Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Molecular mechanisms of...
Methods to detect necroptosis in...
Necroptosis drives death of...
Therapeutic options to target...
Conclusion and prospects
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(2):658-674. doi:10.7150/ijbs.77994 This issue Cite
Review
Targeting Necroptosis: A Novel Therapeutic Option for Retinal Degenerative Diseases
Qi Zhang1,2, Xi-min Hu1, Wen-juan Zhao1, Xiao-xia Ban1, Yan Li1, Yan-xia Huang1, Hao Wan1, Ye He3, Lv-shuang Liao4, Lei Shang5, Bin Jiang6, Guo-ping Qing7,8, Kun Xiong1,2,9 ![]()
1. Department of Anatomy and Neurobiology, School of Basic Medical Sciences, Central South University, Changsha, China.
2. Key Laboratory of Emergency and Trauma, Ministry of Education, College of Emergency and Trauma, Hainan Medical University, Haikou, China.
3. Changsha Aier Eye Hospital, Changsha, China.
4. School of Physical Education, Hunan Institute of Science and Technology, Yueyang, China.
5. Affiliated Eye Hospital of Nanchang University, Jiangxi Research Institute of Ophthalmology and Visual Science, Jiangxi Clinical Research Center for Ophthalmic Disease, Nanchang, China.
6. Department of Ophthalmology, The Second Xiangya Hospital, Central South University, Changsha, China.
7. Beijing Tongren Eye Center, Beijing Tongren Hospital, Capital Medical University, Beijing, China.
8. Beijing Ophthalmology and Visual Sciences Key Laboratory, Beijing, China.
9. Hunan Key Laboratory of Ophthalmology, Changsha, China.
Received 2022-8-13; Accepted 2022-12-15; Published 2023-1-1
Abstract

The discovery of the necroptosis, a form of regulated necrosis that is mediated by receptor-interacting protein kinase 1 (RIPK1), RIPK3, and mixed-lineage kinase domain-like pseudokinase (MLKL), represents a major breakthrough that has dramatically altered the conception of necrosis - traditionally thought of as uncontrolled cell death - in various human diseases. Retinal cell death is a leading cause of blindness and has been identified in most retinal diseases, e.g., age-related macular degeneration, glaucoma, retinal detachment, retinitis pigmentosa, etc. Increasing evidence demonstrates that retinal degenerative diseases also share a common mechanism in necroptosis. Exacerbated necroptotic cell death hinders the treatment for retinal degenerative diseases. In this review, we highlight recent advances in identifying retinal necroptosis, summarize the underlying mechanisms of necroptosis in retinal degenerative diseases, and discuss potential anti-necroptosis strategies, such as selective inhibitors and chemical agents, for treating retinal degenerative diseases.
Keywords: Retinal degenerative diseases, necroptosis, RIPK1, RIPK3, age-related macular degeneration, glaucoma
Introduction
Retinal cell death is a leading cause of vision impairment and blindness in a variety of retinal diseases [1-3]. Severe microenvironmental changes caused by retinal disease and injury can lead to the degeneration of retinal cells [4-6]. These microenvironmental changes include, but are not limited to, oxidative stress, hypoxic/ischemic stress, and excessive glutamate [7-9]. Mutations in genes encoding enzymes and retinoid-binding proteins also result in retinal cell death in inherited retinal diseases [10, 11].
According to different responses to various pathological stresses/genetic defects, cells may die from accidental cell death (ACD, also called necrosis) or from regulated cell death (RCD), a well-controlled process involving a variety of signaling cascades or effectors [12, 13]. Degeneration of retinal cells has been thought to occur through apoptosis, which is the most studied type of RCD mediated by activation of caspases (caspase-3/7/8/9) [14]. Apoptosis is characterized by cell shrinkage, nuclear condensation, and membrane blebbing without release of cellular contents [15]. Thus, apoptosis usually does not elicit an inflammatory response and pathological subsequences [16].
Autophagy is a self-degradative process that contributes to removing damaged cellular components, proteins, and inflammatory cytokines [17]. Excessive rates of autophagy can result in an autophagy-dependent cell death through excessive catabolism [18]. However, the role of autophagy in retinal degenerative diseases is debated. Autophagy in glaucoma promotes autophagic cell death of retinal ganglion cells (RGCs) [19]. In contrast, impairment of autophagy has been reported in patients with age-related macular degeneration (AMD), which causes the retinal pigment epithelium (RPE) cells to fail to sufficiently resist oxidative damage, thereby leading to RPE degeneration [20].
Unlike apoptosis and autophagy, pyroptosis is a pro-inflammatory form of RCD, characterized by cell swelling, membrane rupture, and inflammatory cytokine release [21]. Pyroptosis is initiated by activation of the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome complex [22]. Thereafter, activated caspase-1 from the complex cleaves gasdermin D (GSDMD) to induce the formation of cell membrane pores that facilitate cell rupture and release of intracellular cytokines [23]. Although pyroptosis and apoptosis are both regulated by caspases, caspase inhibition is not sufficient to hinder or prevent retinal cell loss, indicating that other forms of cell death are also involved [24].
Necroptosis is a form of regulated necrotic cell death governed by receptor-interacting protein (RIP) kinases (RIPKs), which contribute to the pathogenesis of neurodegenerative diseases [25, 26]. Emerging evidences indicate that necroptosis is a common mechanism of retinal cell death in addition to apoptosis [14, 27, 28]. Although the clinical manifestation and pathogenesis of each retinal disease differ, the necroptotic signaling pathways seem to be shared among them, at least in part, and inhibition of necroptosis provides efficient neuroprotective effects in the treatment of multiple retinal degenerative disorders [29-31].
In the present review, we summarize the current understanding of necroptotic mechanisms and their roles in retinal diseases and injuries. We also highlight key molecules involved the necroptosis of retinal cells and the respective inhibitors that promise to produce new therapeutic strategies for the management of retinal degenerative diseases.
Molecular mechanisms of necroptosis
RIPK1 dominates which to happen: apoptosis or necroptosis
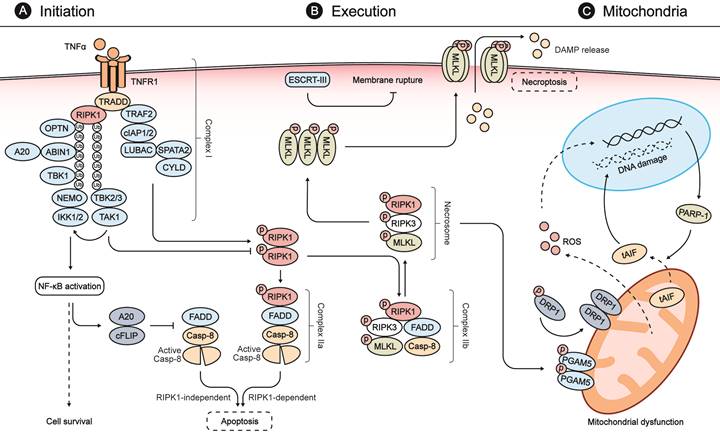
Necroptosis is initiated by diverse stimuli detected by specific death receptors which include, but are not limited to, tumor necrosis factor (TNF)-receptor 1 (TNFR1), FAS receptor, and Toll-like receptors (TLRs) [12, 25]. These death receptors commonly include a death domain (DD) that can recruit RIPK1 by homotypic binding following stimulation by their respective cognate ligands (Fig. 1) [25]. The TNF-mediated signaling pathway is one of the most studied pathways for necroptosis. Following binding of TNF-α to TNFR1, TNFR1-associated DD protein (TRADD) recruits RIPK1 to initiate the formation of a membrane-associated protein complex (complex I) to induce direct pro-inflammatory signaling via the activation of nuclear factor-κB (NF-κB). This inflammatory signaling cascade blocks the formation of cytosolic complex IIa, comprised of FADD and caspase-8, which would otherwise modulate RIPK1-independent apoptosis [25, 32, 33]. If RIPK1 is allowed to dimerize by blocking its ubiquitination, the activated RIPK1 dimer can interact with FADD and caspase-8 to form complex IIa, which mediates apoptosis [25, 33]. Under certain conditions, when cells are defective in activating caspase-8 or when RIPK3 is overexpressed, the activated RIPK1 recruits RIPK3 and MLKL, leading to the formation of complex IIb and subsequent execution of necroptosis [25, 33, 34].
Molecular mechanisms of necroptosis. (A) Stimulation of TNFR1 by TNFα promotes the formation of a membrane-associated protein complex (complex I), which is composed of TRADD, RIPK1, TRAF2, cIAP1/2, LUBAC, TAK1, and the IKK complex. In complex I, cIAP1/2 and LUBAC promote RIPK1 ubiquitination that recruits TAK1, TAB2/3, and the IKK complex composed of IKK1/2 and NEMO. Complex I can induce expressions of both pro-survival and pro-inflammatory genes via NFκB activation. NFκB activation, in turn, blocks the formation of FADD and caspase-8 complex (also called complex IIa-RIA) through A20 and cFLIP to modulate RIPK1-independent apoptosis. When complex I is unstable or the ubiquitination of RIPK1 is inhibited, stimulation of TNFR1 leads to the dimerization and activation of RIPK1, which interacts with FADD and caspase-8 to form complex IIa-RDA and mediates subsequent apoptosis. When the activity of caspase 8 is blocked, the activated RIPK1 recruits RIPK3 and MLKL, leading to the formation of complex IIb and subsequent phosphorylation of MLKL. (B) In the necrosome, RIPK3 phosphorylates MLKL and leads to its activation and oligomerization. Then the oligomerized MLKLs translocate to the plasma membrane to form a pore-forming complex, causing plasma membrane rupture and release of DAMP molecules, which provoke further inflammation and secondary injury. ESCRT-III, a downstream regulator of MLKL, can counteract a limited amount of plasma membrane perforation mediated by MLKL through shedding of the affected segments. (C) During the process of necroptosis, the necrosome can also phosphorylate PGAM5 and lead to a PGAM5-mediated mitochondrial dysfunction. The activated PGAM5 dephosphorylates DRP1 and causes its translocation from the cytosol to the mitochondrion to stimulate mitochondrial fission and ROS generation. The increasing levels of ROS subsequently induce PARP1 activation, which stimulates the release of tAIF from the mitochondria to the cytosol and, from there, to the nucleus, which provokes DNA degradation and subsequent cell damage.
Oligomerization and translocation of MLKL executes necroptosis
Phosphorylation of MLKL by the RIPK3 kinase domain is required for MLKL to function in the execution of necroptosis [35-37]. RIPK3 recruits and phosphorylates MLKL, leading to the latter's oligomerization and subsequent translocation to the plasma membrane, where it assembles into a pore-forming complex and causes loss of plasma membrane integrity [32, 36, 37]. The cell rupture caused by necroptosis can promote inflammation and secondary injury by releasing intracellular damage-associated molecular patterns (DAMPs) such as high-mobility group box 1 (HMGB1) protein [38]. This process can be counteracted by the endosomal sorting complex ESCRT-III, which can facilitate the shedding of sections of MLKL-embedded plasma membrane from intact cells [37, 39].
Mitochondrial mechanisms involving necroptosis
The mitochondria are the intracellular sites where ATP is generated to meet sustained cellular energy demands [40]. Mitochondria are also critical for regulation of apoptosis and necrosis via different mechanisms [41]. Induction of mitochondrial outer membrane permeabilization (MOMP) by BAX/BAK leads to the release of cytochrome c to promote caspase activation and apoptosis [42]. In contrast, opening of the mitochondrial permeability transition pore (mPTP) in the inner mitochondrial membrane (IMM) is a major event of necrosis that shuts down ATP production and leads to cellular energy starvation [18].
Mitochondrial dysfunction is also involved in necroptosis [40]. The RIPK1/RIPK3 necrosome can activate phosphoglycerate mutase family member 5 (PGAM5) [43]. PGAM5 can, in turn, cause the translocation of dynamin-related protein 1 (DRP1) from the cytosol to the mitochondrion to stimulate mitochondrial fission and reactive oxygen species (ROS) generation, both of which are early steps for the execution of the necroptosis [38, 43]. The mitochondrial protein apoptosis-inducing factor (AIF), a protein vital to respiratory chain stability and/or to maintenance of the mitochondrial structure, is also involved in the mitochondrial mechanisms of necroptosis [44]. Poly (ADP-ribose) polymerase 1 (PARP1) activation stimulates the release of truncated AIF from the mitochondria to the cytosol [44, 45]. Truncated AIF then rapidly translocates to the nucleus to provoke DNA degradation and subsequent cell damage. Notably, it was reported that the translocation of AIF could be attenuated by inhibition of RIPK1 during acetaminophen-evoked necroptosis, suggesting a crosstalk between TNFR1- and mitochondria-mediated necroptosis [46]. These key molecules mentioned above have been widely applied in characterizing necroptotic cell death in retinal degenerative diseases and discovering potential targeted therapeutic strategies.
Methods to detect necroptosis in retinal cells
Cell viability assays
The assessment of cell viability is fundamental to in vitro cell stress research [47, 48]. In combination with the treatment of necroptosis inhibitors, cell viability assays are important tools to estimate necroptosis-related injury to retinal cells in in vitro models of retinal degenerative diseases. Such assays include the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay and 4',6-diamidino-2-phenylindole (DAPI)/Hoechst staining [28, 49-51]. The in vitro cell viability of RGCs can also be measured by the release of lactate dehydrogenase, which rapidly increases in cell culture medium upon plasma membrane rupture following retinal ischemia-reperfusion (I/R) injury [28].
Morphological changes
Necroptosis shows similar morphological features to necrosis. Both are characterized by cytoplasmic swelling, plasma membrane rupture, and release of intracellular contents (Fig. 2) [12]. In contrast, apoptosis is marked by plasma membrane blebbing and apoptotic body formation, while autophagy-dependent cell death is characterized by accumulation of double membrane vesicles called autophagosomes [15, 18]. Although pyroptosis exhibits similar morphological changes to necroptosis, the two can be distinguished by specific inhibitors of their respective master regulators [38, 52]. Transmission electron microscopy (TEM) is usually used to identify the necroptotic morphology of retinal cells [14, 53-55]. For example, in retinal tissues from glaucoma patients, ultrastructural changes in RGCs revealed by TEM showed typical necroptotic morphology such as cell swelling and membrane rupture [56].
Detecting the necroptosis in retina. (A) Cell loss occurs widely in different retina injuries/diseases and leads to visual impairment, owing to ischemia/reperfusion, excitotoxicity, and inflammatory reactions. In rat retinas following high intraocular pressure (HIOP) injury (an in vivo animal model of retinal ischemia/reperfusion), hematoxylin and eosin staining indicated significant cell loss in the ganglion cell layer caused by excessive pressure. Cell membrane rupture played a critical role in the cell loss caused by HIOP injury - a notable observation, given that membrane rupture is a typical feature of necroptosis. (B) In vitro observation of R28 cells (a retinal precursor cell line) provides a feasible way to show retinal cell swelling (the other hallmark of necroptosis; upper left) and membrane rupture (upper right) following oxygen-glucose deprivation/recovery injury (an in vitro cell model of retinal ischemia/reperfusion). TEM enables an ultra-high-resolution characterization of retinal cell swelling and membrane rupture of necroptotic cell death (lower right) compared with the normal microscopy (lower left). (C) TNFα/Smac-mimetic/Z-VAD-FMK (TSZ)-induced cell necroptosis is a classical tool to investigate necroptotic signaling. As indicated by flow cytometry with propidium iodide/Annexin V staining, TSZ and TSZ+DMSO treatment showed a large number of necrotic cells (PI positive; upper left and/or right quadrants of each panel) compared with the normal group. Necrostatin-1 (Nec-1), a selective inhibitor of RIPK1 that can reverse necroptotic cell death, is widely used as a positive control to determine the existence of necroptosis by flow cytometry. (D) Upregulation of RIPK1, RIPK3, and MLKL indicated by immunofluorescence or immunohistochemistry are key biomarkers to identify necroptotic cell death. As showed by immunofluorescence assay, the expression of MLKL was significantly enhanced in retinal ganglion cells following HIOP injury, indicating the presence and execution of necroptosis.
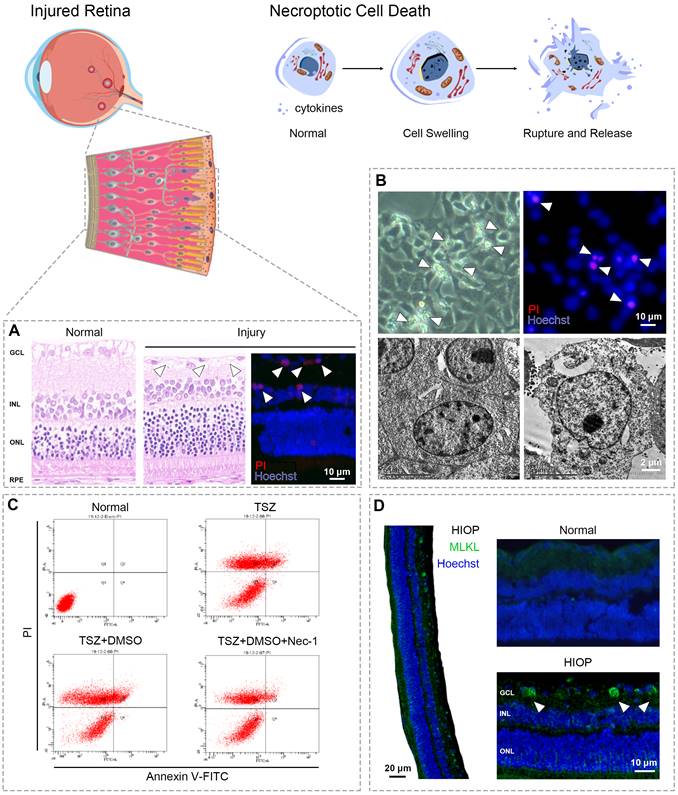
Fluorescence microscopy is also used to identify the plasma membrane rupture typical of necroptosis through propidium iodide (PI) staining in vitro and in vivo [28, 57, 58]. It's important to note that distinguishing necroptosis from necrosis and other PCD via PI and/or Annexin V staining must be paired with specific necroptotic inhibitors [28, 57].
Genetic features
Upregulated phosphorylation of RIPK1/3 and MLKL can also be used as an indicator of necroptosis [59]. In retinal tissue from glaucoma patients, the upregulated expression of RIPK1 in retinal cells was used to assess necroptosis indicated by immunohistochemistry [56]. Notably, the elevated MLKL immunofluorescence was applied as a typical necroptotic marker in AMD human retina sections [60]. The upregulated phosphorylation of RIPK1/3, and MLKL was also considered as key indicator of necroptosis in retinal cells [14, 28, 29]. Of note, RIPK1 plays multiple roles in necroptosis, apoptosis and inflammatory pathways; thus, upregulation of RIPK1 is insufficient to define necroptosis. Rather, detection must be made in combination with RIPK3 and/or MLKL expression, preferably along with inhibitors of other types of RCD [34, 61].
Necroptosis drives death of retinal cells in retinal diseases and injuries
Age-related macular degeneration
Age-related macular degeneration (AMD) is a major cause of irreversible visual loss in elderly people worldwide [20, 62]. The etiology of AMD is complicated, consisting of lipofuscin accumulation, chronic inflammation, and dysfunctional ocular microcirculation, to name just some components [63]. In the retinas of AMD patients, necroptotic retinal cell death was identified by elevated MLKL immunofluorescence [60]. Notably, the immunofluorescent intensity of MLKL was well correlated with the amount of lipofuscin accumulation.
Oxidative stress-induced death of RPE cells is recognized as the leading theory of AMD pathogenesis [64]. Sodium iodate is extensively used to induce oxidative stress in pre-clinical animal models of AMD [49]. Emerging evidence demonstrates that necroptosis is widely involved in sodium iodate-induced RPE and photoreceptor cell death (Table 1) [49, 64, 65]. Following sodium iodate injection, RIPK1 catalytic activity increases in rat retinas, with the thickness of the outer nuclear layer (ONL) being reduced in a time-dependent manner [65]. RIPK3 aggregation is also detected in RPE cells [49]. Moreover, the release of HMGB1 was observed post sodium iodate administration [66]. HMGB1 translocation is thought to activate both innate and adaptive immunity, further stimulating necroptosis and many of the harmful immunologic responses thereto [67]. Interestingly, Ding et al. reported that deletion of thyroid hormone receptors (THRs) significantly suppressed the sodium iodate-induced upregulation of RIPK1/3 and MLKL and, concomitantly, diminished RPE and photoreceptor cell loss [66]. This finding indicates a role of THR signaling in necroptosis and the potential of suppressing THR signaling for treating dry AMD.
Necroptosis in models of retinal degenerative diseases and injuries
| Retinal disorders | Disease models | Cell types | Related molecules | Descriptions | Reference |
|---|---|---|---|---|---|
| AMD | Sodium iodate insult | RPE | RIPK1, RIPK3, MLKL, HMGB1 | Sodium iodate insult induced necroptosis of RPE cells in the damaged and undamaged junction areas in retina, which displayed folding of the outer nuclear layer and shortened outer segment areas. | [49, 64, 66] |
| H2O2/tBHP insult | RPE | RIPK3, HMGB1 | Oxidative stress induced RIPK3-mediated necrosis as a predominant form of RPE cell death, which showed intracellular PI staining and membrane blebbing. | [58] | |
| Hypoxia insult | Microglia | RIPK1, RIPK3, MLKL | Hypoxia activated the RIP1-RIP3-MLKL signaling axis to induce necroptosis of retinal microglia. | [29] | |
| dsRNA induction | Photoreceptors, RPE | RIPK3, HMGB1 | After dsRNA injection, dying photoreceptors and RPE cells exhibited necroptotic morphology accompanied by swollen vacuoles in the inner segments of the retina. | [53] | |
| Glaucoma | High intraocular pressure | RGC | RIPK1, RIPK3, ERK1/2, Arginase 1 | High intraocular pressure induced necroptosis of RGCs and decreased retinal cell numbers in the GCL and INL/ONL. | [69, 70, 195] |
| Systemic hypotension | RGC | RIPK1, RIPK3, Angiotensin II | Systemic hypotension with a normal IOP triggered necroptosis of RGC, which was associated with angiotensin II-related glial cell activation. | [76] | |
| Optic nerve crush | RGC | RIPK1, RIPK3 | Optic nerve crush injury caused necroptosis of RGCs, accompanied by elevated expression of RIPK3. | [27] | |
| Ischemia and ischemia reperfusion insult | RGC | RIPK1, RIPK3, MLKL, RSK3, Calpain, tAIF, Daxx | Ischemia and ischemia reperfusion insult increased formation of RIPK1-RIPK3 complexes and mitochondrial polarization in RGCs to induce necroptotic death. | [28, 78, 82] | |
| Retinal detachment | Subretinal injection | Photoreceptors | RIPK1, RIPK3 | Retinal detachment caused necroptosis of photoreceptors accompanied by increased expression of RIPK1 and RIPK3. | [14, 55, 93] |
| Retinitis pigmentosa | rd10 mutation | Cone photoreceptor | RIPK1, RIPK3 | The retinas of rd10 mice exhibited a typical necrotic morphology and elevated expression of RIPK3 in cone, but not rod photoreceptors. | [103] |
| P23H rhodopsin mutation | Rod photoreceptor | RIPK1, RIPK3, DRP1 | The P23H rhodopsin mutant rat displayed marked upregulation of the RIP1/RIP3/DRP1 axis in rod photoreceptors; cell loss could be rescued by necrostatin-1 treatment. | [105] | |
| Irbp mutation | Photoreceptors | RIPK1, RIPK3 | In the retinas of Irbp-/- mouse, RIPK1 and RIPK3-mediated and TNF-induced necrosis contributed to both cone and rod photoreceptor degeneration. | [104] | |
| retinal degenerative rd1 mice | Microglia | RIPK1, RIPK3, TLR4 | Microglia experienced RIPK1 and RIPK3-dependent necroptosis in the retinal degenerative rd1 mice; Tlr4 deficiency was able to ameliorate microglial necroptosis. | [106] | |
| Retinal light damage | Blue light exposure | RGC | RIPK1, RIPK3, AIF | Blue light insult caused a decrease in the viability of RGC-5 cells, with upregulated expressions of both RIPK1, RIPK3, and AIF. | [51] |
| Blue light exposure | Retinal cell | CaMKII, Drp1, AIF | Blue light insult induced AIF-mediated necroptosis of R28 cells with activation of CaMK II-induced Drp1 phosphorylation. | [30] | |
| White light insult | Photoreceptors | RIP1 | White light insult (5500 lux, 2-4 hours) induced both apoptosis and necrosis in 661W cells; RIPK1 was hyper-phosphorylated when caspase-8 was inhibited. | [116] | |
| White light insult | RGC | RIP1 | White light insult (1000 lux, 48 hours) induced cell death of RGC-5 cells, which could be attenuated by RIPK1 inhibition. | [115] | |
| Ocular blast injury | Overpressure airwave blast | Retinal cell | RIPK1, RIPK3 | Overpressure airwave blast caused retinal cell death in multiple layers of the retina, with increased labeling for RIPK1 and RIPK3. | [121] |
| Repeated primary ocular blast | RGC | RIPK1, RIPK3 | In a repeated primary ocular blast injury mouse model, the RIPK-mediated necroptosis occurred in the GCL, OPL, and IPL of the retina. | [124] | |
| Mild blunt trauma | Retinal cell | RIPK1, RIPK3 | Blast-induced mild blunt trauma caused necroptotic cell death mediated by RIPK1 and RIPK3 in multiple layers of the retina. | [122] | |
| Plastic pellet impact | RGC | RIPK1, MLKL | Following blunt ocular injury caused by plastic pellet impact, MLKL protein expression was upregulated at 48 h in RGCs, inner nuclear cells, and ONL cells. | [123] | |
| Blast-induced open ocular trauma | Retinal cell | RIPK1, RIPK3 | Blast wave exposure caused necroptotic cell death in multiple layers of the retina; RIPK1 increased in the ONL, INL, and Müller glia, while RIPK3 increased in the ONL, INL, IPL, and GCL. | [125] | |
| Cytomegalovirus retinitis | Murine cytomegalovirus infection | Retinal cell | RIPK1, RIPK3 | In murine cytomegalovirus-infected eyes, necroptotic cell death occurred in multiple layers of retina with increased expressions of TNFα, RIP1K, and RIPK3, whereas Bax depletion increased the cleavage of RIPK1. | [133, 136] |
| Leber congenital amaurosis | Rpe65-deficiency | Cone photoreceptor | RIPK1, RIPK3, MLKL, Dio2, RPE65 | In retinas of Leber congenital amaurosis model mice (Rpe65-deficiency), cone cells showed severe degenerations associated with necroptosis signaling; Dio2 deficiency reduced necroptosis activity. | [137] |
| Achromatopsia | pde6cw59 mutant | Cone photoreceptor | RIPK1, RIPK3, pde6c | In pde6cw59 mutant zebrafish, severe degeneration of cone photoreceptors occurred in the retina, with high expressional levels of RIPK1 and RIPK3. | [31] |
| DNA alkylation agent-induced retinal degeneration | Methyl methanesulfonate insult | Photoreceptors | RIPK1, RIPK3 | Alkylation agent-induced dying photoreceptors exhibited necrotic morphology and overexpression of RIPK1 and RIPK3. | [142] |
AMD, age-related macular degeneration; RPE, retinal pigment epithelium; RIPK, receptor-interacting protein kinase; MLKL, mixed lineage kinase domain-like protein; HMGB1, molecule high mobility group box-1; tBHP, tert-butyl hydroperoxide; PI, propidium iodide; FGF2, fibroblast growth factor 2; IL, interleukin; RGC, retinal ganglion cell; ERK, extracellular signal-regulated protein kinase; RSK3, ribosome S6 kinase 3; tAIF, truncated apoptosis-inducing factor; DRP1, dynamin-related protein 1; IRBP, interphotoreceptor retinoid-binding protein; TNF, tumor necrosis factor-alpha; TLR4, Toll-like receptor 4; CaMKII, calcium/calmodulin-dependent protein kinase II; GCL, ganglion cell layer; OPL, outer plexiform layer; IPL, inner plexiform layer; ONL, outer nuclear layer; INL, inner nuclear layer; Dio2, type 2 iodothyronine deiodinase; PDE6C, phosphordiesterase 6C.
In response to oxidative stress induced by H2O2 or tert-butyl hydroperoxide (tBHP), necroptosis regulated by RIPK1/3 was found to be a major type of cell death in RPE cells [58]. Hypoxia insult also induced necroptosis of microglia, inducing explosive release of fibroblast growth factor 2, and causing retinal angiogenesis, which is the main pathological change in wet AMD [29]. The selective deletion of Ripk3 in microglia could significantly reduce retinal neovascularization, suggesting it as a promising anti-angiogenic target for treating retinal neovascular diseases [29].
Necroptosis was also observed in dsRNA (a component of drusen in AMD)-induced RPE degeneration [53]. In this study, knockout of RIPK3 was able to prevent both cell loss and inflammation. A recent study indicated a lipofuscin-triggered necroptosis in AMD retina [60]. Further pharmacological experiments revealed that the necroptotic pathway activated by lipofuscin is distinct from canonical RIPK-dependent signaling and can be blocked with a chemical inhibitor of MLKL instead of RIPK1/3 [60]. Further clarification of the necroptotic pathways and their roles in AMD based on clinico-pathological analysis can aid discovery of new therapeutic target for retinal degenerative diseases.
Glaucoma
Glaucoma is also a leading cause of irreversible visual loss and blindness [68]. Loss of RGCs induced by I/R injury following intraocular pressure (IOP) elevation and reduction is one of the crucial elements in the pathophysiology of glaucoma [28]. The role of necroptosis as a pathogenic factor in glaucoma is documented by multiple studies demonstrating that I/R injury triggers activation of the RIPK signaling pathway, which induces the loss of RGCs [28, 69-71]. In retinal tissues from glaucoma patients, typical necroptotic morphological abnormalities (cell swelling and membrane rupture) and significantly elevated expressions of RIPK1 have been revealed by TEM and immunohistochemistry in RGCs [56]. It was reported that necroptotic signaling, including RIPK1/3 and MLKL, was significantly activated in an in vitro model of retinal I/R injury [72-74]. In vivo studies have also supported that both RIPK1 and RIPK3 are highly expressed in the ganglion cell layer (GCL) of I/R-injured retinas [69, 75]. Interestingly, Jeon et al. demonstrated that systemic hypotension with normal IOP could trigger angiotensin II-associated glial cell activation that subsequently caused RGC necroptosis, suggesting a new underlying cause of RGC loss in glaucoma [76].
RSK3 (ribosome S6 kinase 3, a Ser/Thr kinase) reportedly plays a crucial role in the phosphorylation of RIPK3 in retinal necroptosis after retinal I/R injury [28]. In this report, RSK inhibition with small interfering RNA or chemical inhibitor downregulated the phosphorylation of RIPK3 and reduced necroptosis both in vitro and in vivo. The interaction between extracellular signal-regulated kinase (ERK) and RIPK3 is also associated with I/R-induced retinal necroptosis [77]. Downregulation of ERK phosphorylation was shown to decrease RIPK3 accumulation, whereas RIPK1 accumulation was not affected, indicating a RIPK1-independent pathway [77]. Upon retinal ischemic injury, RIPK3 can phosphorylate DAXX at Ser-668 to induce its translocation from nucleus to cytoplasm, leading to cell death with plasma membrane rupture [78]. In hippocampal neurons, the inhibition of RIPK1 blocked this RIPK3-DAXX interaction and subsequent DAXX translocation to cytoplasm [79]. However, the mechanism by which RIPK1 regulates the interaction between RIPK3 and DAXX upon retinal I/R injury is still unknown.
Calpains are a group of calcium-dependent proteases that are activated by increased cytosolic calcium during cell death [80]. Our own previous studies indicated that in vitro retinal I/R injury induced calpain-regulated necrosis of RGCs [81-83]. Recent research has revealed that the activity of calpain is regulated by peptidyl-prolyl isomerase 1 (PIN1) and calcium/calmodulin-dependent protein kinase II (CaMKII) in the presence of excessive glutamate, which has been proposed to induce excitotoxicity to mediate the loss of RGCs [9, 80, 84]. Retinal I/R injury can trigger a newly identified form of cell death that integrates elements of necroptosis, pyroptosis, and apoptosis [59, 85]. This new combined form of cell death, termed PANoptosis, has been deeply investigated in infectious diseases and is believed to be regulated by upstream master molecules Z-DNA binding protein 1 (ZBP1) and transforming growth factor beta-activated kinase 1 (TAK1) [86]. There is little evidence yet to identify the upstream regulators of PANoptosis during retinal I/R injury, even in whole neural injury. In the future, it is worth identifying the master regulator of PANoptosis to develop inhibitors that target multiple cell death pathways in the retina and wider nervous system.
Glaucoma is also characterized by optic nerve (ON) degeneration, which is thought to precede the death of RGCs or any clinical manifestations [87]. It was found that expression of RIPK1/3 was significantly elevated following ON crush models [27]. Of note, caspase inhibition alone failed to provide protection and, in fact, unexpectedly exacerbated RGC necrosis of in ON injured retinas. In contrast, inhibition of RIPKs in combination with caspase blockade delayed both apoptosis and necroptosis of RGCs [27]. Moreover, inhibition of RIPK1 promoted moderate axon regeneration that was only minimally affected by caspase blockade [27]. These various reports, taken together, indicate RIPK-regulated necroptosis as being an important driver of glaucoma's pathophysiology.
Retinal detachment
Retinal detachment (RD) is defined as the physical separation of the neurosensory retina from the underlying RPE layer [24, 88]. Although the retina can be surgically reattached, the visual acuity of the RD patients is not always restored due to continuous cell death of photoreceptors [89]. It was indicated that the activation of caspases and apoptosis were increased following RD [90]. However, pan-caspase inhibition fails to prevent the RD-induced photoreceptor loss [91]. Further research revealed that necroptotic signaling was activated after RD [14, 92]. Intriguingly, pan-caspase inhibition even shifted RD-induced photoreceptor death from apoptosis to necroptosis [14]. In contrast, inhibition of RIPK1 in combination with caspase blockade significantly prevented RD-induced photoreceptor death and the reduction of retinal thickness and function [14, 54]. Ripk3 deficiency also exhibited a protective effect on photoreceptors following RD [14]. The inhibition of RIPK1 was also able to reduce autophagic cell death in RD when caspases were inhibited [93]. Intriguingly, Ding et al. reported that induction of autophagy in RD reduced necroptosis of photoreceptors and protected the retina [55]. The crosstalk between autophagy and necroptosis was also observed in ischemic stroke and cancer; however, information about this issue in retinal degenerative diseases is still sparse [94-96].
Retinitis pigmentosa
Retinitis pigmentosa (RP) represents one of the most studied inherited retinal degenerative diseases [97]. In RP, vision loss typically begins with night blindness due to rod dysfunction and loss, then further deteriorates due to cone cell death [98-100]. In genetic mutation models of RP, cone photoreceptor loss is widely characterized to be driven by necroptosis, whereas rod photoreceptor loss has been shown to occur through apoptosis [101, 102]. Adaptive optics scanning laser ophthalmoscopy of RP patients' eyes indicated significantly increased necrotic enlargement of cone photoreceptor cells with concomitant release of HMGB1 within the vitreous humor [100].
Inhibition of RIPK1/3 achieved significant preservation of cone cells in the mouse model of RP (rd10 mice) [103]. Sato et al. reported that necroptosis contributed to both cone and rod photoreceptor degeneration in RP mice lacking interphotoreceptor retinoid-binding protein (IRBP) [104]. This was confirmed in RP patients with early and progressive photoreceptor abnormalities [104]. Another study revealed a bystander effect of rod photoreceptor necroptosis based on a P23H rhodopsin mutant rat, a model representing autosomal dominant RP [105]. The study identified that rod photoreceptors principally died through necroptosis, with subsequent release of bioactive molecules (ATP and HMGB1), which may lead to NLRP3 inflammasome activation and drive further bystander cell death of cone photoreceptors.
Microglia also experience necroptosis in the rd1 mouse model of RP [106]. This necroptotic process was associated with the activation of Toll-like receptor-4 (TLR4), which recruited Toll/IL-1R domain-containing adaptor-inducing IFNβ (TRIF) via its homology interaction motif (RHIM) domain, thereby promoting its interaction with RIPKs [106, 107]. Based on these studies, inhibition of necroptosis may be employed as a potential strategy to prevent or delay retinal degeneration in RP.
Retinal light damage
While the normal daily level of light (380-780 nm; 400-700 nm on primate' retinas) acts on the retina to induce vision and modulate circadian rhythms, excessive light has been reported to cause retinal cell death [108-111]. It was shown that light impinging on the retina could cause retinal cell death by interacting with mitochondrial constituents to generate reactive oxygen species (ROS) and trigger apoptosis [112, 113]. Mitochondrially-associated necroptosis of RGCs was also identified in blue light-induced retinal damage [51]. This necroptosis was able to be attenuated by the siRNA-mediated knockdown of Ripk1. Another study demonstrated that blue light could induce an AIF-mediated retinal necroptosis, which was regulated by the phosphorylation of CaMKII-Drp1 cascade [30, 114].
A study compared the retinal cell deaths induced by light and sodium azide, which is an inhibitor of cytochrome oxidase functioning in the mitochondrial electron transport chain [115]. The authors found that sodium azide killed RGCs mainly through apoptosis, whereas light-induced cell death occurs mainly via necroptosis and could be inhibited by RIPK1 blockade. More powerful light stress (5500 lux, fluorescent white light for 2 to 4 hours) activated Fas ligand (FasL)-Fas signaling and induced apoptosis of photoreceptor cells via a paracrine mechanism [116]. However, blocking FasL or caspase-8 failed to improve overall photoreceptor survival because of a subsequent hyper-phosphorylation of RIPK1 and compensatory activation of necroptosis [116]. Hence, efforts to prevent RGC and photoreceptor loss from light stress should pay attention to necroptosis signaling.
Ocular blast injury
Ocular blast injury is a major medical concern for both military and civilian victims of explosions due to poor visual outcomes [117, 118]. According to the Birmingham eye trauma terminology system (BETTS), ocular blast injuries are mainly classified into two groups - closed-globe injuries and open-globe injuries [119, 120]. Closed-globe injury induces retinal cell loss to all layers of the retina followed by nonapoptotic cell death [121]. Increased labeling for RIPK1/3 and MLKL was observed in the retina following blast injury, specifically at the injury site, suggesting that necroptosis contributes to an ongoing neurodegenerative response post blast [121-123]. However, a study based on repeated primary ocular blast injury in mice indicated that intravitreal injection of RIPK1 inhibitor or vehicle both increased vitreous inflammation and aggravated the RGC degeneration at 28 days post injury [124]. The combination of blast injury and intravitreal injection might exacerbate the RGC damage, suggesting intravitreal injection was not an ideal method for anti-necroptotic drug delivery after closed-globe injury.
Open-globe injury refers to perforation or penetration injuries to the eye [125, 126]. Severe posterior injuries occur after open-globe injury, including occasional RDs, large RPE vacuoles, activation of microglia, and severe regional photoreceptor cell death [125, 127-129]. Significant elevated labeling for markers of necroptosis (RIPK1 increased in the ONL, INL, and Müller glia; RIPK3 increased in the ONL, INL, IPL, and GCL), but not apoptosis, was observed in the retina following open-globe injury, indicating that necroptosis was widely activated in the retina after this injury [125]. In addition, open-globe injury can also cause traumatic glaucoma. Thus, early intervention against retinal necroptosis is needed to prevent exacerbation of retinal damage following open-globe injury [130].
Cytomegalovirus retinitis
Cytomegalovirus (CMV; a ubiquitous DNA herpes virus) retinitis is a potentially blinding manifestation in patients with CMV infection and immune dysfunction [131, 132]. Using a mouse model of murine CMV (MCMV) retinitis with retrovirus-induced immunosuppression, Chien and Dix demonstrated that the expression of RIPK1/3 significantly increased in MCMV-infected eyes [133]. Notably, increased RIPK3 expression was observed in RPE cells, microglia/macrophages, and glial cells, but not in RGCs in MCMV-infected eyes [134]. Depletion of RIPK3 not only inhibited cell death via necroptosis during MCMV retinitis, but also significantly attenuated both caspase 3-dependent and AIF-mediated, caspase 3-independent apoptosis [134, 135]. In addition, a study using Bax-/- mice indicated that Bax gene deletion caused more caspase 3-independent cell death of uninfected bystander retinal cells and more cleaved RIPK1, suggesting that BAX might have an important role in the prevention of necroptosis [136]. More studies using chemical inhibitors or genetic ablation of apoptotic and necroptotic molecules will help to characterize the mechanism of interaction between apoptosis and necroptosis during CMV retinitis.
Others
Necroptosis is also linked to cone photoreceptor degeneration in Leber congenital amaurosis (LCA) [11, 137]. Analysis of cell death pathways revealed that expressions of necroptotic factors (Ripk3, Tnf-α, Tnfra1, Mlkl, and Tradd) were increased in the retinas of LCA mice (Rpe65-deficient) [137]. Of note, deficiency of type 2 iodothyronine deiodinase (Dio2) provided a neuroprotective effect against necroptosis in retinas of LCA mice [137].
Achromatopsia is an inherited retinal disease characterized by early loss of cone photoreceptors and later rod photoreceptor loss [31, 138, 139]. In a zebrafish model of human achromatopsia (pde6cw59), cone photoreceptors expressed high levels of RIPK1/3, whereas rod photoreceptors were immunopositive for caspase-3, indicating activation of apoptosis [31]. Furthermore, morpholino-mediated gene knockdown of ripk3 rescued cone photoreceptor cell death and restored visual function, suggesting that targeting the necroptotic signaling pathway could be an effective therapeutic intervention for achromatopsia [31].
BMI1 (B lymphoma Moloney murine leukemia virus insertion region 1) is a component of the polycomb repressive complex 1, which is required for the proliferation of peripheral retinal progenitor cells and radial growth of the retina [140, 141]. Using Bmi1-/- mice, Barabino et al. demonstrated that loss of Bmi1 caused rapid necroptotic degeneration of cone bipolar neurons and cone photoreceptors, but not in rods, during post-natal eye development [8]. However, the question of whether necroptosis is directly or indirectly repressed by BMI1 has yet to be answered.
DNA-alkylating agents are commonly used as cancer chemotherapeutics, but they potentially trigger retinal degeneration [142, 143]. It is reported that necroptosis plays an important role in alkylation-induced photoreceptor cell loss in male mice [142]. Moreover, Ripk3 deletion partially protected photoreceptor cells from alkylation-induced cell death, attenuating necrosis and reducing inflammation [142]. Intriguingly, it was shown that Parp1 knockout conveyed full protection against alkylation-induced photoreceptor degeneration, whereas RIPK3 knockout only had a partially protective effect, suggesting that activation of necroptosis might be secondary to PARP1 activation in alkylation-induced photoreceptor degeneration [142, 144]. The crosstalk between RIPK3 signaling and PARP1 activation remains to be elucidated.
Although necroptosis is now established as an important type of RCD in retinal degenerative diseases, many questions are still to be addressed. Herpes simplex virus (HSV)-1 induces acute retinal necrosis and significant elevation in TNF-α expression [145]. Similarly, elevated expressions of TNF-α and TNF receptors were closely associated with the severity of diabetic retinopathy [146, 147]. Inhibiting TNF-α afforded a protective effect against HSV-1-induced retinitis; however, no one has yet reported an increase in RIPK1/3 or MLKL in these retinopathies, nor has the involvement of necroptosis therein been definitely documented [148]. Moreover, glutamate neurotoxicity has been reported to be crucial to retinal cell death in experimental diabetic retina [149]. Thus, it is worth confirming glutamate-related necroptosis and identifying the role of CaMKII in the diabetic retina for further treatment of diabetic retinopathy.
Therapeutic options to target necroptosis in retina
In this section, we highlight the recently discovered compounds (selective inhibitors, drugs, and chemical agents) regulating the necroptotic cascade pathway in the treatment of diverse retinal disorders.
Compounds targeting RIPK1
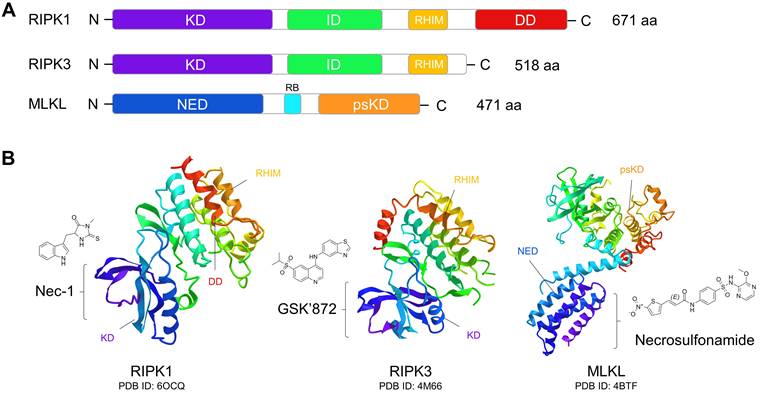
Nec-1 is a selective allosteric inhibitor of RIPK1 that binds to its kinase domain and stabilizes it in an inactive conformation (Fig. 3) [102, 150, 151]. The protective effect of Nec-1 has been widely proven in pre-clinical models of retinal injuries (Table 2). In an AMD mouse model, Nec-1 treatment significantly counteracted necroptosis of RPE and microglial cells, and inhibited retinal neovascularization [29, 49]. Inhibition of pathological angiogenesis by Nec-1 treatment has also been reported in a laser-induced choroidal neovascularization (CNV) model, which was associated with caspase activation and suppression of M2 macrophage polarization [152]. Moreover, necrotic RGC death was attenuated by Nec-1 in rodent models of oxygen and glucose deprivation, optic nerve damage, and high IOP, indicating multiple potential protection effects of Nec-1 on glaucoma [69, 71, 153]. In addition, Nec-1 has been shown to inhibit necroptosis of multiple retinal cell types across diverse models of retinal detachment, retinitis pigmentosa, and retinal light damage (Table 2). Of particular note, treatment with Nec-1 and a pan-caspase inhibitor conferred near-complete protection of RPE cells against cell death induced by tamoxifen, which is widely used in low dosages as an adjuvant therapy for breast cancer [154].
Domain organization of necroptotic signaling proteins and their targeted chemical inhibitors. (A) Functional domains of RIPK1, RIPK3, and MLKL. RIPK1 and RIP3 have very similar domain features, both of them containing the N-terminal serine/threonine kinase domain (KD), intermediate domain (ID), and RIP homotypic interaction motif (RHIM) domain. RHIM mediates the assembly of the RIPK1:RIPK3 complex and is crucial for KD activation and induction of necroptosis. RIPK1 also has a C-terminal death domain (DD), which can bind to the death receptors of TNFR1 and Fas to activate apoptosis. In MLKL, the regulatory brace (RB) domain connects the N-terminal execution domain (NED) and C-terminal pseudokinase domain (psKD). The psKD is usually catalytically inactive and contains an unusual pseudoactive site. The NED is sufficient to form channels that can induce membrane depolarization and cell death. (B) Tertiary structures of RIPK1, RIPK3, and MLKL and their inhibitors. Nec-1 can bind to the kinase domain of RIPK1 to stabilize it in an inactive conformation. GSK'872 can be caged in the kinase domain of RIPK3 to inhibit its kinase activity. Necrosulfonamide can bind to the N-terminal execution domain of MLKL to block its activation.
Anti-necroptosis compounds for treating retinal diseases
| Compounds | Targets | Related diseases | Treatment | Affected cell types | Related molecules | Reference |
|---|---|---|---|---|---|---|
| Nec-1 | RIPK1 | AMD | Pre/post-treatment | RPE, Microglia | RIPK3, MLKL | [29, 49] |
| Glaucoma | Pre/post-treatment | RGC | RIPK3 | [27, 69, 71, 153] | ||
| Retinal detachment | Pre/post-treatment | Photoreceptors | AIF | [14, 54] | ||
| Retinitis pigmentosa | Post-treatment | Photoreceptors, Microglia | RIPK3, DRP1 | [103-106] | ||
| Retinal light damage | Pre/post-treatment | RGC, Photoreceptors | RIPK3, CaMKII, Drp1, AIF | [30, 51, 115, 116] | ||
| Tamoxifen toxicity | Post-treatment | RPE | N.P. | [154] | ||
| Nec-1s | RIPK1 | Ocular blast injury | Post-treatment | RGC | RIPK3, MLKL | [123, 124] |
| Achromatopsia | Post-treatment | Cone photoreceptor | RIPK3 | [31] | ||
| RIC | RIPK1 | AMD | Post-treatment | RPE | N.P. | [65] |
| Glaucoma | Post-treatment | RGC | RIPK3 | [70] | ||
| GSK′872 | RIPK3 | Retinal ischemic stress | Pre-treatment | RGC | MLKL | [72] |
| Necrosulfonamide | MLKL | Achromatopsia | Post-treatment | Cone photoreceptor | RIPK1, RIPK3 | [31] |
| ALLN | Calpain | Glaucoma | Pre-treatment | RGC | AIF | [81, 82] |
| KN-93 | CaMKII | Retinal light damage | Post-treatment | Retinal R28 cells | Drp1, AIF | [30] |
| Mdivi-1 | Drp1 | Retinal light damage | Post-treatment | Retinal R28 cells | CaMKII, AIF | [30] |
| LJH685 | RSK3 | Retinal ischemic stress | Pre-treatment | RGC | RIPK3 | [28] |
| U0126 | ERK | Glaucoma | Post-treatment | RGC | RIPK3 | [77] |
| Melatonin | N.P. | Glaucoma | Post-treatment | RGC | RIPK1, RIPK3 | [184] |
| Anti-thyroid drugs | N.P. | AMD | Pre-treatment | Photoreceptors, RPE | RIPK1, RIPK3, MLKL | [64] |
| LCA | Post-treatment | Cone photoreceptor | RIPK1, RIPK3 | [137] | ||
| Nec-7 | N.P. | AMD | Pre-treatment | RPE | RIPK3 | [58] |
| Rapamycin | N.P. | Retinal detachment | Post-treatment | Photoreceptors | RIPK1, AIF | [55] |
| Trolox | N.P. | Retinal light damage | Post-treatment | Retinal R28 cell | CaMKII, Drp1, AIF | [30] |
| Minocycline | N.P. | Retinal light damage | Pre-treatment | RPE | RIPK3 | [186] |
Nec, Necrostatin; RIPK, receptor-interacting protein kinase; AMD, age-related macular degeneration; RPE, retinal pigment epithelium; MLKL, mixed lineage kinase domain-like protein; RGC, retinal ganglion cell; AIF, apoptosis-inducing factor; CaMKII, calcium/calmodulin-dependent protein kinase II; DRP1, dynamin-related protein 1; N.P., not provided; Nec-1s, Nec-1 stable; RIC, RIPK1-inhibitory compound; AMD, age-related macular degeneration; ALLN, N-acetyl-leucyl-leucyl-norleucinal; DRP1, dynamin-related protein 1; RSK3, ribosome S6 kinase 3; ERK, extracellular signal-regulated protein kinase; LCA, Leber congenital amaurosis.
Nec-1 stable (Nec-1s), a modified derivative of Nec-1, has a higher affinity and specificity for RIPK1 [155, 156]. In rodent models of retinitis pigmentosa, treatment with Nec-1s significantly protected cone and rod photoreceptors from degeneration and reduced cell loss in ONL [104, 105]. Nec-1s also exhibited neuroprotective effects in ocular blast injury [123, 124]. Additionally, Nec-1s delayed cone cell death and preserved the remainder of the outer retina in a zebrafish model of achromatopsia [31]. However, the clinical application of Nec-1 and Nec-1s is limited due to their moderate potency and poor in vivo pharmacokinetic properties [157]. The development of compounds that overcome these limitations would be of significant benefit.
The RIPK1-inhibitory compound (RIC) is another inhibitor of RIPK1 that has a distinct chemical profile compared to Nec-1 [70, 150]. It was reported that RIC effectively suppressed necroptosis of RGC and RPE and had a novel neuroprotective effect in both glaucoma and dry AMD [65, 70]. Additionally, the excellent corneal permeability of RIC offers an appealing advantage for its administration by eye drop, making it a good candidate for clinical therapy [65]. Several other inhibitors of RIPK1, such as GSK'963, RIPA-56, GSK2982772, and DNL747, have been identified in the past decade [61, 158-161]. However, their roles in retinal degenerative diseases are still unknown. Of note, GSK2982772 has been advanced into phase II clinical trials for the treatment of rheumatoid arthritis, plaque psoriasis, and active ulcerative colitis, showing good oral pharmacokinetics and tolerance [162].
Compounds targeting RIPK3
GSK'872, a specific small molecule inhibitor of RIPK3 binding to its kinase domain, plays an important role in suppressing necroptosis and RIPK3-dependent inflammation [163, 164]. It was reported that inhibition of RIPK3 by GSK'872 treatment significantly reduced phosphorylation of MLKL and cytosolic Ca2+ concentrations in retinal cells in vitro following elevated hydrostatic pressure [72, 165]. In addition, the anti-necroptotic effect of GSK'872 has been indicated in osteoclastogenesis and non-alcoholic fatty liver disease in vitro and in vivo [166, 167]. However, the in vivo retinal neuroprotective efficiency of GSK'872 still needs to be confirmed.
HS-1371 and HG-9-91-01 are potent inhibitors of RIPK3 and exhibit significant anti-necroptotic effect against TNF signaling [168, 169]. However, HG-9-91-01 also triggers RIPK1/3 and caspase 1/8-mediated apoptosis and pyroptosis [169]. Recently, Xu et al. discovered a new RIPK3 inhibitor, AZD5423, that effectively protected against cisplatin- and I/R-induced acute kidney injury in mice [170]. Zharp-99 is another recently discovered RIPK3 inhibitor that efficiently blocks TNF-induced necroptosis in both human and mouse cells [171]. Zharp-99 shows promising therapeutic potential, with good in vitro safety profiles and in vivo pharmacokinetic parameters [171]. Further investigations in pre-clinical models of retinal injury using HS-1371, AZD5423, and Zharp-99 will provide crucial insights for developing novel therapies for necroptotic retinopathy.
Compounds targeting MLKL
Necrosulfonamide (NSA) is a selective inhibitor of MLKL that blocks its N-terminal CC domain function [35]. NSA suppresses necroptosis by directly inhibiting necrosome formation, showing protective effects against necroptosis in pulmonary I/R injury, spinal cord injury, and Alzheimer's disease [172-176]. In retinal degenerative disease, NSA showed an ameliorative effect in a pre-clinical animal model of Achromatopsia through alleviating necroptotic degeneration of cone photoreceptor [31]. However, the therapeutic potential of NSA in other retinal disorders is yet to be demonstrated. GW806742X is a potent MLKL inhibitor that binds the pseudokinase domain of MLKL [177]. Treatment with GW806742X and other RIPK1/3 inhibitors significantly blocked necroptosis and inflammation [178, 179]. BI-8925, another potent MLKL inhibitor identified in TNFα-induced necroptosis, works by stabilizing the inactive state of MLKL by an essential π-π stacking interaction [180]. Since GW806742X and BI-8925 are novel inhibitors of MLKL, their potential effects on retinal degenerative diseases need to be addressed in the future.
Others
Ma et al. reported that anti-thyroid drugs (1% sodium perchlorate monohydrate and 0.05% methomazole in drinking water) reduced retinal necroptosis and oxidative stress responses in a mouse model of AMD and LCA [64, 137]. High serum TH levels have been correlated with increased risk of AMD, supporting the clinical therapeutic potential of anti-thyroid treatment [64].
Rapamycin, the first generation of mTOR inhibitors, can also inhibit the necroptosis of photoreceptors in retinal detachment by decreasing the RIPK1 expression and AIF nuclear translocation [55]. However, its clinical application is limited due to poor water solubility and stability. Fortunately, rapamycin has a number of analogs (rapalogs) with improved pharmacokinetic properties [181]. These may be explored in retinal degenerative diseases in the future.
Melatonin is an important hormone that has a wide-ranging neuroprotective effect and can enhance cognitive function in clinical trials [182, 183]. In a model of acute glaucoma, melatonin treatment attenuated necroptosis of RGCs, reduced retinal thinning, and ameliorated retinal dysfunction [184]. Interestingly, apoptosis and pyroptosis have also been inhibited by melatonin, suggesting a multi-protective potential for therapeutic application [184]. Minocycline, another promising clinical neuroprotective agent for acute stroke patients, also exhibited an anti-necroptotic effect on RPE cell loss caused by blue light [185, 186]. Although both of melatonin and minocycline are clinically used and have antioxidant and anti-inflammatory effects, their targeting mechanism in the necroptotic signaling pathway requires further exploration [187, 188].
As mentioned above, compounds that inhibit key effector molecules within the necroptotic signaling pathway also show protective effects on retinal cells, such as KN-93 (CaMKII), Mdivi-1 (DRP1), and LJH685 (RSK) (Table 2) [28, 30]. However, the efficacy and safety of these compounds still need to be evaluated in in vivo pre-clinical models of retinal degenerative diseases.
The human bone marrow-derived mesenchymal stem cell (bmMSC) secretome also exhibits a versatile neuroprotective ability [189]. Among its documented effects are: inhibition of RIPK1/3, MLKL, and apoptotic effectors; modulation of autophagy; and activation of antioxidant machinery, all of which were observed in a model of spontaneous retinal neurodegeneration [190]. Although the neuroprotective effect of MSC-based therapy has been approved for the treatment of retinal neurodegeneration, the specific anti-necroptotic compounds within the bmMSC secretome remain unidentified [191, 192].
Of these compounds that can regulate necroptosis in models of retinal degenerative diseases, some are administered prophylactically (as pre-treatment), while others are used therapeutically (as post-treatment) (Table 2). Since necroptosis, as a process, occurs rapidly once triggered, prophylactic treatments with Nec-1 and GSK'872 are effective approaches to inhibit key kinases near the start of the necroptotic signaling cascade [28, 58]. Excessive oxidative stress and inflammation are triggers for AMD and retinal light damage [193, 194]. Some clinically validated antioxidant and anti-inflammatory compounds, such as anti-thyroid drugs and minocycline, may also mitigate retinal cell death by preventing the triggers, although their specific targets in necroptotic signaling, if any, remain to be elucidated [64, 184].
An effective post-treatment often indicates therapeutic potential of the compound [70, 187]. Because necroptosis can release inflammatory effectors, bystander inflammation may occur for a long time following cell death and lead to subsequent damage and necroptosis of adjacent cells [105]. Therefore, early intervention with anti-necroptosis compounds after the onset of disease is important to alleviate further retinal tissue damage and dysfunction.
Conclusion and prospects
The discovery and characterization of necroptosis represents a major breakthrough in the field of cell death and signaling in the past decade. Extensive and increasing evidence demonstrates that numerous retinal degenerative diseases involve necroptosis mediated by RIPK1/3 and MLKL, occurring in different retinal layers and cell types. Moreover, advances in characterizing mitochondrial dysfunctions and mutations in key regulators - such as CaMKII, DRP1, AIF, and PARP1 - in retinal degenerative diseases involving necroptotic signaling have expanded the understanding of organelle dysfunction and the molecular mechanisms of necroptosis.
Inhibition of these master necroptotic regulators by diverse compounds or genetic interventions could potentially attenuate necroptosis of retinal cells. These strategies have already demonstrated multiple benefits in treating retinal degenerative diseases, such as reducing ocular inflammation, inhibiting retinal neovascularization, and preventing optic nerve damage. Notably, several compounds approved in clinical trials, such as GSK2982772, methomazole, rapalogs, melatonin, and minocycline also exhibited anti-necroptotic abilities in retinal degenerative diseases.
Although we have learned much about necroptosis, its role in necrotizing herpetic and diabetic retinopathy remains enigmatic. Current knowledge about necroptosis in retinal degenerative diseases mainly comes from animal models, with clinical trials of these anti-necroptosis strategies in patients still in their early days. Development of safe necroptosis inhibitors, stem cell-based therapy, and genetic strategies for targeting master regulators of necroptosis remain the key points of future clinical development.
Abbreviations
TNFR1: tumor necrosis factor (TNF)-receptor 1; TRAF2: TNF receptor associated factor 2; cIAP1/2: cellular inhibitor of apoptosis proteins 1 and 2; LUBAC: linear ubiquitin chain assembly complex; TAK1: TGF-activated kinase 1; IKK: IκB kinase; TAB2/3: TAK1-binding protein 2/3; NEMO: NF-κB essential modulator; cFLIP: cellular FLICE-like inhibitory protein; NF-κB: nuclear factor-κB; FADD: Fas-associated protein with death domain; RIPK: receptor-interacting protein (RIP) kinase; complex IIa-RIA: complex IIa-RIPK1-independent apoptosis; complex IIa-RDA: complex IIa-RIPK1-dependent apoptosis; MLKL: mixed-lineage kinase domain-like pseudokinase; DAMP: damage-associated molecular pattern; ESCRT-III: endosomal sorting complexes III; PGAM5: phosphoglycerate mutase family member 5; DRP1: dynamin-related protein 1; ROS: reactive oxygen species; tAIF: truncated apoptosis-inducing factor.
Acknowledgements
The authors would like to acknowledge the language-editing service provided by EditorBar.
Funding
This work was supported by National Natural Science Foundation of China (81971891, 82172196, and 81772134), Key Laboratory of Emergency and Trauma (Hainan Medical University) of Ministry of Education (KLET-202210), and the College Students' Innovation and Entrepreneurship Project (S20210026020013).
Author contributions
KX and QZ were the major contributors to design the study. QZ was the major contributor to manuscript writing, and figures creating. XMH, WJZ, HW, YL, YH and XXB assisted in the data collection and analysis. HW, YH and YXH edited the manuscript. QZ, KX, LS, BJ, GPQ and LSL were major contributors to manuscript revision. All authors have read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Flaxman SR, Bourne RRA, Resnikoff S, Ackland P, Braithwaite T, Cicinelli MV. et al. Global causes of blindness and distance vision impairment 1990-2020: a systematic review and meta-analysis. The Lancet Global health. 2017;5:e1221-e34
2. Nelidova D, Morikawa RK, Cowan CS, Raics Z, Goldblum D, Scholl HPN. et al. Restoring light sensitivity using tunable near-infrared sensors. Science (New York, NY). 2020;368:1108-13
3. Apte RS. Age-Related Macular Degeneration. The New England journal of medicine. 2021;385:539-47
4. Chen M, Luo C, Zhao J, Devarajan G, Xu H. Immune regulation in the aging retina. Progress in retinal and eye research. 2019;69:159-72
5. Cansler SM, Evanson NK. Connecting endoplasmic reticulum and oxidative stress to retinal degeneration, TBI, and traumatic optic neuropathy. Journal of neuroscience research. 2020;98:571-4
6. Goldman D. Müller glial cell reprogramming and retina regeneration. Nature reviews Neuroscience. 2014;15:431-42
7. Wang M, Wan H, Wang S, Liao L, Huang Y, Guo L. et al. RSK3 mediates necroptosis by regulating phosphorylation of RIP3 in rat retinal ganglion cells. Journal of Anatomy. 2020;237:29-47
8. Barabino A, Plamondon V, Abdouh M, Chatoo W, Flamier A, Hanna R. et al. Loss of Bmi1 causes anomalies in retinal development and degeneration of cone photoreceptors. Development (Cambridge, England). 2016;143:1571-84
9. Wang S, Liao L, Wang M, Zhou H, Huang Y, Wang Z. et al. Pin1 Promotes Regulated Necrosis Induced by Glutamate in Rat Retinal Neurons via CAST/Calpain2 Pathway. Frontiers in cellular neuroscience. 2017;11:425
10. Kumaran N, Moore AT, Weleber RG, Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. The British journal of ophthalmology. 2017;101:1147-54
11. Yang F, Ma H, Boye SL, Hauswirth WW, Ding XQ. Overexpression of Type 3 Iodothyronine Deiodinase Reduces Cone Death in the Leber Congenital Amaurosis Model Mice. Advances in experimental medicine and biology. 2018;1074:125-31
12. Hu X-m, Li Z-x, Lin R-h, Shan J-q, Yu Q-w, Wang R-x. et al. Guidelines for Regulated Cell Death Assays: A Systematic Summary, A Categorical Comparison, A Prospective. Frontiers in cell and developmental biology. 2021;9:634690
13. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell research. 2019;29:347-64
14. Trichonas G, Murakami Y, Thanos A, Morizane Y, Kayama M, Debouck CM. et al. Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21695-700
15. Zhang Y, Chen X, Gueydan C, Han J. Plasma membrane changes during programmed cell deaths. Cell research. 2018;28:9-21
16. Xu X, Lai Y, Hua Z-C. Apoptosis and apoptotic body: disease message and therapeutic target potentials. Bioscience Reports. 2019 39
17. Ghafouri-Fard S, Shoorei H, Mohaqiq M, Majidpoor J, Moosavi MA, Taheri M. Exploring the role of non-coding RNAs in autophagy. Autophagy. 2022;18:949-70
18. Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiological Reviews. 2019;99:1765-817
19. Park H-YL, Kim JH, Park CK. Different contributions of autophagy to retinal ganglion cell death in the diabetic and glaucomatous retinas. Scientific reports. 2018;8:13321
20. Zhang Z-Y, Bao X-L, Cong Y-Y, Fan B, Li G-Y. Autophagy in Age-Related Macular Degeneration: A Regulatory Mechanism of Oxidative Stress. Oxidative medicine and cellular longevity. 2020;2020:2896036
21. Xin X, Yang K, Liu H, Li Y. Hypobaric hypoxia triggers pyroptosis in the retina via NLRP3 inflammasome activation. Apoptosis: an international journal on programmed cell death. 2022;27:222-32
22. Swanson KV, Deng M, Ting JPY. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nature Reviews Immunology. 2019;19:477-89
23. Zangiabadi S, Abdul-Sater AA. Regulation of the NLRP3 Inflammasome by Posttranslational Modifications. The Journal of Immunology. 2022;208:286
24. Murakami Y, Notomi S, Hisatomi T, Nakazawa T, Ishibashi T, Miller JW. et al. Photoreceptor cell death and rescue in retinal detachment and degenerations. Progress in retinal and eye research. 2013;37:114-40
25. Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nature reviews Neuroscience. 2019;20:19-33
26. Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt Judy P. et al. Activation of Necroptosis in Multiple Sclerosis. Cell Reports. 2015;10:1836-49
27. Kayama M, Omura K, Murakami Y, Reshef E, Thanos A, Morizane Y. et al. Combined inhibition of apoptosis and necrosis promotes transient neuroprotection of retinal ganglion cells and partial-axon regeneration after optic nerve damage. bioRxiv. 2018: 357566.
28. Wang M, Wan H, Wang S, Liao L, Huang Y, Guo L. et al. RSK3 mediates necroptosis by regulating phosphorylation of RIP3 in rat retinal ganglion cells. Journal of anatomy. 2020;237:29-47
29. He C, Liu Y, Huang Z, Yang Z, Zhou T, Liu S. et al. A specific RIP3+ subpopulation of microglia promotes retinopathy through a hypoxia-triggered necroptotic mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2021;118:e2023290118
30. Yang D, Rong R, Yang R, You M, Wang M, Li H. et al. CaMK II -induced Drp1 phosphorylation contributes to blue light-induced AIF-mediated necroptosis in retinal R28 cells. Biochemical and biophysical research communications. 2021;559:113-20
31. Viringipurampeer IA, Shan X, Gregory-Evans K, Zhang JP, Mohammadi Z, Gregory-Evans CY. Rip3 knockdown rescues photoreceptor cell death in blind pde6c zebrafish. Cell Death & Differentiation. 2014;21:665-75
32. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311-20
33. Shan B, Pan H, Najafov A, Yuan J. Necroptosis in development and diseases. Genes & Development. 2018;32:327-40
34. Mifflin L, Ofengeim D, Yuan J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nature reviews Drug discovery. 2020;19:553-71
35. Sun L, Wang H, Wang Z, He S, Chen S, Liao D. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213-27
36. Hanus J, Anderson C, Wang S. RPE necroptosis in response to oxidative stress and in AMD. Ageing Research Reviews. 2015;24:286-98
37. Tonnus W, Belavgeni A, Beuschlein F, Eisenhofer G, Fassnacht M, Kroiss M. et al. The role of regulated necrosis in endocrine diseases. Nature Reviews Endocrinology. 2021;17:497-510
38. Liu Y, Xu Q, Wang Y, Liang T, Li X, Wang D. et al. Necroptosis is active and contributes to intestinal injury in a piglet model with lipopolysaccharide challenge. Cell Death & Disease. 2021;12:62
39. Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P. et al. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell. 2017;169:286-300.e16
40. Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nature Reviews Molecular Cell Biology. 2020;21:85-100
41. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death & Differentiation. 2018;25:486-541
42. McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V. et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science (New York, NY). 2018;359:eaao6047
43. Dong X-H, Liu H, Zhang M-Z, Zhao P-X, Liu S, Hao Y. et al. Postconditioning with inhaled hydrogen attenuates skin ischemia/reperfusion injury through the RIP-MLKL-PGAM5/Drp1 necrotic pathway. Am J Transl Res. 2019;11:499-508
44. Delavallée L, Cabon L, Galán-Malo P, Lorenzo HK, Susin SA. AIF-mediated caspase-independent necroptosis: A new chance for targeted therapeutics. IUBMB Life. 2011;63:221-32
45. Ozaki T, Yamashita T, Ishiguro S-i. Mitochondrial m-calpain plays a role in the release of truncated apoptosis-inducing factor from the mitochondria. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2009;1793:1848-59
46. Zhang Y-F, He W, Zhang C, Liu X-J, Lu Y, Wang H. et al. Role of receptor interacting protein (RIP)1 on apoptosis-inducing factor-mediated necroptosis during acetaminophen-evoked acute liver failure in mice. Toxicology Letters. 2014;225:445-53
47. Halim AB. Do We have a Satisfactory Cell Viability Assay? Review of the Currently Commercially-Available Assays. Current drug discovery technologies. 2020;17:2-22
48. Zhang H-J, Liu X-B, Chen X-M, Kong Q-H, Liu Y-S, So K-F. et al. Lutein delays photoreceptor degeneration in a mouse model of retinitis pigmentosa. Neural regeneration research. 2022;17:1596-603
49. Hanus J, Anderson C, Sarraf D, Ma J, Wang S. Retinal pigment epithelial cell necroptosis in response to sodium iodate. Cell death discovery. 2016;2:16054
50. Momeni HR, Abnosi MH, Eskandari N. Quantitative evaluation of human sperm viability using MTT assay: A laboratory study. International journal of reproductive biomedicine. 2020;18:983-8
51. Del Olmo-Aguado S, Núñez-Álvarez C, Osborne NN. Blue Light Action on Mitochondria Leads to Cell Death by Necroptosis. Neurochemical research. 2016;41:2324-35
52. Li J, Hao J-H, Yao D, Li R, Li X-F, Yu Z-Y. et al. Caspase-1 inhibition prevents neuronal death by targeting the canonical inflammasome pathway of pyroptosis in a murine model of cerebral ischemia. CNS Neuroscience & Therapeutics. 2020;26:925-39
53. Murakami Y, Matsumoto H, Roh M, Giani A, Kataoka K, Morizane Y. et al. Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death & Differentiation. 2014;21:270-7
54. Dong K, Zhu H, Song Z, Gong Y, Wang F, Wang W. et al. Necrostatin-1 protects photoreceptors from cell death and improves functional outcome after experimental retinal detachment. The American journal of pathology. 2012;181:1634-41
55. Ding J, Yang N, Yan Y, Wang Y, Wang X, Lu L. et al. Rapamycin Inhibited Photoreceptor Necroptosis and Protected the Retina by Activation of Autophagy in Experimental Retinal Detachment. Current eye research. 2019;44:739-45
56. Qin Q, Yu N, Gu Y, Ke W, Zhang Q, Liu X. et al. Inhibiting multiple forms of cell death optimizes ganglion cells survival after retinal ischemia reperfusion injury. Cell Death & Disease. 2022;13:507
57. Crowley LC, Marfell BJ, Scott AP, Waterhouse NJ. Quantitation of Apoptosis and Necrosis by Annexin V Binding, Propidium Iodide Uptake, and Flow Cytometry. Cold Spring Harbor protocols. 2016;2016:953-7
58. Hanus J, Zhang H, Wang Z, Liu Q, Zhou Q, Wang S. Induction of necrotic cell death by oxidative stress in retinal pigment epithelial cells. Cell Death & Disease. 2013;4:e965
59. Yan W-T, Zhao W-J, Hu X-M, Ban X-x, Ning W-Y, Wan H. et al. PANoptosis-like cell death in ischemia/reperfusion injury of retinal neurons. Neural regeneration research. 2023;18:357-63
60. Pan C, Banerjee K, Lehmann GL, Almeida D, Hajjar KA, Benedicto I. et al. Lipofuscin causes atypical necroptosis through lysosomal membrane permeabilization. Proceedings of the National Academy of Sciences of the United States of America. 2021;118:e2100122118
61. Degterev A, Ofengeim D, Yuan J. Targeting RIPK1 for the treatment of human diseases. Proceedings of the National Academy of Sciences of the United States of America. 2019;116:9714-22
62. Papadopoulos Z. Recent Developments in the Treatment of Wet Age-related Macular Degeneration. Current Medical Science. 2020;40:851-7
63. Fleckenstein M, Keenan TDL, Guymer RH, Chakravarthy U, Schmitz-Valckenberg S, Klaver CC. et al. Age-related macular degeneration. Nat Rev Dis Primers. 2021;7:31
64. Ma H, Yang F, Ding X-Q. Inhibition of thyroid hormone signaling protects retinal pigment epithelium and photoreceptors from cell death in a mouse model of age-related macular degeneration. Cell Death & Disease. 2020;11:24
65. Jang KH, Do YJ, Koo TS, Choi JS, Song EJ, Hwang Y. et al. Protective effect of RIPK1-inhibitory compound in in vivo models for retinal degenerative disease. Experimental eye research. 2019;180:8-17
66. Ma H, Yang F, Ding X-Q. Deficiency of thyroid hormone receptor protects retinal pigment epithelium and photoreceptors from cell death in a mouse model of age-related macular degeneration. Cell Death & Disease. 2022;13:255
67. Wen S, Li X, Ling Y, Chen S, Deng Q, Yang L. et al. HMGB1-associated necroptosis and Kupffer cells M1 polarization underlies remote liver injury induced by intestinal ischemia/reperfusion in rats. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2020;34:4384-402
68. Stein JD, Khawaja AP, Weizer JS. Glaucoma in Adults-Screening, Diagnosis, and Management: A Review. Jama. 2021;325:164-74
69. Kim CR, Kim JH, Park HL, Park CK. Ischemia Reperfusion Injury Triggers TNFα Induced-Necroptosis in Rat Retina. Current eye research. 2017;42:771-9
70. Do YJ, Sul JW, Jang KH, Kang NS, Kim YH, Kim YG. et al. A novel RIPK1 inhibitor that prevents retinal degeneration in a rat glaucoma model. Experimental cell research. 2017;359:30-8
71. Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC. et al. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. Journal of neuroscience research. 2010;88:1569-76
72. Liao L, Shang L, Li N, Wang S, Wang M, Huang Y. et al. Mixed lineage kinase domain-like protein induces RGC-5 necroptosis following elevated hydrostatic pressure. Acta Biochim Biophys Sin. 2017;49:879-89
73. Shang L, Ding W, Li N, Liao L, Chen D, Huang J. et al. The effects and regulatory mechanism of RIP3 on RGC-5 necroptosis following elevated hydrostatic pressure. Acta Biochim Biophys Sin. 2017;49:128-37
74. Ding W, Shang L, Huang JF, Li N, Chen D, Xue LX. et al. Receptor interacting protein 3-induced RGC-5 cell necroptosis following oxygen glucose deprivation. BMC neuroscience. 2015;16:49
75. Huang JF, Shang L, Zhang MQ, Wang H, Chen D, Tong JB. et al. Differential neuronal expression of receptor interacting protein 3 in rat retina: involvement in ischemic stress response. BMC neuroscience. 2013;14:16
76. Jeon SJ, Huh J, Jeong E, Park CK, Park HYL. Angiotensin II related glial cell activation and necroptosis of retinal ganglion cells after systemic hypotension in glaucoma. Cell Death & Disease. 2022;13:323
77. Gao S, Andreeva K, Cooper NG. Ischemia-reperfusion injury of the retina is linked to necroptosis via the ERK1/2-RIP3 pathway. Molecular vision. 2014;20:1374-87
78. Lee Y-S, Dayma Y, Park M-Y, Kim KI, Yoo S-E, Kim E. Daxx is a key downstream component of receptor interacting protein kinase 3 mediating retinal ischemic cell death. FEBS Letters. 2013;587:266-71
79. Yang R, Hu K, Chen J, Zhu S, Li L, Lu H. et al. Necrostatin-1 protects hippocampal neurons against ischemia/reperfusion injury via the RIP3/DAXX signaling pathway in rats. Neuroscience Letters. 2017;651:207-15
80. Wang S, Huang Y, Yan Y, Zhou H, Wang M, Liao L. et al. Calpain2 but not calpain1 mediated by calpastatin following glutamate-induced regulated necrosis in rat retinal neurons. Annals of anatomy = Anatomischer Anzeiger: official organ of the Anatomische Gesellschaft. 2019;221:57-67
81. Shang L, Huang JF, Ding W, Chen S, Xue LX, Ma RF. et al. Calpain: a molecule to induce AIF-mediated necroptosis in RGC-5 following elevated hydrostatic pressure. BMC neuroscience. 2014;15:63
82. Chen S, Yan J, Deng HX, Long LL, Hu YJ, Wang M. et al. Inhibition of calpain on oxygen glucose deprivation-induced RGC-5 necroptosis. Journal of Huazhong University of Science and Technology Medical sciences = Hua zhong ke ji da xue xue bao Yi xue Ying De wen ban = Huazhong keji daxue xuebao Yixue Yingdewen ban. 2016;36:639-45
83. Li N, Shang L, Wang SC, Liao LS, Chen D, Huang JF. et al. The Toxic Effect of ALLN on Primary Rat Retinal Neurons. Neurotoxicity research. 2016;30:392-406
84. Wang S, Liao L, Huang Y, Wang M, Zhou H, Chen D. et al. Pin1 Is Regulated by CaMKII Activation in Glutamate-Induced Retinal Neuronal Regulated Necrosis. Frontiers in cellular neuroscience. 2019;13:276
85. Yan W-T, Yang Y-D, Hu X-M, Ning W-Y, Liao L-S, Lu S. et al. Do pyroptosis, apoptosis, and necroptosis (PANoptosis) exist in cerebral ischemia? Evidence from cell and rodent studies. Neural regeneration research. 2022;17:1761-8
86. Malireddi RKS, Kesavardhana S, Kanneganti T-D. ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis). Frontiers in Cellular and Infection Microbiology. 2019;9:406
87. Jonas JB, Aung T, Bourne RR, Bron AM, Ritch R, Panda-Jonas S. Glaucoma. Lancet (London, England). 2017;390:2183-93
88. Wubben TJ, Besirli CG, Zacks DN. Pharmacotherapies for Retinal Detachment. Ophthalmology. 2016;123:1553-62
89. Lo AC, Woo TT, Wong RL, Wong D. Apoptosis and other cell death mechanisms after retinal detachment: implications for photoreceptor rescue. Ophthalmologica Journal international d'ophtalmologie International journal of ophthalmology Zeitschrift fur Augenheilkunde. 2011;226:10-7
90. Nakazawa T, Matsubara A, Noda K, Hisatomi T, She H, Skondra D. et al. Characterization of cytokine responses to retinal detachment in rats. Molecular vision. 2006;12:867-78
91. Hisatomi T, Sakamoto T, Murata T, Yamanaka I, Oshima Y, Hata Y. et al. Relocalization of apoptosis-inducing factor in photoreceptor apoptosis induced by retinal detachment in vivo. The American journal of pathology. 2001;158:1271-8
92. Matsumoto H, Kataoka K, Tsoka P, Connor KM, Miller JW, Vavvas DG. Strain difference in photoreceptor cell death after retinal detachment in mice. Investigative ophthalmology & visual science. 2014;55:4165-74
93. Dong K, Zhu ZC, Wang FH, Ke GJ, Yu Z, Xu X. Activation of autophagy in photoreceptor necroptosis after experimental retinal detachment. International journal of ophthalmology. 2014;7:745-52
94. Ni Y, Gu WW, Liu ZH, Zhu YM, Rong JG, Kent TA. et al. RIP1K Contributes to Neuronal and Astrocytic Cell Death in Ischemic Stroke via Activating Autophagic-lysosomal Pathway. Neuroscience. 2018;371:60-74
95. Goodall ML, Fitzwalter BE, Zahedi S, Wu M, Rodriguez D, Mulcahy-Levy JM. et al. The Autophagy Machinery Controls Cell Death Switching between Apoptosis and Necroptosis. Developmental cell. 2016;37:337-49
96. Wang P, Shao BZ, Deng Z, Chen S, Yue Z, Miao CY. Autophagy in ischemic stroke. Progress in neurobiology. 2018;163-164:98-117
97. Carullo G, Federico S, Relitti N, Gemma S, Butini S, Campiani G. Retinitis Pigmentosa and Retinal Degenerations: Deciphering Pathways and Targets for Drug Discovery and Development. ACS chemical neuroscience. 2020;11:2173-91
98. Wang SK, Xue Y, Rana P, Hong CM, Cepko CL. Soluble CX3CL1 gene therapy improves cone survival and function in mouse models of retinitis pigmentosa. Proceedings of the National Academy of Sciences of the United States of America. 2019;116:10140-9
99. Perdices L, Fuentes-Broto L, Segura F, Cavero A, Orduna-Hospital E, Insa-Sánchez G. et al. Systemic epigallocatechin gallate protects against retinal degeneration and hepatic oxidative stress in the P23H-1 rat. Neural regeneration research. 2022;17:625-31
100. Murakami Y, Ikeda Y, Nakatake S, Tachibana T, Fujiwara K, Yoshida N. et al. Necrotic enlargement of cone photoreceptor cells and the release of high-mobility group box-1 in retinitis pigmentosa. Cell death discovery. 2015;1:15058
101. Chang GQ, Hao Y, Wong F. Apoptosis: final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron. 1993;11:595-605
102. Liu Y, Liu T, Lei T, Zhang D, Du S, Girani L. et al. RIP1/RIP3-regulated necroptosis as a target for multifaceted disease therapy (Review). International journal of molecular medicine. 2019;44:771-86
103. Murakami Y, Matsumoto H, Roh M, Suzuki J, Hisatomi T, Ikeda Y. et al. Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:14598-603
104. Sato K, Li S, Gordon WC, He J, Liou GI, Hill JM. et al. Receptor interacting protein kinase-mediated necrosis contributes to cone and rod photoreceptor degeneration in the retina lacking interphotoreceptor retinoid-binding protein. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:17458-68
105. Viringipurampeer IA, Metcalfe AL, Bashar AE, Sivak O, Yanai A, Mohammadi Z. et al. NLRP3 inflammasome activation drives bystander cone photoreceptor cell death in a P23H rhodopsin model of retinal degeneration. Human molecular genetics. 2016;25:1501-16
106. Huang Z, Zhou T, Sun X, Zheng Y, Cheng B, Li M. et al. Necroptosis in microglia contributes to neuroinflammation and retinal degeneration through TLR4 activation. Cell Death & Differentiation. 2018;25:180-9
107. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805-20
108. Zhao Y, Shen Y. Light-Induced Retinal Ganglion Cell Damage and the Relevant Mechanisms. Cellular and molecular neurobiology. 2020;40:1243-52
109. Organisciak DT, Vaughan DK. Retinal light damage: mechanisms and protection. Progress in retinal and eye research. 2010;29:113-34
110. Tso MO, Woodford BJ. Effect of photic injury on the retinal tissues. Ophthalmology. 1983;90:952-63
111. Contín MA, Benedetto MM, Quinteros-Quintana ML, Guido ME. Light pollution: the possible consequences of excessive illumination on retina. Eye (London, England). 2016;30:255-63
112. King A, Gottlieb E, Brooks DG, Murphy MP, Dunaief JL. Mitochondria-derived reactive oxygen species mediate blue light-induced death of retinal pigment epithelial cells. Photochemistry and photobiology. 2004;79:470-5
113. Osborne NN, Li GY, Ji D, Mortiboys HJ, Jackson S. Light affects mitochondria to cause apoptosis to cultured cells: possible relevance to ganglion cell death in certain optic neuropathies. Journal of neurochemistry. 2008;105:2013-28
114. Cabon L, Galán-Malo P, Bouharrour A, Delavallée L, Brunelle-Navas MN, Lorenzo HK. et al. BID regulates AIF-mediated caspase-independent necroptosis by promoting BAX activation. Cell Death & Differentiation. 2012;19:245-56
115. Ji D, Kamalden TA, del Olmo-Aguado S, Osborne NN. Light- and sodium azide-induced death of RGC-5 cells in culture occurs via different mechanisms. Apoptosis: an international journal on programmed cell death. 2011;16:425-37
116. Chang Q, Peter ME, Grassi MA. Fas ligand-Fas signaling participates in light-induced apoptotic death in photoreceptor cells. Investigative ophthalmology & visual science. 2012;53:3703-16
117. Blanch RJ, Bindra MS, Jacks AS, Scott RA. Ophthalmic injuries in British Armed Forces in Iraq and Afghanistan. Eye (London, England). 2011;25:218-23
118. Liu Y, Feng K, Jiang H, Hu F, Gao J, Zhang W. et al. Characteristics and treatments of ocular blast injury in Tianjin explosion in China. BMC ophthalmology. 2020;20:185
119. Kuhn F, Morris R, Witherspoon CD, Heimann K, Jeffers JB, Treister G. A standardized classification of ocular trauma. Ophthalmology. 1996;103:240-3
120. Scott R. The injured eye. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2011;366:251-60
121. Bricker-Anthony C, Hines-Beard J, Rex TS. Molecular changes and vision loss in a mouse model of closed-globe blast trauma. Investigative ophthalmology & visual science. 2014;55:4853-62
122. Bricker-Anthony C, Rex TS. Neurodegeneration and Vision Loss after Mild Blunt Trauma in the C57Bl/6 and DBA/2J Mouse. PloS one. 2015;10:e0131921
123. Thomas CN, Thompson AM, Ahmed Z, Blanch RJ. Retinal Ganglion Cells Die by Necroptotic Mechanisms in a Site-Specific Manner in a Rat Blunt Ocular Injury Model. Cells. 2019;8:1517
124. Thomas CN, Courtie E, Bernardo-Colón A, Essex G, Rex TS, Ahmed Z. et al. Assessment of necroptosis in the retina in a repeated primary ocular blast injury mouse model. Experimental eye research. 2020;197:108102
125. Bricker-Anthony C, Hines-Beard J, D'Surney L, Rex TS. Exacerbation of blast-induced ocular trauma by an immune response. Journal of neuroinflammation. 2014;11:192
126. Shah MA, Shah SM, Gosai SR, Gupta SS, Khanna RR, Patel KB. et al. Comparative study of visual outcome between open- and closed-globe injuries following surgical treatment of traumatic cataract in children. European journal of ophthalmology. 2018;28:406-11
127. Mir TA, Canner JK, Zafar S, Srikumaran D, Friedman DS, Woreta FA. Characteristics of Open Globe Injuries in the United States From 2006 to 2014. JAMA ophthalmology. 2020;138:268-75
128. Zhang Y, Zhang MN, Jiang CH, Yao Y, Zhang K. Endophthalmitis following open globe injury. The British journal of ophthalmology. 2010;94:111-4
129. Chee YE, Patel MM, Vavvas DG. Retinal detachment after open-globe injury. International ophthalmology clinics. 2013;53:79-92
130. Osman EA. Glaucoma after open globe injury. Saudi journal of ophthalmology: official journal of the Saudi Ophthalmological Society. 2015;29:222-4
131. Shapira Y, Mimouni M, Vishnevskia-Dai V. Cytomegalovirus retinitis in HIV-negative patients - associated conditions, clinical presentation, diagnostic methods and treatment strategy. Acta ophthalmologica. 2018;96:e761-e7
132. Joye A, Gonzales JA. Ocular manifestations of cytomegalovirus in immunocompetent hosts. Current opinion in ophthalmology. 2018;29:535-42
133. Chien H, Dix RD. Evidence for multiple cell death pathways during development of experimental cytomegalovirus retinitis in mice with retrovirus-induced immunosuppression: apoptosis, necroptosis, and pyroptosis. Journal of virology. 2012;86:10961-78
134. Xu J, Mo J, Liu X, Marshall B, Atherton SS, Dong Z. et al. Depletion of the Receptor-Interacting Protein Kinase 3 (RIP3) Decreases Photoreceptor Cell Death During the Early Stages of Ocular Murine Cytomegalovirus Infection. Investigative ophthalmology & visual science. 2018;59:2445-58
135. Zhang M, Covar J, Marshall B, Dong Z, Atherton SS. Lack of TNF-alpha promotes caspase-3-independent apoptosis during murine cytomegalovirus retinitis. Investigative ophthalmology & visual science. 2011;52:1800-8
136. Mo J, Marshall B, Covar J, Zhang NY, Smith SB, Atherton SS. et al. Role of Bax in death of uninfected retinal cells during murine cytomegalovirus retinitis. Investigative ophthalmology & visual science. 2014;55:7137-46
137. Yang F, Ma H, Butler MR, Ding XQ. Deficiency of type 2 iodothyronine deiodinase reduces necroptosis activity and oxidative stress responses in retinas of Leber congenital amaurosis model mice. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2018;32:fj201800484RR
138. Ross M, Ofri R, Aizenberg I, Abu-Siam M, Pe'er O, Arad D. et al. Naturally-occurring myopia and loss of cone function in a sheep model of achromatopsia. Scientific reports. 2020;10:19314
139. Chang B, Dacey MS, Hawes NL, Hitchcock PF, Milam AH, Atmaca-Sonmez P. et al. Cone photoreceptor function loss-3, a novel mouse model of achromatopsia due to a mutation in Gnat2. Investigative ophthalmology & visual science. 2006;47:5017-21
140. Chatoo W, Abdouh M, Duparc RH, Bernier G. Bmi1 distinguishes immature retinal progenitor/stem cells from the main progenitor cell population and is required for normal retinal development. Stem cells (Dayton, Ohio). 2010;28:1412-23
141. Mbefo MK, Arsenijevic Y. The Evaluation of BMI1 Posttranslational Modifications During Retinal Degeneration to Understand BMI1 Action on Photoreceptor Death Execution. Advances in experimental medicine and biology. 2018;1074:359-65
142. Allocca M, Corrigan JJ, Mazumder A, Fake KR, Samson LD. Inflammation, necrosis, and the kinase RIP3 are key mediators of AAG-dependent alkylation-induced retinal degeneration. Science signaling. 2019 12
143. Meira LB, Moroski-Erkul CA, Green SL, Calvo JA, Bronson RT, Shah D. et al. Aag-initiated base excision repair drives alkylation-induced retinal degeneration in mice. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:888-93
144. Calvo JA, Moroski-Erkul CA, Lake A, Eichinger LW, Shah D, Jhun I. et al. Aag DNA glycosylase promotes alkylation-induced tissue damage mediated by Parp1. PLoS genetics. 2013;9:e1003413
145. Zheng M, Atherton SS. Cytokine Profiles and Inflammatory Cells during HSV-1-Induced Acute Retinal Necrosis. Investigative ophthalmology & visual science. 2005;46:1356-63
146. Myśliwiec M, Zorena K, Balcerska A, Myśliwska J, Lipowski P, Raczyńska K. The activity of N-acetyl-beta-d-glucosaminidase and tumor necrosis factor-alpha at early stage of diabetic retinopathy development in type 1 diabetes mellitus children. Clinical Biochemistry. 2006;39:851-6
147. Kuo JZ, Guo X, Klein R, Klein BE, Cui J, Rotter JI. et al. Systemic Soluble Tumor Necrosis Factor Receptors 1 and 2 Are Associated with Severity of Diabetic Retinopathy in Hispanics. Ophthalmology. 2012;119:1041-6
148. Grajewski RS, Li J, Wasmuth S, Hennig M, Bauer D, Heiligenhaus A. Intravitreal treatment with antisense oligonucleotides targeting tumor necrosis factor-α in murine herpes simplex virus type 1 retinitis. Graefe's Archive for Clinical and Experimental Ophthalmology. 2012;250:231-8
149. Gu L, Xu H, Wang F, Xu G, Sinha D, Wang J. et al. Erythropoietin Exerts a Neuroprotective Function Against Glutamate Neurotoxicity in Experimental Diabetic Retina. Investigative ophthalmology & visual science. 2014;55:8208-22
150. Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X. et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nature chemical biology. 2008;4:313-21
151. Xie T, Peng W, Liu Y, Yan C, Maki J, Degterev A. et al. Structural basis of RIP1 inhibition by necrostatins. Structure (London, England: 1993). 2013;21:493-9
152. Ueta T, Ishihara K, Notomi S, Lee J-J, Maidana DE, Efstathiou NE. et al. RIP1 kinase mediates angiogenesis by modulating macrophages in experimental neovascularization. Proceedings of the National Academy of Sciences of the United States of America. 2019;116:23705-13
153. Dvoriantchikova G, Degterev A, Ivanov D. Retinal ganglion cell (RGC) programmed necrosis contributes to ischemia-reperfusion-induced retinal damage. Experimental eye research. 2014;123:1-7
154. Kim LA, Amarnani D, Gnanaguru G, Tseng WA, Vavvas DG, D'Amore PA. Tamoxifen toxicity in cultured retinal pigment epithelial cells is mediated by concurrent regulated cell death mechanisms. Investigative ophthalmology & visual science. 2014;55:4747-58
155. Khoury MK, Gupta K, Franco SR, Liu B. Necroptosis in the Pathophysiology of Disease. The American journal of pathology. 2020;190:272-85
156. Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB. et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death & Disease. 2012;3:e437
157. Jin L, Liu P, Yin M, Zhang M, Kuang Y, Zhu W. RIPK1: A rising star in inflammatory and neoplastic skin diseases. Journal of Dermatological Science. 2020;99:146-51
158. Harris PA, Bandyopadhyay D, Berger SB, Campobasso N, Capriotti CA, Cox JA. et al. Discovery of Small Molecule RIP1 Kinase Inhibitors for the Treatment of Pathologies Associated with Necroptosis. ACS medicinal chemistry letters. 2013;4:1238-43
159. Weisel K, Berger S, Papp K, Maari C, Krueger JG, Scott N. et al. Response to Inhibition of Receptor-Interacting Protein Kinase 1 (RIPK1) in Active Plaque Psoriasis: A Randomized Placebo-Controlled Study. Clinical pharmacology and therapeutics. 2020;108:808-16
160. Weisel K, Berger S, Thorn K, Taylor PC, Peterfy C, Siddall H. et al. A randomized, placebo-controlled experimental medicine study of RIPK1 inhibitor GSK2982772 in patients with moderate to severe rheumatoid arthritis. Arthritis research & therapy. 2021;23:85
161. Beal AM, Bertin J, Reilly MA. Use of RIP1 Kinase Small-Molecule Inhibitors in Studying Necroptosis. In: Ting AT, editor. Programmed Necrosis: Methods and Protocols. New York, NY: Springer New York. 2018 p. 109-24
162. Yang X, Lu H, Xie H, Zhang B, Nie T, Fan C. et al. Potent and Selective RIPK1 Inhibitors Targeting Dual-Pockets for the Treatment of Systemic Inflammatory Response Syndrome and Sepsis. Angewandte Chemie International Edition. 2022;61:e202114922
163. Chen J, Wang S, Fu R, Zhou M, Zhang T, Pan W. et al. RIP3 dependent NLRP3 inflammasome activation is implicated in acute lung injury in mice. Journal of translational medicine. 2018;16:233
164. Guo C, Fu R, Zhou M, Wang S, Huang Y, Hu H. et al. Pathogenesis of lupus nephritis: RIP3 dependent necroptosis and NLRP3 inflammasome activation. Journal of autoimmunity. 2019;103:102286
165. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J. et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nature cell biology. 2014;16:55-65
166. Liang S, Nian Z, Shi K. Inhibition of RIPK1/RIPK3 ameliorates osteoclastogenesis through regulating NLRP3-dependent NF-κB and MAPKs signaling pathways. Biochemical and biophysical research communications. 2020;526:1028-35
167. Zhang H, Zhou L, Zhou Y, Wang L, Jiang W, Liu L. et al. Intermittent hypoxia aggravates non-alcoholic fatty liver disease via RIPK3-dependent necroptosis-modulated Nrf2/NFκB signaling pathway. Life Sciences. 2021;285:119963
168. Park HH, Park SY, Mah S, Park JH, Hong SS, Hong S. et al. HS-1371, a novel kinase inhibitor of RIP3-mediated necroptosis. Experimental & molecular medicine. 2018;50:1-15
169. Huang D, Chen P, Huang G, Sun H, Luo X, He C. et al. Salt-inducible kinases inhibitor HG-9-91-01 targets RIPK3 kinase activity to alleviate necroptosis-mediated inflammatory injury. Cell Death & Disease. 2022;13:188
170. Xu C-h, Wang J-n, Suo X-g, Ji M-l, He X-y, Chen X. et al. RIPK3 inhibitor-AZD5423 alleviates acute kidney injury by inhibiting necroptosis and inflammation. International Immunopharmacology. 2022;112:109262
171. Xia K, Zhu F, Yang C, Wu S, Lin Y, Ma H. et al. Discovery of a Potent RIPK3 Inhibitor for the Amelioration of Necroptosis-Associated Inflammatory Injury. Frontiers in cell and developmental biology. 2020;8:606119
172. Ueda S, Chen-Yoshikawa TF, Tanaka S, Yamada Y, Nakajima D, Ohsumi A. et al. Protective effect of necrosulfonamide on rat pulmonary ischemia-reperfusion injury via inhibition of necroptosis. The Journal of thoracic and cardiovascular surgery. 2021;163:e113-e22
173. Ueda S, Chen-Yoshikawa TF, Mineura K, Yamanashi K, Oda H, Yokoyama Y. et al. Protective Effects of Necrosulfonamide on Ischemia-Reperfusion Injury in Rat Lung. J Heart Lung Transplant. 2020;39:S353
174. Jiao J, Wang Y, Ren P, Sun S, Wu M. Necrosulfonamide Ameliorates Neurological Impairment in Spinal Cord Injury by Improving Antioxidative Capacity. Frontiers in pharmacology. 2019;10:1538
175. Liao D, Sun L, Liu W, He S, Wang X, Lei X. Necrosulfonamide inhibits necroptosis by selectively targeting the mixed lineage kinase domain-like protein. MedChemComm. 2014;5:333-7
176. Motawi TMK, Abdel-Nasser ZM, Shahin NN. Ameliorative Effect of Necrosulfonamide in a Rat Model of Alzheimer's Disease: Targeting Mixed Lineage Kinase Domain-like Protein-Mediated Necroptosis. ACS chemical neuroscience. 2020;11:3386-97
177. Zhong CS, Zeng B, Qiu JH, Xu LH, Zhong MY, Huang YT. et al. Gout-associated monosodium urate crystal-induced necrosis is independent of NLRP3 activity but can be suppressed by combined inhibitors for multiple signaling pathways. Acta pharmacologica Sinica. 2021;43:1324-36
178. Shlomovitz I, Erlich Z, Speir M, Zargarian S, Baram N, Engler M. et al. Necroptosis directly induces the release of full-length biologically active IL-33 in vitro and in an inflammatory disease model. The FEBS journal. 2019;286:507-22
179. Lusthaus M, Mazkereth N, Donin N, Fishelson Z. Receptor-Interacting Protein Kinases 1 and 3, and Mixed Lineage Kinase Domain-Like Protein Are Activated by Sublytic Complement and Participate in Complement-Dependent Cytotoxicity. Frontiers in immunology. 2018;9:306
180. Rübbelke M, Fiegen D, Bauer M, Binder F, Hamilton J, King J. et al. Locking mixed-lineage kinase domain-like protein in its auto-inhibited state prevents necroptosis. Proceedings of the National Academy of Sciences of the United States of America. 2020;117:33272-81
181. Magaway C, Kim E, Jacinto E. Targeting mTOR and Metabolism in Cancer: Lessons and Innovations. Cells. 2019 p. 1584
182. Sumsuzzman DM, Choi J, Jin Y, Hong Y. Neurocognitive effects of melatonin treatment in healthy adults and individuals with Alzheimer's disease and insomnia: A systematic review and meta-analysis of randomized controlled trials. Neuroscience & Biobehavioral Reviews. 2021;127:459-73
183. Alghamdi BS. The neuroprotective role of melatonin in neurological disorders. Journal of neuroscience research. 2018;96:1136-49
184. Ye D, Xu Y, Shi Y, Fan M, Lu P, Bai X. et al. Anti-PANoptosis is involved in neuroprotective effects of melatonin in acute ocular hypertension model. Journal of Pineal Research. 2022: e12828.
185. Malhotra K, Chang JJ, Khunger A, Blacker D, Switzer JA, Goyal N. et al. Minocycline for acute stroke treatment: a systematic review and meta-analysis of randomized clinical trials. Journal of Neurology. 2018;265:1871-9
186. Song W, Zhu R, Gao W, Xing C, Yang L. Blue Light Induces RPE Cell Necroptosis, Which Can Be Inhibited by Minocycline. Frontiers in Medicine. 2022;9:831463
187. Akyuz E, Kullu I, Arulsamy A, Shaikh MF. Melatonin as an Antiepileptic Molecule: Therapeutic Implications via Neuroprotective and Inflammatory Mechanisms. ACS chemical neuroscience. 2021;12:1281-92
188. Aghajani Shahrivar A, Khakpourian Z, Majdi F, Sobhani S, Coleman-Fuller N, Gholami M. et al. Hypothesized neuroprotective effect of minocycline against COVID-19-induced stroke and neurological dysfunction: possible role of matrix metalloprotease signaling pathway. Biologia. 2022;9:1-9
189. Qin C, Bai L, Li Y, Wang K. The functional mechanism of bone marrow-derived mesenchymal stem cells in the treatment of animal models with Alzheimer's disease: crosstalk between autophagy and apoptosis. Stem Cell Research & Therapy. 2022;13:90
190. Usategui-Martín R, Puertas-Neyra K, Galindo-Cabello N, Hernández-Rodríguez LA, González-Pérez F, Rodríguez-Cabello JC. et al. Retinal Neuroprotective Effect of Mesenchymal Stem Cells Secretome Through Modulation of Oxidative Stress, Autophagy, and Programmed Cell Death. Investigative ophthalmology & visual science. 2022;63:27 -
191. Labrador-Velandia S, Alonso-Alonso ML, Di Lauro S, García-Gutierrez MT, Srivastava GK, Pastor JC. et al. Mesenchymal stem cells provide paracrine neuroprotective resources that delay degeneration of co-cultured organotypic neuroretinal cultures. Experimental eye research. 2019;185:107671
192. Labrador-Velandia S, Alonso-Alonso ML, Alvarez-Sanchez S, González-Zamora J, Carretero-Barrio I, Pastor JC. et al. Mesenchymal stem cell therapy in retinal and optic nerve diseases: An update of clinical trials. World journal of stem cells. 2016;8:376-83
193. Abokyi S, To C-H, Lam TT, Tse DY. Central Role of Oxidative Stress in Age-Related Macular Degeneration: Evidence from a Review of the Molecular Mechanisms and Animal Models. Oxidative medicine and cellular longevity. 2020;2020:7901270
194. Amato R, Canovai A, Melecchi A, Pezzino S, Corsaro R, Dal Monte M. et al. Dietary Supplementation of Antioxidant Compounds Prevents Light-Induced Retinal Damage in a Rat Model. Biomedicines. 2021;9:1177
195. Fouda AY, Xu Z, Shosha E, Lemtalsi T, Chen J, Toque HA. et al. Arginase 1 promotes retinal neurovascular protection from ischemia through suppression of macrophage inflammatory responses. Cell Death & Disease. 2018;9:1001
Author contact
![]() Corresponding author: E-mail: xiongkunedu.cn.
Corresponding author: E-mail: xiongkunedu.cn.