Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Apos
3. Role of Apos in...
4. Role of Apos in oxidative...
5. Role of Apos in the foam...
6. Role of Apos in RCT in...
7. Apos as meaningful targeting...
8. Conclusion and outlook
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(14):4493-4510. doi:10.7150/ijbs.86475 This issue Cite
Review
Apolipoproteins as potential communicators play an essential role in the pathogenesis and treatment of early atherosclerosis
Yang Li1, Xinyi Luo1, Zhenglai Hua1, Xiaoxia Xue1, Xiangpeng Wang1, Mingshi Pang1, Tieshan Wang2 ![]() , Aiping Lyu3
, Aiping Lyu3 ![]() , Yuanyan Liu1
, Yuanyan Liu1 ![]()
1. School of Chinese Materia Medica, Beijing University of Chinese Medicine, Beijing 100029, China.
2. Beijing Research Institute of Chinese Medicine, Beijing University of Chinese Medicine, Beijing, China.
3. School of Chinese Medicine, Hong Kong Baptist University, Kowloon, Hong Kong 999077, China.
Received 2023-5-24; Accepted 2023-8-15; Published 2023-8-21
Abstract

Atherosclerosis as the leading cause of the cardiovascular disease is closely related to cholesterol deposition within subendothelial areas of the arteries. Significantly, early atherosclerosis intervention is the critical phase for its reversal. As atherosclerosis progresses, early foam cells formation may evolve into fibrous plaques and atheromatous plaque, ulteriorly rupture of atheromatous plaque increases risks of myocardial infarction and ischemic stroke, resulting in high morbidity and mortality worldwide. Notably, amphiphilic apolipoproteins (Apos) can concomitantly combine with lipids to form soluble lipoproteins that have been demonstrated to associate with atherosclerosis. Apos act as crucial communicators of lipoproteins, which not only can mediate lipids metabolism, but also can involve in pro-atherogenic and anti-atherogenic processes of atherosclerosis via affecting subendothelial retention and aggregation of low-density lipoprotein (LDL), oxidative modification of LDL, foam cells formation and reverse cholesterol transport (RCT) in macrophage cells. Correspondingly, Apos can be used as endogenous and/or exogenous targeting agents to effectively attenuate the development of atherosclerosis. The article reviews the classification, structure, and relationship between Apos and lipids, how Apos serve as communicators of lipoproteins to participate in the pathogenesis progression of early atherosclerosis, as well as how Apos as the meaningful targeting mass is used in early atherosclerosis treatment.
Keywords: Apolipoproteins, Potential communicators of lipoproteins, The pathogenesis of early atherosclerosis, Treatment of early atherosclerosis
1. Introduction
Atherosclerosis is considered to be a chronic inflammatory disease of abnormal accumulation of cholesterol, mainly in the large and medium arteries, leading to high morbidity and mortality worldwide1-3. Atherosclerosis is a long-term and progressive disease that usually progresses from early fatty streak to fibrous plaque and eventually to highly perilous atheromatous plaque. During this development, the early stage of atherosclerosis is a pivotal period for its progression and regression. Without timely intervention, the atherosclerotic lesions will be further aggravated, and the acute rupture of unstable atheromatous plaque may cause sudden thrombotic occlusion of the artery and fatal clinical complications, including myocardial infarction, stroke, pulmonary embolism and peripheral artery disease4-6.
Therapeutic strategies for early atherosclerosis mostly revolve around the reduction of lipids levels. The lipids serve as the source of energy and nutrient for the body, mainly including cholesterol, triglycerides (TG) and phospholipids (PL), which are insoluble in water and cannot exist independently in plasma. More importantly, plasma amphiphilic apolipoproteins (Apos) possess the property of interacting with both lipids and water, and act as indispensable carriers of lipids transport. Lipoproteins can be formed by the bind of lipids and Apos, including chylomicron (CM), very low-density lipoprotein (VLDL), intermediate-density lipoprotein (IDL), low-density lipoprotein (LDL) and high-density lipoprotein (HDL), which can transport lipids to various tissues of the body for metabolism and utilization7. Along with lipids transport function, Apos also act as communicators of lipoproteins to mediate the metabolism of lipids via the interaction with enzymes and cellular receptors. Recently, massive studies pay more attention to atherogenic LDL and anti-atherogenic HDL, which are associated with cardiovascular disease risk and antagonistic effects in atherosclerotic development8,9. The atherogenic property of LDL is primarily attributed to its role as a carrier that transports hepatic cholesterol to peripheral tissues, while HDL enhances reverse cholesterol transport (RCT) to accelerate superfluous cholesterol emigration from peripheral tissues to the liver identified as conferring protection against atherosclerosis8,10. Remarkably, Apos, as functional communicators of lipoproteins, play a crucial role in the interaction between lipoproteins and external objects, so it may participate in the initiation and advance of atherosclerosis.
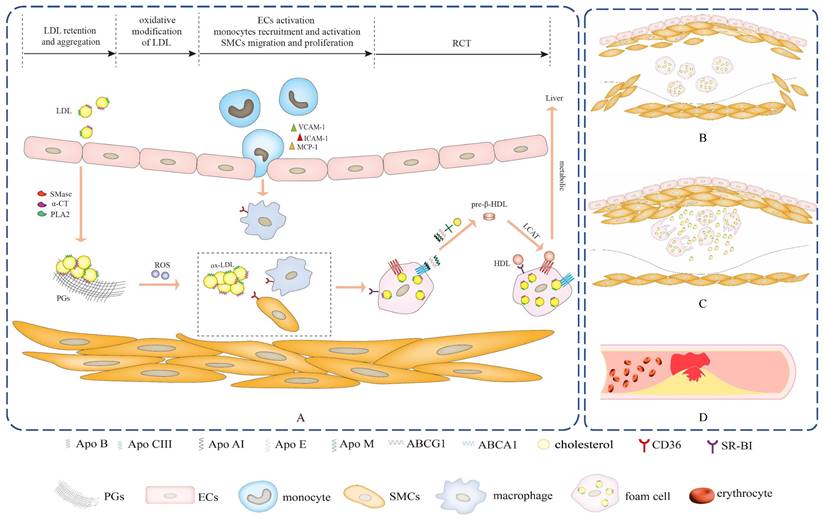
Accumulating evidences demonstrate that Apos are closely associated with atherosclerotic disease and serve vital regulatory roles in a broad spectrum of key events in early atherosclerosis11-15. Atherosclerosis commences with the subendothelial retention and aggregation of cholesterol-rich LDL, which is mediated by the interaction of Apos and extracellular matrix (ECM) components such as proteoglycans (PGs)11,12,16. In addition, Apos act as communicators of lipoproteins to irritate oxidative stress and motivate endothelial cells (ECs) and monocytes activation via initiating related signaling pathways. Apos-mediated the activation of ECs may generate multiple cytokines, such as interleukin (IL)-6, IL-8, intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1) and so on, which incite the recruitment of monocytes and adhesion to ECs17-19. Then, monocytes pass through the endothelial tissue and transform into macrophages. ECs and macrophages can be activated by the induction of Apos, secreting inflammatory cytokines to promote the migration and proliferation of smooth muscle cells (SMCs). Trapped LDL within subendothelial space tends to be oxidized and generate a pro-atherogenic form of oxidized LDL (ox-LDL), which can be taken up unrestrictedly in macrophages and SMCs by binding Apos to cell surface receptors, inducing lipid-laden foam cells formation. A large deposition of foam cells within the subendothelial area is a characteristic feature of early atherosclerosis2,20. Significantly, various cytoprotective Apos, as a defense mechanism of the stress response, generally can be upregulated and accumulated in the lesions and exert manifold conducive effects on the attenuation of atherosclerosis. These conducive effects include the refrain of LDL retention and aggregation, the interference of oxidative stress-related signaling pathways, the inhibiton of ECs and macrophages activation, the modulation of SMCs migration and proliferation and the facilitation of RCT in macrophage cells, which can subsequently reduce foam cells formation and dramatically reverse the progression of atherosclerosis7,21-25.
Apos, as communicators of lipoproteins, play crucial functions in the process of early atherosclerosis. It is worth noting that Apos may be triggered as meaningful targeting mass to ameliorate atherosclerosis via masking the adverse properties of atherogenic Apos and sufficiently amplifying the advantageous properties of anti-atherogenic Apos. Two therapeutic strategies could be taken to synergistically confront the development of early atherosclerosis, one is to regulate endogenous Apos expression and the other is turning to exogenous anti-atherogenic Apos mimetic peptides. In this review, we summarize the classification, structure, and relationship of Apos with lipids, how Apos act as communicators of lipoproteins to affect the development of early atherosclerosis, and even how Apos act as meaningful targeting mass applied for atherosclerosis treatment.
2. Apos
Apos are diverse and possess a variety of biological functions. The amphiphilic nature of Apos enables to interact with lipids and water. Since cholesterol, PL, TG and other lipids are insoluble in water and cannot exist alone in plasma, Apos can combine with lipids to form soluble lipoproteins, and the lipids are transported to body tissues in the form of lipoproteins for metabolism and utilization7. In addition to as carriers of lipids transport, Apos also serve as communicators of lipoproteins to mediate the interaction between lipoproteins and external objects, which are involved in lipid metabolism in lipoproteins through adjusting enzymes activity and binding to cell surface receptors.
2.1 Classification of Apos
Apos are generally classified into five categories, such as A, B, C, D and E, each of which has subcategories, such as AI, AII, AIV and AV in class A, B48 and B100 in class B, CI, CII and CIII in class C, E2, E3 and E4 in class E. So far, more than 20 Apos have been found26-28. In addition to the above mentioned, classical Apos also contain Apo F, Apo H, Apo J, Apo M and so on. In this review, the Apos mainly refers to Apo A, Apo B, Apo C, Apo E, Apo M and Apo J.
Apos are also divided into exchangeable and non-exchangeable Apos based on their movement and exchange properties between different lipoproteins. Exchangeable Apos can be separated from one lipoprotein and recombined with another lipoprotein during circulation, apparently confering lipoproteins orientation and the ability to interact with cell surface receptors29. Exchangeable Apos such as Apo A, Apo C and Apo E must be water-soluble to be exchanged between lipoproteins, while Apo B, as a non-exchangeable apolipoprotein, is highly insoluble in an aqueous medium without interchangeable features with other lipoproteins particles29,30. Non-exchangeable Apo B retains in a single lipoprotein particle from biosynthesis to catabolism. As well, tracer experiments demonstrated that an Apo B molecule remained together with the parent lipoprotein particle throughout its existence31,32.
2.2 The structure of Apos
Exchangeable Apos, sharing unique structure related to their biological functions, are soluble in aqueous solutions and transport between various lipoproteins depending on changes in lipoproteins size and lipids composition29. The representative structural character is owning tandem 11-mer or 22-mer domains, usually starting from proline residues and forming amphiphilic α-helices (AαHs). The exchangeability of exchangeable Apos is largely attributable to the content and/or structure of amphiphilic helical motifs33. AαHs, the secondary structural motif responsible for the reversible lipids binding, are the universal structural character of exchangeable Apos and are vital structural members for these exchangeable Apos functions (Figure 1a). Apos exchange is indispensable for lipoproteins metabolism, and is considered to generate in the lipid-deficient condition that also serves as major receptors for cellular cholesterol and PL. Besides, the exchangeability of Apos requires dynamic Apos structures to accommodate different lipoproteins surface34. Recent findings indicate that the tertiary structures of Apo AI and Apo E are correlated among exchangeable Apos. Apo AI and Apo E are characterized by a series of proline discontinuous AαHs repeats of 11-mer or 22-mer amino acids that exhibit helix-bundle conformation in lipid-free conditions, and their C-terminal domains are primarily associated with lipids binding. Apo C is incapable of constituting helix-bundle conformation attributed to its limited AαHs35.
(a) The 3D structure of exchangeable Apos (PDB:2L7B). AαHs are colored blue and green, and the green AαHs represent the lipids binding domain. The proline residues are rendered in stick representation and are colored yellow. The smooth lines represent the backbone of the exchangeable Apos structure and are colored pink. (b) The 3D structure of nonexchangeable Apos (PDB:6I7S). AαHs are colored blue, and AβS are colored purple and green. The green AβS represent the lipids binding domain. The smooth lines represent the backbone of the non-exchangeable Apos structure and are colored yellow.
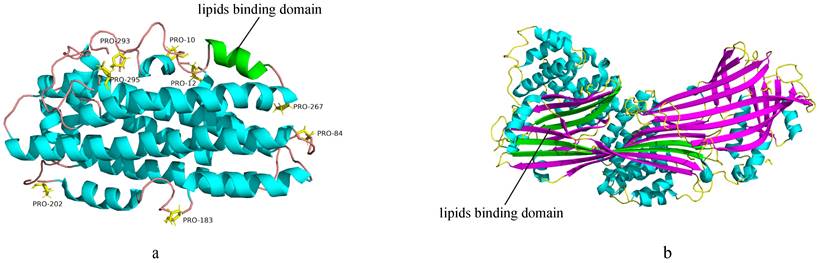
The Apo B, non-exchangeable apolipoprotein, possesses alternative AαHs and amphipathic β-sheet (AβS) domains32,36 (Figure 1b). The N-terminal of Apo B is homologous to lipovitellin and microsomal triglyceride transfer protein37. The AβS domains with a width of approximately 30Å are composed of antiparallel β-sheets, and are essential for lipoprotein assembly and irreversible lipids binding. Apo B48 contains only the β1 domain, while Apo B100 contains both β1 and β2 domains32,38. Moreover, AαHs domains stabilize electrostatic interactions via expansion and constriction of the PL belt, thus maintaining lipoprotein structural integrity39. However, the three-dimensional structure of Apo B remains unclear. Unlike most Apos, Apo M displays a hydrophobic binding pocket similar to lipocalins that enable biological actions. The tertiary structure of Apo M consists of antiparallel β-barrel sheets surrounding the internal ligand binding pocket, flanked by an external α-helix14,40,41.
2.3 Relationship between Apos and lipids
Apo are important serum proteins embedded on the surface of lipoproteins particles, endowing lipids with a soluble form. Apos, as communicators of lipoproteins, are also involve in lipids metabolism through modulating enzymes activity and binding to cellular receptors.
Apos combine with lipids in order to form various densities and sizes of lipoproteins that enable to transport hydrophobic mediators in hydrophilic plasma. Lipoproteins are classified into CM, VLDL, IDL, LDL and HDL in ascending order according to hydrated density42. In Apo A, Apo AI and Apo AII are mainly present in HDL, and Apo AI exists in CM and VLDL as well43,44. Physiologically, the majority of circulating Apo AIV is lipid-free or associated with CM, while the remaining proportions are related to HDL7,45. Apo AV predominately exists in CM, VLDL, and HDL33,46. In Apo B, Apo B48 is present in CM and Apo B100 exists in VLDL, IDL and LDL37. In Apo C, Apo CI, Apo CII and Apo CIII are related to CM, VLDL and HDL. Moreover, Apo CI and Apo CIII are respectively associated with IDL and LDL47-49. Apo E is secreted as a component of CM, VLDL and HDL33. Except for relating to HDL, Apo M is also associated with LDL, VLDL and CM41. Apo J also referred to as clusterin acts extracellularly or intracellularly as a chaperone and is mainly carried by HDL, LDL and VLDL50,51 (Table 1).
Apos exist in CM, VLDL, IDL, LDL and HDL.
| Apos/lipoproteins | CM | VLDL | IDL | LDL | HDL | Reference |
|---|---|---|---|---|---|---|
| Apo AI | + | + | - | - | + | 43, 44 |
| Apo AII | - | - | - | - | + | 43, 44 |
| Apo AIV | + | - | - | - | + | 7, 45 |
| Apo AV | + | + | - | - | + | 33, 46 |
| Apo B48 | + | - | - | - | - | 37 |
| Apo B100 | - | + | + | + | - | 37 |
| Apo CI | + | + | + | + | + | 47, 48, 49 |
| Apo CII | + | + | - | - | + | 47, 48, 49 |
| Apo CIII | + | + | + | + | + | 47, 48, 49 |
| Apo E | + | + | - | - | + | 33 |
| Apo M | + | + | - | + | + | 41 |
| Apo J | - | + | - | + | + | 50, 51 |
Apos act as communicators of lipoproteins to regulate lipid metabolism in lipoproteins by activating or suppressing enzymes. Examples are Apo AI, Apo AIV, Apo AV, Apo CI and Apo E, they may act as activators of lecithin cholesterol acyltransferase (LCAT), which induces free cholesterol (FC) on HDL surface esterified into cholesterol esters (CE) and facilitates CE to get into nascent HDL core to form mature HDL35. However, Apo CIII inhibits LCAT activity. Apo CII works as an obligatory activator for lipoprotein lipase (LPL) that promotes TG hydrolysis in triglyceride-rich lipoproteins (TRL). LPL activity is quite low without the presence of Apo CII48. Apo CI and Apo CIII are recognized as inhibitors of LPL. They are also shown to inhibit hepatic lipase (HL), while Apo E activates HL13,52,53. Apo AIV and Apo E increase the activity of cholesteryl ester transfer protein (CETP), a pivotal alternative approach for transporting CE to the liver, affecting RCT overall rate7,13. Nevertheless, Apo CI levels are inversely correlated with CETP in vitro47.
Apos, as communicators of lipoproteins, may initiate or inhibit lipid uptake from lipoprotein particles by interacting with specific cellular receptors. Apo B as a ligand is essential for LDL binding to LDL receptor (LDLR), facilitating cholesterol ingestion in peripheral tissues and the liver54,55. Apo AI interacts with hepatocellular surface scavenger receptor class B type I (SR-BI) to trigger CE uptake by hepatocytes for further metabolism56. Additionally, as a functional ligand of multiple receptors including LDLR, LDL receptor-related protein (LRP) and heparan sulfate proteoglycans (HSPG), Apo E facilitates the clearance of TRL particles, especially VLDL, VLDL and CM remnant particles, and regulates plasma cholesterol and TG levels 13,57. Besides, Apo E motivates HDL to enter into liver cells for metabolism. In humans, Apo E-containing lipoproteins are cleared faster than those lacking Apo E58. Apo C appears to restrain the clearing function of Apo E. Apo CI could impair remnant clearance via LDLR and LRP mediated by Apo E and block the VLDL binding to its receptor52. As well, Apo CIII retards TRL remnants' catabolism ascribed to its disturbance of interaction of Apo B or Apo E with hepatic receptors15.
3. Role of Apos in subendothelial retention and aggregation of LDL
Apo B acts as a major communicator of LDL can spark subendothelial retention of excessive LDL by interacting with ECM components PGs, which is an initial step of atherosclerosis11,12. Other Apos compositions in LDL, as well as phospholipases and proteases-triggered LDL modification may cause conformational changes in Apo B, which increase the binding of Apo B to PGs and lead to aggregation into large macromolecular complexes, further aggravating the retention of LDL (Figure 2A). As a stress response, several Apos protect against lipoproteins aggregation, exerting anti-atherogenic effects and inhibiting atherosclerotic development.
The progression of atherosclerosis. (A) Apos serve as communicators of lipoproteins to participate in the pathogenesis of fatty streak, which is the early stage of atherosclerosis and is reversible. (B) As the lesion progresses, a large number of SMCs migrate into the intima, and SMCs proliferate and secrete a large amount of extracellular matrix to significantly thicken the intima of the lesion, which is the fibrous plaque stage. (C) The lesion progresses further, foam cells die and disintegrate, releasing their contents, which is the atheromatous plaque stage. (D) Unstable atherosclerotic plaques are prone to rupture and can cause sudden thrombotic occlusion of the artery, blocking blood flow and resulting in fatal clinical complications, including myocardial infarction, stroke, pulmonary embolism and peripheral artery disease.
3.1 Apos trigger subendothelial retention of LDL
The endothelial layer maintains integrity upon the majority of atherosclerotic progression, whereas it is highly permeable to LDL and the exact molecular mechanism promoting this increase in permeability is unclear. Other than LDL particles, the small Apo B-containing lipoproteins with sizes about 70 nm, such as CM remnants, VLDL and IDL, could effectively pass through ECs59. Experimental researches indicate that PGs versican, biglycan and perlecan are ECM components within the subendothelial region and are believed to show a significant role in the process of lipoproteins retention and aggregation59. PGs are macromolecules made up of core proteins and long-side chain glycosaminoglycans (GAGs), composed of duplicated disaccharide units owning negatively charged groups37. The positively charged amino acids in Apo B could bind to PGs via negatively charged GAGs37,60. Eight binding sites in Apo B100 are identified to interact with PGs. Site B (residues 3359-3369) serves as a functional combining site in Apo B100 and others without function possibly bury in the LDL surface lipid layer61. Similar to LDL, smaller particles like CM remnants, VLDL and IDL are easily trapped and accumulated via the electrostatic interactions62. Furthermore, other Apos compositions of LDL affect their combination with PGs. Apo E can mediate the binding of lipoproteins to negatively charged GAGs chains of arterial PGs and the co-localization of Apo E and biglycan in lesions has been testified16. As well, exchangeable Apo CIII content in LDL is related to enhanced affinity with PGs biglycan16,61,63. Unlike Apo B and Apo E, Apo CIII cannot directly combine with PGs due to a lack of positively charged amino acid53. The enhanced combination may be attributed to conformational changes connected with high Apo CIII content, making Apo B or Apo E more approachable to PGs64,65. Moreover, a high Apo CIII/Apo B ratio reduces the lipids composition content of LDL and these changes may contribute to increased membrane fluidity and enable Apo B to form a more conducive conformation for PGs binding53,63.
3.2 Conformational changes in Apos induced by enzymes promote the aggregation of LDL
Following subendothelial retention, lipoproteins are more easily modified by phospholipases and proteases, including sphingomyelinases (SMase), phospholipaseA2 (PLA2) and α-chymotrypsin (α-CT). These enzymes promote the formation of hydrophobic domains in Apo B of lipoproteins, facilitating aggregation and fusion of retained lipoproteins21,64, which form larger lipoprotein-like particles and are hard to leave the arterial walls. As the conformation of Apo B100 on the LDL surface may be related to lipids composition and content as well as LDL particle size, thus other combinative sites could be exposed and play a role in modified LDL16. Site A (residues 3148-3158) in Apo B100 plays a role in PLA2-modified LDL and acts synergistic effect with site B to strengthen the bond to PGs37. The changes in LDL surface lipid monolayer induced by SMase result in significant changes in the conformation of Apo B100, manifesting as decreased α-helical and increased β-sheet content. SMase-induced conformational changes appear to cause exposure of specific hidden regions of Apo B100 and increase surface hydrophobicity of LDL particles that are crucial for aggregation66. α-CT induces Apo B proteolysis and proteolytic fragments released from LDL to stimulate the fusion of LDL21. Apo CIII acts as a SMase activator, which makes Apo CIII enriching in LDL more susceptible to hydrolysis and aggregation by SMase67.
3.3 Apos protect against the aggregation of LDL
Although some Apos accelerate atherosclerotic progression via promoting lipoproteins accumulation within the subendothelial space, several Apos exhibit inhibition of lipoproteins aggregation and anti-atherogenic effects. Apo AI could potently bind LDL surface lipids, thereby interfering with surface lipid distribution and stabilizing the conformation of Apo B100, which interrupts aggregation induced by SMase22. Besides, Apo J may act controlling effect on aggregation. Apo J concentrations do not increase in normal arteries but in the form of LDL binding in atherosclerotic lesions. Apo J-depleted LDL experiments prove that Apo J presents a positive effect on LDL aggregation. In addition to retarding aggregation, Apo J also makes Apo B less sensitive to proteolysis, demonstrating Apo J as a universal mechanism against LDL aggregation21. The mechanism may be associated with the nature of Apo J binding to exposed hydrophobic domains during protein unfolding68, thus preventing the interaction hydrophobic surfaces-mediated interactions and subsequent aggregation of LDL particles67,69.
4. Role of Apos in oxidative modification of subendothelial LDL
LDL-associated Apos act as communicators on oxidative stress-related signaling pathways to instigate reactive oxygen species (ROS) production and lead to the generation of atherogenic ox-LDL that has been widely identified as a major determinant driving the pathogenesis of atherosclerosis. Indeed, ox-LDL and its bioactive oxidized byproducts, such as lipid peroxides, aldehydes, hydroxyl sterols, and lysophosphatidylcholine, exhibit pro-atherogenic properties and contribute to atherogenesis by affecting manifold biological processes, containing the induction of cholesterol accumulation in macrophages as well as potent proinflammatory, proapoptotic and cytotoxic activities70,71. Meanwhile, various Apos accumulate in early atherosclerotic lesions and are considered as a defense mechanism against oxidative stress, which exerts antioxidative effects directly and indirectly through two synergistic manners to restrain atherosclerotic development in vivo. One is involved in oxidative stress-associated signaling pathways that directly remove free radicals and prevent LDL oxidation, and the other interacts with oxidant and LDL oxidation byproducts and blocks their harm in atherosclerosis (Figure 3).
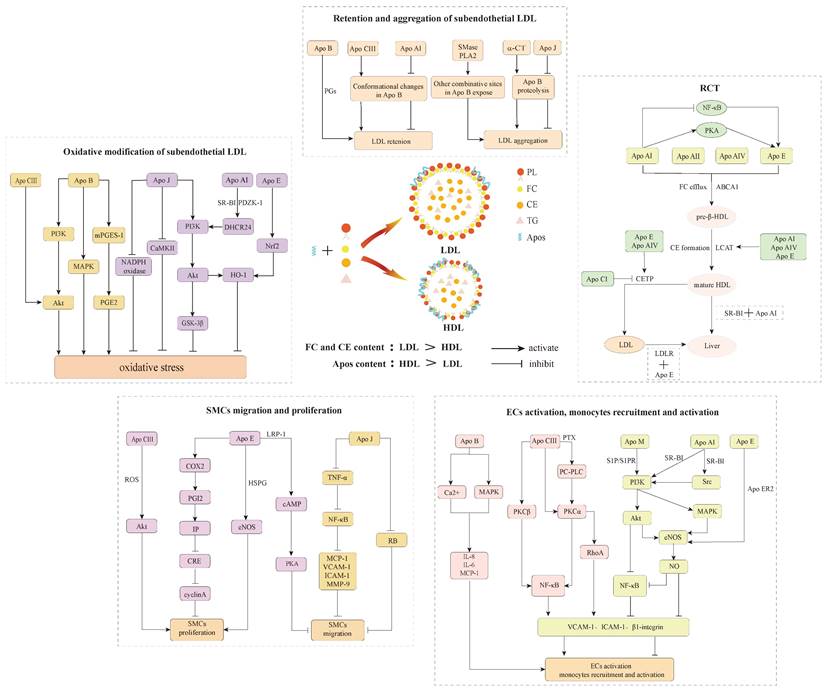
Apos participated processes of retention and aggregation of subendothelial LDL, oxidative modification of subendothelial LDL, ECs activation, monocytes recruitment and activation, SMCs migration and proliferation, as well as RCT.
4.1 Apos promote oxidative stress-related signaling pathways
On most occasions, LDL circulating in plasma is protected from oxidation with the presence of antioxidants72. It is currently acknowledged that oxidative modification of LDL occurs locally within the subendothelial space. The prolonged retention of LDL is mainly through Apo B as a communicator to trigger oxidative stress and induce ox-LDL formation. Apo B is a potent stimulator of the microsomal prostaglandin E synthase-1 (mPGES-1) pathway, contributing to the augmentation of prostaglandin E2 (PGE2) biosynthesis in early atherosclerotic lesions, which enhances vascular oxidative stress to a certain extent17. Apo B also stimulates platelet to interact with leukocytes in a p-selectin-dependent manner through activation of phosphatidylinositol 3-kinase (PI3k)/Akt and mitogen-activated protein kinase (MAPK), increasing leukocyte activation and ROS generation73. Furthermore, the effect of Apo CIII in LDL on ROS production was positive, and elevated ROS and lipid peroxides induced by Apo CIII were detected in aortic lesions in Apo CIII transgenic mice, which may be related to Akt phosphorylation18. The oxidative modification of Apo B mediates recognition of ox-LDL by scavenger receptors, facilitating unrestricted cholesterol uptake and accumulation in macrophages74.
4.2 Apos restrain oxidative stress-related signaling pathways
To maintain the prooxidant-antioxidant balance, several Apos also act as communicators of lipoproteins to synergistically attenuate oxidative damage by inhibiting oxidative stress-related signaling pathways. Apo J is enhanced in atherosclerosis as a protein sensor of oxidative stress, which significantly inhibits ROS production stimulated by ox-LDL through lowering expression related to nicotinamide adenine dinucleotide phosphate (NADPH) oxidase75,76. Apo J against detrimental effects of oxidants appears to be dependent on the phosphorylation of Akt/glycogen synthase kinase-3β (GSK-3β) and PI3K to reduce cellular ROS levels77,78. As well, Apo J exerts a cytoprotective effect against apoptosis caused by ox-LDL. Ma et al.76 found that Apo J conferred cytoprotection against ox-LDL cytotoxicity via interfering with ROS/calmodulin-dependent protein kinase II (CaMKII) pathways. Additionally, the antioxidative properties of Apos are also reflected in the activation of the nuclear factor erythrocyte 2‐related factor 2 (Nrf2) pathway. Nrf2 serves a positive role in regulating gene expression in antioxidative stress protection mechanisms79. Apo E may act as an antioxidative agent in part by interacting with Nrf2, which promotes the activation of downstream heme oxygenase‐1 (HO‐1)23. It has been indicated that HO-1 dramatically lessens atherosclerotic development in mice. Besides, Apo AI increases 3β-hydroxysteroid-Δ24 reductase (DHCR24) level by interacting with SR-BI and PDZ-domain-containing protein 1 (PDZK1), provoking phosphorylation of PI3K/Akt to trigger HO-1 expression to inhibit oxidative stress80.
4.3 Apos interact with oxidant and LDL oxidation byproducts
Except for inhibiting oxidative stress-related signaling pathways mediated by Apos, other antioxidative properties of Apos that interact with oxidant and LDL oxidation byproducts are well established. Apo AI may act directly as an antioxidant owning to contain multiple methionine groups and it suppresses the formation of lipid hydrogen peroxide, oxidized CE and PL, which lowers cytokines levels such as MCP-1 and macrophage colony-stimulating factor, as well as inhibits monocytes adhesion to ECs mediated by oxidative lipids, further blocking deleterious effects of them in atherosclerosis process81,82. Ox-LDL induces monocytes to infiltrate subendothelial and transform into macrophages that secrete myeloperoxidase to catalyze ROS intermediates formation and further aggravate LDL oxidation. Apo AIV could lower macrophage-triggered lipoproteins oxidation and copper ion-induced LDL oxidation is inhibited in a dose-dependent manner by rat Apo AIV. Moreover, Apo AIV accommodates intracellular glutathione redox balance and alleviates apoptosis mediated by oxidant7. Lipoproteins in Apo E-lacking mice are more prone to be oxidized in vitro than in wild-type mice. Evidence of amplified oxidative stress and specific epitopes of LDL oxidation in aortic lesions has been found in Apo E-deficient mice. Although the mechanism is acquainted imperfectly, the antioxidant properties of Apo E are ascribed to bind to copper and iron ions, which may isolate them and thus restrain their involvement in the oxidation process13. Furthermore, the disulfide bond of Apo J may decrease ROS content through its cysteine sulfhydryl groups83. Except for protecting LDL from copper ion-induced oxidation, Apo M may also enable to binding of oxidized phospholipids through its lipocalin structure, making it an effective inhibitor of LDL oxidation14,84. As well, Apo AI, Apo AII and Apo AIV may facilitate the removal of oxidized PL from LDL to attenuate their harmful effects71.
5. Role of Apos in the foam cells formation
Several Apos as communicators of lipoproteins are involved in inflammatory-related signaling pathways to incite robust proinflammatory activity, contributing to the occurrence of several key events involved in foam cells formation, such as ECs activation, monocytes recruitment and activation, as well as SMCs migration and proliferation. Macrophages are mainly derived from activated monocytes and SMCs that migrate from arterial media to intima. Subsequently, macrophages can avidly engulf ox-LDL by the Apos binding to cellular receptors, leading to the formation of lipid-rich foam cells that are typical hallmarks of early atherosclerosis.
5.1 Role of Apos in ECs activation, monocytes recruitment and activation
Apos serve as communicators of lipoproteins to participate in multiple inflammatory responses through motivating ECs-monocytes interactions and exert specific effects on atherogenesis. Research has verified that Apo CIII alone or as a lipoproteins component induced ECs activation and monocytes adhesion in vitro15,19. VLDL and LDL with Apo CIII expedited monocytes adhesion to ECs compared with those without Apo CIII 85,86. Ying et al.87 fed Apo CIII transgenic/LDLR null and LDLR null littermates with an atherogenic diet, and they found that Apo CIII transgenic mice increased aortic inflammation accompanied by enhanced macrophages infiltration and VCAM-1 expression. Under static or flowing conditions, Apo CIII promotes monocytes adhesion to ECs via protein kinase C alpha (PKCα) and RhoA-mediated beta1 (β1)-integrin activation. Besides, ECs-monocytes interactions irritated by Apo CIII may relate to pertussis toxin (PTX)-sensitive G protein and phosphatidylcholine-specific phospholipase C (PC-PLC) that participates in PKCα activation and further activates nuclear factor-kappaB (NF-κB) and upregulates β1-integrin expression in monocytes88. As well, Apo CIII facilitates adhesion molecules expression in ECs such as VCAM-1 and ICAM-1 in the manner of PKCβ and NF-κB activation, causing atherogenesis via ECs activation and monocytes recruitment19. More importantly, Apo B was indicated to activate monocytes and macrophages in ex vivo cultured carotid plaques through evoking MAPK and Ca2+-dependent signaling pathways, which incites the release of multiple cytokines containing IL-6, IL-8 and MCP-1 involved in monocytes recruitment in atherosclerotic lesions17.
The aforesaid findings reveal that some Apos as dangerous signals participate in foam cell formation. Meanwhile, cytoprotective Apos as communicators of lipoproteins may adjust manifold associated signal transduction pathways and actuate resistance to cellular atherogenic response (Figure 3). As a multifunctional protein, Apo E mainly synthesizes in the liver and macrophages, and may function directly in atherosclerotic lesions. Macrophage-derived Apo E shows local cytokine and hormone-like effects on perivascular cells, exerting its atheroprotective effect. For instance, Apo E initiates a signal cascade via interacting with Apo E receptor 2 (Apo ER2), leading to endothelial nitric oxide synthase (eNOS) activation and inducing nitric oxide (NO) generation, and again refraining the expression of VCAM-1 on ECs24,89. Apo E also restrains the T lymphocytes activation and proliferation13. Apo M as a chaperone of sphingosine-1-phosphate (S1P) contributes to the formation of Apo M/S1P complex in HDL, which interacts with S1P receptors (S1PR) to promote vascular barrier function and exert anti‐inflammatory functions90,91. S1PR1, S1PR2 and S1PR3 exist in vascular tissues, as well as S1PR1 is abundant in ECs40,90. The interactions between Apo M/S1P complex and S1PR1 are to maintain tight junctions between ECs and intact endothelial barrier14. Compared with wild-type mice, vascular permeability in lung tissue is enhanced in Apo M-absent mice, demonstrating that Apo M/S1P is necessary for retaining vascular integrity92,93. Apo M/S1P prominently reduces inflammatory and adhesion molecules expression. Zheng et al.94 suggested that Apo M/S1P mediated PI3K/Akt phosphorylation by S1PR2, inhibiting NF-κB translocation and reducing ox-LDL-induced cytokines secretion in ECs, such as VCAM-1, ICAM-1, tumor necrosis factor-α (TNF-α) and IL-1β. Also, Apo M/S1P may activate PI3K/Akt pathway by S1PR1 and S1PR3 to irritate eNOS expression and NO generation, lessening monocytes adhesion to ECs95,96. NO acts as a crucial atheroprotective cytokine regulating leukocyte adherence to ECs. Transgenic mice overexpressing Apo AI refrained aortic fatty streak development, ascribing to activation of tyrosine kinase (Src) and PI3K mediated by Apo AI/SR-BI, which results in parallel activation of Akt and MAPK, as well as triggers eNOS expression and NO generation97. S1P/S1PR2 increases inflammatory and adhesive molecules such as MCP-1, ICAM-1, VCAM-1, TNF-α and IL-1β through activating NF-κB in ECs. SR-BI and S1PR are co-located in ECs and Apo AI/SR-BI may negatively modulate inflammation incited by S1P/S1PR2 through PI3K/Akt/eNOS/NO/NF-κB signal transduction pathway, decreasing the release of inflammatory and adhesive mediators98. Ox-LDL has been reported to activate classical complement cascade to cause leukocyte infiltration and inflammatory response99. Complement complexes can be discovered in atherosclerotic lesions, and ECs exposed to complement are activated and improves the production of IL-6, IL-8 and MCP-1, thereby triggering the adhesion of monocytes to ECs89. Apo J could interact with complement components to prevent ECs activation through complement cascade and exert antiatherosclerotic effect100,101.
5.2 Role of Apos in SMCs migration and proliferation
Excessive migration and proliferation of SMCs from arterial media to intima are also one of the critical factors promoting atherosclerotic foam cells formation. Inflammatory cytokines derived from Apos-mediated activation of ECs and monocytes stimulate SMCs migration and proliferation. SMCs express various cytokines containing fractalkine, ICAM-1, MCP-1, VCAM-1 and matrix metalloproteinase-9 (MMP-9) under pathological conditions102,103, further exacerbating ECs activation, monocytes recruitment, SMCs migration and proliferation, which amplifies pro-inflammatory cascades and form a vicious circle. Apos as communicators of lipoproteins act vital effects in SMCs migration and proliferation by modulating related signaling pathways (Figure 3). Apo CIII may stimulate the proliferation of SMCs through Akt phosphorylation mediated by ROS18. Apo J partially represses the TNF-α/NF-κB signaling pathway and downregulates MMP-9, adhesion mediators and chemokines expression, blocking the migration of SMCs. Except for taking effect on SMCs migration, Apo J also inhibits SMCs proliferation through reducing retinoblastoma protein (RB) phosphorylation to mediate G1 phase cell cycle arrest102. In addition, it has been confirmed that Apo E refrains SMCs migration and proliferation triggered by ox-LDL, serum and platelet-derived growth factor (PDGF) via distinct mechanisms13. Apo E suppresses SMCs proliferation after vascular injury in vivo and its anti-proliferative effects are partly attributed to the inhibition of the cell cycle104. Apo E may trigger cyclooxygenase-2 (COX-2) generation, and promote the synthesis of prostacyclin (PGI2) and the activation of prostacyclin receptor IP, suppressing cyclin A gene expression in cyclic adenosine monophosphate (cAMP)-response element (CRE)-dependent manner and interrupting cycle progression of aortic SMCs105. Simultaneously, other anti-proliferative effects mediated by Apo E involve its binding to HSPG and subsequent induction of NOS activition106,107. Compared to proliferation, the suppression of PDGF-induced SMCs migration by Apo E may be mediated through LRP-1, and their binding stimulates the activation of the cAMP/PKA signaling pathway13,107.
6. Role of Apos in RCT in macrophages or foam cells
It is well established that the continuous influx of cholesterol ultimately exceeds the metabolic capacity of macrophages and thus induces foam cells formation. RCT is responsible for motivating excessive cholesterol efflux from extrahepatic tissues and returns to the liver for further metabolism and excretion56,108. Pieces of evidence support that the anti-atherosclerotic action of HDL is conventionally ascribed to its participation in RCT108,109,110. More importantly, Apos as significant communicators of lipoproteins are involved in RCT and exert specific effects, which not only serve as cholesterol receptors in extrahepatic cells to motivate new HDL particles generation, but also as cholesterol transport carriers to the liver for metabolism. This suggests that Apos-mediated RCT is the prime mechanism of restraining foam cells formation and facilitating atherosclerotic regression (Figure 2A).
6.1 Apos acting as cholesterol receptors facilitate cholesterol efflux from macrophages or foam cells
The initial step of RCT is to mobilize cholesterol efflux from macrophages or foam cells. Lipid-poor Apo AI is the dominant acceptor of cellular FC and PL in an adenosine triphosphate-binding cassette transporter A1 (ABCA1) dependent manner to form nascent discoidal pre-β-HDL that serves as a more active FC acceptor111,112. Other exchangeable Apos including Apo C and Apo E share the amphipathic helical structure and may also function in this regard113,114. Indeed, Apo E owns the competence to promote HDL synthesis by combining with ABCA1 independently of Apo AI. It has been demonstrated that Apo E expression mediated by adenoviral motivates discoidal HDL secretion in mice lacking Apo AI25. Furthermore, human Apo AI is known to be an effective inducer of endogenous Apo E production from lipid-loaded macrophages and independent of ABCA1-mediated cholesterol efflux, relating to NF-κB inhibition and/or PKA pathway activation115. The cholesterol efflux capacity of Apo AII is as efficient as Apo AI and they exert synergistic effects in HDL particles for cholesterol efflux44. Evidence reveals that Apo AIV expedites cholesterol efflux and HDL particles in Apo AIV transgenic mice display a higher capacity to lessen intracellular cholesterol content compared with wild-type mice7. HeLa cells overexpressing human ABCA1 enhanced cholesterol efflux via binding to Apo AIV, implying that Apo AIV was involved in ABCA1-mediated cholesterol efflux116. Besides, Apo AIV was indicated to enhance cholesterol emigration in PL-containing liposomes, comparable to that of Apo AI and Apo E under physiologically relevant concentrations7. Apo CI, Apo CII, and Apo M also serve as cholesterol acceptors to potentially inhibit foam cells formation40,52,117. Whereas Apo CI generation by macrophages does not exert a protective effect in atherosclerotic development despite reducing cholesterol accumulation in macrophages52.
6.2 Apos serving as LCAT activator promote mature HDL formation
In the second step, LCAT catalyzes CE formation on the pre-β-HDL surface via transferring fatty acids from lecithin to FC. Since CE is less soluble than TG, it cannot circulate in exposure to a water environment, thereby it migrates from lipoprotein particles surface to hydrophobic core, rendering discoidal pre-β-HDL into mature spherical HDL118. Mature HDL may further mediate FC efflux via other pathways, containing ABCG1 and SR-BI7. Apo AI is not the only potent activator of LCAT, which could also be activated by Apo AIV, Apo C and Apo E111. In comparison with other Apos, Apo E promotes the enlargement of HDL size ascribed to its interaction with PL polar head groups, contributing to CE enrichment in hydrophobic core119.
6.3 Apos as cellular ligand mediate cholesterol metabolism in the liver
In the final step, HDL is transported to the liver for metabolism. CE on HDL is mainly eliminated through hepatic surface receptor SR-BI112. Apo AI as a ligand interacting with SR-BI mediates CE uptake in HDL, and deficiency of Apo AI induces a reduction in CE selective uptake56,120. Except for Apo AI, Apo E also mediates HDL ingestion into hepatocytes and other cells as a ligand58. While the clearance of HDL may be interfered with Apo CIII. Morton et al.58 emphasized that Apo CIII and Apo E functioned antagonistically on HDL metabolism. As a high-affinity ligand, Apo E mediates whole HDL uptake by hepatocytes and the residence time in the circulation is much shorter than that without Apo E. Whereas Apo CIII coexisting with Apo E on HDL inhibits Apo E binding to receptors and abrogates the benefits of Apo E on HDL clearance. Alternatively, CE from HDL could be moved to Apo B-containing lipoproteins through CETP, subsequently delivered to the liver and taken up by LDLR121. CETP as a lipid transfer protein participates in lipids exchange and transport among various lipoproteins, which is the substitute way for CE delivery and thus influences the overall rate of RCT111. Apo AIV and Apo E increase human plasma CETP activity7,33. Nevertheless, Apo CI is negatively correlated with CETP in vitro37.
7. Apos as meaningful targeting mass for treatment of early atherosclerosis
Apos as communicators of lipoproteins play important roles in the pathogenesis of early atherosclerotic development. Correspondingly, they may serve as meaningful targeting mass to reverse atherosclerosis via covering up their unfavorable features and fully augmenting their beneficial features. There are two main available strategies triggering Apos for the treatment of early atherosclerosis. One reasonable approach is to regulate the expression of endogenous Apos to inhibit atherosclerosis via reducing the level of endogenous atherogenic Apos or mutating them and urging the synthesis of endogenous anti-atherogenic Apos. When the generation of endogenous Apos is insufficient or difficult to reverse the progression of atherosclerosis, the application of exogenous Apos mimetic peptides exhibiting similar biological features to native Apos is considered to be another supplementary and desirable approach for atherosclerotic therapeutic option.
7.1 Endogenous Apos as potential targets for the treatment of early atherosclerosis
Considering that Apos are bound up with the occurrence and progression of early atherosclerosis due to their atherogenic and anti-atherogenic properties, corresponding interventions such as reducing or mutating endogenous atherogenic Apos, as well as elevating endogenous anti-atherogenic Apos levels could retard atherosclerotic development. Remarkably, anti-Apos antisense oligonucleotide (ASO), aiming at Apos mRNA and reducing plasma Apos levels, offers an intriguing therapy for cardiovascular protection. An anti-Apo B ASO effectively lessens hepatic Apo B synthesis and lowers plasma cholesterol, achieving a basic elimination of plasma LDL in LDLR-deficient mice with atherosclerotic lesions. LDL concentrations declined rapidly within 3 days of administration. After 7 days of ASO treatment, the reduction of plasma Apo B concentrations lowered aortic LDL permeability, possibly further reducing LDL retention and aggregation within subendothelial space, which is prior to any detectable changes in foam cells content and size of aortic atherosclerotic plaques122. As well, mutating Apos refrains atherosclerotic development through lessening LDL retention. Skålén et al.123 created heterogeneous transgenic mice expressing recombinant LDL with a mutation in Apo B100, containing tyrosine replaced tryptophan-4369, basic amino acids in Apo B100 binding site (residues 3359-3369) changed to neutral amino acids, lysine 3363 in Apo B100 converted to glutamic acid and 6-GAGs binding site mutation in Apo B100. The results indicated that recombinant LDL with a mutation in Apo B was retained less than in wild-type mice, owning greatly decreased atherogenic potential. Volanesorsen, a second-generation anti-Apo CIII ASO, has been indicated to selectively lower Apo CIII biosynthesis in whole preclinical tested non-human primates and rodents. Additionally, this drug is tested in healthy volunteers in the primary stage of clinical use, displaying a dose-dependent reduction in Apo CIII levels and being well tolerated124,125. A specific antibody against Apo CIII is capable of blocking detrimental pro-atherogenic effects, decreasing the expression of ICAM-1 and VCAM-1 in ECs through inactivating PKCβ and NF-κB, and inhibiting monocyte recruitment49,124. Meanwhile, statin administration effectively inhibits Apo CIII-induced ECs activation and monocyte infiltration by restraining the NF-κB pathway in vivo126.
An upregulator 4010B-30 has been reported to regulate Apo AI gene expression partly through the peroxisome proliferators-activated receptor γ-mediated pathway, contributing to increase cholesterol efflux in macrophage cells and reduce atherosclerosis127. The increase of hepatic Apo AI synthesis by myriocin is associated with the inhibition of extracellular-signal-related kinase phosphorylation128. The anti-atherogenic mechanism of myriocin may be linked to promote cholesterol efflux via this pathway129. RVX-208, composite molecular belonging to the quinazoline family, acts through an epigenetic mechanism by inhibiting the bromodomain and extraterminal protein to upregulate Apo AI, resulting in the stimulation of RCT130. As well, the RVX-208 induced Apo AI messenger ribonucleic acid and protein synthesis in HepG2 cells131. African green monkeys treated with RVX-208 for 63 days elevated plasma Apo AI and HDL cholesterol levels up to 60% and 97%, resulting in increased cholesterol efflux via ABCA1, ABCG1 and SR-BI pathway131. In addition, the increase in Apo AI concentrations could be indirectly realized by modulating its catabolism. Niacin may raise Apo AI levels primarily by preventing the liver from eliminating Apo AI and augmenting RCT132. Elevated circulating endogenous Apo E levels could prevent atherosclerosis in Apo E-absent mice through hepatic gene transfer and Apo E transgenic expression mediated by recombinant adenovirus13. Oleic acid promotes Apo E synthesis and secretion in macrophages originating from monocyte at a locus involving post-translational glycosylation133. Yang et al.134 proved that Apo M was the target for the regulation of lipids by simvastatin. Simvastatin dramatically induces Apo M expression in vitro and in vivo, and its mechanism is related to inhibiting LXRα and expediting hepatocyte nuclear factor-1α. The regulation of Apo M may be a new way for statins to enhance HDL level and facilitate RCT to prevent atherosclerosis (Table 2).
Apos as meaningful targeting mass for treatment of early atherosclerosis.
| Two available strategies | Intervening measure | Mechanisms for treatment early atherosclerosis | Reference |
|---|---|---|---|
| Endogenous Apos as meaningful targeting mass | Anti-Apo B ASO | Lessening hepatic Apo B synthesis; Lowering aortic LDL permeability; Reducing LDL retention and aggregation | 122 |
| Mutating Apo B | Lessening LDL retention | 123 | |
| Anti-Apo CIII ASO | Lessening LDL retention | 124, 125 | |
| A specific antibody against Apo CIII | Decreasing the expression of ICAM-1 and VCAM-1 in ECs through inactivating PKCβ and NF-κB; Inhibiting monocyte recruitment | 49, 124 | |
| Statin administration for Apo CIII | Inhibiting Apo CIII-induced ECs activation and monocyte infiltration by restraining the NF-κB pathway | 126 | |
| 4010B-30 for Apo AI | Regulating Apo AI gene expression partly through the peroxisome proliferators-activated receptor γ-mediated pathway; Stimulating RCT | 127 | |
| Myriocin for Apo AI | Increasing hepatic Apo AI synthesis by inhibiting of extracellular-signal-related kinase phosphorylation; Promoting RCT | 128, 129 | |
| RVX-208 for Apo AI | Upregulating Apo AI by inhibiting the bromodomain and extraterminal protein; Promoting RCT | 130, 131 | |
| Niacin for Apo AI | Preventing hepatic Apo AI clearance; Augmenting RCT | 132 | |
| Niacin for Apo M | Upregulating Apo M gene and protein expression via LXRα regulation | 132 | |
| Simvastatin for Apo M | Inducing Apo M expression through inhibiting LXRα and expediting hepatocyte nuclear factor-1α; Facilitating RCT | 134 | |
| Exogenous Apos mimetic peptides as specific carriers or therapeutic agents | Ac-hE18A-NH2 | Increasing PON-1 activity related to HDL; Decreasing plasma ROS and lipid hydroperoxide levels; Restraining the expression of IL-6, VCAM-1, and MCP-1 induced by lipopolysaccharide in ECs and macrophages; Motivating RCT | 136, 137 |
| ATI-5261 | Prompting RCT | 136 | |
| CS-6253 | Prompting RCT | 136 | |
| Ep1.B | Reducing inflammatory monocytes infiltration; Inducing monocytes to differentiate into dendritic cells at the injured portion | 139 | |
| D-[113-122]Apo J | Reducing LDL retention; Lessening lipoperoxides content in lipoproteins and further sequestering lipoperoxides to prevent the oxidation of LDL | 69, 140 | |
| 4F | Regulating Apo B100 conformation and suppressing SMase-induced LDL aggregation | 22 | |
| D-4F | Exerting antioxidative roles via AMP-activated protein kinase (AMPK)/PKC pathway; Suppressing VSMCs proliferation and migration; Augmenting RCT | 141, 143, 146, 147 | |
| ELK-2A2K2E | Interfering VCAM-1 expression in ECs as well as monocytes activation and infiltration | 148 | |
| 5A | Decreasing ROS production; Reducing ECs adhesion cytokines expression and inflammatory cells infiltration; Inducing RCT | 149, 150 |
7.2 Exogenous Apos mimetic peptides as supplementary targets for the treatment of early atherosclerosis
Apos mimetic peptides derived straightly from the sequence of Apos or amino acid sequence mimicking their AαHs conformation show pleiotropic anti-atherogenic properties, such as inhibiting LDL retention and aggregation, promoting RCT, as well as antioxidative and anti-inflammatory functions, rendering them to be novel and better exogenous agent potential exogenous agent for preventative and therapeutic atherosclerosis owing to the perspective of clinical applications135. Administration of Ac-hE18A-NH2, an Apo E mimetic peptide, to western diet-fed Apo E null mice results in increasing PON-1 activity related to HDL and decreases plasma ROS and lipid hydroperoxide levels136. Ac-hE18A-NH2 restrains the expression of IL-6, VCAM-1 and MCP-1 in lipopolysaccharide-induced ECs and macrophages, and decreases the adhesion of monocytes to ECs. It is also linked to the generation of pre-β-HDL that may be involved in triggering endogenous Apo E secretion in macrophages and motivating cholesterol efflux in an ABCA1-independent manner, thereby inhibiting foam cells formation137. Other Apo E-derived peptides such as ATI-5261 and CS-6253 prompt RCT with analogous efficiency to native Apo E. Human plasma added to ATI-5261 or CS-6253 enhances cholesterol emigration in an ABCA1-dependent way and causes pre-β-HDL formation, as well as mature HDL-ATI-5261 and HDL-CS-6253 motivate SR-BI-mediated hepatic cholesterol uptake. Whereas, ATI-5261 generates muscle toxicity in wild-type mice compared to CS-6253136. Ep1.B, another peptide of Apo E, could inhibit early plaque formation in Apo E null mice, effectively replacing Apo E roles138. The inhibitory effect of Ep1.B was associated with the reduction of inflammatory monocytes infiltration. Besides, Ep1.B induced monocytes to differentiate into dendritic cells at the injured portion, which may be a crucial manner for monocytes to emigrate from atherosclerotic plaques without rendering them into macrophages or foam cells139.
D-[113-122]Apo J derived from native Apo J may bind to hydrophobic domains on lipoproteins surface to avoid interactions between different LDL particles and reduce their retention, as well as lessen lipoperoxides content in lipoproteins and further sequester lipoperoxides to prevent the oxidation of LDL69,140. Similar to Apo E and Apo J mimetics, various synthetic Apo AI peptides containing 4F (L-4F and D-4F), 5A, 6F and ELK-2A2K2E have been designed and certified to be functionally analogous to native Apo AI. 4F interacts strongly with lipids on the surface of LDL and blocks lipid distribution, consequently regulating Apo B100 conformation and suppressing SMase-induced LDL aggregation22. D-4F exerts crucial antioxidative roles in ECs via scavenging ROS and restraining ROS production, which suppresses the activation of NADPH oxidase and the production of ROS via AMP-activated protein kinase (AMPK)/PKC pathway, alleviating oxidative injuries in ECs141. D-4F also promotes ECs repair and ameliorates the oxidative damage of ECs triggered by ox-LDL via Akt/AMPK/eNOS/HO-1 signaling pathway142. In vitro experiments, D-4F suppresses SMCs proliferation and migration induced by ox-LDL in dose-dependent mode. The expression of HO‐1 in SMCs is stimulated by D‐4F and the PI3K/Akt/AMPK pathway is involved in these processes. As well, this pathway mediated by D‐4F alleviates oxidative stress in SMCs via its interaction with ABCA1143. Moreover, 4F activates small G protein and its downstream janus kinase-2 (JAK2)/signal transducers and activators-3 (STAT3) signaling pathway through ABCA1, acting vital roles in macrophage activation144. Activation of STAT3 incites tristetraprolin (TTP) expression that regulates ribonucleic acid stability in macrophages145. Oral D-4F reduces atherosclerosis in Apo E-deficient and LDLR-deficient mice, significantly increasing cholesterol emigration from mouse macrophages by mediating cellular signal transduction146. D-4F may trigger intracellular cAMP release and subsequent PKA-mediated ABCA1 phosphorylation, further improving the interaction with ABCA1 and accelerating cholesterol efflux to form HDL-like particles147. ELK-2A2K2E, another Apo AI mimetic peptide, is reported to restrain foam cells formation through interfering VCAM-1 expression in ECs as well as monocytes activation and infiltration148, which stimulates messenger RNA degradation of TNF-α, MCP-1 and IL-6, and decreases their generation by modulating ABCA1/JAK2/STAT3/TTP signaling pathway144. Beyond that, 5A peptide also exhibits anti-atherogenic effects similar to Apo AI, and antioxidative and anti-inflammatory properties of 5A peptide are mediated mainly by ABCA1 and NF-κB signaling pathways, decreasing ROS production, ECs adhesion cytokines expression and inflammatory cells infiltration, thereby reducing the formation of foam cells149. 5A peptide induces macrophages cholesterol efflux via ABCA1-mediated mechanism both in vivo and vitro, and reduces atherosclerosis in Apo E-absent mice150.
8. Conclusion and outlook
Atherosclerosis is the underlying cause of most cardiovascular diseases and is highly associated with coronary artery disease (CAD), stroke and peripheral artery disease, resulting in high morbidity and mortality worldwide. In this review, we mainly pay attention to the biological characteristics of Apos, the roles of Apos in the pathogenesis of early atherosclerosis, and Apos as the meaningful targeting mass in the treatment of early atherosclerosis. It indicates that Apos serve as critical communicators of lipoproteins for the pro-atherogenic and anti-atherogenic processes of early atherosclerosis through influencing LDL retention and aggregation, oxidative modification of LDL, foam cells formation, and RCT in macrophage cells or foam cells. Correspondingly, Apos may be applied as the meaningful targeting mass in endogenous and/or exogenous manners for the effective treatment of early atherosclerosis.
Although the early intervention of atherosclerosis may achieve the desired therapeutic effect, long-term clinical silence may delay the optimal treatment time because atherosclerosis is a chronic progressive disease. If early treatment is not timely, acute myocardial infarction, ischemic stroke, pulmonary embolism and other severe complication may occur151. Therefore, from a clinical perspective, early detection and diagnosis of atherogenic risk factors are significantly important, which enable to timely intervene before the deterioration and emergence of clinical symptoms. Since increased LDL cholesterol level is a typical hazardous factor for CAD, LDL has traditionally been used for the assessment of risk associated with atherosclerosis and relevant coronary artery disease. While LDL is not a perfect predictive factor because numerous individuals with cholesterol levels within the health-associated reference range also develop atherosclersis152. This is partly attributed to the focusing only on LDL cholesterol and ignoring other important aspects of Apos as communicators of lipoproteins involved in lipid metabolism and atherosclerotic development. Remarkably, if Apos contribute to certain aspects of the pathogenesis of early atherosclerosis, targeting Apos-related indicators could become another potential diagnostic clue for early prevention. Pieces of evidence indicate that Apos measurements as potential communicators of lipoproteins may be better predictors and diagnostic indicators in clinical practice than traditional lipid parameters153. In general, higher concentrations of Apo B, Apo CII and Apo CIII, lower concentrations of Apo AI and Apo E, as well a high Apo B/Apo AI ratio, are identified as independent risk factors for CAD45,126,154. Apo B is superior to non-HDL-cholesterol and LDL-cholesterol as a predictor of cardiovascular risk. Introducing Apo B measurement into routine care could prevent more events than diagnosis and treatment based on non-HDL-cholesterol and LDL-cholesterol levels155,156. Apo B serves as the main transporter of atherogenic lipoproteins, and Apo AI as an anti-atherogenic agent is responsible for the reverse transport of cholesterol. The Apo B/Apo AI ratio reveals the balance between opposite forces of pro-atherosclerosis and anti-atherosclerosis. The higher the Apo B/Apo AI ratio, the more cholesterol is accumulated within the subendothelial space, hence triggering atherosclerotic development and increasing the risk of CAD157. Additionally, epidemiological studies have indicated that plasma Apo CIII levels alone and as a component of lipoproteins are strong and independent predictors of CAD in prospective human cohorts adjusted for classic lipid risk factors126,158. HDL or LDL-containing Apo CIII predicts a higher risk of CAD, and the negative correlation between Apo E in HDL and CAD risk was wholly covered up in Apo CIII-containing HDL58,158. On the contrary, LDL or HDL without Apo CIII could not forecast the occurrence and risk of CAD159. In conclusion, Apos exert a significant role in the diagnosis of atherosclerosis, and further research in this aspect may be a very prospective direction for atherosclerotic therapy in the future.
Abbreviations
Apos: amphiphilic apolipoproteins; LDL: low-density lipoprotein; RCT: reverse cholesterol transport; TG: triglycerides; PL: phospholipids; CM: chylomicron; VLDL: very low-density lipoprotein; IDL: intermediate-density lipoprotein; ECM: extracellular matrix; PGs: proteoglycans; ECs: endothelial cells; ICAM-1: intercellular adhesion molecule-1; VCAM-1: vascular cell adhesion molecule-1; SMCs: smooth muscle cells; ox-LDL: oxidized LDL; AαHs: amphiphilic α-helices; AβS: amphipathic β-sheet; LCAT: lecithin cholesterol acyltransferase; FC: free cholesterol; CE: cholesterol esters; LPL: lipoprotein lipase; TRL: triglyceride-rich lipoproteins; HL: hepatic lipase; CETP: cholesteryl ester transfer protein; LDLR: LDL receptor; LRP: LDL receptor-related protein; HSPG: heparan sulfate proteoglycans; GAGs: glycosaminoglycans; SMase: sphingomyelinases; PLA2: phospholipaseA2; α-CT: α-chymotrypsin; ROS: reactive oxygen species; mPGES-1: microsomal prostaglandin E synthase-1; PGE2: prostaglandin E2; PI3k: phosphatidylinositol 3-kinase; MAPK: mitogen-activated protein kinase; NADPH: nicotinamide adenine dinucleotide phosphate; GSK-3β: glycogen synthase kinase-3β; CaMKII: calmodulin-dependent protein kinase II; Nrf2: nuclear factor erythrocyte 2‐related factor 2; HO‐1: heme oxygenase‐1; DHCR24: 3β-hydroxysteroid-Δ24 reductase; PDZK1: PDZ-domain-containing protein 1; PKCα: protein kinase C alpha; β1: beta1; PTX: pertussis toxin; PC-PLC: phosphatidylcholine-specific phospholipase C; Apo ER2: apo E receptor 2; eNOS: endothelial nitric oxide synthase; NO: nitric oxide; S1P: sphingosine-1-phosphate; S1PR: S1P receptors; TNF-α: tumor necrosis factor-α; MMP-9: matrix metalloproteinase-9; RB: retinoblastoma protein; PDGF: platelet-derived growth factor; COX-2: cyclooxygenase-2; PGI2: prostacyclin; cAMP: cyclic adenosine monophosphate; CRE: cAMP-response element; ABCA1: adenosine triphosphate-binding cassette transporter A1; ASO: antisense oligonucleotide; LXRα: liver x receptor α; AMPK: AMP-activated protein kinase; JAK2: janus kinase-2; STAT3: signal transducers and activators-3; TTP: tristetraprolin; CAD: coronary artery disease.
Acknowledgements
A special thanks for the long-term subsidy mechanism from the Ministry of Finance and the Ministry of Education of PRC (People's Republic of China) for BUCM (Beijing University of Chinese Medicine).
Funding
This work was supported by the Beijing Natural Science Foundation [7202111] and the National Science and Technology Major Project [2018ZX10101001-005-003].
Author contributions
Yang Li and Yuanyan Liu proposed the concept and constructed the framework of the article. Zhenglai Hua, Xinyi Luo, Xiaoxia Xue, Xiangpeng Wang, and Mingshi Pang searched and collected literature. Yang Li and Yuanyan Liu wrote and revised the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Malekmohammad K, Sewell RDE, Rafieian-Kopaei M. Antioxidants and Atherosclerosis: Mechanistic Aspects. Biomolecules. 2019;9:301
2. Hartley A, Haskard D, Khamis R. Oxidized LDL and anti-oxidized LDL antibodies in atherosclerosis - Novel insights and future directions in diagnosis and therapy. Trends Cardiovasc Med. 2019;29:22-26
3. Fatkhullina AR, Peshkova IO, Koltsova EK. The Role of Cytokines in the Development of Atherosclerosis. Biochemistry (Mosc). 2016;81:1358-1370
4. Libby P. The changing landscape of atherosclerosis. Nature. 2021;592:524-533
5. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS. et al. Atherosclerosis. Nat Rev Dis Primers. 2019;5:56
6. Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ Res. 2016;118:535-546
7. Qu J, Ko CW, Tso P, Bhargava A. Apolipoprotein A-IV: A Multifunctional Protein Involved in Protection against Atherosclerosis and Diabetes. Cells. 2019;8:319
8. Ahotupa M. Oxidized lipoprotein lipids and atherosclerosis. Free Radic Res. 2017;51:439-447
9. Wolkowicz P, White CR, Anantharamaiah GM. Apolipoprotein Mimetic Peptides: An Emerging Therapy against Diabetic Inflammation and Dyslipidemia. Biomolecules. 2021;11:627
10. Hu J, Xi D, Zhao J, Luo T, Liu J, Lu H. et al. High-density Lipoprotein and Inflammation and Its Significance to Atherosclerosis. Am J Med Sci. 2016;352:408-415
11. Feig JE. Regression of atherosclerosis: insights from animal and clinical studies. Ann Glob Health. 2014;80:13-23
12. Wilkins JT, Gidding SS, Robinson JG. Can atherosclerosis be cured? Curr Opin Lipidol. 2019;30:477-484
13. Greenow K, Pearce NJ, Ramji DP. The key role of apolipoprotein E in atherosclerosis. J Mol Med (Berl). 2005;83:329-342
14. Christoffersen C, Nielsen LB. Apolipoprotein M: bridging HDL and endothelial function. Curr Opin Lipidol. 2013;24:295-300
15. Luo M, Peng D. The emerging role of apolipoprotein C-III: beyond effects on triglyceride metabolism. Lipids Health Dis. 2016;15:184
16. Gustafsson M, Flood C, Jirholt P, Borén J. Retention of atherogenic lipoproteins in atherogenesis. Cell Mol Life Sci. 2004;61:4-9
17. Ketelhuth DF, Rios FJ, Wang Y, Liu H, Johansson ME, Fredrikson GN. et al. Identification of a danger-associated peptide from apolipoprotein B100 (ApoBDS-1) that triggers innate proatherogenic responses. Circulation. 2011;124:2433-2443 1-7
18. Li H, Han Y, Qi R, Wang Y, Zhang X, Yu M. et al. Aggravated restenosis and atherogenesis in ApoCIII transgenic mice but lack of protection in ApoCIII knockouts: the effect of authentic triglyceride-rich lipoproteins with and without ApoCIII. Cardiovasc Res. 2015;107:579-589
19. Kawakami A, Aikawa M, Alcaide P, Luscinskas FW, Libby P, Sacks FM. Apolipoprotein CIII induces expression of vascular cell adhesion molecule-1 in vascular endothelial cells and increases adhesion of monocytic cells. Circulation. 2006;114:681-687
20. Wang D, Yang Y, Lei Y, Tzvetkov NT, Liu X, Yeung AWK. et al. Targeting Foam Cell Formation in Atherosclerosis: Therapeutic Potential of Natural Products. Pharmacol Rev. 2019;71:596-670
21. Martínez-Bujidos M, Rull A, González-Cura B, Pérez-Cuéllar M, Montoliu-Gaya L, Villegas S. et al. Clusterin/apolipoprotein J binds to aggregated LDL in human plasma and plays a protective role against LDL aggregation. FASEB J. 2015;29:1688-1700
22. Nguyen SD, Javanainen M, Rissanen S, Zhao H, Huusko J, Kivelä AM. et al. Apolipoprotein A-I mimetic peptide 4F blocks sphingomyelinase-induced LDL aggregation. J Lipid Res. 2015;56:1206-1221
23. Dai N, Tang C, Liu H, Huang S. Effect of electroacupuncture on inhibition of inflammatory response and oxidative stress through activating ApoE and Nrf2 in a mouse model of spinal cord injury. Brain Behav. 2021;11:e2328
24. Stannard AK, Riddell DR, Sacre SM, Tagalakis AD, Langer C, von Eckardstein A. et al. Cell-derived apolipoprotein E (ApoE) particles inhibit vascular cell adhesion molecule-1 (VCAM-1) expression in human endothelial cells. J Biol Chem. 2001;276:46011-46016
25. Kypreos KE, Zannis VI. Pathway of biogenesis of apolipoprotein E-containing HDL in vivo with the participation of ABCA1 and LCAT. Biochem J. 2007;403:359-367
26. Ren L, Yi J, Li W, Zheng X, Liu J, Wang J. et al. Apolipoproteins and cancer. Cancer Med. 2019;8:7032-7043
27. Gianazza E, Zoanni B, Mallia A, Brioschi M, Colombo GI, Banfi C. Proteomic studies on apoB-containing lipoprotein in cardiovascular research: A comprehensive review. Mass Spectrom Rev. 2021
28. Huebbe P, Rimbach G. Evolution of human apolipoprotein E (APOE) isoforms: Gene structure, protein function and interaction with dietary factors. Ageing Res Rev. 2017;37:146-161
29. Mitsche MA, Small DM. Surface pressure-dependent conformation change of apolipoprotein-derived amphipathic α-helices. J Lipid Res. 2013;54:1578-1588
30. Davidsson P, Hulthe J, Fagerberg B, Camejo G. Proteomics of apolipoproteins and associated proteins from plasma high-density lipoproteins. Arterioscler Thromb Vasc Biol. 2010;30:156-163
31. Tian Y, Wen H, Qi X, Mao X, Shi Z, Li J. et al. Analysis of apolipoprotein multigene family in spotted sea bass (Lateolabrax maculatus) and their expression profiles in response to Vibrio harveyi infection. Fish Shellfish Immunol. 2019;92:111-118
32. Segrest JP, Jones MK, De Loof H, Dashti N. Structure of apolipoprotein B-100 in low density lipoproteins. J Lipid Res. 2001;42:1346-1367
33. Wu CL, Zhao SP, Yu BL. Intracellular role of exchangeable apolipoproteins in energy homeostasis, obesity and non-alcoholic fatty liver disease. Biol Rev Camb Philos Soc. 2015;90:367-376
34. Gursky O. Apolipoprotein structure and dynamics. Curr Opin Lipidol. 2005;16:287-294
35. Saito H, Lund-Katz S, Phillips MC. Contributions of domain structure and lipid interaction to the functionality of exchangeable human apolipoproteins. Prog Lipid Res. 2004;43:350-380
36. Johs A, Hammel M, Waldner I, May RP, Laggner P, Prassl R. Modular structure of solubilized human apolipoprotein B-100. Low resolution model revealed by small angle neutron scattering. J Biol Chem. 2006;281:19732-19739
37. Jiang ZG, Gantz D, Bullitt E, McKnight CJ. Defining lipid-interacting domains in the N-terminal region of apolipoprotein B. Biochemistry. 2006;45:11799-11808
38. Hussain MM, Shi J, Dreizen P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J Lipid Res. 2003;44:22-32
39. Hamilton RT, Asatryan L, Nilsen JT, Isas JM, Gallaher TK, Sawamura T. et al. LDL protein nitration: implication for LDL protein unfolding. Arch Biochem Biophys. 2008;479:1-14
40. Yao Mattisson I, Christoffersen C. Apolipoprotein M and its impact on endothelial dysfunction and inflammation in the cardiovascular system. Atherosclerosis. 2021;334:76-84
41. Arkensteijn BW, Berbée JF, Rensen PC, Nielsen LB, Christoffersen C. The apolipoprotein m-sphingosine-1-phosphate axis: biological relevance in lipoprotein metabolism, lipid disorders and atherosclerosis. Int J Mol Sci. 2013;14:4419-4431
42. Dominiczak MH, Caslake MJ. Apolipoproteins: metabolic role and clinical biochemistry applications. Ann Clin Biochem. 2011;48:498-515
43. Koike T, Koike Y, Yang D, Guo Y, Rom O, Song J. et al. Human apolipoprotein A-II reduces atherosclerosis in knock-in rabbits. Atherosclerosis. 2021;316:32-40
44. Wang Y, Niimi M, Nishijima K, Waqar AB, Yu Y, Koike T. et al. Human apolipoprotein A-II protects against diet-induced atherosclerosis in transgenic rabbits. Arterioscler Thromb Vasc Biol. 2013;33:224-231
45. Dittrich J, Beutner F, Teren A, Thiery J, Burkhardt R, Scholz M. et al. Plasma levels of apolipoproteins C-III, A-IV, and E are independently associated with stable atherosclerotic cardiovascular disease. Atherosclerosis. 2019;281:17-24
46. Li G, Luo H, Sun G, Wu G, Wu G, Wang Y. et al. Cloning and characterization of a novel apolipoprotein gene, apolipoprotein AV, in tree shrews. Mol Biol Rep. 2013;40:5429-5438
47. Gautier T, Deckert V, Aires V, Le Guern N, Proukhnitzky L, Patoli D. et al. Human apolipoprotein C1 transgenesis reduces atherogenesis in hypercholesterolemic rabbits. Atherosclerosis. 2021;320:10-18
48. Gao M, Yang C, Wang X, Guo M, Yang L, Gao S. et al. ApoC2 deficiency elicits severe hypertriglyceridemia and spontaneous atherosclerosis: A rodent model rescued from neonatal death. Metabolism. 2020;109:154296
49. Jin JL, Guo YL, Li JJ. Apoprotein C-III: A review of its clinical implications. Clin Chim Acta. 2016;460:50-54
50. Banfi C, Brioschi M, Barcella S, Wait R, Begum S, Galli S. et al. Proteomic analysis of human low-density lipoprotein reveals the presence of prenylcysteine lyase, a hydrogen peroxide-generating enzyme. Proteomics. 2009;9:1344-1352
51. Seo JA, Kang MC, Ciaraldi TP, Kim SS, Park KS, Choe C. et al. Circulating ApoJ is closely associated with insulin resistance in human subjects. Metabolism. 2018;78:155-166
52. Westerterp M, Van Eck M, de Haan W, Offerman EH, Van Berkel TJ, Havekes LM. et al. Apolipoprotein CI aggravates atherosclerosis development in ApoE-knockout mice despite mediating cholesterol efflux from macrophages. Atherosclerosis. 2007;195:e9-16
53. D'Erasmo L, Di Costanzo A, Gallo A, Bruckert E, Arca M. ApoCIII: A multifaceted protein in cardiometabolic disease. Metabolism. 2020;113:154395
54. Walldius G, Jungner I. Apolipoprotein B and apolipoprotein A-I: risk indicators of coronary heart disease and targets for lipid-modifying therapy. J Intern Med. 2004;255:188-205
55. Walldius G, Jungner I. The apoB/apoA-I ratio: a strong, new risk factor for cardiovascular disease and a target for lipid-lowering therapy-a review of the evidence. J Intern Med. 2006;259:493-519
56. Xu X, Song Z, Mao B, Xu G. Apolipoprotein A1-Related Proteins and Reverse Cholesterol Transport in Antiatherosclerosis Therapy: Recent Progress and Future Perspectives. Cardiovasc Ther. 2022;2022:4610834
57. Reardon CA. Apolipoprotein E mimetic is more effective than apolipoprotein A-I mimetic in reducing lesion formation in older female apo E null mice: a commentary. Atherosclerosis. 2012;225:39-40
58. Morton AM, Koch M, Mendivil CO, Furtado JD, Tjønneland A, Overvad K. et al. Apolipoproteins E and CIII interact to regulate HDL metabolism and coronary heart disease risk. JCI Insight. 2018;3:e98045
59. Borén J, Williams KJ. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol. 2016;27:473-483
60. Ketelhuth DF, Hansson GK. Adaptive Response of T and B Cells in Atherosclerosis. Circ Res. 2016;118:668-678
61. Gustafsson M, Borén J. Mechanism of lipoprotein retention by the extracellular matrix. Curr Opin Lipidol. 2004;15:505-514
62. Sandesara PB, Virani SS, Fazio S, Shapiro MD. The Forgotten Lipids: Triglycerides, Remnant Cholesterol, and Atherosclerotic Cardiovascular Disease Risk. Endocr Rev. 2019;40:537-557
63. Hiukka A, Ståhlman M, Pettersson C, Levin M, Adiels M, Teneberg S. et al. ApoCIII-enriched LDL in type 2 diabetes displays altered lipid composition, increased susceptibility for sphingomyelinase, and increased binding to biglycan. Diabetes. 2009;58:2018-2026
64. Borén J, Packard CJ, Taskinen MR. The Roles of ApoC-III on the Metabolism of Triglyceride-Rich Lipoproteins in Humans. Front Endocrinol (Lausanne). 2020;11:474
65. Flood C, Gustafsson M, Pitas RE, Arnaboldi L, Walzem RL, Borén J. Molecular mechanism for changes in proteoglycan binding on compositional changes of the core and the surface of low-density lipoprotein-containing human apolipoprotein B100. Arterioscler Thromb Vasc Biol. 2004;24:564-570
66. Sneck M, Nguyen SD, Pihlajamaa T, Yohannes G, Riekkola ML, Milne R. et al. Conformational changes of apoB-100 in SMase-modified LDL mediate formation of large aggregates at acidic pH. J Lipid Res. 2012;53:1832-1839
67. Ruuth M, Nguyen SD, Vihervaara T, Hilvo M, Laajala TD, Kondadi PK. et al. Susceptibility of low-density lipoprotein particles to aggregate depends on particle lipidome, is modifiable, and associates with future cardiovascular deaths. Eur Heart J. 2018;39:2562-2573
68. Yerbury JJ, Poon S, Meehan S, Thompson B, Kumita JR, Dobson CM. et al. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 2007;21:2312-2322
69. Rivas-Urbina A, Rull A, Montoliu-Gaya L, Pérez-Cuellar M, Ordóñez-Llanos J, Villegas S. et al. Low-density lipoprotein aggregation is inhibited by apolipoprotein J-derived mimetic peptide D-[113-122]apoJ. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865:158541
70. Hansel B, Kontush A, Bonnefont-Rousselot D, Bruckert E, Chapman MJ. Alterations in lipoprotein defense against oxidative stress in metabolic syndrome. Curr Atheroscler Rep. 2006;8:501-509
71. Elsøe S, Ahnström J, Christoffersen C, Hoofnagle AN, Plomgaard P, Heinecke JW. et al. Apolipoprotein M binds oxidized phospholipids and increases the antioxidant effect of HDL. Atherosclerosis. 2012;221:91-97
72. Osterud B, Bjorklid E. Role of monocytes in atherogenesis. Physiol Rev. 2003;83:1069-1112
73. Assinger A, Wang Y, Butler LM, Hansson GK, Yan ZQ, Söderberg-Nauclér C. et al. Apolipoprotein B100 danger-associated signal 1 (ApoBDS-1) triggers platelet activation and boosts platelet-leukocyte proinflammatory responses. Thromb Haemost. 2014;112:332-341
74. Markakis KP, Koropouli MK, Grammenou-Savvoglou S, van Winden EC, Dimitriou AA, Demopoulos CA. et al. Implication of lipoprotein associated phospholipase A2 activity in oxLDL uptake by macrophages. J Lipid Res. 2010;51:2191-2201
75. Yang N, Qin Q. Apolipoprotein J: A New Predictor and Therapeutic Target in Cardiovascular Disease? Chin Med J (Engl). 2015;128:2530-2534
76. Ma Y, Gong Z, Nan K, Qi S, Chen Y, Ding C. et al. Apolipoprotein-J blocks increased cell injury elicited by ox-LDL via inhibiting ROS-CaMKII pathway. Lipids Health Dis. 2019;18:117
77. Foster EM, Dangla-Valls A, Lovestone S, Ribe EM, Buckley NJ. Clusterin in Alzheimer's Disease: Mechanisms, Genetics, and Lessons From Other Pathologies. Front Neurosci. 2019;13:164
78. Jun HO, Kim DH, Lee SW, Lee HS, Seo JH, Kim JH. et al. Clusterin protects H9c2 cardiomyocytes from oxidative stress-induced apoptosis via Akt/GSK-3β signaling pathway. Exp Mol Med. 2011;43:53-61
79. Shen B, Zhao C, Wang Y, Peng Y, Cheng J, Li Z. et al. Aucubin inhibited lipid accumulation and oxidative stress via Nrf2/HO-1 and AMPK signalling pathways. J Cell Mol Med. 2019;23:4063-4075
80. Wu BJ, Chen K, Shrestha S, Ong KL, Barter PJ, Rye KA. High-density lipoproteins inhibit vascular endothelial inflammation by increasing 3β-hydroxysteroid-Δ24 reductase expression and inducing heme oxygenase-1. Circ Res. 2013;112:278-288
81. Feig JE, Feig JL, Dangas GD. The role of HDL in plaque stabilization and regression: basic mechanisms and clinical implications. Coron Artery Dis. 2016;27:592-603
82. Feig JE, Shamir R, Fisher EA. Atheroprotective effects of HDL: beyond reverse cholesterol transport. Curr Drug Targets. 2008;9:196-203
83. Lee YN, Shim YJ, Kang BH, Park JJ, Min BH. Over-expression of human clusterin increases stress resistance and extends lifespan in Drosophila melanogaster. Biochem Biophys Res Commun. 2012;420:851-856
84. Christoffersen C, Jauhiainen M, Moser M, Porse B, Ehnholm C, Boesl M. et al. Effect of apolipoprotein M on high density lipoprotein metabolism and atherosclerosis in low density lipoprotein receptor knock-out mice. J Biol Chem. 2008;283:1839-1847
85. Norata GD, Tsimikas S, Pirillo A, Catapano AL. Apolipoprotein C-III: From Pathophysiology to Pharmacology. Trends Pharmacol Sci. 2015;36:675-687
86. Kawakami A, Aikawa M, Libby P, Alcaide P, Luscinskas FW, Sacks FM. Apolipoprotein CIII in apolipoprotein B lipoproteins enhances the adhesion of human monocytic cells to endothelial cells. Circulation. 2006;113:691-700
87. Yingchun H, Yahong M, Jiangping W, Xiaokui H, Xiaohong Z. Increased inflammation, endoplasmic reticulum stress and oxidative stress in endothelial and macrophage cells exacerbate atherosclerosis in ApoCIII transgenic mice. Lipids Health Dis. 2018;17:220
88. Kawakami A, Aikawa M, Nitta N, Yoshida M, Libby P, Sacks FM. Apolipoprotein CIII-induced THP-1 cell adhesion to endothelial cells involves pertussis toxin-sensitive G protein- and protein kinase C alpha-mediated nuclear factor-kappaB activation. Arterioscler Thromb Vasc Biol. 2007;27:219-225
89. Sacre SM, Stannard AK, Owen JS. Apolipoprotein E (apoE) isoforms differentially induce nitric oxide production in endothelial cells. FEBS Lett. 2003;540:181-187
90. Galvani S, Sanson M, Blaho VA, Swendeman SL, Obinata H, Conger H. et al. HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci Signal. 2015;8:ra79
91. Del Gaudio I, Rubinelli L, Sasset L, Wadsack C, Hla T, Di Lorenzo A. Endothelial Spns2 and ApoM Regulation of Vascular Tone and Hypertension Via Sphingosine-1-Phosphate. J Am Heart Assoc. 2021;10:e021261
92. Borup A, Christensen PM, Nielsen LB, Christoffersen C. Apolipoprotein M in lipid metabolism and cardiometabolic diseases. Curr Opin Lipidol. 2015;26:48-55
93. Christoffersen C, Obinata H, Kumaraswamy SB, Galvani S, Ahnström J, Sevvana M. et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci U S A. 2011;108:9613-9618
94. Zheng Z, Zeng Y, Zhu X, Tan Y, Li Y, Li Q. et al. ApoM-S1P Modulates Ox-LDL-Induced Inflammation Through the PI3K/Akt Signaling Pathway in HUVECs. Inflammation. 2019;42:606-617
95. De Palma C, Meacci E, Perrotta C, Bruni P, Clementi E. Endothelial nitric oxide synthase activation by tumor necrosis factor alpha through neutral sphingomyelinase 2, sphingosine kinase 1, and sphingosine 1 phosphate receptors: a novel pathway relevant to the pathophysiology of endothelium. Arterioscler Thromb Vasc Biol. 2006;26:99-105
96. Nofer JR, van der Giet M, Tölle M, Wolinska I, von Wnuck Lipinski K, Baba HA. et al. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J Clin Invest. 2004;113:569-581
97. Mineo C, Shaul PW. HDL stimulation of endothelial nitric oxide synthase: a novel mechanism of HDL action. Trends Cardiovasc Med. 2003;13:226-231
98. Ren K, Lu YJ, Mo ZC, -Liu X, Tang ZL, Jiang Y. et al. ApoA-I/SR-BI modulates S1P/S1PR2-mediated inflammation through the PI3K/Akt signaling pathway in HUVECs. J Physiol Biochem. 2017;73:287-296
99. Yin C, Ackermann S, Ma Z, Mohanta SK, Zhang C, Li Y. et al. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med. 2019;25:496-506
100. Urbich C, Fritzenwanger M, Zeiher AM, Dimmeler S. Laminar shear stress upregulates the complement-inhibitory protein clusterin: a novel potent defense mechanism against complement-induced endothelial cell activation. Circulation. 2000;101:352-355
101. Schwarz M, Spath L, Lux CA, Paprotka K, Torzewski M, Dersch K. et al. Potential protective role of apoprotein J (clusterin) in atherogenesis: binding to enzymatically modified low-density lipoprotein reduces fatty acid-mediated cytotoxicity. Thromb Haemost. 2008;100:110-118
102. Kim HJ, Yoo EK, Kim JY, Choi YK, Lee HJ, Kim JK. et al. Protective role of clusterin/apolipoprotein J against neointimal hyperplasia via antiproliferative effect on vascular smooth muscle cells and cytoprotective effect on endothelial cells. Arterioscler Thromb Vasc Biol. 2009;29:1558-1564
103. Kim HJ, Park KG, Yoo EK, Kim YH, Kim YN, Kim HS. et al. Effects of PGC-1alpha on TNF-alpha-induced MCP-1 and VCAM-1 expression and NF-kappaB activation in human aortic smooth muscle and endothelial cells. Antioxid Redox Signal. 2007;9:301-307
104. Zhu B, Kuhel DG, Witte DP, Hui DY. Apolipoprotein E inhibits neointimal hyperplasia after arterial injury in mice. Am J Pathol. 2000;157:1839-1848
105. Kothapalli D, Fuki I, Ali K, Stewart SA, Zhao L, Yahil R. et al. Antimitogenic effects of HDL and APOE mediated by Cox-2-dependent IP activation. J Clin Invest. 2004;113:609-618
106. Swertfeger DK, Hui DY. Apolipoprotein E receptor binding versus heparan sulfate proteoglycan binding in its regulation of smooth muscle cell migration and proliferation. J Biol Chem. 2001;276:25043-25048
107. Zhu Y, Hui DY. Apolipoprotein E binding to low density lipoprotein receptor-related protein-1 inhibits cell migration via activation of cAMP-dependent protein kinase A. J Biol Chem. 2003;278:36257-36263
108. Ouimet M, Barrett TJ, Fisher EA. HDL and Reverse Cholesterol Transport. Circ Res. 2019;124:1505-1518
109. Rye KA, Barter PJ. Cardioprotective functions of HDLs. J Lipid Res. 2014;55:168-179
110. Choi HY, Hafiane A, Schwertani A, Genest J. High-Density Lipoproteins: Biology, Epidemiology, and Clinical Management. Can J Cardiol. 2017;33:325-333
111. Tosheska Trajkovska K, Topuzovska S. High-density lipoprotein metabolism and reverse cholesterol transport: strategies for raising HDL cholesterol. Anatol J Cardiol. 2017;18:149-154
112. Uehara Y, Ando S, Yahiro E, Oniki K, Ayaori M, Abe S. et al. FAMP, a novel apoA-I mimetic peptide, suppresses aortic plaque formation through promotion of biological HDL function in ApoE-deficient mice. J Am Heart Assoc. 2013;2:e000048
113. Fisher EA, Feig JE, Hewing B, Hazen SL, Smith JD. High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arterioscler Thromb Vasc Biol. 2012;32:2813-2820
114. Remaley AT, Thomas F, Stonik JA, Demosky SJ, Bark SE, Neufeld EB. et al. Synthetic amphipathic helical peptides promote lipid efflux from cells by an ABCA1-dependent and an ABCA1-independent pathway. J Lipid Res. 2003;44:828-836
115. Niculescu LS, Robciuc MR, Sanda GM, Sima AV. Apolipoprotein A-I stimulates cholesteryl ester transfer protein and apolipoprotein E secretion from lipid-loaded macrophages; the role of NF-κB and PKA signaling pathways. Biochem Biophys Res Commun. 2011;415:497-502
116. Remaley AT, Stonik JA, Demosky SJ, Neufeld EB, Bocharov AV, Vishnyakova TG. et al. Apolipoprotein specificity for lipid efflux by the human ABCAI transporter. Biochem Biophys Res Commun. 2001;280:818-823
117. Wolska A, Dunbar RL, Freeman LA, Ueda M, Amar MJ, Sviridov DO. et al. Apolipoprotein C-II: New findings related to genetics, biochemistry, and role in triglyceride metabolism. Atherosclerosis. 2017;267:49-60
118. Cooke AL, Morris J, Melchior JT, Street SE, Jerome WG, Huang R. et al. A thumbwheel mechanism for APOA1 activation of LCAT activity in HDL. J Lipid Res. 2018;59:1244-1255
119. Settasatian N, Barter PJ, Rye KA. Remodeling of apolipoprotein E-containing spherical reconstituted high-density lipoproteins by phospholipid transfer protein. J Lipid Res. 2008;49:115-126
120. Temel RE, Walzem RL, Banka CL, Williams DL. Apolipoprotein A-I is necessary for the in vivo formation of high-density lipoprotein competent for scavenger receptor BI-mediated cholesteryl ester-selective uptake. J Biol Chem. 2002;277:26565-26572
121. Getz GS, Reardon CA. Apoprotein E and Reverse Cholesterol Transport. Int J Mol Sci. 2018;19:3479
122. Bartels ED, Christoffersen C, Lindholm MW, Nielsen LB. Altered metabolism of LDL in the arterial wall precedes atherosclerosis regression. Circ Res. 2015;117:933-942
123. Skålén K, Gustafsson M, Rydberg EK, Hultén LM, Wiklund O, Innerarity TL. et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002;417:750-754
124. Dib I, Khalil A, Chouaib R, El-Makhour Y, Noureddine H. Apolipoprotein C-III and cardiovascular diseases: when genetics meet molecular pathologies. Mol Biol Rep. 2021;48:875-886
125. Graham MJ, Lee RG, Bell TA 3rd, Fu W, Mullick AE, Alexander VJ. et al. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res. 2013;112:1479-1490
126. Zheng C, Azcutia V, Aikawa E, Figueiredo JL, Croce K, Sonoki H. et al. Statins suppress apolipoprotein CIII-induced vascular endothelial cell activation and monocyte adhesion. Eur Heart J. 2013;34:615-624
127. Du Y, Wang L, Si S, Yang Y, Hong B. A novel compound 4010B-30 upregulates apolipoprotein A-I gene expression through activation of PPARγ in HepG2 cells. Atherosclerosis. 2015;239:589-598
128. Glaros EN, Kim WS, Garner B. Myriocin-mediated up-regulation of hepatocyte apoA-I synthesis is associated with ERK inhibition. Clin Sci (Lond). 2010;118:727-736
129. Glaros EN, Kim WS, Quinn CM, Jessup W, Rye KA, Garner B. Myriocin slows the progression of established atherosclerotic lesions in apolipoprotein E gene knockout mice. J Lipid Res. 2008;49:324-331
130. Nikolic D, Rizzo M, Mikhailidis DP, Wong NC, Banach M. An evaluation of RVX-208 for the treatment of atherosclerosis. Expert Opin Investig Drugs. 2015;24(10):1389-1398
131. Bailey D, Jahagirdar R, Gordon A, Hafiane A, Campbell S, Chatur S. et al. RVX-208: a small molecule that increases apolipoprotein A-I and high-density lipoprotein cholesterol in vitro and in vivo. J Am Coll Cardiol. 2010;55:2580-2589
132. Yang L, Li T, Zhao S, Zhang S. Niacin regulates apolipoprotein M expression via liver X receptor-α. Mol Med Rep. 2019;20:3285-3291
133. Huang ZH, Gu D, Mazzone T. Oleic acid modulates the post-translational glycosylation of macrophage ApoE to increase its secretion. J Biol Chem. 2004;279:29195-29201
134. Yang L, Zhao S. Effect of simvastatin on the expression and regulation mechanism of apolipoprotein M. Int J Mol Med. 2012;29:510-514
135. Recio C, Maione F, Iqbal AJ, Mascolo N, De Feo V. The Potential Therapeutic Application of Peptides and Peptidomimetics in Cardiovascular Disease. Front Pharmacol. 2017;7:526
136. Valanti EK, Chroni A, Sanoudou D. The future of apolipoprotein E mimetic peptides in the prevention of cardiovascular disease. Curr Opin Lipidol. 2019;30:326-341
137. Datta G, White CR, Dashti N, Chaddha M, Palgunachari MN, Gupta H. et al. Anti-inflammatory and recycling properties of an apolipoprotein mimetic peptide, Ac-hE18A-NH(2). Atherosclerosis. 2010;208:134-141
138. Bocksch L, Rider BJ, Stephens T, Dai E, Liu L, Diao H. et al. C-terminal apolipoprotein E-derived peptide, Ep1.B, displays anti-atherogenic activity. Atherosclerosis. 2007;194:116-124
139. Llodrá J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci U S A. 2004;101:11779-11784
140. Rivas-Urbina A, Rull A, Aldana-Ramos J, Santos D, Puig N, Farre-Cabrerizo N. et al. Subcutaneous Administration of Apolipoprotein J-Derived Mimetic Peptide d-[113-122]apoJ Improves LDL and HDL Function and Prevents Atherosclerosis in LDLR-KO Mice. Biomolecules. 2020;10:829
141. Guo Y, Li W, Qian M, Jiang T, Guo P, Du Q. et al. D-4F Ameliorates Contrast Media-Induced Oxidative Injuries in Endothelial Cells via the AMPK/PKC Pathway. Front Pharmacol. 2021;11:556074
142. Liu D, Ding Z, Wu M, Xu W, Qian M, Du Q. et al. The apolipoprotein A-I mimetic peptide, D-4F, alleviates ox-LDL-induced oxidative stress and promotes endothelial repair through the eNOS/HO-1 pathway. J Mol Cell Cardiol. 2017;105:77-88
143. Liu D, Wu M, Du Q, Ding Z, Qian M, Tong Z. et al. The apolipoprotein A-I mimetic peptide, D-4F, restrains neointimal formation through heme oxygenase-1 up-regulation. J Cell Mol Med. 2017;21:3810-3820
144. Wang JL, Gong D, Hu XY, Wu S, Zheng XL, Wu J. et al. ApoA-1 Mimetic Peptide ELK-2A2K2E Decreases Inflammatory Factor Levels Through the ABCA1-JAK2-STAT3-TTP Axis in THP-1-Derived Macrophages. J Cardiovasc Pharmacol. 2018;72:60-67
145. Yin K, Tang SL, Yu XH, Tu GH, He RF, Li JF. et al. Apolipoprotein A-I inhibits LPS-induced atherosclerosis in ApoE(-/-) mice possibly via activated STAT3-mediated upregulation of tristetraprolin. Acta Pharmacol Sin. 2013;34:837-846
146. Navab M, Anantharamaiah GM, Reddy ST, Hama S, Hough G, Grijalva VR. et al. Apolipoprotein A-I mimetic peptides. Arterioscler Thromb Vasc Biol. 2005;25:1325-1331
147. Xie Q, Zhao SP, Li F. D-4F, an apolipoprotein A-I mimetic peptide, promotes cholesterol efflux from macrophages via ATP-binding cassette transporter A1. Tohoku J Exp Med. 2010;220:223-228
148. Ditiatkovski M, D'Souza W, Kesani R, Chin-Dusting J, de Haan JB, Remaley A. et al. An apolipoprotein A-I mimetic peptide designed with a reductionist approach stimulates reverse cholesterol transport and reduces atherosclerosis in mice. PLoS One. 2013;8:e68802
149. Tabet F, Remaley AT, Segaliny AI, Millet J, Yan L, Nakhla S. et al. The 5A apolipoprotein A-I mimetic peptide displays antiinflammatory and antioxidant properties in vivo and in vitro. Arterioscler Thromb Vasc Biol. 2010;30:246-252
150. Amar MJ, D'Souza W, Turner S, Demosky S, Sviridov D, Stonik J. et al. 5A apolipoprotein mimetic peptide promotes cholesterol efflux and reduces atherosclerosis in mice. J Pharmacol Exp Ther. 2010;334:634-641
151. Dai T, He W, Yao C, Ma X, Ren W, Mai Y. et al. Applications of inorganic nanoparticles in the diagnosis and therapy of atherosclerosis. Biomater Sci. 2020;8:3784-3799
152. Kilgore M, Muntner P, Woolley JM, Sharma P, Bittner V, Rosenson RS. Discordance between high non-HDL cholesterol and high LDL-cholesterol among US adults. J Clin Lipidol. 2014;8:86-93
153. Huang F, Yang Z, Xu B, Bi Y, Xu M, Xu Y. et al. Both serum apolipoprotein B and the apolipoprotein B/apolipoprotein A-I ratio are associated with carotid intima-media thickness. PLoS One. 2013;8:e54628
154. Czeck MA, Northrop EF, Evanoff NG, Dengel DR, Rudser KD, Kelly AS. et al. Relationship of Apolipoproteins with Subclinical Cardiovascular Risk in Youth. J Pediatr. 2020;227:199-203.e1
155. Pischon T, Girman CJ, Sacks FM, Rifai N, Stampfer MJ, Rimm EB. Non-high-density lipoprotein cholesterol and apolipoprotein B in the prediction of coronary heart disease in men. Circulation. 2005;112:3375-383
156. Sniderman AD, Williams K, Contois JH, Monroe HM, McQueen MJ, de Graaf J. et al. A meta-analysis of low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B as markers of cardiovascular risk. Circ Cardiovasc Qual Outcomes. 2011;4:337-345
157. Behbodikhah J, Ahmed S, Elyasi A, Kasselman LJ, De Leon J, Glass AD. et al. Apolipoprotein B and Cardiovascular Disease: Biomarker and Potential Therapeutic Target. Metabolites. 2021;11:690
158. Mendivil CO, Rimm EB, Furtado J, Chiuve SE, Sacks FM. Low-density lipoproteins containing apolipoprotein C-III and the risk of coronary heart disease. Circulation. 2011;124:2065-2072
159. Gaudet D, Drouin-Chartier JP, Couture P. Lipid Metabolism and Emerging Targets for Lipid-Lowering Therapy. Can J Cardiol. 2017;33:872-882
Author contact
![]() Corresponding authors: Tieshan Wang: Wangtieshan2011com; Aiping Lyu: aipingluedu.hk; Yuanyan Liu: yyliu_1980com.
Corresponding authors: Tieshan Wang: Wangtieshan2011com; Aiping Lyu: aipingluedu.hk; Yuanyan Liu: yyliu_1980com.