Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Regulation of protein activation...
Regulation of protein activation...
Regulation of protein stability...
Regulation of protein activation...
Conclusions and perspectives
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(16):5245-5256. doi:10.7150/ijbs.86404 This issue Cite
Review
Roles of Protein Post-Translational Modifications During Adipocyte Senescence
Min-Seon Hwang1, Jingyeong Park2, Yunha Ham2, In Hye Lee2, ![]() , Kyung-Hee Chun1,
, Kyung-Hee Chun1, ![]()
1. Department of Biochemistry & Molecular Biology, Graduate School of Medical Science, Brain Korea 21 Project, Institute of Genetic Science, Yonsei University College of Medicine, 50-1 Yonsei-ro, Seodaemun-gu, Seoul, 03722, Republic of Korea.
2. Department of Life Science, College of Natural Science, Ewha Womans University, 52 Ewhayeodae-Gil, Seodaemun-gu, Seoul, 03760, Republic of Korea.
Received 2023-5-22; Accepted 2023-9-27; Published 2023-10-16
Abstract

Adipocytes are adipose tissues that supply energy to the body through lipids. The two main types of adipocytes comprise white adipocytes (WAT) that store energy, and brown adipocytes (BAT), which generate heat by burning stored fat (thermogenesis). Emerging evidence indicates that dysregulated adipocyte senescence may disrupt metabolic homeostasis, leading to various diseases and aging. Adipocytes undergo senescence via irreversible cell-cycle arrest in response to DNA damage, oxidative stress, telomere dysfunction, or adipocyte over-expansion upon chronic lipid accumulation. The amount of detectable BAT decreases with age. Activation of cell cycle regulators and dysregulation of adipogenesis-regulating factors may constitute a molecular mechanism that accelerates adipocyte senescence. To better understand the regulation of adipocyte senescence, the effects of post-translational modifications (PTMs), is essential for clarifying the activity and stability of these proteins. PTMs are covalent enzymatic protein modifications introduced following protein biosynthesis, such as phosphorylation, acetylation, ubiquitination, or glycosylation. Determining the contribution of PTMs to adipocyte senescence may identify new therapeutic targets for the regulation of adipocyte senescence. In this review, we discuss a conceptual case in which PTMs regulate adipocyte senescence and explain the mechanisms underlying protein regulation, which may lead to the development of effective strategies to combat metabolic diseases.
Keywords: Adipocyte, Senescence, Post-translational modification, Metabolic disease, Metabolic homeostasis
Introduction
Adipogenesis is a key process during cell differentiation from pre-adipocytes to mature adipocytes. Adipocytes proliferate to form adipose tissue, where the energy harvested from food that exceeds the energy expenditure is stored as lipids. Pre-adipocytes develop in four stages: growth arrest, mitotic clonal expansion, early differentiation, and terminal differentiation into mature adipocytes [1]. In this process, pre-adipocytes accumulate lipids by increasing the quantities of enzymes needed for triglyceride (TG) synthesis and the accumulation of TG. Increased expression of transcription factors, such as peroxisome proliferator-activated receptor γ (PPARγ), CCAAT/enhancer binding protein α (C/EBPα), and CCAAT/enhancer binding protein β (C/EBPβ), is essential for adipocyte differentiation [2] (Figure 1A). PPARγ is a significant regulator of adipogenesis, and the C/EBP family (α and β) is one of the most critical downstream targets of PPARγ. These proteins are also necessary for the transcription and expression of insulin receptors, adiponectin, adipocyte protein 2 (aP2), and adipokines in mature adipocytes [3, 4].
Overview of differentiation and cellular senescence in adipocytes. (A) Adipogenesis, (B) senescence, and (C) induction of disease. Pre-adipocytes differentiate into mature adipocytes during adipogenesis. PPARγ and C/EBPα/β are regulators of adipogenesis, and their expression is increased. Also, insulin receptors, adiponectin, aP2, and adipokines are highly expressed in mature adipocytes. Stressors such as hyper-proliferation, DNA damage, oxidative stress, metabolic stress, and telomere shortening activate p53, CDK inhibitors, and RB, which induce senescence. During the accumulation of senescent cells, cell size increases, and free fatty acids (FFAs) are released at high levels with increased inflammation and collagen. Decreased adiponectin and poor insulin sensitivity also occur. These factors are related to cellular dysfunction and cause various diseases such as obesity, diabetes, chronic inflammation, and fibrosis.
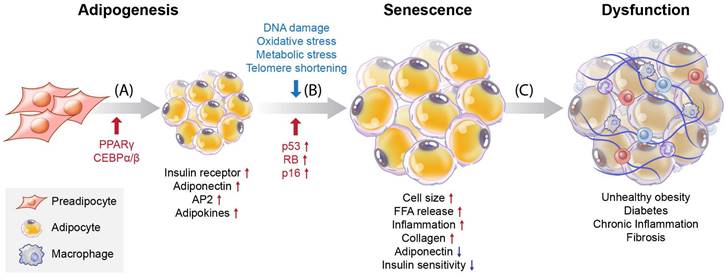
Understanding the processes involved in adipocyte senescence and their regulation is essential. Adipocyte senescence represents an irreversible cell cycle arrest in response to various stressors, including DNA damage, oxidative stress, metabolic stress, and telomere shortening [5, 6] (Figure 1B). These stressors transmit signals through multiple pathways, most of which activate the cell cycle inhibitor p53. These pathways converge upon the activation of the cyclin-dependent kinase (CDK) inhibitors p16, p21, p27, and p15. Eventually, they activate retinoblastoma protein (RB), causing senescence [7]. Unrepaired DNA damage and the loss of repair capacity can induce senescence [8]. In adipose tissue, p53 contributes to insulin resistance in age-associated metabolic diseases. Excessive caloric intake exacerbates oxidative stress and increases the production of p53 and pro-inflammatory cytokines in adipose tissue. In contrast, p53 suppression in adipose tissue ameliorates senescence-like changes by reducing pro-inflammatory cytokines and improving insulin sensitivity [9]. Interestingly, PPARγ controls adipocyte differentiation, inhibits cellular proliferation, and promotes cellular senescence [10, 11]. PPARγ induces the expression of p16INK4α (CDKN2A), a cell cycle inhibitor that promotes senescence, increases senescence-associated-β-galactosidase (SA-β-gal) levels, and triggers G1 arrest [11]. Cellular senescence causes functional disorders of adipogenesis and lipid storage in adipose-derived stromal/progenitor cells [12] (Figure 1C).
Browning (or beiging) of adipose tissue refers to the conversion of white adipocytes (WAT) into brown-like adipocytes, such as beige or brite cells (Figure 2). Brown adipocytes (BAT) are thermogenic, meaning they produce heat by burning stored fat. Adipose tissue browning is typically associated with increased energy expenditure and improved metabolic health. BAT, which are responsible for energy production, contain more mitochondria than do WAT which provide energy storage [13]. Developmentally, in mice, BAT originates from a myogenic factor 5 (MYF5)-positive mesoderm lineage [14] (Figure 2A). Transcriptional control of the BAT-specific thermogenic program is mediated by PPAR-γ coactivator 1-alpha (PGC-1α) and PR domain-containing 16 (PRDM16) [15] (Figure 2B). It is well established that BAT rely on mitochondrial function for maintaining intracellular metabolism. In addition, BAT mitochondria are functionalized by uncoupling protein-1 (UCP1), which allows the translocation of protons to dissipate energy during non-shivering thermogenesis [16, 17] (Figure 2B). Forkhead box protein A3 (FOXA3) expression is increased in visceral fat during aging and has been reported to reduce BAT mass and the beiging capacity of WAT [18, 19]. In the context of aging, it has been reported that mitochondrial enzyme expression is reduced in adipose tissue from old mice, yet little is known regarding the mechanisms that mediate these changes [20, 21]. Similarly, it is well-established that human WAT mitochondrial function, as measured by tissue oxygen consumption, is reduced in both obesity and aging [21]. Consistent with this, Foxa3-knockout mice are long-lived, have increased BAT activity late in life, and are protected from age-related insulin resistance and high-fat diet-induced increases in visceral adiposity [19] (Figure 2C).
Processes deciding adipocyte fate during differentiation and senescence, and their regulators in adipose tissues. Adipose tissue can be divided into white adipose tissue (WAT) and brown adipose tissue (BAT). (A) Through the differentiation of adipocytes, WAT and BAT develop from precursor cells, and Myf5 is expressed from the precursor cells. Pre-adipocytes turn into WAT and BAT during maturation, with transcription factors such as PPARγ, C/EBPs, and PRDM16 regulating this process. Browning of the adipose tissue leads to increased energy expenditure and improved metabolic health. Compared with WAT, BAT contains more mitochondria, which are related to energy production. The increased expression of PPARγ, C/EBPγ, and C/EBPγ is associated with WAT differentiation. (B) BAT is regulated by PGC-1α and PRDM16. Additionally, BAT mitochondria are functionalized by UCP1. With increased lipotoxicity, mitochondrial dysfunction, ROS, and DNA damage, adipocytes become senescent. (C) During senescence, FOXA3 expression is increased concomitant with reduced BAT mass and beiging capacity of WAT. (d) During BAT senescence, mitochondrial enzyme expression is reduced, as is UCP1 expression. In contrast, the expression levels of p16 and p21 are increased.
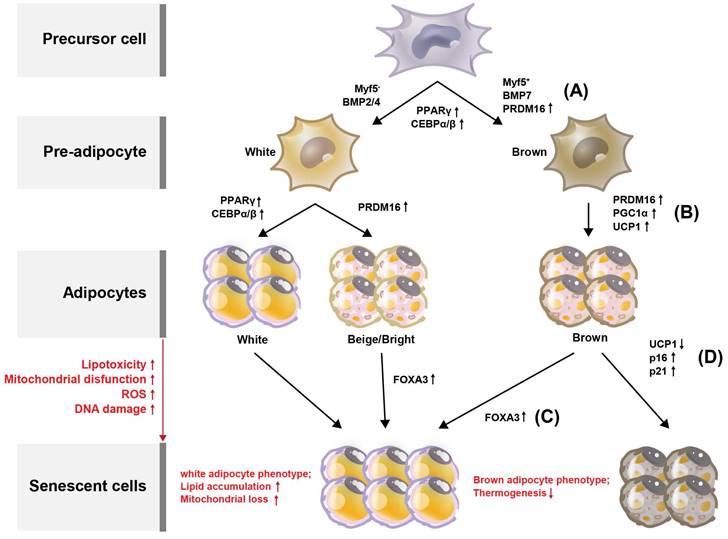
Evidence suggests that replicative capacity and UCP1 expression in BAT are significantly reduced during aging [16, 17]. In particular, the proliferative capacity and UCP1 expression in response to cold stimuli appear to be abolished in aged BAT [22]. Increasing age of brite adipocytes progressively leads to the development of a WAT phenotype, which prevents adipocyte browning in older mice and humans [23, 24]. Conversely, the levels of the senescence markers p16 and p21 are highly increased during the senescence process in BAT [13, 25]. BAT thermogenesis declines with aging [26] (Figure 2D), consistent with the reduction in the thermogenic factor UCP1, whereas UCP1 levels in BAT are stabilized by SIRT5 desuccinylation. Sirt5 deficiency in BAT increases the succinylation of UCP1, resulting in decreased UCP1 stability and function, which impairs mitochondrial homeostasis and alters BAT-mediated thermogenesis [27]. This may thus be one of the mechanisms regulating BAT senescence.
Some detrimental effects of senescent cell accumulation have been reported, including inflammation, insulin resistance in adipose tissue, underlying obesity, and type 2 diabetes [6, 7, 9, 28]. The senescence-associated secretory phenotype (SASP), which includes pro-inflammatory cytokines, is produced via autocrine and paracrine pathways to enhance and diffuse senescent cell influence and induce chronic inflammation in adipose tissues [29]. Under stress stimulation conditions, these interactive signaling pathways converge toward the activation of a transcriptional program managed by nuclear factor kappa B (NF-κB) and C/EBPβ, the core effectors that initiate and maintain SASP gene expression [30]. Removing senescent adipocytes mitigates inflammation and ameliorates insulin resistance in adipose tissue [29]. Therefore, the proper control of adipocyte senescence is critical for the management of various diseases.
The mechanisms controlling cellular senescence can be divided into three categories. These include transcriptional modifications, such as DNA methylation at the genetic and epigenetic levels; mRNA-level regulatory mechanisms, such as mRNA stabilization or degradation; and post-translational modifications (PTMs), such as ubiquitination [31]. Here, we focused on PTMs that regulate protein stability and activation. PTMs such as phosphorylation, acetylation, ubiquitination, and glycosylation alter the chemical properties and functions of proteins [32]. Abnormal PTMs can cause biological dysfunction and lead to various diseases. For example, PTMs significantly affect the structure and function of proteins that regulate adipocyte senescence. Understanding the role of PTMs in adipocyte senescence may guide the discovery of new therapeutic targets to modulate adipocyte senescence.
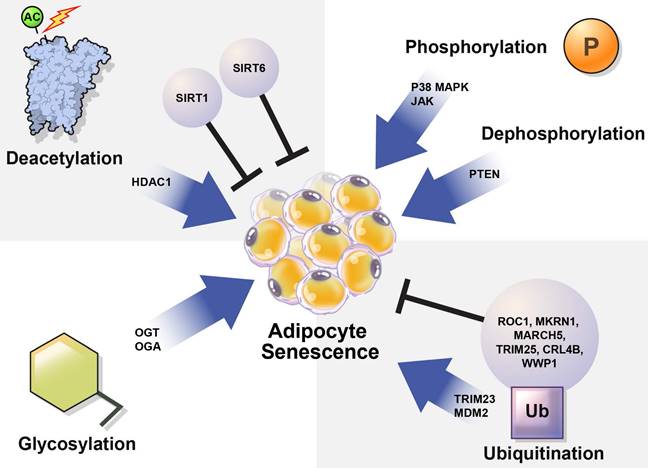
In this review, we summarize how the stability and activation of proteins involved in the induction of adipocyte senescence are regulated by various PTMs (Table 1) and provide new insights regarding the regulation of adipocyte senescence (Figure 3).
The summary of Post-translational Modification (PTM) in adipocyte senescence
| PTM | Substrate | Pathway | Action site | |
|---|---|---|---|---|
| Phosphorylation | JAK[34, 36, 37] | SASP | JAK/STAT pathway | |
| P38 MAPK[40-42] | IRS-2 | p38 MAPK signaling pathway | ||
| Dephosphorylation | PTEN[49-51] | PIP3 | PI3K/AKT signaling pathway | D3 |
| Deacetylation | SIRT1[57,58,62] | PPARγ | Lipogenesis | Lys268 and Lys293 |
| SIRT6[64] | p27Kip1 | Adipocyte differentiation | Lys100 | |
| HDAC1[70, 72] | H3K27 | Thermogenesis | Lys27 | |
| Ubiquitination | MKRN1[83-85] | p14ARF, AMPKα | Fatty acid oxidation | |
| TRIM 23[86] | PPARγ | Adipocyte differentiation | ||
| TRIM 25[87] | ||||
| MARCH 5[88] | Glycolysis and basal mitochondrial respiration | |||
| CRL4B[90] | PPARγ | Adipocyte differentiation | ||
| MDM2[92] | STEAP4 | HIF1-α/PKM2 signaling pathway | Lys18 and Lys161 | |
| WWP1[94] | p27Kip1 | Adipocyte differentiation | ||
| O-GlcNAcylation | OGT, OGA[97, 100,102] | PPARγ, | Adipocyte differentiation | Thr54 of the N-terminal activation function-1 domain |
| C/EBPβ | Ser180 and Ser181 | |||
| HBP flux[103,104] | AMPKα | Fatty acid oxidation | - |
Regulation of cellular senescence in adipocytes by post-translational modification (PTM). PTM regulates adipocyte senescence through various physiological pathways, including phosphorylation, deacetylation, ubiquitination, and glycosylation. Inhibition of p38, JAK, and PTEN can alleviate senescence in adipocytes. Deacetylation also regulates adipocyte senescence. SIRT1 and SIRT6 are deacetylases that play protective roles against adipocyte senescence. SIRT1 negatively regulates HDAC1, which is highly expressed in senescent cells. TRIM23 and MDM2 are E3 ubiquitin ligases that induce adipocyte senescence. However, ROC1 suppress cellular senescence. The E3 ubiquitin ligases MKRN1, MARCH 5, TRIM25, CRL4B, and WWP1 are also potential regulators of adipocyte senescence. OGT and OGA regulate O-GlcNAcylation. Abnormal protein O-GlcNAcylation causes senescence-related diseases.
Regulation of protein activation by phosphorylation
Adipocyte senescence is tightly regulated to maintain energy and metabolic homeostasis [26]. Senescent cells accumulate in aging fat in response to replicative, cytokine-induced, and metabolic stresses [33]. Janus kinase (JAK), p38, and the phosphatase and tensin homolog on chromosome 10 (PTEN) upregulate adipocyte senescence under various cellular stress conditions. Cellular phosphorylation is known to potentiate or downregulate adipocyte senescence; however, the underlying mechanisms require further investigation.
JAK
An increase in cellular senescence in response to intrinsic or extrinsic stresses and the broadly related SASP promote organismal aging and adipose tissue dysfunction [26, 34, 35]. Inhibition of the JAK/signal transducer and activator of transcription (JAK/STAT) pathway, which plays a significant role in adipose tissue development and function and regulates SASP, can partially inhibit SASP secretion [34, 36]. The JAK1/2 inhibitor ruxolitinib decreases the pro-inflammatory SASP in vitro and in vivo and enhances insulin sensitivity in aging mice [34]. Aging cell improvement enhances adipogenesis and metabolism [37, 38]. Activin A secreted by senescent cells, blocks adipogenesis. Treatment of aged mice and primary human senescent fat progenitor cells with a JAK inhibitor reduced activin A levels and restored lipid accumulation and expression of critical adipogenic markers. JAK inhibitors also reduce lipotoxicity and increase insulin sensitivity [37]. The inhibition of JAK activity is a strategy used to alleviate adipocyte cellular senescence by ameliorating senescent adipose progenitors and stem cells (APSCs).
p38 MAPK
SASP is potentiated by the activation of p38 mitogen-activated protein kinase (MAPK), which is induced by increased NF-κB transcriptional activity. p38 MAPK inhibition markedly reduced the secretion of most SASP factors [34, 39]. SASP contributes to dysfunction in aged organs. Age-related adipose tissue changes increase pro-inflammatory cytokines, such as tumor necrosis factor α (TNF-α), promoting adipocyte senescence [3, 35, 40], which reduces brown adipogenic differentiation and promotes insulin resistance in BATs. During this cellular event, the serine residues of insulin receptor substrate 2 (IRS-2) are phosphorylated by p38 MAPK [40-42]. The inhibition of p38 MAPK decreases SASP secretion, which may ameliorate adipocyte senescence.
PTEN
The insulin signaling response displays heightened sensitivity to cellular aging in adipocytes, thereby decreasing the insulin response [6, 26, 43, 44]. PTEN is a phosphatidylinositol phosphate phosphatase that plays an essential role in various cellular processes, including genome maintenance, DNA repair, cell cycle control, proliferation, metabolism, migration, tumorigenesis, and senescence. One of the critical roles of PTEN is to inhibit insulin signaling [45-48]. PTEN negatively regulates insulin signaling by dephosphorylating phosphatidylinositol-3,4,5-triphosphate (PIP3), resulting in decreased phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) signaling [49]. Pten deficiency enhances energy expenditure in brown adipose tissue. PTEN loss and activation of PI3K/AKT signaling lead to an improved ability to handle metabolic stress in mice [50]. PTEN downregulation increased the proliferation of stromal vascular fraction (SVF) cells, including adipocyte progenitor cells, in adipose tissues. Pten deficiency restores the differentiation capacity of high-passage SVF cells and increases adipogenesis. In contrast, PTEN expression is upregulated during cellular senescence [51]. The regulation of PTEN activity and expression levels is a strategy for controlling adipocyte cellular senescence.
Regulation of protein activation by deacetylation
Senescent adipocytes accumulate in aging fat in response to cytokines and metabolic stress [26, 33]. Adipocyte senescence is regulated in response to cold exposure-stimulated thermogenesis. The mitochondrial function and activity of UCP-1, a thermogenesis-related mitochondrial protein in adipocytes, decrease with cellular aging. Additionally, the pro-inflammatory capacity of BAT increases with age [26, 35, 52]. Histone deacetylases (HDACs) such as sirtuin 1 (SIRT1), SIRT 6, and HDAC1 are essential regulators of adipocyte senescence under conditions of cellular stress [26, 53, 54]. Although SIRTs and HDAC1 are deacetylases, their roles in adipocyte senescence differ. SIRT1 and SIRT6 prevent adipocyte senescence, whereas HDAC1 potentiates adipocyte senescence. The number of brown and beige adipocytes decreases with age. Cellular deacetylation events regulate adipocyte senescence. However, the underlying mechanisms remain unclear.
SIRT1
A reduction in beige adipocyte formation has been detected in aging adipose tissues [26]. SIRT1, a nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase, drives beige adipocyte generation in WAT [26]. In addition, NAD+ levels, SIRT1 activity, and SIRT1 expression decrease with age and cellular senescence progression [10, 55, 56]. PPARγ directly interacts with and negatively regulates SIRT1 expression [10]. PPARγ acetylation, which is correlated with Sirt1 deficiency, increases during cellular senescence [10]. SIRT1 limits pre-adipocyte hyperplasia through c-Myc deacetylation, improves insulin sensitivity, reduces inflammation, and suppresses lipid accumulation by inhibiting PPARγ [57]. The proto-oncoprotein zinc finger and BTB domain-containing 7C (ZBTB7C) negatively regulate Sirt1 transcript levels. Its expression level is increased in the WAT of aging mice. ZBTB7C, a potent SIRT1 repressor, increases PPARγ acetylation [58]. Expression of nicotinamide phosphoribosyltransferase (NAMPT), which recycles NAD+, increases during cellular senescence. NAMPT activity promotes pro-inflammatory SASP [34, 59-61]. Age-related reduction in SIRT1 activity may be a critical mechanism in the loss of beige adipose tissue as well as in age-associated thermogenic impairment [26]. Upregulation of SIRT1 potentiates brown remodeling of subcutaneous WAT by deacetylation of PPARγ at Lys268 and Lys293 [62]. SIRT1 regulation is likely a key mechanism controlling adipocyte senescence.
SIRT6
The essential roles of SIRT6 in adipocytes are the regulation of lipid metabolism and the prevention of inflammation. SIRT6 stimulates lipolysis, enhances adipose tissue browning, and ameliorates adipose tissue inflammation, thereby improving insulin action in the peripheral tissues [63]. SIRT6 promotes cell proliferation and antagonizes cellular senescence; however, SIRT6 expression decreases during cellular senescence [64]. Moreover, SIRT6 suppresses p27Kip1 (p27) expression during cellular senescence [64]. SIRT6 mediates the polyubiquitination of p27, directing its degradation by the proteasome and thereby regulating the acetylation status of p27. Thus, SIRT6 delays cellular senescence [64]. Aging and excessive caloric intake, which are two major risk factors for obesity and diabetes, lead to decreased SIRT6 levels [65]. Sirt6 deficiency in pre-adipocytes blocks adipogenesis and regulates mitotic clonal expansion [66]. Sirt6 deletion in adipose tissue impairs the thermogenic function of BAT, causing morphological ''whitening'' of brown fat, reduced oxygen consumption, obesity, decreased core body temperature, and cold sensitivity. Fat Sirt6-deleted mice exhibit increased blood glucose levels, severe insulin resistance, and hepatic steatosis. Moreover, Sirt6 deficiency inhibits WAT browning following cold exposure or β3-agonist treatment [67]. Taken together, these results indicate that SIRT6 plays a protective role against adipocyte senescence.
HDAC1
Inhibition of signaling pathways that induce SASP using HDAC inhibitors, including trichostatin A (TSA), suppresses senescence. At low concentrations, TSA acts as a pan-SASP blocker [34, 68]. TSA may decrease PPARγ expression [53, 69], potentiating cellular senescence. HDAC inhibitors are also involved in regulating thermogenic adipocyte differentiation, adaptive thermogenesis, and metabolic disorder pathogenesis [70]. HDAC1 is highly expressed in senescent cells [71]. HDAC1 negatively regulates the thermogenic program in BAT. Repression of HDAC1 promotes acetylation and prevents methylation of histone H3K27, which increases the expression of BAT-specific genes such as UCP1, PGC-1α, and PRDM16 [70, 72]. SIRT1 negatively regulates HDAC1 function [54, 73]. SIRT1 is degraded and downregulated during cellular senescence [56, 74], indicating that Sirt1 deficiency may increase HDAC1 activity in senescent cells [54]. Thus, regulation of HDAC1 function may improve adipocyte senescence.
Regulation of protein stability by ubiquitination: E3 ubiquitin ligases
Another regulatory mechanism that influences physiological cellular senescence is the post-translational modification of cellular proteins through ubiquitination. The ubiquitin-proteasome pathway (UPP) regulates the differentiation of various cell types. Alterations in the UPP in mature adipocytes can potentially modulate adipose function during adipocyte aging [75]. Ubiquitin is activated by E1 and transferred to E2 or Ub conjugase. In turn, the E2 enzyme transfers ubiquitin to E3 or directly ubiquitinates the target protein in conjunction with E3 [72, 73]. Proteasome activity decreases during senescence, which may be associated with aging and age-associated diseases [75]. E3 ubiquitin-protein ligases are crucial factors in the regulation of senescence by ubiquitination. Knockdown of regulator of cullins-1 (ROC1), a component of the SKP, Cullin, F-box (SCF) E3 ubiquitin ligases, suppresses the growth of several human cell lines by inducing senescence [76]. In addition, the inactivation of the von Hippel-Lindau (VHL) tumor suppressor gene, which encodes a subunit of an E3 ubiquitin ligase, causes a senescence-like phenotype in human cancer cell lines [77]. Therefore, E3 ubiquitin ligase activity appears to be essential for regulating adipocyte senescence. Several E3 ligases have recently been found to be present in adipocytes, including seven in absentia homolog 2 (SIAH2), makorin ring finger protein 1 (MKRN1), tripartite motif protein 23 (TRIM23), and neural precursor cell-expressed developmentally downregulated protein 4 (NEDD4) [78]. The ubiquitination pathway regulates p53 tumor suppressor stability, localization, and functions in normal cells [79]. E3 ubiquitin ligases, including murine double minute 2 (MDM2) and MKRN1, predominantly regulate p53 expression levels and activities under various physiological conditions [79, 80]. Overexpression of MDM2 and CDK4 can induce human telomere reverse transcriptase (hTERT) overexpression and p53 degradation in human 2H transgenic bone marrow-mesenchymal stem cells (BM-MSCs), increase cell proliferation and migration, and suppress the adipogenic differentiation potential in vitro [81].
MKRN1
In telomerase-positive cells, overexpression of MKRN1, an E3 ligase, promotes hTERT degradation, decreases telomerase activity, and subsequently decreases telomere length [82]. MKRN1 knockdown induces senescence by stabilizing p14ARF. MKRN1 also regulates SASP [83]. MKRN1 negatively regulates PPARγ via ubiquitin-mediated proteasomal degradation, with PPARγ2 and MKRN1 interacting directly [84]. MKRN1 also ubiquitinates and degrades AMP-activated protein kinase alpha (AMPKα) subunits, whereas MKRN1 depletion promotes glucose consumption and suppresses lipid accumulation via AMPK stabilization and activation [85]. These results suggest that the E3 ligase MKRN1 potentially regulates adipocyte senescence, warranting further studies.
TRIM23 and TRIM25
TRIM23 is an E3 ligase that can regulate PPARγ protein stability and mediate abnormal polyubiquitin conjugation [86]. TRIM23 is required to form late enhanceosomes and recruit Pol II during late adipogenic differentiation, whereas treatment with the proteasome inhibitor MG132 inhibits the reduction of PPARγ in TRIM23-knockdown cells [86]. The 26S proteasome does not readily recognize PPARγ aberrantly ubiquitinated by TRIM23, resulting in its protection from proteasomal degradation [86]. However, TRIM25 directly induces PPARγ ubiquitination and its proteasome-dependent degradation [87]. TRIM25 decreases PPARγ expression and inhibits 3T3-L1 adipocyte differentiation [87]. Therefore, TRIM25 expression is negatively correlated with PPARγ expression.
MARCH 5
The E3 ubiquitin ligase, membrane-associated RING-CH-type finger 5 (MARCH 5) regulates mitochondrial dynamics and is in turn regulated by PPARγ in adipocytes undergoing adipogenesis [88]. MARCH 5 depletion increases glycolysis and basal mitochondrial respiration [88]. MARCH5-deficient cells display mitochondrial elongation and phenotypic changes owing to increased SA-β-Gal expression caused by cellular senescence [89].
CRL4B
The aryl hydrocarbon receptor (AhR) reduces PPARγ protein stability through a proteasome-dependent mechanism [90]. Overexpression of AhR in 3T3-L1 cells induced a decrease in endogenous PPARγ, which was reversed by treatment with MG132 [90]. AhR serves as a substrate receptor in the Cullin 4B-RING E3 ubiquitin ligase (CRL4B) AhR complex to induce polyubiquitination of PPARγ [90].
MDM2
MDM2, an E3 ubiquitin ligase, regulates adipogenesis by initiating adipocyte differentiation through the promotion of cAMP-mediated transcriptional activation of cAMP response element-binding proteins (CREB) and the induction of C/EBPδ expression [91]. High-fat diet (HFD)-fed Mdm2-adipocyte-specific knock-in (Mdm2-AKI) mice display epididymal white adipose tissue (eWAT) dysfunction, including senescence [92]. MDM2 suppresses six-transmembrane epithelial antigen of prostate 4 (STEAP4) expression via ubiquitin modification [92]. Revival of STEAP4 rescued MDM2-induced adipose dysfunction in eWAT of HFD-fed Mdm2-AKI mice [92].
WWP1
Obesity upregulates WW domain-containing E3 ubiquitin protein ligase 1 (WWP1) in WAT. WWP1, which belongs to the NEDD4-like family of E3 ubiquitin ligases, is upregulated in obese WAT [93]. WWP1 induces p27Kip1 degradation via ubiquitination and inhibits p27Kip1-mediated replicative senescence [94]. WWP1 overexpression decreases reactive oxygen species (ROS) levels in 3T3-L1 cells, and WWP1 protects against obesity-associated oxidative stress in adipocytes and WAT [95].
Regulation of protein activation by O-GlcNAcylation
O-linked-N-acetylglucosamination (O-GlcNAcylation), a post-translational glycosylation event, occurs on proteins in the nucleus, cytoplasm, and mitochondria, regulates cell signaling, and is associated with several pathological conditions [96]. O-GlcNAcylation is the single-sugar addition of O-linked-β-N-acetylglucosamine (O-GlcNAc) to the hydroxyl groups of the serine or threonine residues of target proteins [97]. The attachment and removal of O-GlcNAc from proteins are processed by O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA) [98]. O-GlcNAcylation is highly responsive to glucose, and insulin resistance enhances O-GlcNAcylation [99]. Increased levels of O-GlcNAcylation have been observed in response to the induction of adipocyte differentiation [100]. Post-translational modifications regulate OGT activity, and OGT activation occurs when tyrosine phosphorylation of OGT increases following insulin stimulation in 3T3-L1 cells [101]. Abnormal protein O-GlcNAcylation is essential for the development and progression of senescence-related diseases [102].
O-GlcNAcylation levels increased significantly on and after day 5 of 3T3-L1 differentiation induction [100]. Simultaneously, C/EBPα and adiponectin expression, and lipid droplet size increased [100]. O-GlcNAcylation of vimentin, long-chain fatty acid-CoA, and pyruvate carboxylase increases with adipocyte differentiation [100]. Treatment with 6-diazo-5-oxo-norleucine (DON), a glutamine:fructose-6-phosphate amidotransferase (GFAT) inhibitor, blocked adipocyte differentiation at the stage of C/EBPα expression, which was associated with an overall increase in O-GlcNAcylation [100]. Therefore, it can be assumed that O-GlcNAcylation partially participates in adipogenesis.
In 3T3-L1 adipocytes, O-GlcNAc causes post-transcriptional modification of PPARγ. The primary O-GlcNAc site of PPARγ is threonine 54 of the N-terminal activation function-1 domain [97]. In 3T3-L1 cells, an increase in O-GlcNAc modification mediated by the OGA inhibitor NButGT decreases PPARγ transcriptional activity and terminal adipocyte differentiation [97].
C/EBPβ is also modified by O-GlcNAc, which is present in nucleocytoplasmic proteins. GlcNAcylation sites (Ser180 and Ser181) are located in the regulatory domain and are extremely close to the phosphorylation sites (Thr188, Ser184, and Thr179) that regulate DNA-binding activity [102]. GlcNAcylation of Ser180 and Ser181 blocks the phosphorylation of Thr188, Ser184, and Thr179, thereby delaying adipocyte differentiation [102]. In contrast, the mutation of Ser180 and Ser181 to Ala increased C/EBPβ transcriptional activity [102]. Therefore, GlcNAcylation and phosphorylation appear to modulate the function of C/EBPβ by alternately occupying adjacent sites.
The hexosamine biosynthesis pathway (HBP) flux functions as a nutrient sensor and induces O-GlcNAc modification of the AMPK α subunit in both immortal and primary murine adipocytes [103]. O-linked glycosylation via HBP flux regulates AMPK activation and induces fatty acid oxidation in 3T3-L1 adipocytes [104] whereas removal of O-GlcNAc by hexosaminidase reduces AMPK activity [104]. Thus, HBP correlates with O-GlcNAc and is likely to affect adipocyte senescence.
Conclusions and perspectives
Adipogenesis is the process of differentiation of pre-adipocytes into mature adipocytes. There are two main types of fat cells that contain lipids: WAT, which store energy, and BAT, which produces heat. Pre-adipocytes develop in four stages: growth arrest, mitotic clone expansion, early differentiation, and terminal differentiation into mature adipocytes [1]. In this process, increased expression of transcription factors is essential for adipocyte differentiation [2] (Figure 1A).
Upon DNA damage, oxidative stress, metabolic stress, or telomere shortening, adipocytes undergo senescence and irreversible cell cycle arrest, thereby inhibiting adipogenesis [5, 6]. These cellular stressors activate p53 and induce CDK inhibitors and RB, which cause senescence [7]. In addition, the modification of adipogenesis regulators such as PPAR and C/EBPα/β results in adipocyte senescence. Moreover, mitochondrial UCP1 expression has been reported to decrease in old mouse adipocytes; however, little is known about this mechanism [16, 17, 27]. As UCP1 levels decrease during the aging of BAT, the levels of aging markers p16 and p21 increase significantly [13, 25]. In addition, the expression of UCP1, a thermogenesis factor, in BAT is stabilized by SIRT5 desuccinylation. This may thus be a mechanism by which BAT senescence is regulated; however, this possibility requires further investigation.
Once lipids continuously accumulate in the adipose tissue, the adipocyte senescence rate increases and insulin sensitivity decreases, resulting in adipose tissue dysfunction. SASP induces chronic inflammation in adipose tissue. Cellular senescence causes lipid storage dysfunction. Thus, the appropriate control of fat cell aging is a viable strategy for preventing aging-related diseases. Although the underlying mechanisms remain poorly understood, adipocyte senescence is essential for diverse physiological processes, including metabolism and various age-related diseases. To better understand the processes of adipocyte senescence, it is important to identify the modulators of adipogenic factors, including PPARγ, and their regulatory molecular mechanisms, such as PTMs. PTMs are associated with oxidative stress, inflammation, and aging, thereby influencing aging characteristics [105]. Some PTMs participate in healthy aging, suggesting that they are essential regulators and predictive markers of the senescence process [106]. PTMs significantly affect aging by targeting epigenetic and non-epigenetic pathways. Therefore, understanding the role of PTMs in cellular senescence may advance the development of targeted therapies for age-related diseases.
p38, JAK, and PTEN influenced fat cell senescence through phosphorylation under stress (Figure 3). Inhibition of the JAK/STAT pathway inhibits SASP secretion and Pten deficiency increases adipogenesis [34, 36-39].
SIRT1 induces beige adipocyte production in WAT [107]. PPARγ interacts directly with SIRT1 to negatively regulate SIRT1 expression [10]. NAMPT activity that rescues NAD+ promotes SASP. SIRT6 also inhibits p27 expression during cell aging, thereby slowing this process [34, 59-61]. SIRT1 and SIRT6 regulation is expected to be a key mechanism in controlling fat cell aging [63-65, 67]. Suppression of the SASP pathway inhibits aging. HDAC1 is highly expressed in older cells. Regulation of HDAC1 function may retard adipocyte aging (Figure 3).
The inactivation of tumor suppressor genes can result in phenotypes similar to those observed during aging [77]. Therefore, E3 ubiquitin ligase activity appears to be essential for the regulation of adipocyte aging. MKRN1 may negatively regulate PPARγ through ubiquitination [84]. Because TRIM25 reduces PPARγ expression and suppresses the differentiation of 3T3-L1 adipocytes, TRIM25 correlates negatively with PPARγ expression [87]. MARCH 5 depletion increases glycolysis and basal mitochondrial respiration [88]. In addition, components of E3 ubiquitin ligases such as ROC1, TRIM23, CRL4B, WWP1, and MDM2 inhibit adipocyte senescence [76, 86, 90, 92, 95].
O-GlcNacylation regulates cell signaling and plays an essential role in the development and progression of age-related diseases. OGT and OGA regulate the attachment and removal of O-GlcNAc. In 3T3-L1 cells, these enzymes induce adipocyte senescence. We predict that new treatments for adipocyte senescence can be developed by understanding the involvement of PTMs in fat-cell aging (Figure 3).
In particular, a clearer understanding is needed regarding how PTMs, including acetylation/deacetylation, phosphorylation, ubiquitination, and glycosylation, regulate the function of necessary adipogenic factors, such as PPAR (Figure 3). Finally, it is important to establish the degree to which prevention of adipocyte senescence mediates beneficial effects on adipose tissue in various disease conditions. Future research on the regulatory mechanisms underlying adipocyte senescence will likely provide critical insights regarding the new and complex networks involved in human biological processes, including aging and metabolism.
Abbreviations
AhR: aryl hydrocarbon receptor; AMPK: AMP-activated protein kinase; aP2: adipocyte protein 2; BAT: brown adipocyte; CDK: cyclin-dependent kinase; C/EBPα/b: CCAAT/enhancer binding protein α/b; CREB: cAMP-mediated transcriptional activation of cAMP response element-binding proteins; CRL4B: Cullin 4B-RING E3 ubiquitin ligase; eWAT: epididymal white adipose tissue; FOXA3: forkhead box protein A3; HBP: hexosamine biosynthesis pathway; HDAC: histone deacetylase; HFD: high-fat diet; hTERT: human telomere reverse transcriptase; JAK/STAT: Janus kinase/signal transducer and activator of transcription; MAPK: mitogen-activated protein kinase; MARCH5: membrane-associated RING-CH-type finger 5; MDM2: murine double minute 2; MKRN1: makorin ring finger protein 1; MYF5: myogenic factor 5; NAD: nicotinamide adenine dinucleotide; NAMPT: nicotinamide phosphoribosyltransferase; NEDD4: neural precursor cell-expressed developmentally downregulated protein 4; NF-κB: nuclear factor kappa B; OGA: O-GlcNAcase; O-GlcNAc: O-linked-β-N-acetylglucosamine; O-GlcNAcylation: O-linked-N-acetylglucosamination; OGT: O-GlcNAc transferase; p27: p27Kip1; PGC-1α: PPAR-g coactivator 1-alpha; PI3K/AKT: phosphatidylinositol 3-kinase/protein kinase B; PPARγ: peroxisome proliferator-activated receptor γ; PRDM16: PR domain-containing 16; PTEN: phosphatase and tensin homolog on chromosome 10; PTM: post-translational modifications; RB: retinoblastoma protein; ROC1: regulator of cullins-1; SA-β-gal: senescence-associated-β-galactosidase; SASP: senescence-associated secretory phenotype; SIRT: sirtuin; STEAP4: six-transmembrane epithelial antigen of prostate 4; SVF: stromal vascular fraction; TG: triglyceride; TRIM23: tripartite motif protein 23; TSA: trichostatin A; UCP1: uncoupling protein-1; UPP: ubiquitin-proteasome pathway; WAT: white adipocyte; WWP1: WW domain-containing E3 ubiquitin protein ligase 1; ZBTB7C: zinc finger and BTB domain-containing 7C.
Acknowledgements
This study was supported by a grant from the National Research Foundation of Korea (grant number NRF-2021R1A2C1091259), the Bio & Medical Technology Development Program of NRF funded by Korean government (MSIT) (NRF-2022M3A9G8082639), and Korea Drug Development Fund funded by Ministry of Science and ICT, Ministry of Trade, Industry, and Energy, and Ministry of Health and Welfare (HN21C0153). MID (Medical Illustration & Design), as a member of the Medical Research Support Services of Yonsei University College of Medicine, providing excellent support with medical illustration.
Author Contributions
I. H. L. and K. C. conceived the study. All the authors wrote the manuscript and prepared the figures and table. All the authors reviewed the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gregoire FM, Smas CM, Sul HS. Understanding adipocyte differentiation. Physiological reviews. 1998;78:783-809
2. Vishwanath D, Srinivasan H, Patil MS, Seetarama S, Agrawal SK, Dixit M, Dhar K. Novel method to differentiate 3T3 L1 cells in vitro to produce highly sensitive adipocytes for a GLUT4 mediated glucose uptake using fluorescent glucose analog. Journal of cell communication and signaling. 2013;7:129-40
3. Mancuso P, Bouchard B. The impact of aging on adipose function and adipokine synthesis. Frontiers in endocrinology. 2019;10:137
4. Lefterova MI, Zhang Y, Steger DJ, Schupp M, Schug J, Cristancho A. et al. PPARγ and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes & development. 2008;22:2941-52
5. Frasca D, Blomberg BB. Adipose tissue, immune aging, and cellular senescence. Seminars in immunopathology: Springer. 2020 p. 573-87
6. Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H. et al. Fat tissue, aging, and cellular senescence. Aging cell. 2010;9:667-84
7. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nature reviews Molecular cell biology. 2014;15:482-96
8. Nassour J, Martien S, Martin N, Deruy E, Tomellini E, Malaquin N. et al. Defective DNA single-strand break repair is responsible for senescence and neoplastic escape of epithelial cells. Nature communications. 2016;7:10399
9. Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T. et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nature medicine. 2009;15:1082-7
10. Han L, Zhou R, Niu J, McNutt MA, Wang P, Tong T. SIRT1 is regulated by a PPARγ-SIRT1 negative feedback loop associated with senescence. Nucleic acids research. 2010;38:7458-71
11. Gan Q, Huang J, Zhou R, Niu J, Zhu X, Wang J. et al. PPARγ accelerates cellular senescence by inducing p16INK4α expression in human diploid fibroblasts. Journal of cell science. 2008;121:2235-45
12. Mitterberger MC, Lechner S, Mattesich M, Zwerschke W. Adipogenic differentiation is impaired in replicative senescent human subcutaneous adipose-derived stromal/progenitor cells. Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences. 2014;69:13-24
13. Schosserer M, Grillari J, Wolfrum C, Scheideler M. Age-induced changes in white, brite, and brown adipose depots: a mini-review. Gerontology. 2018;64:229-36
14. Sanchez-Gurmaches J, Guertin DA. Adipocyte lineages: tracing back the origins of fat. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2014;1842:340-51
15. Becerril S, Gómez-Ambrosi J, Martín M, Moncada R, Sesma P, Burrell MA, Frühbeck G. Role of PRDM16 in the activation of brown fat programming. Relevance to the development of obesity. 2013
16. Zoico E, Rubele S, De Caro A, Nori N, Mazzali G, Fantin F. et al. Brown and beige adipose tissue and aging. Frontiers in endocrinology. 2019;10:368
17. Nicholls DG. Fifty years on: How we uncovered the unique bioenergetics of brown adipose tissue. Acta Physiologica. 2023;237:e13938
18. Ma X, Xu L, Mueller E. Calorie hoarding and thrifting: Foxa3 finds a way. Adipocyte. 2015;4:325-8
19. Xu L, Panel V, Ma X, Du C, Hugendubler L, Gavrilova O. et al. The winged helix transcription factor Foxa3 regulates adipocyte differentiation and depot-selective fat tissue expansion. Molecular and cellular biology. 2013;33:3392-9
20. Soro-Arnaiz I, Li QOY, Torres-Capelli M, Melendez-Rodriguez F, Veiga S, Veys K. et al. Role of Mitochondrial Complex IV in Age-Dependent Obesity. Cell Rep. 2016;16:2991-3002
21. Boudina S, Graham TE. Mitochondrial function/dysfunction in white adipose tissue. Exp Physiol. 2014;99:1168-78
22. Reinisch I, Schreiber R, Prokesch A. Regulation of thermogenic adipocytes during fasting and cold. Molecular and cellular endocrinology. 2020;512:110869
23. Goncalves LF, Machado TQ, Castro-Pinheiro C, de Souza NG, Oliveira KJ, Fernandes-Santos C. Ageing is associated with brown adipose tissue remodelling and loss of white fat browning in female C57BL/6 mice. Int J Exp Pathol. 2017;98:100-8
24. Wu CL, Yu P, Sun RX. Adipose tissue and age-dependent insulin resistance: New insights into WAT browning (Review). Int J Mol Med. 2021 47
25. Feng X, Wang L, Zhou R, Zhou R, Chen L, Peng H. et al. Senescent immune cells accumulation promotes brown adipose tissue dysfunction during aging. Nature Communications. 2023;14:3208
26. Ou M-Y, Zhang H, Tan P-C, Zhou S-B, Li Q-F. Adipose tissue aging: mechanisms and therapeutic implications. Cell death & disease. 2022;13:1-10
27. Wang G, Meyer JG, Cai W, Softic S, Li ME, Verdin E. et al. Regulation of UCP1 and mitochondrial metabolism in brown adipose tissue by reversible succinylation. Molecular cell. 2019;74:844-57 e7
28. Markowski DN, Thies HW, Gottlieb A, Wenk H, Wischnewsky M, Bullerdiek J. HMGA2 expression in white adipose tissue linking cellular senescence with diabetes. Genes & nutrition. 2013;8:449-56
29. Lee G, Kim YY, Jang H, Han JS, Nahmgoong H, Park YJ. et al. SREBP1c-PARP1 axis tunes anti-senescence activity of adipocytes and ameliorates metabolic imbalance in obesity. Cell metabolism. 2022;34:702-18 e5
30. Watanabe S, Kawamoto S, Ohtani N, Hara E. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer science. 2017;108:563-9
31. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends in cell biology. 2018;28:436-53
32. Chen L, Kashina A. Post-translational modifications of the protein termini. Frontiers in cell and developmental biology. 2021;9:719590
33. de Magalhães JP, Passos JF. Stress, cell senescence and organismal ageing. Mechanisms of ageing and development. 2018;170:2-9
34. Birch J, Gil J. Senescence and the SASP: many therapeutic avenues. Genes & development. 2020;34:1565-76
35. Di Micco R, Krizhanovsky V, Baker D, d'Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nature reviews Molecular cell biology. 2021;22:75-95
36. Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T. et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proceedings of the National Academy of Sciences. 2015;112:E6301-E10
37. Xu M, Palmer AK, Ding H, Weivoda MM, Pirtskhalava T, White TA. et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. elife. 2015;4:e12997
38. Von Bank H, Kirsh C, Simcox J. Aging adipose: Depot location dictates age-associated expansion and dysfunction. Ageing research reviews. 2021;67:101259
39. Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. The EMBO journal. 2011;30:1536-48
40. Lorenzo M, Fernández-Veledo S, Vila-Bedmar R, Garcia-Guerra L, De Alvaro C, Nieto-Vazquez I. Insulin resistance induced by tumor necrosis factor-α in myocytes and brown adipocytes. Journal of animal Science. 2008;86:E94-E104
41. Graja A, Schulz TJ. Mechanisms of aging-related impairment of brown adipocyte development and function. Gerontology. 2015;61:211-7
42. Tchkonia T, Pirtskhalava T, Thomou T, Cartwright MJ, Wise B, Karagiannides I. et al. Increased TNFα and CCAAT/enhancer-binding protein homologous protein with aging predispose preadipocytes to resist adipogenesis. American Journal of Physiology-Endocrinology and Metabolism. 2007;293:E1810-E9
43. Bluher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299:572-4
44. Katic M, Kennedy AR, Leykin I, Norris A, McGettrick A, Gesta S. et al. Mitochondrial gene expression and increased oxidative metabolism: role in increased lifespan of fat-specific insulin receptor knock-out mice. Aging cell. 2007;6:827-39
45. Ho J, Cruise ES, Dowling RJ, Stambolic V. PTEN nuclear functions. Cold Spring Harbor Perspectives in Medicine. 2020 10
46. Lee Y-R, Chen M, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nature reviews Molecular cell biology. 2018;19:547-62
47. Li YZ, Di Cristofano A, Woo M. Metabolic role of PTEN in insulin signaling and resistance. Cold Spring Harbor Perspectives in Medicine. 2020 10
48. Gericke A, Munson M, Ross AH. Regulation of the PTEN phosphatase. Gene. 2006;374:1-9
49. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960-76
50. Chen C-Y, Chen J, He L, Stiles BL. PTEN: tumor suppressor and metabolic regulator. Frontiers in endocrinology. 2018;9:338
51. Kirstein AS, Kehr S, Nebe M, Hanschkow M, Barth LA, Lorenz J. et al. PTEN regulates adipose progenitor cell growth, differentiation, and replicative aging. Journal of Biological Chemistry. 2021 297
52. Cedikova M, Kripnerová M, Dvorakova J, Pitule P, Grundmanova M, Babuska V. et al. Mitochondria in white, brown, and beige adipocytes. Stem cells international. 2016. 2016
53. Lv X, Qiu J, Hao T, Zhang H, Jiang H, Tan Y. HDAC inhibitor Trichostatin A suppresses adipogenesis in 3T3-L1 preadipocytes. Aging (Albany NY). 2021;13:17489
54. Willis-Martinez D, Richards HW, Timchenko NA, Medrano EE. Role of HDAC1 in senescence, aging, and cancer. Experimental gerontology. 2010;45:279-85
55. Lee IH. Mechanisms and disease implications of sirtuin-mediated autophagic regulation. Experimental & molecular medicine. 2019;51:1-11
56. Xu C, Wang L, Fozouni P, Evjen G, Chandra V, Jiang J. et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nature cell biology. 2020;22:1170-9
57. Majeed Y, Halabi N, Madani AY, Engelke R, Bhagwat AM, Abdesselem H. et al. SIRT1 promotes lipid metabolism and mitochondrial biogenesis in adipocytes and coordinates adipogenesis by targeting key enzymatic pathways. Scientific reports. 2021;11:8177
58. Choi W-I, Yoon J-H, Choi S-H, Jeon B-N, Kim H, Hur M-W. Proto-oncoprotein Zbtb7c and SIRT1 repression: implications in high-fat diet-induced and age-dependent obesity. Experimental & Molecular Medicine. 2021;53:917-32
59. Ma C, Pi C, Yang Y, Lin L, Shi Y, Li Y. et al. Nampt expression decreases age-related senescence in rat bone marrow mesenchymal stem cells by targeting Sirt1. PloS one. 2017;12:e0170930
60. Nacarelli T, Lau L, Fukumoto T, Zundell J, Fatkhutdinov N, Wu S. et al. NAD+ metabolism governs the proinflammatory senescence-associated secretome. Nature cell biology. 2019;21:397-407
61. Stromsdorfer KL, Yamaguchi S, Yoon MJ, Moseley AC, Franczyk MP, Kelly SC. et al. NAMPT-mediated NAD+ biosynthesis in adipocytes regulates adipose tissue function and multi-organ insulin sensitivity in mice. Cell reports. 2016;16:1851-60
62. Qiang L, Wang L, Kon N, Zhao W, Lee S, Zhang Y. et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Pparγ. Cell. 2012;150:620-32
63. Bae EJ, Park B-H. Multiple Roles of Sirtuin 6 in Adipose Tissue Inflammation. Diabetes & Metabolism Journal. 2023;47:164-72
64. Zhao G, Wang H, Xu C, Wang P, Chen J, Wang P. et al. SIRT6 delays cellular senescence by promoting p27Kip1 ubiquitin-proteasome degradation. Aging (Albany NY). 2016;8:2308
65. Kuang J, Chen L, Tang Q, Zhang J, Li Y, He J. The role of Sirt6 in obesity and diabetes. Frontiers in physiology. 2018;9:135
66. Chen Q, Hao W, Xiao C, Wang R, Xu X, Lu H. et al. SIRT6 is essential for adipocyte differentiation by regulating mitotic clonal expansion. Cell reports. 2017;18:3155-66
67. Yao L, Cui X, Chen Q, Yang X, Fang F, Zhang J. et al. Cold-inducible SIRT6 regulates thermogenesis of brown and beige fat. Cell reports. 2017;20:641-54
68. Vizioli MG, Liu T, Miller KN, Robertson NA, Gilroy K, Lagnado AB. et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes & development. 2020;34:428-45
69. Lagace DC, Nachtigal MW. Inhibition of histone deacetylase activity by valproic acid blocks adipogenesis. Journal of Biological Chemistry. 2004;279:18851-60
70. Ong BX, Brunmeir R, Zhang Q, Peng X, Idris M, Liu C, Xu F. Regulation of thermogenic adipocyte differentiation and adaptive thermogenesis through histone acetylation. Frontiers in Endocrinology. 2020;11:95
71. Soliman MA, Berardi P, Pastyryeva S, Bonnefin P, Feng X, Colina A. et al. ING1a expression increases during replicative senescence and induces a senescent phenotype. Aging cell. 2008;7:783-94
72. Li F, Wu R, Cui X, Zha L, Yu L, Shi H, Xue B. Histone deacetylase 1 (HDAC1) negatively regulates thermogenic program in brown adipocytes via coordinated regulation of histone H3 lysine 27 (H3K27) deacetylation and methylation. Journal of Biological Chemistry. 2016;291:4523-36
73. Binda O, Nassif C, Branton P. SIRT1 negatively regulates HDAC1-dependent transcriptional repression by the RBP1 family of proteins. Oncogene. 2008;27:3384-92
74. Sasaki T, Maier B, Bartke A, Scrable H. Progressive loss of SIRT1 with cell cycle withdrawal. Aging cell. 2006;5:413-22
75. Dasuri K, Zhang L, Ebenezer P, Fernandez-Kim SO, Bruce-Keller AJ, Szweda LI, Keller JN. Proteasome alterations during adipose differentiation and aging: links to impaired adipocyte differentiation and development of oxidative stress. Free Radical Biology and Medicine. 2011;51:1727-35
76. Jia L, Soengas MS, Sun Y. ROC1/RBX1 E3 ubiquitin ligase silencing suppresses tumor cell growth via sequential induction of G2-M arrest, apoptosis, and senescence. Cancer research. 2009;69:4974-82
77. Young AP, Schlisio S, Minamishima YA, Zhang Q, Li L, Grisanzio C. et al. VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nature cell biology. 2008;10:361-9
78. Brunmeir R, Xu F. Functional regulation of PPARs through post-translational modifications. International journal of molecular sciences. 2018;19:1738
79. Sane S, Rezvani K. Essential roles of E3 ubiquitin ligases in p53 regulation. International journal of molecular sciences. 2017;18:442
80. Pan M, Blattner C. Regulation of p53 by E3s. Cancers. 2021;13:745
81. Damerell V, Pepper MS, Prince S. Molecular mechanisms underpinning sarcomas and implications for current and future therapy. Signal transduction and targeted therapy. 2021;6:246
82. Kim JH, Park S-M, Kang MR, Oh S-Y, Lee TH, Muller MT, Chung IK. Ubiquitin ligase MKRN1 modulates telomere length homeostasis through a proteolysis of hTERT. Genes & development. 2005;19:776-81
83. Kotla S, Le N-T, Vu HT, Ko KA, Gi YJ, Thomas TN. et al. Endothelial senescence-associated secretory phenotype (SASP) is regulated by Makorin-1 ubiquitin E3 ligase. Metabolism. 2019;100:153962
84. Kim J, Park K, Lee E, Jang W, Seo J, Shin S. et al. Suppression of PPARγ through MKRN1-mediated ubiquitination and degradation prevents adipocyte differentiation. Cell Death & Differentiation. 2014;21:594-603
85. Lee M-S, Han H-J, Han SY, Kim IY, Chae S, Lee C-S. et al. Loss of the E3 ubiquitin ligase MKRN1 represses diet-induced metabolic syndrome through AMPK activation. Nature communications. 2018;9:3404
86. Watanabe M, Takahashi H, Saeki Y, Ozaki T, Itoh S, Suzuki M. et al. The E3 ubiquitin ligase TRIM23 regulates adipocyte differentiation via stabilization of the adipogenic activator PPARγ. Elife. 2015;4:e05615
87. Lee JM, Choi SS, Lee YH, Khim KW, Yoon S, Kim B-g. et al. The E3 ubiquitin ligase TRIM25 regulates adipocyte differentiation via proteasome-mediated degradation of PPARγ. Experimental & molecular medicine. 2018;50:1-11
88. Bond ST, Moody SC, Liu Y, Civelek M, Villanueva CJ, Gregorevic P. et al. The E3 ligase MARCH5 is a PPARγ target gene that regulates mitochondria and metabolism in adipocytes. American Journal of Physiology-Endocrinology and Metabolism. 2019;316:E293-E304
89. Park Y-Y, Lee S, Karbowski M, Neutzner A, Youle RJ, Cho H. Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin-related protein 1 and mitofusin 1. Journal of cell science. 2010;123:619-26
90. Dou H, Duan Y, Zhang X, Yu Q, Di Q, Song Y. et al. Aryl hydrocarbon receptor (AhR) regulates adipocyte differentiation by assembling CRL4B ubiquitin ligase to target PPARγ for proteasomal degradation. Journal of Biological Chemistry. 2019;294:18504-15
91. Hallenborg P, Feddersen S, Francoz S, Murano I, Sundekilde U, Petersen R. et al. Mdm2 controls CREB-dependent transactivation and initiation of adipocyte differentiation. Cell Death & Differentiation. 2012;19:1381-9
92. Zhao W, Xu Q, Yang J, Xie X, Li C, Zhang W. et al. Murine double minute 2 aggravates adipose tissue dysfunction through ubiquitin-mediated six-transmembrane epithelial antigen of prostate 4 degradation. Iscience. 2022;25:104544
93. Sudol M, Hunter T. NeW wrinkles for an old domain. Cell. 2000;103:1001-4
94. Cao X, Xue L, Han L, Ma L, Chen T, Tong T. WW Domain-containing E3 ubiquitin protein ligase 1 (WWP1) delays cellular senescence by promoting p27Kip1 degradation in human diploid fibroblasts. Journal of Biological Chemistry. 2011;286:33447-56
95. Kobayashi M, Hoshino S, Abe T, Okita N, Tagawa R, Nagai W. et al. Identification of WWP1 as an obesity-associated E3 ubiquitin ligase with a protective role against oxidative stress in adipocytes. Biochemical and biophysical research communications. 2019;508:117-22
96. Banerjee PS, Lagerlöf O, Hart GW. Roles of O-GlcNAc in chronic diseases of aging. Molecular aspects of medicine. 2016;51:1-15
97. Ji S, Park SY, Roth J, Kim HS, Cho JW. O-GlcNAc modification of PPARγ reduces its transcriptional activity. Biochemical and biophysical research communications. 2012;417:1158-63
98. Butkinaree C, Park K, Hart GW. O-linked β-N-acetylglucosamine (O-GlcNAc): extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochimica et Biophysica Acta (BBA)-General Subjects. 2010;1800:96-106
99. Lefebvre T, Dehennaut V, Guinez C, Olivier S, Drougat L, Mir A-M. et al. Dysregulation of the nutrient/stress sensor O-GlcNAcylation is involved in the etiology of cardiovascular disorders, type-2 diabetes and Alzheimer's disease. Biochimica Et Biophysica Acta (BBA)-General Subjects. 2010;1800:67-79
100. Ishihara K, Takahashi I, Tsuchiya Y, Hasegawa M, Kamemura K. Characteristic increase in nucleocytoplasmic protein glycosylation by O-GlcNAc in 3T3-L1 adipocyte differentiation. Biochemical and biophysical research communications. 2010;398:489-94
101. Whelan SA, Lane MD, Hart GW. Regulation of the O-linked β-N-acetylglucosamine transferase by insulin signaling. Journal of Biological Chemistry. 2008;283:21411-7
102. Li X, Molina H, Huang H, Zhang Y-y, Liu M, Qian S-w. et al. O-linked N-acetylglucosamine modification on CCAAT enhancer-binding protein β: role during adipocyte differentiation. Journal of Biological Chemistry. 2009;284:19248-54
103. Teo CF, Wollaston-Hayden EE, Wells L. Hexosamine flux, the O-GlcNAc modification, and the development of insulin resistance in adipocytes. Molecular and cellular endocrinology. 2010;318:44-53
104. Luo B, Parker GJ, Cooksey RC, Soesanto Y, Evans M, Jones D, McClain DA. Chronic hexosamine flux stimulates fatty acid oxidation by activating AMP-activated protein kinase in adipocytes. Journal of Biological Chemistry. 2007;282:7172-80
105. Ebert T, Tran N, Schurgers L, Stenvinkel P, Shiels PG. Ageing-Oxidative stress, PTMs and disease. Molecular Aspects of Medicine. 2022;86:101099
106. Santos AL, Lindner AB. Protein posttranslational modifications: roles in aging and age-related disease. Oxidative Medicine and Cellular Longevity. 2017. 2017
107. Ou M-Y, Zhang H, Tan P-C, Zhou S-B, Li Q-F. Adipose tissue aging: Mechanisms and therapeutic implications. Cell death & disease. 2022;13:300
Author contact
![]() Corresponding authors: In Hye Lee Ph.D., Department of Life Science, College of Natural Science, Ewha Womans University, 52 Ewhayeodae-Gil, Seodaemun-gu, Seoul, 03760, Republic of Korea. Tel: +82-2-3277-3032, E-mail: lih3026ac.kr. Kyung-Hee Chun Ph.D., Department of Biochemistry & Molecular Biology, Yonsei University College of Medicine, 50-1 Yonsei-ro, Seodaemun-gu, Seoul 03722, Republic of Korea. Tel: +82-2-2228-1699, Fax: +82-2-312-5041, E-mail: khchunac.
Corresponding authors: In Hye Lee Ph.D., Department of Life Science, College of Natural Science, Ewha Womans University, 52 Ewhayeodae-Gil, Seodaemun-gu, Seoul, 03760, Republic of Korea. Tel: +82-2-3277-3032, E-mail: lih3026ac.kr. Kyung-Hee Chun Ph.D., Department of Biochemistry & Molecular Biology, Yonsei University College of Medicine, 50-1 Yonsei-ro, Seodaemun-gu, Seoul 03722, Republic of Korea. Tel: +82-2-2228-1699, Fax: +82-2-312-5041, E-mail: khchunac.