Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(9):2711-2724. doi:10.7150/ijbs.81918 This issue Cite
Research Paper
Synergism of FAK and ROS1 inhibitors in the treatment of CDH1-deficient cancers mediated by FAK-YAP signaling
Jiaming Gao1*, Yunying Yao1*, Chenxuan Liu1, Xi Xie1, Donghe Li1, Ping Liu1, Zaiqi Wang3 ![]() , Baoyuan Zhang1
, Baoyuan Zhang1 ![]() , Ruibao Ren1,2
, Ruibao Ren1,2 ![]()
1. Shanghai Institute of Hematology, State Key Laboratory for Medical Genomics, National Research Center for Translational Medicine at Shanghai, International Center for Aging and Cancer, Collaborative Innovation Center of Hematology, Ruijin Hospital affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, China.
2. International Center for Aging and Cancer, Hainan Medical University, Haikou, Hainan Province, China.
3. InxMed (Shanghai) Co., Ltd, Shanghai, China.
* These authors contributed equally to this study.
Received 2022-12-16; Accepted 2023-5-5; Published 2023-5-15
Abstract

CDH1 deficiency is common in diffuse gastric cancer and triple negative breast cancer patients, both of which still lack effective therapeutics. ROS1 inhibition results in synthetic lethality in CDH1-deficient cancers, but often leads to adaptive resistance. Here, we demonstrate that upregulation of the FAK activity accompanies the emergence of resistance to ROS1 inhibitor therapy in gastric and breast CDH1-deficient cancers. FAK inhibition, either by FAK inhibitors or by knocking down its expression, resulted in higher cytotoxicity potency of the ROS1 inhibitor in CDH1-deficient cancer cell lines. Co-treatment of mice with the FAK inhibitor and ROS1 inhibitors also showed synergistic effects against CDH1-deficient cancers. Mechanistically, ROS1 inhibitors induce the FAK-YAP-TRX signaling, decreasing oxidative stress-related DNA damage and consequently reducing their anti-cancer effects. The FAK inhibitor suppresses the aberrant FAK-YAP-TRX signaling, reinforcing ROS1 inhibitor's cytotoxicity towards cancer cells. These findings support the use of FAK and ROS1 inhibitors as a combination therapeutic strategy in CDH1-deficient triple negative breast cancer and diffuse gastric cancer patients.
Keywords: FAK Inhibitor, ROS1 inhibitor, Combination therapy, drug resistance, YAP-TRX axis
Introduction
Deficiency for E-cadherin protein caused by mutations in the cadherin 1 (CDH1) gene is a major driver of tumorigenesis in diffuse gastric cancer (DGC) and in breast cancer [1, 2]. Patients with CDH1 deficiency generally show the worst prognosis and shortest overall survival time in DGC and triple-negative breast cancer (TNBC) [3, 4]. Due to the lack of effective targeted therapies, conventional chemotherapy continues to serve as the standard of care for treating DGC and TNBC. However, clinical outcomes of conventional chemotherapy are typically poor, with substantial toxicity mitigating further benefits of treatment [5, 6]. Hence, novel regimens targeting CDH1 deficiency are urgently needed.
Targeting the receptor tyrosine kinase, ROS proto-oncogene 1 (ROS1), can lead to synthetic lethality in cancers associated with CDH1 deficiency [7], and several ROS1 inhibitors have been approved to treat non-small cell lung cancers by the US Food and Drug Administration (US FDA) due to their strong clinical responses [8, 9]. In particular, clinical trials of the ROS1 inhibitors crizotinib and entrectinib continue in CDH1-deficient breast cancer and DGC patients [10, 11]. However, the development of resistance to ROS1 inhibitors remains an urgent focus of clinical research, since the only remaining clinical interventions for ROS1 inhibitor-resistant cancers are chemotherapy or locally ablative therapy [12]. Notably, rational combination strategies incorporating ROS1 inhibitors with a non-overlapping mechanism may facilitate some degree of success in overcoming drug resistance [13].
A growing body of evidence supports a close association between the effects of CDH1 deficiency and focal adhesion kinase (FAK) signaling. In DGC and TNBC patients, FAK significantly promote self-renewing cancer stem cells and correlates with malignant potential [14-16]. Moreover, FAK inhibitors have been shown to attenuate tumor growth in Cdh1-deficient mice and show potent cytotoxic effects in DGC and TNBC cell lines [17, 18]. FAK also functions as an informative biomarker of cellular stress and plays a key role in promoting resistance to chemotherapy and various targeted therapies [19]. Co-treatment with FAK inhibitors, including inhibitors of the B-Raf proto-oncogene (BRAF), mitogen-activated protein kinase (MEK), and KRAS-G12C, is currently well-accepted as a viable potential strategy for overcoming adaptive resistance to chemotherapy, radiotherapy, or targeted therapies [20-24], supported by evidence emerging from a series of clinical trials evaluating FAK inhibitor combination regimens [25]. In light of these findings, we hypothesized that ROS1 inhibitors used in combination with FAK inhibitors could provide durable benefits for DGC and TNBC patients with CDH1 deficiency.
In the present study, we tested this hypothesis and found that co-treatment with ROS1 inhibitors and FAK inhibitors conferred synergistic anti-tumor effects in CDH1-deficient mice model of DGC in vitro and in vivo. Additional experiments indicated that FAK-YAP-thioredoxin (TRX) signaling was enhanced by administration of a ROS1 inhibitor, sequentially decreasing the oxidative stress-related DNA damage, which compromised the effects of the ROS1 inhibitor. Treatment with the FAK inhibitor, IN10018, which has been granted fast-track designation by the US FDA, led to enhanced effects of ROS1 inhibition by rescuing aberrant FAK-YAP-TRX signaling. Since there are currently no highly selective ROS1 inhibitors available (e.g., crizotinib targets ROS1, anaplastic lymphoma kinase (ALK) and mesenchymal epithelial transition factor receptor (MET) kinase), we also investigated whether the observed anti-tumor effects were indeed achieved through ROS1 inhibition. Silencing of each predicted target in NUGC-4 cells showed that ROS1, but not other targets, was critical for the synergistic effects of combination therapy with IN10018. These findings suggest that co-administration of FAK inhibitors and ROS1 inhibitors may be an effective potential treatment option for CDH1-deficient cancer patients and warrant further testing in clinical trials.
Materials and Methods
Reagents
Crizotinib, entrectinib, and VS-4718 were purchased from MedChemExpress (MCE). IN10018 was provided by InxMed. All the siRNAs used in the study were synthesized by Genepharma (siRNA sequences given in Table S1 of the Supplementary materials). The antibodies used in the study are listed in Table S2 of the Supplementary materials.
Cell Culture
DGC cell lines were cultured in RPMI-1640 medium with 10% fetal calf serum and 1% penicillin-streptomycin (Life Technologies). MDA-MB-231 cells were cultured in DMEM supplemented with 10% fetal calf serum and 1% penicillin/streptomycin (Life Technologies). Hs-578t cells were cultured in DMEM supplemented with 10% fetal calf serum, 0.01 mg/mL insulin, and 1% penicillin/streptomycin (Life Technologies). The crizotinib-resistant cell line was continuously cultured with crizotinib at a concentration that increased stepwise for 6 months.
Cell Viability Assay
All cells were seeded in 96-well plates at a density of 3,000 cells per well. After cells were cultured for 12 h, drugs were added to the medium. Following treatment for 72 h, cell viability was determined using the CellTiter-Glo luminescent assay (Promega) according to the manufacturer's protocol. Viability was detected by a microplate plate reader (Thermo Fisher Scientific). Dose-response curves were generated using GraphPad Prism.
Cell Clonogenic Assay
All cells were seeded in 12-well plates at a density of 1,000 cells per well. After cells were cultured for 24 h, drugs were added to the medium. The cells were maintained for 2 weeks with drug treatments. Colonies were fixed at 4% paraformaldehyde (Sigma-Aldrich) for 15 min and stained with crystal violet (0.5% w/v, Sigma-Aldrich) for the collection of images.
Western Blotting
Cells were lysed with RIPA buffer containing 1% protease/phosphatase inhibitors (Roche), and protein levels were quantified using the Pierce™ Rapid Gold BCA Protein Assay Kit (Thermo Fisher Scientific). Proteins in lysates were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to nitrocellulose membranes (0.2 μM, Amersham Protran). Blocking and antibody incubation were performed using 5% non-fat powdered milk, followed by washing in Tris-buffered saline with 0.1% Tween 20 (TBS-T). Primary antibodies were incubated overnight at 4°C and secondary antibodies (Cell Signaling Technology) were incubated at room temperature for 1 h.
In Vivo Efficacy
All experiments were approved by the Ethics Committee of Shanghai Jiao Tong University School of Medicine, and all animal studies were performed following AAALAC guidance. Balb/c nude mice and NOD scid mice were purchased from GemPharmatech Co., Ltd. and maintained under 14-h light/10-h dark cycles (dark 20:00-6:00). The use of the SNU-668 and NUGC-4 cell line-derived xenografts was approved by the Institutional Animal Care and Use Committee (IACUC) of Shanghai WuXi AppTec. Experiments involving DGC cancer PDX models 051009 and 0501013 were approved by IACUC of Nanjing Personal Oncology Biotechnology. Mice developed tumors reaching a volume of 100-200 mm3. Six mice in each group received oral administration once per day of one of the following drug regimens: (1) control vehicle, 25 mg/kg (0.5% Natrosol 250 HX in distilled water); (2) IN10018, 25 mg/kg; (3) crizotinib or entrectinib, 25 mg/kg; or (4) IN10018 plus (S)-crizotinib, 25 mg/kg. Tumor volumes were measured by caliper and calculated. At the appropriate end point, mice were humanely killed, and NUGC-4 tumors were harvested for Western blot analysis.
RNA-Seq
Total RNA was extracted following the instructions accompanying the TRIzol reagent (Thermo Scientific). DNA libraries were prepared from samples by using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England Biolabs). Transcriptome RNA sequencing (RNA-Seq) was performed using Illumina high-throughput RNA sequencing (HiSeq 2500 sequencer). Differential expression analysis of RNA-Seq data for any two groups was performed using DESeq2 [26]. Genes with Benjamini-Hochberg-adjusted P values <0.05 and absolute log2 fold changes >1 were considered to be differentially expressed. Gene Ontology (GO) and pathway enrichment analysis using Kyoto Encyclopedia of Genes and Genomes (KEGG) for the differentially expressed genes were performed using the R package clusterProfiler, with a threshold value of P < 0.05 [27, 28]. Data were visualized using the ggplot2 R package (https://ggplot2.tidyverse.org/).
Detection of ROS
Cells were co-cultured with diacetyldichlorofluorescein diacetate (DCFH-DA; Abbkine, KTB1910) at a final DCFH-DA concentration of 10 mM for 30 min. The intracellular levels of ROS were analyzed by flow cytometry.
Comet assay
The assay was performed using the Comet Assay Kit from Abcam (ab238544) as described by the manufacturer. Briefly, cells in RPMI medium were mixed with comet agarose 1:10 (v/v) and immediately pipetted onto agarose pre-coated slides. The slides were transferred to a lysis buffer and then to an alkaline solution for 30 mins at 4ºC in the dark. The slides were analyzed by electrophoresis. The cells were stained with Vista Green DNA Dye and observed using a fluorescence microscope with an image analysis system.
Immunofluorescence Assay and Immunohistochemistry
Cells were fixed in 4% paraformaldehyde for 20 min, and then washed four times with phosphate-buffered saline (PBS). Cells were incubated with blocking buffer (0.1% Triton X-100 and 5% bovine serum albumin in Tris-buffered saline [TBS]) for 2 h and then washed three times with PBS containing 0.1% Tween 20 (Sigma-Aldrich). Cells were incubated with the first antibody overnight at 4°C and then incubated for 1 h with fluorescent-conjugated secondary antibodies. After being washed three times for 5 min each time with PBS containing 0.1% Tween 20, the cells were observed using a laser scanning confocal microscope (Lecia Microsystems TCS 7300).
The Immunohistochemistry was performed as described [24]. The primary Abs included p-FAK (1:300), YAP (1:500), γH2AX (1:400), and Cleaved Caspase 3 (1:300). Then, DAB Substrate Kit (abcam, ab64238) was used to detect immunoactivity. Hematoxylin was used for nuclear staining.
RNA Extraction and Quantitative real-time PCR analysis
Total RNA of NUGC-4 cell samples was isolated by RNA iso plus reagent (Takara). PrimeScript RT Master Mix Kit (Takara, RR036A) was used to synthesize complementary DNA (cDNA) from 1μg RNA. In addition, quantitative PCR (qPCR) was conducted by the SYBR-green super mix kit (Bio-Rad) and the Quantstudio 7 Flex Real-Time PCR system 12 (Thermo Scientific, MA, USA). Relative expression levels were calculated by the 2-ΔΔCT method. For the primers sequences which used in the experiment were listed in Table S2.
Statistical analysis
GraphPad Prism, version 5.0 (GraphPad Software), was used for all data analysis. Unpaired student's two-tailed t-tests were used to assess the differences between samples. Data are shown as the mean ± SEM. Statistically significant differences of p < 0.05, p < 0.01 and p < 0.001 are noted with *, **, and ***, respectively.
Results
FAK signaling is upregulated during resistance to ROS1 inhibitor therapies in CDH1-deficient cancer cells
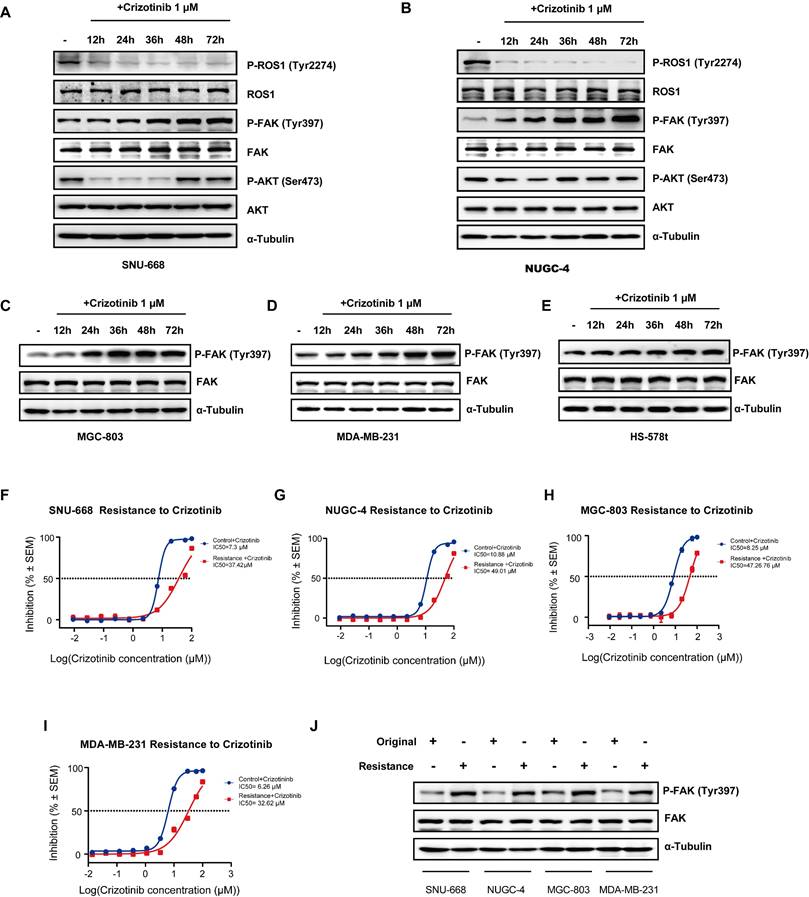
In order to investigate whether ROS1 inhibitors affect FAK signaling to overcome drug resistance in CDH1-deficiency cancers, such as gastric and breast cancers, we first used Western blot analysis to determine ROS1 and FAK signaling activity levels by examining phosphorylation of their downstream targets in five cancer cell lines (SNU-668, NUGC-4, MGC-803, MDA-MB-231, and Hs-578t) across 12 hours of treatment with crizotinib/entrectinib. We found that levels of p-ROS1 significantly decreased, while p-AKT levels remained stable and p-FAK levels markedly increased, during 24 hours of treatment with either crizotinib (Figure 1 A-B) or entrectinib (Figure S1 A-B) in SNU-668 and NUGC-4 DGC cell lines. The p-FAK levels also increased with treatment time in MGC-803 cell, and TNBC cell lines MDA-MB-231, and Hs-578t (Figure 1 C-E). To determine if this observed activation of p-FAK was associated with crizotinib resistance, we established drug-resistant cell lines derived from SNU-668, NUGC-4, MGC-803, and MDA-MB-231 cells by continuous exposure to crizotinib for 6 months. A resistant phenotype was confirmed by comparing the cell killing effects of crizotinib between the original and derived cells (Figure 1 F-I). Subsequent Western blot analysis showed that p-FAK Y397 levels appeared higher in the resistant cell lines than in the original cell lines (Figure 1 J). These findings confirmed that the upregulation of FAK activity accompanies the emergence of resistance to ROS1 inhibitor therapies in gastric and breast CDH1-deficient cancers.
ROS1 inhibitors increase FAK signaling in CDH1-deficient cancer cell lines. (A-E) FAK signaling and downstream marker expression levels assessed by Western blot analysis at various times after crizotinib treatment. (F-I) Original and crizotinib-resistant cancer cells were treated with various concentrations of crizotinib for 72 h and assessed for viability by using the CellTiter-Glo luminescent assay. (J) Western blot analysis assessing FAK signaling in original and crizotinib-resistant cell lines.
FAK inhibition plus ROS1 inhibitors combination treatment synergistically inhibits growth of CDH1-deficient cancer cells with or without crizotinib resistance
Based on our above findings of p-FAK upregulation under ROS1 inhibitor resistance, we next tested whether inhibiting FAK activity could restore the negative impact of ROS1 inhibitors on cancer cell proliferation in vitro using CTG assays in the five CDH1-deficient cell lines. We found that monotherapy treatments with the FAK inhibitors IN10018 and VS-4718 resulted in moderate cancer cell killing effects (Figure S2 A-E). Further tests of these FAK inhibitors (3 or 5 μM) combined with crizotinib or entrectinib (30 μM to 4.572 nM) resulted in the dose-dependent inhibition of cancer cell growth in non-resistant CDH1-deficient cell lines that was stronger than either monotherapy (Figure 2 A-F, Figure S2 F-U). The corresponding IC50 values for each cell survival curve were summarized in Table S3. These apparently synergistic effects of the combined treatment were evaluated using Bliss scores. To test the long-term (10-day) effects of the crizotinib plus IN10018 drug combination, colony formation assays were also conducted with non-drug-resistant CDH1-deficient cell lines. Consistent with the aforementioned results in short-term assays, significantly fewer colonies were consistently observed in the combined treatment group of each cell line compared with that in the monotherapy groups (Figure 2 G-H). Furthermore, FAK knockdown by siRNA in both NUGC-4 and MDA-MB-231 cell lines resulted in higher anti-tumor potency of the crizotinib monotherapy (Figure 2 I and J).
Co-treatment with FAK inhibition plus crizotinib synergistically inhibits growth of CDH1-deficient cancer cells. (A-E) Cells were treated with various concentrations of crizotinib with or without IN10018 (3 μM or 5 μM) for 72 h. Cell viability was determined by the CellTiter-Glo luminescent assay. Data represent mean ± SEM; n ≥ 3. (F) SynergyFinder 2.0 software was used for Bliss model analysis, with a Bliss score >10 suggesting synergistic effects for the drug combination. (G) Cell clonogenic assay results for co-treatment with crizotinib plus IN10018. Representative images of single-cell clone proliferation, stained with crystal violet. (H) Quantification of the results (G). Data are presented as the mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with control cells. (I, J) Knockdown of FAK results in enhanced inhibition of cell viability for NUGC-4 and MDA-MB-231 cells treated with crizotinib. Cells were transfected with control or FAK siRNA, and crizotinib was added 24 h later. Cell viability was evaluated 72 h later. Data represent mean ± SEM; n = 4.
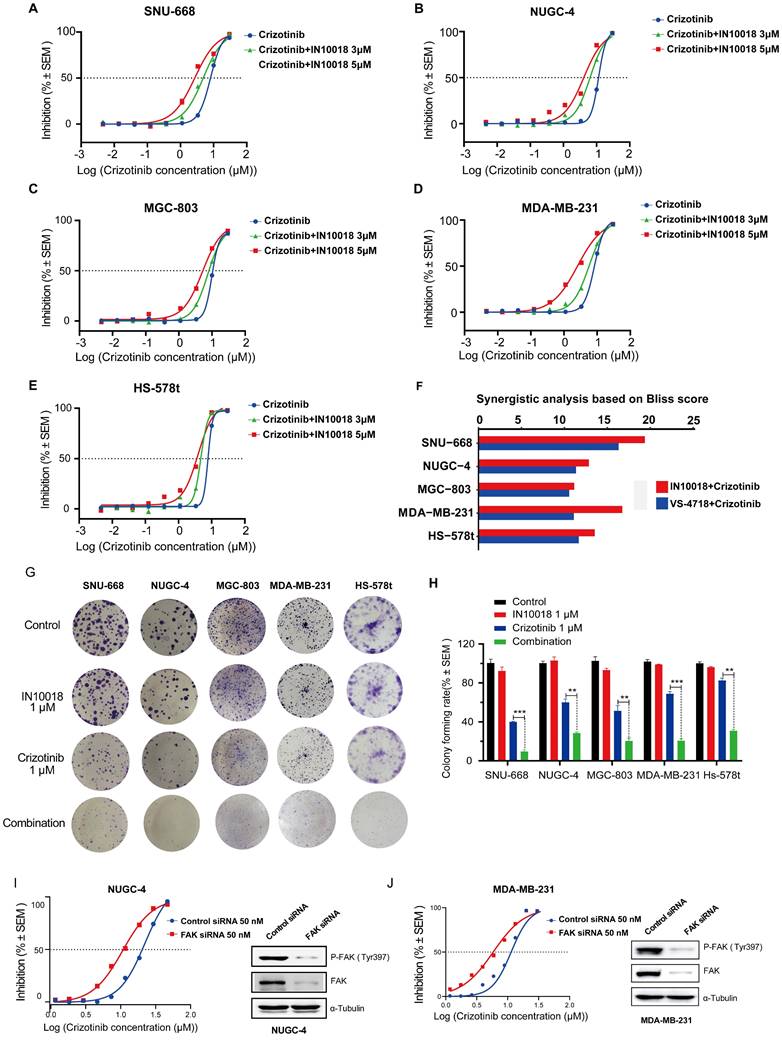
We then examined whether the IN10018 and crizotinib combination also showed enhanced effects in the crizotinib-resistant cell lines. CellTiter-Glo Luminescent Cell Viability (CTG) assays similarly showed significantly greater cell killing following combination treatment in crizotinib-resistant lines compared with that induced by monotherapies, suggesting that the addition of IN10018 restored sensitivity to crizotinib in resistant cells (Figure S3 A-H). Collectively, these data indicated that FAK activation was related to ROS1 inhibitor (crizotinib)-resistance in CDH1-deficient cancer cells, and that FAK inhibition could thus overcome this drug resistance, ultimately leading to synergistic therapeutic effects in these cancers.
FAK inhibitor enhances the anti-tumor effects of ROS1 inhibitor in CDH1-deficient cancers in vivo
In light of the above in vitro synergistic anti-tumor effects of the combination treatment, we next generated two human CDH1-deficient cell line (SNU-668 and NUGC-4)-derived xenograft (CDX) models in mice to evaluate the potential in vivo therapeutic effects of the ROS1 inhibitor-IN10018 combination. Administration of either crizotinib, entrectinib, or IN10018 monotherapies (25 mg/kg, p.o., daily) resulted in relatively limited tumor inhibition compared with that following administration of the drug combinations, which led to significantly attenuated tumor development (Figure 3 A-D). Furthermore, inhibition of tumor growth was sustained for 2 weeks beyond the end of the dosing period under the combination regimen (Figure 2 A, C), and entrectinib showed comparable anti-tumor effects in the NUGC-4 and SNU-668 CDX models (Figure S4 A-D). All treatments were well tolerated in this study (Figure 3 A-D and Figure S4 A-D). After completion of the treatment period, Western blot analysis of tumor samples harvested from NUGC-4 CDX model mice showed that p-FAK was upregulated to significantly higher levels in the crizotinib monotherapy group compared with that in the combination treatment group (Figure 3 E), suggesting that FAK inhibition by IN10018 administration could suppress the activation of FAK signaling associated with crizotinib.
In vivo effects of IN10018 plus crizotinib on CDH1-deficient cell line-derived xenograft (CDX) and patient-derived xenograft (PDX) models. (A-D) Tumor growth and mouse body weight changes in mice with SNU-668 and NUGC-4 xenografts treated with the indicated agents. Mice were orally administered vehicle control (0.5% Natrosol 250 HX), 25 mg/kg of IN10018, or 25 mg/kg of crizotinib once daily. Tumor sizes and mouse body weights were recorded twice per week. Data represent mean ± SEM; n ≥ 5. Comparisons were conducted using unpaired student's T-tests. *P < 0.05, **P < 0.01, and ***P < 0.001. Synergy P value is shown. (E) Western blot analysis for FAK signaling in tumor tissue of the NUGC-4 CDX model. (F-I) Tumor growth and mouse body weight changes in the 0501013 and 051009 PDX models. (J, K) P-FAK (Tyr 397) IHC staining for the 051009 tumors. Scale bar = 50 µm. (K) Quantification of the expression levels of P-FAK (Tyr 397) in 051009 tumors from (J). (Data represent Mean ± SEM, n≥12). Statistics analysis was done using unpaired student's T-test. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
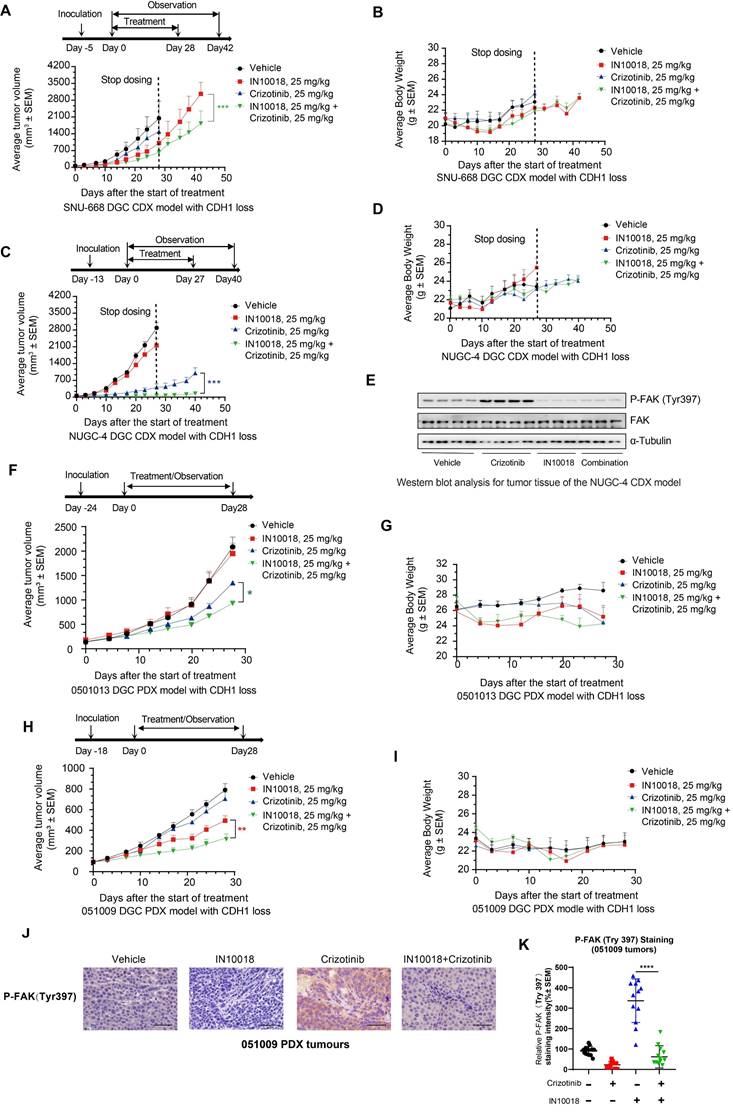
We then evaluated the in vivo effects of the co-administration of crizotinib with a FAK inhibitor in two CDH1-deficient DGC patient-derived xenograft (PDX) models, 0501013 and 051009. Similar to the results obtained in CDX model mice, tumor volumes in the group treated with crizotinib (25 mg kg-1, p.o.) plus IN10018 (25 mg kg-1, p.o., daily) for 4 weeks were significantly smaller than those in either the monotherapy or vehicle control groups (Figure 3F-I). Mice displayed good tolerance for all treatments during the drug dosing period (Figure 3 F-I). IHC analysis of tumor samples collected from the 051009 PDX model at the end of therapy showed that P-FAK expression levels were significantly higher in the crizotinib monotherapy compared to the combination therapy, which was consistent with the results obtained in the NUGC-4 tumors (Figure 3 J, K). These cumulative findings indicated that targeting ROS1 with either crizotinib or entrectinib, in conjunction with IN10018-mediated FAK inhibition, enhanced the in vivo anti-tumor effects of either monotherapy in both CDX and PDX CDH1-deficient mouse models.
Inhibiting FAK enhances the oxidative stress-induced DNA damage caused by crizotinib via regulation of the YAP-TRX axis
To explore which pathways might be responsible for the synergistic effects of FAK/ROS1 inhibition, we conducted whole transcriptome profiling (RNA-seq) in NUGC-4 cells treated for 24 h with crizotinib, IN10018, the crizotinib-IN10018 combination, or vehicle control. Gene Ontology and KEGG analysis revealed that genes differentially expressed between crizotinib monotherapy and the combination therapy were primarily enriched in pathways related to DNA damage, oxidative stress, and Hippo signaling (Figure 4 A, B and Figure S5 A, B). To verify the involvement of these processes, we performed diacetyldichlorofluorescein diacetate (DCF) fluorescence flow cytometry, comet assays, and γH2AX expression assays to evaluate ROS accumulation and DNA damage. The results of these assays showed that oxidative stress and DNA damage were greater in the combined treatment group compared with the crizotinib monotherapy group (Figure 4 C-G and Figure S5 C).
Reactive oxygen species (ROS) accumulation and DNA damage is enhanced by addition of IN10018 with crizotinib vs crizotinib alone. (A, B) GO and KEGG enrichment analysis of significantly up-regulated and down-regulated genes between crizotinib monotherapy group and combination treatment group. The Y-axis represents the name of the pathway, and the X-axis represents the P value with the log2-transformed in enriched pathways, the different colors of bar represent pathways that were significantly enriched for significantly up-regulated and down-regulated genes in two groups. (C) Intracellular ROS production as assessed by DCFH-DA fluorescence flow cytometry. Quantitative histograms of the mean fluorescence intensity (M.F.I.) values (D) of n ≥ 5 independent FACS experiments are shown (Data represent mean ± SEM). The NUGC-4 cells were treated with 3 μM crizotinib with or without 3 μM IN10018 (as indicated) for 6 h. (D, E) DNA damage in NUGC-4 cells as assessed by the comet assay following treatment with 3 μM crizotinib with or without 3 μM IN10018 (as indicated) for 6 h. (E) Quantification of the results (D) (Data are presented as the mean ± SEM, n=100. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with crizotinib-treated cells). (F, G) DNA damage as detected by γH2AX immunofluorescence following treatment with 3 μM crizotinib with or without 3 μM IN10018 (as indicated) for 6 h. Scale bars = 25 μm. (G) Quantification of the results (F), n=100. Data are presented as the mean ± SEM of the number of γH2AX foci per nucleus from three separate experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with crizotinib-treated cells. (H) YAP immunofluorescence image following treatment with 3 μM crizotinib with or without 3 μM IN10018 (as indicated) for 48 h. Scale bars = 46.2 μm. (I) Western blot analysis of YAP/TRX signaling and DNA damage markers following treatment with 3 μM crizotinib with or without 3 μM IN10018 (as indicated) for 6 h. (J) Western blot analysis of YAP immunofluorescence and DNA damage following treatment with 3 μM crizotinib for 6 h. (K) Inhibition of cell growth in NUGC-4 cells with YAP knockdown treated with crizotinib. (L) Western blot analysis for YAP signaling in tumor tissue of the NUGC-4 CDX model. (M) YAP, γH2AX and Cleaved Caspase 3 IHC staining for the 051009 tumors. Scale bar = 50 µm.
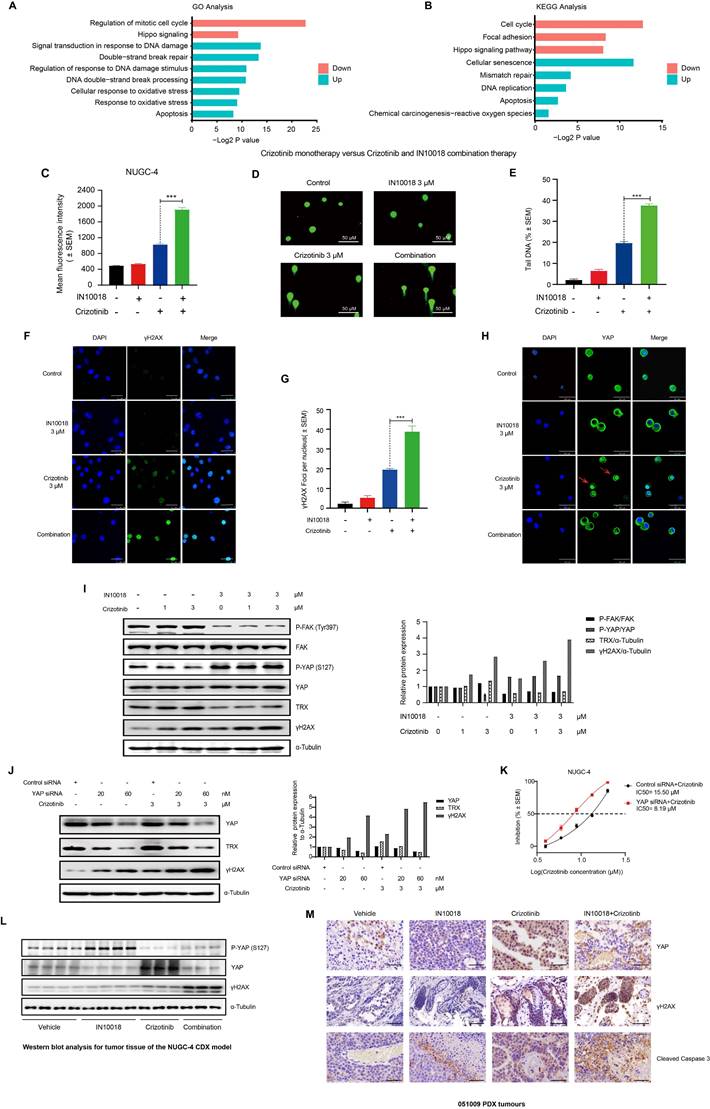
YAP is an essential transcriptional co-factor for Hippo pathway signaling, and aberrant regulation of YAP activity has been linked to the occurrence of drug resistance in cancer treatments [29]. FAK controls YAP signaling by regulating its translocation to the nucleus and subsequent activation [30]. Further examination of our RNA-seq data revealed that FAK-mediated YAP signaling could potentially participate in the mechanism responsible for crizotinib resistance. We also used RT-qPCR and western blot analysis to test the expression of YAP signaling downstream genes in NUGC-4 cells. Crizotinib monotherapy induced activation of the YAP signaling pathway, which can be significantly suppressed by combination therapy (Figure S5 D, E). Notably, the nuclear localization of YAP was significantly increased at 48 h of exposure to crizotinib, suggesting that the regulation of YAP signaling may be abnormal (Figure 4 H and Figure S5 F). Treatment with the FAK inhibitor IN10018 combined with crizotinib resulted in obvious YAP export from the nucleus (Figure 4 H and Figure S5 F). Detection of the ROS scavenger TRX, a downstream biomarker of YAP [31], showed its concomitant downregulation in the group treated with IN10018 (Figure 4 I), which could explain the heightened accumulation of ROS in the combined treatment group compared with that in the crizotinib monotherapy group. Crizotinib has been shown to trigger ROS accumulation, which can in turn directly induce DNA damage [32]. Consistent with this effect, we observed that exposure to a ROS1 inhibitor induced γH2AX expression, which was further increased in cells treated with the combination therapy in a crizotinib dose-dependent manner (Figure 4 I). We then assessed whether the increased DNA damage was dependent on YAP-TRX expression in cells treated with crizotinib. Knockdown of YAP by siRNA also led to reduced TRX expression, further supporting the role of YAP-regulated TRX expression. Reduced TRX expression coincided with elevated γH2AX levels under YAP knockdown (Figure 4 J). Moreover, cell killing and DNA damage were both markedly increased under crizotinib treatment in NUGC-4 cells with YAP knocked down (Figure 4 K and Figure S5 G, H).
To further validate the role of the mechanistic results in vitro, we evaluated the YAP signalling and DNA damage in vivo. IHC staining and western blot analysis of tumor samples from NUGC-4 showed that crizotinib group tumours exhibited high level expression of YAP in nuclear, which was significantly suppressed in the combination therapy tumours. (Figure 4 L and Figure S6 A,B).
We also note that more DNA damage and apoptosis was induced by the combination therapy than by the crizotinib monotherapy (Figure 4 L and Figure S5 A,B). The IHC staining of the 051009 tumors showed comparable results of the NUGC-4 tumors (Figure 4 M and Figure S6 C). Taken together, these results suggested that FAK inhibition led to increased DNA damage induced by crizotinib through regulation of the FAK-YAP axis in vitro and in vivo.
Exogenous N-acetylcysteine (NAC) or YAP activation mitigates the anti-tumor effects of FAK/ROS1 inhibition
To confirm whether the cell killing effects of the combination therapy were dependent on oxidative stress, we assessed whether exogenous application of the ROS scavenger NAC protected NUGC-4 cells from the oxidative damage induced by the co-treatment of IN10018 and crizotinib in vitro. Cells pre-treated for 12 h with NAC (5 mM) and then exposed to the crizotinib-IN10018 combination therapy showed significantly lower cell death than those in the combination treatment group without pretreatment, further supporting a role of oxidative stress in the cell killing effects of the combination therapy (Figure 5 A-C).
Antioxidant N-acetylcysteine (NAC) and activation of YAP rescue cell killing induced by co-treatment of crizotinib and IN10018. (A) NUGC-4 cells were pretreated with 5 mM NAC for 24 h before the addition of crizotinib, IN10018, or both for 48 h. (B, C) Intracellular production of reactive oxygen species (ROS) assessed by DCFH-DA fluorescence cytometry. NUGC-4 cells were pretreated with NAC for 24 h. After removal of NAC, cells were treated with 3 μM crizotinib or 3 μM IN10018 or both (as indicated) for 12 h. (C) Quantification of the results(B) (Data represent mean ± SEM of three independent experiments; n = 4). (D, E) Knockdown of LATS1/2 in NUGC-4 cells enhances cell resistance to treatment with crizotinib alone or with IN10018. Cells were transfected with control siRNA or LATS1/2 siRNA. After 48 h, crizotinib alone or plus IN10018 was added. Cell viability was evaluated 72 h later (Data represent mean ± SEM; n = 4). (F, G) Intracellular production of ROS assessed by DCFH-DA fluorescence cytometry. LATS1/2 was knocked down in NUGC-4 cells, which were then treated with 3 μM crizotinib, 3 μM IN10018, or both (as indicated) for 12 h. (G) Quantification of the results (F) (Data represent mean ± SEM of three independent experiments, n=4). (H) Knockdown of LATS1/2 in NUGC-4 cells. Western blot analysis for YAP/TRX signaling and DNA damage after treatment with 3 μM crizotinib for 6 h.
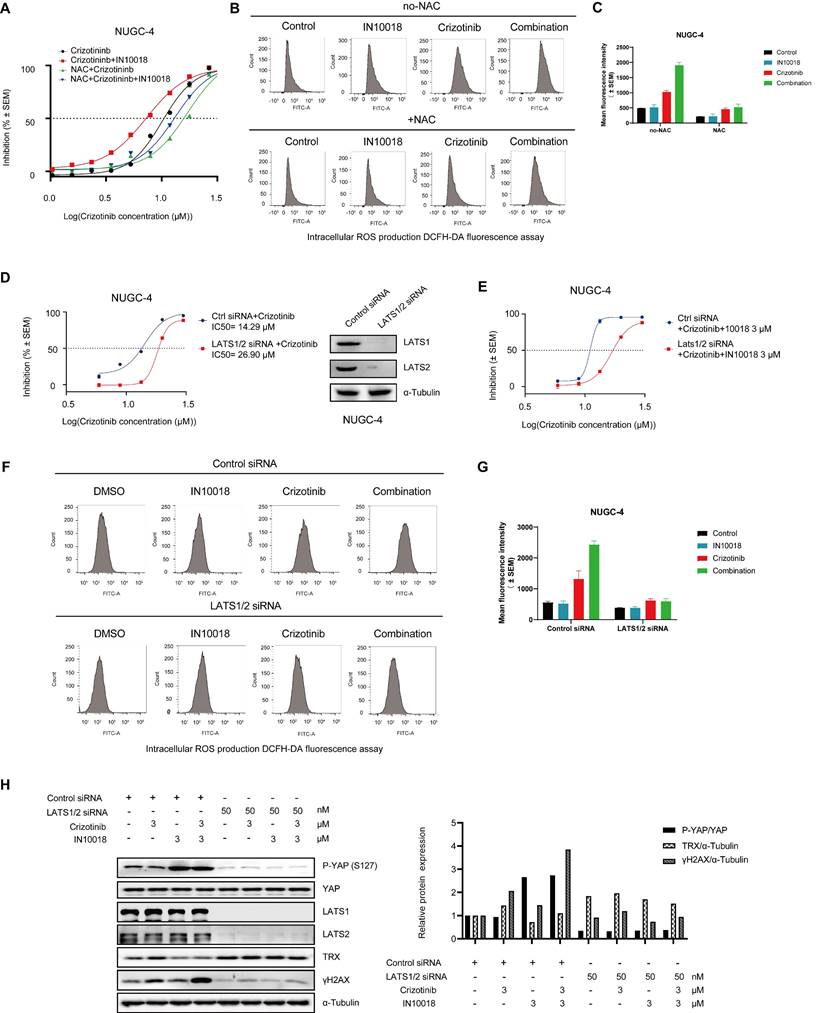
Because LATS1/2 phosphorylates YAP in the cytoplasm to block its localization to the nucleus and consequently downregulate its activity, we hypothesized that suppressing LATS1/2 expression would activate YAP signaling in the nucleus by limiting its retention in the cytoplasm. To test this hypothesis, we generated siRNA-induced LATS1/2 knockdown in the NUGC-4 cell line (Figure 5 D). The resulting cells displayed significantly less cell death and ROS accumulation in response to the combination therapy than the scrambled siRNA control cells under the same conditions (Figure 5 D-G). Knockdown of LATS1/2 by siRNA also led to upregulated TRX expression and induced a lower level of γH2AX, which coincided with the inhibition of cell death (Figure 5 H). These results further supported the likelihood that suppression of oxidative stress response by inhibiting the FAK/YAP/TRX pathway and ROS1 inhibition was indeed responsible for the anti-tumor effects of the combination treatment.
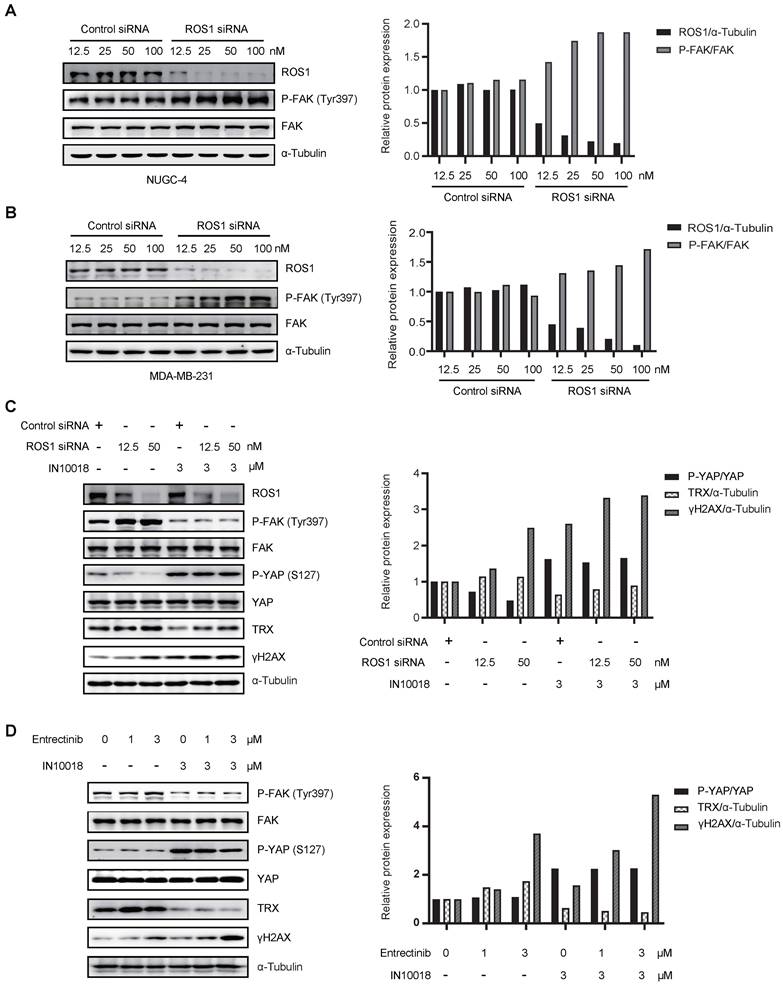
ROS1-dependent effects of FAK signaling activation
Crizotinib and entrectinib are non-selective tyrosine kinase inhibitors, and both have been shown to target ALK and ROS1. To exclude the potential influence of off-target effects, we generated siRNA-induced ROS1 and ALK knockdown cell lines on the NUGC-4 genetic background. Western blot analysis with band density quantification showed that ROS1 knockdown, but not ALK knockdown, induced hyperactivation of FAK (Figure 6 A-B and Figure S7 A-B). Moreover, cells with ROS1 knockdown treated with IN10018 exhibited higher levels of γH2AX than cells with ROS1 knockdown without FAK inhibitor treatment, which was consistent with the results obtained following ROS1 inhibitor treatment (Figure 6 C and Figure S7 C). In addition, we observed that DNA damage was substantially increased in cells treated with entrectinib plus a FAK inhibitor (Figure 6 D). These results thus demonstrated that crizotinib- or entrectinib-induced activation of FAK signaling and the subsequent enhanced DNA damage depend on ROS1, but not on ALK, inhibition in CDH1-deficient cells.
Activation of FAK signaling and enhanced DNA damage depend on ROS1, not on anaplastic lymphoma kinase (ALK). (A, B) Western blot analysis for FAK signaling and downstream markers of ROS1 after siRNA treatment of NUGC-4 and MDA-MB-231 cells. (C) Western blot analysis for YAP signaling and DNA damage in NUGC-4 cells after treatment with ROS1 siRNA. (D) Western blot analyzes for YAP/TRX signaling and DNA damage following treatment with 3 μM entrectinib, 3 μM IN10018, or both (as indicated) for 6 h.
Discussion
DGC and TNBC remain aggressive diseases without effective targeted therapies [5, 6]. Both of these cancers feature CDH1 gene deficiency, and synthetic lethality between ROS1 and CDH1 has already been established, providing a potential clinical treatment strategy for patients with these types of cancer [7]. On the basis of preclinical findings, two clinical trials, the ROLO study (NCT03620643) and the ROSALINE study (NCT04551495), are being conducted to assess the efficacy of co-administering a ROS1 inhibitor with either crizotinib or entrectinib in advanced DGC and TNBC featured with CDH1 deficiency [10, 11]. However marked and durable the initial response may be to treatment with crizotinib, the vast majority of patients with non-small cell lung cancer will develop resistance within a few years [12]. Therefore, it is necessary to explore a therapeutic regimen to overcome ROS1 inhibitor-resistance in DGC and TNBC.
As a key coordinator of cellular responses to environmental stresses, FAK is an attractive target supporting tumorigenesis processes and resistance mechanisms. The inducible product of FAK tyrosine phosphorylation is enhanced by therapeutic stress to resist the deleterious effects of tumor therapy, including MEK inhibitors and KRAS-G12C inhibitors [23, 24]. In the present study, hyperactivation of FAK signaling was also found to be related to the adaptive resistance to a ROS1 inhibitor in the treatment of CDH1-deficient cancers. In addition, administration of a FAK inhibitor promoted the anti-tumor effect of ROS1 inhibitors for both original and crizotinib-resistant CDH1-deficient cancer cells.
Rational combination therapy may enhance therapeutic efficacy by overcoming primary and acquired resistance. Combination therapy that includes a FAK inhibitor has shown synthetic anti-tumor effects and has overcome drug resistance in preclinical studies [33, 34]. IN10018 (formerly known as BI 853520) is a specific FAK kinase inhibitor that is currently under evaluation in a number of phase 1/2 clinical trials [35, 36]. Considering the synergistic effects of the drug combinations tested in the present study in vitro, we assessed the combination therapy of IN10018 with ROS1 inhibitors in vivo. Co-treatment with IN10018 and ROS1 inhibitors significantly reduced the tumor size in CDX models and PDX models with CDH1 deficiency. We also observed that monotherapy with crizotinib induced FAK activation in the NUGC-4 CDX model. These results suggested a potential clinical benefit from the co-administration of a FAK inhibitor with the clinically available crizotinib or entrectinib for patients with CDH1-deficient cancer.
Previous reports have indicated that crizotinib reduces cell growth through the accumulation of intracellular oxidative stress and DNA damage in gastric cancer and in human liver cells [32, 37]. Adaptively enhanced anti-oxidative signaling is one mechanism whereby anti-tumor effects may be compromised. TRX has been suggested for use in chemotherapy to overcome oncogene-induced senescence by preventing ROS accumulation [38, 39]. Transcriptome profiling results have suggested that IN10018 in combination with crizotinib induces stronger oxidative stress-related DNA damage and affects the activity of the Hippo pathway. The FAK-YAP signaling axis is a well-known pathway in the development of cancer [40]. YAP is located upstream of antioxidant enzymes, especially for TRX [31, 41]. We thus considered whether FAK-YAP signaling further enhanced TRX activity to decrease the effect of the ROS1 inhibitor. Indeed, ROS1 inhibition by either crizotinib or entrectinib increased FAK-YAP activity and enhanced TRX activity, which decreased oxidative stress-related DNA damage to compromise the anti-tumor effect of the drug treatment. This mechanism was confirmed by our rescue studies with the antioxidant NAC and by transfection studies with knockdown of LATS1/2, which acted as negative regulator of YAP signaling. Taken together, our findings indicated that FAK inhibition enhanced the effects of ROS1 inhibitors through regulation of the FAK-YAP-TRX signaling axis.
Crizotinib and entrectinib, as multi-target tyrosine kinase inhibitors, similarly induce FAK activation and have two identical targets: ROS1 and ALK. To determine whether ALK, ROS1, or both contributed to FAK activation, ALK and ROS1 were knocked down using their respective siRNAs. We found that knockdown of ROS1, but not of ALK, specifically enhanced the phosphorylation of FAK. Furthermore, the combination of ROS1 siRNA with IN10018 effectively attenuated YAP-TRX signaling, promoting oxidative stress-related DNA damage.
In conclusion, our results indicate that ROS1 inhibition through the administration of either crizotinib or entrectinib compromised cancer treatment outcomes by blunting oxidative stress-related DNA damage through the stimulation of the FAK-YAP-TRX signaling pathway. Co-administration of the FAK inhibitor IN10018 enhances the anti-tumor effect of ROS1 inhibitors in the treatment of CDH1-deficient cancers.
Abbreviations
DGC: Diffuse gastric cancer; TNBC: Triple-negative breast cancer; FAK: Focal adhesion kinase; ROS1: ROS proto-oncogene 1 receptor tyrosine kinase; TRX: Thioredoxin; ROS: Reactive oxygen species; NAC: N-acetylcysteine.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The authors thank B.J for assistance with the manuscript submission and L.Y for preparing figures. We thank State Key Laboratory for Medical Genomics of Ruijin Hospital for technical support.
Funding
This work was supported by the Key Project of National Natural Science Foundation of China (No. 82230088 to R.R.), National Natural Science Foundation of China (No. 81870112 to R.R.), Shanghai Science and Technology Development Funds (No. 20Z11900200 to R.R.), Shanghai Collaborative Innovation Program on Regenerative Medicine and Stem Cell Research (No. 2019CXJQ01 to R.R.), the Samuel Waxman Cancer Research Foundation (to R.R.) and the Innovative Research Team of High-level Local Universities in Shanghai (to R.R.).
Availability of data and materials
All data generated or analyzed during this study are available from the corresponding author on reasonable request.
Ethics Committee approval
Animal experiments were approved by the Ethics Committee of Shanghai Jiao Tong University School of Medicine.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kaurah P, MacMillan A, Boyd N, Senz J, De Luca A, Chun N. et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. Jama. 2007;297:2360-72
2. Desmedt C, Zoppoli G, Gundem G, Pruneri G, Larsimont D, Fornili M. et al. Genomic Characterization of Primary Invasive Lobular Breast Cancer. J Clin Oncol. 2016;34:1872-81
3. Oliveira C, Pinheiro H, Figueiredo J, Seruca R, Carneiro F. Familial gastric cancer: genetic susceptibility, pathology, and implications for management. Lancet Oncol. 2015;16:e60-e70
4. Bareche Y, Venet D, Ignatiadis M, Aftimos P, Piccart M, Rothe F. et al. Unravelling triple-negative breast cancer molecular heterogeneity using an integrative multiomic analysis. Ann Oncol. 2018;29:895-902
5. Blair VR, McLeod M, Carneiro F, Coit DG, D'Addario JL, van Dieren JM. et al. Hereditary diffuse gastric cancer: updated clinical practice guidelines. Lancet Oncol. 2020;21:e386-e97
6. Korde LA, Somerfield MR, Carey LA, Crews JR, Denduluri N, Hwang ES. et al. Neoadjuvant Chemotherapy, Endocrine Therapy, and Targeted Therapy for Breast Cancer: ASCO Guideline. J Clin Oncol. 2021;39:1485-505
7. Bajrami I, Marlow R, van de Ven M, Brough R, Pemberton HN, Frankum J. et al. E-Cadherin/ROS1 Inhibitor Synthetic Lethality in Breast Cancer. Cancer Discov. 2018;8:498-515
8. Shaw AT, Ou S-HI, Bang Y-J, Camidge DR, Solomon BJ, Salgia R. et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371:1963-71
9. Frampton JE. Entrectinib: A Review in NTRK+ Solid Tumours and ROS1+ NSCLC. Drugs. 2021;81:697-708
10. Trust RMNF. Crizotinib in Lobular Breast, Diffuse Gastric and Triple Negative Lobular Breast Cancer or CDH1-mutated Solid Tumours (ROLo). 2018.
11. Institute JB. Neoadjuvant Study of Targeting ROS1 in Combination With Endocrine Therapy in Invasive Lobular Carcinoma of the Breast (ROSALINE) (ROSALINE). 2020.
12. Lin JJ, Choudhury NJ, Yoda S, Zhu VW, Johnson TW, Sakhtemani R. et al. Spectrum of Mechanisms of Resistance to Crizotinib and Lorlatinib in Fusion-Positive Lung Cancer. Clin Cancer Res. 2021;27:2899-909
13. Wang Y, Tian P, Xia L, Li L, Han R, Zhu M. et al. The clinical efficacy of combinatorial therapy of EGFR-TKI and crizotinib in overcoming MET amplification-mediated resistance from prior EGFR-TKI therapy. Lung Cancer. 2020;146:165-73
14. Thiagarajan PS, Sinyuk M, Turaga SM, Mulkearns-Hubert EE, Hale JS, Rao V. et al. Cx26 drives self-renewal in triple-negative breast cancer via interaction with NANOG and focal adhesion kinase. Nat Commun. 2018;9:578
15. Pan MR, Hou MF, Ou-Yang F, Wu CC, Chang SJ, Hung WC. et al. FAK is Required for Tumor Metastasis-Related Fluid Microenvironment in Triple-Negative Breast Cancer. J Clin Med. 2019 8
16. Peng K, Li S, Li Q, Zhang C, Yuan Y, Liu M. et al. Positive Phospho-Focal Adhesion Kinase in Gastric Cancer Associates With Poor Prognosis After Curative Resection. Front Oncol. 2022;12:953938
17. Zhang H, Schaefer A, Wang Y, Hodge RG, Blake DR, Diehl JN. et al. Gain-of-Function Mutations Promote Focal Adhesion Kinase Activation and Dependency in Diffuse Gastric Cancer. Cancer Discov. 2020;10:288-305
18. Xu Q, Zhang J, Telfer BA, Zhang H, Ali N, Chen F. et al. The extracellular-regulated protein kinase 5 (ERK5) enhances metastatic burden in triple-negative breast cancer through focal adhesion protein kinase (FAK)-mediated regulation of cell adhesion. Oncogene. 2021;40:3929-41
19. Dawson JC, Serrels A, Stupack DG, Schlaepfer DD, Frame MC. Targeting FAK in anticancer combination therapies. Nat Rev Cancer. 2021;21:313-24
20. Le Large TYS, Bijlsma MF, El Hassouni B, Mantini G, Lagerweij T, Henneman AA. et al. Focal adhesion kinase inhibition synergizes with nab-paclitaxel to target pancreatic ductal adenocarcinoma. J Exp Clin Cancer Res. 2021;40:91
21. Pietilä EA, Gonzalez-Molina J, Moyano-Galceran L, Jamalzadeh S, Zhang K, Lehtinen L. et al. Co-evolution of matrisome and adaptive adhesion dynamics drives ovarian cancer chemoresistance. Nature communications. 2021;12:1-19
22. Shinde R, Terbuch A, Little M, Caldwell R, Kurup R, Riisnaes R. et al. Abstract CT143: Phase I study of the combination of a RAF-MEK inhibitor CH5126766 and FAK inhibitor defactinib in an intermittent dosing schedule with expansions in KRAS mutant cancers. Cancer Research. 2020;80:CT143-CT
23. Paradis JS, Acosta M, Saddawi-Konefka R, Kishore A, Gomes F, Arang N. et al. Synthetic Lethal Screens Reveal Cotargeting FAK and MEK as a Multimodal Precision Therapy for -Driven Uveal Melanoma. Clin Cancer Res. 2021;27:3190-200
24. Zhang B, Zhang Y, Zhang J, Liu P, Jiao B, Wang Z. et al. Focal Adhesion Kinase (FAK) Inhibition Synergizes with KRAS G12C Inhibitors in Treating Cancer through the Regulation of the FAK-YAP Signaling. Adv Sci (Weinh). 2021;8:e2100250
25. Mohanty A, Pharaon RR, Nam A, Salgia S, Kulkarni P, Massarelli E. FAK-targeted and combination therapies for the treatment of cancer: an overview of phase I and II clinical trials. Expert Opin Investig Drugs. 2020;29:399-409
26. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550
27. Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb). 2021;2:100141
28. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284-7
29. Dey A, Varelas X, Guan KL. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat Rev Drug Discov. 2020;19:480-94
30. Song X, Xu H, Wang P, Wang J, Affo S, Wang H. et al. Focal adhesion kinase (FAK) promotes cholangiocarcinoma development and progression via YAP activation. J Hepatol. 2021;75:888-99
31. Yuan T, Rafizadeh S, Azizi Z, Lupse B, Gorrepati KDD, Awal S. et al. Proproliferative and antiapoptotic action of exogenously introduced YAP in pancreatic cells. JCI Insight. 2016;1:e86326
32. Ji J, Chen W, Lian W, Chen R, Yang J, Zhang Q. et al. (S)-crizotinib reduces gastric cancer growth through oxidative DNA damage and triggers pro-survival akt signal. Cell Death Dis. 2018;9:660
33. Diaz Osterman CJ, Ozmadenci D, Kleinschmidt EG, Taylor KN, Barrie AM, Jiang S. et al. FAK activity sustains intrinsic and acquired ovarian cancer resistance to platinum chemotherapy. Elife. 2019 8
34. Jiang H, Liu X, Knolhoff BL, Hegde S, Lee KB, Jiang H. et al. Development of resistance to FAK inhibition in pancreatic cancer is linked to stromal depletion. Gut. 2020;69:122-32
35. InxMed (Shanghai) Co. L. IN10018 Monotherapy and Combination Therapy for Metastatic Melanoma. 2019
36. InventisBio Co. L. Study to Evaluate D-1553 in Combination With IN10018 in Subjects With Solid Tumors. 2022
37. Guo L, Gong H, Tang T-L, Zhang B-K, Zhang L-Y, Yan M. Crizotinib and Sunitinib Induce Hepatotoxicity and Mitochondrial Apoptosis in L02 Cells via ROS and Nrf2 Signaling Pathway. Front Pharmacol. 2021;12:620934
38. Zhang J, Li X, Han X, Liu R, Fang J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharmacol Sci. 2017;38:794-808
39. Idelchik M, Begley U, Begley TJ, Melendez JA. Mitochondrial ROS control of cancer. Semin Cancer Biol. 2017;47:57-66
40. Feng X, Arang N, Rigiracciolo DC, Lee JS, Yeerna H, Wang Z. et al. A Platform of Synthetic Lethal Gene Interaction Networks Reveals that the GNAQ Uveal Melanoma Oncogene Controls the Hippo Pathway through FAK. Cancer Cell. 2019 35
41. Shao D, Zhai P, Del Re DP, Sciarretta S, Yabuta N, Nojima H. et al. A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nature Communications. 2014;5:3315
Author contact
![]() Corresponding authors: Dr. Ruibao Ren, rbrenedu.cn; Dr. Baoyuan Zhang, yb57651edu.mo; Dr. Zaiqi Wang, zaiqi.wangcom.
Corresponding authors: Dr. Ruibao Ren, rbrenedu.cn; Dr. Baoyuan Zhang, yb57651edu.mo; Dr. Zaiqi Wang, zaiqi.wangcom.