Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(3):981-993. doi:10.7150/ijbs.79852 This issue Cite
Research Paper
Galectin-9 blockade synergizes with ATM inhibition to induce potent anti-tumor immunity
Shuang Zheng1,2#, Jiaming Song1#, Dongli Linghu1,3#, Riyao Yang4,5, Boning Liu1, Zhen Xue1,2, Qihui Chen1, Chengjie Liu1, Diansheng Zhong1,2 ![]() , Mien-Chie Hung6,5
, Mien-Chie Hung6,5 ![]() , Linlin Sun1
, Linlin Sun1 ![]()
1. Tianjin Key Laboratory of Lung Cancer Metastasis and Tumor Microenvironment, Lung Cancer Institute, Tianjin Medical University General Hospital, Tianjin 300052, P. R. China.
2. Department of Medical Oncology, Tianjin Medical University General Hospital, Tianjin 300052, P. R. China.
3. Department of Lung Cancer Surgery, Tianjin Medical University General Hospital, Tianjin 300052, P.R. China.
4. Antibody Therapeutics, Inc., Hayward, CA, USA.
5. Department of Molecular and Cellular Oncology, The University of Texas M. D. Anderson Cancer Center, Houston, Texas 77030, USA.
6. Graduate Institute of Biomedical Sciences and Center for Molecular Medicine, China Medical University, Taichung 40402, Taiwan.
#These authors contributed equally to this work.
Received 2022-10-13; Accepted 2023-1-1; Published 2023-1-16
Abstract

Although current cancer immunotherapies that target PD-1/PD-L1 immune checkpoint to reinvigorate exhausted T cells have achieved impressive clinical outcomes, only a small proportion of patients respond. New therapeutic targets are therefore needed to be identified to further unleash the anti-tumor potential of T cells and benefit more patients. Galectin-9 (Gal-9), initially identified as a ligand for TIM-3 to induce T cell death, acts as an immunosuppressive regulator in the tumor microenvironment (TME) but its potential as a therapeutic target remains largely elusive. Here we show that antibody neutralization of Gal-9, in combination with inhibition of Ataxia telangiectasia mutated (ATM), a kinase essential for DNA damage response (DDR), is a promising modality for cancer immunotherapy. Genetic depletion of ATM in tumors markedly potentiated anti-Gal-9 therapy in a syngeneic mouse model. Mechanistically, ATM inhibition greatly upregulated Gal-9 expression and secretion in a variety of human and murine tumor cells via the cGAS-STING-interferon β (IFNβ) innate immune pathway. Combination of Gal-9 inhibition with AZD1390, a selective ATM inhibitor currently evaluated in clinical trials, significantly suppressed tumor growth and prolonged survival in multiple syngeneic mouse models, including the poorly-immunogenic LLC lung tumors that do not respond to PD-1/PD-L1 blockade, concomitant with increased T cell infiltration. These results reveal Gal-9 induction via STING/IFNβ signaling as an important mechanism mediating tumor immune escape that could be targeted for cancer immunotherapies, and unveil a novel anti-Gal-9-based combination strategy for cancer immunotherapies in a wide variety of malignancies, including those resistant to PD-1/PD-L1 blockade.
Keywords: Cancer immunotherapy, DNA damage response, cGAS-STING, galectin
Introduction
Therapeutic blockade of the programmed cell death protein 1 (PD-1) and the cytotoxic T-lymphocyte associated protein 4 (CTLA-4) immune checkpoints has emerged as a powerful strategy for cancer treatment in multiple cancer types by reinvigorating exhausted T cells [1,2]. However, the remarkable responses are currently limited to a minority of patients, highlighting the need for exploring new therapeutic targets to further unleash the anti-tumor potential of T cells and benefit more patients. Galectin-9 (Gal-9), a member of the galectin family, was initially identified as a ligand for TIM-3 to induce T cell death [3]. Elevated expression of Gal-9 is associated with a more terminally exhausted T cell phenotype [4]. Gal-9 protein has two conserved distinct carbohydrate-recognition domains (CRD) for binding β-galactosides, capable of crosslinking glycoproteins and modulating cell signaling [5]. Gal-9 primarily facilitates immunosuppression in the tumor microenvironment (TME) by interacting with multiple cell surface receptors on immune cells, such as TIM-3, Dectin-1 and CD44 [3,6,7]. It is constitutively expressed in antigen-presenting cells (APCs), and can be induced in tumor cells by interferon (IFN) β and γ [8]. The expression of Gal-9 is aberrantly elevated in multiple cancer types and largely correlates with poor patient survival [9-12]. However, the regulation of Gal-9 in the context of cancer therapies and its potential as a therapeutic target remain largely elusive.
The defect in DNA repair is a hallmark of cancer. DNA damage response (DDR) pathway is essential for the maintenance of genomic integrity [13,14]. The deficiency of DDR has been shown to be a critical contribution to tumor immunogenicity. Mounting evidence supports that DDR-targeted therapies can induce innate immunity and promote antitumor immune response [15]. Ataxia telangiectasia mutated (ATM) is an important DDR kinase which senses and mediates the repair of DNA double-strand breaks (DSBs) by homologous recombination (HR) [16,17]. Clinically, hereditary mutations of ATM result in ataxia telangiectasia (A-T), a syndrome characterized with neurodegeneration, immunodeficiency, radiation sensitivity and cancer predisposition [18]. Somatic mutations or deletions of ATM are commonly found in a variety of cancers [19]. Inhibition of ATM results in reduced HR and compromised DSB repair, leading to cell cycle arrest or cell death. ATM inhibitors have been extensively evaluated in clinical trials [20] and found to sensitize tumors to chemotherapy, radiation, and other DDR inhibitors in multiple cancer types [19]. Moreover, prior study indicates that ATM deficiency activates cGAS/STING through promoting cytoplasmic leakage of mitochondrial DNA, and subsequently induces type I interferon (T1IFN)-mediated innate immune response in certain cancer types [21,22]. Although ATM inhibition has been shown to potentiate anti-PD-1 therapy [22], the therapeutic efficacy of their combination is moderate, suggesting that there might be additional unknown mechanisms that mediate immune escape. Better understanding of the immunological effect of ATM inhibition on the TME will help to identify new targets to enhance its efficacy.
In this study, we demonstrate that Gal-9 is induced by ATM inhibition through cGAS-STING-IFNβ pathway in a variety of tumor cells. Importantly, Gal-9 blockade combined with genetic depletion or pharmaceutical inhibition of ATM synergistically induces potent anti-tumor immunity and markedly reduces tumor growth in syngeneic mouse models. These findings highlight Gal-9 as a promising therapeutic target in ATM-silenced tumors, and provide a rationale for combining ATM inhibition with anti-Gal-9 as novel strategy for cancer immunotherapy.
Materials and Methods
Cell lines and reagents
All cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA), independently validated by short tandem repeat (STR) DNA fingerprinting, and tested negative for mycoplasma infection. NCI-H157, A549, NCI-H1299, HeLa, BT549, MDA-MB-231, CT26, B16-F10, MC-38, LLC and 293T cells were cultured in RPMI 1640 or DMEM media, supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C in a humidified atmosphere with 5% CO2. Inhibitors for ATM (KU-60019, AZD1390, AZD0156), STING (H151), p-TBK1 (GSK8612), p-STAT1 (fludarabine) were purchased from Selleck (Houston, TX, USA). Human IFNβ and Galectin-9 ELISA assay Kits were purchased from R&D SYSTEMS (Minneapolis, MN, USA). Anti-human Gal-9 antibody was purchased from BioRad (Hercules, CA, USA). Anti-mouse Gal-9 antibody and IgG isotype control were purchased from BioXCell (Lebanon, NH, USA). Antibodies against cGAS, STING, p-STING(Ser366), p-TKB1(Ser172), TKB1, p-STAT1(Tyr701), STAT1 and Actin were purchased from Cell Signaling Technology (Cambridge, MA, USA). Anti-mouse fluorochrome-conjugated antibodies including FITC-CD4, Percp/Cy5.5-CD8, AF700-CD3, APC/Cy7-CD45, PE/Cy7-IFNγ antibodies were purchased from BioLegend (San Diego, CA, USA).
Real-time PCR
Real-time PCR analysis was performed as described previously [23]. In brief, total RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA). cDNA was prepared using HiScript III 1st Strand cDNA Synthesis Kit (Vazyme, Nanjing, China) with 1 μg of total RNA according to the manufacture's protocol. Real-time PCR reactions were then performed in a mixture containing 1×SYBR Green Mix (Vazyme, Nanjing, China), specific primers and cDNA template using the Applied Biosystem 7900 system (USA). The relative expression levels of target genes were normalized to that of GAPDH. Primers used for different target genes are listed in Supplemental Table 1.
Immunoblot analysis
Immunoblot analysis was performed as described previously [24]. In brief, proteins were resolved by SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Burlington, MA, USA). The membranes were blocked in Tris-buffered saline containing 0.2% Tween 20 and 5% fat-free dry milk, and then incubated with primary antibodies at 4 °C overnight, and horseradish peroxidase-conjugated secondary antibodies at room temperature for 1 hour. After washes, specific protein bands were visualized using chemiluminescence detection reagent (Millipore, Billerica, MA, USA) according to the manufacturer's instructions (Pierce Biotechnology, Waltham, MA, USA).
ELISA
ELISA analysis was performed as described previously [8]. Briefly, Nunc Maxisorp ELISA Plates (BioLegend) were coated with capture antibody in PBS at 4 °C overnight according to the manufacture's protocol (R&D SYSTEMS, Minneapolis, MN, USA). The coated plates were then washed, blocked with BSA and incubated with conditional media. This was followed by incubation first with the detection antibody and then with HRP-labeled IgG antibodies. After washes, the plates were incubated with TMB substrate solution for 20 mins, and the reaction was terminated by the addition of H2SO4. Absorbances at 450 nm with a reference wavelength of 570 nm was then measured using a microplate reader (SpectraMax M5, Molecular Devices, CA, USA).
Crystal violet staining
Cells were seeded in appropriate dilutions to form colonies in 1-3 weeks. Colonies were fixed with 4% paraformaldehyde, stained with 0.5% crystal violet staining solution at room temperature for 30 min, and then washed three times with deionized water.
Cell proliferation assay
Cell proliferation was determined using Cell Counting Kit-8 (CCK8) assay (Bimake.cn, Shanghai, China). Cells were seeded in 96-well plates to grow for 2-4 days, and then incubated with CCK8 reagent at 37 °C for 1-3 hours. The absorbance at 450 nm was measured with a microplate reader (SpectraMax M5, Molecular Devices, CA, USA).
Generation of stable knockdown cells by lentiviral transduction
The plasmids encoding shRNA targeting human STING, IFNAR1 or murine ATM were obtained from Shanghai Genechem Co., Ltd. n. The sequences for different shRNAs are listed in Supplemental Table 2. To generate stable knockdown cells, 293T cells were co-transfected with 4 μg shRNA expressing construct, 3 μg pCMV-dR8.2 and 1 μg pCMV-VSVG helper construct using Lipofectamine 3000 reagent (Life Technologies, Carlsbad, CA, USA). The virus stocks were harvested from the culture medium after 2 days and then filtered to remove non-adherent 293T cells. To select the cells that stably express the specific shRNA, cells were plated at subconfluent densities and transduced with a cocktail containing 1 ml virus stock, 1 ml regular medium and 8 μg/ml polybrene, and then selected with 2 μg/ml of puromycin 48-72 hours after lentiviral transduction.
Analysis of TILs by flow cytometry
Approximately 5×105 CT26 cells were inoculated subcutaneously into the left flank of BALB/c mice and treated with isotype control, AZD1390, anti-Gal-9 antibody (BioXCell, Lebanon, NH, USA), or the combination AZD1390 and anti-Gal-9 antibody. Tumors were excised on day 14 after tumor inoculation, and then mechanically minced and incubated in DNase I (50ug/mL, Solarbio) and collagenase IV (3 mg/mL, Solarbio) for 20 minutes at 37 °C. The dissociated cells were then passed through a 70 μm cell strainer (BD). The filtered cells were stained with Zombie Violet™ Fixable Viability Kit (BioLegend), blocked with FcR Blocking Reagent (Miltenyi), and stained with the indicated surface antibodies in staining buffer (PBS containing 1% BSA and 0.1% NaN3) for 20 minutes on ice. Intracellular antibodies were added after fixation and permeabilization according to the manufacturer's instructions (Thermo Fisher Scientific). The stained cells were analyzed by Flow Cytometer (BD Biosciences, CA, USA).
Animal models
All experiments were performed in accordance with the guidelines for the Care and Use of Laboratory Animals of Tianjin Medical University Hospital (Tianjin, P.R. China). 6-8-week-old male C57BL/6J mice and female BALB/c mice were purchased from Beijing Vital River Laboratory Animal Technology Co. Ltd and housed in laminar flow cabinets under specific pathogen-free conditions. For CT26 subcutaneous model, a total of 5×105 CT26 cells were injected subcutaneously into left flank of the BALB/c mice. For MC-38 subcutaneous model, a total of 5×105 MC-38 cells were injected subcutaneously into left flank of C57BL/6J mice. For LLC subcutaneous model, a total of 5×105 LLC cells were injected subcutaneously into left flank of C57BL/6J mice. After tumors were palpable, mice were randomized into treatment cohorts as isotype control, AZD1390 (5 mg/kg for 9 times), anti-Gal-9 antibody (100 μg/dose for 3 times) (BioXCell, Lebanon, NH, USA), or the combination AZD1390 and anti-Gal-9 antibody. Tumor size and body weight were measured every 2-3 days. Tumor volumes were calculated using the formula: ab2/2 (a: length, b: width). According to animal ethics, the mice were sacrificed when tumors reached 2000 mm3 in size.
For ATM-silenced CT26 subcutaneous model, a total of 1×106 vector control or ATM KD CT26 cells were injected subcutaneously into right flank of the BALB/c mice (female, 6-8 weeks old). After the tumors were palpable, mice were randomized and treated with anti-Gal-9 (100 μg/dose) or IgG isotype control (BioXCell, Lebanon, NH, USA) intraperitoneally every 3 days for 3 times. Tumor size and body weight were measured every 2-3 days. Tumor volumes were calculated using the formula: ab2/2 (a: length, b: width). According to animal ethics, the mice were sacrificed when tumors reached 2000 mm3 in size.
Statistical analysis
Statistical analysis was performed using the GraphPad Prism software. All data are presented as mean ± standard deviation (S.D.). Unpaired two-tailed t tests or one-way ANOVA were used for comparison between two groups. P values < 0.05 were considered to be statistically significant.
Results
Genetic depletion of ATM sensitizes tumors to anti-Gal-9 therapy
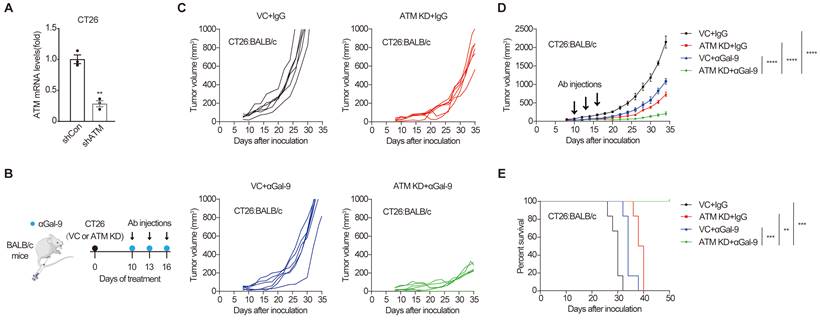
Analysis of ATM mutation frequency in The Cancer Genome Atlas (TCGA) cohort using c-Bioportal [25,26] indicated that ATM is frequently mutated in multiple human cancers, with a higher mutation frequency of 40% in mantle cell lymphoma, and approximately 20-35% in cutaneous, uterine, lung and colorectal cancers (Fig. S1A). To examine the efficacy of anti-Gal-9 therapy in ATM-silenced tumors, we first established an ATM knockdown (KD) cancer cell line by transducing the murine CT26 colon cancer cells with lentiviruses encoding shRNA for ATM. Substantially reduced ATM expression was verified using real-time PCR (Fig. 1A). ATM depletion did not affect the growth of tumor cells in vitro, as demonstrated by crystal violet staining and CCK-8 assay (Fig. S1B and C), suggesting that there were no cell-intrinsic growth defects in ATM KD cells. We then inoculated syngeneic BALB/c mice with control or ATM KD CT26 tumor cells, and found that ATM-depleted cells formed smaller tumors compared with vector control (VC) (Fig. S1D), indicating a tumor-suppressive effect of ATM depletion. Interestingly, while anti-Gal9 (RG-1) treatment (100 μg/dose for 3 times) partially suppressed the growth of control tumors, it resulted in almost complete growth suppression in ATM-KD tumors, correlated with markedly prolonged mouse survival (Fig. 1B-E), suggesting a remarkable synergy between Gal-9 inhibition and ATM depletion. Together, these results indicated that genetic depletion of ATM significantly potentiates anti-Gal-9 therapy in a syngeneic mouse model.
ATM depletion sensitizes tumors to anti-Gal-9 therapy in CT26 syngeneic mouse model. A. RT-qPCR analysis of ATM mRNA in vector control and ATM-KD CT26. B. A diagram of treatment strategy. BALB/c mice were inoculated with vector control (VC) or ATM-KD CT26 cells and then treated with isotype or anti-Gal-9 antibody (100μg /dose for 3 times). C. Tumor growth curves of individual BALB/c mice inoculated with VC or ATM-KD CT26 cells and subjected to indicated treatment (n = 6 mice/group). D. Average tumor growth of mice inoculated with VC or ATM-KD CT26 cells and subjected to indicated treatments. Error bars represent SEM of the means. Anti-Gal-9 antibody treatment schedule is indicated by arrows. E. Survival curves of BALB/c mice inoculated with VC or ATM-KD CT26 cells and subjected to the indicated treatments (n = 6 mice/group). n.s., not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
ATM inhibition induces Gal-9 expression and secretion in cancer cells
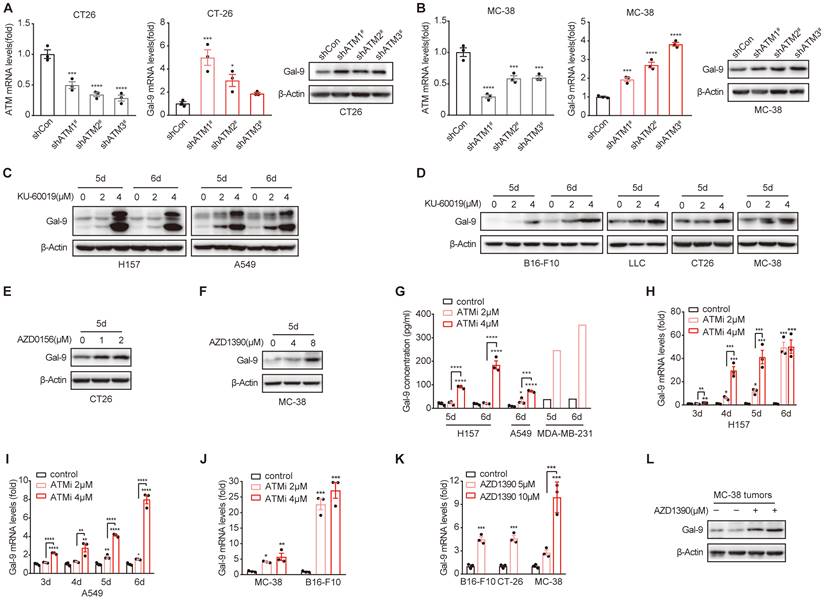
Next, we sought to investigate the molecular mechanisms underlying the potent efficacy of anti-Gal-9 in ATM-depleted tumors. Given that high levels of intratumoral Gal-9 in the TME is supposed to predict the response to anti-Gal-9 therapy, we hypothesized that ATM depletion might result in Gal-9 induction and thus render these tumors highly sensitive to anti-Gal-9 therapy. To test the possibility, we evaluated the expression of Gal-9 in VC and ATM-KD CT26 cells by RT-PCR and western blot. Indeed, we found Gal-9 mRNA and protein levels were increased in ATM KD CT26 cells compared with those of VC (Fig. 2A). Similar results were found in ATM-silenced MC-38 cells (Fig. 2B). These results suggested that genetic inhibition of ATM leads to Gal-9 induction in tumor cells. To further validate our findings, we treated a panel of human and murine cancer cell lines with a potent and selective ATM kinase inhibitor, Ku-60019, and analyzed Gal-9 protein levels by western blot. Similar to ATM KD, we found that Gal-9 levels were substantially enhanced in response to ATM inhibition (ATMi) in H157 and A549 human non-small cell lung cancer (NSCLC) cells, MDA-MB-231 human triple-negative breast cancer (TNBC) cells, B16-F10 murine melanoma cells, LLC murine lung tumor cells, CT26 and MC-38 murine colon tumor cells (Fig. 2C-D, Fig. S2A). Consistently, Gal-9 was also upregulated in response to AZD0156 and AZD1390, two another ATM inhibitors that have been evaluated in clinical trials (Fig. 2E-F). Given that Gal-9 is a secretory protein, we subsequently evaluated the secretion levels of Gal-9 by ELISA. In agreement, ATM inhibition resulted in a significant increase in the secretion of Gal-9 in H157, A549 and MDA-MB-231 cells (Fig. 2G). To investigate whether ATMi-mediated Gal-9 upregulation occurs at the transcriptional level, we evaluated Gal-9 mRNA levels with or without ATMi treatment. Consistently, Ku-60019 caused an increase in Gal-9 mRNA levels in multiple cancer cells (H157, A549, B16-F10 and MC-38), in a time and dose-dependent manner; Notably, Gal-9 mRNA levels were remarkably elevated on day 6 of Ku-60019 treatment (Fig. 2H-J), which, in comparison, only resulted in slightly upregulated PD-L1 mRNA levels (Fig. S2B). Similarly, AZD1390 treatment also resulted in an increase in Gal-9 mRNA in murine B16-F10, MC38 and CT26 cells (Fig. 2K), further suggesting that ATMi treatment promotes Gal-9 transcription. In addition, Gal-9 expression was found to be increased in ATMi-treated MC38 tumors harvested from C57BL/6J syngeneic mice (Fig. 2L, Fig. S2C). These results indicated that inhibition of ATM results in Gal-9 induction in tumor cells both in vitro and in vivo.
ATM KD or inhibition results in increased Gal-9 expression and secretion in a variety of tumor cells. A. RT-qPCR and immunoblot analysis of ATM and Gal-9 in vector control and ATM-KD CT26. B. RT-qPCR and immunoblot analysis of ATM and Gal-9 in vector control and ATM-KD MC-38 cells. C-F. Immunoblot analysis of Gal-9 in H157, A549, B16-F10, LLC, CT26 and MC-38 cells treated with KU-60019 (2 μmol/L or 4 μmol/L), AZD0516 (1 μmol/L or 2 μmol/L), AZD1390 (4 μmol/L or 8 μmol/L) or DMSO (vehicle control) for indicated time. G. ELISA analysis of Gal-9 in H157, A549 and MDA-MB-231 cancer cells treated with KU-60019 (2 μmol/L or 4 μmol/L) or DMSO (vehicle control) for indicated time. H and I. RT-qPCR analysis of Gal-9 mRNA levels in H157 and A549 cancer cells in response to KU-60019 (2 μmol/L or 4 μmol/L) or DMSO (vehicle control) for indicated time. J. RT-qPCR analysis of Gal-9 mRNA in MC-38 and B16-F10 cells in response to KU-60019 (2 μmol/L or 4 μmol/L) or DMSO (vehicle control) for 6 days. K. RT-qPCR analysis of Gal-9 mRNA in MC-38, B16-F10 and CT26 cells in response to AZD1390 (5 μmol/L or 10 μmol/L) or DMSO (vehicle control) for 6 days. L. Immunoblot analysis of Gal-9 in AZD1390-treated MC38 tumors harvested from C57BL/6J syngeneic mice. n.s., not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
ATM inhibition activates cGAS-STING-IFNβ signaling pathway in multiple cancer types
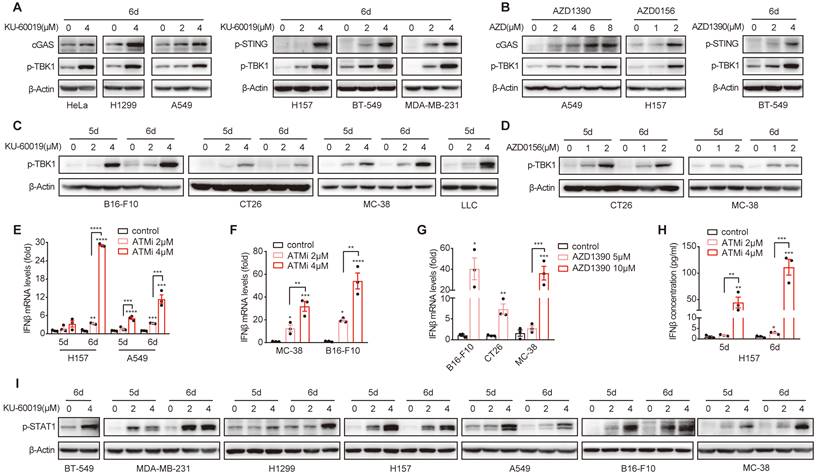
Next, we investigated the signaling pathway that could mediate ATMi-induced Gal-9 expression. Given the important role of IFNβ in upregulating Gal-9 expression [8], we thus evaluated whether STING-IFNβ pathway is activated in response to ATM inhibition in these cancer cell lines. Indeed, we found that inhibition of ATM led to a robust increase in the expression of cGAS, the phosphorylation of STING and TBK1 in a variety of human cancer cell lines, including HeLa, H157, H1299, A549, MDA-MB-231 and BT549 (Fig. 3A, Fig. S3A and B). Similar results were observed by treating cells with two another ATM inhibitors, AZD1390 and AZD0156 (Fig. 3B). Increased p-TBK1 levels was also found in various murine tumor cells including B16-F10, MC-38 and CT26 and LLC in response to ATM inhibition (Fig. 3C and D). In addition, the expression and secretion of IFNβ was elevated in response to ATM inhibition in various cancer cell lines (H157, A549, MC-38, CT26, and B16-F10) (Fig. 3E-H). Furthermore, the phosphorylation of STAT1, a key transcription factor downstream of IFNβ pathway that contributes to the activation of IFN-stimulated genes, was also enhanced upon ATM inhibition in a panel of human and murine cancer cells (BT549, MDA-MB-231, H1299, H157, A549, B16-F10 and MC-38) (Fig. 3I, Fig. S3C). Together, these results demonstrated that ATM inhibition leads to cGAS-STING activation and IFNβ signaling in multiple cancer types.
ATM inhibition activates cGAS-STING-IFNβ signaling in multiple cancer types. A and B. Immunoblot analysis of cGAS, p-STING and p-TBK1 levels in HeLa, H1299, A549, H157, BT549 and MDA-MB-231 cells in response to KU-60019, AZD1390, AZD0516 or DMSO (vehicle control) for 6 days. C and D. Immunoblot analysis of p-TBK1 levels in B16-F10, CT26, MC-38 and LLC cells in response to KU-60019 (2 μmol/L or 4 μmol/L), AZD0156 (1 μmol/L or 2 μmol/L) or DMSO (vehicle control) for 6 days. E. RT-qPCR analysis of IFNβ mRNA in H157 and A549 cancer cells in response to KU-60019 (2 μmol/L or 4 μmol/L) or DMSO (vehicle control) for indicated time. F and G. RT-qPCR analysis of IFNβ mRNA in in MC-38, B16-F10 and CT26 cells in response to KU-60019 (2 μmol/L or 4 μmol/L), AZD1390 (5 μmol/L or 10 μmol/L) or DMSO (vehicle control) for 6 days. H. ELISA analysis of IFNβ in H157 cancer cells treated with KU-60019 (2 μmol/L or 4 μmol/L) or DMSO (vehicle control) for indicated time. I. Immunoblot analysis of p-STAT1 levels in BT549, MDA-MB-231, H1299, A549, H157, B16-F10 and MC-38 cells in response to KU-60019 (2 μmol/L or 4 μmol/L) or DMSO (vehicle control) for indicated time. n.s., not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Gal-9 induction by ATM inhibition is mediated by STING-IFNβ pathway
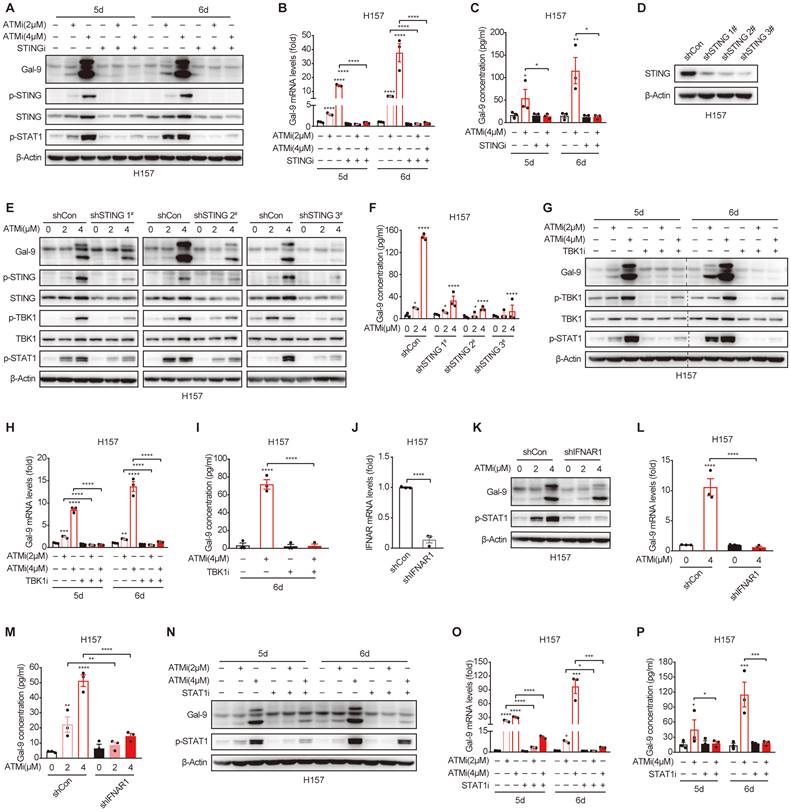
Based on the results above, we hypothesized that STING/IFNβ activation might contribute to ATMi-induced Gal-9 upregulation. To test the possibility, we pretreated the cells with H151, a specific STING inhibitor, and found that the induction of Gal-9 protein in response to ATM inhibition was completely suppressed in the presence of H151 (Fig. 4A). Similarly, the increase in Gal-9 mRNA and secretion was also abrogated upon H151 pretreatment in H157 and A549 cells, as determined by real-time PCR (Fig. 4B, Fig. S4A) and ELISA (Fig. 4C, Fig. S4B), concomitant with a decrease in IFNβ (Fig. S4C-E). To further validate the results, we generated STING stable knockdown H157 cells (Fig. 4D), and confirmed that STING depletion significantly prevented the increase in Gal-9 protein expression and secretion following ATM inhibition (Fig. 4E and F). In addition, inhibition of TBK1, a critical downstream kinase of STING, abrogated the induction of Gal-9 following ATM inhibition, as determined by levels of Gal-9 mRNA, protein and secretion (Fig. 4G-I). These results suggest that STING-TBK1 pathway is required for ATMi-induced Gal-9 upregulation. Moreover, knockdown of IFNAR1 (Fig. 4J), one of the two subunits of IFNβ receptor, resulted in a significant abrogation of increase in Gal-9 protein in response to ATM inhibition in H157 cells (Fig. 4K). Similarly, the upregulation of Gal-9 mRNA expression and secretion upon ATMi treatment was also prevented upon IFNAR1 depletion (Fig. 4L and M). Consistently, pharmacological inhibition of STAT1 led to a marked attenuation in the upregulation of Gal-9 mRNA, protein and secretion in response to ATM inhibition (Fig. 4N-P, Fig. S4F), indicating the importance of IFNβ signaling for Gal-9 induction by ATMi. Together, these results demonstrated that ATM inhibition enhances Gal-9 in a STING-TKB1-IFNβ dependent manner.
Gal-9 induction by ATM inhibition is mediated by STING-IFNβ pathway. A-C. Immunoblot, RT-qPCR and ELISA analysis of Gal-9 in H157 cells concurrently treated with KU-60019 (2 μmol/L or 4 μmol/L) and STING inhibitor (H151, 10 μmol/L) for indicated time. D. Immunoblot analysis of Gal-9 in vector control and STING-KD H157 cells expressing three different STING shRNAs. E-F. Immunoblot and ELISA analysis of Gal-9 in vector control and STING-KD H157 cells treated with KU-60019 (2 μmol/L or 4 μmol/L) or DMSO (vehicle control) for 5 days. G-I. Immunoblot, RT-qPCR and ELISA analysis of Gal-9 in H157 cells concurrently treated with KU-60019 (2 μmol/L or 4 μmol/L) and p-TKB1 inhibitor (GSK8612, 5 μmol/L) for indicated time. J. RT-qPCR analysis of IFNAR1 in vector control and IFNAR1-KD H157 cells. K-M. Immunoblot, RT-qPCR and ELISA analysis of Gal-9 in vector control and IFNAR1-KD H157 cells treated with KU-60019 (2 μmol/L or 4 μmol/L) or DMSO (vehicle control) for 5 days. N-P. Immunoblot, RT-qPCR and ELISA analysis of Gal-9 in H157 cells concurrently treated with KU-60019 (2 μmol/L or 4 μmol/L) and p-STAT1 inhibitor (Fludarabine, 0.25 μmol/L) for indicated time. n.s., not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Gal-9 blockade combined with ATM inhibition potently suppresses tumor growth in multiple syngeneic mouse models
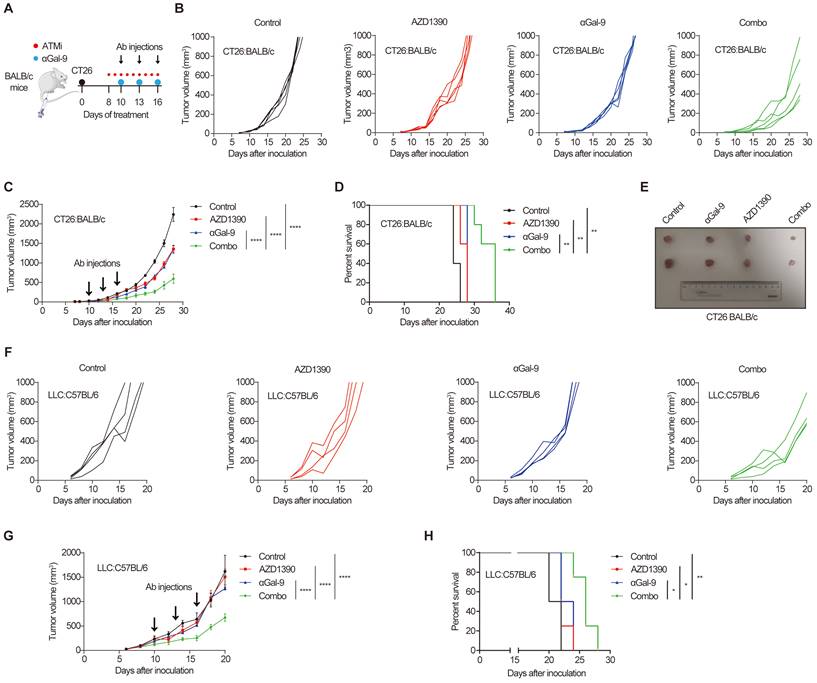
The remarkable synergy of Gal-9 blockade and ATM depletion in tumor growth suppression (Fig. 1) prompted us to investigate the translational value of this observation by combining anti-Gal-9 therapy with AZD1390, a potent ATM inhibitor currently evaluated in clinical trials [27]. As shown in Fig. 5A, BALB/c mice bearing CT26 tumors were treated with isotype control, ATM inhibitor AZD1390 (5 mg/kg for 9 times) alone, anti-Gal-9 antibody (100 μg/dose for 3 times) alone, or a combination of AZD1390 and anti-Gal-9 antibody. We found that while ATMi or anti-Gal-9 alone only moderately decreased tumor growth, their combination markedly improved tumor growth control and enhanced survival in CT26 syngeneic mouse model even with only three injections of anti-Gal-9 antibody (Fig. 5B-E). No significant body weight loss was observed in the combinational treatment group, suggesting the therapeutic doses used in for ATMi plus Gal-9 antibody were safe for animals (Fig. S5A). To confirm the efficacy of the combinational therapy, we used MC-38 murine colon tumor as an independent syngeneic model. Similarly, although the antitumor effects of anti-Gal-9 or ATMi alone were transient and minimal in MC-38 mouse model, their combination resulted in significantly greater inhibition of tumor growth than either single agent and prolonged overall survival (Fig. S5B-F). Next, we sought to assess whether ATM inhibition could enhance anti-Gal-9 therapy in the poorly immunogenic LLC murine lung cancer model [28] that is resistant to current immune checkpoint blockade [29]. While AZD1390 or anti-Gal-9 alone had negligible tumor-suppressive effect in LLC tumors, their combination significantly enhanced the antitumor efficacy and extended the survival of mice (Fig. 5F-H). Therefore, our results from three murine tumor models strongly suggested that anti-Gal-9 therapy combined with ATM inhibition significantly reduces tumor growth and enhances the survival of host mice, even in poorly immunogenic tumor models that are refractory to current immune checkpoint therapy.
Gal-9 blockade in combination with ATM inhibition potently suppresses tumor growth and prolongs mice survival. A. A diagram of treatment strategy. BALB/c mice were inoculated with CT26 cells and then treated with isotype control, ATM inhibitor AZD1390 (5mg/kg for 9 times) alone, anti-Gal-9 antibody (100μg /dose for 3 times) alone, or their combination. B. Tumor growth curves of individual CT26-bearing BALB/c mice treated as indicated (n=5 mice/group). C. Average tumor growth of mice inoculated with CT26 tumor cells and subjected to the indicated treatments. Error bars represent SEM of the means. Anti-Gal-9 antibody treatment schedule is indicated by arrows. D. Survival curves of mice inoculated with CT26 tumors and subjected to the indicated treatments. E. The size of tumors harvested from individual CT26-bearing BALB/c mice on Day 14 after inoculation. F. Tumor growth curves of individual of LLC-bearing C57BL/6 mice treated with isotype control, ATM inhibitor AZD1390 (5mg/kg for 9 times) alone, anti-Gal-9 antibody (100μg /dose for 3 times) alone, or their combination (n=4 mice/group). G. Average tumor growth of mice inoculated with LLC tumor cells and subjected to the indicated treatments. Error bars represent SEM of the means. Anti-Gal-9 antibody treatment schedule is indicated by arrows. H. Survival curves of mice inoculated with LLC tumors and subjected to the indicated treatments. n.s., not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Combination of anti-Gal-9 therapy with ATM inhibition increases T cell infiltration
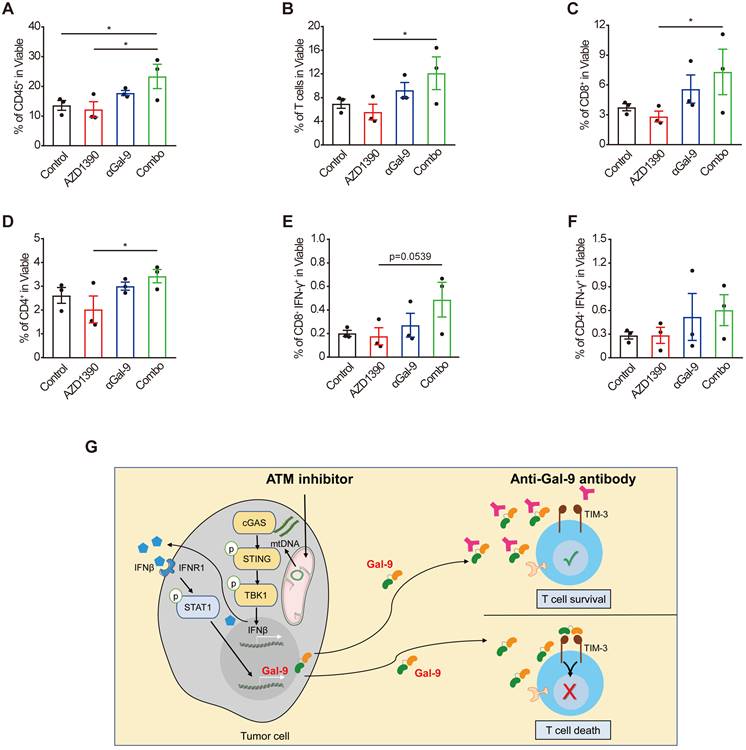
To investigate the alterations of tumor-infiltrating T cells upon the combination treatment that underlie the therapeutic efficacy, we performed flow cytometric analysis of CT26 tumor cells harvested from BALB/c mice on day14 after inoculation. The gating strategy for flow analysis is shown in Supplementary Fig.S6. Given that Gal-9 is known to bind to TIM-3 to induce T cell death, we posited that the Gal-9-neutralizing antibody is supposed to increase the numbers of T cells through preventing Gal-9 from binding to TIM3 to increase T cell survival [30]. Indeed, anti-Gal-9 monotherapy caused a moderate increase in tumor infiltration of T cells, while ATMi alone did not have a detectable impact. In contrast, we observed a significant increase in the number of intratumoral CD45+ immune cells (Fig. 6A), total T-cells (Fig. 6B), CD8+ cytotoxic T-cell (Fig. 6C), and CD4+ T cells (Fig. 6D) in the combination treatment group, compared with single-agent ATMi or anti-Gal-9 treatment. Furthermore, we found increased percentage of IFN-γ positive CD8+ T (Fig. 6E) and CD4+ T cells (Fig. 6F) (although a statistical significance was not reached), suggesting that the combination treatment tended to promote T cell activation. These results indicate that the combination of anti-Gal-9 therapy and ATM inhibition synergistically increases T cell infiltration and thus promotes anti-tumor immunity.
Combination of Gal-9 blockade with ATM inhibition increases T cell infiltration. Average frequency of tumor-infiltrating CD45+ cells (A), total T cells (B), CD8+T cells (C), CD4+ T cells (D), IFN-γ+CD8+ T cells (E) and IFN-γ+CD8+ T cells (F) in CT26 tumors harvested from BALB/c mice treated with isotype control, AZD1390 alone, anti-Gal-9 antibody (RG-1) alone, or their combination (n = 3 mice/group). Flow cytometric analysis was performed on day 14 after inoculation with CT26 tumor cells. Data represent the mean ± SEM. n.s., not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001. (G) Graphical summary of key findings. ATM inhibition triggers mitochondrial DNA (mtDNA) leakage and activates cGAS/STING/IFNβ innate immune pathway in tumor cells. Secreted IFNβ binds to IFNR1, activates the downstream STAT1 signaling and leads to Gal-9 induction. Gal-9 secreted from tumor cells interacts with TIM3 on T cells and induces T cell death. However, Gal-9 neutralizing antibody prevents Gal-9 from binding to TIM3 to increase T cell survival. Therefore, the combination of ATM inhibition and anti-Gal-9 increases T cell infiltration and induces anti-tumor immunity.
Discussion
Although anti-PD-1/PD-L1 immunotherapy has revolutionized the field of cancer treatment in multiple tumor types, only a small subset of patients benefits from such therapies, partially due to the “coldness” of the tumors, characterized by limited infiltration of lymphocytes (TILs) [31], particularly cytotoxic T cells. DDR-targeted therapies including ATM inhibitors have been shown to convert the “cold tumor” into “hot tumor” in some cancer types, through inducing cGAS/STING-mediated innate immunity. Consistent with this, herein, we show that inhibition of ATM induces the activation of cGAS-STING-IFNβ ubiquitously in multiple human and murine cancer cell lines. IFNβ plays a pivotal role in cancer immunosurveillance and anti-tumor immune response through activation of CD8+T cells, maturation of dendritic cells, and inhibition of regulatory T cells, etc. [32]. However, increasing evidence suggests that persistent IFNβ production can lead to immunosuppressive effects that promote cancer [33,34]. In this study we reveal Gal-9 upregulation via the STING-IFNβ pathway as a factor that limits the anti-tumor efficacy of ATM inhibition (Fig. 6G). Tumors highly expressing Gal-9 are thus likely to be T cell-inflamed “hot” tumors, which might underlie the positive correlation of Gal-9 with clinical outcome in some tumors [35]. Although our previous study showed that Gal-9 could be upregulated by exogenous IFNβ in tumor cells [8], the role of STING signaling-induced endogenous IFNβ in Gal-9 regulation remains largely unknown. We demonstrate that Gal-9 expression can be increased by tumor cell-intrinsic IFNβ in response to ATM inhibition in a STING-dependent manner. Therefore, our and others' studies suggest that ATM inhibition acts as a double-edged sword to modulate immune response. On the one hand, it activates cGAS/STING to boost tumor immunogenicity, on the other hand, it induces the expression of immunosuppressive regulators, such as PD-L1 [21] and Gal-9, to inhibit T cell activation and elicit T cell death, etc. Intriguingly, although we found that PD-L1 could also be upregulated in response to ATM inhibition, the extent of its alteration is minimal compared with that of Gal-9, suggesting that Gal-9, but not PD-L1, likely plays a predominant role in mediating the immunomodulation by ATMi. This study thus uncovers induction of tumoral Gal-9 as another important mechanism mediating immune escape that could be targeted for cancer immunotherapy. Our findings thus deepen the understanding of the immunological effects of DDR-targeted therapies and provide a rationale for a novel combinational modality to enhance the efficacy of the ATM inhibitors that are currently undergoing in a number of clinical trials [20,27].
Gal-9 is emerging as a promising therapeutic target for cancer immunotherapy recently. Anti-Gal-9 antibody in combination with an agonist antibody to the T cell co-stimulatory receptor GITR (glucocorticoid-induced tumor necrosis factor receptor-related protein) significantly outperforms monotherapy with either agent in suppressing tumor growth and improving survival in the mouse models of colon cancer and TNBC [8]. In a pancreatic mouse model, the combination of Gal-9 neutralization and PD-1 blockade slows tumor progression and extends mouse survival [6]. An anti-Gal-9 antibody, LYT-200, is currently evaluated in a clinical trial (NCT04666688), alone and in combination with chemotherapy or anti-PD-1 in patients with metastatic solid tumors [36]. Therefore, Gal-9 neutralizing antibody has a great potential for cancer immunotherapy when combined with other modalities. Here, we demonstrate that anti-Gal-9 therapy in combination with ATM inhibition synergically suppresses tumor growth and prolongs survival in three syngeneic mouse models, including colon tumor and the poorly-immunogenic LLC lung tumor which generally lacks TILs and is unresponsive to anti-PD-1/PD-L1 therapy [37]. These results envisage a novel anti-Gal-9-based combination strategy for a variety of human cancers, including those resistant to current anti-PD-1 immunotherapies. Based on our and other findings that ATM inhibition sensitize tumors to both anti-PD-1 [21,22] and anti-Gal9 therapies, it is conceivable that triple combination of ATMi with anti-PD-1 and anti-Gal-9 would produce an even more potent anti-tumor effect. It will be intriguing to investigate these combination modalities in preclinical and clinical investigations in the future. Importantly, given that ATM has been shown to be essential for T cell and B cell maturation and function [38,39] it is conceivable that ATMi will have undesirable effect on immune cells, which should be taken into consideration for long-term treatment. This might partially explain why anti-Gal-9 showed lower efficacy in mice with ATMi treatment than those with depletion of tumoral ATM. As DNA damage in general primes the type I IFN system via the STING pathway [40], our work raises the exciting possibility that anti-Gal-9 plus other DDR-targeted therapies, such as PARP inhibition, also enhance antitumor immune responses leading to durable antitumor immunity.
Our study reveals some potential biomarkers that may predict the response to anti-Gal-9 treatment, such as the “hotness” of TME, and high levels of intratumoral Gal-9, which is induced by ATM deficiency. Germline mutations of ATM result in A-T syndrome, a cancer-predisposing disorder. ATM loss/mutation is commonly found in a range of sporadic cancers [19]. For example, the deficiency of ATM protein evaluated by immunohistochemistry (IHC) assay is observed in over 40% of lung adenocarcinomas [41]. Our findings thus are clinically important by revealing Gal-9 as a promising therapeutic target for A-T Syndrome and ATM-deficient tumors. Therefore, ATM loss/mutation represent as a predictive biomarker to stratified patients who would response to Gal-9 blockade. Moreover, as ATM inhibitors have already been extensively evaluated in clinical trials, our work underscores Gal-9-targeted cancer immunotherapy as a promising modality that can be evaluated in future clinical trials in combination with these ATM inhibitors.
Consistent with Gal-9 being a TIM-3 ligand that induces T cell death, we found that Gal-9 blockade led to an increase in the infiltration of T cells. Notably, the combination of anti-Gal-9 with ATMi synergistically induces significantly higher numbers of intratumoral T cells, compared with single-agent ATMi or anti-Gal-9 treatment. Intriguingly, we also found a trend of increase in the numbers of IFNγ-positive CD4+ and CD8+ cells in either anti-Gal-9 or the combination treatment group, suggesting that Gal-9 blockade may act to reinvigorate T cell activity in an unknown mechanism. Although TIM-3 is a key receptor that mediates Gal-9 function, other TIM-3-independent mechanisms of Gal-9 actions have also been reported, such as Treg expansion and differentiation [7,42], macrophage polarization [6] and natural killer cell function [43]. Thus, given the pleotropic roles of Gal-9 in multiple immunomodulatory pathways, further experiments are warranted to investigate the impact of anti-Gal-9 therapy combined with ATM inhibition on other immune cells in the TME.
In summary, the study uncovers an intriguing link between DDR and an “immune checkpoint” protein Gal-9. Induction of Gal-9 via STING/IFNβ may represent a novel mechanism for tumors to invade immunosurveillance, thereby limiting the anti-tumor efficacy of DDR-targeted therapies. The findings have important clinical implications by highlighting Gal-9 as a promising therapeutic target for A-T Syndrome and ATM-deficient tumors and revealing combining ATM inhibition with anti-Gal-9 therapy as a novel strategy for cancer treatment. Our study thus provides a strong rationale for the use of ATM as both a predictive biomarker and a therapeutic target to enhance anti-Gal-9 therapy in future clinical trials. These findings will open a new line of immunotherapies for a wide variety of malignancies, including those with primary or adaptive resistance to PD-1/PD-L1 blockade.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was funded by the following: National Natural Science Foundation of China (82172687 and 81972186 to L.L.S., 82272672 to D.S.Z.); Natural Science Foundation of Tianjin (20JCYBJC00360 to L.L.S.); Health Commission of Tianjin (TJWJ2021MS002 to L.L.S.); Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-061B); Ministry of Science and Technology in Taiwan (110-2639-B-039-001-ASP to M.C.H.).
Author Contributions
Conceptualization, L.L.S., R.Y.Y., D.S.Z, and M.C.H.; Methodology, S.Z., J.M.S, D.L.LH., and B.N.L; Investigation, S.Z., J.M.S, D.L.LH., B.N.L, Z.X, Q.H.C, and C.J.L; Data Curation, S.Z., J.M.S., R.Y.Y.; Writing-Original Draft, L.L.S.; Writing-Review & Editing, L.L.S., R.Y.Y and M.C.H; Funding Acquisition, L.L.S.,D.S.Z, and M.C.H; Supervision, L.L.S., D.S.Z, and M.C.H.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H. et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027-34
2. de Miguel M, Calvo E. Clinical Challenges of Immune Checkpoint Inhibitors. Cancer Cell. 2020;38:326-33
3. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ. et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245-52
4. Okoye I, Xu L, Motamedi M, Parashar P, Walker JW, Elahi S. Galectin-9 expression defines exhausted T cells and impaired cytotoxic NK cells in patients with virus-associated solid tumors. J Immunother Cancer. 2020 8
5. Thiemann S, Baum LG. Galectins and Immune Responses-Just How Do They Do Those Things They Do? Annu Rev Immunol. 2016;34:243-64
6. Daley D, Mani VR, Mohan N, Akkad N, Ochi A, Heindel DW. et al. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat Med. 2017;23:556-67
7. Wu C, Thalhamer T, Franca RF, Xiao S, Wang C, Hotta C. et al. Galectin-9-CD44 interaction enhances stability and function of adaptive regulatory T cells. Immunity. 2014;41:270-82
8. Yang R, Sun L, Li CF, Wang YH, Yao J, Li H. et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat Commun. 2021 12
9. Yuan F, Ming H, Wang Y, Yang Y, Yi L, Li T. et al. Molecular and clinical characterization of Galectin-9 in glioma through 1,027 samples. J Cell Physiol. 2020;235:4326-34
10. Seifert AM, Reiche C, Heiduk M, Tannert A, Meinecke AC, Baier S. et al. Detection of pancreatic ductal adenocarcinoma with galectin-9 serum levels. Oncogene. 2020;39:3102-13
11. Enninga EAL, Nevala WK, Holtan SG, Leontovich AA, Markovic SN. Galectin-9 modulates immunity by promoting Th2/M2 differentiation and impacts survival in patients with metastatic melanoma. Melanoma Res. 2016;26:429-41
12. Kikushige Y, Miyamoto T, Yuda J, Jabbarzadeh-Tabrizi S, Shima T, Takayanagi SI. et al. A TIM-3/Gal-9 Autocrine Stimulatory Loop Drives Self-Renewal of Human Myeloid Leukemia Stem Cells and Leukemic Progression. Cell Stem Cell. 2015;17:341-52
13. Pilié PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol. 2019;16:81-104
14. Sun LL, Yang RY, Li CW, Chen MK, Shao B, Hsu JM. et al. Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing. Am J Cancer Res. 2018;8:1307-16
15. Chabanon RM, Rouanne M, Lord CJ, Soria JC, Pasero P, Postel-Vinay S. Targeting the DNA damage response in immuno-oncology: developments and opportunities. Nat Rev Cancer. 2021 0123456789
16. Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell. 2017;66:801-17
17. Geuting V, Reul C, Löbrich M. ATM Release at Resected Double-Strand Breaks Provides Heterochromatin Reconstitution to Facilitate Homologous Recombination. PLoS Genet. 2013;9:1-14
18. Shiloh Y. ATM and related protein kinases: Safeguarding genome integrity. Nat Rev Cancer. 2003;3:155-68
19. Choi M, Kipps T, Kurzrock R. ATM mutations in cancer: Therapeutic implications. Mol Cancer Ther. 2016;15:1781-91
20. Lavin MF, Yeo AJ. Clinical potential of ATM inhibitors. Mutat Res - Fundam Mol Mech Mutagen. 2020;821:111695
21. Zhang Q, Green MD, Lang X, Lazarus J, Parsels JD, Wei S. et al. Inhibition of ATM increases interferon signaling and sensitizes pancreatic cancer to immune checkpoint blockade therapy. Cancer Res. 2019;79:3940-51
22. Hu M, Zhou M, Bao X, Pan D, Jiao M, Liu X. et al. ATM inhibition enhances cancer immunotherapy by promoting mtDNA leakage and cGAS/STING activation. J Clin Invest. 2021 131
23. Xue Z, Zheng S, Linghu D, Liu B, Yang Y, Chen MK. et al. PD-L1 deficiency sensitizes tumor cells to DNA-PK inhibition and enhances cGAS-STING activation. Am J Cancer Res. 2022;12:2363-75
24. Liang X mei, Qin Q, Liu B ning, Li X qing, Zeng L li, Wang J. et al. Targeting DNA-PK overcomes acquired resistance to third-generation EGFR-TKI osimertinib in non-small-cell lung cancer. Acta Pharmacol Sin. 2021;42:648-54
25. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013 6
26. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4
27. Durant ST, Zheng L, Wang Y, Chen K, Zhang L, Zhang T. et al. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci Adv. 2018 4
28. Lechner MG, Karimi SS, Barry-Holson K, Angell TE, Murphy KA, Church CH. et al. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J Immunother. 2013;36:477-89
29. Li S, Simoni Y, Zhuang S, Gabel A, Ma S, Chee J. et al. Characterization of neoantigen-specific T cells in cancer resistant to immune checkpoint therapies. Proc Natl Acad Sci U S A. 2021;118:1-10
30. Yang R, Sun L, Li CF, Wang YH, Xia W, Liu B. et al. Development and characterization of anti-galectin-9 antibodies that protect T cells from galectin-9-induced cell death. J Biol Chem. 2022;298:101821
31. Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17:e542-51
32. Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: Implications for cancer therapy. Nat Rev Cancer. 2016;16:131-44
33. Patel SA, Minn AJ. Combination Cancer Therapy with Immune Checkpoint Blockade: Mechanisms and Strategies. Immunity. 2018;48:417-33
34. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C. et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell. 2016;167:1540-1554.e12
35. Zhou X, Sun L, Jing D, Xu G, Zhang J, Lin L. et al. Galectin-9 expression predicts favorable clinical outcome in solid tumors: A systematic review and meta-analysis. Front Physiol. 2018 9
36. Sharma M, Mahipal A, Bolen J, Elenko E, Korth C. abstract 1551: First in class immunotherapy Atargeting Galectin-9 promotes T-cell activation and anti-tumor response against pancreatic cancer and other solid tumors. in Immunology, pp. 1551-1551, American Association for Cancer Research, 10.1158/. 2021;9:4666688
37. Li HY, McSharry M, Bullock B, Nguyen TT, Kwak J, Poczobutt JM. et al. The tumor microenvironment regulates sensitivity of murine lung tumors to PD-1/PD-L1 antibody blockade. Cancer Immunol Res. 2017;5:767-77
38. Weitering TJ, Takada S, Weemaes CMR, van Schouwenburg PA, van der Burg M. ATM: Translating the DNA Damage Response to Adaptive Immunity. Trends Immunol. 2021;42:350-65
39. Riabinska A, Lehrmann D, Jachimowicz RD, Knittel G, Fritz C, Schmitt A. et al. ATM activity in T cells is critical for immune surveillance of lymphoma in vivo. Leukemia. 2020;34:771-86
40. Härtlova A, Erttmann SF, Raffi FAM, Schmalz AM, Resch U, Anugula S. et al. DNA Damage Primes the Type I Interferon System via the Cytosolic DNA Sensor STING to Promote Anti-Microbial Innate Immunity. Immunity. 2015;42:332-43
41. Villaruz LC, Jones H, Dacic S, Abberbock S, Kurland BF, Stabile LP. et al. ATM protein is deficient in over 40% of lung adenocarcinomas. Oncotarget. 2016;7:57714-25
42. Madireddi S, Eun SY, Mehta AK, Birta A, Zajonc DM, Niki T. et al. Regulatory T Cell-Mediated Suppression of Inflammation Induced by DR3 Signaling Is Dependent on Galectin-9. J Immunol. 2017;199:2721-8
43. Golden-Mason L, McMahan RH, Strong M, Reisdorph R, Mahaffey S, Palmer BE. et al. Galectin-9 Functionally Impairs Natural Killer Cells in Humans and Mice. J Virol. 2013;87:4835-45
Author contact
![]() Corresponding authors: Linlin Sun, Tianjin Key Laboratory of Lung Cancer Metastasis and Tumor Microenvironment, Lung Cancer Institute, Tianjin Medical University General Hospital, Anshan Road 154, Heping District, Tianjin 300052, P. R. China. Tel: +22-60362409, Fax: +22-60362306, E-mail: lsun1edu.cn. Mien-Chie Hung, Graduate Institute of Biomedical Sciences and Center for Molecular Medicine, China Medical University, Taichung 40402, Taiwan. Taiwan. Tel: 886-4-22057153 Fax: 886 -4-22060248, E-mail: mhungedu.tw. Diansheng Zhong, Department of Medical Oncology, Tianjin Medical University General Hospital, Anshan Road 154, Heping District, Tianjin 300052, P. R. China. Tel: +22-60817009, Fax: +22-60817009, Email: dzhongedu.cn.
Corresponding authors: Linlin Sun, Tianjin Key Laboratory of Lung Cancer Metastasis and Tumor Microenvironment, Lung Cancer Institute, Tianjin Medical University General Hospital, Anshan Road 154, Heping District, Tianjin 300052, P. R. China. Tel: +22-60362409, Fax: +22-60362306, E-mail: lsun1edu.cn. Mien-Chie Hung, Graduate Institute of Biomedical Sciences and Center for Molecular Medicine, China Medical University, Taichung 40402, Taiwan. Taiwan. Tel: 886-4-22057153 Fax: 886 -4-22060248, E-mail: mhungedu.tw. Diansheng Zhong, Department of Medical Oncology, Tianjin Medical University General Hospital, Anshan Road 154, Heping District, Tianjin 300052, P. R. China. Tel: +22-60817009, Fax: +22-60817009, Email: dzhongedu.cn.