Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(13):4327-4339. doi:10.7150/ijbs.86614 This issue Cite
Research Paper
Sirtuin-3 activates the mitochondrial unfolded protein response and reduces cerebral ischemia/reperfusion injury
Xie Xiaowei1,2, Xu Qian3, Zhou Dingzhou1 ![]()
1. Department of Neurosurgery, Hunan Provincial People' s Hospital (The First-Affiliated Hospital of Hunan Normal University), Changsha 410005, Hunan Province, People's Republic of China.
2. Clinical Nursing Teaching and Research Section, The Second Xiangya Hospital, Central South University, Changsha 410011, Hunan Province, People's Republic of China.
3. Department of Neurology, Haikou City People' s Hospital, Xiangya School of Medicine, Central South University, Haikou 570100, Hainan Province, People's Republic of China.
Received 2023-5-29; Accepted 2023-8-6; Published 2023-8-21
Abstract

Sirtuin-3 (Sirt3) deacetylates several mitochondrial proteins implicated into cerebral ischemia/reperfusion (I/R) injury. The mitochondrial unfolded protein response (UPRmt) favors mitochondrial proteostasis during various stressors. Here, we used Sirt3 transgenic mice and a transient middle cerebral artery occlusion model to evaluate the molecular basis of Sirt3 on the UPRmt during brain post-ischemic dysfunction. The present study illustrated that Sirt3 abundance was suppressed in the brain after brain ischemic abnormalities. Overexpression of Sirt3 in vivo suppressed the infarction size and attenuated neuroinflammation after brain I/R injury. Sirt3 overexpression restored neural viability by reducing mitochondrial ROS synthesis, maintaining the mitochondrial potential and improving mitochondrial adenosine triphosphate synthesis. Sirt3 overexpression protected neuronal mitochondria against brain post-ischemic malfunction via eliciting the UPRmt by the forkhead box O3 (Foxo3)/sphingosine kinase 1 (Sphk1) pathway. Inhibiting either the UPRmt or the Foxo3/Sphk1 pathway relieved the favorable influence of Sirt3 on neural function and mitochondrial behavior. In contrast, Sphk1 overexpression was sufficient to reduce the infarction size, attenuate neuroinflammation, sustain neuronal viability and prevent mitochondrial abnormalities during brain post-ischemia dysfunction. Thus, the UPRmt protects neural viability and mitochondrial homeostasis, and the Sirt3/Foxo3/Sphk1 pathway is a promosing therapeutic candidate for ischemic stroke.
Keywords: Sirt3, UPRmt, cerebral I/R injury, Foxo3, Sphk1, mitochondria
Introduction
Cerebral ischemia/reperfusion (I/R) injury results from the restoration of blood flow to the ischemic brain after a stroke, cardiac arrest or traumatic brain injury [1, 2]. I/R injury is initiated in the ischemic phase, mainly as a result of deficiency in oxygen or nutrients supply to neural cells [3]. However, when blood flow is restored during the reperfusion phase, the ischemic zone of the brain can undergo oxidative stress, inflammatory responses and disordered energy metabolism [4], resulting in blood-brain barrier disruption, neuronal death [5], brain edema and hemorrhaging [6]. Considering that cerebral I/R injury has been associated with motor dysfunction, cognitive impairment and even death, it is important to determine the pathological alterations underlying brain I/R.
Recent data reported that mitochondrial abnormality as a key player in brain post-ischemic injury, for several reasons [7, 8]. First, interrupted oxidative phosphorylation due to mitochondrial dysfunction can induce neural energy failure [9, 10]. Second, mitochondrial damage can cause the release of damage-associated molecular patterns [11], thus triggering an inflammatory response, activating microglia/astrocytes and further exacerbating brain damage. Third, disrupted mitochondrial function increases the generation of ROS, thereby contributing to oxidative injury in the brain [12, 13]. Last but not least, poorly structured mitochondria transmit pro-apoptotic signal [14, 15], thus augmenting I/R-induced neuronal death.
Regarding mitochondrial damage, the mitochondrial unfolded protein response (UPRmt) is elicited as an endogenous protective system to modify protein folding and prevent abnormal protein accumulation/expression within mitochondria [16, 17]. The protective effects of the UPRmt on the brain have been confirmed in a rat traumatic brain injury model [18]. Moreover, activation of the UPRmt has been shown to promote mitochondrial proteostasis during adult neurogenesis [19], and to prevent oxidative stress-related neurodegenerative diseases during aging [20]. However, the mechanisms of the UPRmt in brain post-ischemic damage are not yet completely investigated [21, 22].
Sirtuin-3 (Sirt3) is a mitochondrial deacetylase that modifies several mitochondrial proteins involved in energy metabolism, ROS production and apoptosis [23, 24]. Sirt3 was reported to be involved in the UPRmt in cancer cells [25], and increased Sirt3 expression was associated with the upregulation of UPRmt-related genes such as the caseinolytic mitochondrial matrix peptide proteolytic subunit (CLPP), 60-kDa heat shock protein (HSP60) and Lon peptidase 1 (LONP1) in breast cancer [26]. In the heart, Sirt3 was found to trigger the mRNA expression of UPRmt-related genes by the AMPK pathway, thus improving metabolic remodeling and reducing cardiac hypertrophy [27]. Considering that Sirt3 activity is beneficial during brain post-ischemic dysfunction [28, 29], we wondered whether Sirt3 overexpression restrain reperfusion-caused brain abnormalities through eliciting the UPRmt.
Sphingosine kinase 1 (Sphk1) is the rate-limiting enzyme in the conversion of sphingosine into sphingosine-1-phosphate, an important regulator of mitochondrial lipid metabolism. Following mitochondrial stress, Sphk1 translocates to the outer mitochondrial membrane, where it increases sphingosine-1-phosphate production and ultimately activates the UPRmt [30, 31]. The protein stability of Sphk1 is determined by forkhead box O3 (Foxo3), and the degradation of Foxo3a is prevented by deacetylation [32]. Given that Sirt3 is a mitochondrial deacetylase, in this study we investigated whether Sirt3 could induce the UPRmt through the Foxo3a/Sphk1 pathway in order to reduce cerebral I/R injury.
Materials and Methods
Animals and models
Sphk1Tg and Sirt3Tg mice were established as previously described [33]. WT, Sphk1Tg and Sirt3Tg mice were treated with 10% chloral hydrate (350 mg/kg, intraperitoneally), and then were subjected to transient MCAO on the right side with a nylon filament. The internal and external carotid arteries were detached, and the middle cerebral artery was blocked with a 4-0 monofilament fiber. The sham surgery was performed similarly, but without MCAO [14]. After two hours of ischemia, the fiber was removed for reperfusion, and the mice were monitored for successful model generation based on their observed symptoms. After 24 hours of reperfusion, the mice were euthanized and their brain tissues were collected. The infarction size in the brain was observed using 2% TTC (Sigma) [34].
Cellular experiments
The N2a cell line was obtained from ATCC (CCL-131TM). DMEM containing 10% fetal bovine serum was used to treat N2a cells. To induce N2a cell I/R injury, cells were cultured in an airtight chamber containing 95% N2 and 5% CO2 for 30 minutes to induce oxygen-glucose deficiency, and then the chamber was sealed with normal oxygen for four hours to induce reoxygenation injury [35]. AEBSF (3 μM, cat. no. S7378, Selleck) was used to inhibit the UPRmt in N2a cells. Carbenoxolone (5 mM, cat. no. S4368, Selleck) was used to inhibit the Foxo3 pathway in N2a cells.
Histology
Fresh brain tissues were firstly treated using 4% paraformaldehyde, followed by embedding using paraffin. Then, 5-μm sections of brain tissues were stained H&E and Nissl staining [36].
qRT-PCR and western blot analysis
The primers used in the present experiments were showed in supplemental information. Protein extracts were obtained from cells or tissues, quantified with a bicinchoninic acid assay. The separated proteins were moved to polyvinylidene difluoride membranes and then treated with antibodies at 4 ℃ overnight [37]. The samples were washed and treated with the corresponding secondary antibodies for one hour [38]. After another washing, the membranes were treated with enhanced chemiluminescence reagents for imaging. ImageJ was used to analyze the relative expression of targeted protein bands. The primary antibodies were as follows: Foxo3 (1:1000, ab23683, Abcam), Sphk1 (1:1000, #3297, Cell Signaling Technology) and GAPDH (1:1000, ab8245, Abcam).
ELISAs
Cells were collected after OGD/R, and ELISAs were performed to analyze the activity of caspase-3 (cat. no. 62218, ThermoFisher), caspase-9 (cat. no. NBP2-75042, Novus), ATP (Mouse Adenosine Triphosphate [ATP] ELISA Kit, cat. no. MOEB2556, AssayGenie), GSH (Mouse Glutathione ELISA Kit, cat. no. MOEB2568, AssayGenie), SOD (Mouse Super Oxidase Dimutase ELISA Kit, cat. no. CSB-E08556m, Cusabio), Complex I (Complex I Enzyme Activity Microplate Assay Kit, cat. no. ab109712, Abcam) and Complex III (cat. no. ab287844, Abcam), according to the manufacturers' instructions and as described previously [39].
Adenovirus transfection
The Sirt3 and Sphk1 adenoviruses were purchased from Vectro Biolabs [40]. AD-293T cells were treated using the adenoviruses (2 plaque-forming units/cell) in serum-free DMEM for one hour. After cell reached 90% confluence, cells were completely lysed (typically after 72 hours). The cells would be destroyed and the maximum number of virions would be freed [41]. A multiplicity of infection of 300 in 1 mL of medium was used to infect N2a cells.
Cell viability determination
Cells at 40% confluency in 24-well plates were transfected with the indicated adenovirus for 48 hours, and then were subjected to OGD/R. MTT assays (Sigma-Aldrich) were performed on a SmartReaderTM 96 (Accuris Instruments) at 570 nm to assess cell viability [39]. The levels of LDH were measured with a Mouse Lactate Dehydrogenase (LDH) ELISA Kit (cat. no. MODL00786, AssayGenie) [42].
Immunofluorescence staining
N2a cells were transfected with the adenovirus and then fixed using 4% paraformaldehyde/PBS, followed by incubation with antibodies (Foxo3, 1:1000, ab23683, Abcam; Sphk1, 1:1000, #3297, Cell Signaling Technology) for two hours. Images were observed under an inverted-phase contrast microscope (Olympus) [38].
Mitochondrial membrane potential and mitochondrial ROS staining
The membrane-permeant JC-1 dye was applied to evaluate the changes in the mitochondrial potential [22]. In brief, JC-1 (5 μM, cat. no. T3168, ThermoFisher) was used to incubate with N2a cells and then observed under an inverted-phase contrast microscope (Olympus) [40]. To capture the production of mitochondrial ROS, N2a cells were treated with MitoSOXTM (10 μM, cat. no. M36008, ThermoFisher) for 20 minutes and then observed under an inverted-phase contrast microscope (Olympus) [43].
Statistical analysis
Data are shown as the mean ± SEM and all the experiments were treated at least three times. Student's t-tests for comparisons of two group and one-way analysis of variance for comparisons of multiple groups were performed using GraphPad Prism 7 software (GraphPad Software, San Diego, CA, USA). P values < 0.05 were considered statistically significant.
Results
Sirt3 overexpression attenuates brain I/R injury
To illuminate the molecular basis of Sirt3 on brain I/R injury, Sirt3 expression was assessed by western blotting in the brain in a mouse middle cerebral artery occlusion (MCAO) model. Results exhibited that the transcription of Sirt3 in the brain was markedly suppressed by MCAO (Figure 1A). ELISA further confirmed that Sirt3 activity in the brain was reduced after MCAO treatment.
Sirt3 overexpression attenuates cerebral I/R injury. WT and Sirt3Tg mice were subjected to transient MCAO. A, B. TTC staining was used to observe the infarction size in the brain after MCAO. C. H&E staining was used to detect histological alterations in the brain after MCAO. D, E. Nissl staining was used to observe the number of Nissl bodies in brain tissues after MCAO. F-I. RNA was isolated from brain tissues, and the levels of IL-6, CRP, TNFα and MCP1 were analyzed. J, K. ELISAs were used to detect GSH and SOD activity levels in brain tissues after MCAO. *p<0.05 vs. sham+WT group, #p<0.05 vs. MCAO+WT group.
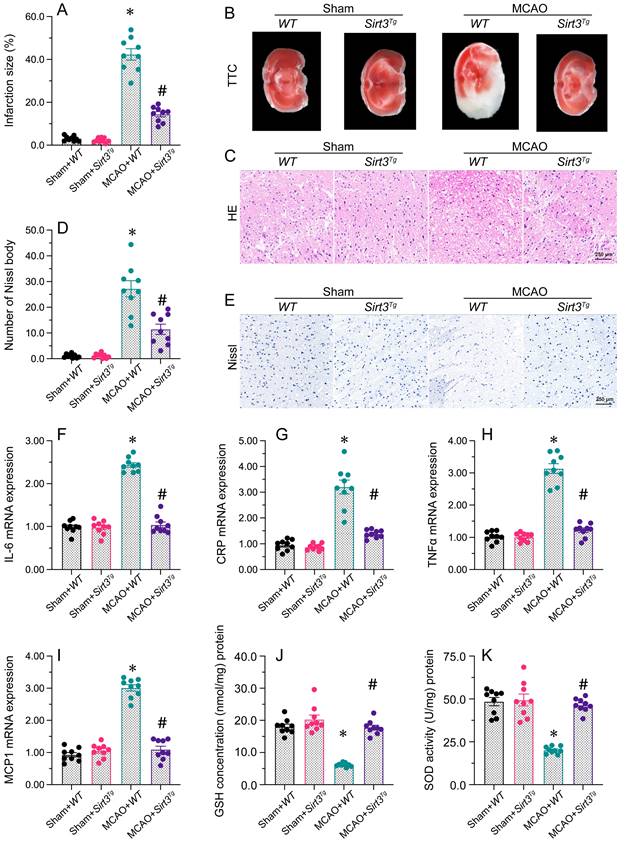
To determine whether normalizing Sirt3 expression could reduce the vulnerability of the brain to I/R injury, we next subjected Sirt3 transgenic (Sirt3Tg) mice to MCAO, and compared their infarct sizes with those of wild-type (WT) mice. Staining with TTC revealed that MCAO drastically promoted infarction in the brain tissues of WT mice, but not of Sirt3Tg mice (Figure 1A). H&E staining of the infarct zone showed that the WT tissues had a porous appearance, and the neuronal nuclei were deeply stained. Moreover, the penumbral tissue was swollen, microglial cells were more prevalent and vascular hyperplasia was detected in WT brain tissues following MCAO. Interestingly, these histological alterations were not detectable in Sirt3Tg mice subjected to MCAO. In addition, Nissl staining of brain tissues demonstrated that Nissl bodies were rapidly reduced after MCAO in WT mice. The above phenotypic alterations were relieved in Sirt3Tg mice.
Next, we performed qRT-PCR analyses, which detected that MCAO elevated the mRNA expressions of IL-6, CRP, TNFα and MCP1 in WT brain tissues. However, overexpression of Sirt3 prevented this pro-inflammatory response. Moreover, the activities of deoxidation enzymes were significantly downregulated after MCAO in WT brain tissues, while Sirt3 overexpression increased the anti-oxidative capacity of the brain following MCAO. Thus, Sirt3 overexpression was sufficient to ameliorate cerebral I/R injury.
Sirt3 maintains neuronal viability and reduces OGD/R-caused cell death
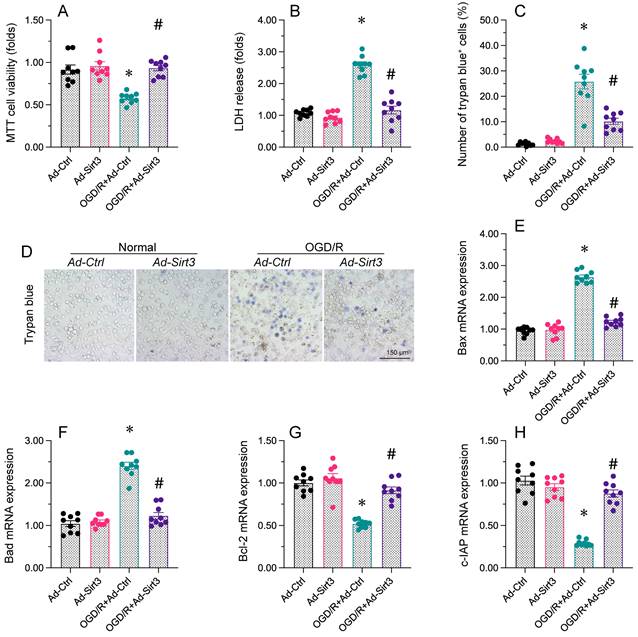
A drop in cell viability and an increased rate of apoptosis are thought to augment the brain post-ischemic dysfunction. Therefore, next work is to figure out Sirt3 overexpression could increase neural viability. Neuro-2a (N2a) cells were transfected with a Sirt3 overexpression-adenovirus (Ad-Sirt3) or control overexpression-adenovirus (Ad-Ctrl), and then were treated with oxygen-glucose deprivation and reoxygenation (OGD/R). A MTT assay illuminated that cell viability was reduced by OGD/R, whereas Ad-Sirt3 transfection maintained cell viability following OGD/R. Similarly, a LDH release experiment demonstrated that OGD/R increased LDH leakage from Ad-Ctrl cells, whereas Ad-Sirt3 treatment reversed this change. Trypan blue staining indicated that OGD/R elevated the ratio of apoptotic N2a cells in the Ad-Ctrl group; this increase was lessened in the Ad-Sirt3 group. An ELISA revealed that caspase-3 activity was rapidly elevated by OGD/R in Ad-Ctrl cells, but was attenuated to near-physiological condtions in Ad-Sirt3 cells. We also observed that OGD/R enhanced the transcription of fatal genes such as Bax and Bad, while it markedly reduced the levels of pro-survival genes such as Bcl-2 and c-IAP in Ad-Ctrl cells. Transfection of Ad-Sirt3 suppressed Bax/Bad transcription and increased Bcl-2/c-IAP levels following OGD/R. Thus, Sirt3 overexpression maintained cell viability and prevented apoptosis in neurons.
Sirt3 stabilizes mitochondrial function in neurons
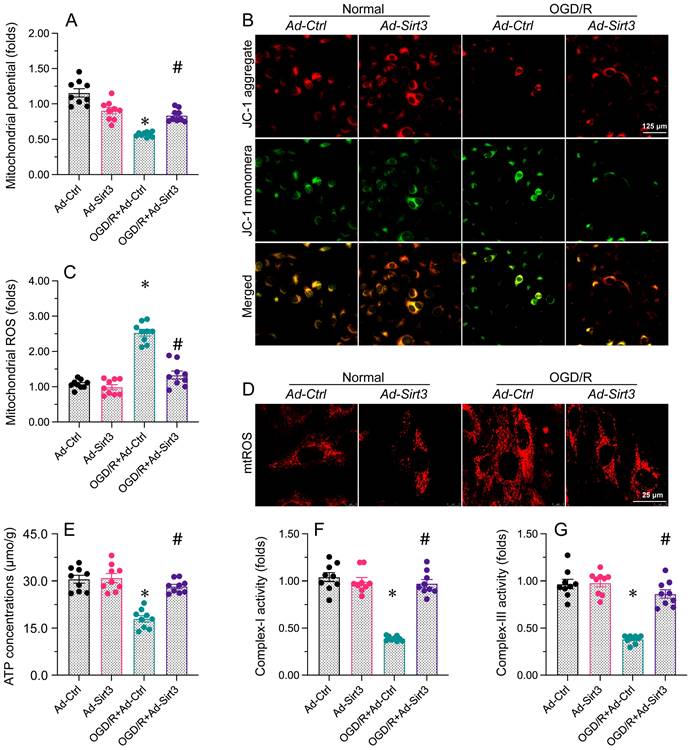
Since mitochondrial dysfunction is regarded as a contributor to neural apoptosis after brain I/R injury [44, 45], we next investigated whether Sirt3 was able to favor mitochondrial performance during brain I/R injury. In vitro, the mitochondrial potential was rapidly inhibited in N2a cells exposed to OGD/R; however, Sirt3 overexpression sustained the mitochondrial potential despite treatment with OGD/R. OGD/R also induced mitochondrial ROS production, whereas this change was attenuated in Ad-Sirt3-transfected cells. Considering the importance of mitochondria for adenosine triphosphate (ATP) production, we used an ELISA to measure ATP levels. We found that OGD/R interrupted ATP synthesis in Ad-Ctrl-transfected N2a cells, but not in Ad-Sirt3-transfected N2a cells. Cellular ATP production depends upon mitochondrial respiration. OGD/R notably repressed the content of mitochondrial respiratory complexes I and III, while Ad-Sirt3 transfection enhanced them in OGD/R-treated cells.
Sirt3 maintains neuronal viability and reduces OGD/R-induced cell death. N2a cells were transfected with Ad-Ctrl or Ad-Sirt3, and then were subjected to OGD/R to simulate cerebral I/R injury in vitro. A. Cell viability was determined with an MTT assay. B. An LDH release assay was used to measure LDH levels in media from N2a cells. C, D. Trypan blue staining was used to observe the number of apoptotic cells after OGD/R. E-H. RNA was isolated from N2a cells, and the levels of Bax, Bad, Bcl-2 and c-IAP were measured. *p<0.05 vs. Ad-Ctrl group, #p<0.05 vs. OGD/R+Ad-Ctrl group.
Sirt3 stabilizes mitochondrial function in neurons. N2a cells were transfected with Ad-Ctrl or Ad-Sirt3, and then were subjected to OGD/R to simulate cerebral I/R injury in vitro. A, B. JC-1 probes were used to analyze the mitochondrial membrane potential in N2a cells after OGD/R injury. The red-to-green fluorescence ratio was used to quantify the mitochondrial membrane potential. C, D. Immunofluorescence was used to measure the production of mitochondrial ROS. E. The concentration of ATP in N2a cells was detected with an ELISA. F, G. ELISAs were used to determine complex I and complex III activity levels in N2a cells. *p<0.05 vs. Ad-Ctrl group, #p<0.05 vs. OGD/R+Ad-Ctrl group.
Sirt3 overexpression protects neuronal viability and mitochondrial function through the UPRmt
To assess whether Sirt3 protected neuronal mitochondria by activating the UPRmt, we next measured the mRNA expression of UPRmt-related genes in brain tissues from WT or Sirt3Tg mice. Compared with the sham treatment, MCAO treatment reduced the mRNA expression of mtDNAj, ClpP, Lonp1 and Hsp10 in WT mice (Figure 4A); however, these differences were not observed in Sirt3Tg mice. Similar results were obtained in N2a cells in vitro: OGD/R inhibited the mRNA expression of mtDNAj, ClpP, Lonp1 and Hsp10, whereas Ad-Sirt3 transfection reversed this effect.
Sirt3 overexpression protects neuronal viability and mitochondrial function by activating the UPRmt. WT and Sirt3Tg mice were subjected to transient MCAO. N2a cells were transfected with Ad-Ctrl or Ad-Sirt3, and then were subjected to OGD/R to simulate cerebral I/R injury in vitro. A-D. RNA was collected from brain tissues, and mtDNAj, Lonp1, ClpP and Hsp10 levels were measured using qPCR. E-H. The mRNA levels of mtDNAj, Lonp1, ClpP and Hsp10 were evaluated using qRT-PCR in N2a cells subjected to OGD/R. I. AEBSF was used to inhibit the UPRmt, and cell viability was determined with an MTT assay. J. AEBSF was used to inhibit the UPRmt, and then an LDH release assay was used to measure LDH levels in media from N2a cells. K. AEBSF was used to inhibit the UPRmt, and then the concentration of ATP in N2a cells was detected with an ELISA. *p<0.05 vs. Ad-Ctrl group or sham+WT group, #p<0.05 vs. OGD/R+Ad-Ctrl group or MCAO+WT group, @p<0.05 vs. OGD/R+Ad-Sirt3 group.
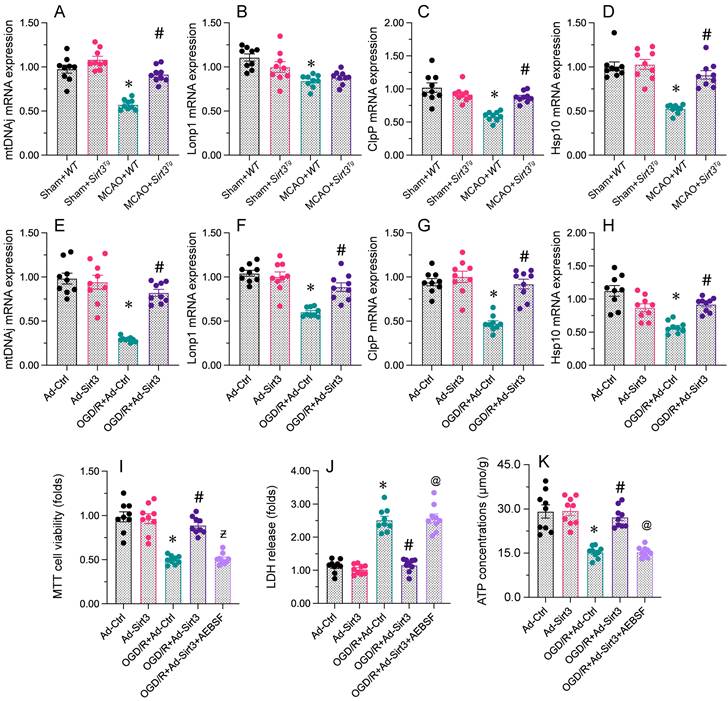
To confirm that the UPRmt was responsible for the defensive impacts of Sirt3 on N2a cell viability and mitochondrial capacity, we incubated N2a cells with AEBSF, an inhibitor of the UPRmt, before transfecting the cells with Ad-Sirt3. A CCK-8 assay indicated that AEBSF abrogated the protective effects of Ad-Sirt3 on N2a cell viability following OGD/R. An LDH release assay demonstrated that AEBSF elevated LDH release from Ad-Sirt3-transfected OGD/R-treated N2a cells. Regarding mitochondrial function, AEBSF also reduced ATP synthesis following OGD/R in Ad-Sirt3-transfected N2a cells.
Sirt3 activates the UPRmt in neurons through the Foxo3a/Sphk1 pathway
To understand the mechanism through which Sirt3 activates the UPRmt, we focused on the Foxo3a/Sphk1 pathway. Western blotting of brain tissues revealed that Foxo3a and Sphk1 levels were significantly reduced in MCAO-treated WT mice, but were returned to physiological state in MCAO-treated Sirt3Tg mice. In consonance with the above evidence, immunofluorescence analyses illuminated that OGD/R treatment inhibited Foxo3a and Sphk1 expression in N2a cells, whereas Ad-Sirt3 transfection reversed these effects.
Although the above results demonstrated that Sirt3 activates the Foxo3a/Sphk1 pathway, further investigation was needed to determine whether the Foxo3a/Sphk1 pathway participates in Sirt3-induced UPRmt activation. We used carbenoxolone, a Foxo3 inhibitor, to suppress the Foxo3a/Sphk1 pathway. Carbenoxolone prevented Sirt3 overexpression from upregulating mtDNAj, ClpP, Lonp1 and Hsp10 in N2a cells during OGD/R treatment. These findings suppored that Sirt3 activates the UPRmt in neurons through the Foxo3a/Sphk1 pathway.
Sphk1 overexpression suppresses brain I/R injury
To assess whether restoration of the Foxo3/Sphk1 pathway would be sufficient to reduce brain I/R injury, we established an MCAO model in Sphk1 transgenic (Sphk1Tg) mice. The MCAO-induced infarction of brain tissue was attenuated in Sphk1Tg mice compared with WT mice. Furthermore, H&E histological analysis demonstrated that neuronal nuclei around the infarction were pyknotic and deeply stained after MCAO treatment in WT mice, while these structural abnormalities were alleviated in neurons from Sphk1Tg mice. Similarly, Nissl staining of brain tissues revealed that MCAO significantly elevated the number of Nissl bodies in WT mice whereas the above changes seem to be imperceptible in Sphk1Tg mice.
MCAO also upregulated the gene expression of IL-6, CRP, TNFα and MCP1 in brain tissues from WT mice. However, the above gene transcriptions were ameliorated in Sphk1Tg mice. Additionally, MCAO treatment reduced the concentrations of oxidation-suppressing factors (GSH and GPX) in WT brains, whereas Sphk1 overexpression attenuated this effect. These results demonstrated that restoring Sphk1 expression was sufficient to attenuate brain I/R injury.
Sirt3 activates the UPRmt in neurons through the Foxo3a/Sphk1 pathway. WT and Sirt3Tg mice were subjected to transient MCAO. N2a cells were transfected with Ad-Ctrl or Ad-Sirt3, and then were subjected to OGD/R to simulate cerebral I/R injury in vitro. A-C. Proteins were isolated from brain tissues, and Western blotting was used to measure the expression of Foxo3 and Sphk1. D-F. Immunofluorescence was used to observe changes in Foxo3 and Sphk1 levels in N2a cells after OGD/R. G-J. Carbenoxolone (CBX) was used to inhibit the Foxo3/Sphk1 pathway, and then qRT-PCR was performed to measure mtDNAj, Lonp1, ClpP and Hsp10 mRNA levels in N2a cells subjected to OGD/R. *p<0.05 vs. Ad-Ctrl group or sham+WT group, #p<0.05 vs. OGD/R+Ad-Ctrl group or MCAO+WT group, @p<0.05 vs. OGD/R+Ad-Sirt3 group.
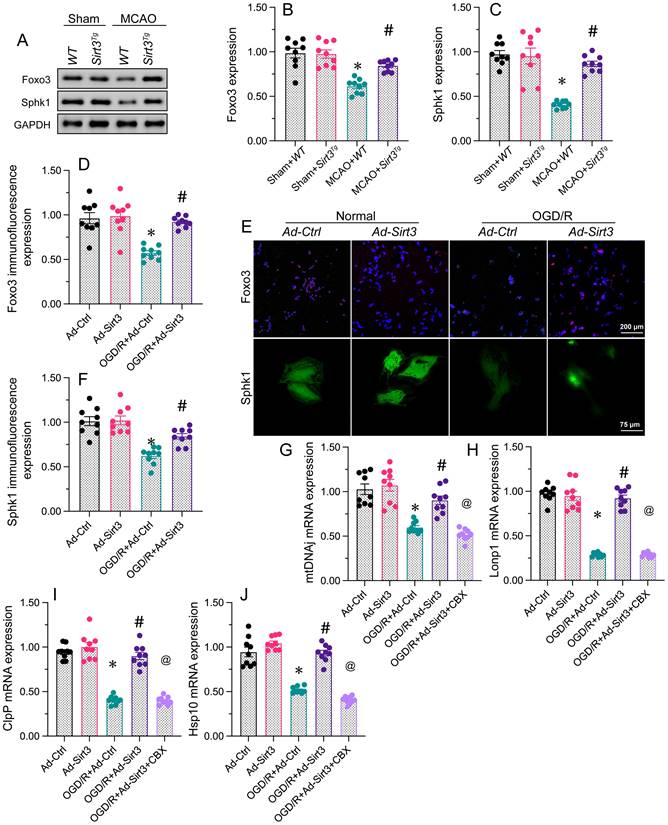
Sphk1 overexpression protects the brain against I/R injury. WT and Sphk1Tg mice were subjected to transient MCAO. A, B. TTC staining was used to observe the infarction size in the brain after MCAO. C. H&E staining was used to detect histological alterations in the brain after MCAO. D, E. Nissl staining was used to observe the number of Nissl bodies in brain tissues after MCAO. F-I. RNA was isolated from brain tissues, and the levels of IL-6, CRP, TNFα and MCP1 were measured. J, K. ELISAs were used to detect changes in GSH and SOD activity in brain tissues after MCAO. *p<0.05 vs. sham+WT group, #p<0.05 vs. MCAO+WT group.
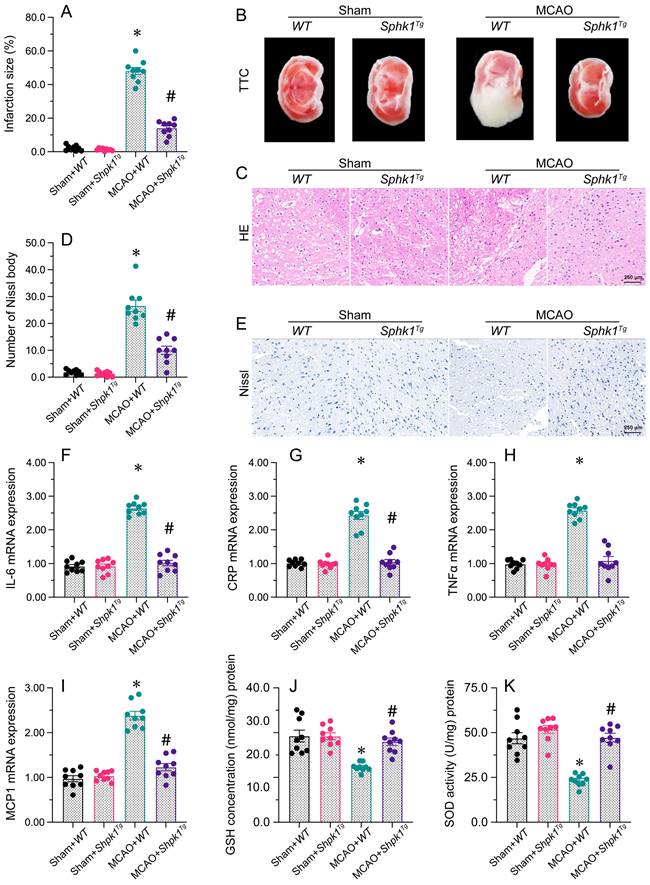
Impediment of the UPRmt reduces Sphk1 overexpression-caused protection on neurons and mitochondria
Next, we evaluated whether Sphk1 overexpression reduces brain I/R dysfunction by normalizing the UPRmt. The present data exhibited that adenoviral overexpression of Sphk1 (Ad-Sphk1) impede OGD/R-mediated cell viability decrease. However, treatment with the UPRmt inhibitor AEBSF abolished the pro-survival effects of Ad-Sphk1. Ad-Sphk1 also suppressed LDH release from OGD/R-treated N2a cells, while AEBSF prevented this effect. Regarding mitochondrial function, Ad-Sphk1 sustained the mitochondrial membrane potential in N2a cells under OGD/R stress, while AEBSF administration abrogated the protective effects of Ad-Sphk1. Moreover, Ad-Sphk1 transfection repressed mitochondrial ROS production in OGD/R-challenged N2a cells, but AEBSF nullified the anti-oxidative capacities of Ad-Sphk1. Lastly, ELISAs showed that caspase-3 and caspase-9 activity levels were augmented upon OGD/R treatment and reduced to near-normal levels following Ad-Sphk1 transfection; however, in N2a cells treated with AEBSF, Ad-Sphk1 failed to inhibit caspase-3/9 activation. Thus, inhibiting the UPRmt neutralized the beneficial impacts of Sphk1 overexpression on neural function and mitochondrial integrity.
Inhibiting the UPRmt abolishes the protective effects of Sphk1 overexpression on neurons and their mitochondria. N2a cells were transfected with Ad-Ctrl or Ad-Sphk1, and then were subjected to OGD/R to simulate cerebral I/R injury in vitro. A. Cell viability was determined with an MTT assay. B. An LDH release assay was used to measure LDH levels in media from N2a cells. C, D. JC-1 probes were used to analyze the mitochondrial membrane potential in N2a cells after OGD/R injury. The red-to-green fluorescence ratio was used to quantify the mitochondrial membrane potential. E. Immunofluorescence was used to measure the production of mitochondrial ROS. F, G. The activity levels of caspase-3 and caspase-9 were determined using ELISAs in N2a cells. *p<0.05 vs. Ad-Ctrl group, #p<0.05 vs. OGD/R+Ad-Ctrl group, @p<0.05 vs. OGD/R+Ad-Sphk1 group.
Discussion
Our work highlighted that Sirt3 can attenuate brain I/R injury via normalizing the UPRmt ; and this function is highly relied on the Foxo3/Sphk1 cascade. This observation had several novel findings. First, Sirt3 was depressed by brain post-ischemic injury, and reduced Sirt3 expression was associated with an augmented infarction size in the brain, suggesting that Sirt3 is an endogenous protector against ischemic stroke. Second, overexpression of Sirt3 restored neural viability by normalizing mitochondrial homeostasis, indicating that Sirt3 prevents ischemic stroke by protecting mitochondria. Third, inhibiting the UPRmt abolished the beneficial impacts of Sirt3 overexpression on neural function and mitochondrial performance during brain I/R injury, suggesting that inducing the UPRmt could be an unconventional medicinal way to diminish the vulnerability of mitochondria during brain I/R injury. Fourth, Sirt3 promoted the UPRmt by upregulating the Foxo3/Sphk1 pathway, and enhancing Sphk1 expression was sufficient to protect neurons and their mitochondria against brain I/R injury. This investigation demonstrates that the UPRmt is a critical protective mechanism to maintain neural viability and mitochondrial homeostasis, and suggest that the Sirt3/Foxo3/Sphk1 pathway is a valuable medicinal objective for patients with ischemic brain damage in clinical practice.
The involvement of mitochondrial dysfunction in brain ischemic dysfunction is broadly reported [17, 43, 46-48]. MCAO was found to upregulate mitochondrial fission-related proteins, increase mitochondrial fission, augment mitochondrial ROS production and activate mitochondrial apoptosis in the brain [49]. Similarly, transmission electron microscopy revealed that MCAO induced mitochondrial fragmentation in brain tissues [50]. Disrupted mitochondrial homeostasis was associated with increased NLR family pyrin domain-containing 3 inflammasome levels in the brain through an undefined mechanism during brain ischemic dysfunction [51]. Brain post-ischemic damage was also found to induce optic atrophy 1 cleavage at the S1 site, thus contributing to mitochondrial cristae remodeling and mitochondrial membrane rupture in neurons [52].
Considering that mitochondrial dysfunction accelerates cerebral I/R injury, several pharmacological interventions have been introduced to attenuate these pathological effects. Ligustilide was found to mitigate brain ischemic dysfunction by activating Parkin-induced mitophagy [53]. Danhong injection, a classical Chinese medicinal approach, was proved to alleviate brain post-ischemic abnormalities by improving mitochondria-dependent neural metabolism [9]. Garciesculenxanthone B was explained to reduce limit brain post-ischemic malfunction by enhancing PTEN-induced kinase 1/Parkin-dependent mitophagy [54].
In addition to these pharmacological approaches, several careful studies have examined non-pharmacological therapies to restore mitochondrial homeostasis during cerebral I/R injury. Mitochondrial transplantation surgery was used to deliver new, well-organized mitochondria to the infarcted brain [55]. This technique was reported to alleviate mitochondrial DNA damage and increase the mitochondrial membrane potential, thus ameliorating neurobehavioral deficits and reducing the infarct size after cerebral I/R injury [55]. Consistent with these findings, delivery of placental mitochondria was revealed to diminish transient brain ischemic anomaly via boosting mitochondrial oxidation-suppressing enzymes levels [56].
The UPRmt maintains mitochondrial proteostasis in response to various cellular stressors [16, 17, 57]. The UPRmt is stirred if the mitochondrial protein folding machinery is overwhelmed or impaired, leading to the accumulation of misfolded or unfolded proteins [58]. Upon activation, the UPRmt augments the abundance of various molecular chaperones, proteases and other proteins that help to restore proteostasis and promote mitochondrial function [59, 60]. The UPRmt is regulated by several transcription factors, including stress-activated transcription factor 1 and hypoxia inducible factor 1α [61, 62], which translocate from the mitochondria to the nucleus where UPRmt genes expression are rapidly elicited.
The beneficial effects of the UPRmt on cerebral diseases have been widely described. Activation of the UPRmt was reported to restore the concentrations of mitochondrial complex I and thus prevent the progression of Alzheimer's disease [63]. In subarachnoid hemorrhage, overexpression of GrpE-like 1 was found to enhance mtHSP70 expression, thus activating the UPRmt and accelerating the degradation of abnormal proteins [64]. During traumatic brain injury, activation of the UPRmt was shown to maintain the mitochondrial membrane potential, reduce the neuroinflammatory response and inhibit mitochondrial apoptosis [18]. Injection of honokiol seem to relieve cognitive impairment in APP/PS2 mice by eliciting the UPRmt [65].
In this work, our data showed that Sirt3 restored UPRmt activity through the Foxo3/Sphk1 pathway. The beneficial impact of Sirt3 on the brain has been described in several previous studies. Upregulation of Sirt3 through melatonin administration was found to ameliorate brain I/R injury via reducing mitochondrial oxidative stress [29]. Similarly, treatment with genipin elevated Sirt3 expression and thus improved uncoupling protein 2-dependent mitochondrial metabolism during cerebral I/R injury [66]. Sirt3 overexpression activated Wnt/β-catenin and blocked mitochondrial fission, thereby reducing cerebral I/R injury [67]. In another study, Sirt3 overexpression inhibited the TLR4/NF-κB pathway and activated the Nrf2/Keap1 pathway, ultimately normalizing the redox response and reducing neuroinflammation during brain ischemic dysfunction [68]. On the basis of these observations of our study, Sirt3 downregulation seems to be a biomarker of brain post-ischemic abnormalities, so Sirt3 overexpression is a bright medicative method to prevent brain post-ischemic anomaly.
In sum, our study illustrated the beneficial effects of Sirt3 overexpression during brain I/R injury. Sirt3 overexpression preserved the UPRmt by inducing the Foxo3/Sphk1 pathway in neurons, thereby improving mitochondrial function and reducing neuronal apoptosis. On the basis of our results, augmentation of Sirt3 expression and activation of the UPRmt are encouraging medicative tool for ischemic stroke.
Supplementary Material
Supplementary primers.
Acknowledgements
Funding
This work was supported by National Natural Science Foundation of China (82101540), Natural Science Foundation of Hunan Province (2021JJ40298) and Scientific research project of Hunan Provincial Health Commission (202112051206, 202304049105).
Data availability
All data generated or analyzed during this study are included in this published article.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wu M, Gu X, Ma Z. Mitochondrial Quality Control in Cerebral Ischemia-Reperfusion Injury. Mol Neurobiol. 2021;58:5253-71
2. Kapanova G, Tashenova G, Akhenbekova A, Tokpınar A, Yılmaz S. Cerebral ischemia reperfusion injury: from public health perspectives to mechanisms. Folia Neuropathol. 2022;60:384-9
3. Sharma D, Maslov LN, Singh N, Jaggi AS. Remote ischemic preconditioning-induced neuroprotection in cerebral ischemia-reperfusion injury: Preclinical evidence and mechanisms. Eur J Pharmacol. 2020;883:173380
4. Yang H, Qi C, Su F, Shan W, Guo A, Wu J. et al. Cerebral Ischemia/Reperfusion Injury and Pharmacologic Preconditioning as a Means to Reduce Stroke-induced Inflammation and Damage. Neurochem Res. 2022;47:3598-614
5. Winquist RJ, Kerr S. Cerebral ischemia-reperfusion injury and adhesion. Neurology. 1997;49:S23-6
6. Gu Y, Zhou C, Piao Z, Yuan H, Jiang H, Wei H. et al. Cerebral edema after ischemic stroke: Pathophysiology and underlying mechanisms. Front Neurosci. 2022;16:988283
7. Vongsfak J, Pratchayasakul W, Apaijai N, Vaniyapong T, Chattipakorn N, Chattipakorn SC. The Alterations in Mitochondrial Dynamics Following Cerebral Ischemia/Reperfusion Injury. Antioxidants (Basel). 2021 10
8. Huang J, Chen L, Yao ZM, Sun XR, Tong XH, Dong SY. The role of mitochondrial dynamics in cerebral ischemia-reperfusion injury. Biomed Pharmacother. 2023;162:114671
9. Zeng M, Zhou H, He Y, Wang Z, Shao C, Yin J. et al. Danhong injection alleviates cerebral ischemia/reperfusion injury by improving intracellular energy metabolism coupling in the ischemic penumbra. Biomed Pharmacother. 2021;140:111771
10. Wang P, Cui Y, Liu Y, Li Z, Bai H, Zhao Y. et al. Mitochondrial ferritin alleviates apoptosis by enhancing mitochondrial bioenergetics and stimulating glucose metabolism in cerebral ischemia reperfusion. Redox Biol. 2022;57:102475
11. Ji ZJ, Shi Y, Li X, Hou R, Yang Y, Liu ZQ. et al. Neuroprotective Effect of Taohong Siwu Decoction on Cerebral Ischemia/Reperfusion Injury via Mitophagy-NLRP3 Inflammasome Pathway. Front Pharmacol. 2022;13:910217
12. He R, Jiang Y, Shi Y, Liang J, Zhao L. Curcumin-laden exosomes target ischemic brain tissue and alleviate cerebral ischemia-reperfusion injury by inhibiting ROS-mediated mitochondrial apoptosis. Mater Sci Eng C Mater Biol Appl. 2020;117:111314
13. Zhong H, Song R, Pang Q, Liu Y, Zhuang J, Chen Y. et al. Propofol inhibits parthanatos via ROS-ER-calcium-mitochondria signal pathway in vivo and vitro. Cell Death Dis. 2018;9:932
14. Gong L, Tang Y, An R, Lin M, Chen L, Du J. RTN1-C mediates cerebral ischemia/reperfusion injury via ER stress and mitochondria-associated apoptosis pathways. Cell Death Dis. 2017;8:e3080
15. Lai Y, Lin P, Chen M, Zhang Y, Chen J, Zheng M. et al. Restoration of L-OPA1 alleviates acute ischemic stroke injury in rats via inhibiting neuronal apoptosis and preserving mitochondrial function. Redox Biol. 2020;34:101503
16. Zhu L, Zhou Q, He L, Chen L. Mitochondrial unfolded protein response: An emerging pathway in human diseases. Free radical biology & medicine. 2021;163:125-34
17. Wang Y, Jasper H, Toan S, Muid D, Chang X, Zhou H. Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox Biol. 2021;45:102049
18. Sun GW, Ding TY, Wang M, Hu CL, Gu JJ, Li J. et al. Honokiol Reduces Mitochondrial Dysfunction and Inhibits Apoptosis of Nerve Cells in Rats with Traumatic Brain Injury by Activating the Mitochondrial Unfolded Protein Response. J Mol Neurosci. 2022;72:2464-72
19. Zhou Q, Zhu L, Qiu W, Liu Y, Yang F, Chen W. et al. Nicotinamide Riboside Enhances Mitochondrial Proteostasis and Adult Neurogenesis through Activation of Mitochondrial Unfolded Protein Response Signaling in the Brain of ALS SOD1(G93A) Mice. Int J Biol Sci. 2020;16:284-97
20. Muñoz-Carvajal F, Sanhueza M. The Mitochondrial Unfolded Protein Response: A Hinge Between Healthy and Pathological Aging. Front Aging Neurosci. 2020;12:581849
21. Sun D, Wang J, Toan S, Muid D, Li R, Chang X. et al. Molecular mechanisms of coronary microvascular endothelial dysfunction in diabetes mellitus: focus on mitochondrial quality surveillance. Angiogenesis. 2022;25:307-29
22. Ma L, Zou R, Shi W, Zhou N, Chen S, Zhou H. et al. SGLT2 inhibitor dapagliflozin reduces endothelial dysfunction and microvascular damage during cardiac ischemia/reperfusion injury through normalizing the XO-SERCA2-CaMKII-coffilin pathways. Theranostics. 2022;12:5034-50
23. Shen Y, Wu Q, Shi J, Zhou S. Regulation of SIRT3 on mitochondrial functions and oxidative stress in Parkinson's disease. Biomed Pharmacother. 2020;132:110928
24. Anamika Khanna A, Acharjee P Acharjee A, Trigun SK. Mitochondrial SIRT3 and neurodegenerative brain disorders. J Chem Neuroanat. 2019;95:43-53
25. Papa L, Germain D. SirT3 regulates the mitochondrial unfolded protein response. Mol Cell Biol. 2014;34:699-710
26. Chen H, Zhang DM, Zhang ZP, Li MZ, Wu HF. SIRT3-mediated mitochondrial unfolded protein response weakens breast cancer sensitivity to cisplatin. Genes Genomics. 2021;43:1433-44
27. Xu M, Xue RQ, Lu Y, Yong SY, Wu Q, Cui YL. et al. Choline ameliorates cardiac hypertrophy by regulating metabolic remodelling and UPRmt through SIRT3-AMPK pathway. Cardiovasc Res. 2019;115:530-45
28. Cai H, Bian X, Chen L, Zhang N, Li L, Tang W. et al. Selective intra-arterial brain cooling induces cerebral protection against ischemia/reperfusion injury through SENP1-Sirt3 signaling. Free Radic Biol Med. 2021;171:272-83
29. Liu L, Chen H, Jin J, Tang Z, Yin P, Zhong D. et al. Melatonin ameliorates cerebral ischemia/reperfusion injury through SIRT3 activation. Life Sci. 2019;239:117036
30. Kim S, Sieburth D. FSHR-1/GPCR Regulates the Mitochondrial Unfolded Protein Response in Caenorhabditis elegans. Genetics. 2020;214:409-18
31. Kim S, Sieburth D. Sphingosine Kinase Activates the Mitochondrial Unfolded Protein Response and Is Targeted to Mitochondria by Stress. Cell Rep. 2018;24:2932-45.e4
32. Bigarella CL, Rimmele P, Liang R, Azodi Y, Izac B, d'Escamard V. et al. Acetylation Is Critical for Foxo3 Protein Stability and Activity in Hematopoietic Stem and Progenitor Cells. Blood. 2012;120:1198 -
33. Takuwa N, Ohkura S, Takashima S, Ohtani K, Okamoto Y, Tanaka T. et al. S1P3-mediated cardiac fibrosis in sphingosine kinase 1 transgenic mice involves reactive oxygen species. Cardiovasc Res. 2010;85:484-93
34. Wang J, Zhu P, Toan S, Li R, Ren J, Zhou H. Pum2-Mff axis fine-tunes mitochondrial quality control in acute ischemic kidney injury. Cell Biol Toxicol. 2020;36:365-78
35. Cai Y, Yang E, Yao X, Zhang X, Wang Q, Wang Y. et al. FUNDC1-dependent mitophagy induced by tPA protects neurons against cerebral ischemia-reperfusion injury. Redox Biol. 2021;38:101792
36. Zhou H, Toan S, Zhu P, Wang J, Ren J, Zhang Y. DNA-PKcs promotes cardiac ischemia reperfusion injury through mitigating BI-1-governed mitochondrial homeostasis. Basic Res Cardiol. 2020;115:11
37. Tan Y, Zhang Y, He J, Wu F, Wu D, Shi N. et al. Dual specificity phosphatase 1 attenuates inflammation-induced cardiomyopathy by improving mitophagy and mitochondrial metabolism. Mol Metab. 2022;64:101567
38. Zou R, Shi W, Qiu J, Zhou N, Du N, Zhou H. et al. Empagliflozin attenuates cardiac microvascular ischemia/reperfusion injury through improving mitochondrial homeostasis. Cardiovasc Diabetol. 2022;21:106
39. Zou R, Tao J, Qiu J, Lu H, Wu J, Zhu H. et al. DNA-PKcs promotes sepsis-induced multiple organ failure by triggering mitochondrial dysfunction. J Adv Res. 2022;41:39-48
40. Wang S, Zhu H, Li R, Mui D, Toan S, Chang X. et al. DNA-PKcs interacts with and phosphorylates Fis1 to induce mitochondrial fragmentation in tubular cells during acute kidney injury. Sci Signal. 2022;15:eabh1121
41. Zhou H, Dai Z, Li J, Wang J, Zhu H, Chang X. et al. TMBIM6 prevents VDAC1 multimerization and improves mitochondrial quality control to reduce sepsis-related myocardial injury. Metabolism. 2023;140:155383
42. Wang J, Wang X, Du W, Xue Z, Huang W, Guan Z. et al. BI-1 ameliorates myocardial injury by activating the mitochondrial unfolded protein response and FUNDC1-related mitophagy in cardiorenal syndrome type 3. Cell Signal. 2022;91:110218
43. Zhu H, Tan Y, Du W, Li Y, Toan S, Mui D. et al. Phosphoglycerate mutase 5 exacerbates cardiac ischemia-reperfusion injury through disrupting mitochondrial quality control. Redox Biol. 2021;38:101777
44. Chang X, Toan S, Li R, Zhou H. Therapeutic strategies in ischemic cardiomyopathy: Focus on mitochondrial quality surveillance. EBioMedicine. 2022;84:104260
45. Chang X, Li Y, Cai C, Wu F, He J, Zhang Y. et al. Mitochondrial quality control mechanisms as molecular targets in diabetic heart. Metabolism. 2022;137:155313
46. Zhu H, Toan S, Mui D, Zhou H. Mitochondrial quality surveillance as a therapeutic target in myocardial infarction. Acta Physiol (Oxf). 2021;231:e13590
47. Zhou H, Ren J, Toan S, Mui D. Role of mitochondrial quality surveillance in myocardial infarction: From bench to bedside. Ageing Res Rev. 2021;66:101250
48. Chang X, Lochner A, Wang HH, Wang S, Zhu H, Ren J. et al. Coronary microvascular injury in myocardial infarction: perception and knowledge for mitochondrial quality control. Theranostics. 2021;11:6766-85
49. Liu JF, Su G, Chen LX, Zhou JP, Gao J, Zhang JJ. et al. Irisin Attenuates Apoptosis Following Ischemia-Reperfusion Injury Through Improved Mitochondria Dynamics and ROS Suppression Mediated Through the PI3K/Akt/mTOR Axis. Mol Neurobiol. 2023
50. Xie W, Zhu T, Zhou P, Xu H, Meng X, Ding T. et al. Notoginseng leaf triterpenes ameliorates mitochondrial oxidative injury via the NAMPT-SIRT1/2/3 signaling pathways in cerebral ischemic model rats. J Ginseng Res. 2023;47:199-209
51. Gong Z, Pan J, Shen Q, Li M, Peng Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J Neuroinflammation. 2018;15:242
52. Li X, Li H, Xu Z, Ma C, Wang T, You W. et al. Ischemia-induced cleavage of OPA1 at S1 site aggravates mitochondrial fragmentation and reperfusion injury in neurons. Cell Death Dis. 2022;13:321
53. Mao Z, Tian L, Liu J, Wu Q, Wang N, Wang G. et al. Ligustilide ameliorates hippocampal neuronal injury after cerebral ischemia reperfusion through activating PINK1/Parkin-dependent mitophagy. Phytomedicine. 2022;101:154111
54. Wu M, Lu G, Lao YZ, Zhang H, Zheng D, Zheng ZQ. et al. Garciesculenxanthone B induces PINK1-Parkin-mediated mitophagy and prevents ischemia-reperfusion brain injury in mice. Acta Pharmacol Sin. 2021;42:199-208
55. Xie Q, Zeng J, Zheng Y, Li T, Ren J, Chen K. et al. Mitochondrial Transplantation Attenuates Cerebral Ischemia-Reperfusion Injury: Possible Involvement of Mitochondrial Component Separation. Oxid Med Cell Longev. 2021;2021:1006636
56. Nakamura Y, Lo EH, Hayakawa K. Placental Mitochondria Therapy for Cerebral Ischemia-Reperfusion Injury in Mice. Stroke. 2020;51:3142-6
57. Inigo JR, Chandra D. The mitochondrial unfolded protein response (UPR(mt)): shielding against toxicity to mitochondria in cancer. J Hematol Oncol. 2022;15:98
58. O'Malley J, Kumar R, Inigo J, Yadava N, Chandra D. Mitochondrial Stress Response and Cancer. Trends Cancer. 2020;6:688-701
59. Vögtle FN. Open questions on the mitochondrial unfolded protein response. Febs j. 2021;288:2856-69
60. Wang G, Fan Y, Cao P, Tan K. Insight into the mitochondrial unfolded protein response and cancer: opportunities and challenges. Cell Biosci. 2022;12:18
61. Suárez-Rivero JM, Pastor-Maldonado CJ, Povea-Cabello S, Álvarez-Córdoba M, Villalón-García I, Talaverón-Rey M. et al. Activation of the Mitochondrial Unfolded Protein Response: A New Therapeutic Target? Biomedicines. 2022 10
62. Zhu L, Luo X, Fu N, Chen L. Mitochondrial unfolded protein response: A novel pathway in metabolism and immunity. Pharmacol Res. 2021;168:105603
63. Trushina E, Trushin S, Hasan MF. Mitochondrial complex I as a therapeutic target for Alzheimer's disease. Acta Pharm Sin B. 2022;12:483-95
64. Ma C, Gao B, Wang Z, You W, Yu Z, Shen H. et al. GrpEL1 regulates mitochondrial unfolded protein response after experimental subarachnoid hemorrhage in vivo and in vitro. Brain Res Bull. 2022;181:97-108
65. Hou M, Bao W, Gao Y, Chen J, Song G. Honokiol improves cognitive impairment in APP/PS1 mice through activating mitophagy and mitochondrial unfolded protein response. Chem Biol Interact. 2022;351:109741
66. Zhao B, Sun LK, Jiang X, Zhang Y, Kang J, Meng H. et al. Genipin protects against cerebral ischemia-reperfusion injury by regulating the UCP2-SIRT3 signaling pathway. Eur J Pharmacol. 2019;845:56-64
67. Zhao H, Luo Y, Chen L, Zhang Z, Shen C, Li Y. et al. Sirt3 inhibits cerebral ischemia-reperfusion injury through normalizing Wnt/β-catenin pathway and blocking mitochondrial fission. Cell Stress Chaperones. 2018;23:1079-92
68. Gao J, Chen N, Li N, Xu F, Wang W, Lei Y. et al. Neuroprotective Effects of Trilobatin, a Novel Naturally Occurring Sirt3 Agonist from Lithocarpus polystachyus Rehd, Mitigate Cerebral Ischemia/Reperfusion Injury: Involvement of TLR4/NF-κB and Nrf2/Keap-1 Signaling. Antioxid Redox Signal. 2020;33:117-43
Author contact
![]() Corresponding author: Zhou Dingzhou, Email: zhoudzedu.cn.
Corresponding author: Zhou Dingzhou, Email: zhoudzedu.cn.