Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
1. About NAFLD and NASH
2. Current advances of liver...
3. Customized liver organoids...
4. Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(11):3595-3613. doi:10.7150/ijbs.85145 This issue Cite
Review
Customized liver organoids as an advanced in vitro modeling and drug discovery platform for non-alcoholic fatty liver diseases
Dong Wook Han1,2,3,4* ![]() , KangHe Xu5*, Zhe-Long Jin1,2,4*, Yong-Nan Xu1,2, Ying-Hua Li1,2, Lin Wang3, Qilong Cao3, Kee-Pyo Kim6, DongHee Ryu5, Kwonho Hong7
, KangHe Xu5*, Zhe-Long Jin1,2,4*, Yong-Nan Xu1,2, Ying-Hua Li1,2, Lin Wang3, Qilong Cao3, Kee-Pyo Kim6, DongHee Ryu5, Kwonho Hong7 ![]() , Nam-Hyung Kim1,2,3,4
, Nam-Hyung Kim1,2,3,4 ![]()
1. Guangdong Provincial Key Laboratory of Large Animal Models for Biomedicine, School of Biotechnology and Health Sciences, Wuyi University, Jiangmen, China.
2. International Healthcare Innovation Institute (Jiangmen), Jianghai, Jiangmen, Guangdong Province, China.
3. Research and Development, Qingdao Haier Biotech Co. Ltd, Qingdao, China.
4. Guangdong ORGANOID Biotechnology Co. Ltd, Jiangmen, China.
5. Department of Surgery, College of Medicine, Chungbuk National University, Cheongju, Republic of Korea.
6. Department of Life Sciences, College of Medicine, The Catholic University of Korea, Seoul, Republic of Korea.
7. Department of Stem Cell and Regenerative Biotechnology, The institute of advanced regenerative science, Konkuk University, Seoul, Republic of Korea.
*These authors contributed equally to this work.
Received 2023-4-10; Accepted 2023-6-12; Published 2023-7-9
Abstract

Non-alcoholic fatty liver disease (NAFLD) and its progressive form non-alcoholic steatohepatitis (NASH) have presented a major and common health concern worldwide due to their increasing prevalence and progressive development of severe pathological conditions such as cirrhosis and liver cancer. Although a large number of drug candidates for the treatment of NASH have entered clinical trial testing, all have not been released to market due to their limited efficacy, and there remains no approved treatment for NASH available to this day. Recently, organoid technology that produces 3D multicellular aggregates with a liver tissue-like cytoarchitecture and improved functionality has been suggested as a novel platform for modeling the human-specific complex pathophysiology of NAFLD and NASH. In this review, we describe the cellular crosstalk between each cellular compartment in the liver during the pathogenesis of NAFLD and NASH. We also summarize the current state of liver organoid technology, describing the cellular diversity that could be recapitulated in liver organoids and proposing a future direction for liver organoid technology as an in vitro platform for disease modeling and drug discovery for NAFLD and NASH.
Keywords: non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), liver organoids, disease modeling, drug discovery
Introduction
The liver is the largest solid organ in the human body and plays critical roles in a wide range of physiological functions, including metabolism, detoxification, and protein production (1, 2). The liver is composed of several cell types, including (1) hepatocytes, the parenchymal cells of the liver (2); cholangiocytes, the epithelial cells of the bile ducts (3); Kupffer cells, the resident macrophages (4); hepatic stellate cells (HSCs) that store vitamin A and produce extracellular matrix (ECM); and (5) highly specialized endothelial cells known as liver sinusoidal endothelial cells (LSECs) (1, 3). The cell-to-cell communication among these cell types is essential for maintaining functional homeostasis of liver (4).
The liver is susceptible to many types of damage, and long-term liver damage can lead to chronic liver diseases (5). Among chronic liver diseases, non-alcoholic fatty liver disease (NAFLD), one of the most common liver diseases (6, 7), is characterized by the excessive accumulation of triglycerides in liver cells exceeding 5% of the liver's weight (8). While NAFLD can be a simple steatosis, it can potentially develop into non-alcoholic steatohepatitis (NASH), the most severe form of NAFLD (9). NASH can further develop into cirrhosis, liver cancer, and liver failure, and becomes a leading indication for liver transplantation (10). The global prevalence of NAFLD has been rapidly increasing and its current global prevalence is estimated to be 25% (11). Thus, NAFLD and NASH have become a major global health concern.
For the past couple of decades, many pharmaceutical companies have undertaken tremendous efforts for developing new drugs for NAFLD and NASH (12). However, these efforts have failed to achieve novel and effective treatment for NAFLD and NASH (13). Although animal models have enhanced our fundamental understanding of the pathogenesis of NAFLD and NASH, a growing body of evidence has suggested that animal models are not sufficient for translating scientific findings from animals into humans due to the fundamental genetic and physiological differences between animals and humans, necessitating the development of a human-specific model system (14, 15). Primary human hepatocytes (PHHs), a gold standard for hepatic research, have also faced large hurdles, such as their limited accessibility and rapid loss of functionality upon in vitro culture, impeding their industrial and clinical application (16, 17). As an alternative to PHHs, 2D hepatocyte-like cells generated from human pluripotent stem cells (hPSCs) and directly converted induced hepatocytes (iHeps) with defined factors have been also suggested (18-23). However, the relatively low functionality and proliferative capacity of these 2D hepatocyte-like cells has also hampered the translation of these cell types into the human setting (24). Furthermore, previous 2D cell types including PHH and 2D hepatocyte-like cells lack pro-fibrotic and pro-inflammatory cell types, which are critical for initiation and progression of NAFLD and NASH (15). Therefore, developing a human-specific model with liver tissue-like cell type composition and cytoarchitecture is direly needed for closely mirroring the pathogenesis of NAFLD and NASH.
To overcome the aforementioned issues of previous model systems, recent organoid technology was utilized for generating 3D liver tissue-like organoids (liver organoids) with improved structural and functional features. The stably expandable liver organoids displaying structural and functional similarities with liver tissue could be robustly generated from both liver biopsy and hPSCs (14, 25, 26). Moreover, recent advances have also described the presence of not only parenchymal cell types including both hepatocytes and cholangiocytes but also non-parenchymal cell types including HSCs and Kupffer cells (27, 28) with a functional bile canaliculi network in liver organoids (29). However, liver organoids generated using different protocols exhibit quite diverse structural and functional features with distinct cellular makeups. Considering the roles of each cell type (hepatocytes, cholangiocytes, HSCs, Kupffer cells, and LSECs) in the initiation and progression of NAFLD and NASH, the development of liver organoid models with in vivo-like cell type diversity is a prerequisite for future research examining in vitro modeling and drug discovery for NAFLD and NASH.
In the current study, we describe the role of each liver cell type and the cellular crosstalk among these diverse cell types in the pathogenesis of NAFLD and NASH. We next summarize current technical advances of liver organoid technology and compare the structural and functional features of liver organoids generated using distinct protocols. We also discuss the potential usefulness as well as the limitation of liver organoid technology as an in vitro modeling and drug screening platform for NAFLD and NASH. Finally, we propose a novel concept for utilizing customized liver organoids that properly recapitulate key inter-cellular, inter-tissue, and inter-organ communications for the pathogenesis of NAFLD and NASH. The distinct types of customized liver organoids such as monocellular liver organoids, multi-tissue liver organoids, and multi-organ liver organoids might represent a novel and suitable in vitro model system for unveiling the underlying mechanism of NAFLD and NASH and for discovering novel therapeutics.
1. About NAFLD and NASH
NAFLD is the most common liver disease worldwide, affecting approximately 25% of the world's population (30). The incidence has increased by 7.5% per year over 10 years, especially in young adults (<45 years) (31). Indeed, among adults aged 18 to 39 years, the prevalence of NAFLD has increased by about seven-fold (32). A total of 6% to 30% of individuals diagnosed with NAFLD by ultrasound could see their disease develop into NASH with biopsy confirmation (30), eventually leading to severe liver fibrosis, cirrhosis, and hepatocellular carcinoma (HCC) (10). Due to the high prevalence of NAFLD, the incidence of HCC associated with NASH has also been increasing and, subsequently, HCC has become the fourth leading cause of cancer death worldwide (33). Indeed, patients with NAFLD or NASH show a significantly higher incidence of HCC compared to unaffected individuals (30, 34). Thus, the number of registered patients requiring liver transplantation due to NASH has been rapidly increasing (35).
NAFLD is a spectrum of liver diseases characterized by hepatic steatosis without liver damage or inflammation, and it is usually associated with obesity (36). In contrast, NASH is a more severe form of NAFLD, with the main pathological features including steatosis, inflammation, and fibrosis (37). The pathogenesis of NAFLD without the influence of alcohol is complex and associated with multiple factors including genetic and epigenetic factors, metabolism, and the gut-liver axis, among others (38). Excessive lipid accumulation leads to hepatocyte lipotoxicity, which is an important link in the development of NAFLD (37). It was demonstrated that insulin resistance promotes the release of free fatty acids (FFA) from adipose tissue into the blood and that insulin induces the production of new FFA in the liver through new lipogenesis, where these FFA recombine to form triglycerides, the main component of fat accumulated in the liver (39). Insulin resistance also promotes adipose tissue dysfunction, which alters the production and secretion of adipokines and inflammatory cytokines (40). The damaged hepatocytes also secrete inflammatory cytokines and chemokines and their cellular contents, which activate Kupffer cells, resulting in a pro-inflammatory response and cellular immune infiltration, leading to the development of NASH (41). Once NAFLD has progressed to NASH, it further promotes insulin resistance in adipose tissue and the liver, which leads to a harmful cycle of insulin resistance, liver fat accumulation, and inflammation (39). The accumulation of fat in the liver in the form of triglycerides also leads to mitochondrial dysfunction, activation of oxidative stress, reactive oxygen species (ROS) production, and endoplasmic reticulum (ER) stress-related mechanisms, ultimately leading to hepatocyte death (41).
A growing body of evidence suggests that the cellular crosstalk among distinct cellular compartments in the liver is crucial for the initiation and progression of NAFLD and NASH. Although the underlying mechanism of NASH pathogenesis remains largely elusive, each non-parenchymal liver cell compartment, including Kupffer cells, HSCs, and LSECs, is known to trigger hepatocyte injury, inflammation, fibrosis, and vascular dysfunction. Here, we describe the role of each non-parenchymal cell type in the pathogenesis of NAFLD and NASH (Figure 1).
Role of each cell type in the liver during the pathogenesis of NAFLD and NASH. In a healthy liver, hepatocytes, cholangiocytes, Kupffer cells, HSCs, and LSECs play distinct roles in maintaining both structural and functional homeostasis. Upon injury, the cellular crosstalk among distinct hepatic cellular compartments leads to drastic structural and phenotypic alterations and to activation of cholangiocytes, LSECs, Kupffer cells, and HSCs into their relevant pathological states, triggering the pathogenesis of NAFLD and NASH.
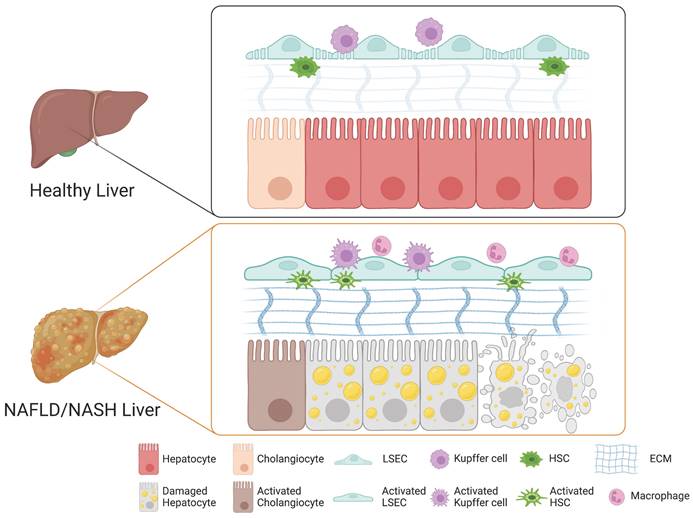
1) Role of Kupffer cells in NASH
Liver macrophages consist mainly of two populations, Kupffer cells and monocyte-derived macrophages (42). Kupffer cells are the resident macrophages in the lumen of hepatic sinusoids and account for 80-90% of colonized macrophages in the human body (43). Kupffer cells play a crucial role in regulating and maintaining immunity in the liver (43). Upon different stimuli, macrophages including Kupffer cells and monocyte-derived macrophages undergo phenotypic differentiation into either classically activated M1 macrophages or alternatively activated M2 macrophages (44). M1 macrophages with a high antigen presentation capacity produce diverse pro-inflammatory cytokines including TNFα, IL-1B, CCL2, and CCL5 and promote macrophage-mediated tissue damage. In contrast, M2 macrophages are involved in anti-inflammatory responses and tissue repair via balancing the activity of M1 macrophages (45). In NAFLD, the balance between pro-inflammatory M1 Kupffer cells and anti-inflammatory M2 Kupffer cells is critical for modulating the initiation and progression of liver injury (46). Indeed, a previous study demonstrated that the selective cell death of M1 Kupffer cells leads to M2 Kupffer cell-mediated protection of alcoholic liver injury (47). Therefore, balancing pro-inflammatory M1 Kupffer cells and anti-inflammatory M2 Kupffer cells would be an alternative strategy for blocking further progression of NAFLD (47).
In the case of NASH, damaged hepatocytes lead to activation of Kupffer cells and infiltration of circulating monocyte-derived macrophages (Figure 1) (48). The activated macrophages produce pro-inflammatory cytokines that induce the activation and transdifferentiation of HSCs and also influence the physiological functions of other cellular compartments such as LSECs and other immune cells (49, 50). The crosstalk between Kupffer cells and hepatocytes is bidirectional (41). The damaged hepatocytes release their cellular contents including damage-associated molecular patterns (DAMPs), contributing the activation of Kupffer cells (51). The activated Kupffer cells produce diverse cytokines, contributing to hepatic lipid deposition and hepatocyte death (52). On the other hand, Kupffer cells also exert their roles in the elimination of apoptotic hepatocytes via efferocytosis, by which DAMP-mediated inflammation can be ameliorated by clearing DMAP-producing damaged hepatocytes (53). An increasing body of evidence suggests the crucial roles of Kupffer cells in the progression and regression of NASH.
2) Role of HSCs in NASH
HSCs represent 5-8% of all liver cells (54). HSCs reside in the space of Disse, a thin perisinusoidal area between sinusoidal endothelial cells and hepatocytes (55). Under normal physiological conditions, HSCs are in a quiescent state and store vitamin A in lipid droplets (56). Upon liver injury, quiescent HSCs undergo phenotypic switch toward activated HSCs, which are proliferative, migrative, contractile, and fibrogenic, with progressive loss of their vitamin A-storing activity (57). Although HSCs play a central role in the deposition of extracellular matrix (ECM) (58), an intricate macromolecular structural network forming a scaffold for adhesion (59), the deposition and remodeling of ECM in the space of Disse, a key factor for liver fibrosis, is rather orchestrated by cellular crosstalk among multiple cell types (60). Kupffer cells and Kupffer cell-derived cytokines and chemokines play a crucial role in the activation of HSCs and transdifferentiation of these cells into myofibroblasts, subsequently leading to liver fibrosis (61). The release of TGF-β, the most important fibrogenic cytokine, from Kupffer cells activates HSCs via multiple SMAD proteins (62). The Sonic hedgehog pathway is another driving force for HSC activation (63). The accumulation of fat as a form of triglyceride, in the liver causes mitochondrial dysfunction, activation of oxidative stress, production of ROS, and an ER-stress-related mechanism, all contributing to the activation of HSCs (41). Besides these intracellular signaling pathways, other extracellular factors including nutrients, alcohol, and other toxic compounds delivered with portal blood flow to the space of Disse are also known to be involved in HSC activation (64).
In a healthy liver, quiescent HSCs produce collagen IV and VI into the space of Disse, contributing to ECM homeostasis by providing the proper scaffold for architecture and function (60). During liver injury, quiescent HSCs become activated and transdifferentiated into myofibroblasts, with loss of lipid droplets, increased cellular proliferation, and development of mature rough ER to support the production of ECM fibers and matrix remodeling enzymes (65). Activated HSCs start to produce excessive amounts of ECM, mostly collagens I and III, affecting the mechanical characteristics of tissue and the stiffness of the ECM, characteristically encountered during liver fibrosis (Figure 1) (66). The remodeling of ECM impairs the exchange of nutrients and activates the immune system (67). The overproduction of ECM further activates HSCs and contributes to loss of endothelial fenestrations of LSECs, further exacerbating liver fibrosis (68). Thus, the balance between deposition and remodeling of ECM is the key driver for progression of liver fibrosis. Progressive liver fibrosis is known to contribute to the scarring of the liver, which can further develop into cirrhosis, an end-stage liver disease (69).
3) Role of LSECs in NASH
In contrast to other blood vessels, the hepatic sinusoids are specialized vascular structures that lack a basement membrane and instead are lined by fenestrated (porous) endothelial cells, so-called LSECs (70). LSECs have both sinusoidal and abluminal sides and can communicate with hepatocytes and HSCs through the abluminal side (71). Under normal physiological conditions, the fenestrae on LSECs mediate the exchange of plasma, nutrients, lipids, and lipoproteins between the sinusoidal lumen and the space of Disse, allowing only particles smaller than the fenestrae to reach the parenchymal cells (72). Moreover, in a healthy liver, LSECs prevent HSC activation and promote reversion to a quiescent state through VEGF-stimulated nitric oxide (NO) production (73). Upon liver injury, however, fenestrae on LSECs undergo drastic changes in their structural and functional properties (74). Defenestration, a reduction in the number and diameter of fenestrae and formation of a continuous basement membrane of LSECs, so-called capillarization, are characteristics typical of chronic liver diseases (Figure 1) (60). While the structural modification of fenestrae can protect the liver from further damage by restricting toxins approaching the parenchymal cells, the defenestration on LSECs influences hepatocytes by creating a microenvironment lacking nutrients and oxygen (75). The lack of nutrients and oxygen from the blood flow impacts the physiological function of hepatocytes and could lead to further progression of liver injury (75). LSECs also indirectly promote liver fibrosis by secreting pro-inflammatory and pro-fibrotic factors (76). Defenestration, the main characteristic of activated LSECs, precedes fibrogenesis in the liver (77). Defenestration and capillarization of LSECs due to liver injury promote the activation of HSCs, contributing to liver fibrosis through loss of VEGF-stimulated NO production (60). A previous study showed that capillarization of LSECs blocks the transfer of chylomicron remnants to hepatocytes, leading to cholesterol and triglyceride synthesis, promoting steatosis (78). Thus, both remodeling of ECM and structural changes of LSECs such as defenestration and capillarization induce hepatocyte apoptosis, promoting the activation of the immune system and chronic inflammation (Figure 1). Subsequently, chronic inflammation could lead to fibrosis, cirrhosis, and even end-stage HCC (79).
4) Role of cholangiocytes in NASH
Hepatocytes and cholangiocytes are two main cell types in the liver. While cholangiocytes were mainly known to modulate bile secretion, they have become increasingly recognized for their impact on biliary and liver diseases (80). A previous study has suggested that steatosis can promote biliary senescence and liver fibrosis during cholestasis (81). Cholestasis is a liver disease caused by the reduction or stoppage of bile flow from the liver into the bile duct, leading to hepatic bile acid accumulation and subsequent damage (82). Under normal physiological conditions, cholangiocytes are mitotically quiescent (83). Under pathological conditions, however, they become proliferative, pro-inflammatory, pro-fibrotic, or senescent in response to damage such as cholestasis (84). Senescent cholangiocytes are known to play an important role in liver inflammation and fibrosis during cholestatic liver injury via secretion of cytokines and fibrotic factors such as TGF-β1 and IL-6 (80). TGF-β1 is known to activate HSCs and induce their transdifferentiation into myofibroblasts, contributing to liver fibrosis (62). It was previously described that cellular senescence and damage to cholangiocytes in patients with NAFLD and NASH increase with the progression of hepatic steatosis (85). Thus, senescence of cholangiocytes may be an important factor in the progression of NAFLD and NASH.
2. Current advances of liver organoid technology
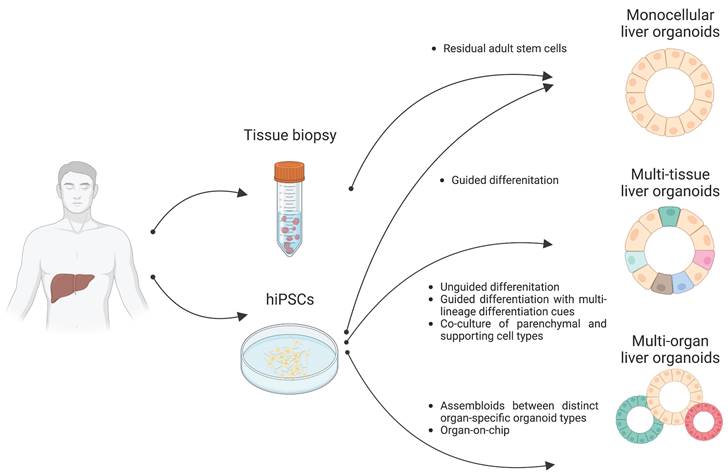
Organoids are 3D multicellular aggregates derived from diverse in vitro and in vivo sources including pluripotent stem cells, multipotent tissue-specific stem cells, and tissue biopsy containing adult stem cells or differentiated cells via cell-to-cell and cell-to-matrix interactions (86, 87). Organoids exhibit structural and functional features comparable to their tissue of origin (24, 88). Early liver organoids typically comprised a monocellular epithelial type such as parenchymal cells (e.g., hepatocytes or cholangiocytes) (14, 25, 26). Recently, multi-tissue liver organoids containing both parenchymal cell types and non-parenchymal supporting cell types such as Kupffer cells and HSCs have been also described (27, 28). Furthermore, recent progress has reported the production of multi-organ organoids interconnecting distinct organ domains in an individual organoid structure, which may be a useful source for understanding liver organogenesis (89) (Figure 2).
Distinct types of liver organoids. Monocellular liver organoids such as HOs and COs can be generated from either liver tissue or hPSCs. Multi-tissue liver organoids containing both parenchymal and non-parenchymal cell types can be generated by multiple ways such as by unguided differentiation, guided differentiation with multi-lineage differentiation cues, and coculture of parenchymal- and non-parenchymal-supporting cell types. Multi-organ liver organoids with proper inter-organ crosstalk can also be generated by either assembloid or organ-on-chip technology.
As we discussed in the previous section, the pathogenesis and progression of NAFLD and NASH are mediated by a tight cellular crosstalk among distinct liver cell types. Therefore, to establish liver organoid-based human-specific models for NAFLD and NASH, we should clearly understand the current technical situation of liver organoid technology. In this section, we revisit current protocols for producing monocellular epithelial liver organoids, multi-tissue liver organoids, and multi-organ liver organoids in terms of their origins, cell types, self-renewal capacity, and functionality.
1) Liver organoids from primary tissues
Following the breakthrough report demonstrating the successful production of intestinal organoids (90, 91), studies have reported the production of liver epithelial organoids from primary human liver tissues (14, 92). Minced tissue fragments or even single cell-dissociated primary hepatocytes and cholangiocytes have successfully produced liver epithelial organoids representing the tissue-specific characteristics of either hepatocytes or cholangiocytes, resulting in the establishment of self-renewing hepatocyte organoids (HOs) and cholangiocyte organoids (COs), respectively (14, 93) (Figure 2).
COs can be further segregated into intrahepatic COs (ICOs) and extrahepatic COs (ECOs) based on their tissue of origin (94-96). Organoids typically closely resemble the molecular, functional, and morphological features of the tissues from which they are derived (97). However, COs display more dynamic cellular plasticity than organoids from other tissues (95). Indeed, mouse ICOs express not only cholangiocyte markers but also markers for progenitors and hepatocytes, suggesting their bipotential characteristics (92). Like mouse ICOs, human ICOs exhibit the concomitant expression of progenitor, hepatocyte, and cholangiocyte markers (14). Upon further differentiation, human ICOs acquire mature hepatocyte features, as evidenced by a series of in vitro functionality assays such as the secretion of albumin and bile acid, glycogen storage activity, and potential for detoxification and drug metabolism (14). In contrast to ICOs, ECOs could not activate hepatocyte-specific transcriptional program even with the same culture condition that can induce the transdifferentiation of ICOs into a hepatic state, showing the distinct cellular plasticity of COs based on their tissue of origin (94, 96, 98).
The establishment of stably expandable HOs from both primary mouse and human liver cells has also been demonstrated (99). HOs from both mouse and human exhibit morphology and gene expression patterns typical of hepatocytes but not cholangiocytes (93). Like ICOs, mouse HOs exhibit bipotential transdifferentiation capacity into either a hepatocyte or cholangiocyte state based upon the culture conditions (100). In contrast to human COs and mouse HOs, HOs from human liver cells show limited self-renewal capacity, necessitating the development of a new culture condition supporting the long-term expansion of human HOs (93). Nevertheless, HOs from both mouse and human are functionally mature, as shown by morphology typical of HOs, in vitro functionality, and the presence of bile canaliculi network (93).
2) Liver organoids from hPSCs
Due to the limited accessibility of primary tissue, pluripotent stem cells (PSCs) including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) have been highlighted as alternative sources for organoid production (101, 102). Indeed, the production of organoids with structural and functional similarities to distinct human organs have been well documented over the past decade. Furthermore, in contrast to adult tissue-derived organoids, PSC-derived organoids hold great benefits for patient-specific disease modeling and drug discovery (101).
A. Monocellular liver organoids from PSCs
There are also several protocols available for differentiating PSCs into HOs (25, 28, 29, 103, 104). In contrast to HOs from primary liver tissue, PSC-derived HOs are morphologically similar to COs from primary tissues, as they grow in epithelial cyst form (25, 103-105). PSC-derived HOs are stably expandable in vitro, and display key hepatic functions upon maturation (25, 103-105). Furthermore, their usefulness as an in vitro disease modeling platform has been validated for steatosis and citrullinemia, a rare autosomal recessive genetic disorder (25, 105). Using a novel 3D protocol, Guan et al. showed the production of HOs with interestingly diverse morphologies (104). Their HOs are composed of either hepatocytes, cholangiocytes, or a mixture of both hepatocytes and cholangiocytes with self-renewal capacity. The HOs are functionally mature, as evidenced by glycogen storage, drug metabolism, and secretion of albumin and bile acids (103). Moreover, this previous work employed the researchers' HOs for modeling Alagille syndrome, a rare genetic disorder characterized by paucity of bile ducts, and patient-derived HOs were found to exhibit fewer duct-like structures as in the patients' liver (104).
The differentiation of PSCs including both ESCs and iPSCs into COs has also been described (106-108). PSC-derived COs are morphologically and functionally similar to COs from primary tissue and also form branched tubular structures (106). PSC-derived COs with key cholangiocyte functions could be used for modeling genetic diseases that cause hepatobiliary complications, such as cystic fibrosis and Alagille syndrome (109, 110). Furthermore, pathological phenotypes could be rescued by pharmacological intervention, suggesting that liver organoids are a promising tool for not only disease modeling but also drug screening.
Recent studies have successfully recapitulated the functional interconnection between hepatocytes and cholangiocytes (29, 111). These recent works utilized pre-differentiated hepatoblasts as a starting population to achieve the robust and homogeneous production of HOs. The generated HOs displayed a unique structural feature in which a dense hepatic core is surrounded by multiple biliary cysts. Both hepatocytes and cholangiocytes in HOs were found to be functional, as shown by the secretion of albumin and apolipoprotein B and activity of gamma glutamyl transferase and alkaline phosphatase, respectively. Furthermore, both the hepatic and bile duct parts are functionally interconnected through a bile canaliculi network, and this unique and advanced structure is suitable for modeling drug-induced cholestasis (29).
Collectively, different types of liver organoids (e.g., HOs, COs, and even HOs with functional interconnection between hepatic and biliary structures) could be generated from PSCs, and each type of liver organoid represents a suitable in vitro model system for studying distinct liver diseases.
B. Multi-tissue liver organoids from PSCs
The first step of liver organogenesis is the formation of the liver bud, a condensed structure in which primitive hepatic endoderm cells from the foregut endodermal sheet delaminate and invade the septum transversum mesenchyme, which is the source of HSCs as well as LSECs that begin to form vessels (112-114). These dynamic morphogenetic changes are orchestrated by nascent endothelial cells and adjunct cardiac mesoderm. Using classical-guided differentiation protocols, PSCs normally produce cell types from a singular germ layer, despite their pluripotency (115). In contrast, unguided differentiation protocols based on intrinsic differentiation signals allow for differentiation of PSCs into relatively diverse cell types from multiple germ layers (116). The differentiation procedure of liver organoids is highly defined and composed of multiple differentiation steps tightly guided by signal pathways (117). Both hepatocytes and cholangiocytes originate from the same source, hepatoblasts, that could be differentiated from definitive endoderm (118, 119). However, mesoderm is the common origin of non-parenchymal cells such as Kupffer cells, HSCs, and LSECs (120-122). Thus, achieving multi-tissue liver organoids containing both endoderm-derived parenchymal cells (e.g., hepatocytes and cholangiocytes) and mesoderm-derived non-parenchymal cells (e.g., Kupffer cells, HSCs, and LSECs) is theoretically and technically challenging.
By recapitulating organogenetic interactions between endothelial and mesenchymal cells in vitro, Takebe et al. (123) have successfully demonstrated the generation of multi-tissue liver organoids, so-called liver bud containing both endoderm- and mesoderm-derived cell types by coculturing iPSC-derived hepatic endodermal cells with human umbilical vein endothelial cells and mesenchymal stem cells (Figure 2). This in vitro reconstructed liver bud displays similarity with the in vivo liver bud in terms of gene expression patterns and vascularized structures. Upon transplantation into a cranial window model, the human vasculatures in the engrafted liver bud could functionally be connected to the host vessels, contributing to the functional maturation of the engrafted liver bud.
Instead of coculturing parenchymal cells and non-parenchymal supporting cell types, Ouchi et al. induced the concomitant differentiation of PSCs into multiple germ layers (28). Although the researchers' protocol is based on guided differentiation, they tried to implement a mesodermal differentiation cue by adding retinoic acid, which plays dual roles for both parenchymal and non-parenchymal cell specification (Figure 2). The resultant multi-tissue liver organoids contain not only parenchymal hepatocytes and cholangiocytes but also non-parenchymal supporting cells such as Kupffer cells and HSCs under the same culture conditions. Interestingly, both Kupffer cells and HSCs in these multi-tissue liver organoids are functional, as demonstrated by proper immune response against inflammatory stimuli and vitamin A storage activity, respectively. Furthermore, both the activation of HSCs and excessive production of ECM could be observed under steatosis conditions. Taken together, these results show that multi-tissue liver organoids containing non-parenchymal supporting cell types may be an advanced platform for closely mirroring the in vivo scenario, allowing for more precise disease modeling and drug screening for liver diseases.
C. Multi-organ liver organoids from PSCs
Close interactions among distinct organs are essential for maintaining the diverse vital functions of the human body. For example, the liver is tightly linked to the pancreas, and the aberrant interactions between the two organs may lead to dysregulated glucose levels and metabolic disorders such as type 2 diabetes mellitus (124). Thus, recapitulating inter-organ interactions in vitro is important for closely simulating the real in vivo situation as well as discovering potential therapeutics for diseases such as type 2 diabetes mellitus mediated by altered inter-organ interactions. To this end, several previous studies have attempted to reproduce the in vivo interactions between distinct tissue compartments using organoid technology. Indeed, recent advances in organoid technology have successfully demonstrated the generation of assembloids between distinct brain organoids representing different parts of brain tissues (125). Moreover, the potential usefulness of assembloid technology has been well described in previous studies (126, 127) wherein pathological outputs that could be mediated by the interactions among distinct parts of brain were successfully recapitulated in patient-derived assembloids.
Recently, the generation of multi-organ liver organoids with functional interconnection to biliary and pancreatic domains was also demonstrated by fusing anterior gut spheroids with posterior gut spheroids derived from human PSCs (89). Previous research conducted by Koike et al. involved the fusion of anterior and posterior spheroids to generate boundary organoids containing multi-endoderm domains. Through cell-to-cell communication, hepato-biliary-pancreatic (HBP) progenitors expressing HHEX and PDX1 emerged at the interface of the fused spheroids. The specification of HBP progenitors in the fused spheroids was found to be critically influenced by retinoic acid signals. Isolated HBP progenitor domains from the fused spheroids, when cultured for an extended period, developed into mature organoids known as HBP organoids. The long-term culture of HBP organoids demonstrated the presence of multiple organ domains comprising hepato-biliary-pancreatic domains, with an interconnected functioning between the pancreas and bile duct. It is worth noting that the abolishment of HES1, a transcription factor regulating the posterior foregut lineage (128, 129), could lead to the conversion of biliary tissue into pancreatic tissue in mice (130, 131), with an increased number of pancreatic structures observed in HES1-deficient HBP organoids, suggesting the potential utility of multi-organ liver organoids for closely recapitulating human organogenesis and facilitating in vitro disease modeling (89).
Combined with state-of-art tissue engineering technologies, such as organ-on-chip technology, inter-organ interactions could also be recapitulated in liver organoids. Indeed, a recent study has successfully demonstrated the recapitulation of human liver-pancreatic islet axis using a microfluidic multi-organoid system (132). Dynamic interaction between two organoids, as evidenced by glucose-stimulated insulin secretion from islet organoids and altered glucose utilization in liver organoids, suggests the potential usefulness of multi-organ liver organoids for both studying the complex pathogenesis of metabolic disorders and developing new therapeutics (132). Further efforts for recapitulating more complex and mature inter-organ crosstalk in liver organoids are required for an advanced platform for in vitro disease modeling and drug discovery.
3. Customized liver organoids for distinct therapeutic targets
For the study of NAFLD and NASH, animal models, particularly murine models, have been extensively employed to elucidate the underlying mechanisms of pathogenesis. However, the use of larger animal models such as rabbits, minipigs, and monkeys, which are more similar to humans, has been limited due to ethical concerns, difficulties in handling, time requirements, and high costs (133, 134). Three primary types of murine models, namely genetic, dietary, and combination models, have been widely used for replicating the pathophysiology of human NAFLD and NASH (Table 1) (135). An ideal animal model for NAFLD/NASH should exhibit not only steatosis (accumulation of fat in the liver) but also steatohepatitis phenotypes, including inflammation and fibrosis (136). Furthermore, considering the close association between metabolic disorders and NAFLD/NASH, an ideal animal model should replicate the metabolic abnormalities observed in NAFLD/NASH patients (137-140). Genetic models, such as SREBP-1c transgenic mice and PTEN null mice, exhibit phenotypes charactertistic of both steatosis and steatohepatitis (141-143). On the other hand, genetic models like ob/ob mice, db/db mice, and KK-Ay mice primarily display steatosis phenotypes, without progressing to steatohepatitis in the absence of additional factors like a high-fat diet (144-148). This discrepancy in pathological patterns among different animal models suggests that the diverse outcomes may hinder the translation of findings from animal models to clinical applications. Dietary animal models, on the other hand, succeed in relatively replicating steatohepatitis phenotypes (149), although the specific pathological outcomes vary depending on species, strain, and gender (150). To overcome these challenges, combination models, which involve feeding specific diets to genetic murine models, have been employed and have demonstrated close resemblance to human diseases (135, 151, 152). Nevertheless, it is important to acknowledge the significant species-specific differences between human and animal livers, which may impede the clinical translation of discoveries made in animal models. Furthermore, animal models often fail to fully recapitulate the entire spectrum of NAFLD and NASH observed in humans (135, 153, 154).
Comparison of animal models, 2D monolayer cell culture models, and 3D liver organoid models.
| Model | Species | Type | Phenotype reproduced | Advantages | Disadvantages |
|---|---|---|---|---|---|
| Animal models | Animal | SREBP-1c transgenic mice (141, 212) | Steatosis, Insulin resistance, Inflammation, Fibrosis | -Physiological environment -Immune system -Multiple cell types -Functionality -Structure -Metabolism | -Non-human species -Low-throughput -High cost -Heterogeneous phenotypes (species, strain, gender) |
| PTEN null mice (142, 143) | Steatosis, Fibrosis | ||||
| Ob/ob mice (144, 213, 214) Db/db mice (146, 147, 215) | Obesity, Steatosis, Insulin resistance, (Inflammation, Fibrosis) | ||||
| MAT1A null mice (216-218) | Steatosis, Fibrosis | ||||
| High-fat diet mice (150, 219, 220) | Obesity, Steatosis, Hepatic insulin resistance, Oxidative stress, Inflammation, Fibrosis | ||||
| Methionine- and choline-deficient diet mice (221-223) | Steatosis, Hepatic Insulin resistance, Inflammation, Oxidative stress, Mitochondrial damage, Apoptosis, Fibrosis | ||||
| Cholesterol and cholate diet mice (224) | Steatosis, Hepatic Insulin resistance, Inflammation, Oxidative stress, Fibrosis | ||||
| 2D monolayer cell culture models | Human | PHHs (155, 225) | Steatosis, ER stress, Apoptosis | -Low cost -Easy handling -High-throughput compatibility -Easy downstream application | -Limited physiological environment -Absence of non-parenchymal cell types - Absence of cell-to-cell and cell-to-matrix interactions -Low hepatic maturity |
| HepG2 (159, 226, 227) | Steatosis, Apoptosis, | ||||
| HuH 7 (159, 160, 228) | Steatosis, ER stress, Apoptosis, | ||||
| HepaRG (161, 229, 230) | Steatosis, Oxidative stress | ||||
| PSC-hepatocyte-like cells (162, 231) | Steatosis | ||||
| PSC-HSCs (232) | Inflammation, Activation of HSCs | ||||
| 3D liver organoid models | Human | Spheroids (3D coculture) (233-235) | Steatosis, Activation of HSCs, Oxidative stress, Apoptosis, Inflammation, Expression of profibrotic markers, Mitochondrial dysfunction | -Complex structural organization -Long-term expansion -Multiple cell types -High/mid-throughput compatibility -Semi-physiological environment -Immune system | -Complicated differentiation steps -Relatively high cost -Heterogeneity (batch variation) -Limited hepatic maturity |
| Monocellular liver organoids (from primary tissue) (176, 236, 237) | Mainly steatosis | ||||
| Multi-tissue liver organoids (198, 199, 238) | Steatosis, HSC activation, Ductular reaction, Oxidative stress, Bile canaliculi disruption, Expression of profibrotic markers, Collagen secretion and deposition |
For in vitro modeling of NAFLD and NASH, the 2D monolayer cell culture system using singular hepatic cell types has been widely adopted due to its relatively low cost, easy handling, and high- compatibility with high-throughput applications (Table 1). Due to the limited availability and rapid loss of functionality of PHHs, alternative cell sources, including immortalized cell lines (HepG2, HuH-7, and HepaRG) and hPSC-derived 2D hepatocyte-like cells, have been proposed as substitutes for PHHs. Previous studies (155-162) have successfully demonstrated that the 2D monolayer cell culture system could successfully recapitulate hallmarks of NAFLD and NASH, including cytoplasmic accumulation of triglycerides in hepatocytes, ER stress, inflammation, and cell death. Consequently, the 2D monolayer cell culture system has been used for evaluating drug-induced hepatotoxicity and the efficacy of therapeutic compounds. However, the 2D monolayer cell culture models have limitations, as they lack non-parenchymal cell types, which play a key role in the pathogenesis of NAFLD and NASH (163). Additionally, altered metabolic functionality has been another challenging issue of 2D monolayer cell culture models (164). Furthermore, the inability to replicate cell-to-cell and cell-to-matrix interactions as well as biomolecular gradients present in tissues, hinders the accurate representation of physiological conditions in the 2D monolayer cell culture system (24). Therefore, more sophisticated culture systems that mimic in vivo-like cellular compartments and spatial organization are required. Coculturing hepatocytes with the missing non-parenchymal cell types has shown significant improvements in the functional aspects of in vitro NAFLD and NASH models (165-167). However, further efforts are needed for obtaining a sufficient number of highly pure and functional non-parenchymal cell populations, as well as optimizing the coculture condition for maintaining distinct cell types using a singular medium condition, before coculture models can be fully established.
As we discussed in the first section, the cellular crosstalk among distinct cellular compartments including both parenchymal and non-parenchymal cell types is critical for the pathogenesis of NAFLD and NASH. Indeed, each cell type plays crucial and diverse roles for the induction and progression of liver diseases. For this reason, PHHs and 2D hepatocyte-like cells are insufficient for reproducing the cellular crosstalk, as they lack pro-inflammatory and pro-fibrotic cell types, which are critical for initiation and progression of NAFLD and NASH (24, 168). Animal models also show fundamental genetic and physiological differences with humans (169). Therefore, recent liver organoids that exhibit structural and functional similarity to liver tissue may represent an advanced and attractive human-specific model system (27, 28). However, as we described in the second section, substantial differences in current liver organoid technologies contribute to the variability of generated liver organoids in terms of origin, cellular compartment, functionality, self-renewal capacity, and most importantly capacity for reproducing inter-cellular, inter-tissue, and inter-organ communications. Although this variability may be a hurdle to overcome for achieving the standardized production of highly uniform organoids, it also provides a great opportunity for utilizing customized liver organoids with distinct inter-cellular, inter-tissue, and inter-organ communications as a novel in vitro model system for precisely predicting the efficacy of diverse drug candidates targeting distinct therapeutic targets. In this last section, we discuss how to utilize distinct types of liver organoids including monocellular epithelial liver organoids, multi-tissue liver organoids, and multi-organ liver organoids for unveiling the mechanism underlying the pathogenesis of NAFLD and NASH as well as for evaluating the efficacy of drug candidates targeting distinct therapeutic mechanisms.
1) Driving force to NAFLD/NASH and related therapeutic targets
Currently, lifestyle modification has been the first-line treatment for preventing and controlling NAFLD and NASH (170). However, lifestyle intervention is not a viable treatment option in patients with advanced fibrosis or cirrhosis (171). Thus, the development of novel treatments that could effectively and safely reverse NASH symptoms including fibrosis are urgently demanded. Although lipogenesis, by the increased delivery of FFAs from diet and adipose tissue into the liver or the increased de novo lipogenesis, has been considered as a major factor for the pathogenesis of NAFLD, growing evidence suggests that NASH is rather mediated by the synergistic interaction among multiple concomitant factors such as genetic variants, metabolic disorders, oxidative stress, altered immune response, and even the disruption of the gut-liver axis (41). Considering this complex mechanism underlying the development of NASH, diverse treatment strategies for NASH with distinct therapeutic targets are currently under preclinical and clinical trial testing.
2) Customized organoids for each therapeutic target
As the outcomes of current liver organoid technology look quite diverse in terms of cellular compartments, appropriately generated liver organoids containing the right cell types with proper inter-cellular, inter-tissue, and inter-organ crosstalk are prerequisites for evaluating the efficacy of each drug candidate targeting distinct pathological mechanisms. Here we suggest that different types of liver organoids are theoretically suitable for evaluating current drug candidates in different stages of clinical trials based on their target mechanism such as de novo lipogenesis, metabolism, cellular stress, inflammation, and fibrosis. Based on this, we suggest a concept of utilizing customized liver organoids with distinct inter-cellular, inter-tissue, or inter-organ interactions, which might have a great potential for evaluating drug candidates targeting distinct mechanisms underlying the development and progression of NAFLD and NASH.
A. Therapeutic targets requiring monocellular epithelial liver organoids
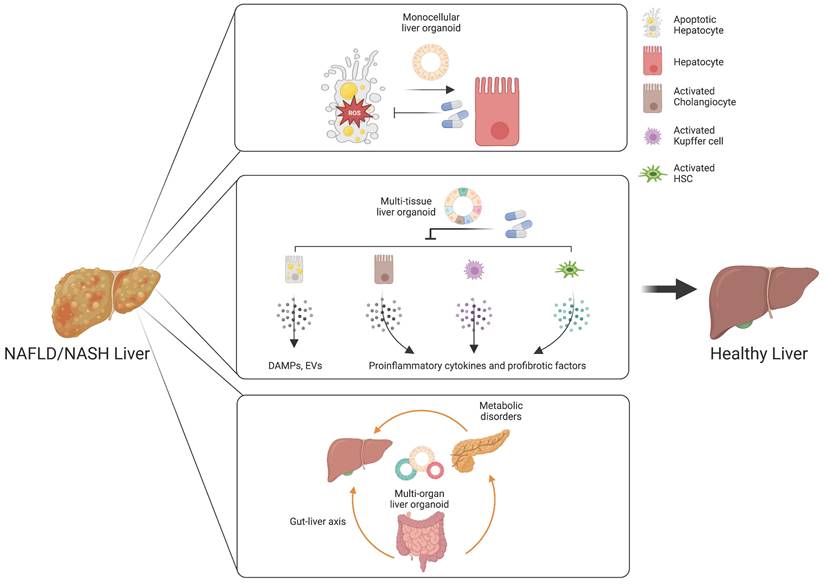
Monocellular epithelial liver organoids, such as HOs and COs both from tissue biopsy and PSCs, consist mostly of a single cell type (14, 25, 26). Although epithelial HOs and COs recapitulate the structural features of liver tissue in a relatively limited way compared with multi-tissue liver organoids, they can self-renew and become fully functional upon further maturation (25). Despite the limited cellular diversity of monocellular epithelial liver organoids, these organoids can be a useful model for efficacy evaluation for some drug candidates targeting de novo lipogenesis, anti-cellular stress, and hepatic cell death (Figure 3) (172).
Customized liver organoids for evaluating the efficacy of drugs targeting distinct therapeutic targets. Monocellular liver organoids can be a useful model for drug candidates targeting de novo lipogenesis, anti-cellular stress, and hepatic cell death. Multi-tissue liver organoids are suitable for drugs targeting an inflammatory response and hepatocyte-derived DAMPS or EVs. Multi-organ liver organoids may serve as a great model for understanding the influences of the gut-liver axis and metabolic disorders in the pathogenesis of NAFLD and NASH.
De novo lipogenesis is the primary factor associated with fatty liver (173). Indeed, increased de novo lipogenesis is observed in 20% to ~30% of patients with NAFLD and NASH compared to unaffected individuals (174). Both key transcription factors such as SREBP-1c (sterol regulatory element-binding protein 1c) and ChREBP (carbohydrate regulatory element-binding protein) and enzymes including acetyl-CoA carboxylase (ACC) and fatty acid synthase are involved in de novo lipogenesis and serve as potential therapeutic targets (173). Currently, diverse drug candidates are in both preclinical and clinical trial testing; these candidates inhibit: 1) ATP-citrate lyase (ACLY), a cytoplasmic enzyme responsible for the generation of acetyl-coenzyme A (acetyl-CoA); 2) ACC, which converts acetyl-CoA to malonyl-CoA; 3) FAS, a rate-controlling enzyme that converts malonyl-CoA into palmitic acid during de novo lipogenesis; 4) SCD1 (stearoyl coenzyme A desaturase 1), an enzyme that catalyzes the rate-limiting step in the formation of monosaturated fatty acids; 5) SREBP-1c, an insulin-sensitive transcription factor that plays a key role in the induction of lipogenic genes in the liver; and 6) SREBP2, a key transcription factor regulating expression of genes involved in cholesterol biosynthesis (175). Although all the inhibitors aim at distinct targets during lipogenesis, they all require a liver organoid model by which the distinct steps of lipogenesis can be effectively inhibited for reducing hepatic de novo lipogenesis and steatosis. Thus, monocellular epithelial liver organoids such as HOs and COs may be a suitable liver organoid model for drug candidates targeting lipogenesis. Indeed, monocellular epithelial liver organoids from human fetal liver have recently been used for in vitro modeling of steatosis and for finding potential drug candidates for NAFLD. In a study by Hendriks et al. (176), 17 candidate NAFLD drugs from recent drug development programs were screened using monocellular epithelial liver organoids. Among them, inhibitors of ACC, FAS, DGAT2 (diacylglycerol O-acyltransferase 2), FXR (Farnesoid X receptor) agonists, and recombinant FGF19 were found to effectively reduce the steatosis phenotype, thus supporting the concept that monocellular epithelial liver organoids serve as a suitable model for drug candidates targeting de novo lipogenesis.
Oxidative stress, a condition by which the generation of highly toxic ROS exceeds the capacity of antioxidants to detoxify, is also a major factor in the pathogenesis of several chronic diseases including NAFLD (177). The accumulation of lipids leads to overproduction of ROS and stress in mitochondria and ER, contributing to inflammation, cellular injury, and cell death (178). Several enzymes involved in the detoxification process of ROS and diverse antioxidants including vitamin C (ascorbic acid), vitamin A (retinol), and vitamin E (tocopherol) may be a therapeutic target for reducing ROS (179). Currently, several antioxidants including vitamin E are under investigation to address their therapeutic effects for NAFLD and NASH (180). Therefore, monocellular epithelial liver organoids including HOs and COs may also be a desirable model for evaluating the efficacy of antioxidants as drug candidates for NAFLD and NASH.
NASH is also characterized by hepatocyte injury, and thus cell death seems to be an important factor in the progression of NAFLD and NASH (181). It has also been suggested that distinct cell death mechanisms including apoptosis, necroptosis, pyroptosis, ferroptosis, and autophagy are associated with progression of NAFLD and NASH (182). Among the distinct cell death pathways, apoptosis is the most common and best-characterized cell death pathway in NASH (183). Indeed, diverse anti-apoptotic agents including Emricasan, Selonsertib, and Rapamycin attenuate inflammation and reverse fibrosis during preclinical studies (184-186). Besides apoptosis, a growing body of evidence suggests that necroptosis, pyroptosis, and ferroptosis may play a crucial role in the development of NAFLD and NASH, thus expanding the potential therapeutic targets to diverse cell death pathways (182). Again, monocellular epithelial liver organoids may be an appropriate model for evaluating anti-cell death agents.
B. Therapeutic targets requiring multi-tissue liver organoids
An inflammatory response is a prerequisite for the initiation and progression of NAFLD and NASH (187). Immune cells and pro-inflammatory cytokines play crucial roles in the pathogenesis of NAFLD and NASH (188). As we already described in the first section, Kupffer cells are the key cell type for regulating and maintaining immunity in the liver. Upon liver injury, the damaged hepatocytes lead to activation of Kupffer cells and infiltration of circulating monocyte-derived macrophages (189). The activated macrophages produce a plethora of pro-inflammatory cytokines, resulting in the activation of HSCs (61). Activated HSCs are also known to produce pro-inflammatory chemokines that attract monocytes into the injured liver (190). In pathological conditions, cholangiocytes also play an important role in liver inflammation via secretion of cytokines and fibrotic factors (191). Hepatocyte-derived DAMPs are also known to be a major mediator for immune response (192). Moreover, the tight crosstalk among distinct cellular compartments is critical for the ignition and further progression of NAFLD and NASH, as previously discussed. Taken together, these findings show that the inflammatory response produced during the initiation and progression of NAFLD and NASH is orchestrated by multiple cellular compartments and their tight crosstalk in the liver. In this case, multi-tissue liver organoids containing major pro-inflammatory cell types including DAMP-producing hepatocytes, activated cholangiocytes, activated Kupffer cells, and activated HSCs would be beneficial for recapitulating the in vivo pro-inflammatory immune response and for evaluating the therapeutic efficacy of drug candidates (Figure 3).
The damaged hepatocytes and activated Kupffer cells lead to the activation and transdifferentiation of HSCs into ECM-producing myofibroblasts (193). Thus, multiple drug candidates either directly affecting ECM synthesis and turnover or indirectly influencing HSC activation via inhibition of TGF-β, the most potent fibrotic factor, are currently under preclinical and clinical investigation (194). In this case, multi-tissue liver organoids containing Kupffer cells and HSCs might also be a useful model.
Extracellular vesicles (EVs) have been highlighted as a novel therapeutic target for NAFLD and NASH (195). EVs play an important role in intercellular communication as a signaling mediator between liver and other organs by carrying various bioactive molecules including lipids, proteins, DNA, and RNAs (195). The increased level of hepatocyte-derived EVs correlates with the severity of NASH, and thus the protein and mRNA composition of these EVs may be a useful biomarker for the diagnosis of NAFLD and NASH (196). In NAFLD and NASH, EVs lead to lipid accumulation and activation of macrophages and HSCs, promoting inflammation and fibrosis (197). Therefore, EVs may serve as new targets for the treatment of NAFLD and NASH. In this sense, multi-tissue liver organoids containing both donors and recipients of EVs would be a suitable model for unveiling the role of EVs in the pathogenesis of NAFLD and NASH.
Recently, Ouchi et al. (198) established a multi-tissue liver organoid-based steatosis model using either FFA exposure or Wolman disease patient-derived iPSCs and successfully replicated typical symptoms of NAFLD and NASH, including lipid accumulation, triglyceride production, hepatocyte ballooning, increased stiffness, proliferation of HSCs, production of inflammatory cytokine, recruitment of immune cells, and enhanced fibrosis. Treatment with recombinant FGF19 was found to signicantly reduce the pathological hallmarks of NASH in the multi-tissue liver organoid-based steatosis model, such as lipid accumulation, hepatocyte damage, stiffness, and ROS production. Another study by Guan et al. (199) used similar multi-tissue liver organoids to model autosomal recessive polycystic kidney disease (ARPKD), a monogenic disorder that causes liver fibrosis. Severe fibrosis was observed in ARPKD liver organoids, mediated by the activation of HSCs, and was efficiently ameliorated by treatment with PDGFR tyrosine kinase inhibitors (Crenolanib, Sunitinib, Imatinib). These previous reports strongly support the notion that multi-tissue liver organoids provide a suitable model for replicating typical symptoms of NAFLD and NASH, as well as for evaluating the efficacy of drug candidates targeting therapeutic mechanisms mediated by inter-tissue interactions.
C. Therapeutic targets requiring multi-organ liver organoids
The gut-liver axis supports bidirectional interactions between the liver and gastrointestinal tract, where trillions of microorganisms form the gut microbiota (200). The bile acids synthesized from the liver influence the composition and function of the gut microbiota in the gastrointestinal tract (201). On the other side, the gut microbiota and its metabolites regulate the synthesis of bile acids and hepatic lipid metabolism (202). The gastrointestinal tract, the largest mammalian-microbial interface, regulates symbiotic interactions between the host and microorganisms (203). In a physiologically healthy condition, the gastrointestinal epithelial barrier supports digestion, immunity, and metabolic function (204). Another important role of the intestinal barrier entails preventing the entrance of harmful intestinal bacteria and their metabolites into the circulation (205). However, increased epithelial permeability mediated by disruption of the gastrointestinal epithelial barrier leads to bacterial translocation, which promotes hepatic inflammation and oxidative stress, contributing to the pathogenesis of NAFLD and NASH (206). Multi-organ liver organoids may serve as a great in vitro model for reproducing the gut-liver axis and for developing a novel class of drugs targeting a disturbed gut-liver axis (Figure 3).
NAFLD and NASH are closely associated with metabolic disorders including type 2 diabetes mellitus (207). The global prevalence of NAFLD and NASH in patients with type 2 diabetes mellitus surpasses 55% (208). Currently, diverse antidiabetic drugs for NAFLD and NASH are in clinical trial testing and are exhibiting promising efficacy (209). Similarly, multi-organ organoids that reproduce the pathology of both metabolic disorders and chronic liver diseases may be a useful model for evaluating the efficacy of antidiabetic drug candidates.
4. Conclusion
The scientific society has reached a consensus that liver organoid technology is an advanced and suitable human-specific model system for closely recapitulating the liver in vitro. Nevertheless, many hurdles remain to be addressed before translating liver organoid technology into the clinic and industrial settings. First, many protocols have failed to fully reproduce all the cellular compartments in the liver. Although recent advances have described the generation of multi-tissue liver organoids containing relatively diverse cell types (27, 28), the presence of functional LSECs with their proper structural and functional features has yet to be reported. Moreover, creating liver organoids containing all the parenchymal and non-parenchymal cell types in a ratio similar to liver tissue is paramount for recapitulating the cellular crosstalk that occurs in vivo. Second, there is no standardized protocol for liver organoid production, perhaps appropriating much time and valuable resources of many laboratories for simply reproducing some leading protocols. Indeed, the resultant liver organoids generated by different labs display quite distinct features, even with similar differentiation guiding cues, bringing the issue of reproducibility to the forefront. Third, as with other types of organoids, the variation between batches of liver organoids or even between individual liver organoids is a critical consideration impeding the translation of liver organoid technology. Fourth, the unique structure of the liver has not been demonstrated in vitro. Although multiple studies have successfully described the structural similarity of liver organoids with liver tissue by demonstrating the presence of functional bile canaliculi networks within liver organoids (25, 29, 103, 104), the production of liver organoids showing the typical hexagonal hepatic lobules consisting of a portal triad remains challenging. Nevertheless, liver organoids represent an alternative technology with the potential for addressing and overcoming the diverse limitations of preexisting animal models and 2D cell culture systems toward an enhanced understanding of the mechanisms underlying liver diseases as well as drug discovery and efficacy and safety testing (24).
Of course, a future direction of liver organoid technology should aim to fully reproduce the inter-cellular, inter-tissue, and even inter-organ communications that occur in the human body. Microfluidic-based organ-on-chip technology, together with standardized liver organoid production technology, may facilitate this concept (210, 211). However, current liver organoid technology is able to recapitulate only relatively limited crosstalk across distinct cell types, tissues, and organs. We therefore suggest to utilize distinct types of liver organoids such as monocellular epithelial organoids, multi-tissue organoids, and multi-organ organoids that reflect the distinct causes of NAFLD and NASH to facilitate our fundamental understanding of underlying disease mechanisms as well as accelerate the development of effective treatments for NAFLD and NASH.
Acknowledgements
This work was supported by Scientific Research of High-level Talents of Wuyi University, China (2019TP007), Guangdong Provincial Key Laboratory of Large Animal Models of Biomedicine, China (2021B1212040016), and also by the Department of Education of Guangdong Province, China (2021ZDZX2046). All the figures were created with BioRender.com.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Trefts E, Gannon M, Wasserman DH. The liver. Curr Biol. 2017;27(21):R1147-R51
2. Halpern KB, Shenhav R, Matcovitch-Natan O, Toth B, Lemze D, Golan M. et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature. 2017;542(7641):352-6
3. StemBook. Cambridge (MA)2008
4. Kmiec Z. Cooperation of liver cells in health and disease. Adv Anat Embryol Cell Biol. 2001;161:III-XIII 1-151
5. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol. 2021;18(3):151-66
6. Shetty A, Giron F, Divatia MK, Ahmad MI, Kodali S, Victor D. Nonalcoholic Fatty Liver Disease after Liver Transplant. J Clin Transl Hepatol. 2021;9(3):428-35
7. Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM. et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology. 2015;148(3):547-55
8. Kneeman JM, Misdraji J, Corey KE. Secondary causes of nonalcoholic fatty liver disease. Therap Adv Gastroenterol. 2012;5(3):199-207
9. Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol. 2013;10(11):656-65
10. Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332(6037):1519-23
11. Younossi ZM, Golabi P, Paik JM, Henry A, Van Dongen C, Henry L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): a systematic review. Hepatology. 2023
12. Reimer KC, Wree A, Roderburg C, Tacke F. New drugs for NAFLD: lessons from basic models to the clinic. Hepatol Int. 2020;14(1):8-23
13. Baran B, Akyuz F. Non-alcoholic fatty liver disease: what has changed in the treatment since the beginning? World J Gastroenterol. 2014;20(39):14219-29
14. Huch M, Gehart H, van Boxtel R, Hamer K, Blokzijl F, Verstegen MM. et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell. 2015;160(1-2):299-312
15. Kruitwagen HS, Oosterhoff LA, Vernooij I, Schrall IM, van Wolferen ME, Bannink F. et al. Long-Term Adult Feline Liver Organoid Cultures for Disease Modeling of Hepatic Steatosis. Stem Cell Reports. 2017;8(4):822-30
16. Schwartz RE, Fleming HE, Khetani SR, Bhatia SN. Pluripotent stem cell-derived hepatocyte-like cells. Biotechnol Adv. 2014;32(2):504-13
17. Elaut G, Henkens T, Papeleu P, Snykers S, Vinken M, Vanhaecke T. et al. Molecular mechanisms underlying the dedifferentiation process of isolated hepatocytes and their cultures. Curr Drug Metab. 2006;7(6):629-60
18. Hannan NR, Segeritz CP, Touboul T, Vallier L. Production of hepatocyte-like cells from human pluripotent stem cells. Nat Protoc. 2013;8(2):430-7
19. Kim JH, Wang M, Lee J, Park HJ, Han C, Hong HS. et al. Prediction of hepatotoxicity for drugs using human pluripotent stem cell-derived hepatocytes. Cell Biol Toxicol. 2018;34(1):51-64
20. Huang P, Zhang L, Gao Y, He Z, Yao D, Wu Z. et al. Direct reprogramming of human fibroblasts to functional and expandable hepatocytes. Cell Stem Cell. 2014;14(3):370-84
21. Sekiya S, Suzuki A. Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature. 2011;475(7356):390-3
22. Lim KT, Lee SC, Gao Y, Kim KP, Song G, An SY. et al. Small Molecules Facilitate Single Factor-Mediated Hepatic Reprogramming. Cell Rep. 2016;15(4):814-29
23. Lim KT, Kim J, Hwang SI, Zhang L, Han H, Bae D. et al. Direct Conversion of Mouse Fibroblasts into Cholangiocyte Progenitor Cells. Stem Cell Reports. 2018;10(5):1522-36
24. Prior N, Inacio P, Huch M. Liver organoids: from basic research to therapeutic applications. Gut. 2019;68(12):2228-37
25. Mun SJ, Ryu JS, Lee MO, Son YS, Oh SJ, Cho HS. et al. Generation of expandable human pluripotent stem cell-derived hepatocyte-like liver organoids. J Hepatol. 2019;71(5):970-85
26. Wang S, Wang X, Tan Z, Su Y, Liu J, Chang M. et al. Human ESC-derived expandable hepatic organoids enable therapeutic liver repopulation and pathophysiological modeling of alcoholic liver injury. Cell Res. 2019;29(12):1009-26
27. Guan Y, Enejder A, Wang M, Fang Z, Cui L, Chen SY. et al. A human multi-lineage hepatic organoid model for liver fibrosis. Nat Commun. 2021;12(1):6138
28. Ouchi R, Togo S, Kimura M, Shinozawa T, Koido M, Koike H. et al. Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 2019;30(2):374-84 e6
29. Ramli MNB, Lim YS, Koe CT, Demircioglu D, Tng W, Gonzales KAU. et al. Human Pluripotent Stem Cell-Derived Organoids as Models of Liver Disease. Gastroenterology. 2020;159(4):1471-86 e12
30. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73-84
31. Kanwal F, Kramer JR, Duan Z, Yu X, White D, El-Serag HB. Trends in the Burden of Nonalcoholic Fatty Liver Disease in a United States Cohort of Veterans. Clin Gastroenterol Hepatol. 2016;14(2):301-8 e1-2
32. Allen AM, Therneau TM, Larson JJ, Coward A, Somers VK, Kamath PS. Nonalcoholic fatty liver disease incidence and impact on metabolic burden and death: A 20 year-community study. Hepatology. 2018;67(5):1726-36
33. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394-424
34. Wild SH, Walker JJ, Morling JR, McAllister DA, Colhoun HM, Farran B. et al. Cardiovascular Disease, Cancer, and Mortality Among People With Type 2 Diabetes and Alcoholic or Nonalcoholic Fatty Liver Disease Hospital Admission. Diabetes Care. 2018;41(2):341-7
35. Shingina A, DeWitt PE, Dodge JL, Biggins SW, Gralla J, Sprague D. et al. Future Trends in Demand for Liver Transplant: Birth Cohort Effects Among Patients With NASH and HCC. Transplantation. 2019;103(1):140-8
36. Lazo M, Hernaez R, Eberhardt MS, Bonekamp S, Kamel I, Guallar E. et al. Prevalence of nonalcoholic fatty liver disease in the United States: the Third National Health and Nutrition Examination Survey, 1988-1994. Am J Epidemiol. 2013;178(1):38-45
37. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24(7):908-22
38. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. 2016;65(8):1038-48
39. Marjot T, Moolla A, Cobbold JF, Hodson L, Tomlinson JW. Nonalcoholic Fatty Liver Disease in Adults: Current Concepts in Etiology, Outcomes, and Management. Endocr Rev. 2020 41(1)
40. Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9(5):367-77
41. Kim KH, Lee MS. Pathogenesis of Nonalcoholic Steatohepatitis and Hormone-Based Therapeutic Approaches. Front Endocrinol (Lausanne). 2018;9:485
42. Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. 2014;60(5):1090-6
43. Chen J, Deng X, Liu Y, Tan Q, Huang G, Che Q. et al. Kupffer Cells in Non-alcoholic Fatty Liver Disease: Friend or Foe? Int J Biol Sci. 2020;16(13):2367-78
44. Zhou D, Yang K, Chen L, Wang Y, Zhang W, Xu Z. et al. Macrophage polarization and function: new prospects for fibrotic disease. Immunol Cell Biol. 2017;95(10):864-9
45. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate Immunity and Inflammation in NAFLD/NASH. Dig Dis Sci. 2016;61(5):1294-303
46. Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol. 2013;3(2):785-97
47. Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F. et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology. 2014;59(1):130-42
48. Horst AK, Tiegs G, Diehl L. Contribution of Macrophage Efferocytosis to Liver Homeostasis and Disease. Front Immunol. 2019;10:2670
49. Ioannou GN, Subramanian S, Chait A, Haigh WG, Yeh MM, Farrell GC. et al. Cholesterol crystallization within hepatocyte lipid droplets and its role in murine NASH. J Lipid Res. 2017;58(6):1067-79
50. Li H, Zhou Y, Wang H, Zhang M, Qiu P, Zhang M. et al. Crosstalk Between Liver Macrophages and Surrounding Cells in Nonalcoholic Steatohepatitis. Front Immunol. 2020;11:1169
51. Ibrahim SH, Hirsova P, Gores GJ. Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut. 2018;67(5):963-72
52. Oates JR, McKell MC, Moreno-Fernandez ME, Damen M, Deepe GS Jr, Qualls JE. et al. Macrophage Function in the Pathogenesis of Non-alcoholic Fatty Liver Disease: The Mac Attack. Front Immunol. 2019;10:2893
53. Mihm S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int J Mol Sci. 2018 19(10)
54. Wang M, Li L, Xu Y, Du J, Ling C. Roles of hepatic stellate cells in NAFLD: From the perspective of inflammation and fibrosis. Front Pharmacol. 2022;13:958428
55. Tacke F, Weiskirchen R. Update on hepatic stellate cells: pathogenic role in liver fibrosis and novel isolation techniques. Expert Rev Gastroenterol Hepatol. 2012;6(1):67-80
56. Mallat A, Lotersztajn S. Cellular mechanisms of tissue fibrosis. 5. Novel insights into liver fibrosis. Am J Physiol Cell Physiol. 2013;305(8):C789-99
57. Zisser A, Ipsen DH, Tveden-Nyborg P. Hepatic Stellate Cell Activation and Inactivation in NASH-Fibrosis-Roles as Putative Treatment Targets? Biomedicines. 2021 9(4)
58. Karin D, Koyama Y, Brenner D, Kisseleva T. The characteristics of activated portal fibroblasts/myofibroblasts in liver fibrosis. Differentiation. 2016;92(3):84-92
59. Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK. Extracellular matrix structure. Adv Drug Deliv Rev. 2016;97:4-27
60. Sanz-García C, Fernández-Iglesias A, Gracia-Sancho J, Arráez-Aybar LA, Nevzorova YA, Cubero FJ. The Space of Disse: The Liver Hub in Health and Disease. Livers. 2021
61. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14(7):397-411
62. Dewidar B, Meyer C, Dooley S, Meindl-Beinker AN. TGF-beta in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells. 2019 8(11)
63. Li H, Yang L, Sun Y, Zhang Y, Chai J, Liu B. et al. Silencing of CD147 inhibits hepatic stellate cells activation related to suppressing aerobic glycolysis via hedgehog signaling. Cytotechnology. 2021;73(2):233-42
64. Acharya P, Chouhan K, Weiskirchen S, Weiskirchen R. Cellular Mechanisms of Liver Fibrosis. Front Pharmacol. 2021;12:671640
65. Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev. 2017;121:27-42
66. Lee UE, Friedman SL. Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol. 2011;25(2):195-206
67. Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15(12):786-801
68. Natarajan V, Harris EN, Kidambi S. SECs (Sinusoidal Endothelial Cells), Liver Microenvironment, and Fibrosis. Biomed Res Int. 2017;2017:4097205
69. Lo RC, Kim H. Histopathological evaluation of liver fibrosis and cirrhosis regression. Clin Mol Hepatol. 2017;23(4):302-7
70. Ni Y, Li JM, Liu MK, Zhang TT, Wang DP, Zhou WH. et al. Pathological process of liver sinusoidal endothelial cells in liver diseases. World J Gastroenterol. 2017;23(43):7666-77
71. Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D. et al. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J Hepatol. 2017;66(1):212-27
72. Szafranska K, Kruse LD, Holte CF, McCourt P, Zapotoczny B. The wHole Story About Fenestrations in LSEC. Front Physiol. 2021;12:735573
73. Deleve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology. 2008;48(3):920-30
74. Warren A, Bertolino P, Benseler V, Fraser R, McCaughan GW, Le Couteur DG. Marked changes of the hepatic sinusoid in a transgenic mouse model of acute immune-mediated hepatitis. J Hepatol. 2007;46(2):239-46
75. Cai J, Hu M, Chen Z, Ling Z. The roles and mechanisms of hypoxia in liver fibrosis. J Transl Med. 2021;19(1):186
76. Lafoz E, Ruart M, Anton A, Oncins A, Hernandez-Gea V. The Endothelium as a Driver of Liver Fibrosis and Regeneration. Cells. 2020 9(4)
77. DeLeve LD. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology. 2015;61(5):1740-6
78. Hammoutene A, Rautou PE. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J Hepatol. 2019;70(6):1278-91
79. Dhar D, Baglieri J, Kisseleva T, Brenner DA. Mechanisms of liver fibrosis and its role in liver cancer. Exp Biol Med (Maywood). 2020;245(2):96-108
80. Meadows V, Baiocchi L, Kundu D, Sato K, Fuentes Y, Wu C. et al. Biliary Epithelial Senescence in Liver Disease: There Will Be SASP. Front Mol Biosci. 2021;8:803098
81. Kennedy L, Meadows V, Sybenga A, Demieville J, Chen L, Hargrove L. et al. Mast Cells Promote Nonalcoholic Fatty Liver Disease Phenotypes and Microvesicular Steatosis in Mice Fed a Western Diet. Hepatology. 2021;74(1):164-82
82. Reichel C, Meier-Abt PJ. [Cholestatic liver diseases]. Ther Umsch. 1997;54(11):639-44
83. Alpini G, Ueno Y, Tadlock L, Glaser SS, LeSage G, Francis H. et al. Increased susceptibility of cholangiocytes to tumor necrosis factor-alpha cytotoxicity after bile duct ligation. Am J Physiol Cell Physiol. 2003;285(1):C183-94
84. Ferreira-Gonzalez S, Lu WY, Raven A, Dwyer B, Man TY, O'Duibhir E. et al. Paracrine cellular senescence exacerbates biliary injury and impairs regeneration. Nat Commun. 2018;9(1):1020
85. Natarajan SK, Ingham SA, Mohr AM, Wehrkamp CJ, Ray A, Roy S. et al. Saturated free fatty acids induce cholangiocyte lipoapoptosis. Hepatology. 2014;60(6):1942-56
86. Huch M, Koo BK. Modeling mouse and human development using organoid cultures. Development. 2015;142(18):3113-25
87. Simian M, Bissell MJ. Organoids: A historical perspective of thinking in three dimensions. J Cell Biol. 2017;216(1):31-40
88. Kretzschmar K, Clevers H. Organoids: Modeling Development and the Stem Cell Niche in a Dish. Dev Cell. 2016;38(6):590-600
89. Koike H, Iwasawa K, Ouchi R, Maezawa M, Giesbrecht K, Saiki N. et al. Modelling human hepato-biliary-pancreatic organogenesis from the foregut-midgut boundary. Nature. 2019;574(7776):112-6
90. Fordham RP, Yui S, Hannan NR, Soendergaard C, Madgwick A, Schweiger PJ. et al. Transplantation of expanded fetal intestinal progenitors contributes to colon regeneration after injury. Cell Stem Cell. 2013;13(6):734-44
91. Mustata RC, Vasile G, Fernandez-Vallone V, Strollo S, Lefort A, Libert F. et al. Identification of Lgr5-independent spheroid-generating progenitors of the mouse fetal intestinal epithelium. Cell Rep. 2013;5(2):421-32
92. Huch M, Dorrell C, Boj SF, van Es JH, Li VS, van de Wetering M. et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature. 2013;494(7436):247-50
93. Hu H, Gehart H, Artegiani B, C LO-I, Dekkers F, Basak O. et al. Long-Term Expansion of Functional Mouse and Human Hepatocytes as 3D Organoids. Cell. 2018;175(6):1591-606 e19
94. Verstegen MMA, Roos FJM, Burka K, Gehart H, Jager M, de Wolf M. et al. Human extrahepatic and intrahepatic cholangiocyte organoids show region-specific differentiation potential and model cystic fibrosis-related bile duct disease. Sci Rep. 2020;10(1):21900
95. Sampaziotis F, Muraro D, Tysoe OC, Sawiak S, Beach TE, Godfrey EM. et al. Cholangiocyte organoids can repair bile ducts after transplantation in the human liver. Science. 2021;371(6531):839-46
96. Rimland CA, Tilson SG, Morell CM, Tomaz RA, Lu WY, Adams SE. et al. Regional Differences in Human Biliary Tissues and Corresponding In vitro-Derived Organoids. Hepatology. 2021;73(1):247-67
97. Marsee A, Roos FJM, Verstegen MMA, Consortium HPBO, Gehart H, de Koning E. et al. Building consensus on definition and nomenclature of hepatic, pancreatic, and biliary organoids. Cell Stem Cell. 2021;28(5):816-32
98. Spence JR, Lange AW, Lin SC, Kaestner KH, Lowy AM, Kim I. et al. Sox17 regulates organ lineage segregation of ventral foregut progenitor cells. Dev Cell. 2009;17(1):62-74
99. Peng WC, Logan CY, Fish M, Anbarchian T, Aguisanda F, Alvarez-Varela A. et al. Inflammatory Cytokine TNFalpha Promotes the Long-Term Expansion of Primary Hepatocytes in 3D Culture. Cell. 2018;175(6):1607-19 e15
100. Wang X, Ni C, Jiang N, Wei J, Liang J, Zhao B. et al. Generation of liver bipotential organoids with a small-molecule cocktail. J Mol Cell Biol. 2020;12(8):618-29
101. Azar J, Bahmad HF, Daher D, Moubarak MM, Hadadeh O, Monzer A. et al. The Use of Stem Cell-Derived Organoids in Disease Modeling: An Update. Int J Mol Sci. 2021 22(14)
102. McCauley HA, Wells JM. Pluripotent stem cell-derived organoids: using principles of developmental biology to grow human tissues in a dish. Development. 2017;144(6):958-62
103. Wu F, Wu D, Ren Y, Huang Y, Feng B, Zhao N. et al. Generation of hepatobiliary organoids from human induced pluripotent stem cells. J Hepatol. 2019;70(6):1145-58
104. Guan Y, Xu D, Garfin PM, Ehmer U, Hurwitz M, Enns G. et al. Human hepatic organoids for the analysis of human genetic diseases. JCI Insight. 2017 2(17)
105. Akbari S, Sevinc GG, Ersoy N, Basak O, Kaplan K, Sevinc K. et al. Robust, Long-Term Culture of Endoderm-Derived Hepatic Organoids for Disease Modeling. Stem Cell Reports. 2019;13(4):627-41
106. Dianat N, Dubois-Pot-Schneider H, Steichen C, Desterke C, Leclerc P, Raveux A. et al. Generation of functional cholangiocyte-like cells from human pluripotent stem cells and HepaRG cells. Hepatology. 2014;60(2):700-14
107. Banales JM, Masyuk TV, Gradilone SA, Masyuk AI, Medina JF, LaRusso NF. The cAMP effectors Epac and protein kinase a (PKA) are involved in the hepatic cystogenesis of an animal model of autosomal recessive polycystic kidney disease (ARPKD). Hepatology. 2009;49(1):160-74
108. Lee SO, Masyuk T, Splinter P, Banales JM, Masyuk A, Stroope A. et al. MicroRNA15a modulates expression of the cell-cycle regulator Cdc25A and affects hepatic cystogenesis in a rat model of polycystic kidney disease. J Clin Invest. 2008;118(11):3714-24
109. Ogawa M, Ogawa S, Bear CE, Ahmadi S, Chin S, Li B. et al. Directed differentiation of cholangiocytes from human pluripotent stem cells. Nat Biotechnol. 2015;33(8):853-61
110. Sampaziotis F, de Brito MC, Madrigal P, Bertero A, Saeb-Parsy K, Soares FAC. et al. Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation. Nat Biotechnol. 2015;33(8):845-52
111. Tanimizu N, Ichinohe N, Sasaki Y, Itoh T, Sudo R, Yamaguchi T. et al. Generation of functional liver organoids on combining hepatocytes and cholangiocytes with hepatobiliary connections ex vivo. Nat Commun. 2021;12(1):3390
112. Asahina K, Zhou B, Pu WT, Tsukamoto H. Septum transversum-derived mesothelium gives rise to hepatic stellate cells and perivascular mesenchymal cells in developing mouse liver. Hepatology. 2011;53(3):983-95
113. Collardeau-Frachon S, Scoazec JY. Vascular development and differentiation during human liver organogenesis. Anat Rec (Hoboken). 2008;291(6):614-27
114. Ober EA, Lemaigre FP. Development of the liver: Insights into organ and tissue morphogenesis. J Hepatol. 2018;68(5):1049-62
115. Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132(4):661-80
116. Watabe T, Miyazono K. Roles of TGF-beta family signaling in stem cell renewal and differentiation. Cell Res. 2009;19(1):103-15
117. Thompson WL, Takebe T. Human liver model systems in a dish. Dev Growth Differ. 2021;63(1):47-58
118. Olgasi C, Cucci A, Follenzi A. iPSC-Derived Liver Organoids: A Journey from Drug Screening, to Disease Modeling, Arriving to Regenerative Medicine. Int J Mol Sci. 2020 21(17)
119. Gordillo M, Evans T, Gouon-Evans V. Orchestrating liver development. Development. 2015;142(12):2094-108
120. Matsumoto K, Yoshitomi H, Rossant J, Zaret KS. Liver organogenesis promoted by endothelial cells prior to vascular function. Science. 2001;294(5542):559-63
121. Tasnim F, Xing J, Huang X, Mo S, Wei X, Tan MH. et al. Generation of mature kupffer cells from human induced pluripotent stem cells. Biomaterials. 2019;192:377-91
122. Coll M, Perea L, Boon R, Leite SB, Vallverdu J, Mannaerts I. et al. Generation of Hepatic Stellate Cells from Human Pluripotent Stem Cells Enables In vitro Modeling of Liver Fibrosis. Cell Stem Cell. 2018;23(1):101-13 e7
123. Takebe T, Sekine K, Suzuki Y, Enomura M, Tanaka S, Ueno Y. et al. Self-organization of human hepatic organoid by recapitulating organogenesis in vitro. Transplant Proc. 2012;44(4):1018-20
124. Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med. 2017;23(7):804-14
125. Sloan SA, Andersen J, Pasca AM, Birey F, Pasca SP. Generation and assembly of human brain region-specific three-dimensional cultures. Nat Protoc. 2018;13(9):2062-85
126. Bagley JA, Reumann D, Bian S, Levi-Strauss J, Knoblich JA. Fused cerebral organoids model interactions between brain regions. Nat Methods. 2017;14(7):743-51
127. Miura Y, Li MY, Birey F, Ikeda K, Revah O, Thete MV. et al. Generation of human striatal organoids and cortico-striatal assembloids from human pluripotent stem cells. Nat Biotechnol. 2020;38(12):1421-30
128. Spence JR, Lange AW, Lin S-CJ, Kaestner KH, Lowy AM, Kim I. et al. Sox17 regulates organ lineage segregation of ventral foregut progenitor cells. Developmental cell. 2009;17(1):62-74
129. Thamm K, Seaver EC. Notch signaling during larval and juvenile development in the polychaete annelid Capitella sp I. Developmental biology. 2008;320(1):304-18
130. Fukuda A, Kawaguchi Y, Furuyama K, Kodama S, Horiguchi M, Kuhara T. et al. Ectopic pancreas formation in Hes1-knockout mice reveals plasticity of endodermal progenitors of the gut, bile duct, and pancreas. The Journal of clinical investigation. 2006;116(6):1484-93
131. Sumazaki R, Shiojiri N, Isoyama S, Masu M, Keino-Masu K, Osawa M. et al. Conversion of biliary system to pancreatic tissue in Hes1-deficient mice. Nature genetics. 2004;36(1):83-7
132. Tao T, Deng P, Wang Y, Zhang X, Guo Y, Chen W. et al. Microengineered Multi-Organoid System from hiPSCs to Recapitulate Human Liver-Islet Axis in Normal and Type 2 Diabetes. Adv Sci (Weinh). 2022;9(5):e2103495
133. Harding J, Roberts RM, Mirochnitchenko O. Large animal models for stem cell therapy. Stem cell research & therapy. 2013;4(2):1-9
134. Ramos MJ, Bandiera L, Menolascina F, Fallowfield JA. In vitro models for non-alcoholic fatty liver disease: Emerging platforms and their applications. Iscience. 2022;25(1):103549
135. Takahashi Y, Soejima Y, Fukusato T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World journal of gastroenterology: WJG. 2012;18(19):2300
136. Arab JP, Arrese M, Trauner M. Recent insights into the pathogenesis of nonalcoholic fatty liver disease. Annual Review of Pathology: Mechanisms of Disease. 2018;13:321-50
137. Ratziu V, Giral P, Charlotte F, Bruckert E, Thibault V, Theodorou I. et al. Liver fibrosis in overweight patients. Gastroenterology. 2000;118(6):1117-23
138. Wanless IR, Lentz JS. Fatty liver hepatitis (steatohepatitis) and obesity: an autopsy study with analysis of risk factors. Hepatology. 1990;12(5):1106-10
139. Marchesini G, Brizi M, Morselli-Labate AM, Bianchi G, Bugianesi E, McCullough AJ. et al. Association of nonalcoholic fatty liver disease with insulin resistance. The American journal of medicine. 1999;107(5):450-5
140. Gaggini M, Morelli M, Buzzigoli E, DeFronzo RA, Bugianesi E, Gastaldelli A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients. 2013;5(5):1544-60
141. Nakayama H, Otabe S, Ueno T, Hirota N, Yuan X, Fukutani T. et al. Transgenic mice expressing nuclear sterol regulatory element-binding protein 1c in adipose tissue exhibit liver histology similar to nonalcoholic steatohepatitis. Metabolism. 2007;56(4):470-5
142. Sato W, Horie Y, Kataoka E, Ohshima S, Dohmen T, Iizuka M. et al. Hepatic gene expression in hepatocyte-specific Pten deficient mice showing steatohepatitis without ethanol challenge. Hepatology Research. 2006;34(4):256-65
143. Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J. et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. The Journal of clinical investigation. 2004;113(12):1774-83
144. Brix AE, Elgavish A, Nagy TR, Gower BA, Rhead WJ, Wood PA. Evaluation of liver fatty acid oxidation in the leptin-deficient obese mouse. Molecular genetics and metabolism. 2002;75(3):219-26
145. Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proceedings of the National Academy of Sciences. 1997;94(6):2557-62
146. Wortham M, He L, Gyamfi M, Copple BL, Wan Y-JY. The transition from fatty liver to NASH associates with SAMe depletion in db/db mice fed a methionine choline-deficient diet. Digestive diseases and sciences. 2008;53:2761-74
147. Sahai A, Malladi P, Pan X, Paul R, Melin-Aldana H, Green RM. et al. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: role of short-form leptin receptors and osteopontin. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2004;287(5):G1035-G43
148. Okumura K, Ikejima K, Kon K, Abe W, Yamashina S, Enomoto N. et al. Exacerbation of dietary steatohepatitis and fibrosis in obese, diabetic KK-Ay mice. Hepatology research. 2006;36(3):217-28
149. Ibrahim SH, Hirsova P, Malhi H, Gores GJ. Animal models of nonalcoholic steatohepatitis: eat, delete, and inflame. Digestive diseases and sciences. 2016;61:1325-36
150. Lieber CS, Leo MA, Mak KM, Xu Y, Cao Q, Ren C. et al. Model of nonalcoholic steatohepatitis. The American journal of clinical nutrition. 2004;79(3):502-9
151. Ip E, Farrell GC, Robertson G, Hall P, Kirsch R, Leclercq I. Central role of PPARα-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology. 2003;38(1):123-32
152. Kashireddy PV, Rao MS. Lack of peroxisome proliferator-activated receptor α in mice enhances methionine and choline deficient diet-induced steatohepatitis. Hepatology Research. 2004;30(2):104-10
153. Golding H, Khurana S, Zaitseva M. What is the predictive value of animal models for vaccine efficacy in humans? The importance of bridging studies and species-independent correlates of protection. Cold Spring Harbor perspectives in biology. 2018;10(4):a028902
154. Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W. et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proceedings of the National Academy of Sciences. 2013;110(9):3507-12
155. Wanninger J, Neumeier M, Hellerbrand C, Schacherer D, Bauer S, Weiss TS. et al. Lipid accumulation impairs adiponectin-mediated induction of activin A by increasing TGFbeta in primary human hepatocytes. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids. 2011;1811(10):626-33
156. Gomez-Lechon MJ, Donato MT, Martínez-Romero A, Jiménez N, Castell JV, O'Connor J-E. A human hepatocellular in vitro model to investigate steatosis. Chemico-biological interactions. 2007;165(2):106-16
157. Cui W, Chen SL, Hu K-Q. Quantification and mechanisms of oleic acid-induced steatosis in HepG2 cells. American journal of translational research. 2010;2(1):95
158. Green CJ, Parry SA, Gunn PJ, Ceresa CD, Rosqvist F, Piché M-E. et al. Studying non-alcoholic fatty liver disease: the ins and outs of in vivo, ex vivo and in vitro human models. Hormone Molecular Biology and Clinical Investigation. 2018;41(1):20180038
159. Gunn PJ, Green CJ, Pramfalk C, Hodson L. In vitro cellular models of human hepatic fatty acid metabolism: differences between Huh7 and HepG2 cell lines in human and fetal bovine culturing serum. Physiological Reports. 2017;5(24):e13532
160. Takahara I, Akazawa Y, Tabuchi M, Matsuda K, Miyaaki H, Kido Y. et al. Toyocamycin attenuates free fatty acid-induced hepatic steatosis and apoptosis in cultured hepatocytes and ameliorates nonalcoholic fatty liver disease in mice. PloS one. 2017;12(3):e0170591
161. Michaut A, Le Guillou D, Moreau C, Bucher S, McGill MR, Martinais S. et al. A cellular model to study drug-induced liver injury in nonalcoholic fatty liver disease: application to acetaminophen. Toxicology and applied pharmacology. 2016;292:40-55
162. Lyall MJ, Cartier J, Thomson JP, Cameron K, Meseguer-Ripolles J, O'Duibhir E. et al. Modelling non-alcoholic fatty liver disease in human hepatocyte-like cells. Philosophical Transactions of the Royal Society B: Biological Sciences. 2018;373(1750):20170362
163. van Son KC, Verschuren L, Hanemaaijer R, Reeves H, Takkenberg RB, Drenth JP. et al. Non-Parenchymal Cells and the Extracellular Matrix in Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease. Cancers. 2023;15(4):1308
164. Russell S, Wojtkowiak J, Neilson A, Gillies RJ. Metabolic Profiling of healthy and cancerous tissues in 2D and 3D. Scientific reports. 2017;7(1):15285
165. Barbero-Becerra VJ, Giraudi PJ, Chavez-Tapia NC, Uribe M, Tiribelli C, Rosso N. The interplay between hepatic stellate cells and hepatocytes in an in vitro model of NASH. Toxicology in vitro. 2015;29(7):1753-8
166. Zhong S, Zhao L, Wang Y, Zhang C, Liu J, Wang P. et al. Cluster of differentiation 36 deficiency aggravates macrophage infiltration and hepatic inflammation by upregulating monocyte chemotactic protein-1 expression of hepatocytes through histone deacetylase 2-dependent pathway. Antioxidants & redox signaling. 2017;27(4):201-14
167. Chen Y, Ma K. NLRC4 inflammasome activation regulated by TNF-α promotes inflammatory responses in nonalcoholic fatty liver disease. Biochemical and biophysical research communications. 2019;511(3):524-30
168. Strobel S, Kostadinova R, Fiaschetti-Egli K, Rupp J, Bieri M, Pawlowska A. et al. A 3D primary human cell-based in vitro model of non-alcoholic steatohepatitis for efficacy testing of clinical drug candidates. Sci Rep. 2021;11(1):22765
169. Martignoni M, Groothuis GM, de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol. 2006;2(6):875-94
170. Raza S, Rajak S, Upadhyay A, Tewari A, Anthony Sinha R. Current treatment paradigms and emerging therapies for NAFLD/NASH. Front Biosci (Landmark Ed). 2021;26(2):206-37
171. Ahmed IA, Mikail MA, Mustafa MR, Ibrahim M, Othman R. Lifestyle interventions for non-alcoholic fatty liver disease. Saudi J Biol Sci. 2019;26(7):1519-24
172. Harrison SP, Baumgarten SF, Verma R, Lunov O, Dejneka A, Sullivan GJ. Liver Organoids: Recent Developments, Limitations and Potential. Front Med (Lausanne). 2021;8:574047
173. Sanders FW, Griffin JL. De novo lipogenesis in the liver in health and disease: more than just a shunting yard for glucose. Biol Rev Camb Philos Soc. 2016;91(2):452-68
174. Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146(3):726-35
175. Xu X, Poulsen KL, Wu L, Liu S, Miyata T, Song Q. et al. Targeted therapeutics and novel signaling pathways in non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH). Signal Transduct Target Ther. 2022;7(1):287
176. Hendriks D, Brouwers JF, Hamer K, Geurts MH, Luciana L, Massalini S. et al. Engineered human hepatocyte organoids enable CRISPR-based target discovery and drug screening for steatosis. Nature Biotechnology. 2023:1-15
177. Cichoz-Lach H, Michalak A. Oxidative stress as a crucial factor in liver diseases. World J Gastroenterol. 2014;20(25):8082-91
178. Arroyave-Ospina JC, Wu Z, Geng Y, Moshage H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants (Basel). 2021 10(2)
179. Miazek K, Beton K, Sliwinska A, Brozek-Pluska B. The Effect of beta-Carotene, Tocopherols and Ascorbic Acid as Anti-Oxidant Molecules on Human and Animal In vitro/In vivo Studies: A Review of Research Design and Analytical Techniques Used. Biomolecules. 2022 12(8)
180. Delli Bovi AP, Marciano F, Mandato C, Siano MA, Savoia M, Vajro P. Oxidative Stress in Non-alcoholic Fatty Liver Disease. An Updated Mini Review. Front Med (Lausanne). 2021;8:595371
181. Overi D, Carpino G, Franchitto A, Onori P, Gaudio E. Hepatocyte Injury and Hepatic Stem Cell Niche in the Progression of Non-Alcoholic Steatohepatitis. Cells. 2020 9(3)
182. Shojaie L, Iorga A, Dara L. Cell Death in Liver Diseases: A Review. Int J Mol Sci. 2020 21(24)
183. Hirsova P, Gores GJ. Death Receptor-Mediated Cell Death and Proinflammatory Signaling in Nonalcoholic Steatohepatitis. Cell Mol Gastroenterol Hepatol. 2015;1(1):17-27
184. Shan L, Wang F, Zhai D, Meng X, Liu J, Lv X. New Drugs for Hepatic Fibrosis. Front Pharmacol. 2022;13:874408
185. Yoon YC, Fang Z, Lee JE, Park JH, Ryu JK, Jung KH. et al. Selonsertib Inhibits Liver Fibrosis via Downregulation of ASK1/ MAPK Pathway of Hepatic Stellate Cells. Biomol Ther (Seoul). 2020;28(6):527-36
186. Wang W, Yan J, Wang H, Shi M, Zhang M, Yang W. et al. Rapamycin ameliorates inflammation and fibrosis in the early phase of cirrhotic portal hypertension in rats through inhibition of mTORC1 but not mTORC2. PLoS One. 2014;9(1):e83908
187. Albhaisi S, Noureddin M. Current and Potential Therapies Targeting Inflammation in NASH. Front Endocrinol (Lausanne). 2021;12:767314
188. Moayedfard Z, Sani F, Alizadeh A, Bagheri Lankarani K, Zarei M, Azarpira N. The role of the immune system in the pathogenesis of NAFLD and potential therapeutic impacts of mesenchymal stem cell-derived extracellular vesicles. Stem Cell Res Ther. 2022;13(1):242
189. Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol. 2021;18(1):45-56
190. Weiskirchen R, Tacke F. Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg Nutr. 2014;3(6):344-63
191. Pinto C, Giordano DM, Maroni L, Marzioni M. Role of inflammation and proinflammatory cytokines in cholangiocyte pathophysiology. Biochim Biophys Acta Mol Basis Dis. 2018;1864(4 Pt B):1270-8
192. Gong J, Tu W, Liu J, Tian D. Hepatocytes: A key role in liver inflammation. Front Immunol. 2022;13:1083780
193. Kershenobich Stalnikowitz D, Weissbrod AB. Liver fibrosis and inflammation. A review. Ann Hepatol. 2003;2(4):159-63
194. Zhang J, Liu Q, He J, Li Y. Novel Therapeutic Targets in Liver Fibrosis. Front Mol Biosci. 2021;8:766855
195. Srinivas AN, Suresh D, Santhekadur PK, Suvarna D, Kumar DP. Extracellular Vesicles as Inflammatory Drivers in NAFLD. Front Immunol. 2020;11:627424
196. Ipsen DH, Tveden-Nyborg P. Extracellular Vesicles as Drivers of Non-Alcoholic Fatty Liver Disease: Small Particles with Big Impact. Biomedicines. 2021 9(1)
197. Dorairaj V, Sulaiman SA, Abu N, Abdul Murad NA. Extracellular Vesicles in the Development of the Non-Alcoholic Fatty Liver Disease: An Update. Biomolecules. 2020 10(11)
198. Ouchi R, Togo S, Kimura M, Shinozawa T, Koido M, Koike H. et al. Modeling steatohepatitis in humans with pluripotent stem cell-derived organoids. Cell metabolism. 2019;30(2):374-84 e6
199. Guan Y, Enejder A, Wang M, Fang Z, Cui L, Chen S-Y. et al. A human multi-lineage hepatic organoid model for liver fibrosis. Nature communications. 2021;12(1):6138
200. Ding JH, Jin Z, Yang XX, Lou J, Shan WX, Hu YX. et al. Role of gut microbiota via the gut-liver-brain axis in digestive diseases. World J Gastroenterol. 2020;26(40):6141-62
201. Shao JW, Ge TT, Chen SZ, Wang G, Yang Q, Huang CH. et al. Role of bile acids in liver diseases mediated by the gut microbiome. World J Gastroenterol. 2021;27(22):3010-21
202. Ramirez-Perez O, Cruz-Ramon V, Chinchilla-Lopez P, Mendez-Sanchez N. The Role of the Gut Microbiota in Bile Acid Metabolism. Ann Hepatol. 2017;16(Suppl 1):S21-S6
203. Parker A, Lawson MAE, Vaux L, Pin C. Host-microbe interaction in the gastrointestinal tract. Environ Microbiol. 2018;20(7):2337-53
204. Vancamelbeke M, Vermeire S. The intestinal barrier: a fundamental role in health and disease. Expert Rev Gastroenterol Hepatol. 2017;11(9):821-34
205. Ghosh S, Whitley CS, Haribabu B, Jala VR. Regulation of Intestinal Barrier Function by Microbial Metabolites. Cell Mol Gastroenterol Hepatol. 2021;11(5):1463-82
206. Nicoletti A, Ponziani FR, Biolato M, Valenza V, Marrone G, Sganga G. et al. Intestinal permeability in the pathogenesis of liver damage: From non-alcoholic fatty liver disease to liver transplantation. World J Gastroenterol. 2019;25(33):4814-34
207. Dharmalingam M, Yamasandhi PG. Nonalcoholic Fatty Liver Disease and Type 2 Diabetes Mellitus. Indian J Endocrinol Metab. 2018;22(3):421-8
208. Younossi ZM, Golabi P, de Avila L, Paik JM, Srishord M, Fukui N. et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J Hepatol. 2019;71(4):793-801
209. Jin D, Cui Z, Jin S, Zhou T, Guo B, Gao P. et al. Comparison of efficacy of anti-diabetics on non-diabetic NAFLD: A network meta-analysis. Front Pharmacol. 2022;13:1096064
210. Kanabekova P, Kadyrova A, Kulsharova G. Microfluidic Organ-on-a-Chip Devices for Liver Disease Modeling In vitro. Micromachines (Basel). 2022 13(3)
211. Panwar A, Das P, Tan LP. 3D Hepatic Organoid-Based Advancements in LIVER Tissue Engineering. Bioengineering (Basel). 2021 8(11)
212. Shimomura I, Hammer RE, Richardson JA, Ikemoto S, Bashmakov Y, Goldstein JL. et al. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue: model for congenital generalized lipodystrophy. Genes & development. 1998;12(20):3182-94
213. Bray GA, York DA. Hypothalamic and genetic obesity in experimental animals: an autonomic and endocrine hypothesis. Physiological reviews. 1979;59(3):719-809
214. Diehl AM. Lessons from animal models of NASH. Hepatology Research. 2005;33(2):138-44
215. Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ. et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell. 1996;84(3):491-5
216. London RM, George J. Pathogenesis of NASH: animal models. Clinics in liver disease. 2007;11(1):55-74
217. Lu SC, Alvarez L, Huang Z-Z, Chen L, An W, Corrales FJ. et al. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proceedings of the National Academy of Sciences. 2001;98(10):5560-5
218. Martinez-Chantar M. Spontaneous oxidative stress and liver tumors in mice lacking methionine adenosyltransferase 1A. The FASEB Journal. 2002
219. Zou Y, Li J, Lu C, Wang J, Ge J, Huang Y. et al. High-fat emulsion-induced rat model of nonalcoholic steatohepatitis. Life sciences. 2006;79(11):1100-7
220. Ito M, Suzuki J, Tsujioka S, Sasaki M, Gomori A, Shirakura T. et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatology Research. 2007;37(1):50-7
221. Leclercq IA, Farrell GC, Field J, Bell DR, Gonzalez FJ, Robertson GR. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. The Journal of clinical investigation. 2000;105(8):1067-75
222. Chowdhry S, Nazmy MH, Meakin PJ, Dinkova-Kostova AT, Walsh SV, Tsujita T. et al. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radical Biology and Medicine. 2010;48(2):357-71
223. dela Peña A, Leclercq I, Field J, George J, Jones B, Farrell G. NF-κB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology. 2005;129(5):1663-74
224. Matsuzawa N, Takamura T, Kurita S, Misu H, Ota T, Ando H. et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology. 2007;46(5):1392-403
225. Sharma S, Mells JE, Fu PP, Saxena NK, Anania FA. GLP-1 analogs reduce hepatocyte steatosis and improve survival by enhancing the unfolded protein response and promoting macroautophagy. PloS one. 2011;6(9):e25269
226. Lin Y, Ding D, Huang Q, Liu Q, Lu H, Lu Y. et al. Downregulation of miR-192 causes hepatic steatosis and lipid accumulation by inducing SREBF1: Novel mechanism for bisphenol A-triggered non-alcoholic fatty liver disease. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids. 2017;1862(9):869-82
227. Green CJ, Johnson D, Amin HD, Sivathondan P, Silva MA, Wang LM. et al. Characterization of lipid metabolism in a novel immortalized human hepatocyte cell line. American Journal of Physiology-Endocrinology and Metabolism. 2015;309(6):E511-E22
228. Khamphaya T, Chukijrungroat N, Saengsirisuwan V, Mitchell-Richards KA, Robert ME, Mennone A. et al. Nonalcoholic fatty liver disease impairs expression of the type II inositol 1, 4, 5-trisphosphate receptor. Hepatology. 2018;67(2):560-74
229. Tolosa L, Gómez-Lechón MJ, Jiménez N, Hervás D, Jover R, Donato MT. Advantageous use of HepaRG cells for the screening and mechanistic study of drug-induced steatosis. Toxicology and applied pharmacology. 2016;302:1-9
230. Luckert C, Braeuning A, De Sousa G, Durinck S, Katsanou ES, Konstantinidou P. et al. Adverse outcome pathway-driven analysis of liver steatosis in vitro: a case study with cyproconazole. Chemical research in toxicology. 2018;31(8):784-98
231. Graffmann N, Ring S, Kawala M-A, Wruck W, Ncube A, Trompeter H-I. et al. Modeling nonalcoholic fatty liver disease with human pluripotent stem cell-derived immature hepatocyte-like cells reveals activation of PLIN2 and confirms regulatory functions of peroxisome proliferator-activated receptor alpha. Stem cells and development. 2016;25(15):1119-33
232. Coll M, Perea L, Boon R, Leite SB, Vallverdú J, Mannaerts I. et al. Generation of hepatic stellate cells from human pluripotent stem cells enables in vitro modeling of liver fibrosis. Cell stem cell. 2018;23(1):101-13 e7
233. Leite SB, Roosens T, El Taghdouini A, Mannaerts I, Smout AJ, Najimi M. et al. Novel human hepatic organoid model enables testing of drug-induced liver fibrosis in vitro. Biomaterials. 2016;78:1-10
234. Bell CC, Hendriks DF, Moro SM, Ellis E, Walsh J, Renblom A. et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Scientific reports. 2016;6(1):25187
235. Feaver RE, Cole BK, Lawson MJ, Hoang SA, Marukian S, Blackman BR. et al. Development of an in vitro human liver system for interrogating nonalcoholic steatohepatitis. JCI insight. 2016 1(20)
236. Kruitwagen HS, Oosterhoff LA, Vernooij IG, Schrall IM, van Wolferen ME, Bannink F. et al. Long-term adult feline liver organoid cultures for disease modeling of hepatic steatosis. Stem Cell Reports. 2017;8(4):822-30
237. Haaker MW, Kruitwagen HS, Vaandrager AB, Houweling M, Penning LC, Molenaar MR. et al. Identification of potential drugs for treatment of hepatic lipidosis in cats using an in vitro feline liver organoid system. Journal of veterinary internal medicine. 2020;34(1):132-8
238. Ramli MNB, Lim YS, Koe CT, Demircioglu D, Tng W, Gonzales KAU. et al. Human pluripotent stem cell-derived organoids as models of liver disease. Gastroenterology. 2020;159(4):1471-86 e12
Author contact
![]() Corresponding authors: Dong Wook Han (dwhan.stemcom), Kwonho Hong (hongkac.kr), and Nam-Hyung Kim (namhyungkimcom); Tel.: +86-07503299840, Wuyi University, Jiangmen 529020, China.
Corresponding authors: Dong Wook Han (dwhan.stemcom), Kwonho Hong (hongkac.kr), and Nam-Hyung Kim (namhyungkimcom); Tel.: +86-07503299840, Wuyi University, Jiangmen 529020, China.