression of mitochondrial DNA coding genes is inhibited, and mitochondrial ATP is reduced [75]. In addition, expression of the mitochondrial biosynthetic proteins PGC-1α and NRF1 is significantly inhibited in the mouse cardiac hypertrophy models [76]. This confirms the involvement of MQC in myocardial hypertrophy. Therefore, the interaction of both PGC1a and Nrf1/2 may have important regulatory effects on mitochondrial biogenesis and mitochondrial homeostasis. And the two can further affect the development and maturation of cardiomyocytes and the synthesis of mitochondrial mtDNA, and regulate the life process of cardiomyocytes.
MQC disorders not only lead to cardiomyocyte hypertrophy, but also contribute to myocardial fibrosis [77]. Studies have shown that the level of mitochondrial fragmentation is increased in cardiac fibroblasts treated with TGF-β1. In addition, expression of mitochondrial fission-related proteins Mff\Fis1 and Drp1 is significantly up-regulated, and levels of the fusion-related proteins opa1 and mfn1 is decreased. Inhibition of mitochondrial division significantly reduces TGF-β1- induced activation of cardiac fibroblasts. TGF-β1 also increases cellular levels of ROS and triggers Pink1-mediated mitophagy, and this effect is weakened by application of antioxidants. Mitochondrial fission also increases glycolysis, which plays a key role in the activation of cardiac fibroblasts [78]. Similar results are seen in primary mouse aortic adventitial fibroblasts. In those cells, Drp1 dephosphorylation induces mitochondrial fission and stimulates the proliferation, migration and phenotypic transition of AngII-treated outer membrane fibroblasts. HSP90 inhibit Drp1 dephosphorylation and significantly reverses Ang-II induced mitochondrial fission and fibroblast proliferation and migration [79]. The AngII-induced damage to myocardial mitochondria is related to an imbalance of mitochondrial redox. By comparing the effect of overexpression of mitochondrial targeted catalase (mCAT) with overexpression of peroxisome targeted catalase (pCAT) in mice, it can be shown that AngII-induced cardiac hypertrophy, fibrosis and mitochondrial damage is somewhat less in mCAT-overexpessing mice than WT mice [80].
MQC and cardiomyocyte inflammation
Cardiomyocyte inflammatory response plays an important regulatory role in the pathological mechanism of ischemic cardiomyopathy. It can directly affect the apoptosis or necroptosis program of cardiomyocytes and cause severe myocardial damage. We have previously reported the important regulatory role of inflammasome NLRP3 and mitochondria in ICM. In a mouse model of heart failure with preserved ejection fraction (HFpEF), mitochondria exhibit increased acetylation, and NLPR3 inflammasome assembly is increased on the hyperacetylated mitochondria. In addition, excessive production of IL-1β/IL-18 and severe myocardial fibrosis also appeared. Increases in β‑hydroxybutyric acid levels can attenuate the NLPR3 inflammasome formation, mitochondrial dysfunction and myocardial fibrosis caused by overexpression of pro-inflammatory cytokines. The interaction of mitochondrial hyperacetylation and inflammation is a key driver of the pathogenesis of HFpEF [81].
Studies have also shown that mitochondrial DNA released from autophagic cardiomyocytes promotes inflammatory responses mediated via Toll-like receptor (TLR-9) and can induce severe myocardial damage. Consistent with that finding, the myocardium of DNase II-deficient mice showed inflammatory cell infiltration and increased expression of inflammatory cytokine messenger RNA, and autolysosomes within the myocardium show deposition of mitochondrial DNA. TLR-9 knockout reduces the severity of the myocardial inflammation seen in DNase II-deficient mice [82].
MQC also indirectly affects inflammatory responses in rat cardiomyocytes. During LPS‑induced inflammation, STE20-like protein kinase 2 (MST2) mediates the up-regulation the mitochondrial fission genes Drp1, Mff and Fis1. This effect was associated with leading to reductions in mitochondrial ATP production, dysregulation of mitochondrial respiratory chain, increased mitochondrial oxidative stress, loss of membrane potential, and increased mitochondrial fragmentation. In addition, expression levels of IL-2, IL-8, MMP9, and Caspase-3/9 mRNA and protein are all increased. Also contributing to these effects was the interaction between Drp1 and Fis1 can alter mitochondrial dynamics within cardiomyocytes. Pretreatment with the MST2 inhibitor XMU-MP1 or inhibition of the Drp1-Fis1 interaction significantly reduces the mitochondrial fission and release of inflammatory factors, enabling mitochondrial homeostasis and cardiomyocyte activity to be maintained [83].
The studies summarized above indirectly confirm the regulatory function of MQC in cardiomyocytes under inflammatory stress, though the regulatory functions of mitophagy and the UPRmt under those conditions have not yet been elucidated. In a preliminary study, we found that mitophagy and the UPRmt enable mitochondrial energy metabolism and respiratory chain function to be maintained under stress, ensuring an energy supply for cardiomyocytes. It has also been observed that in LPS-treated cardiomyocytes, urolithin A increases mitophagy, reduces mitochondrial fragmentation, restores the mitochondrial membrane potential, inhibits excessive production of mitochondrial ROS, and significantly reduces the inflammatory injury of cardiomyocytes [84]. After FUNDC1 knockout, however, mitophagy is inhibited, and the protective effect of urolithin A is blocked.
Interestingly, enhancement of endogenous UPRmt by oligomycin reduces the mitochondrial injury and myocardial dysfunction. After FUNDC1 knockdown using targeted siRNA, mitophagy was inhibited, but UPRmt was not blocked. We therefore suggest that endogenous UPRmt is a downstream regulator of mitotic phagocytosis, serving as a supplementary regulator to maintain mitochondrial homeostasis and suppress myocardial inflammatory injury when mitophagy is inhibited. Mitophagy and UPRmt may thus play synergistic regulatory roles after inflammatory injury to cardiomyocytes and serve as complementary regulators protecting mitochondrial structure and function in cardiomyocytes under inflammatory stress [85].
Mitochondrial biogenesis may also play a regulatory role after inflammatory injury to cardiomyocytes. Sirtuin-3 (SIRT3) and AMPK indirectly stimulate mitochondrial biogenesis, thereby increasing mitochondrial renewal and cardiomyocyte functionality. In LPS-treated cardiomyocytes, the inflammatory responses are associated with decreases in SIRT3 and AMPK levels, which are followed by mitochondrial respiratory chain dysfunction, mitochondrial oxidative stress, and abnormal opening of mPTPs, as well as inhibition of PGC1α, Tfam and NRF2 expression and activation of caspase‑3. Conversely, overexpression of SIRT3 activates AMPK, improves mitochondrial redox balance and respiratory chain function, and increases expression of PGC1α, Tfam and NRF2 [86]. These findings suggest MQC is a key mediator of myocardial cell damage in cardiomyocytes subjected to inflammatory stress. In another study we found that PGC-1α knockout exacerbated neuropathy, whereas overexpression of TFAM had a protective effect on mitochondria. It can be seen that PGC-1α and TFAM can jointly regulate mitochondrial biosynthesis and turnover, which in turn affects mtDNA replication and mtDNA depletion [87].
MQC in cardiomyocyte oxidative stress and calcium homeostasis
In hears affected by ICM, cardiomyocytes produce large amounts of extracellular matrix and ROS. The oxidative stress caused by the excessive accumulation of ROS leads to alterations in cell membrane permeability and, in turn, Ca2+ overload, expression of inflammatory factors, and cell apoptosis. Because the myocardium is rich in mitochondria and mitochondria are the main producers of ROS [88]. ROS produced by mitochondria are important contributors to cardiomyocyte injury, and are a principle factor affecting mitophagy and mitochondrial fusion/fission [89].
As shown in Figure 4, during prolonged mitochondrial dysfunction, the damaged mitochondria are eliminated through mitophagy, and an imbalance in the mitophagic pathway can lead to a redox dysregulation with excessive ROS production and reductions in cell energy metabolism and ATP production. Members of the mitochondrial respiratory chain, including the NOX family and NADPH oxidase, are the main sources of ROS in cardiomyocytes. Interactions among several MQC mechanisms affect the mitochondrial oxidative stress response and become the main regulatory mechanism leading to cardiomyocytes death [37, 89].
Mechanisms of mitochondrial calcium overload-mediated oxidative stress injury and dysregulated mitochondrial. The mitochondrial calcium homeostasis mechanism is controlled by the mitochondrial calcium uptake and release system. Sarcoplasmic/Endoplasmic Reticulum Ca 2+ -ATPase (SERCA) is a pump that transports calcium ions from the cytoplasm to the ER. The activation of SERCA can inhibit the excessive release of calcium, thereby inhibiting the excessive production of mitochondrial ROS and improving mitochondrial function. At the same time, mitochondrial homeostasis disorder caused by mitochondrial calcium overload will further induce mitochondrial PTP activation and mitochondrial energy metabolism dysfunction, leading to excessive mitochondrial division and mitochondrial quality control network disorder.
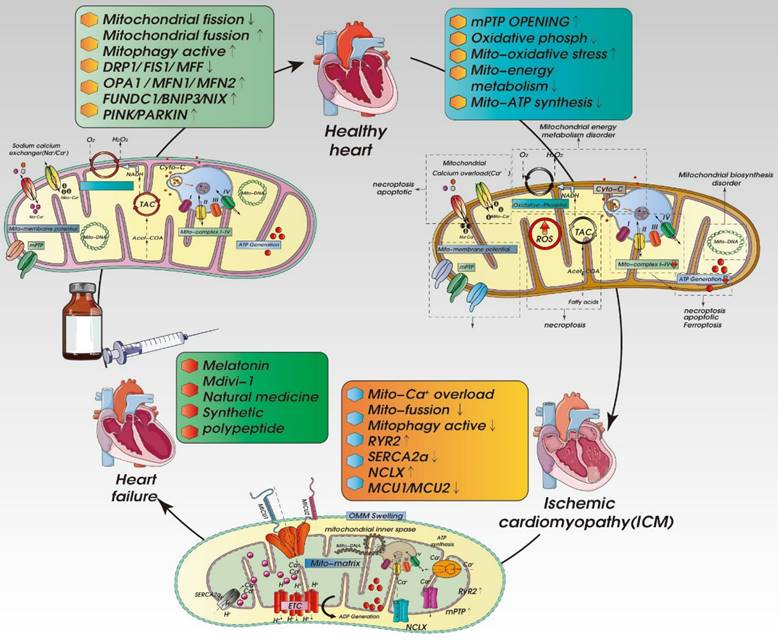
In patients with late stage acute myocardial ischemia as well as mice with heart failure induced by TAC, AMPK2α is converted to AMPK1α. In addition, PINK-mediated reductions in mitophagy and mitochondrial function are accompanied by oxidative stress damage and cardiomyocyte apoptosis. By increasing PINK-mediated mitophagy and improving mitochondrial function, cardiac overexpression of AMPK2α prevents the development of TAC-induced heart failure. By contrast, AMPK2α-/- mutant mice showed early progression of TAC-induced heart failure that was worsened by reducing cardiac mitophagy. Increasing cardiac mitophagy resulted in elimination of damaged mitochondria, improved mitochondrial function, and reductions in ROS production and cardiomyocyte apoptosis. It appears that AMPK2α- and PINK1-mediated mitophagy is a crucial regulatory mechanism that inhibits the progression of heart failure [90].
After 8 minutes of KCl-induced cardiac arrest in C57BL/6 wild-type mice, Drp1 translocates to the mitochondrial membrane, resulting in increased mitochondrial fission, and lactate and ROS production, and oxidative damage. Administration of Mdivi-1 during cardiopulmonary resuscitation can inhibit Drp1 activation, preserve mitochondrial morphology, reduce oxidative damage, and enhance myocardial function after spontaneous circulation is restored [91]. It thus appears that Drp1- and Mfn-mediated mitochondrial division and fusion directly and indirectly affect cardiomyocyte oxidative damage, and their dual regulation plays a leading role in mitochondrial homeostasis.
It also appears that insulin-like growth factor II (IGF-II), acting via its receptor IGF-IIR on cardiomyocytes, can also trigger mitochondrial dysfunction, mitochondrial ROS accumulation, oxidative stress damage, and mitochondrial disruption that leads to the formation of autophagosomes. The loss of mitochondrial contents increases levels of Parkin-dependent mitophagy and initiates the cell death program via the cytochrome c pathway [92]. This suggests that while excessive mitophagy can cause oxidative stress damage to cardiomyocytes, moderately upregulated mitophagy can protect cardiomyocytes from oxidative stress-induced damage. In HL-1 mouse atrial cardiomyocytes and AC16 human ventricular cells subjected to antimycin A-induced mitochondrial oxidative stress damage there was accumulation of mitochondrial ROS, and mitochondrial respiratory dysfunction. In addition, the nuclear DNA experienced oxidative damage, which triggered the cell death program. However, the mTOR inhibitor rapamycin, which suppresses antimycin A-induced protein ubiquitination induced mitophagy and promoted removal of some structurally damaged mitochondria, protecting the cells from oxidative stress [93].
MQC is also involved in the regulation of Ca2+ homeostasis. It is known that increases in intracellular Ca2+ can lead to activation of phospholipases, degradation of membrane phospholipids and increases membrane permeability that enable extracellular Ca2+ to flow into the cell down its concentration gradient and exacerbate Ca2+ overload [63]. Moreover, the mitochondrial dysfunction would decrease ATP production, suppressing the ATP-dependent activity of the plasma membrane Ca2+ pump and SERCA, thereby inhibiting removal of the excessively high cytosolic Ca2+ [61, 63].
The distribution of Mfn2 within cells is not limited to the outer mitochondrial membrane, as it has also been detected in the ER membrane. The dual positioning of Mfn1 in the ER and mitochondrial membranes is thought to facilitate Ca2+ transfer from the ER to the mitochondria, which could potentially cause mitochondrial Ca2+ overload [94]. Mfn2 knockdown using targeted siRNA disrupts ER morphology and the interaction between the ER and mitochondria, reducing the efficiency of mitochondrial Ca2+ uptake. This suggests that Mfn2 is a key mediator of Ca2+ transport between the ER and mitochondria and that Mfn2 is also an important regulator involved in maintaining Ca2+ homeostasis in cardiomyocytes under stress, which may explain why under some conditions Mfn2 mediates Ca2+ overload and cell death. Mfn2-deficient mice exhibit moderate cardiac hypertrophy with slight deterioration in cardiac function. However, the lack of Mfn2 increases the likelihood of Ca2+ overload, abnormal opening of mPTPs, changes in mitochondrial structure or morphology, and cell death [95].
MQC and cardiomyocyte longevity regulation
Cell death is the process by which multicellular organisms clear infected, damaged, or dysfunctional cells, and a way for the body to renew itself. This process can help a healthy myocardium clear damaged cells. However, the stressed myocardium, MQC disorders (such as excessive mitophagy) also can activate the cardiomyocyte death program [96, 97]. The mitochondrial cell death pathway starts from permeabilization of the mitochondrial outer membrane. Proteins such as cytochrome c and other molecules are released from the gap between the inner and outer mitochondrial membranes into the cytosol, which activates caspase-3/-9/-12 and leads to rapid cell death. Through MQC, mitophagy, fusion/fission, and mitochondrial biogenesis are all involved in regulating the fate of cardiomyocytes [98].
MQC is able to fully participate in the process of cardiomyocyte death (including apoptosis or necrosis). Under stressful conditions such as ischemia/hypoxia intracellular ROS will be overproduced and Ca2+ overload may occur. The balance between mitochondrial fusion and fission is disrupted, and Drp1 drives mitochondrial fission. In cardiomyocyte-specific Drp1 KO mice, mitochondria are elongated and there is accumulation of damaged mitochondria due to a decrease in mitophagy, which leads to increases in apoptosis. The severe mitochondrial dysfunction is also accompanied by left ventricular dysfunction, with some mice dying within 13 weeks [99].
It was also found that the specific deletion of Mfn2 from cardiomyocytes leads to accumulation of autophagic lysosomes and fusion of autophagosomes and lysosomes, while cardiac-specific Mfn2 KO mice exhibit dysregulation of mitochondrial energy metabolism. In addition, Mfn2 knockdown using targeted siRNA prevented autophagic lysosomal fusion in cardiomyocytes and increased the cells' susceptibility to ischemia-reperfusion. This suggests that not only does Mfn2 regulate mitochondrial fusion/fission, it also supports mitophagy in cells subjected to ischemic stress by mediating mitophagic lysosome fusion [100].
In addition to disrupting mitochondrial structure and function, hypoxia/ischemia induces AMPK activation. Stimulation of AMPK activity using the AMP analog AICAR increases mitophagy and blocks the mitochondrial apoptosis pathway. Conversely, blocking AMPK activation suppresses mitophagy and increase of cardiomyocyte apoptosis [101]. Under conditions of hypoxic/ischemic stress mitochondria will reorganize its structure and function to repair/remove damaged mtDNA and dysfunctional mitochondria, as mitochondrial fusion and fission directly affects the distributions of mitochondrial proteins and DNA. When mitochondria undergo fission into two daughter mitochondria. One has a high membrane potential and high fusion affinity, while the other has a lower membrane potential and low fusion affinity [102]. Investigation of mitochondrial fusion in the heart tube of Drosophila melanogaster revealed that heart-specific deletion of MARF [tinc Δ 4gal4 directional expression of mitochondrial assembly regulator] and OPA1 increases the heterogeneity of mitochondrial morphology and promotes systolic dysfunction. Moreover, inhibition of mitochondrial fusion increases compensatory expression of mitochondrial nuclear coding genes and the level of mitochondrial biogenesis [103].
We previously showed that FUNDC1 is a mitophagy receptor that interacts with LC3II to mediate mitophagy and regulate the number and quality of mitochondria in response to hypoxia‑induced cardiomyocyte injury. Under hypoxic stress, FUNDC1 can triggers mitophagy in platelets from wild-type mice. In cardiac-specific FUNDC1 KO mice, cardiac function is reduced and infarcted areas are larger following myocardial infarction. Hypoxic preconditioning can induce FUNDC1-dependent mitophagy and suppress cardiomyocyte death [104]. We also found that by regulating MQC, the serine/threonine kinase casein kinase 2α (CK2α) and DUSP1 can alter mitochondrial structure and function and promote or block cardiomyocyte apoptosis or necrosis via the mitochondrial pathway. CK2α induces FUNDC1 inactivation through a post-transcriptional modification at Ser13, thereby inhibiting mitophagy, disrupting mitochondrial homeostasis, promoting cardiomyocyte death and expansion of myocardial infarction area, and cardiac insufficiency. It can also lead to dysregulation of the mitochondrial gene transcriptome, inhibition of the electron transport chain, and suppression of mitochondrial biogenesis. With the resultant accumulation of ROS and abnormal opening of mPTPs, Drp1- and Fis1- mediated mitochondrial fission is up-regulated and Mfn- and OPA1-mediated mitochondrial fusion is inhibited, increasing the numbers of fragmented mitochondria. In addition, mitochondrial DNA is destroyed, which ultimately leads to mitochondrial apoptosis [105]. However, deletion of CK2α increases FUNDC1-mediated mitophagy, restores balance to mitochondrial fusion/fission, increases the level of mitochondrial biogenesis and the copy number/transcription of mtDNA, and inhibits cardiomyocyte apoptosis [105].
DUSP1 expression is down-regulated after acute cardiac ischemia/reperfusion injury. Compared to wild-type mice, DUSP1 transgenic mice also show a smaller infarct size and better cardiac function. DUSP1 deficiency promotes JNK activation, which enhances expression of Mff and promotes phosphorylation/activation of Bnip3. This increases mitophagy levels, and a large number of mitochondria to be cleared, resulting in insufficient mitochondrial ATP production and activation of caspase-3/-9-mediated cardiomyocyte apoptosis. Interestingly, reintroduction of DUSP1 attenuates Mff/Bnip3 activation, inhibit the mitochondrial fission and mitophagy, and reduce cardiomyocyte apoptosis [106].
MQC in vascular degeneration, stiffness and fibrosis
During the onset of ICM, the structure and function of coronary blood vessels undergo pathological changes that include thickening and hardening of arterial walls, dilatation of the vascular lumen, thickening of the arterial intima, and vascular calcification [107]. Under ischemic/hypoxic stress, the vascular collagen content increases while the elastin content decreases, which decreases the stretchability of the arterial wall. Eventually, the arterial blood supply function is reduced, and the myocardium becomes ischemic/hypoxic, which causes a decline of heart function and the occurrence of heart failure. Vascular endothelial dysfunction and arteriosclerosis are independently associated with an increased risk of ICM. These are therefore major therapeutic targets for ICM prevention and treatment [108, 109].
Under physiological conditions, vascular cells, and especially vascular endothelial cells, are a dynamic balance of regeneration, growth, degradation and death. In the process of cardiovascular disease or aging, blood vessels are degenerating and dying, and the ultrastructure of vascular endothelial cells are significantly altered. Aging vascular endothelial cells become flattened and widened, cell gaps also widen, intracellular ROS production increases, cell permeability increases, and macromolecular substances enter the intima from the plasma, resulting in abnormal endothelial cell function and impaired endothelium-dependent vasodilation, which eventually leads to vasospasm, thrombosis, macrophage infiltration, and inflammatory responses [110].
Mitochondria are present in all vascular cell types, where they play important roles regulating tissue repair, apoptosis, and a variety of physiological and pathological processes. As shown in Figure 5, in cardiomyocytes, excessive accumulation of mitochondrial ROS, impaired MQC and mitochondrial Ca2+ overload can lead to vascular injury [111]. However, different from mitochondria in cardiomyocytes, where there is high energy demand, the content of mitochondria in vascular endothelial cells accounts for only 2%-6% of the cytoplasmic volume, and their main function is to transmit the cells' responses to environmental signals [112]. In the pathogenesis of ICM, vascular endothelial dysfunction is closely related to mitochondrial structural and functional dysfunction. MQC is involved in the occurrence and development of vascular aging, arteriosclerosis and arterial fibrosis [112]. These processes interact with metabolism and energy demand in endothelial cells.
Mitochondrial and myocardial/coronary microvascular endothelial cell injury. Mitochondrial injury is accompanied by reduced ATP production and oxidative stress during coronary microvascular injury in end-stage IM and ICM. This stimulates increased levels of mitochondrial fragmentation. Excessive stress stimulation affects mitochondrial function and also leads to mitochondrial DNA damage, which can damage the transcription and translation of mitochondrial electron transport chain proteins and induce mitochondrial fusion dysfunction. Excessive mitochondrial damage and stress will promote mitochondrial division, thus transforming the reticular mitochondrial network into point and fragment mitochondria. However, FUNDC1 and PINK/Parkin mediated mitophagy cannot completely remove fragmented mitochondria, which will lead to dysfunction of mitochondrial energy metabolism, which will reduce ATP production and affect the life of myocardial cells. Furthermore, PGC1α- and Tfam-mediated mitochondrial biosynthesis dysfunction also affects mitochondrial neogenesis. lead to the death of coronary microvascular endothelial cells.
Activation of mitochondrial fission mediated by Drp1 and excessive production ROS in endothelial cells cause oxidative stress and endothelial dysfunction [113, 114]. With activation of Drp1 and excessive mitochondrial production of ROS, the level of mitochondrial fragmentation is increased. However, Drp1 knockdown using targeted siRNA or treatment with a mitochondrial fission inhibitor improves endothelial cell function, enables mitochondrial structure and redox balance to be maintained, and improves endothelial cell activity [115]. This makes regulation of mitochondrial fusion/fission key to the maintenance of endothelial cell function. Similar findings were also made in the vascular endothelium of spontaneously hypertensive rats, the thoracic aortas of which exhibit increased medial thickness and elevated expression of IL-6/TNF-α and Drp1. After administration of Mdivi‑1, however, inhibition of excessive mitochondrial fission reduces inflammatory responses and protects the vascular endothelium [116].
Selective mitophagy can reverse endothelial-dependent relaxation dysfunction that increases arterial stiffness in aging blood vessels by altering the total redox state of the vessels through inhibition the ROS production. Mitophagy inhibition exacerbates the vascular injury caused by oxidative stress, and interaction of fibrosis with mitochondrial fission inhibition is a key factor leading to vascular remodeling [117]. Protective mitophagy can delay the progression of vascular calcification, while lactate can induce expression of NR4A1 and activation of DNA-PKCs/p53. Inducing Drp1 to migrate to mitochondria in human aortic smooth muscle cells increases mitochondrial fission and fragmentation, inhibits BNIP3-mediated mitophagy, disrupts mitochondrial energy metabolism, and leads to abnormal opening of mPTPs and loss of mitochondrial membrane potential. These effects promote apoptosis among aortic smooth muscle cells and accelerates the Ca2+ deposition and vascular calcification. We also observed that in WT mouse endothelial cells, NR4A1 activates CK2α, promotes Mff phosphorylation, and enhances translocation of Drp1 to mitochondria, leading to excessive mitochondrial cleavage and disruption of mitochondrial structure and function and, ultimately, accumulation of damaged mitochondria and apoptosis. Deleting NR4A1 reverses these effects, enabling stable mitochondrial structure and function to be maintained and protecting against acute microvascular injury, which enabled vascular function and endothelial cell activity to be maintained [118]. These findings further confirm the important protective regulatory roles of MSQ, mitochondrial fusion/fission, mitophagy and mitochondrial biogenesis in vascular endothelial cells.
MQC in cardiac microvascular endothelial cell regeneration, proliferation, mobilization and paracrine function
The cardiac microvasculature is composed of coronary arteries with diameters ranging from 100-400 μm and capillaries with diameters less than 10 μm [119]. Unlike the main coronary blood vessels in the heart, coronary microvessels cannot be examined with coronary angiography. However, because microvessels account for 95% of the coronary circulation, and microvascular damage or disorder cannot be detected by early coronary angiography, coronary microvascular disease is clinically regarded as an “underwater iceberg” and a non-negligible contributor to the pathogenesis of ICM [120]. The primary function of the cardiac microvascular system is to distribute blood flow and play an important auxiliary role in regulating blood circulation. As the end of coronary efferent circulation, coronary microvessels can directly affect myocardial perfusion. Indeed, the impact of coronary microvascular dysfunction on myocardial ischemia is now receiving clinical attention [119], as coronary microvascular disorders cannot be resolved through percutaneous coronary intervention (PCI) or coronary artery bypass graft, and there are no drugs that specifically target coronary microvascular ischemia or dysfunction. When patients experience persistent chest pains or other ischemic symptoms without obvious coronary atherosclerosis or continue to show slow blood flow after PCI, these symptoms are currently believed to be related to coronary microvascular disease [121].
In ICM patients, microvascular damage generally occurs in necrotic or infarcted areas within the myocardium, so re-establishing blood flow may not yield improvement of the patient's condition. However, restoring flow can enable transmission of nutrients to the necrotic area, speeding up healing, reducing infarct expansion, delaying left ventricular remodeling, and promoting formation of new microvessels [122]. Moreover, mechanical stimulation of the vascular wall and vasoactive substances in the blood, and release to make stress response to external stimulation and unknown source information.
Microvascular endothelial cells cover the inner wall of blood vessel and are in direct contact with the bloodstream. They are usually in a differentiated and static state but under the action of angiogenic factors, they change from a differentiated to an undifferentiated state. The functions of microvascular endothelial cells include reducing vascular permeability, regulating material exchange between tissue and blood, maintaining blood fluidity by balancing anticoagulant/fibrinolytic systems and antiplatelet functions, and regulating vasoconstriction/relaxation by releasing vasoconstrictor and diastolic factors [123]. The energy demand in endothelial cells is relatively low, and glycolysis is the main source of ATP production. In cultured endothelial cells, at least 75% of ATP synthesis depends on glycolysis, and only a small part depends on mitochondria. Nevertheless, PGC-1α is abundantly expressed in endothelial cells, where it participates in mitochondrial biogenesis, exerts anti-apoptotic and anti-inflammatory effects, and improves the bioavailability of NO. Overexpression of PGC-1α increases expression of uncoupling protein 2 (UCP2), MnSOD, CAT and other antioxidant enzymes [124, 125]. In that way, PGC-1α contributes to the maintenance of mitochondria and repair of oxidative damage in microvascular endothelial cells [126]. Also involved are mitochondrial fusion\fission, mitophagy and other regulatory mechanisms.
In an earlier literature summary, we explained that RIPK3-mediated apoptosis is the primary factor contributed to the pathogenesis of myocardial ischemia-reperfusion injury. RIPK3 is also an important regulator endothelial cell apoptosis. Following ischemia-reperfusion, vascular endothelial expression of RIPK3, IP3R, and XO is enhanced, which leads to oxidative stress injury, Ca2+ overload, and mitochondrial swelling/deformation, and eventually vascular endothelial cell apoptosis initiated via the mitochondrial pathway [127].
In an experimental study of myocardial ischemic injury, we confirmed that RIPK3 migrates to SR, causing in stress injury in cardiomyocytes. It is therefore likely that the endothelial Ca2+ overload is related to RIPK3-mediated ER stress. Deletion of RIPK3 reduces expression of adhesion molecules and various inflammatory factors and reduces endothelial swelling and apoptosis, which enables the patency of the microvascular system to be maintained [127]. In addition, we showed that overexpression of SERCA can normalize the ratio between eNOS and ET-1 levels in cardiac microvascular endothelial cells, restore intracellular Ca2+ homeostasis, and inhibit abnormal activation of the MCU and opening of mPTPs [128]. This significantly reduced vascular wall edema and lumenal stenosis. Moreover, SERCA overexpression enhanced Mfn2- and OPA1‑mediated mitochondrial fusion, parkin-dependent mitophagy, PGC1‑α‑mediated mitochondrial biogenesis, and the level of mitochondrial DNA synthesis, which corrected the MQC imbalance and protected microvascular endothelial cells following ischemia/reperfusion [129].
Regulation of MQC for Management of ICM
Lifestyle modification, exercise, and smoking cessation
For ICM patients, poor lifestyle is an important factor that affects the pathological mechanism and pathogenesis of the disease. As mentioned before, sedentary lifestyles, smoking and other bad lifestyles can to varying degrees directly and indirectly cause vascular damage or blockage, becoming important pathogenic factors for ICM [130]. The recent decline in ischemic heart disease mortality is directly related to lifestyle changes. Research has found that about 40% of the decline in ICM deaths can be attributed to medical interventions and lifestyle interventions. Because many of these interventions have not yet reached their full potential in terms of the universality of the intervention protocol and acceptance by the public, it is expected that the mortality rate from ICM will continue to decline over the next few decades [131]. However, for this to occur it will be necessary for ICM patients to change their exercise habits, though the persistence of myocardial ischemia is determinant to optimization of exercise plans. Exercise training is an important auxiliary treatment for cardiac rehabilitation of ICM patients, which can reduce ICM mortality, morbidity and re-hospitalization rates, and can alleviate the development of myocardial hypertrophy and heart failure at later ICM stages [132].
From the perspective of mitochondrial redox balance, exercise or training can increase the activity of antioxidant enzymes and improve the ability of GSH, GST and GSH-Px to scavenge free radicals [133]. However, increasing exercise intensity and extending exercise duration can potentially cause a rapid increase of ROS production and resulting lipid peroxidation, which would be damage to the body, so care must be taken [134]. Studies have also shown that exercise inhibits and delays the progression of mitochondrial dysfunction by improving mitochondrial metabolism, biosynthesis, and respiratory chain function. Moreover, exercise affects post-translational modification of MQC-related proteins, enhances the function of mitochondrial kinetic regulatory proteins, and indirectly modifies the regulation of the mitochondrial fission/fusion balance and mitophagy [135].
Exercise has beneficial effects on age-related damage to mitochondrial biogenesis and dynamics and reductions of mitophagy. As a result, exercise may have an impact on crucial cell signaling pathways involved in maintaining mitochondria quality and quantity in the elderly. Although several exercise programs are known to improve mitochondrial structure and function, further research is needed to better understand the cell signaling pathways involved in the control of mitochondrial quality and quantity. Several exercise programs are known to modify mitochondrial activity and turnover, but additional study is needed to precisely determine the interaction mechanism between mitochondrial function, aging and physical activity, as well as the factors affecting mitochondrial function [136].
From the perspective of MQC, exercise maintains mitochondrial function and cardiomyocyte activity by promoting the clearance of dysfunctional mitochondria through mitochondrial biogenesis and mitophagy. Moderate exercise can cause mitochondrial oxidative stress and promote the mitophagy to remove damaged or dysfunctional mitochondria and maintain cell homeostasis. Moderate and heavy exercise have a two-way regulatory effect on mitochondrial fusion and fission [137]. During strenuous exercise in rats, Mfn1 and Mfn2 mRNA transcription and protein expression gradually decreases, while Fis1 expression and mitochondrial fragmentation increase. This may be due to the strong cellular demand for ATP during vigorous or prolonged exercise. Under those conditions, mitochondria may divide to produce more mitochondria to meet demand, but mitochondria with a split structure not only cannot produce enough ATP, but also affect the energy metabolism of the normal mitochondria [135, 138]. Exercise is thus a complex stimulus, and it may be possible to achieve two-way regulation of cardiac function by regulating the MQC governing myocardial mitochondria. Appropriate low- and medium-intensity exercise improve myocardial mitochondrial respiratory function as well as myocardial cell energy metabolism by up-regulating the MQC system. High-intensity exhaustive exercise may cause autophagy and even apoptosis through the MQC, reducing the energy metabolism level of cardiomyocytes [139].
Pharmacological intervention
In recent years, research has provided variety of drugs, such as the mitochondrially targeted antioxidant MitoQ, that exert protective effects on cardiomyocytes and endothelial cells by affecting the regulation MQC and mitochondrial homeostasis and mitigating myocardial injury after infarction [140]. Studies have shown that MitoQ has beneficial regulatory effects on vascular endothelial cells in elderly mice. Administration of MitoQ to elderly mice with primary aging-related endothelial dysfunction improves endothelial function and increases mitochondrial levels of PGC1a\mtSOD and COX-IV, while also reducing susceptibility to mitochondrial damage [141]. Another compound, the hormone melatonin, has been shown to significantly reduce infarct area and improve heart function and blood flow restoration after myocardial infarction. Melatonin inhibits Drp1‑dependent mitochondrial fission through AMP-activated protein kinase α (AMPKα); inhibits oligomerization of voltage‑dependent anion channel 1 (VDAC1), release of hexokinase 2, and abnormal opening of mPTP; improves the activity of mouse cardiac microcirculation endothelial cells; and reduces infiltration by inflammatory cells and endothelial damage. In addition, in both acute myocardial infarction patients and ischemia/reperfusion injury model mice, melatonin is able to mitigate ischemia/reperfusion injury. However, deletion of AMPKα eliminates the beneficial effects of melatonin on the quality of cardiomyocytes and endothelial cells [142].
Melatonin is also able to influence MQC and SR homeostasis through BAP31 and reduce LPS-mediated myocardial inflammatory damage. It does this by supporting mitochondrial function and inhibiting ER/SR stress, which improves cardiomyocyte viability. On the other hand, inhibition of the ERK pathway and BAP31 reduces melatonin's ability to affect mitochondrial function and ER/SR homeostasis under LPS stress [143]. Melatonin also suppresses myocardial damage caused by cardiorenal syndrome type 3 (CRS-3) following acute kidney injury. In the kidney, ischemia/reperfusion injury has the same adverse effects in the kidney as it does in the heart. That is, it inhibits the quality control of mitochondrial function, resulting in loss of mitochondrial membrane potential, mitochondrial fission/fusion balance and Ca2+ homeostasis, which disrupts energy metabolism. Interestingly, MCU activation eliminates the cardioprotection provided by melatonin, which results in disruption of mitochondrial structure and function in cardiomyocytes. It thus appears the negative effects of renal ischemia/reperfusion injury on myocardial vitality and cardiac function are related to IP3R phosphorylation, MCU upregulation, and Ca2+ overload. Melatonin protects heart function from CRS-3 by inhibiting IP3R-MCU signaling [144].
Several drugs used to treat or prevent the myocardial microvascular injury target the MQC. These include empagliflozin, a sodium-glucose cotransporter 2 (SGLT2) inhibitor that has been shown to have a cardioprotective effect. We observed that empagliflozin improves the structure and function of the diabetic myocardium, in part by inhibiting cardiac microvascular endothelial cell senescence as well as the production of mitochondrial ROS and the resultant oxidative stress, thereby protecting the barrier function of cardiac microvascular endothelial cells. Empagliflozin also supports eNOS phosphorylation and endothelial-dependent relaxation and improves microvessel density and perfusion. What's more, it triggers AMPK activation, inhibits Drp1 S616 phosphorylation and increases Drp1 S637 phosphorylation, which ultimately inhibits mitochondrial fission and restores mitochondrial function [145].
In addition, the mitochondrial fission inhibitor Mdivi-1 protects the myocardium by affecting the regulation of the mitochondrial fission/fusion. By inhibiting excessive mitochondrial fission during ischemia/reperfusion injury, Mdivi-1 preserves the integrity of mitochondrial structure and function in cardiomyocytes and reduces infarct area after myocardial infarction [146]. Mdivi-1 also reduces Drp1 expression, increases Mfn2 expression, suppresses excessive inhibition of autophagy, and preserves mitochondrial respiratory chain function and oxidative phosphorylation, thereby protecting cardiomyocytes [147]. Moreover, Mdivi-1 directly interacts with ion channels to maintain the stability of the intracellular electrophysiological environment in cardiomyocytes. in HL-1 cells, for example, Mdivi-1 treatment leads to increased production of spontaneous action potentials [148].
We also observed that quercetin, an active ingredient in various natural medicines, protects human cardiomyocytes subjected to hypoxia/reoxygenation. Quercetin pretreatment inhibits H/R‑mediated ROS production and oxidative stress. In addition, quercetin increases mitophagy, up-regulates transmembrane BAX inhibitor-1 motif containing 6 (TMBIM6) mRNA and protein expression, which preserves mitochondrial energy metabolism and suppresses the SR stress in human cardiomyocytes. These protective effects of quercetin are blocked by SIRT1 knockdown using targeted siRNA [149].
Future Perspectives and Translational issues for mito-protective agents in ICM treatment
In this review, we summarized the clinical symptoms, disease manifestations at different stages, and epidemiological characteristics of ICM. The incidence of ICM is related to unhealthy lifestyle habits, including smoking, lack of exercise, and a high-fat/high-sugar diet. At present, substantial progress has been made in our understanding of ICM and mitochondrial. Although the medical community has some understanding of the mechanism underlying the abnormal mitochondrial contributing to the occurrence and development of acute myocardial injury after myocardial infarction or ischemia/reperfusion, the pathological mechanism of coronary microvascular/microcirculation injury or dysfunction after acute ischemia/reperfusion has not yet been clarified, and there is a need for further research into relevant targeted therapeutic drugs. It will involve both animal and cell experiments and translational studies that explain the mechanism underlying the clinical features of coronary microvascular injury. This highly important, as coronary microvascular injury is a crucial driver of the occurrence and development of ICM. In addition, the roles of mitochondrial biosynthesis and the mitochondrial unfolded protein response in ICM is not well understood. It will therefore be necessary to conduct in-depth exploration of the mitochondrial quality detection system and directly connect ICM to ICM-related vascular injury. This could potentially serve as the basis for developing mitochondrially targeted strategies to improve myocardial and vascular function and provide a theoretical basis for the prevention and treatment of ICM and other vascular injury diseases.
Summary and Conclusion
In this review, we endeavored to summarize mitochondrial quality monitoring and the pathological mechanisms responsible for the development and progression of ICM. The important contribution of abnormal MQC to the occurrence and development of vascular aging and aging-related cardiovascular diseases is now well established. At present, however, research mainly involves animal and cell experiments, though a few translational studies have shown the clinical relevance of these mechanisms to humans. In addition, little is known about the role of changes in mitochondrial dynamics in vascular aging. It is therefore necessary carry out a more in-depth exploration to establish the precise linkage between changes in MQC and vascular aging and aging-related cardiovascular diseases. It is anticipated this will provide a theoretical basis for the development of mitochondrially targeted strategies to improve vascular function as well as to prevent and treat ICM.
Funding
This study is supported by the National Natural Science Foundation of China (No. 81902011)
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kirvalidze I, Jorbenadze T, Khetsuriani R. Etiopathogenesis of cardiomyopathies: a review. Georgian medical news 2009(174):104-108.
2. Hearse DJ. Myocardial ischaemia: can we agree on a definition for the 21st century? Cardiovascular research. 1994;28(12):1737-1744 discussion 1745-1736
3. Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN. et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation. 2020;141(9):e139-e596
4. Dong Y, Chen H, Gao J, Liu Y, Li J, Wang J. Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. Journal of molecular and cellular cardiology. 2019;136:27-41
5. Krukenkamp IB. Energy metabolism and oxygen consumption during myocardial ischemia. A review. Scandinavian journal of thoracic and cardiovascular surgery Supplementum. 1993;41:31-37
6. Heusch G. Myocardial Ischemia: Lack of Coronary Blood Flow or Myocardial Oxygen Supply/Demand Imbalance? Circulation research. 2016;119(2):194-196
7. Braunwald E, Sarnoff SJ, Case RB, Stainsby WN, Welch GH Jr. Hemodynamic determinants of coronary flow: effect of changes in aortic pressure and cardiac output on the relationship between myocardial oxygen consumption and coronary flow. The American journal of physiology. 1958;192(1):157-163
8. Elgendy IY, Mahtta D, Pepine CJ. Medical Therapy for Heart Failure Caused by Ischemic Heart Disease. Circulation research. 2019;124(11):1520-1535
9. Kaski JC, Crea F, Gersh BJ, Camici PG. Reappraisal of Ischemic Heart Disease. Circulation. 2018;138(14):1463-1480
10. Bairey Merz CN, Pepine CJ, Walsh MN, Fleg JL. Ischemia and No Obstructive Coronary Artery Disease (INOCA): Developing Evidence-Based Therapies and Research Agenda for the Next Decade. Circulation. 2017;135(11):1075-1092
11. Wolf D, Ley K. Immunity and Inflammation in Atherosclerosis. Circulation research. 2019;124(2):315-327
12. Prabhu SD, Frangogiannis NG. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circulation research. 2016;119(1):91-112
13. Ong SB, Hernández-Reséndiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek XY, Cabrera-Fuentes HA, Hausenloy DJ. Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacology & therapeutics. 2018;186:73-87
14. Dick SA, Epelman S. Chronic Heart Failure and Inflammation: What Do We Really Know? Circulation research. 2016;119(1):159-176
15. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circulation research. 2010;107(9):1058-1070
16. Niccoli G, Scalone G, Crea F. Acute myocardial infarction with no obstructive coronary atherosclerosis: mechanisms and management. European heart journal. 2015;36(8):475-481
17. Ruparelia N, Chai JT, Fisher EA, Choudhury RP. Inflammatory processes in cardiovascular disease: a route to targeted therapies. Nature reviews Cardiology. 2017;14(3):133-144
18. Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circulation research. 2014;114(2):266-282
19. Sager HB, Hulsmans M, Lavine KJ, Moreira MB, Heidt T, Courties G, Sun Y, Iwamoto Y, Tricot B, Khan OF. et al. Proliferation and Recruitment Contribute to Myocardial Macrophage Expansion in Chronic Heart Failure. Circulation research. 2016;119(7):853-864
20. Kingery JR, Hamid T, Lewis RK, Ismahil MA, Bansal SS, Rokosh G, Townes TM, Ildstad ST, Jones SP, Prabhu SD. Leukocyte iNOS is required for inflammation and pathological remodeling in ischemic heart failure. Basic research in cardiology. 2017;112(2):19
21. Liu Q, Zhang D, Hu D, Zhou X, Zhou Y. The role of mitochondria in NLRP3 inflammasome activation. Molecular immunology. 2018;103:115-124
22. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nature reviews Immunology. 2019;19(8):477-489
23. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221-225
24. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153(2):348-361
25. Mattera R, Benvenuto M, Giganti MG, Tresoldi I, Pluchinotta FR, Bergante S, Tettamanti G, Masuelli L, Manzari V, Modesti A. et al. Effects of Polyphenols on Oxidative Stress-Mediated Injury in Cardiomyocytes. Nutrients. 2017 9(5)
26. Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, Dong W. ROS and ROS-Mediated Cellular Signaling. Oxidative medicine and cellular longevity. 2016;2016:4350965
27. Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends in biochemical sciences. 2011;36(1):30-38
28. D'Autréaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nature reviews Molecular cell biology. 2007;8(10):813-824
29. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiological reviews. 2014;94(3):909-950
30. Cadenas S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochimica et biophysica acta Bioenergetics. 2018;1859(9):940-950
31. Peoples JN, Saraf A, Ghazal N, Pham TT, Kwong JQ. Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med. 2019;51(12):1-13
32. Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. The Journal of clinical investigation. 2018;128(9):3716-3726
33. Afridi I, Kleiman NS, Raizner AE, Zoghbi WA. Dobutamine echocardiography in myocardial hibernation. Optimal dose and accuracy in predicting recovery of ventricular function after coronary angioplasty. Circulation. 1995;91(3):663-670
34. Kim RJ, Wu E, Rafael A, Chen EL, Parker MA, Simonetti O, Klocke FJ, Bonow RO, Judd RM. The use of contrast-enhanced magnetic resonance imaging to identify reversible myocardial dysfunction. The New England journal of medicine. 2000;343(20):1445-1453
35. Maes AF, Borgers M, Flameng W, Nuyts JL, van de Werf F, Ausma JJ, Sergeant P, Mortelmans LA. Assessment of myocardial viability in chronic coronary artery disease using technetium-99m sestamibi SPECT. Correlation with histologic and positron emission tomographic studies and functional follow-up. J Am Coll Cardiol. 1997;29(1):62-68
36. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. 2021;18(5):1106-1121
37. Zhu P, Hu S, Jin Q, Li D, Tian F, Toan S, Li Y, Zhou H, Chen Y. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: A mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox biology. 2018;16:157-168
38. Zhu H, Tan Y, Du W, Li Y, Toan S, Mui D, Tian F, Zhou H. Phosphoglycerate mutase 5 exacerbates cardiac ischemia-reperfusion injury through disrupting mitochondrial quality control. Redox biology. 2021;38:101777
39. Koshinuma S, Miyamae M, Kaneda K, Kotani J, Figueredo VM. Combination of necroptosis and apoptosis inhibition enhances cardioprotection against myocardial ischemia-reperfusion injury. Journal of anesthesia. 2014;28(2):235-241
40. Corsetti G, Chen-Scarabelli C, Romano C, Pasini E, Dioguardi FS, Onorati F, Knight R, Patel H, Saravolatz L, Faggian G. et al. Autophagy and Oncosis/Necroptosis Are Enhanced in Cardiomyocytes from Heart Failure Patients. Medical science monitor basic research. 2019;25:33-44
41. Lichý M, Szobi A, Hrdlička J, Neckář J, Kolář F, Adameová A. Programmed Cell Death in the Left and Right Ventricle of the Late Phase of Post-Infarction Heart Failure. Int J Mol Sci. 2020 21(20)
42. Gyöngyösi M, Winkler J, Ramos I, Do QT, Firat H, McDonald K, González A, Thum T, Díez J, Jaisser F. et al. Myocardial fibrosis: biomedical research from bench to bedside. European journal of heart failure. 2017;19(2):177-191
43. González A, Schelbert EB, Díez J, Butler J. Myocardial Interstitial Fibrosis in Heart Failure: Biological and Translational Perspectives. J Am Coll Cardiol. 2018;71(15):1696-1706
44. Talman V, Ruskoaho H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell and tissue research. 2016;365(3):563-581
45. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cellular and molecular life sciences: CMLS. 2014;71(4):549-574
46. Guo Y, Gupte M, Umbarkar P, Singh AP, Sui JY, Force T, Lal H. Entanglement of GSK-3β, β-catenin and TGF-β1 signaling network to regulate myocardial fibrosis. Journal of molecular and cellular cardiology. 2017;110:109-120
47. Ma ZG, Yuan YP, Wu HM, Zhang X, Tang QZ. Cardiac fibrosis: new insights into the pathogenesis. International journal of biological sciences. 2018;14(12):1645-1657
48. Jain M, Rivera S, Monclus EA, Synenki L, Zirk A, Eisenbart J, Feghali-Bostwick C, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling. J Biol Chem. 2013;288(2):770-777
49. Gorski PA, Ceholski DK, Hajjar RJ. Altered myocardial calcium cycling and energetics in heart failure-a rational approach for disease treatment. Cell metabolism. 2015;21(2):183-194
50. Dewenter M, von der Lieth A, Katus HA, Backs J. Calcium Signaling and Transcriptional Regulation in Cardiomyocytes. Circulation research. 2017;121(8):1000-1020
51. Eisner DA, Caldwell JL, Kistamás K, Trafford AW. Calcium and Excitation-Contraction Coupling in the Heart. Circulation research. 2017;121(2):181-195
52. Fleckenstein-Grün G, Frey M, Thimm F, Hofgärtner W, Fleckenstein A. Calcium overload-an important cellular mechanism in hypertension and arteriosclerosis. Drugs. 1992;44(Suppl 1):23-30
53. Dridi H, Kushnir A, Zalk R, Yuan Q, Melville Z, Marks AR. Intracellular calcium leak in heart failure and atrial fibrillation: a unifying mechanism and therapeutic target. Nature reviews Cardiology. 2020;17(11):732-747
54. Gao P, Yang W, Sun L. Mitochondria-Associated Endoplasmic Reticulum Membranes (MAMs) and Their Prospective Roles in Kidney Disease. Oxid Med Cell Longev. 2020;2020:3120539
55. Gustafsson AB, Gottlieb RA. Heart mitochondria: gates of life and death. Cardiovascular research. 2008;77(2):334-343
56. Orrenius S, Gogvadze V, Zhivotovsky B. Calcium and mitochondria in the regulation of cell death. Biochem Biophys Res Commun. 2015;460(1):72-81
57. Görlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: A mutual interplay. Redox biology. 2015;6:260-271
58. Merry TL, Chan A, Woodhead JST, Reynolds JC, Kumagai H, Kim SJ, Lee C. Mitochondrial-derived peptides in energy metabolism. American journal of physiology Endocrinology and metabolism. 2020;319(4):E659-e666
59. Saks V, Monge C, Guzun R. Philosophical basis and some historical aspects of systems biology: from Hegel to Noble - applications for bioenergetic research. Int J Mol Sci. 2009;10(3):1161-1192
60. Jonckheere AI, Smeitink JA, Rodenburg RJ. Mitochondrial ATP synthase: architecture, function and pathology. Journal of inherited metabolic disease. 2012;35(2):211-225
61. Zhou H, Ren J, Toan S, Mui D. Role of mitochondrial quality surveillance in myocardial infarction: From bench to bedside. Ageing research reviews. 2021;66:101250
62. Fernie AR, Carrari F, Sweetlove LJ. Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Current opinion in plant biology. 2004;7(3):254-261
63. Boyman L, Karbowski M, Lederer WJ. Regulation of Mitochondrial ATP Production: Ca(2+) Signaling and Quality Control. Trends in molecular medicine. 2020;26(1):21-39
64. Valcarcel-Jimenez L, Gaude E, Torrano V, Frezza C, Carracedo A. Mitochondrial Metabolism: Yin and Yang for Tumor Progression. Trends in endocrinology and metabolism: TEM. 2017;28(10):748-757
65. Kolwicz SC Jr, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circulation research. 2013;113(5):603-616
66. Eiyama A, Okamoto K. PINK1/Parkin-mediated mitophagy in mammalian cells. Current opinion in cell biology. 2015;33:95-101
67. Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, Ikeda S, Shirakabe A, Sadoshima J. Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circulation research. 2019;124(9):1360-1371
68. Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW 2nd. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science (New York, NY). 2015;350(6265):aad2459
69. Roberts DJ, Tan-Sah VP, Ding EY, Smith JM, Miyamoto S. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Molecular cell. 2014;53(4):521-533
70. Tan VP, Smith JM, Tu M, Yu JD, Ding EY, Miyamoto S. Dissociation of mitochondrial HK-II elicits mitophagy and confers cardioprotection against ischemia. Cell death & disease. 2019;10(10):730
71. Pasdois P, Parker JE, Halestrap AP. Extent of mitochondrial hexokinase II dissociation during ischemia correlates with mitochondrial cytochrome c release, reactive oxygen species production, and infarct size on reperfusion. J Am Heart Assoc. 2012;2(1):e005645
72. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nature reviews Cardiology. 2018;15(7):387-407
73. Nadruz W. Myocardial remodeling in hypertension. Journal of human hypertension. 2015;29(1):1-6
74. Wijnker PJM, Sequeira V, Kuster DWD, Velden JV. Hypertrophic Cardiomyopathy: A Vicious Cycle Triggered by Sarcomere Mutations and Secondary Disease Hits. Antioxid Redox Signal. 2019;31(4):318-358
75. Rosca MG, Tandler B, Hoppel CL. Mitochondria in cardiac hypertrophy and heart failure. Journal of molecular and cellular cardiology. 2013;55:31-41
76. Wu W, Ziemann M, Huynh K, She G, Pang ZD, Zhang Y, Duong T, Kiriazis H, Pu TT, Bai RY. et al. Activation of Hippo signaling pathway mediates mitochondria dysfunction and dilated cardiomyopathy in mice. Theranostics. 2021;11(18):8993-9008
77. Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovascular research. 2009;81(3):449-456
78. Gao QY, Zhang HF, Tao J, Chen ZT, Liu CY, Liu WH, Wu MX, Yin WY, Gao GH, Xie Y. et al. Mitochondrial Fission and Mitophagy Reciprocally Orchestrate Cardiac Fibroblasts Activation. Front Cell Dev Biol. 2020;8:629397
79. Huang G, Cong Z, Wang X, Yuan Y, Xu R, Lu Z, Wang X, Qi J. Targeting HSP90 attenuates angiotensin II-induced adventitial remodelling via suppression of mitochondrial fission. Cardiovascular research. 2020;116(5):1071-1084
80. Dai DF, Rabinovitch P. Mitochondrial oxidative stress mediates induction of autophagy and hypertrophy in angiotensin-II treated mouse hearts. Autophagy. 2011;7(8):917-918
81. Deng Y, Xie M, Li Q, Xu X, Ou W, Zhang Y, Xiao H, Yu H, Zheng Y, Liang Y. et al. Targeting Mitochondria-Inflammation Circuit by β-Hydroxybutyrate Mitigates HFpEF. Circulation research. 2021;128(2):232-245
82. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K. et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485(7397):251-255
83. Tian Y, Song H, Qin W, Ding Z, Zhang Y, Shan W, Jin D. Mammalian STE20-Like Kinase 2 Promotes Lipopolysaccharides-Mediated Cardiomyocyte Inflammation and Apoptosis by Enhancing Mitochondrial Fission. Frontiers in physiology. 2020;11:897
84. Wang Y, Jasper H, Toan S, Muid D, Chang X, Zhou H. Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox biology. 2021;45:102049
85. Ng MYW, Wai T, Simonsen A. Quality control of the mitochondrion. Developmental cell. 2021;56(7):881-905
86. Xin T, Lu C. SirT3 activates AMPK-related mitochondrial biogenesis and ameliorates sepsis-induced myocardial injury. Aging (Albany NY). 2020;12(16):16224-16237
87. Chandrasekaran K, Anjaneyulu M, Choi J, Kumar P, Salimian M, Ho CY, Russell JW. Role of mitochondria in diabetic peripheral neuropathy: Influencing the NAD(+)-dependent SIRT1-PGC-1α-TFAM pathway. Int Rev Neurobiol. 2019;145:177-209
88. Bhatti JS, Bhatti GK, Reddy PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders - A step towards mitochondria based therapeutic strategies. Biochimica et biophysica acta Molecular basis of disease. 2017;1863(5):1066-1077
89. Zhou H, Wang J, Hu S, Zhu H, Toanc S, Ren J. BI1 alleviates cardiac microvascular ischemia-reperfusion injury via modifying mitochondrial fission and inhibiting XO/ROS/F-actin pathways. Journal of cellular physiology. 2019;234(4):5056-5069
90. Wang B, Nie J, Wu L, Hu Y, Wen Z, Dong L, Zou MH, Chen C, Wang DW. AMPKα2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circulation research. 2018;122(5):712-729
91. Sharp WW, Beiser DG, Fang YH, Han M, Piao L, Varughese J, Archer SL. Inhibition of the mitochondrial fission protein dynamin-related protein 1 improves survival in a murine cardiac arrest model. Critical care medicine. 2015;43(2):e38-47
92. Huang CY, Kuo WW, Ho TJ, Chiang SF, Pai PY, Lin JY, Lin DY, Kuo CH, Huang CY. Rab9-dependent autophagy is required for the IGF-IIR triggering mitophagy to eliminate damaged mitochondria. Journal of cellular physiology. 2018;233(9):7080-7091
93. Dutta D, Xu J, Kim JS, Dunn WA Jr, Leeuwenburgh C. Upregulated autophagy protects cardiomyocytes from oxidative stress-induced toxicity. Autophagy. 2013;9(3):328-344
94. Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(17):E2174-2181
95. Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O'Shea KM, Riley DD, Lugus JJ, Colucci WS, Lederer WJ. et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Molecular and cellular biology. 2011;31(6):1309-1328
96. Campos JC, Bozi LH, Bechara LR, Lima VM, Ferreira JC. Mitochondrial Quality Control in Cardiac Diseases. Frontiers in physiology. 2016;7:479
97. Uygur A, Lee RT. Mechanisms of Cardiac Regeneration. Developmental cell. 2016;36(4):362-374
98. Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR, Giorgi C, Pinton P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell calcium. 2018;69:62-72
99. Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M. et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circulation research. 2015;116(2):264-278
100. Zhao T, Huang X, Han L, Wang X, Cheng H, Zhao Y, Chen Q, Chen J, Cheng H, Xiao R. et al. Central role of mitofusin 2 in autophagosome-lysosome fusion in cardiomyocytes. J Biol Chem. 2012;287(28):23615-23625
101. Zhang H, Liu B, Li T, Zhu Y, Luo G, Jiang Y, Tang F, Jian Z, Xiao Y. AMPK activation serves a critical role in mitochondria quality control via modulating mitophagy in the heart under chronic hypoxia. International journal of molecular medicine. 2018;41(1):69-76
102. Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell death and differentiation. 2013;20(1):31-42
103. Dorn GW 2nd, Clark CF, Eschenbacher WH, Kang MY, Engelhard JT, Warner SJ, Matkovich SJ, Jowdy CC. MARF and Opa1 control mitochondrial and cardiac function in Drosophila. Circulation research. 2011;108(1):12-17
104. Zhang W, Siraj S, Zhang R, Chen Q. Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and protects the heart from I/R injury. Autophagy. 2017;13(6):1080-1081
105. Zhou H, Zhu P, Wang J, Zhu H, Ren J, Chen Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell death and differentiation. 2018;25(6):1080-1093
106. Jin Q, Li R, Hu N, Xin T, Zhu P, Hu S, Ma S, Zhu H, Ren J, Zhou H. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox biology. 2018;14:576-587
107. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovascular research. 2018;114(4):590-600
108. Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circulation research. 2015;116(7):1231-1244
109. Crafts TD, Jensen AR, Blocher-Smith EC, Markel TA. Vascular endothelial growth factor: therapeutic possibilities and challenges for the treatment of ischemia. Cytokine. 2015;71(2):385-393
110. Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature. 2005;438(7070):937-945
111. Zhang W, Huang Q, Zeng Z, Wu J, Zhang Y, Chen Z. Sirt1 Inhibits Oxidative Stress in Vascular Endothelial Cells. Oxid Med Cell Longev. 2017;2017:7543973
112. Caja S, Enríquez JA. Mitochondria in endothelial cells: Sensors and integrators of environmental cues. Redox biology. 2017;12:821-827
113. Vásquez-Trincado C, García-Carvajal I, Pennanen C, Parra V, Hill JA, Rothermel BA, Lavandero S. Mitochondrial dynamics, mitophagy and cardiovascular disease. The Journal of physiology. 2016;594(3):509-525
114. Dumont A, Lee M, Barouillet T, Murphy A, Yvan-Charvet L. Mitochondria orchestrate macrophage effector functions in atherosclerosis. Molecular aspects of medicine. 2021;77:100922
115. Huang M, Wei R, Wang Y, Su T, Li P, Chen X. The uremic toxin hippurate promotes endothelial dysfunction via the activation of Drp1-mediated mitochondrial fission. Redox biology. 2018;16:303-313
116. Liu X, Tan H, Liu X, Wu Q. Correlation between the expression of Drp1 in vascular endothelial cells and inflammatory factors in hypertension rats. Experimental and therapeutic medicine. 2018;15(4):3892-3898
117. Zhu Y, Han XQ, Sun XJ, Yang R, Ma WQ, Liu NF. Lactate accelerates vascular calcification through NR4A1-regulated mitochondrial fission and BNIP3-related mitophagy. Apoptosis. 2020;25(5-6):321-340
118. Zhou H, Wang J, Zhu P, Zhu H, Toan S, Hu S, Ren J, Chen Y. NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting Mff-required mitochondrial fission by CK2α. Basic research in cardiology. 2018;113(4):23
119. Chang X, Lochner A, Wang HH, Wang S, Zhu H, Ren J, Zhou H. Coronary microvascular injury in myocardial infarction: perception and knowledge for mitochondrial quality control. Theranostics. 2021;11(14):6766-6785
120. Zhou H, Ma Q, Zhu P, Ren J, Reiter RJ, Chen Y. Protective role of melatonin in cardiac ischemia-reperfusion injury: From pathogenesis to targeted therapy. J Pineal Res. 2018 64(3)
121. Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. 2015;131(6):550-559
122. Dikalova A, Mayorov V, Xiao L, Panov A, Amarnath V, Zagol-Ikapitte I, Vergeade A, Ao M, Yermalitsky V, Nazarewicz RR. et al. Mitochondrial Isolevuglandins Contribute to Vascular Oxidative Stress and Mitochondria-Targeted Scavenger of Isolevuglandins Reduces Mitochondrial Dysfunction and Hypertension. Hypertension (Dallas, Tex: 1979). 2020;76(6):1980-1991
123. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nature reviews Immunology. 2007;7(10):803-815
124. Li J, Geng XY, Cong XL. PGC-1α ameliorates AngiotensinII-induced eNOS dysfunction in human aortic endothelial cells. Vascular pharmacology. 2016;83:90-97
125. Sawada N, Jiang A, Takizawa F, Safdar A, Manika A, Tesmenitsky Y, Kang KT, Bischoff J, Kalwa H, Sartoretto JL. et al. Endothelial PGC-1α mediates vascular dysfunction in diabetes. Cell metabolism. 2014;19(2):246-258
126. Valle I, Alvarez-Barrientos A, Arza E, Lamas S, Monsalve M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovascular research. 2005;66(3):562-573
127. Zhou H, Wang J, Zhu P, Hu S, Ren J. Ripk3 regulates cardiac microvascular reperfusion injury: The role of IP3R-dependent calcium overload, XO-mediated oxidative stress and F-action/filopodia-based cellular migration. Cellular signalling. 2018;45:12-22
128. Li C, Ma Q, Toan S, Wang J, Zhou H, Liang J. SERCA overexpression reduces reperfusion-mediated cardiac microvascular damage through inhibition of the calcium/MCU/mPTP/necroptosis signaling pathways. Redox biology. 2020;36:101659
129. Tan Y, Mui D, Toan S, Zhu P, Li R, Zhou H. SERCA Overexpression Improves Mitochondrial Quality Control and Attenuates Cardiac Microvascular Ischemia-Reperfusion Injury. Molecular therapy Nucleic acids. 2020;22:696-707
130. Barbaresko J, Rienks J, Nöthlings U. Lifestyle Indices and Cardiovascular Disease Risk: A Meta-analysis. American journal of preventive medicine. 2018;55(4):555-564
131. Goldman L, Cook EF. The decline in ischemic heart disease mortality rates. An analysis of the comparative effects of medical interventions and changes in lifestyle. Annals of internal medicine. 1984;101(6):825-836
132. Magherini F, Fiaschi T, Marzocchini R, Mannelli M, Gamberi T, Modesti PA, Modesti A. Oxidative stress in exercise training: the involvement of inflammation and peripheral signals. Free Radic Res. 2019;53(11-12):1155-1165
133. Pingitore A, Lima GP, Mastorci F, Quinones A, Iervasi G, Vassalle C. Exercise and oxidative stress: potential effects of antioxidant dietary strategies in sports. Nutrition (Burbank, Los Angeles County, Calif). 2015;31(7-8):916-922
134. Powers SK, Radak Z, Ji LL. Exercise-induced oxidative stress: past, present and future. The Journal of physiology. 2016;594(18):5081-5092
135. Tanaka T, Nishimura A, Nishiyama K, Goto T, Numaga-Tomita T, Nishida M. Mitochondrial dynamics in exercise physiology. Pflugers Archiv: European journal of physiology. 2020;472(2):137-153
136. Moreira OC, Estébanez B, Martínez-Florez S, de Paz JA, Cuevas MJ, González-Gallego J. Mitochondrial Function and Mitophagy in the Elderly: Effects of Exercise. Oxid Med Cell Longev. 2017;2017:2012798
137. Joseph AM, Adhihetty PJ, Leeuwenburgh C. Beneficial effects of exercise on age-related mitochondrial dysfunction and oxidative stress in skeletal muscle. The Journal of physiology. 2016;594(18):5105-5123
138. Casuso RA, Huertas JR. The emerging role of skeletal muscle mitochondrial dynamics in exercise and ageing. Ageing research reviews. 2020;58:101025
139. Fiorenza M, Gunnarsson TP, Hostrup M, Iaia FM, Schena F, Pilegaard H, Bangsbo J. Metabolic stress-dependent regulation of the mitochondrial biogenic molecular response to high-intensity exercise in human skeletal muscle. The Journal of physiology. 2018;596(14):2823-2840
140. Xiao L, Xu X, Zhang F, Wang M, Xu Y, Tang D, Wang J, Qin Y, Liu Y, Tang C. et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox biology. 2017;11:297-311
141. Gioscia-Ryan RA, LaRocca TJ, Sindler AL, Zigler MC, Murphy MP, Seals DR. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. The Journal of physiology. 2014;592(12):2549-2561
142. Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q, Jin Q, Cao F, Tian F, Chen Y. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. Journal of pineal research. 2017 63(1)
143. Zhang J, Wang L, Xie W, Hu S, Zhou H, Zhu P, Zhu H. Melatonin attenuates ER stress and mitochondrial damage in septic cardiomyopathy: A new mechanism involving BAP31 upregulation and MAPK-ERK pathway. Journal of cellular physiology. 2020;235(3):2847-2856
144. Wang J, Toan S, Li R, Zhou H. Melatonin fine-tunes intracellular calcium signals and eliminates myocardial damage through the IP3R/MCU pathways in cardiorenal syndrome type 3. Biochemical pharmacology. 2020;174:113832
145. Zhou H, Wang S, Zhu P, Hu S, Chen Y, Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox biology. 2018;15:335-346
146. Maneechote C, Palee S, Kerdphoo S, Jaiwongkam T, Chattipakorn SC, Chattipakorn N. Differential temporal inhibition of mitochondrial fission by Mdivi-1 exerts effective cardioprotection in cardiac ischemia/reperfusion injury. Clin Sci (Lond). 2018;132(15):1669-1683
147. Aishwarya R, Alam S, Abdullah CS, Morshed M, Nitu SS, Panchatcharam M, Miriyala S, Kevil CG, Bhuiyan MS. Pleiotropic effects of mdivi-1 in altering mitochondrial dynamics, respiration, and autophagy in cardiomyocytes. Redox biology. 2020;36:101660
148. So EC, Hsing CH, Liang CH, Wu SN. The actions of mdivi-1, an inhibitor of mitochondrial fission, on rapidly activating delayed-rectifier K⁺ current and membrane potential in HL-1 murine atrial cardiomyocytes. European journal of pharmacology. 2012;683(1-3):1-9
149. Chang X, Zhang T, Meng Q, Shiyuan Wang, Yan P, Wang X, Luo D, Zhou X, Ji R. Quercetin Improves Cardiomyocyte Vulnerability to Hypoxia by Regulating SIRT1/TMBIM6-Related Mitophagy and Endoplasmic Reticulum Stress. Oxid Med Cell Longev. 2021;2021:5529913
150. Klinge CM. Estrogenic control of mitochondrial function. Redox biology. 2020;31:101435
Author contact
![]() Corresponding authors: Hao Zhou, Senior Department of Cardiology, The Sixth Medical Centre of People's Liberation Army General Hospital, Beijing, China; E-mail: zhouhaoorg. Pingjun Zhu, Department of Respiratory and Critical Care Medicine, The Second Medical Center & National Clinical Research Center for Geriatric Diseases, Chinese PLA General Hospital, Beijing, China; Email: zhupingjuncom. Ruxiu Liu, Guang'anmen Hospital of China Academy of Chinese Medical Sciences, Beijing, China; E-mail: liuruxiu1com.
Corresponding authors: Hao Zhou, Senior Department of Cardiology, The Sixth Medical Centre of People's Liberation Army General Hospital, Beijing, China; E-mail: zhouhaoorg. Pingjun Zhu, Department of Respiratory and Critical Care Medicine, The Second Medical Center & National Clinical Research Center for Geriatric Diseases, Chinese PLA General Hospital, Beijing, China; Email: zhupingjuncom. Ruxiu Liu, Guang'anmen Hospital of China Academy of Chinese Medical Sciences, Beijing, China; E-mail: liuruxiu1com.