Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(4):1228-1240. doi:10.7150/ijbs.81100 This issue Cite
Research Paper
Transmembrane BAX inhibitor motif containing 6 suppresses presenilin-2 to preserve mitochondrial integrity after myocardial ischemia-reperfusion injury
Li Ma1 ![]() , Lihan Liao1, Na Zhou1, Huikang Tao1, Hao Zhou2, Ying Tan2, Weidan Chen1, Fan Cao1, Xinxin Chen1
, Lihan Liao1, Na Zhou1, Huikang Tao1, Hao Zhou2, Ying Tan2, Weidan Chen1, Fan Cao1, Xinxin Chen1 ![]()
1. Guangdong Provincial Key Laboratory of Research in Structural Birth Defect Disease, Heart Center, Guangzhou Women and Children's Medical Center, Guangzhou Medical University, Guangzhou, China
2. Department of Cardiology, Chinese PLA General Hospital, Medical School of Chinese PLA, Beijing, 100037, China
Received 2022-11-21; Accepted 2023-1-21; Published 2023-2-13
Abstract

Myocardial ischemia-reperfusion (I/R) damage is characterized by mitochondrial damage in cardiomyocytes. Transmembrane BAX inhibitor motif containing 6 (TMBIM6) and presenilin-2 (PS2) participate in multiple mitochondrial pathways; thus, we investigated the impact of these proteins on mitochondrial homeostasis during an acute reperfusion injury. Myocardial post-ischemic reperfusion stress impaired myocardial function, induced structural abnormalities and promoted cardiomyocyte death by disrupting the mitochondrial integrity in wild-type mice, but not in TMBIM6 transgenic mice. We found that TMBIM6 bound directly to PS2 and promoted its post-transcriptional degradation. Knocking out PS2 in mice reduced I/R injury-induced cardiac dysfunction, inflammatory responses, myocardial swelling and cardiomyocyte death by improving the mitochondrial integrity. These findings demonstrate that sufficient TMBIM6 expression can prevent PS2 accumulation during cardiac I/R injury, thus suppressing reperfusion-induced mitochondrial damage. Therefore, TMBIM6 and PS2 are promising therapeutic targets for the treatment of cardiac reperfusion damage.
Keywords: TMBIM6, PS2, mitochondria, cardiac ischemia/reperfusion injury
Introduction
During the pathology underlying myocardial infarction, coronary artery occlusion reduces fresh blood and oxygen to the cardiac myocyte [1]. Although reperfusion and revascularization are the standard treatments to reduce myocardial ischemic injury caused by myocardial infarction, reperfusion itself can damage the heart and therefore myocardial ischemia-reperfusion (I/R) damage seems to be a complication induced by revascularization treatments [2]. Clinical evidence has revealed that myocardial reperfusion challenge is closely linked to the degree of perioperative complications following myocardial infarction [3]. However, there are no effective approaches to alleviate the additional damage induced by myocardial revascularization stress, since the molecular pathways underlying I/R-induced myocardial dysfunction are not fully understood.
Recent observations depicted the importance of mitochondria in the pathogenesis of myocardial revascularization stress [4-6]. Although mitochondria are metabolic centers that determine the rate of oxidative phosphorylation in cardiomyocytes, they also manage numerous extracellular and intercellular signals, including those involved in the inflammatory response, calcium homeostasis, metabolic reprogramming, autophagy, oxidative stress, endoplasmic reticulum (ER) function and cell death [7-12]. Mild mitochondrial injury promotes oxidative stress and impairs adenosine triphosphate (ATP) metabolism, thus reducing the contraction/relaxation capacities of cardiomyocytes [13-15]. Severe mitochondrial dysfunction leads to cardiomyocyte death, followed by pro-inflammatory cell recruitment and abnormal inflammatory response activation [16, 17]. Therefore, mitochondria have been regarded as potential drug targets during cardiac I/R injury.
Presenilin-2 (PS2) is a component of the γ-secretase complex, which was originally reported to cleave amyloid precursor protein. Now, ample evidence showed that PS2 is linked to Alzheimer's disease [18], and mutations in PS2 are considered to be reliable genetic markers of Alzheimer's disease [19]. Recent studies have also indicated that PS2 disrupts mitochondrial homeostasis by altering mitochondrial calcium input [20], mitochondria-ER coupling [21], mitochondrial phenotypes [22], the mitochondrial oxidative capacity [23], mitochondrial oxygen consumption, the mitochondrial membrane potential [24] and mitochondria-induced cell death [25]. More importantly, mutations in PS2 have been linked with the development of dilated cardiomyopathy and heart failure [26]. PS2 protein expression was found to increase significantly in low-glucose- or hypoxia-treated cardiomyocytes [27], and knocking out PS2 was reported to increase cardiomyocyte contraction by enhancing the peak amplitudes of calcium transients [28]. However, the influence of PS2 on myocardial revascularization stress has not been determined.
Transmembrane BAX inhibitor motif containing 6 (TMBIM6) is a calcium channel-like protein that is primarily localized on the surface of the ER [29]. TMBIM6 is also termed as Bax inhibitor-1, since it was originally found to prevent Bax-induced mitochondrial membrane hyper-permeability and apoptosis [30]. TMBIM6 has subsequently been reported to influence mitochondrial bioenergetics [31], the mPTP opening rate [32] and mitochondrial morphology [33]. Genetic overexpression of TMBIM6 was recently shown to reduce myocardial revascularization stress by preserving the mitochondrial integrity [34], although the downstream effectors of TMBIM6 in cardiomyocytes have not been defined. In this study, we investigated whether PS2 is a downstream signal of TMBIM6 and thus disturbs the mitochondrial integrity in the setting of myocardial revascularization stress.
Materials and Methods
Animals and I/R model
TMBIM6 transgenic (TMBIM6Tg) mice and PS2 knockout (PS2KO) mice (Jackson Laboratory) were genotyped via PCR analysis of mouse tail DNA. For the I/R experiments, ischemia was achieved through occlusion of the LAD through a 7.0 silk suture for 45 min. Then, removal of the silk suture and then restore the fresh blood of LAD to induce the reperfusion for 4 hrs, while the re-opened ligature was left in place to facilitate future analysis of the infarcted tissue [35]. Mice treated with the same procedures without LAD occlusion and reperfusion were used as the sham group. TTC staining was performed to assess the infarcted area. Specifically, the mouse was anesthetized, the chest was re-opened, the LAD was re-occluded to promote the TTC perfusion into the myocardium [36]. During contraction, the dye circulated and was distributed through the heart. Then, we isolated the heart which was following rinsed with cold PBS, and sliced at 1-mm intervals. Afterwards, the sections were incubated in a 1% TTC (Sigma-Aldrich) solution to visualize the infarcts and viable myocardium [37]. The nonischemic area and infarcted area, were determined using computerized planimetry. These areas were comprehensively analyzed in serial sections from each mouse using ImageJ software. Mice that died within 24 hours of surgery were treated as technical errors and excluded from subsequent analyses. Mice in which the open ligatures were lost after the end of reperfusion were also excluded to ensure the accuracy of the TTC measurements [38].
Histology
The hearts of the mice were placed in Hank's balanced solution, and then treated with 4% paraformaldehyde. Subsequently, the samples were dehydrated and treated with paraffin. Finally, an 8-µm sections of heart tissue were generated through a Microtome [6]. Subsequently, HE staining was used to stain these heart sections to further observe the myocardial fiber in the presence of reperfusion stress according to a standard protocol. and then were mounted. For each group, a representative image was selected to show the average or median level of the group based on histological features and observed under a Hamamatsu NanoZoomer 2.0-HT Slide Scanner [39].
Immunofluorescence
Mouse hearts were placed in Hanks' balanced solution and then treated with 4% paraformaldehyde. Subsequently, samples were saturated in 5, 10, 15 and 20% sucrose in PBS. Then, a 6-µm sections of heart tissues were generated through the Leica CM 3050S Cryostat (Leica Microsystems) [40]. After blockade with 10% donkey serum, the sections were treated with primary antibodies including Gr1 (1:1000, Abcam, #ab25377), caspase-12 (1:1000, Abcam, #ab235180), caspase-9 (1:1000, Abcam, #ab222231) or PS2 (1:1000, Abcam, #ab51249) overnight at 4 °C. Subsequently, sections were treated with 0.1% PBST three times at room temperature and then reacted with secondary antibodies. To validate antibody specificity, the IgG isoform control from the same species was used instead of the primary antibody, and immunofluorescence was evaluated as described above [39]. To distinguish genuine target staining from the background, secondary antibody-only controls were performed without the addition of the respective primary antibodies. Immunofluorescence pictures were observed under a Zeiss LSM 880 Airy Scan Confocal Microscope [41]. The representative pictures were captured to show the average or median level of the group based on the fluorescence features.
Cell culture and transfection
HL-1 cells were purchased from the Chinese Academy of Sciences (Shanghai, China). In vitro, HL-1 cells were subjected to hypoxic conditions for 45 min, followed by normal oxygen conditions for two hours, as previously described [42]. Cell survival rate was determined using a CCK-8 assay based on our previous studies. HL-1 cells were transfected with siRNA to knowkout the expression of PS2 in the presence of hypoxia/reoxygenation condition using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA). The TMBIM6 adenovirus was transfected into HL-1 cells based on a previous report [43].
ELISA
ELISAs from MyBioSource, Inc. were applied to observe the changes of caspase-9 activities (Mouse Caspase 9 ELISA Kit, catalog #MBS451593), caspase-12 activities (Mouse Caspase-12 ELISA Kit, catalog #MBS9425959), ATP concentrations (Mouse Adenosine Triphosphate ELISA Kit, catalog #MBS724442), mitochondrial respiratory complex I activities (Mouse Mitochondrial Respiratory Chain Complex I ELISA Kit, catalog #MBS912812), mitochondrial respiratory complex II activities (Mouse Mitochondrial Respiratory Chain Complex II ELISA Kit, catalog #MBS108909) and mitochondrial respiratory complex III (Mouse Mitochondrial Respiratory Chain Complex III ELISA Kit, catalog #MBS108907). LDH content was also detected using an ELISA (CyQUANTTM LDH Cytotoxicity Assay Kit, catalog #C20300, ThermoFisher) [34].
Mitochondrial membrane potential and cellular oxidative stress
The relative immunofluorescence of a JC-1 probes (#ab113850, Abcam) was applied to analyze the mitochondrial potential, based on a previous study [40]. Intracellular ROS levels were assessed with a Total Reactive Oxygen Species Assay Kit (#88-5930-74, ThermoFisher, Inc.) [41].
mPTP opening detection and TUNEL staining
The mPTP opening rate was analyzed using an ELISA Kit (#ab239704, Abcam) [35]. Cell death rate was detected using a TUNEL assay (#25879, CST) [6].
Western blots
Proteins were extracted according to the conventional method [44]. In brief, after blocked by 5% milk, the membranes were incubated overnight at 4℃ with PS2 antibody (#ab51249, Abcam). The protein signals were visualized using Tanon Image software (version 5100; Tanon, Shanghai, China). The signal intensity of the target protein was normalized against GAPDH, and then the fold change was calculated relative to the control group.
Immunoprecipitation
For crosslinking immunoprecipitation, cells were co-treated with antibodies and dithiobis (succinimidyl propionate) for 40 min and then treated with 10% neutral buffered formalin (Sigma-Aldrich) [45]. Before co-immunoprecipitation, siRNA or Ad-TMBIM6 were transfected into HL1 cells for 48 hours. The cells were then lysed in an immunoprecipitation buffer. The immunoprecipitation was then performed as described previously [45].
RNA extraction, reverse transcription and qPCR
Total RNA was extracted using TRIzol™ reagent (Invitrogen) and reverse-transcribed using a Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics, Risch-Rotkreuz, Switzerland) [46]. Then, qPCR was conducted through the Fast Start Universal SYBR® Green Master Mix (Roche Diagnostics). The mRNA levels detected in each sample were normalized to GAPDH levels. The following primers were used: TNFα (Forward, 5'-AGATGGAGCAACCTAAGGTC-3'; Reverse, 5'-GCAGACCTCGCTGTTCTAGC-3'), IL-6 (Forward, 5'-CAGACTCGCGCCTCTAAGGAGT-3'; Reverse, 5'-GATAGCCGATCCGTCGAA-3'), MCP1 (Forward, 5'-GGATGGATTGCACAGCCATT-3'; Reverse, 5'-GCGCCGACTCAGAGGTGT-3').
Statistical analysis
Data were presented as the mean ± SEM and analyzed using GraphPad Prism 9.0 software. For the analysis of in vivo experiments, data with sample sizes < 6 were subjected to nonparametric tests, as a normal distribution cannot be assessed accurately with a low sample size. The tests applied to assess significance are described in each figure legend, and the precise p-values of significant changes are indicated on the graphs. P<0.05 was significant.
Results
TMBIM6 overexpression reduces cardiomyocyte death upon H/R treatment
To investigate the impact of TMBIM6 on cardiac reperfusion dysfunction, we transfected HL-1 cells with a TMBIM6 adenovirus (Ad-TMBIM6) and then subjected the cells to hypoxia-reoxygenation (H/R) injury. Subsequently, a CCK-8 assay showed that cellular viability was lower in the H/R injury group than in the control group (Figure 1A). Accordingly, a lactate dehydrogenase (LDH) release assay indicated that LDH release from HL-1 cells into the medium was greater in the H/R injury group than in the control group (Figure 1B). TMBIM6 overexpression protected cardiomyocyte viability (Figure 1A) and prevented LDH leakage (Figure 1B) following H/R injury.
TMBIM6 overexpression reduces cardiomyocyte death upon H/R treatment. HL-1 cells were transfected with Ad-TMBIM6 before H/R treatment. A. Cell viability was determined with a CCK-8 assay. B. LDH release was determined with an ELISA. C, D. The activities of caspase-12 and caspase-9 were measured with ELISAs. E-G. Double immunofluorescence analysis of caspase-12 and caspase-9 in cardiomyocytes after H/R injury. #p<0.05.
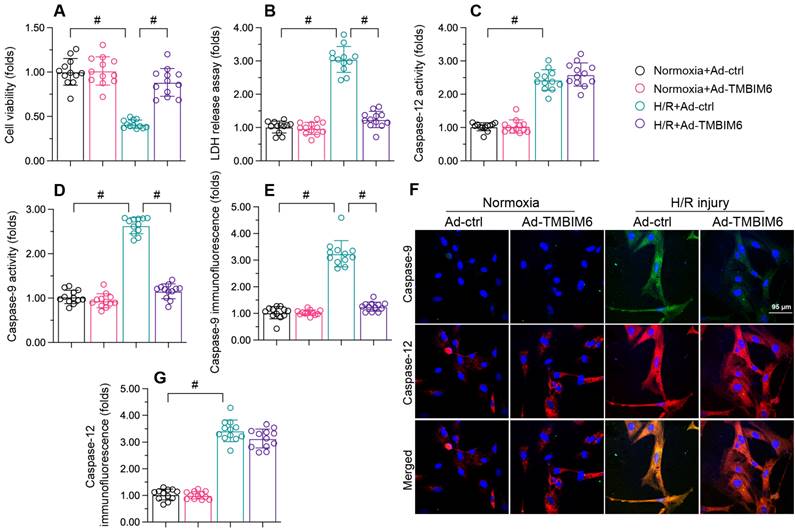
At the molecular level, cardiomyocyte death primarily occurs via caspase-9-induced mitochondrial apoptosis or caspase-12-induced endoplasmic reticular apoptosis. To determine whether TMBIM6 overexpression improved cardiomyocyte survival by inhibiting one of these pathways, we used enzyme-linked immunosorbent assays (ELISAs) to evaluate caspase-9 and caspase-12 activity levels in HL-1 cells. The activities of caspase-9/12 were both significantly elevated upon H/R injury exposure; however, Ad-TMBIM6 transfection mainly prevented caspase-9 upregulation after H/R, suggesting that TMBIM6 inhibits mitochondrial apoptosis (Figure 1C and D).
To validate these results, we performed immunofluorescence assays. As shown in Figure 1E-G, the immunofluorescence signals of caspase-9 and caspase-12 in HL-1 cells were significantly elevated upon H/R injury. TMBIM6 overexpression reduced caspase-9 levels following H/R injury, but had no influence on caspase-12 levels (Figure 1E-G). These data confirmed that TMBIM6 suppresses mitochondria-induced cardiomyocyte death during cardiac post-ischemic dysfunction.
TMBIM6 overexpression attenuates myocardial I/R damage
To translate our in vitro findings, we subjected TMBIM6 transgenic (TMBIM6Tg) or control (TMBIM6flox) mice to myocardial I/R damage or a sham operation in vivo. As shown in Figure 2A, I/R injury augmented the infarction area in TMBIM6flox control mice, but not in TMBIM6Tg mice. TUNEL staining showed that reperfusion elevated the ratio of dysfunctional cells in control heart tissues, but not in TMBIM6Tg heart tissues (Figure 2B and C).
TMBIM6 overexpression attenuates myocardial I/R injury. TMBIM6Tg mice and TMBIM6flox control mice were subjected to cardiac I/R injury. A. Myocardial infarction size was determined by TTC staining. B-C. TUNEL staining was used to analyze the number of apoptotic cells. D. HE staining was used to analyze changes in the myocardial structure in response to I/R injury. E. Electron microscopy was used to observe ultrastructural alterations in the myocardium and mitochondria after I/R injury. F, G. A double immunofluorescence assay was used to visualize the accumulation of Gr1-positive pro-inflammatory cells within the myocardium after I/R injury. #p<0.05.
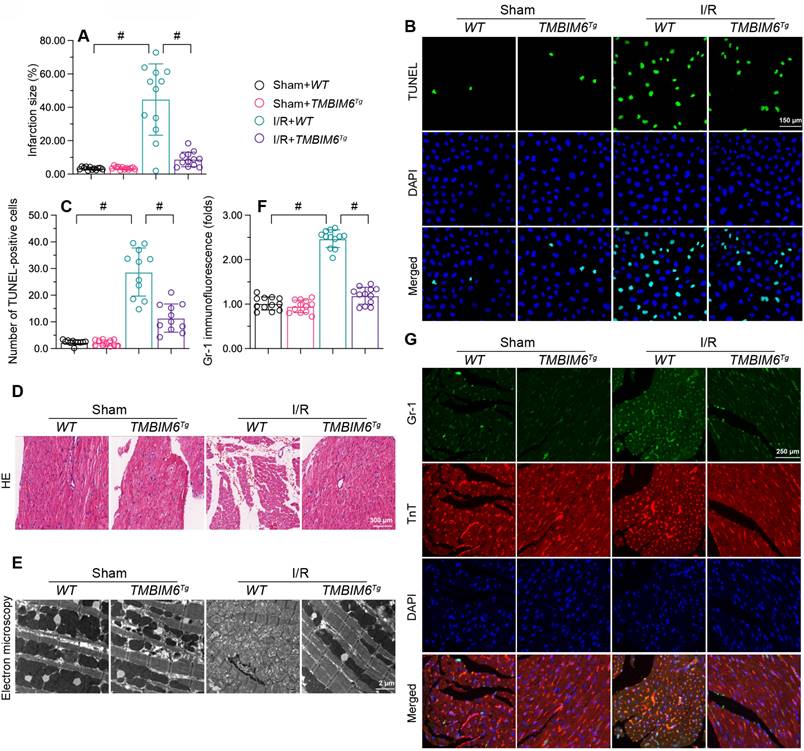
Next, we used HE staining to observe changes underlying the myocardium. Compared with the sham-operated group, I/R-treated TMBIM6flox mice exhibited disorganized and swollen myocardial fibers, while this change was not observed in I/R-treated TMBIM6Tg mice (Figure 2D). We then used electron microscopy to detect ultrastructural changes in the myocardium. In TMBIM6flox control mice, I/R injury induced mitochondrial swelling and rupture (Figure 2E), followed by cytoplasmic vacuolization; however, these structural alterations were not noted in I/R-treated TMBIM6Tg mice.
Excessive cardiomyocyte death and myocardial fiber swelling can induce a cardiac inflammatory response; thus, we used immunofluorescence to evaluate inflammatory cell accumulation in the myocardium. I/R promoted Gr1-positive cells recruitment within the myocardium in TMBIM6flox mice, but not in TMBIM6Tg mice (Figure 2F and G). These results indicated that TMBIM6 overexpression reduced cardiomyocyte death, improved the myocardial structure and repressed the cardiac inflammatory response during I/R injury.
TMBIM6 overexpression maintains heart function
Myocardial infarction often reduces cardiac contraction/relaxation; thus, we used echocardiography to assess cardiac function in hearts. In control TMBIM6flox mice, reperfusion disrupted the cardiac systolic capacity, as evidenced by a lower ejection fraction (EF), impaired fractional shortening (FS) and augmented left ventricular systolic dimension (LVSd) (Figure 3A-C). I/R injury also impaired the myocardial diastolic capacity in these mice, as evidenced by an increased ratio of early to late transmitral flow velocities (E/A), elevated ratio of mitral peak velocity of early filling to early diastolic mitral annular velocity (E/e') and amplified left ventricular diastolic dimension (LVDd) (Figure 3D-F). However, TMBIM6 overexpression normalized the cardiac systolic and diastolic capacities following reperfusion damage (Figure 3A-F).
TMBIM6 overexpression maintains heart function. TMBIM6Tg mice and TMBIM6flox control mice were subjected to cardiac I/R injury. A-F. Echocardiography was used to assess cardiac function after I/R injury. G-L. Cardiomyocytes were isolated from the mice, and their contraction properties were analyzed. #p<0.05.
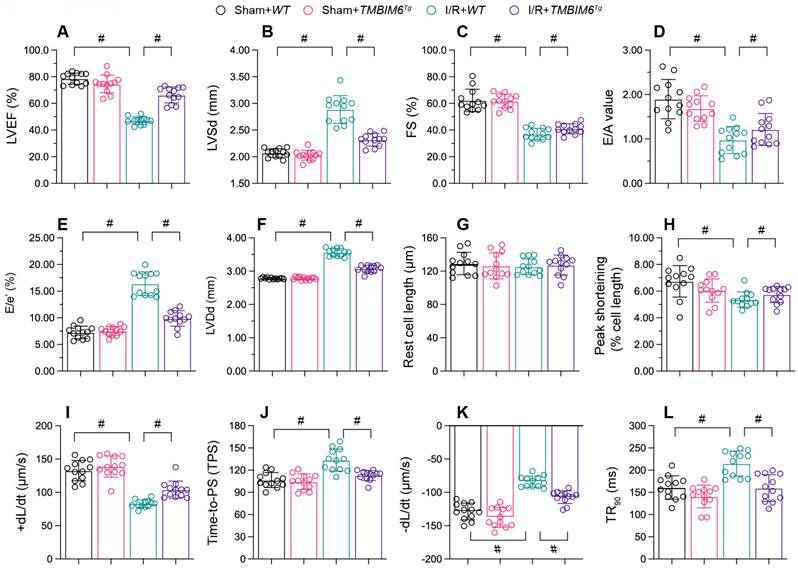
Besides, we conducted ex vivo analyses of cardiomyocytes isolated from the mice after reperfusion model. I/R injury or TMBIM6 overexpression negatively affected on the resting lengths of cardiomyocytes (Figure 3G). In cardiomyocytes from TMBIM6flox control mice, I/R injury reduced the peak shortening (PS), impaired the maximal velocity of shortening (+dL/dt), repressed the time to peak shortening (TPS), blunted the maximal velocity of relengthening (-dL/dt) and elevated the time to 90% relengthening (TR90) (Figure 3H-L). However, these functional abnormalities were alleviated in cardiomyocytes isolated from I/R-treated TMBIM6Tg mice (Figure 3H-L).
TMBIM6 overexpression sustains the mitochondrial integrity of cardiomyocytes
Mitochondrial dysfunction is an important contributor to cardiac I/R injury [47-50]; thus, we investigated whether TMBIM6 overexpression could normalize mitochondrial function in HL-1 cells during H/R injury. We first measured ATP production, which is primarily carried out by the mitochondrial respiratory complexes. H/R treatment reduced the ATP content in HL-1 cardiomyocytes (Figure 4A). Moreover, ELISAs indicated that H/R injury significantly repressed mitochondrial respiretory complex activity in these cells (Figure 4B-D). However, TMBIM6 overexpression maintained mitochondrial respiratory complex activity (Figure 4B-D) and therefore enhanced ATP production (Figure 4A) in HL-1 cells following H/R injury.
TMBIM6 overexpression sustains the mitochondrial integrity of cardiomyocytes. HL-1 cells were transfected with Ad-TMBIM6 before H/R treatment. A. ATP production was detected with an ELISA. B-D. The activities of the mitochondrial respiratory complexes were determined with ELISAs. E, F. The mitochondrial membrane potential was analyzed with an immunofluorescence assay using a JC-1 probe. G, H. Cellular ROS production was detected using an immunofluorescence assay in cardiomyocytes after H/R injury. I. The mPTP opening rate was recorded in cardiomyocytes transfected with Ad-TMBIM6 and subjected to H/R injury. #p<0.05.
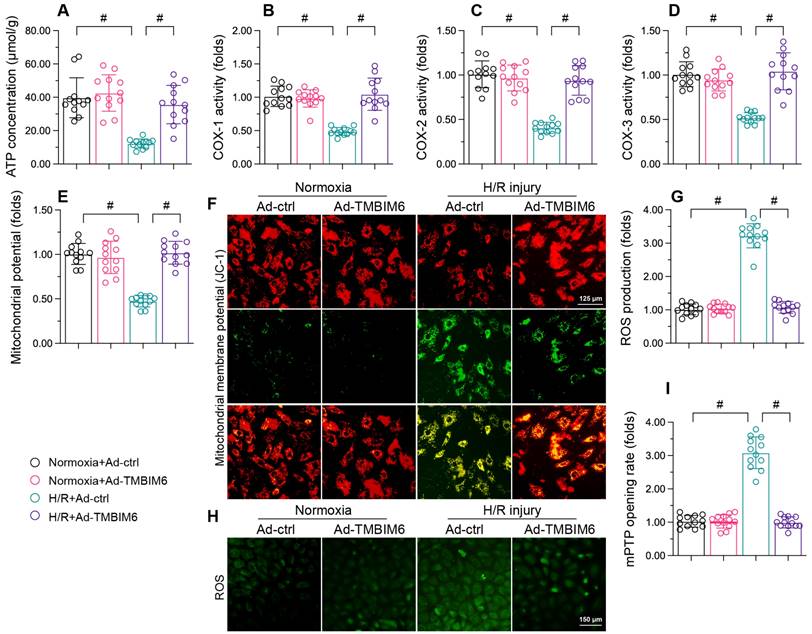
We then performed immunofluorescence assays, which revealed that reoxygenation stress dissipated the mitochondrial potential in HL-1 cells, whereas TMBIM6 overexpression reversed this effect (Figure 4E and F). TMBIM6 overexpression also neutralized H/R-induced cellular oxidative stress (Figure 4G and H). Moreover, H/R injury augmented the mPTP opening rate in HL-1 cells, while TMBIM6 overexpression repressed it (Figure 4I). These results demonstrated that TMBIM6 overexpression normalized mitochondrial homeostasis in the setting of cardiac revascularization stress.
TMBIM6 binds directly to PS2 to promote its degradation
To determine the molecular mechanism whereby TMBIM6 preserved cardiomyocytes and their mitochondria function against reoxygenation stress, we focused on protein-protein interactions. First, we used the inBio Discover platform (https://inbio-discover.com) to analyze the potential protein network underlying TMBIM6 (Figure 5A). PS2, one of the potential interactive proteins, has been regarded as a regulator of mitochondrial homeostasis. Therefore, we assessed whether TMBIM6 could bind directly to PS2 to preserve mitochondrial function and integrity. A molecular docking analysis revealed several possible binding sites between TMBIM6 and PS2 (Figure 5B-D). The crosslinks between TMBIM6 and PS2 were validated through co-immunoprecipitation analyses (Figure 5E).
TMBIM6 binds directly to PS2 and promotes its degradation. HL-1 cells were transfected with Ad-TMBIM6 before H/R treatment. A. The potential protein network underlying TMBIM6 was analyzed on the inBio Discover platform. B-D. Molecular docking analysis of TMBIM6 and PS2. E. A co-immunoprecipitation assay was used to analyze the binding between TMBIM6 and PS2 following I/R injury. F. RNA was collected, and the transcription of PS2 was analyzed via qPCR. G. Proteins were collected from cardiomyocytes, and TMBIM6 protein expression was determined via Western blotting. H, I. Immunofluorescence analysis of PS2 in cardiomyocytes exposed to H/R injury or Ad-TMBIM6. #p<0.05.
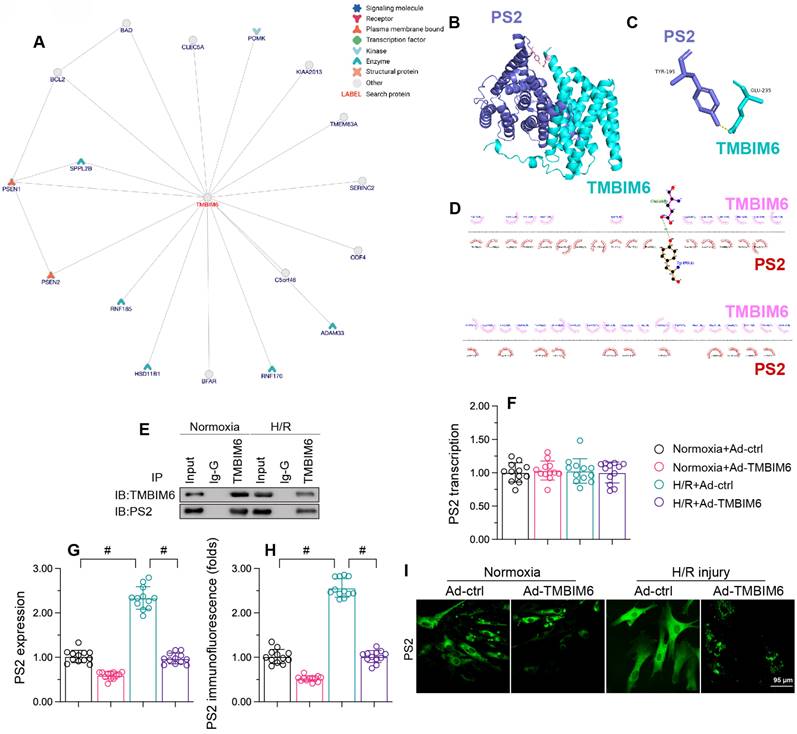
Next, we evaluated the effects of TMBIM6 overexpression on PS2 in HL-1 cells. Quantitative real-time PCR (qPCR) analyses demonstrated that neither reoxygenation stress nor TMBIM6 overexpression altered PS2 transcription (Figure 5F). However, Western blotting indicated that reoxygenation stress elevated PS2 protein content, whereas TMBIM6 overexpression inhibited this effect (Figure 5G). Immunofluorescence analyses confirmed that reoxygenation stress rapidly upregulated PS2 content in HL-1 cells, whereas TMBIM6 overexpression suppressed this increase (Figure 5H and I). These results illustrated that TMBIM6 binds directly to PS2 and promotes its degradation at the protein level.
PS2 deficiency sustains heart function during reperfusion dysfunction
To figure out the action of PS2 on cardiac reperfusion dysfunction, we used echocardiography to compare the myocardial function of PS2 knockout (PS2KO) and wild-type (WT) mice. As shown in Figure 6A-C, I/R injury reduced the EF, suppressed the FS and augmented the LVSd in WT mice; however, these phenotypic alterations were lessened in PS2KO mice. Similarly, I/R injury impaired the diastolic capacity of the heart (E/A, E/e' and LVDd) in WT mice, but not in PS2KO mice (Figure 6D-F).
PS2 deficiency sustains heart function during I/R injury. PS2KO and WT mice were subjected to cardiac I/R injury. A-F. Echocardiography was used to evaluate cardiac function after I/R injury. G-L. Cardiomyocytes were isolated from the mice, and their contraction properties were analyzed. #p<0.05.
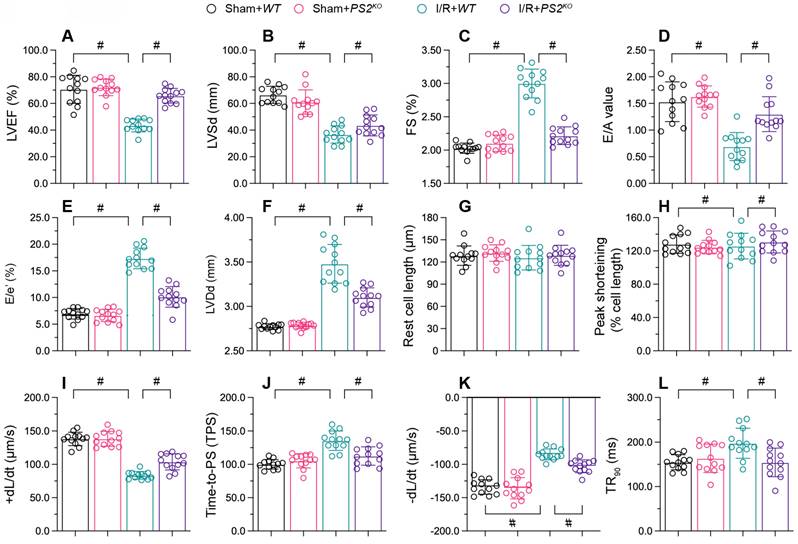
We then isolated cardiomyocytes from I/R-treated WT and PS2KO mice to evaluate their contraction properties. I/R injury blunted the PS, +dL/dt and TPS values in cardiomyocytes from WT mice, but not from PS2KO mice (Figure 6G-L). Moreover, cardiomyocytes isolated from PS2KO mice could maintain normal relaxation function after I/R injury, in contrast to those from WT mice (Figure 6G-L). These results demonstrated that knocking out PS2 normalized heart function during I/R injury.
Knocking out PS2 attenuates I/R-induced damage in the heart
We then assessed the effects of PS2 deficiency on cardiac damage after reperfusion dysfunction. HE assay revealed that reperfusion caused myocardial fiber swelling in WT mice, whereas this structural change was significantly ameliorated in PS2KO mice (Figure 7A). Electron microscopy indicated that I/R injury led to mitochondrial morphological disorder in myocardia from WT mice, but not from PS2KO mice (Figure 7B). Moreover, qPCR analysis of pro-inflammatory cytokines demonstrated that IL-6, MCP1 and TNFα were significantly elevated in the myocardium following I/R injury (Figure 7C-E), while loss of PS2 prevented pro-inflammatory cytokine activation.
PS2 deficiency attenuates I/R-induced damage in cardiomyocytes. PS2KO and WT mice were subjected to cardiac I/R injury. A. HE staining was used to analyze changes in the myocardial structure following I/R injury. B. Electron microscopy was used to observe ultrastructural alterations in the myocardium and mitochondria after I/R injury. C-E. RNA was collected, and qPCR was used to analyze the transcription of IL-6, MCP1 and TNFα. F. HL-1 cells were transfected with siRNA against PS2. Then, cell viability was determined with a CCK-8 assay. G, H. ELISAs were used to analyze the activities of caspase-12 and caspase-9. p<0.05.

In vitro, small interfering RNA (siRNA) against PS2 was transfected into HL-1 cells prior to H/R injury. A CCK-8 assay demonstrated that reoxygenation suppressed the viability of HL-1 cells, while PS2-siRNA negated this effect (Figure 7F). Although H/R injury promoted the activation of capsase-9 and caspase-12, PS2-siRNA treatment prevented caspase-9 activation (Figure 7G and H). These results confirmed that PS2 inhibition could alleviate I/R-induced cardiac injury.
PS2 silencing maintains mitochondrial function in H/R-treated cardiomyocytes
Finally, to determine whether PS2 inhibition protected mitochondria during cardiac I/R injury, we further analyzed mitochondrial function in HL-1 cells following PS2-siRNA treatment. As shown in Figure 8A, H/R injury repressed ATP production in cardiomyocytes, while PS2-siRNA treatment preserved ATP synthesis. PS2-siRNA also prevented mitochondrial respiratory complex inactivation (Figure 8B-D) and sustained the mitochondrial potential in reoxygenation-treated cardiomyocytes (Figure 8E and F). Moreover, PS2 inhibition markedly suppressed cellular oxidative stress (Figure 8G and H) and prevented mPTP opening (Figure 8I) following H/R treatment. Therefore, we concluded that PS2 silencing normalized mitochondrial function in H/R-treated cardiomyocytes.
PS2 silencing maintains mitochondrial function in H/R-treated cardiomyocytes. HL-1 cells were transfected with PS2-siRNA before H/R treatment. A. ATP production was detected with an ELISA. B-D. The activities of the mitochondrial respiratory complexes were determined using ELISAs. E, F. The mitochondrial membrane potential was analyzed with an immunofluorescence assay using the JC-1 probe. G, H. Cellular ROS production was detected with an immunofluorescence assay in cardiomyocytes after H/R injury. I. The mPTP opening rate was recorded in cardiomyocytes transfected with PS2-siRNA and subjected to H/R injury. #p<0.05.

Discussion
The molecular mechanisms underlying cardiac reperfusion dysfunction were fully delineated, so there are not yet effective treatment approaches for cardiac reperfusion dysfunction in clinical practice. Our present study had three main findings: 1) TMBIM6 overexpression exerts cardioprotective effects by normalizing mitochondrial function and cardiomyocyte viability during myocardial I/R injury; 2) abnormal PS2 upregulation seems to augment reperfusion-motivated cardiac damage by disrupting the mitochondrial integrity and promoting cardiomyocyte death; 3) TMBIM6 downregulates PS2 by binding directly to it, thereby preserving the mitochondrial integrity and cardiac function. Our findings identified TMBIM6/PS2 as a novel signaling pathway in the pathogenesis of cardiac reperfusion dysfunction. Thus, stabilization of TMBIM6 expression, inhibition of PS2 activation and preservation of mitochondrial function are promising therapeutic strategies to reduce reperfusion-caused cardiomyocyte dysfunction and heart failure.
Mitochondrial dysfunction is well known to induce or exacerbate cardiac I/R injury [39, 49-52]. I/R injury causes excessive mitochondrial fission, and the resulting fragmented mitochondria exhibit reduced ATP production [39, 53, 54]. I/R injury also impairs mitochondrial function by inhibiting mitochondrial autophagy, thus triggering cardiomyocyte death and cardiac dysfunction [39, 47, 55-57]. A reduced mitochondrial potential as well as augmented mitochondria-derived ROS generation lead to cardiomyocyte oxidative stress [58]. In addition, excessive mitochondrial calcium uptake interrupts mitochondrial oxidative phosphorylation and promotes the opening of the mPTP, an early marker of cardiomyocyte necrosis [59]. Therefore, mitochondria seem to be a key treatment target during myocardial reperfusion dysfunction [60]. Herein, we found that reperfusion promoted mitochondrial ROS overloading, reduced the mitochondrial membrane potential, inactivated the mitochondrial respiratory complexes and suppressed ATP production. These effects worked together to induce mitochondrial dysfunction in the reperfused heart.
Our results identified PS2 as a novel inducer of mitochondrial damage, as increased PS2 expression was associated with reduced mitochondrial integrity. Accordingly, previous research described the mitochondrial involvement of PS2 in Alzheimer's Disease [61]. Mutations in PS2 were found to promote the accumulation of amyloid-β [62]. PS2 deficiency was reported to induce mitochondria-ER interactions and facilitate the transfer of calcium from the mitochondria into the ER [20]. Abnormal calcium signaling in mitochondria due to PS2 was found to alter the mitochondrial morphology and activate mitochondrial apoptosis [63]. PS2 was also shown to enhance mitochondria-ER contact by binding to the mitochondrial fusion protein mitofusin 2 [64].
In the present study, we used PS2 knockout mice to investigate the influence of PS2 on myocardial reperfusion dysfunction. We found that loss of PS2 improved the mitochondrial integrity and favored cardiomyocyte viability during I/R injury. PS2 deficiency maintained the mitochondrial potential, reduced mitochondria-derived ROS production and inhibited mitochondria-induced cardiomyocyte death, thus enhancing the viability of cardiomyocytes and elevating their resistance to reperfusion injury. To date, this is the first exploration to demonstrate that PS2 induces mitochondrial dysfunction during I/R injury.
Our results also showed that TMBIM6 is an upstream inhibitor that binds to and post-transcriptionally downregulates PS2. Through molecular docking and protein-protein interaction analyses, we demonstrated that TMBIM6 can prevent PS2 accumulation in cardiomyocytes during I/R injury. However, in our reperfusion model, TMBIM6 was markedly downregulated in the myocardium, while PS2 was upregulated. Overexpression of TMBIM6 was able to reduce PS2 expression, thereby preserving the mitochondrial integrity and enhancing cardiac function upon I/R injury.
Previous studies have had similar findings regarding TMBIM6 [65]. Zhou et al. reported that myocardial I/R injury suppressed TMBIM6 expression by upregulating the DNA-dependent protein kinase catalytic subunit, which recognizes double-stranded DNA damage in cardiomyocytes [34]. Sufficient TMBIM6 expression was identified as a prerequisite for the activation of mitochondrial autophagy and the mitochondrial adaptive stress response, two mitochondrial quality control mechanisms that alleviate mitochondrial injury [66]. Ample TMBIM6 expression was found to enhance calcium-related mitochondrial bioenergetics [31] and mitochondrial glucose metabolism [67]. Moreover, TMBIM6 was shown to inhibit Bax-induced mitochondrial apoptosis [68]. These results illustrate that TMBIM6 can protect the heart by attenuating mitochondrial damage and preserving myocardial function.
Overall, our data demonstrated that TMBIM6 downregulation and PS2 upregulation are pathological contributors to mitochondrial damage during cardiac I/R injury. Restoring sufficient TMBIM6 expression can prevent PS2 accumulation, thus interrupting reperfusion-induced mitochondrial damage for the benefit of the heart. Therefore, TMBIM6 and PS2 are potential therapeutic targets in patients with cardiac I/R injury.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92-100
2. Frank A, Bonney M, Bonney S, Weitzel L, Koeppen M, Eckle T. Myocardial ischemia reperfusion injury: from basic science to clinical bedside. Semin Cardiothorac Vasc Anesth. 2012;16:123-32
3. Valikeserlis I, Athanasiou AA, Stakos D. Cellular mechanisms and pathways in myocardial reperfusion injury. Coron Artery Dis. 2021;32:567-77
4. Marin W, Marin D, Ao X, Liu Y. Mitochondria as a therapeutic target for cardiac ischemia-reperfusion injury (Review). Int J Mol Med. 2021;47:485-99
5. Wang J, Zhou H. Mitochondrial quality control mechanisms as molecular targets in cardiac ischemia-reperfusion injury. Acta Pharm Sin B. 2020;10:1866-79
6. Tan Y, Mui D, Toan S, Zhu P, Li R, Zhou H. SERCA Overexpression Improves Mitochondrial Quality Control and Attenuates Cardiac Microvascular Ischemia-Reperfusion Injury. Mol Ther Nucleic Acids. 2020;22:696-707
7. Ramachandra CJA, Hernandez-Resendiz S, Crespo-Avilan GE, Lin YH, Hausenloy DJ. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine. 2020;57:102884
8. He S, Wang X, Chen A. Myocardial ischemia/reperfusion injury: the role of adaptor proteins Crk. Perfusion. 2017;32:345-9
9. Mui D, Zhang Y. Mitochondrial scenario: roles of mitochondrial dynamics in acute myocardial ischemia/reperfusion injury. J Recept Signal Transduct Res. 2021;41:1-5
10. Wang J, Toan S, Zhou H. Mitochondrial quality control in cardiac microvascular ischemia-reperfusion injury: New insights into the mechanisms and therapeutic potentials. Pharmacol Res. 2020;156:104771
11. Wang J, Toan S, Zhou H. New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury. Angiogenesis. 2020;23:299-314
12. Gao X, Li H, Zhang W, Wang X, Sun H, Cao Y. et al. Photobiomodulation Drives MiR-136-5p Expression to Promote Injury Repair after Myocardial Infarction. Int J Biol Sci. 2022;18:2980-93
13. Huang J, Li R, Wang C. The Role of Mitochondrial Quality Control in Cardiac Ischemia/Reperfusion Injury. Oxid Med Cell Longev. 2021;2021:5543452
14. Buja LM. Myocardial ischemia and reperfusion injury. Cardiovasc Pathol. 2005;14:170-5
15. Wang S, Wang Y, Cheng H, Zhang Q, Fu C, He C. et al. The Networks of Noncoding RNAs and Their Direct Molecular Targets in Myocardial Infarction. Int J Biol Sci. 2022;18:3194-208
16. Xu Z, Zhou J. Zinc and myocardial ischemia/reperfusion injury. Biometals. 2013;26:863-78
17. Morciano G, Bonora M, Campo G, Aquila G, Rizzo P, Giorgi C. et al. Mechanistic Role of mPTP in Ischemia-Reperfusion Injury. Adv Exp Med Biol. 2017;982:169-89
18. Fedeli C, Filadi R, Rossi A, Mammucari C, Pizzo P. PSEN2 (presenilin 2) mutants linked to familial Alzheimer disease impair autophagy by altering Ca(2+) homeostasis. Autophagy. 2019;15:2044-62
19. Nikolac Perkovic M, Pivac N. Genetic Markers of Alzheimer's Disease. Adv Exp Med Biol. 2019;1192:27-52
20. Zampese E, Fasolato C, Kipanyula MJ, Bortolozzi M, Pozzan T, Pizzo P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc Natl Acad Sci U S A. 2011;108:2777-82
21. Rossini M, García-Casas P, Filadi R, Pizzo P. Loosening ER-Mitochondria Coupling by the Expression of the Presenilin 2 Loop Domain. Cells. 2021 10
22. Contino S, Suelves N, Vrancx C, Vadukul DM, Payen VL, Stanga S. et al. Presenilin-Deficient Neurons and Astrocytes Display Normal Mitochondrial Phenotypes. Front Neurosci. 2020;14:586108
23. Contino S, Porporato PE, Bird M, Marinangeli C, Opsomer R, Sonveaux P. et al. Presenilin 2-Dependent Maintenance of Mitochondrial Oxidative Capacity and Morphology. Front Physiol. 2017;8:796
24. Behbahani H, Shabalina IG, Wiehager B, Concha H, Hultenby K, Petrovic N. et al. Differential role of Presenilin-1 and -2 on mitochondrial membrane potential and oxygen consumption in mouse embryonic fibroblasts. J Neurosci Res. 2006;84:891-902
25. Xu X, Shi YC, Gao W, Mao G, Zhao G, Agrawal S. et al. The novel presenilin-1-associated protein is a proapoptotic mitochondrial protein. J Biol Chem. 2002;277:48913-22
26. Li D, Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S. et al. Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am J Hum Genet. 2006;79:1030-9
27. Mohuczy D, Qian K, Phillips MI. Presenilins in the heart: presenilin-2 expression is increased by low glucose and by hypoxia in cardiac cells. Regul Pept. 2002;110:1-7
28. Takeda T, Asahi M, Yamaguchi O, Hikoso S, Nakayama H, Kusakari Y. et al. Presenilin 2 regulates the systolic function of heart by modulating Ca2+ signaling. Faseb j. 2005;19:2069-71
29. Philippaert K, Roden M, Lisak D, Bueno D, Jelenik T, Radyushkin K. et al. Bax inhibitor-1 deficiency leads to obesity by increasing Ca(2+)-dependent insulin secretion. J Mol Med (Berl). 2020;98:849-62
30. Robinson KS, Clements A, Williams AC, Berger CN, Frankel G. Bax inhibitor 1 in apoptosis and disease. Oncogene. 2011;30:2391-400
31. Sano R, Hou YC, Hedvat M, Correa RG, Shu CW, Krajewska M. et al. Endoplasmic reticulum protein BI-1 regulates Ca²⁺-mediated bioenergetics to promote autophagy. Genes Dev. 2012;26:1041-54
32. Lee GH, Lee HY, Li B, Kim HR, Chae HJ. Bax inhibitor-1-mediated inhibition of mitochondrial Ca2+ intake regulates mitochondrial permeability transition pore opening and cell death. Sci Rep. 2014;4:5194
33. Oka T, Sayano T, Tamai S, Yokota S, Kato H, Fujii G. et al. Identification of a novel protein MICS1 that is involved in maintenance of mitochondrial morphology and apoptotic release of cytochrome c. Mol Biol Cell. 2008;19:2597-608
34. Zhou H, Toan S, Zhu P, Wang J, Ren J, Zhang Y. DNA-PKcs promotes cardiac ischemia reperfusion injury through mitigating BI-1-governed mitochondrial homeostasis. Basic Res Cardiol. 2020;115:11
35. Zhou H, Wang J, Hu S, Zhu H, Toanc S, Ren J. BI1 alleviates cardiac microvascular ischemia-reperfusion injury via modifying mitochondrial fission and inhibiting XO/ROS/F-actin pathways. J Cell Physiol. 2019;234:5056-69
36. Zhou H, Zhu P, Wang J, Zhu H, Ren J, Chen Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2alpha-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018;25:1080-93
37. Xu H, Wan XD, Zhu RR, Liu JL, Liu JC, Zhou XL. Keap-NRF2 signaling contributes to the Notch1 protected heart against ischemic reperfusion injury via regulating mitochondrial ROS generation and bioenergetics. Int J Biol Sci. 2022;18:1651-62
38. Wu JW, Hu H, Hua JS, Ma LK. ATPase inhibitory factor 1 protects the heart from acute myocardial ischemia/reperfusion injury through activating AMPK signaling pathway. Int J Biol Sci. 2022;18:731-41
39. Zhu H, Tan Y, Du W, Li Y, Toan S, Mui D. et al. Phosphoglycerate mutase 5 exacerbates cardiac ischemia-reperfusion injury through disrupting mitochondrial quality control. Redox Biol. 2021;38:101777
40. Wang J, Zhu P, Li R, Ren J, Zhou H. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. 2020;30:101415
41. Wang J, Zhu P, Toan S, Li R, Ren J, Zhou H. Pum2-Mff axis fine-tunes mitochondrial quality control in acute ischemic kidney injury. Cell Biol Toxicol. 2020;36:365-78
42. Zhu H, Jin Q, Li Y, Ma Q, Wang J, Li D. et al. Melatonin protected cardiac microvascular endothelial cells against oxidative stress injury via suppression of IP3R-[Ca(2+)]c/VDAC-[Ca(2+)]m axis by activation of MAPK/ERK signaling pathway. Cell Stress Chaperones. 2018;23:101-13
43. Wang Y, Jasper H, Toan S, Muid D, Chang X, Zhou H. Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox Biol. 2021;45:102049
44. Zhou H, Zhu P, Wang J, Toan S, Ren J. DNA-PKcs promotes alcohol-related liver disease by activating Drp1-related mitochondrial fission and repressing FUNDC1-required mitophagy. Signal Transduct Target Ther. 2019;4:56
45. Wang S, Zhu H, Li R, Mui D, Toan S, Chang X. et al. DNA-PKcs interacts with and phosphorylates Fis1 to induce mitochondrial fragmentation in tubular cells during acute kidney injury. Sci Signal. 2022;15:eabh1121
46. Zou R, Tao J, Qiu J, Lu H, Wu J, Zhu H. et al. DNA-PKcs promotes sepsis-induced multiple organ failure by triggering mitochondrial dysfunction. Journal of Advanced Research. 2022
47. Sun D, Wang J, Toan S, Muid D, Li R, Chang X. et al. Molecular mechanisms of coronary microvascular endothelial dysfunction in diabetes mellitus: focus on mitochondrial quality surveillance. Angiogenesis. 2022;25:307-29
48. Chang X, Toan S, Li R, Zhou H. Therapeutic strategies in ischemic cardiomyopathy: Focus on mitochondrial quality surveillance. EBioMedicine. 2022;84:104260
49. Zhu H, Toan S, Mui D, Zhou H. Mitochondrial quality surveillance as a therapeutic target in myocardial infarction. Acta Physiol (Oxf). 2021;231:e13590
50. Zhou H, Ren J, Toan S, Mui D. Role of mitochondrial quality surveillance in myocardial infarction: From bench to bedside. Ageing Res Rev. 2021;66:101250
51. Chang X, Li Y, Cai C, Wu F, He J, Zhang Y. et al. Mitochondrial quality control mechanisms as molecular targets in diabetic heart. Metabolism. 2022;137:155313
52. Zhu H, Zhou H. Novel Insight into the Role of Endoplasmic Reticulum Stress in the Pathogenesis of Myocardial Ischemia-Reperfusion Injury. Oxid Med Cell Longev. 2021;2021:5529810
53. Jin Q, Li R, Hu N, Xin T, Zhu P, Hu S. et al. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol. 2018;14:576-87
54. Chang X, Lochner A, Wang HH, Wang S, Zhu H, Ren J. et al. Coronary microvascular injury in myocardial infarction: perception and knowledge for mitochondrial quality control. Theranostics. 2021;11:6766-85
55. Tan Y, Xi D, Cai C, Jiang X, Chen S, Hu R. et al. DUSP1 overexpression attenuates septic cardiomyopathy through reducing VCP phosphorylation and normalizing mitochondrial quality control. Acta Pharmaceutica Sinica B. 2022
56. Ma L, Zou R, Shi W, Zhou N, Chen S, Zhou H. et al. SGLT2 inhibitor dapagliflozin reduces endothelial dysfunction and microvascular damage during cardiac ischemia/reperfusion injury through normalizing the XO-SERCA2-CaMKII-coffilin pathways. Theranostics. 2022;12:5034-50
57. Zou R, Shi W, Qiu J, Zhou N, Du N, Zhou H. et al. Empagliflozin attenuates cardiac microvascular ischemia/reperfusion injury through improving mitochondrial homeostasis. Cardiovasc Diabetol. 2022;21:106
58. Lahnwong S, Palee S, Apaijai N, Sriwichaiin S, Kerdphoo S, Jaiwongkam T. et al. Acute dapagliflozin administration exerts cardioprotective effects in rats with cardiac ischemia/reperfusion injury. Cardiovasc Diabetol. 2020;19:91
59. Gong Y, Lin J, Ma Z, Yu M, Wang M, Lai D. et al. Mitochondria-associated membrane-modulated Ca(2+) transfer: A potential treatment target in cardiac ischemia reperfusion injury and heart failure. Life Sci. 2021;278:119511
60. Yang M, Linn BS, Zhang Y, Ren J. Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochim Biophys Acta Mol Basis Dis. 2019;1865:2293-302
61. Eckert A, Schulz KL, Rhein V, Götz J. Convergence of amyloid-beta and tau pathologies on mitochondria in vivo. Mol Neurobiol. 2010;41:107-14
62. De Strooper B, Scorrano L. Close encounter: mitochondria, endoplasmic reticulum and Alzheimer's disease. Embo j. 2012;31:4095-7
63. Zampese E, Fasolato C, Pozzan T, Pizzo P. Presenilin-2 modulation of ER-mitochondria interactions: FAD mutations, mechanisms and pathological consequences. Commun Integr Biol. 2011;4:357-60
64. Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P. Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cell Rep. 2016;15:2226-38
65. Zhou H, Dai Z, Li J, Wang J, Zhu H, Chang X. et al. TMBIM6 prevents VDAC1 multimerization and improves mitochondrial quality control to reduce sepsis-related myocardial injury. Metabolism. 2023;140:155383
66. Wang J, Wang X, Du W, Xue Z, Huang W, Guan Z. et al. BI-1 ameliorates myocardial injury by activating the mitochondrial unfolded protein response and FUNDC1-related mitophagy in cardiorenal syndrome type 3. Cell Signal. 2021;91:110218
67. Lee GH, Yan C, Shin SJ, Hong SC, Ahn T, Moon A. et al. BAX inhibitor-1 enhances cancer metastasis by altering glucose metabolism and activating the sodium-hydrogen exchanger: the alteration of mitochondrial function. Oncogene. 2010;29:2130-41
68. Chae HJ, Kim HR, Xu C, Bailly-Maitre B, Krajewska M, Krajewski S. et al. BI-1 regulates an apoptosis pathway linked to endoplasmic reticulum stress. Mol Cell. 2004;15:355-66
Author contact
![]() Corresponding author: Dr. Li Ma, E-mail: 2019761008edu.cn. Guangdong Provincial Key Laboratory of Research in Structural Birth Defect Disease, Heart Center, Guangzhou Women and Children's Medical Center, Guangzhou Medical University, Guangzhou, China. Dr. Xinxin Chen, E-mail: xinxinchengzcom. Guangdong Provincial Key Laboratory of Research in Structural Birth Defect Disease, Heart Center, Guangzhou Women and Children's Medical Center, Guangzhou Medical University, Guangzhou, China.
Corresponding author: Dr. Li Ma, E-mail: 2019761008edu.cn. Guangdong Provincial Key Laboratory of Research in Structural Birth Defect Disease, Heart Center, Guangzhou Women and Children's Medical Center, Guangzhou Medical University, Guangzhou, China. Dr. Xinxin Chen, E-mail: xinxinchengzcom. Guangdong Provincial Key Laboratory of Research in Structural Birth Defect Disease, Heart Center, Guangzhou Women and Children's Medical Center, Guangzhou Medical University, Guangzhou, China.