Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Tumor initiation and TME...
Cellular components in glioma
Functional importance of TME...
Glioma mouse models
Single-cell RNA-seq in glioma
Future clinical application
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(13):4311-4326. doi:10.7150/ijbs.83531 This issue Cite
Review
The Evolution of Tumor Microenvironment in Gliomas and Its Implication for Target Therapy
Yang Hu*,1, Zhixing Li*,1, Yichi Zhang*,1, Yuzheng Wu1, Zihao Liu1, Jianhao Zeng3, Zhexue Hao1, Jin Li1 ![]() , Jiaoyan Ren2
, Jiaoyan Ren2 ![]() , Maojin Yao1
, Maojin Yao1 ![]() #
#
1. The First Affiliated Hospital of Guangzhou Medical University, Guangzhou Institute of Respiratory Disease & China State Key Laboratory of Respiratory Disease, Guangzhou, 510182, China.
2. School of Food Sciences and Engineering, South China University of Technology, Guangzhou, 510641, China.
3. Department of Microbiology, Immunology, and Cancer Biology, University of Virginia Health System, Charlottesville, VA 22908, USA.
* These authors contributed equally to this work.
#Leading corresponding author.
Received 2023-2-14; Accepted 2023-8-3; Published 2023-8-21
Abstract

Gliomas develop in unique and complicated environments that nourish tumor cells. The tumor microenvironment (TME) of gliomas comprises heterogeneous cells, including brain-resident cells, immune cells, and vascular cells. Reciprocal interactions among these cells are involved in the evolution of the TME. Moreover, the study of attractive therapeutic strategies that target the TME is transitioning from basic research to the clinic. Mouse models are indispensable tools for dissecting the processes and mechanisms leading to TME evolution. In this review, we overview the paradoxical roles of the TME, as well as the recent progress of mouse models in TME research. Finally, we summarize recent advances in TME-targeting therapeutic strategies.
Keywords: Glioma, Tumor microenvironment, Tumor evolution, Targeted therapy, Mouse model
Introduction
Tumors are complex tissues that are composed of malignant cells and multiple distinct niche cells [1]. In solid tumors, the tumor microenvironment (TME) comprises vasculature, extracellular matrix (ECM), non-malignant cells surrounding the tumor (including stromal cells, fibroblasts, and immune cells), and environmental molecules (such as growth factors, cytokines, chemokines, and exosomes). Bidirectional communication between tumor cells and their microenvironment has been revealed to be actively involved in tumor initiation, progression and even impede the efficacy of therapy. To better understand the evolution of the TME, the “seed and soil” theory was first proposed by Stephen Paget in 1863 to delineate the tropism of tumor metastases to specific organs [2]. Since then, increasing evidence has confirmed the essential role of the TME in tumorigenesis [1]. The TME provides a nourishing environment for every stage of cancer development, including primary tumor growth, metastatic dissemination and survival in the periphery, as well as secondary outgrowth [3].
For cancer treatments to be effective, the drugs must reach the tumor tissue at adequate concentrations. However, abnormal vasculature can limit the efficacy of drug delivery and treatment [4], and alteration of stromal cells may contribute to both inherent and acquired resistance [5]. Hence, drugs targeting TME cells have been developed and clinically tested. Unlike tumor cells, stromal cells within the TME are genetically and/or epigenetically stable, and thus, targeting the TME is an even more attractive option with minimal risk of therapeutic resistance.
Brain tumors are one of the leading causes of cancer-associated mortality, with limited treatment options. According to the World Health Organization (WHO), primary brain tumors can be classified into more than 100 types based on phenotypic and genotypic parameters [6]. Approximately 80.9% of primary malignant brain tumors are gliomas, of which glioblastoma multiforme (GBM) is the most aggressive type. Relative survival estimates for GBM are poor, with 5.6% of patients surviving five years post-diagnosis [7], and the median overall survival (OS) is only 14.6 months after receiving combination therapy of surgery, temozolomide chemotherapy and radiotherapy [8]. Consequently, clinical treatment of brain tumors, especially GBM, remains a significant challenge.
The TME has critical effects on all stages of tumorigenesis, recurrence and therapeutic response. Tumor cells produce and secrete a variety of cytokines and growth factors, which promote the infiltration of non-neoplastic cells and alter their physiology toward pro-tumorigenic features. Compared with the TME in other cancers, the brain TME is distinctive because it contains several unique tissue-resident cell types, including microglia, astrocytes and neurons. In addition, the blood-brain barrier (BBB) or blood-brain tumor barrier (BBTB), formed by endothelial cells, pericytes and astrocytic endfeet, shields the brain from immune cell entry and/or attack [9]. The majority of non-neoplastic cells within gliomas are tumor-associated myeloid cells (TAMs), accounting for up to 30% of the tumor mass, and can originate from both brain-resident microglia and bone marrow-derived macrophages (BMDMs) [10]. In addition, astrocytes [11] and neurons [12] are both brain-specific cell types that are tightly involved in the initiation and progression of gliomas. Due to its crucial role in gliomas, the TME may be a therapeutic target to improve outcomes for patients. Most importantly, tumor cells co-evolve with TME cells from tumor initiation to progression along with the increased complexity. In this review, we will discuss the roles of these components and the potential reciprocal interactions involved in TME evolution.
Tumor initiation and TME development
In the last century, it has been well recognized that cumulative somatic mutations drive tumorigenesis. Fearon and Vogelstein [13] first hypothesized a genetic model for colorectal tumorigenesis in which mutations occur, accumulate, and contribute to the transformation of normal epithelial cells to malignant cells. Hanahan and Weinberg [1, 14] further conceived that eight essential alterations in cell physiology serve as hallmarks of cancer. However, ostensible niche cells recruited by cancer cells contribute to the acquisition of these hallmarks and exert influences on cancer development by creating the TME [1].
Tumor initiation might require a distinct microenvironment since mutant cells can constantly shape the surrounding niche. Cancers are not just clones of mutant and malignant cells but complex abnormal organs, to which multiple normal cell types are recruited by malignant cells under changes that promote tumor development. Bidirectional communication between cancer cells and their surrounding environment creates and ignites the evolution of the TME. The steps of tumor formation involve the co-evolution of neoplastic cells and components of the surrounding stroma. However, the mechanisms underlying the infiltration of tumor cells into the TME and the interaction of TME components with tumor cells remain unclear. The following sections aim to elucidate the cellular and molecular mechanisms in gliomas.
Cellular components in glioma
Tumor-associated myeloid cells (microglia/macrophages)
The predominant immune cellular components in gliomas are TAMs (Figure 1A), which account for up to 30%-50% of the tumor mass [15]. TAMs can be subdivided ontogenetically into at least two major populations: (1) tissue-resident microglia (TRMs) of yolk sac origin and (2) BMDMs. As tissue-resident macrophages, microglia act as the first and main form of active immune surveillance in the central nervous system (CNS). Once inflammation is triggered, microglia can rapidly migrate toward the brain lesion, where they phagocytose and remove debris and release cytotoxic molecules such as proinflammatory cytokines, reactive oxygen intermediates and proteinases. However, this antitumor effect can be attenuated when microglia are reprogrammed in the setting of glioma. In the presence of microglia, the migration and invasiveness of glioma cells was increased in vitro [16]. TAMs depletion by infusion of ganciclovir in CD11b-HSVTK mice largely attenuated glioma expansion [17]. Several factors, including STI1, EGF, CSF-1, CCL2, IL-6, TGF-β and TGF-β2, are released from TAMs and can promote glioma proliferation and/or migration [10]. Some flow cytometry experiments have demonstrated that the majority of TAMs in gliomas are derived from monocyte-derived macrophages but not resident microglia [18]. Another study using irradiation chimeras revealed that intrinsic microglia are the main source of brain tumors [19]. Elucidation of the cellular origin of TAMs could provide new therapeutic strategies aiming to inhibit the recruitment of monocytes from blood. Moreover, redirecting their phagocytosis function to sustain their “defender” antitumor activity was proposed as a new strategy to target TAMs [20]. Of note, Rao et al. showed that microglia and macrophages rather than CD8+ T cells mediated the therapeutic effect of PD-1 blockade [21]. Understanding the precise nature of TAMs is critical for not only improving the understanding of the role of TAMs in cancer biology but also identifying ways to better exploit macrophage/microglia-based therapy in the clinic.
Cell components and their significant roles in the glioma microenvironment. The brain TME comprises cells of the immune system, including various specialized organ-resident cell types. Tumor formation involves the co-evolution of neoplastic cells and surrounding non-transformed cells. The TME is both a cause and consequence of tumorigenesis. A) Tumor cells release factors to recruit TAMs into the microenvironment and polarize TAMs to an M2-like phenotype, and TAMs in turn promote glioma progress. B) Glioma secretes factors to inhibit DC differentiation and subsequently causes a lack of CD8+ T cell priming and activation, and an increase in the number of Tregs results in the development of the TME of glioma. C) NETs mediate the crosstalk between glioma cells and neutrophils by regulating the HMGB1/RAGE/IL-8 axis. D) The cytotoxic capacity of NK cells is reduced due to the suppression of HLA-G from GBM cells. CD8+ and CD4+ T cells highly express genes involved in cell exhaustion, creating an immunosuppressive microenvironment that favors tumor growth, which is regulated by regulator T cells. E) Tumor cells release cytokines to activate astrocytes and TAAs, which in turn promote glioma growth and invasion by releasing pro-tumor factors and increasing ECM degradation. F) Neuronal activity-dependent secretion of the synaptic NLGN3, cleaved by ADAM10 sheddase, promotes tumor cell proliferation by activating the PI3K-mTOR pathway. Figures were created with biorender.com.
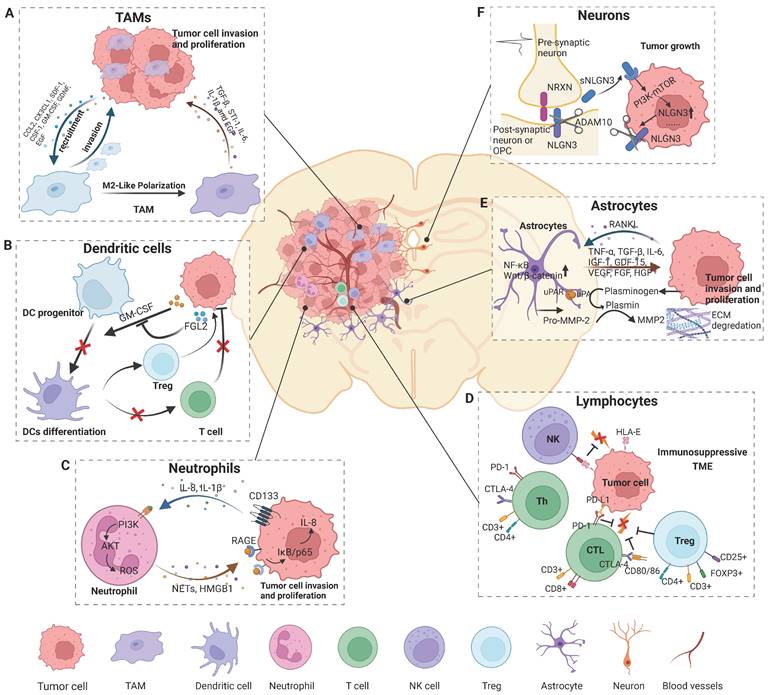
Dendritic cells
Dendritic cells (DCs) are professional antigen-presenting cells and play a critical role in both the innate and adaptive immune responses (Figure 1B). DCs can be divided into two distinct subsets: myeloid DCs (mDCs) and plasmacytoid DCs (pDCs). pDCs promote glioma progression in a murine model, while the depletion of pDCs increases the survival of mice bearing glioma by decreasing the number and suppressive function of regulatory T cells (Tregs) [22]. Furthermore, TGFβ and IL-10 secreted by glioma cells can disrupt the normal function of DCs. In glioblastoma, FGL2, secreted from tumor cells, blocks GM-CSF-induced CD103+ DCs development by suppressing NF-κB, STAT1/5, and p38 activation. Subsequently, CD8+ T cells are not primed or activated, which results in GBM progression [23].
Neutrophils
Neutrophils account for 50% to 70% of circulating leukocytes and are involved in almost all processes in tumor development (Figure 1C). Human glioma tissues have a high amount of neutrophil infiltration, and neutrophils are an important prognostic factor in glioma [24]. Wang et al. found that glioma patients with higher levels of neutrophils or neutrophil to lymphocyte ratios were more likely to have poor prognosis [25]. Neutrophil extracellular traps (NETs), produced by tumor-infiltrating neutrophils (TINs), are an oncogenic marker of high-grade gliomas (HGGs) and promote tumor cell proliferation and invasion [26]. Furthermore, neutrophil count is a prognostic indicator for patients with IDH wild-type GBM treated with temozolomide [25]. Increased neutrophil infiltration into tumors during anti-VEGF therapy promotes glioma progression and treatment resistance by increasing glioblastoma-initiating cell proliferation and migration, which is mediated by S100A4 [27]. Therefore, agents targeting neutrophils and S100A4 combined with standard antiangiogenetic therapy may inhibit glioma progression and diminish antiangiogenic therapy resistance. A growing number of studies have focused on the mechanism of neutrophil recruitment. Glioma cells ectopically expressing CD133 increase neutrophil recruitment via the IL-1 signaling pathway both in vitro and in vivo, indicating that CD133-positive tumor-initiating cells might generate a unique TME through co-evolution with neutrophils [28]. IL8 is also an effective chemoattractant that recruits neutrophils at the tumor site and facilitates the proliferation of glioma cells [26]. TINs have been found to mediated NETs formation and HMGB1 production via the PI3K/AKT/ROS axis. HMGB1, an important component of NETs, binds to RAGE and stimulates the NF-κB signaling pathway in tumor cells, thus promoting IL8 secretion and glioma progression [26]. Neutrophils have an inherent ability to traverse the BBB/BBTB and penetrate the glioma site. Surgical tumor removal leads to the release of inflammatory factors, such as IL-8, which activate neutrophils and cause them to migrate to the glioma. Therefore, neutrophil-mediated drug delivery systems are an ideal treatment approach due to their enhanced therapeutic specificity and efficacy in glioma. Xue et al. reported a novel preclinical antitumor therapy strategy to eliminate residual glioma cells by delivering drugs into tumor lesions using a system mediated by neutrophils and paclitaxel-loaded liposomes [29].
Lymphocytes
Lymphocytes primarily comprise CD4+ and CD8+ T cells, a small proportion of B cells and natural killer (NK) cells (Figure 1D). CD8+ T cells are considered critical for tumor clearance, and increased tumor-infiltrating CD3+ and CD8+ T cell numbers are associated with prolonged patient survival in glioblastoma [30]. During tumor evolution, brain cancer cells develop strategies to escape T-cell antitumor responses. Glioma cells inhibit immune activation by secreting TGF-β as well as IL-10 and downregulating MHC class II expression on monocytes [31]. PD-L1 is one of the most prominent inhibitory regulators of lymphocytes. Higher expression of PD-L1 predicts a poor prognosis in glioma patients [32]. Glioma cells can upregulate PD-L1 expression in circulating monocytes and TAMs through autocrine/paracrine IL-10 signaling [33]. CTLA-4 is another immune checkpoint protein that negatively regulates T-cell activation. Higher CTLA-4 expression is detected in high-grade glioma samples and is caused by a lack of CD80/86 co-stimulatory molecule expression [34]. Chongsathidkiet et al. found that the number of circulating T cells in patients with glioma was less than 1/3 of that of normal people, and the T cells that were lacking from circulation were found sequestered in bone marrow. After hindering S1P1 internalization, T-cell sequestration is restored [35]. This may partly explain why most gliomas are “cold tumors”, with less T cell infiltration in and around the tumor lesion. CD4+CD25+FoxP3+ Tregs are a pro-tumorigenic subpopulation because of their highly immunosuppressive effects in many cancer types (Figure 1D). Factors such as proliferating and dying tumor cell self-antigens can recruit Tregs around tumor lesions. Of note, tumor cell- and/or DC-derived TGF-β can greatly induce the enrichment of Tregs. Soluble factors, such as CCL22 or CCL2, secreted by glioblastoma cells attract Tregs to the local tumor environment [36, 37]. The accumulation of Tregs is an important immune escape mechanism for gliomas. Heimberger AB et al. found that different amounts of Tregs do not seem to be related to patient outcome [38]. However, another study showed that increased Treg frequencies were associated with early tumor recurrence and reduced survival rates in glioblastoma patients [39]. Treg activity can be modulated to combat immunosuppression and enhance the anti-tumor immune response in these patients. Systemic anti-CD25 administration hindered the suppressive function of CD25+ Tregs, while it greatly enhanced the CD8+ cytotoxic T-cell response [40].
NK cells are another kind of cytotoxic lymphocyte that can identify and kill tumor cells and infiltrate different gliomas (Figure 1D). Research has shown that NK cells can recognize and eradicate stem cell-like human glioblastoma cells that express low amounts of HLA-I molecules and express several ligands recognized by activating NK receptors [41]. Although NK cells have potent cytotoxic effects on medulloblastoma cells through the NKG2D/MICA-ULBP-2 interaction, high expression of HLA-I can protect medulloblastoma cells from the cytotoxicity of NK cells [42]. Most studies to date have focused on the interaction of NKG2D with its ligand HLA-E and methods for restoring the NK cell-mediated anti-tumor immune responses.
Vasculature
The BBB and angiogenesis
The blood vessels are mainly lined with tightly packed endothelial cells surrounded by pericytes. In the brain, these cells together with astrocytes form the BBB. The BBB functions as a selective physical barrier between the brain and the periphery that distinguishes the brain from other organs [43]. As it restricts the transportation of large, hydrophilic molecules, the BBB protects the brain from toxins, pathogens and inflammatory cells. This characteristic also hinders the delivery of therapeutic agents into the brain, which is the factor limiting clinical use. However, the integrity of the BBB is often disrupted during tumor progression, resulting in intravascular leakiness. Recent research has shown that invasive glioma cells can displace astrocytic endfeet from endothelial or vascular smooth muscle cells, causing local damage to the BBB [44]. However, not all brain tumor types or subtypes show the same degree of BBB impairment. For example, compared with SHH-medulloblastoma, WNT-medulloblastoma has a more permeable vasculature which enables the accumulation of chemotherapeutic drugs within the tumor lesion [45].
The tumor-associated vasculature within the brain has abnormal endothelial walls, pericyte coverage and basement membranes [46]. The leakiness of tumor vessels causes an abnormal blood flow, increased interstitial fluid pressure and edema, which block effective delivery of therapeutic agents within tumors. Vascular endothelial growth factor (VEGF) is highly expressed in gliomas and promotes vascular permeability, extracellular matrix degeneration and vascular endothelial cell migration, proliferation, and angiogenesis. VEGF expression can be upregulated via hypoxia-dependent mechanisms and hypoxia-independent mechanisms such as PI3K-Akt, MAPK and Raf signaling. High VEGF leads to immature, dysfunctional vessels and an impaired BBB. Targeting blood vessel-related factors, particularly targeting VEGF and VEGFR, has been an attractive strategy in gliomas.
Lymphatic vessels
In the past, it was commonly believed that the lymphatic drainage system was notably absent from the brain. However, recent research has indicated that the meninges have a lymphatic vascular network, that is, the meningeal lymphatic system. Meningeal lymphatic vessels (MLVs) are connected to the deep cervical lymph nodes (CLNs) and efficiently drain fluid, soluble molecules and immune cells from the cerebrospinal fluid into CLNs [47, 48]. Recently, study showed that the presence and distribution of new lymphatic vessels in glioma samples via immunohistochemical staining for the marker molecules CD31, Prox1, and LYVE-1 [49]. Intracranial tumors in mice induce extensive remodeling of dorsal MLVs [50]. Disruption of dorsal MLVs blocks DC trafficking from brain tumors to deep CLNs and reduces the efficacy of combined anti-PD-1/CTLA-4 checkpoint therapy [50]. Furthermore, VEGF-C, an important lymphangiogenic factor, may potentiate checkpoint therapy. Mice bearing tumors overexpressing VEGF-C respond better to anti-PD-1/CTLA-4 combination therapy, and these effects are abolished by either dorsal MLV ablation or CCL21/CCR7 blockade [50]. Hence, dorsal MLVs might be a potential target in glioma immunotherapy.
Astrocytes
Astrocytes account for approximately half of all brain cells and play an important role in the CNS (Figure 1E). These cells are components of the BBB and are responsible for mediating ionic and transmitter homeostasis, modulating synaptic activity and plasticity, and responding to CNS damage [11, 51]. Immunohistochemical studies of human glioma biopsies have revealed that reactive astrocytes are located around gliomas [52]. These reactive astrocytes may interact with glioma cells and contribute to tumor growth and aggression (Figure 1). Tumoral RANKL activates astrocytes via the NF-κB signaling pathway, leading to an increase in tumor-associated astrocytes (TAAs). These TAAs in turn secrete various factors, such as TGF-β, which promote glioma cell invasion [53]. In the same way, glioma cells significantly activate astrocytes by upregulating Wnt/β-catenin signaling, and then astrocytes increase the degradation of ECM to promote tumor invasiveness. [54]. TAAs can secrete TNF-α, TGF-β, IL-6, IGF-1, GDF-15, VEGF, FGF, HGF, and EGF to enhance the proliferation of GBM cells [11, 51]. Moreover, TAAs might also participate in the adaptation of GBM cells to a hypoxic microenvironment through CCL20/CCR6 signaling, thus promoting angiogenesis and tumor cell invasion [11]. In addition, astrocytes have been shown to be involved in resistance to radiotherapy or chemotherapy [55]. Multiple factors, such as tenascin-C, IL-10, IFN-γ, IL-6, STAT-3, GDF-15 and PD-L1, secreted by TAAs protect GBM cells against anticancer immune reactions [51]. Overall, modulating underlying TAA-associated signaling might be a promising approach for GBM treatment.
Neurons
Neurons, a brain-specific cell type, are a crucial component of the brain TME and may contribute to the initiation and progression of tumors (Figure 1F). Active neurons exert a mitogenic effect on normal neural precursor and oligodendrocyte precursor cells (OPCs), the putative cells of origin of HGG [56-58]. Optogenetic control of cortical neuronal activity promotes HGG cell proliferation and growth via the secretion of the activity-regulated mitogen synaptic protein neuroligin-3 (NLGN3) in a patient-derived pediatric HGG orthotopic xenograft model [59]. Soluble NLGN3, released from post-synaptic neurons, promotes feedforward increases in the expression of NLGN3 through activation of the PI3K-mTOR pathway in glioma cells [59]. Further research has demonstrated that the ADAM10 sheddase cleaves NLGN3 from both neurons and OPCs [60]. Administration of an ADAM10 inhibitor robustly blocks HGG xenograft growth by preventing the release of NLGN3 into the TME in mice [60]. Synapses exist between neurons, normal OPCs and other subpopulations in gliomas that closely resemble OPCs [61, 62]. Monje et al. further found that structural synapses formed between glioma cells and neurons in the HGG microenvironment [63]. Neuronal activity evoked glioma excitatory postsynaptic currents mediated by AMPAR and non-synaptic prolonged glioma potassium currents amplified by gap junction-coupled, and then induced glioma cell membrane depolarization [63]. Glioma membrane depolarization induced by optogenetics promoted glioma xenograft cell proliferation, and pharmacological or genetic blockade of electrochemical signaling inhibited glioma xenograft growth [63]. These findings illustrate the unexplored potential of targeting neural circuits for therapy and may provide promising new approaches for the development of therapeutics for the treatment of this devastating group of neural cancers.
Cancer stem cell
Many researchers have identified and isolated glioma stem cells (GSCs) in GBM. GSCs promote tumor angiogenesis through elevated expression of VEGF [64]. These GSCs are associated with vascular niches and interact with endothelial cells to regulate their self-renewal and tumorigenicity [65]. Recently, Shideng Bao et al. showed that GSCs transdifferentiate into pericytes to support vessel function and tumor growth in xenograft models [66]. GSCs are recruited to endothelial cells by the SDF-1/CXCR4 axis and generate vascular pericytes by TGFβ signaling [66]. Selective elimination of GSC-derived pericytes (G-pericytes) by HsvTK-induced ganciclovir toxicity disrupts tumor vascular structure and function, and inhibits GBM growth [66]. Moreover, targeting G-pericytes disrupts the blood-tumor barrier (BTB) and increases vascular permeability by impairing BTB tight junctions, which enhances drug delivery to improve GBM chemotherapy efficacy [67]. BMX is an essential factor for maintaining G-pericytes. Inhibiting BMX with ibrutinib disrupts G-pericytes and the BTB to increase vascular permeability, effectively improves GBM treatment efficacy and extends the survival of tumor-bearing mice [67].
Functional importance of TME cell types for glioma progression
Many studies have revealed that TAMs are crucial for glioma progression (Figure 2). Chemoattractants such as MCP-1 (CCL2), MCP-3 (CCL7), SDF-1 (CXCL12), CX3CL1, POSTN, GM-CSF, M-CSF (CSF-1) and EGF produced by glioma cells actively recruit TAMs and thus promote tumor growth [68]. CCL2 is one of the most important chemoattractants and was originally isolated from a human glioblastoma cell line. Elevated expression of CCL2 contributes to the accumulation of TAMs and promotes tumor aggressiveness in human glioma tissue [69]. However, a recent study showed that CCL7 has a stronger correlation than CCL-2 with TAM infiltration in human gliomas [70]. In a murine astrocytoma model, the secretion of SDF-1 by glioma cells specifically promotes tropism toward hypoxic tumor areas [71]. CSF-1 is a chemokine that mobilizes monocytes toward sites of inflammation. The production of CSF-1 by glioblastoma cells attracts microglia and transforms them into a pro-tumorigenic phenotype, and microglia in turn stimulate glioblastoma cell invasion via EGFR activation [72]. In summary, these chemoattractants secreted by glioma cells have potential as future therapeutic targets.
TAMs reciprocally interact and co-evolve with tumor cells. Tumor cell-derived factors recruit and polarize TAMs into an anti-inflammatory and pro-tumorigenic M2-like phenotype, and TAMs substantially promote tumor growth, tumor cell migration, and invasion in diverse manners. TAM-derived factors can facilitate the destruction of the ECM and glioma vascularization. Moreover, M2-polarized TAMs contribute to an immunosuppressive tumor microenvironment by secreting soluble factors and via direct cell-cell interactions. Figures were created with biorender.com.
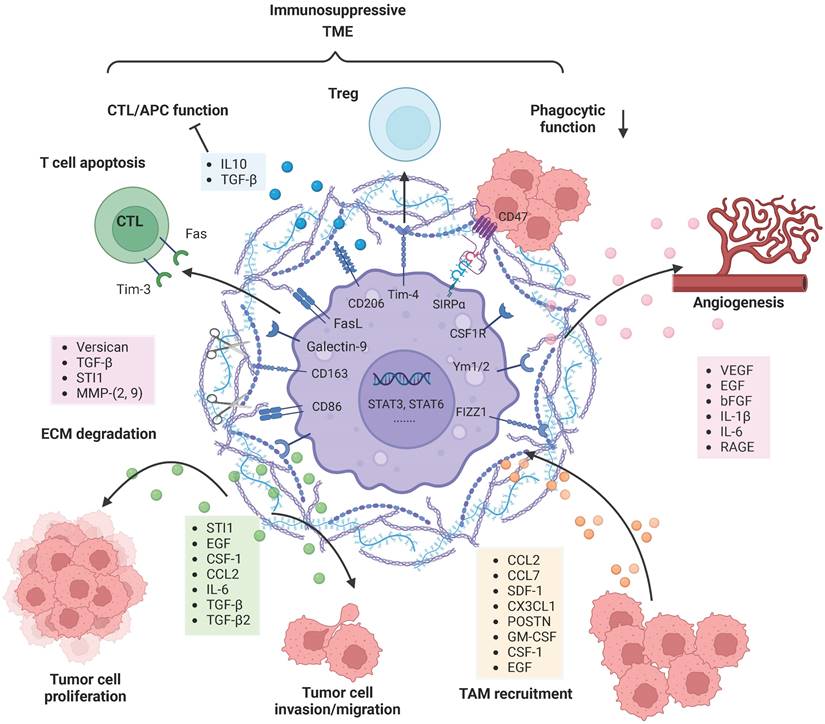
Once TAMs are recruited to the glioma milieu, they undergo significant in situ reprogramming in the glioma microenvironment, and unique gene expression patterns are acquired. For instance, genes encoding cell cycle-related proteins, Th17 immune response-related proteins, complement-related factors, ECM components, proteases, lipid metabolism mediators and clotting factors were upregulated in both BMDMs and microglia. Re-educated TAMs promote glioma cell invasion and tumor growth in diverse manners (Figure 2). TGF-β is mainly derived from TAMs and can actively facilitate glioma cell invasion. Induction of MMP-2 and MMP-9 expression by TGF-β promotes the invasive properties of glioma cells, possibly via degradation of the ECM [73]. STI1 synthesized and secreted by microglial cells has been shown to promote tumor cell proliferation and migration through MMP-9 [74]. Versican is a modular proteoglycan released from glioma cells and is involved in the control of cellular growth and differentiation [75]. TLR2 signaling in TAMs is substantially activated by glioma-derived versican, which leads to increased microglial MT1-MMP expression [76]. Upregulated MT1-MMP expression triggers MMP protein release and enables the degradation of the ECM to promote glioma invasion.
Other studies have also demonstrated that TAM-derived factors such as VEGF, EGF, bFGF, IL-1β and IL-6 are able to facilitate glioma vascularization (Figure 2). Notably, the interaction between RAGE and its ligands is crucial during angiogenesis. Chen et al. revealed that RAGE signaling in TAMs drives angiogenesis and that RAGE ablation results in decreased expression of IL6 and VEGF and abrogates tumor angiogenesis [77]. Furthermore, selective depletion of resident microglia induces similar effects, suggesting that resident microglia rather than peripheral macrophages are responsible for the pro-angiogenic effects [78]. Overall, many TAM-derived substances are able to support and facilitate glioma maintenance.
In addition, TAMs contribute to an immunosuppressive TME through a variety of mechanisms (Figure 2). Although educated TAMs have high levels of antigen presentation-related gene expression, they do not exert antitumor effects by activating effector T cells. SIRPα is expressed on TAMs and activated by CD47, an anti-phagocytic “don't eat me” surface protein expressed on cancer cells, particularly cancer stem cells. The phagocytic function of TRM-derived TAMs is attenuated due to increased SIRPα-CD47 signaling [79]. The immune checkpoint receptor PD-1 and its ligand PD-L1 play an important role in inhibiting the immune response. TAMs are the predominant source of PD-L1, and they greatly impair anti-tumor immunity. In addition, Rao Ganesh et al. revealed that the anti-PD-1 therapy may be mediated by cells of the innate immune system, especially TAMs, rather than CD8+ T cells [21]. Another relevant immune checkpoint receptor is T-cell immunoglobulin and mucin-domain-containing molecule 3 (Tim-3), which is expressed on T cells, NK cells and DCs, and its binding to Galectin-9 results in the apoptosis of these cells. Feng Yuan et al. have found a strong correlation between Galectin-9 expression and M2 TAMs levels, suggesting the role of TAMs in regulating Tim-3 signaling [80]. FasL is expressed on almost all TAMs and can induce activated T-cell apoptosis by binding to the Fas receptor [81]. TAMs as well as DCs are able to increase STAT3 activity and thus stimulate the secretion of IL-10, TGFβ and other anti-inflammatory molecules [80]. Meanwhile, the production of IFN-γ and IL-2 is significantly decreased, leading to effector T-cell unresponsiveness [82]. Tregs are able to inhibit the actions of effector lymphocytes. Friederike et al. revealed that MHC class II(+)CD40(dim)CD86(dim)IL-10(+) microglia are potent inducers of Foxp3+ Tregs in a murine model [83]. In addition, TIM4 is a newly described molecule expressed on TAMs. TIM4-expressing macrophages phagocytose apoptotic T cells in glioma and gain immune tolerogenic properties, further facilitating the development of Tregs [84]. Further research is required to uncover the complex multidirectional interactions in the glioma microenvironment.
Recognition of these concepts will benefit our understanding of cancer biology and lead to the development of alternative strategies to treat human cancer.
Glioma mouse models
Mouse models are the indispensable tools for understanding the composition of TME and the influence on disease, and developing effective therapies targeted to TME. Most common gliomas experimental models can be divided into xenograft transplantation models and genetically engineered models. Next, we discuss existing types of gliomas models, as well as the advantages and drawbacks of these models, to aid the choice of an appropriate animal model for specific experimental purposes.
Transplantation models
The existing in vivo models include models generated from transplantation of established glioma cell lines or cells isolated from patient samples, representing the simplest approach to generating tumors and the least technically challenging. There are two types of transplantation models: allograft and xenograft models. Tumor growth and drug efficacy can be measured in models in which tumor cells are implanted subcutaneously. However, these models lack the brain TME. Consequently, orthotopic implantation is a superior approach for glioma research. Xenograft models are generated via intracranial injection of human glioma cells (e.g., U87-MG, LN18, or U251 cells) or patient-derived cells into immunocompromised mice, and these models retain human GBM features and can be used in therapy studies. Nevertheless, these models lack the proper immune environment; hence, tumor immunology research in such models cannot be performed. The syngeneic glioma transplantation model retains the authenticity of an immunocompetent host. Established mouse or rat C6, 9L and GL261 glioma cells that originate from tumors induced by methylnitrosourea or methylcholanthrene are injected into C57/B6 mice or rats, and the models are used to study the biology of glioma or new therapeutic agents. However, the established glioma cell line transplantation models do not reflect the heterogeneity of gliomas or the pathogenesis of the disease in human.
Genetically engineered models
Molecular analyses of human gliomas have identified several mutations and alterations in the human glioma genome. The genetics and histology of xenograft models do not precisely reflect those of human glioma. Retroviral or transgenic glioma models involve manipulation of the mouse genome at the molecular level to create the molecular subtypes seen clinically. Approaches to generating mouse models of glioma include induction of loss-of-function (loss or inhibition of key tumor suppressors) or gain-of-function (expression of oncogenes) and somatic gene transfer by viral vectors. P53, PTEN, Nf1, and Rb are typically targeted tumor suppressors for the generation of mouse models of gliomas. V-Src, RAS and EGFRvIII are the most common oncogenes that have activating mutations and abnormal expression in gliomas.
In the Ctv-a mouse model, the transfer of PDGF-B to OPCs induces the formation of gliomas that resemble human WHO grade II oligodendroglioma [85]. Mice transgenic for S100b-verbB and with p53 deletion can develop oligodendrogliomas with expression signatures similar to those of OPCs [86]. The injection of VSV-G pseudotyped PDGF-IRES-Cre retrovirus into the rostral subcortical white matter of transgenic mice with floxed Pten and p53 induces proneural-type GBM [87]. Mature neurons and astrocytes can also give rise to GBM tumors in mice. Injections of shNF1-shp53 virus into the cortex of synapsin I-Cre transgenic mice causes expression of Cre specifically in neurons, leading to the formation of gliomas [88]. Similarly, CamK2a-Cre mice injected with the same virus, which targets mature neurons, develop tumors [88]. In the hippocampus, subventricular zone, striatum or cortex, the majority of cells expressing GFAP are mostly differentiated astrocytes. When H-RasV12-shp53 virus is injected into these areas, all GFAP-Cre mice develop tumors [88]
Many genetically engineered glioma models have been established based on gene knock-in or knock-out in embryonic stem cells. Experiments models with enhanced expression of potential oncogenes driven by a designated promoter can reveal their roles in tumor formation. GFAP promoter-driven overexpression of PDGFB in the brain lead to the development of glioblastoma in Trp53-null mice [89]. S100β is glial-specific expressed and is expressed in both oligodendroglia and astrocytes early in brain development. All grades of oligodendrogliomas overexpress EGFR, and only high-grade tumors are found to have ink4a/arf deletion. Transgenic mice expressing v-erbB, a transforming allele of EGFR driven by the S100β promoter, develop low-grade oligodendroglioma, and mice with ink4a/arf or p53 heterozygous mutations develop high-grade tumors [90]. However, the tumors of genetically engineered models based on embryonic stem cells greatly differ from human tumors, and the genetic manipulation can lead to significant developmental aberrations and can even be lethal to the animal before the tumor form.
Targeted expression of gene mutations is usually achieved through the Cre-lox system, which can be employed for knockout or forced expression of the target gene. Most commonly, transgenic Cre mice express the recombinase driven under the control of the GFAP promoter or Nestin (expressed in the neural progenitor cells) promoter. Transgenic mice harboring heterozygous loss-of-function mutations in Nf1 and p53 driven by GFAP-cre develop variable grade astrocytoma at 5-10 months. Mutation of PTEN causes accelerated tumor formation in this model and increases the grade of tumors. Between 15 to 40 weeks of age, 73% of the hGFAP-Cre; p53lox/lox; Ptenlox/+ mice harbored grade III astrocytomas and GBMs [91]. HGG exhibits strong RAS activation in humans. Activation of oncogenic K-ras in mouse glioneuronal precursor cells and adult subventricular zone cells with GFAP-Cre resulted in intermediate-grade, infiltrating glioma with 100% penetrance at 3 months. Cre recombinase-modified estrogen receptor ligand binding domain fusion protein (cre-ERT) translocates to the nucleus and initiates recombination after administration of tamoxifen. Inducible nestin-creERT mice to incorporate the tumor suppressor Nf1flox/+; p53flox/flox; Ptenflox/+ or Nf1flox/flox; p53flox/flox developed high grade astrocytomas after tamoxifen treatment [92].
How tumor cells co-evolve with the TME during tumorigenesis and tumor development (from the first gene mutation to tumorigenesis to tumor development) remains to be resolved, and one of the reasons is the lack of relevant models. Mosaic analysis with double markers (MADM) is a mouse genetic system that can be used to explore the dynamic interactions between tumor cells and other non-malignant cells in the TME. By design, MADM allows simultaneous gene knockout and fluorescent labeling of different genotype cells with different colors via Cre/loxP mediated mitotic recombination between two homologous chromosomes. MADM simultaneously and permanently generates sparse mutant cells with green fluorescent protein (GFP) and wild-type (WT) cells with red fluorescent protein (RFP) which are derived from the same precursor cell. The sibling red WT cells serves as an ideal internal control, facilitating detailed analyses of cellular aberrations of all lineages in the native environment [58]. With these well-designed features, the MADM system has been applied to trace the cell origin of tumors at single-cell resolution. hGFAP-Cre/Nestin-Cre-induced and MADM-mediated sporadic concurrent inactivation of p53 and NF1 in neural stem cell transgenic mice lead to 100% high-grade astrocytoma/GBM at 5 months [58]. The results indicated that OPCs are the cell of origin for glioma in this model. MADM-based genetic model in which sporadic Ptch1+/-, p53+/- granule neuron precursors (GNPs) generated by Math1-Cre develop SHH-subtype medulloblastoma in two months [93]. Math1-Cre faithfully labeled GNPs, enabling to study the establishment and evolution of the TME throughout tumorigenesis [93]. Sparse tumor GNPs transdifferentiate into TuAstrocytes, which secrete IL-4 to polarize microglia and induce IGF1 secretion, which is critical for tumor progression [93].
Single-cell RNA-seq in glioma
Single-cell RNA sequencing (scRNA-seq) enables the examination of the heterogeneity of tumor cells and the TME, shedding light on the interaction of the TME with tumor cells. The scRNA-seq of gliomas measures gene expression at the single-cell level, thereby uncovering the heterogeneity and evolutionary trajectory of the TME and tumor cells and providing molecular insight into precision therapy. We next briefly discuss what scRNA-seq reveals about the heterogeneity of tumor cells, TME cells and their phenotypic alterations in the TME.
Single-cell RNA-seq of tumor cells
In recent years, scRNA-seq has been used in studies such as The Cancer Genome Atlas (TCGA) to map the genetic landscape and expression signature of tumors and TME cells, thereby identifying driver mutations and tumor subtypes on specific transcriptional profiles. A large cohort study with single cell analysis provides a general framework for understanding the genotype, phenotype and TME composition of coupled tumor cells.
The scRNA-seq profiles were extracted from IDH-mutant astrocytoma (IDH-A) and oligodendroglioma (IDH-O) to decipher the differences in tumor cell genotypes and phenotypes between the two tumor subtypes. The IDH-A and IDH-O share a similar developmental hierarchy and spectrum. Tumor cell morphological and histopathologic differences between the two subtypes can be explained by distinct genetic events and TME composition [94]. Tumor pathological grade is associated with increased proliferation of glioma cells, suppressed differentiation of tumor cells, and alignment with highly polarized tumor associated macrophages/microglia [94].
GBM can be classified into proneural, neural, classical, and mesenchymal subtypes based on bulk gene expression data [95]. By leveraging the cellular resolution of the technique, scRNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Analysis shows that four subtypes could be observed in individual patient tumors [96]. Another scRNA-seq analysis also demonstrated that malignant cells in each glioblastoma sample exist in four main cellular states including (1) neural-progenitor-like (NPC-like), (2) oligodendrocyte-progenitor like (OPC-like), (3) astrocyte-like (AC-like), and (4) mesenchymal-like (MES-like) states, and exhibit distinct patterns of distribution in the TME [97]. The relative proportion of each cellular type is attributed to genetic alterations in CDK4, NF1, EGFR, and PDGFRA [97]. ScRNA-seq of GBM revealed extensive genetic heterogeneity at the transcriptional level. Expression and/or mutational heterogeneity of RTKs exists among individual glioblastoma cells and may affect treatments targeting receptor immunogenicity or RTK signaling [96]. In addition, intratumoral heterogeneity is associated with prognosis. In the proneural subtype, increased intratumoral heterogeneity is relevant to decreased survival [96]. By comparing the differences before and after treatment, scRNA-seq could also help us understand previous therapeutic failures and explore potential therapeutic targets.
Single-cell RNA-seq of the TME
Molecular and genetic variation in cancer cells may translate to phenotypic and functional variation in the TME. Emerging evidence suggests the close association between the molecular profile of glioma tumor cells and their surrounding microenvironment, and thus this heterogeneity in molecular state may also be imprinted into microenvironmental heterogeneity within tumors.
Combined flow cytometry sorting and scRNA-seq analysis were used to investigate the immune and non-immune components of GBM with single-cell resolution, and populations were analyzed longitudinally during initiation and progression. As GBM progresses, the immune cell composition changes. The immune microenvironment during the early stages of GBM tumorigenesis involves the transformation of "M1"-like pro-inflammatory microglia into centrally infiltrating "M2"-like tumor-promoting macrophages during the later stages [98]. These transitions coincide with the collapse of the BBB and the explosive growth of EGFR+ cancer cells [98]. ScRNA-seq was used to distinguish blood-derived macrophages and microglia under malignant and non-malignant conditions. Müller et al. identified phenotypic differences among TAMs of different lineages by scRNA-seq analysis of IDH-wt GBM [99]. TAMs derived from blood preferentially express immunosuppressive cytokines compared with microglia. A significant amount of blood-derived TAMs infiltrated pretreatment gliomas, which varied with tumor subtype and compartment [99]. Since blood-derived TAMs are more closely associated with poor prognosis, specifically targeting TAMs from immunosuppressive blood sources appears to be a better therapeutic strategy.
The scRNA-seq of endothelial cells (ECs) from human GBM tumor core and adjacent tissues provides a molecular perspective on the heterogeneity of the human BBB and its metabolic alterations in glioblastoma. The ECs in tumors are associated with altered metabolic transcriptome profiles characterized by increased expression of genes involved in glycolysis, citric acid cycle and oxidative phosphorylation [100]. Understanding the heterogeneity of the BBB phenotype is also crucial to develop rational treatment regimens and optimize drug delivery.
Multifocal samples are considered excellent models for studying the natural evolution of GBM, as they may represent different stages of tumor development. Wang et al. conducted a comprehensive analysis of eight multifocal IDH wild-type primary GBMs by sequencing 61,062 single cells [101]. By analyzing 4 pairs of GBM lesions in different regions, two lesions were found to be of the same origin but with different copy number changes. There is an underlying evolutionary sequence order between the two lesions. Further single-cell sequencing analysis revealed that the natural evolution of GBM may be accompanied by widespread gene activation and the acquisition of a module consisting of 12 genes, and the genes in the module were considered a natural evolutionary signature (NES) [101]. NES is significantly associated with tumor development, clinical outcome, and tumor cell status. Integrating scRNA-seq of multifocal GBMs identified FOSL2 as one of the hallmarks of higher expression in older lesions that may be involved in tumor evolution [101]. Furthermore, the hypoxic response was significantly associated with NES, possibly involved in NES transition through activation of the HIF1A-FOSL2 axis [101]. NES-high tumor cells recruit and polarize BMDMs by activating the FOSL2-ANXA1 axis to suppress T cell activity [101]. Polarized TAMs can also upregulate CCL2 to induce tumor cell migration [101]. This study further highlights the significance of single cell sequencing in investigating the evolution of glioma.
Using single-cell sequencing data, we are able to identify different cell subtypes and further analyze the evolutionary relationships between each subtype of tumor cells as well as TME cells. This is crucial for the successful development of precise therapy for glioma.
Future clinical application
Although there has been great progress in multimodal comprehensive treatment for gliomas, including surgery, radiotherapy, and systemic therapy (chemotherapy, targeted therapy), the outcomes are still not satisfactory. The immune cells of malignant glioma can “harmonize” with tumor cells and other components of the TME, thus forming a microenvironment conducive to the proliferation of glioma cells. Targeting tumor cells and their “partners” has become an attractive strategy in precision medicine (Table 1).
Clinical trial of microenvironment-targeting therapies in brain tumors
| Drug | Target | Phase | Tumor types | Study Identifier |
|---|---|---|---|---|
| BLZ945 | CSF-1R inhibitor | I/II | GBM/rGBM | NCT02829723 |
| PLX3397 | CSF-1R inhibitor | II | rGBM | NCT01349036 |
| Nivolumab | Anti-PD-1 | II/III | rGBM | NCT02017717 |
| Pembrolizumab | Anti-PD-1 | II | rGBM | NCT02337491 |
| Ipilimumab | anti-CTLA-4 | I | rGBM | NCT03233152 |
| DCVax-L | dendritic cell vaccine | III | GBM/rGBM | NCT00045968 |
| Bevacizumab | anti-VEGF-A | approved | Recurrent glioma | |
| Bevacizumab | anti-VEGF-A | III | Newly diagnosed GBM | NCT00884741 |
| Cediranib | inhibits VEGFR | II/III | rGBM | NCT00777153 |
TAM therapy
Targeting TAMs is a promising efficacious therapeutic strategy. Currently, approaches include inhibition of TAM recruitment, TAM depletion and TAM re-education.
The first approach is to inhibit the accumulation of macrophages or microglia in tumors by targeting chemoattractants and/or their receptors. Preclinical data show that a CCL2-neutralizing antibody prolongs the survival in mouse and human glioma xenograft models by inhibiting TAM infiltration [102]. In addition, blockade of CCL2/CCR2 signaling enhances the efficacy of temozolomide [102] or immune checkpoint inhibitors [103] in GBM mouse models.
Alternative strategies include reducing the population of immunosuppressive TAMs or converting TAMs from a tumor-promoting phenotype to a tumor-inhibiting phenotype. CSF-1/CSF-1R is the essential signaling pathway for macrophage survival and proliferation, but the CSF-1R inhibitor BLZ945 suppresses glioma growth and progression by altering macrophage polarization, but not by depleting macrophages [104], and improves the survival rate of glioma mice [105]. However, resistance to long-term BLZ945 treatment ultimately develops in a manner driven by the reprogramming of TAMs in the TME through the IGF-1/PI3K pathway [105]. PLX3397 (another CSF-1R inhibitor) markedly suppresses glioma cell proliferation and invasion and reduces the grade of malignancy in a PDGF-B-driven proneural glioma mouse model [106]. In addition, PLX3397 enhances the sensitivity of glioma cells to tyrosine kinase inhibitors by re-educating TAMs in preclinical combination trials [106]. Surprisingly, PLX3397 shows no survival benefit in clinical trials of GBM patient, despite showing good tolerability and adequate BBB penetration [107]. The SIRPα-CD47 signaling axis is important for macrophage phagocytosis. Recently, research demonstrated that anti-CD47 strategies show promising antitumor effects in various gliomas, including GBM [108] and pediatric glioma [109], by increasing TAM-mediated phagocytosis of macrophages.
Immune checkpoint blockade therapy
Glioma cells can evade immune surveillance through activation of immune checkpoint ligands such as PD-1, CTLA-4, and IDO. Numerous studies over the past decade have reported that preclinical trials have succeeded in the treatment of gliomas. CTLA-4 blockade prolongs survival in 80% of treated animals and enhances antitumor immunity by increasing the proliferation of CD4+ T cells without affecting Treg suppressive function [110]. The combination of PD-1 blockade and localized radiation therapy increases tumor infiltration by cytotoxic T cells and decreases Tregs, which results in long-term survival in mice with orthotopic glioma [111]. In a glioma mouse model, 100% of mice survive long-term following IDO, CTLA-4, and PD-L1 blockade therapy, and tumor-infiltrating of Tregs is decreased [112]. However, in CheckMate 143 (NCT02017717), the first clinical trial of PD pathway inhibition in GBM, nivolumab failed to prolong the OS of recurrent GBM patients compared with the anti-VEGF antibody bevacizumab [113]. Surprisingly, a recent clinical trial showed that anti-PD-1 immunotherapy induced local and systemic immunomodulatory treatment effects in recurrent glioblastoma [114]. In addition, a phase II study (NCT02337491) including 80 patients with recurrent glioblastoma reported that pembrolizumab (another PD-1 monoclonal antibody) alone or with bevacizumab therapy does not significantly improve OS [115]. Intracerebral administration of nivolumab and ipilimumab (CTLA-4 monoclonal antibody) was well-tolerated and associated with encouraging OS in recurrent glioblastoma patients with maximal safe resection [116].
Dendritic cell vaccines
Dendritic cell vaccines (DCVs) are also gaining substantial clinical attention because they have considerable promise in the treatment of glioma, and several preclinical and clinical trials are underway. The first two studies used DCVs to treat glioma patients, and this treatment safely induced immune responses [117] and enhanced the median survival of patients by improving tumor-specific T-cell activation, infiltration, and/or function [118]. In addition, 31 other studies have been published from 2001 to 2018 detailing the treatment of GBM with DCVs. Currently, there are three phase III trials using DCV treatment for GBM registered at ClinicalTrials.gov (#NCT00045968, #NCT01759810, # NCT02546102). Recent studies have also sought to identify factors that enhance the efficacy of DCVs. One study reported that combining a DCV with temozolomide could enhance antitumor immunity by increasing tumor-specific immune responses in glioma [119]. Gene therapy targeted to Flt3L and HSV1-TK in combination with a DCV can improve antitumor immunity, therapeutic efficacy and long-term survival by modifying the brain TME in a syngeneic glioma model [120]. Another study identified that the AKT inhibitor MK2206, the DNA-PK inhibitor NU7441, and the MEK inhibitor trametinib increase the ability of a DCV to activate and expand tumor-reactive T cells and augment antitumor activity in glioblastoma mice [121]. Pre-conditioning the vaccine site with a potent recall antigen (e.g., tetanus/diphtheria toxoid) significantly enhances DC migration bilaterally and improves the survival of glioblastoma patients, and this phenomenon is dependent on the chemokine CCL3 in murine models [122]. The combination of DC immunotherapy with hypericin-based photodynamic therapy to induce immunogenic cell death (ICD) enhances anti-tumor responses and improves OS in preclinical glioma models [123].
Targeting the vasculature
Angiogenesis is a hallmark of glioblastoma and remains an important therapeutic target in its treatment. Targeting blood vessel-related factors, particularly VEGF and VEGFR, has been an attractive strategy in gliomas. Bevacizumab, a humanized monoclonal antibody against VEGF, is currently approved for recurrent glioblastoma. In one study, after treatment with bevacizumab combined with other chemotherapy agents, edema was decreased and progression-free survival (PFS) and OS were increased in patients with recurrent glioma [124]. However, the use of bevacizumab does not improve OS and PFS in newly diagnosed glioblastoma [125]. This class of therapy causes adverse events, including hypertension, proteinuria, poor wound healing, thrombosis, convulsion, neutropenia, and reduced cognitive function, which impair the patient's quality of life over time.
An abnormal vasculature creates an abnormal microenvironment and blocks the effective delivery of therapeutic agents to tumors. Vascular normalization therapy supports physiological angiogenesis to improve the TME (reduce hypoxia, improve perfusion and drug delivery) and thus may be an alternative strategy for tumor therapy. AZD2171 (cediranib), a pan-VEGF receptor tyrosine kinase inhibitor, normalizes the structure and function of the tumor vasculature, alleviating edema and increasing tumor perfusion, which is associated with prolonged PFS and OS in recurrent glioblastoma patients [126]. These data identify potential new targets for recurrent glioblastoma treatment.
Conclusion
Gliomas are among the leading causes of cancer-related deaths and have poor prognosis and a high rate of recurrence. In recent years, tumors have been recognized as complex tissues that recruit TME cells that also play key roles in every aspect of tumor development. Increasing evidence indicates that tumor-TME cell interactions lead to the co-evolution of tumor cells and the surrounding environment, which is critical in glioma initiation and development. The development of appropriate mouse models could improve the understanding of the mechanisms underlying these interactions and the identification of new therapeutic targets for treatment. Considering the high abundance and genetic stability of cells in the TME, TME-targeting therapy has emerged as a promising strategy. TAM-directed therapies combined with immune checkpoint blockade therapy and/or DCVs could enhance the antitumor effect of tumor-infiltrating lymphocytes and then phenotypically shift the TME from a “cold” to a “hot” environment. Recently, the results of an encouraging phase 3 prospective externally controlled cohort trial (NCT00045968) indicated that an autologous tumor lysate-loaded DCV combined with temozolomide chemotherapy extended OS in both newly diagnosed and recurrent GBM [127]. In the future, clinical translation of treatment strategies to re-educate or manipulate factors in the abnormal TME in gliomas should be achieved.
Abbreviations
ADAM10: a disintegrin and metalloproteinase domain-containing protein 10; AMPAR: α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors; BBB: blood-brain barrier; BBTB: blood-brain tumor barrier; BMDMs: bone marrow-derived macrophages; BMX: bone marrow and X-linked; CAMK2A: calcium/calmodulin dependent protein kinase II alpha; CLNs: cervical lymph nodes; CNS: central nervous system; CSF-1: colony stimulating factor-1; CTLA-4: cytotoxic T lymphocyte 4; DCs: dendritic cells; DCVs: dendritic cell vaccines; ECM: extracellular matrix; EGF: epidermal growth factor; FGF: fibroblast growth factor; FGL2: fibrinogen-like protein 2; GBM: glioblastoma multiforme; GDF-15: growth differentiation factor 15; GFAP: glial fibrillary acidic protein; GFP: green fluorescent protein; GNPs: granule neuron precursors; GSCs: glioma stem cells; HGF: hepatocyte growth factor; HGG: high-grade gliomas; HMGB1: high mobility group box 1; IDO: Indoleamine 2,3-dioxygenase; IFN-γ: Interferon gamma; IGF-1: insulin-like growth factor 1; IRES: internal-ribosomal entry sites; LYVE-1: lymphatic endothelial hyaluronic acid receptor 1; MADM: Mosaic Analysis with Double Markers; MAPK: mitogen-activated protein kinases; MHC: major histocompatibility complex; MICA: class I-related chains A; MLV: Meningeal lymphatic vessels; MMP-2: matrix metalloproteinase-2; NETs: neutrophil extracellular traps; NF1: Neurofibromatosis type 1; NF-κB: nuclear factor kappa-B; NK: natural killer; NKG2D: NK group 2 member D; NLGN3: neuroligin-3; OPCs: oligodendrocyte precursor cells; OS: overall survival; PD-1: programmed cell death protein 1; PDGF-B: platelet-derived growth factor B; PD-L1: programmed cell death ligand 1; PFS: progression-free survival; PI3K: phosphatidylinositol-3-kinase; Prox1: prospero-related homeobox 1; PTEN: phosphatase and tensin homolog; RAGE: the receptor for advanced glycation end-products; RANKL: NF-κB ligand; RFP: red fluorescent protein; S1P1: Sphingosine-1-phosphate receptor 1; scRNA-seq: single-cell RNA sequencing; SDF-1: stromal-cell derived factor-1; SHH: Sonic hedgehog; SIRPα: Signal regulatory protein α; STAT-3: signal transducer and activator of transcription 3; STI1: stress-inducible protein 1; TAAs: tumor-associated astrocytes; TAMs: tumor associated myeloid cells; TGF-β: transforming growth factor-β; Tim-3: mucin-domain-containing molecule 3; TINs: tumor-infiltrating neutrophils; TLR2: Toll-like receptor 2; TME: tumor microenvironment; TNF-α: tumor necrosis factor; Tregs: regulatory T cells; TRM: tissue-resident microglia; ULPB-2: UL16 binding protein; VEGF: vascular endothelial growth factor; VSVG: virus G glycoprotein; WHO: World Health Organization; WT: wild-type.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant number 82072787), Guangdong Basic and Applied Basic Research Foundation (grant number 202102010368), Department of Science and Technology of Guangdong Province International Cooperation Project (grant number 2022A0505050012), Tertiary Education Scientific research project of Guangzhou Municipal Education Bureau (202235421) and Natural Science Foundation of Guangdong Province (grant number 2020KZDZX1160).
Author contributions
Maojin Yao and Yang Hu conceived the review, designed and produced illustrative material, and wrote the manuscript. Jin Li, Jiaoyan Ren, Zhixing Li and Yichi Zhang supervised and revised the manuscript. Yuzheng Wu, Zihao Liu, Jianhao Zeng and Zhexue Hao participated in data organization and discussion. All authors approved the final version.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
2. Stephen, Paget. THE DISTRIBUTION OF SECONDARY GROWTHS IN CANCER OF THE BREAST
3. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nature medicine. 2013;19:1423-37
4. Fukumura D, Jain RK. Tumor microenvironment abnormalities: causes, consequences, and strategies to normalize. Journal of cellular biochemistry. 2007;101:937-49
5. Chen F, Zhuang X, Lin L, Yu P, Wang Y, Shi Y. et al. New horizons in tumor microenvironment biology: challenges and opportunities. BMC medicine. 2015;13:45
6. Louis DN, Perry A, Wesseling P. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro-oncology. 2021;23:1231-51
7. Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011-2015. Neuro-oncology. 2018;20:iv1-iv86
8. Stupp R, Mason WP, Van dB, M J, Weller M, Fisher B, Taphoorn MJB. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. 9: 196-7.
9. Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol. 2017
10. Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nature neuroscience. 2016;19:20-7
11. Brandao M, Simon T, Critchley G, Giamas G. Astrocytes, the rising stars of the glioblastoma microenvironment. Glia. 2018;67:779-90
12. Johung T, Monje M. Neuronal activity in the glioma microenvironment. Current Opinion in Neurobiology. 2017;47:156-61
13. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759-67
14. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57-70
15. Morantz RA, Wood GW, Foster M, Clark M, Gollahon K. Macrophages in experimental and human brain tumors. Part 2: studies of the macrophage content of human brain tumors. J Neurosurg. 1979;50:305-11
16. Bettinger I, Thanos S, Paulus W. Microglia promote glioma migration. Acta neuropathologica. 2002;103:351-5
17. Markovic DS, Vinnakota K, Chirasani S, Synowitz M, Raguet H, Stock K. et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12530-5
18. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J. et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. 2015;17:170-82
19. Müller A, Brandenburg S, Turkowski K, Müller S, Vajkoczy P. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int J Cancer. 2015;137:278-88
20. Tzetzo SL, Abrams SI. Redirecting macrophage function to sustain their "defender" antitumor activity. Cancer cell. 2021;39:734-7
21. Rao G, Latha K, Ott M, Sabbagh A, Marisetty A, Ling X. et al. Anti-PD-1 Induces M1 Polarization in the Glioma Microenvironment and Exerts Therapeutic Efficacy in the Absence of CD8 Cytotoxic T Cells. 2020; 26: 4699-712.
22. Dey M, Chang AL, Miska J, Wainwright DA, Ahmed AU, Balyasnikova IV. et al. Dendritic Cell-Based Vaccines that Utilize Myeloid Rather than Plasmacytoid Cells Offer a Superior Survival Advantage in Malignant Glioma. Journal of immunology. 2015;195:367-76
23. Yan J, Zhao Q, Gabrusiewicz K, Kong LY, Xia X, Wang J. et al. FGL2 promotes tumor progression in the CNS by suppressing CD103(+) dendritic cell differentiation. Nature communications. 2019;10:448
24. Fossati G, Ricevuti G, Edwards SW, Walker C, Dalton A, Rossi ML. Neutrophil infiltration into human gliomas. Acta Neuropathol. 1999;98:349-54
25. Wang Z, Zhong L, Li G, Huang R, Wang Q, Wang Z. et al. Pre-treatment neutrophils count as a prognostic marker to predict chemotherapeutic response and survival outcomes in glioma: a single-center analysis of 288 cases. American journal of translational research. 2020;12:90-104
26. Zha C, Meng X, Li L, Mi S, Qian D, Li Z. et al. Neutrophil extracellular traps mediate the crosstalk between glioma progression and the tumor microenvironment via the HMGB1/RAGE/IL-8 axis. 2020; 17: 154-68.
27. Liang J, Piao Y, Holmes L, Fuller GN, Henry V, Tiao N. et al. Neutrophils promote the malignant glioma phenotype through S100A4. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:187-98
28. Lee SY, Kim JK, Jeon HY, Ham SW, Kim H. CD133 Regulates IL-1beta Signaling and Neutrophil Recruitment in Glioblastoma. Molecules and cells. 2017;40:515-22
29. Xue J, Zhao Z, Zhang L, Xue L, Shen S, Wen Y. et al. Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nature nanotechnology. 2017;12:692-700
30. Kmiecik J, Poli A, Brons NH, Waha A, Eide GE, Enger PO. et al. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. Journal of neuroimmunology. 2013;264:71-83
31. Perng P, Lim M. Immunosuppressive Mechanisms of Malignant Gliomas: Parallels at Non-CNS Sites. Frontiers in oncology. 2015;5:153
32. Nduom EK, Wei J, Yaghi NK, Huang N, Kong LY, Gabrusiewicz K. et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro-oncology. 2016;18:195-205
33. Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res. 2013;19:3165-75
34. Liu F, Huang J, Liu X, Cheng Q, Luo C, Liu Z. CTLA-4 correlates with immune and clinical characteristics of glioma. Cancer cell international. 2020;20:7
35. Chongsathidkiet P, Jackson C, Koyama S. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. 2018; 24: 1459-68.
36. Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D. et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer research. 2016;76:5671-82
37. Crane CA, Ahn BJ, Han SJ, Parsa AT. Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: implications for immunotherapy. Neuro-oncology. 2012;14:584-95
38. Heimberger AB, Abou-Ghazal M, Reina-Ortiz C, Yang DS, Sun W, Qiao W. et al. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:5166-72
39. Sayour EJ, McLendon P, McLendon R, De Leon G, Reynolds R, Kresak J. et al. Increased proportion of FoxP3+ regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer Immunology, Immunotherapy. 2015;64:419-27
40. Fecci PE, Sweeney AE, Grossi PM, Nair SK, Learn CA, Mitchell DA. et al. Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:4294-305
41. Avril T, Vauleon E, Hamlat A, Saikali S, Etcheverry A, Delmas C. et al. Human glioblastoma stem-like cells are more sensitive to allogeneic NK and T cell-mediated killing compared with serum-cultured glioblastoma cells. Brain pathology. 2012;22:159-74
42. Fernández L, Portugal R, Valentín J, Martín R, Maxwell H, González-Vicent M. et al. In vitro Natural Killer Cell Immunotherapy for Medulloblastoma. Frontiers in oncology. 2013 3
43. O'Brown NM, Pfau SJ, Gu C. Bridging barriers: a comparative look at the blood-brain barrier across organisms. Genes Dev. 2018;32:466-78
44. van Tellingen O, Yetkin-Arik B, de Gooijer MC, Wesseling P, Wurdinger T, de Vries HE. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat. 2015;19:1-12
45. Phoenix TN, Patmore DM, Boop S, Boulos N, Jacus MO, Patel YT. et al. Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer cell. 2016;29:508-22
46. Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nature reviews Neuroscience. 2007;8:610-22
47. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD. et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337-41
48. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M. et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. The Journal of experimental medicine. 2015;212:991-9
49. Meng FW, Liu FS, Liu WH, Li L, Jie LL. Formation of new lymphatic vessels in glioma: An immunohistochemical analysis. Neuropathology: official journal of the Japanese Society of Neuropathology. 2020
50. Hu X, Deng Q, Ma L, Li Q, Chen Y, Liao Y. et al. Meningeal lymphatic vessels regulate brain tumor drainage and immunity. Cell research. 2020;30:229-43
51. Zhang H, Zhou Y, Cui B, Liu Z, Shen H. Novel insights into astrocyte-mediated signaling of proliferation, invasion and tumor immune microenvironment in glioblastoma. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2020;126:110086
52. Nagashima G, Suzuki R, Asai J, Fujimoto T. Immunohistochemical analysis of reactive astrocytes around glioblastoma: an immunohistochemical study of postmortem glioblastoma cases. Clin Neurol Neurosurg. 2002;104:125-31
53. Kim JK, Jin X, Sohn YW, Jin X, Jeon HY, Kim EJ. et al. Tumoral RANKL activates astrocytes that promote glioma cell invasion through cytokine signaling. Cancer letters. 2014;353:194-200
54. Lu P, Wang Y, Liu X, Wang H, Zhang X, Wang K. et al. Malignant gliomas induce and exploit astrocytic mesenchymal-like transition by activating canonical Wnt/beta-catenin signaling. Medical oncology (Northwood, London, England). 2016;33:66
55. Chen W, Wang D, Du X, He Y, Chen S, Shao Q. et al. Glioma cells escaped from cytotoxicity of temozolomide and vincristine by communicating with human astrocytes. Medical oncology (Northwood, London, England). 2015;32:43
56. Barres BA, Raff MC. Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature. 1993;361:258-60
57. Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS. et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science. 2014;344:1252304
58. Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H. et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011;146:209-21
59. Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S. et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell. 2015;161:803-16
60. Venkatesh HS, Tam LT, Woo PJ, Lennon J, Nagaraja S, Gillespie SM. et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature. 2017;549:533-7
61. Bergles DE, Roberts JD, Somogyi P, Jahr CE. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature. 2000;405:187-91
62. Lin SC, Bergles DE. Synaptic signaling between GABAergic interneurons and oligodendrocyte precursor cells in the hippocampus. Nat Neurosci. 2004;7:24-32
63. Venkatesh HS, Morishita W, Geraghty AC, Silverbush D, Gillespie SM, Arzt M. et al. Electrical and synaptic integration of glioma into neural circuits. Nature. 2019;573:539-45
64. Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB. et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer research. 2006;66:7843-8
65. Thirant C, Galan-Moya EM, Dubois LG, Pinte S, Chafey P, Broussard C. et al. Differential proteomic analysis of human glioblastoma and neural stem cells reveals HDGF as a novel angiogenic secreted factor. Stem cells. 2012;30:845-53
66. Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK. et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013;153:139-52
67. Zhou W, Chen C, Shi Y, Wu Q, Gimple RC, Fang X. et al. Targeting Glioma Stem Cell-Derived Pericytes Disrupts the Blood-Tumor Barrier and Improves Chemotherapeutic Efficacy. Cell Stem Cell. 2017;21:591-603.e4
68. Roesch S, Rapp C, Dettling S, Herold-Mende C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. International journal of molecular sciences. 2018 19
69. Platten M, Kretz A, Naumann U, Aulwurm S, Egashira K, Isenmann S. et al. Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Annals of neurology. 2003;54:388-92
70. Okada M, Saio M, Kito Y, Ohe N, Yano H, Yoshimura S. et al. Tumor-associated macrophage/microglia infiltration in human gliomas is correlated with MCP-3, but not MCP-1. International journal of oncology. 2009;34:1621-7
71. Wang SC, Hong JH, Hsueh C, Chiang CS. Tumor-secreted SDF-1 promotes glioma invasiveness and TAM tropism toward hypoxia in a murine astrocytoma model. Laboratory investigation; a journal of technical methods and pathology. 2012;92:151-62
72. Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH. et al. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Molecular medicine. 2012;18:519-27
73. Wick W, Platten M, Weller M. Glioma cell invasion: regulation of metalloproteinase activity by TGF-beta. Journal of neuro-oncology. 2001;53:177-85
74. Fonseca AC, Romão L, Amaral RF, Assad Kahn S, Lobo D, Martins S. et al. Microglial stress inducible protein 1 promotes proliferation and migration in human glioblastoma cells. Neuroscience. 2012;200:130-41
75. Naso MF, Zimmermann DR, Iozzo RV. Characterization of the complete genomic structure of the human versican gene and functional analysis of its promoter. The Journal of biological chemistry. 1994;269:32999-3008
76. Hu F, Dzaye O, Hahn A, Yu Y, Scavetta RJ, Dittmar G. et al. Glioma-derived versican promotes tumor expansion via glioma-associated microglial/macrophages Toll-like receptor 2 signaling. Neuro-oncology. 2015;17:200-10
77. Chen X, Zhang L, Zhang IY, Liang J, Wang H, Ouyang M. et al. RAGE expression in tumor-associated macrophages promotes angiogenesis in glioma. Cancer research. 2014;74:7285-97
78. Brandenburg S, Müller A, Turkowski K, Radev YT, Rot S, Schmidt C. et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta neuropathologica. 2016;131:365-78
79. Hutter G, Theruvath J, Graef CM, Zhang M. Microglia are effector cells of CD47-SIRPα antiphagocytic axis disruption against glioblastoma. 2019; 116: 997-1006.
80. Yuan F, Ming H, Wang Y, Yang Y, Yi L, Li T. et al. Molecular and clinical characterization of Galectin-9 in glioma through 1,027 samples. Journal of cellular physiology. 2020;235:4326-34
81. Didenko VV, Ngo HN, Minchew C, Baskin DS. Apoptosis of T lymphocytes invading glioblastomas multiforme: a possible tumor defense mechanism. Journal of neurosurgery. 2002;96:580-4
82. Dieckmann D, Plottner H, Berchtold S, Berger T, Schuler G. Ex vivo isolation and characterization of CD4(+)CD25(+) T cells with regulatory properties from human blood. The Journal of experimental medicine. 2001;193:1303-10
83. Ebner F, Brandt C, Thiele P, Richter D, Schliesser U, Siffrin V. et al. Microglial activation milieu controls regulatory T cell responses. Journal of immunology. 2013;191:5594-602
84. Xu L, Xiao H, Xu M, Zhou C, Yi L, Liang H. Glioma-derived T cell immunoglobulin- and mucin domain-containing molecule-4 (TIM4) contributes to tumor tolerance. The Journal of biological chemistry. 2011;286:36694-9
85. Lindberg N, Kastemar M, Olofsson T, Smits A, Uhrbom L. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene. 2009;28:2266-75
86. Persson AI, Petritsch C, Swartling FJ, Itsara M, Sim FJ, Auvergne R. et al. Non-stem cell origin for oligodendroglioma. Cancer cell. 2010;18:669-82
87. Lei L, Sonabend AM, Guarnieri P, Soderquist C, Ludwig T, Rosenfeld S. et al. Glioblastoma models reveal the connection between adult glial progenitors and the proneural phenotype. PloS one. 2011;6:e20041
88. Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O. et al. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012;338:1080-4
89. Hede SM, Hansson I, Afink GB, Eriksson A, Nazarenko I, Andrae J. et al. GFAP promoter driven transgenic expression of PDGFB in the mouse brain leads to glioblastoma in a Trp53 null background. Glia. 2009;57:1143-53
90. Weiss WA, Burns MJ, Hackett C, Aldape K, Hill JR, Kuriyama H. et al. Genetic determinants of malignancy in a mouse model for oligodendroglioma. Cancer research. 2003;63:1589-95
91. Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ. et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129-33
92. Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK. et al. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer cell. 2009;15:45-56
93. Yao M, Ventura PB, Jiang Y, Rodriguez FJ, Wang L, Perry JSA. et al. Astrocytic trans-Differentiation Completes a Multicellular Paracrine Feedback Loop Required for Medulloblastoma Tumor Growth. Cell. 2020;180:502-20.e19
94. Venteicher AS, Tirosh I. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. 2017; 355.
95. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD. et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell. 2010;17:98-110
96. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396-401
97. Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ. et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. 2019;178:835-49.e21
98. Yeo AT, Rawal S, Delcuze B, Christofides A, Atayde A, Strauss L. et al. Single-cell RNA sequencing reveals evolution of immune landscape during glioblastoma progression. 2022; 23: 971-84.
99. Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A. et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. 2017; 18: 234.
100. Xie Y, He L, Lugano R, Zhang Y, Cao H, He Q. et al. Key molecular alterations in endothelial cells in human glioblastoma uncovered through single-cell RNA sequencing. JCI insight. 2021 6
101. Wu L, Wu W. Natural Coevolution of Tumor and Immunoenvironment in Glioblastoma. 2022; 12: 2820-37.
102. Zhu X, Fujita M, Snyder LA, Okada H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. Journal of neuro-oncology. 2011;104:83-92
103. Flores-Toro JA, Luo D, Gopinath A, Sarkisian MR, Campbell JJ, Charo IF. et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. 2020; 117: 1129-38.
104. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF. et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nature medicine. 2013;19:1264-72
105. Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT. et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science. 2016;352:aad3018
106. Yan D, Kowal J, Akkari L, Schuhmacher AJ, Huse JT, West BL. et al. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene. 2017;36:6049-58
107. Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY. et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-oncology. 2016;18:557-64
108. Zhang M, Hutter G, Kahn SA, Azad TD, Gholamin S, Xu CY. et al. Anti-CD47 Treatment Stimulates Phagocytosis of Glioblastoma by M1 and M2 Polarized Macrophages and Promotes M1 Polarized Macrophages In vivo. PloS one. 2016;11:e0153550
109. Gholamin S, Mitra SS. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. 2017; 9.
110. Fecci PE, Ochiai H, Mitchell DA, Grossi PM, Sweeney AE, Archer GE. et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res. 2007;13:2158-67
111. Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J. et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86:343-9
112. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A. et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. 2014;20:5290-301
113. Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A. et al. Effect of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA oncology. 2020;6:1003-10
114. Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, Idoate MA. et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. 2019; 25: 470-6.
115. Nayak L, Molinaro AM, Peters K, Clarke JL. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. 2021; 27: 1048-57.
116. Duerinck J, Schwarze JK. Intracerebral administration of CTLA-4 and PD-1 immune checkpoint blocking monoclonal antibodies in patients with recurrent glioblastoma: a phase I clinical trial. 2021; 9.
117. Kikuchi T, Akasaki Y, Irie M, Homma S, Abe T, Ohno T. Results of a phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer Immunol Immunother. 2001;50:337-44
118. Yu JS, Wheeler CJ, Zeltzer PM, Ying H, Finger DN, Lee PK. et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001;61:842-7
119. Park SD, Kim CH, Kim CK, Park JA, Sohn HJ, Hong YK. et al. Cross-priming by temozolomide enhances antitumor immunity of dendritic cell vaccination in murine brain tumor model. Vaccine. 2007;25:3485-91
120. Mineharu Y, King GD, Muhammad AK, Bannykh S, Kroeger KM, Liu C. et al. Engineering the brain tumor microenvironment enhances the efficacy of dendritic cell vaccination: implications for clinical trial design. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:4705-18
121. Guo J, Muse E, Christians AJ, Swanson SJ, Davila E. An Anticancer Drug Cocktail of Three Kinase Inhibitors Improved Response to a Dendritic Cell-Based Cancer Vaccine. Cancer immunology research. 2019;7:1523-34
122. Mitchell DA, Batich KA, Gunn MD, Huang M-N, Sanchez-Perez L, Nair SK. et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519:366-9
123. Garg AD, Vandenberk L, Koks C, Verschuere T, Boon L, Van Gool SW. et al. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci Transl Med. 2016;8:328ra27
124. Vredenburgh JJ, Desjardins A, Herndon JE 2nd, Dowell JM, Reardon DA, Quinn JA. et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2007;13:1253-9
125. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA. et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. The New England journal of medicine. 2014;370:699-708
126. Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS. et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer cell. 2007;11:83-95
127. Liau LM, Ashkan K, Brem S, Campian JL, Trusheim JE, Iwamoto FM. et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination With Extension of Survival Among Patients With Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA oncology. 2022
Author contact
![]() Corresponding authors: Maojin Yao, yaomaojincn. Jiaoyan Ren, jyrenedu.cn (J. Ren); Jin Li, JinLedu.cn.
Corresponding authors: Maojin Yao, yaomaojincn. Jiaoyan Ren, jyrenedu.cn (J. Ren); Jin Li, JinLedu.cn.