Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Epidemiology
Definition of TKI resistance
Mechanisms of TKI resistance
Innovative strategies
Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(1):175-181. doi:10.7150/ijbs.86305 This issue Cite
Review
Mechanisms underlying therapeutic resistance of tyrosine kinase inhibitors in chronic myeloid leukemia
Jingnan Sun1, Ruiping Hu1, Mengyuan Han1, Yehui Tan1 ![]() , Mengqing Xie1,3
, Mengqing Xie1,3 ![]() , Sujun Gao1
, Sujun Gao1 ![]() , Ji-Fan Hu1,2
, Ji-Fan Hu1,2 ![]()
1. Hematology Department, First hospital of Jilin University, Changchun, Jilin, 130021, P.R. China.
2. Stanford University Medical School, Palo Alto Veterans Institute for Research, Palo Alto, CA94304, USA.
3. Oncology Department, Cancer hospital Chinese Academy of Medical Sciences, Langfang District, 065001, P.R. China.
Received 2023-5-18; Accepted 2023-9-27; Published 2024-1-1
Abstract

Chronic myeloid leukemia (CML) is a malignant clonal disease involving hematopoietic stem cells that is characterized by myeloid cell proliferation in bone marrow and peripheral blood, and the presence of the Philadelphia (Ph) chromosome with BCR-ABL fusion gene. Treatment of CML has dramatically improved since the advent of tyrosine kinase inhibitors (TKI). However, there are a small subset of CML patients who develop resistance to TKI. Mutations in the ABL kinase domain (KD) are currently recognized as the leading cause of TKI resistance in CML. In this review, we discuss the concept of resistance and summarize recent advances exploring the mechanisms underlying CML resistance. Overcoming TKI resistance appears to be the most successful approach to reduce the burden of leukemia and enhance cures for CML. Advances in new strategies to combat drug resistance may rapidly change the management of TKI-resistant CML and expand the prospects for available therapies.
Keywords: chronic myeloid leukemia, TKI Resistance, ABL mutation, mechanism
Introduction
Chronic myeloid leukemia is a myeloproliferative disease that originates from pluripotent stem cells carrying a characteristic reciprocal translocation between an Abelson leukemia virus (ABL) oncogene from the long arm of chromosome 9 and the breakpoint cluster region (BCR) from the long arm of chromosome 22. The fusion protein encoded by the BCR-ABL fusion gene has abnormally tyrosine kinase activity, leading to increased cell proliferation through its downstream signaling pathways. As the cause of CML, the resulting BCR-ABL fusion protein, is an ideal therapeutic target. The emergence of first and second-generation tyrosine kinase inhibitors (TKI) has led to overall survival (OS) rate of 82%-95% for CML. However, about 20%-30% of the patients ultimately develop resistance to TKI[1]. Here, we summarize recent advances on the mechanism of TKI resistance and the characteristics of mutations in the ABL kinase domain.
Epidemiology
The incidence of CML is (1.6-2)/100,000 worldwide, accounting for 15% of all patients with leukemia[2]. In Western countries, patients older than 70 years comprise more than 20% of CML patients, while children and adolescents account for < 5% of all cases, with the median age of CML patients about 57 years[1] . In Asia and Africa, the median age at diagnosis is less than 50 years[1]. At the end of the 1990s, imatinib mesylate (IM) was successfully used to treat CML, ushering in the era of molecularly targeted cancer therapy. According to the International Collaborative Research Group, the overall survival rate of IM treatment at 5 years is 90%-95%, and the Progression-free survival (PFS) at 5 years is nearly 80%-90%[3, 4]. At 10 years, the OS is stil 82%-85%, but the resistance rate to IM in first-line patients is 10%-15%, and the resistance rate to second-generation TKI is < 10%[5]. Summarizing multiple studies worldwide, mutations in the ABL kinase domain accounts for about 22.4%-54.46% of resistance CML patients, with p-loop mutations accounting for about one-fourth[6, 7].
Definition of TKI resistance
Primary resistance refers to the lack of hematologic, cytogenetic, or molecular response to TKI in the early stages of treatment. Secondary resistance refers to the loss of response after a patient has gained a certain degree of therapeutic response[8].
The latest European Leukemia Network (ELN) treatment guidelines and the National Comprehensive Cancer Network (NCCN) CML Clinical Practice Guidelines consider clinical responses to include "best", "failure", and "warning"[1, 9]. TKI resistance is defined as "failure" in the evaluation of CML treatment response, referring to the definition of ELN failure in 2020 recommendations, with BCR-ABLIS > 10% if confirmed within 1-3 months, BCR-ABLIS > 10% at 6 months of TKI treatment, BCR -ABLIS > 1% at 12 months of treatment, or BCR-ABLIS >1% at any time after 12 months of treatment, with resistance mutations, the emergence of high-risk additional chromosome abnormalities (ACA)[10, 11]. The same definitions are recommended for second-line treatment[1].
BCR-ABL fusion genes are necessary for CML to develop; however, the BCR-ABL oncogene alone is not sufficient to explain disease progression[11, 12]. In fact, BCR-ABL transcript levels increase with disease progression, promoting a secondary molecular, chromosomal-level hit and ultimately leading to the expansion of malignant cell clones[13]. Once a second strike is obtained, TKI therapy that inhibits BCR-ABL alone tends to fail[14]. Although the ultimate source of disease progression from these additional strikes remains BCR-ABL, it also suggests that the causes of disease progression include pathways other than ABL-dependent mechanisms.
Mechanisms of TKI resistance
TKI resistance can be driven by ABL-dependent and independent mechanisms, depending on whether they are associated with the ABL kinase domain. Both mechanisms can induce significant clinical resistance, but secondary resistance usually involves ABL-dependent pathways, such as BCR-ABL mutations, gene amplification, or increased expression[15, 16]. ABL-independent resistance is more commonly seen in primary resistance, such as genomically unstable, quiescence leukemia stem cells, or individual differences in IM blood concentrations due to differences in oral bioavailability, the high affinity of serum proteins for IM, and cellular influx/efflux transporters[14, 17, 18] (Figure 1).
Mechanisms of TKI resistance in CML patients.
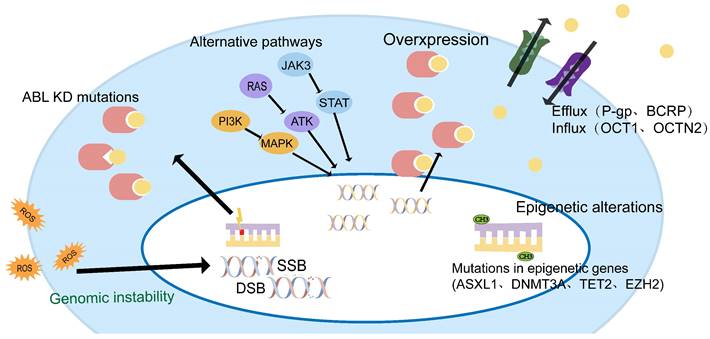
CML-chronic phase (CMP-CP) progression to CML-blastic phase (CML-BP) is a multifactorial, multi-step process. It is believed that disease progression may be triggered by a series of different but equivalent events[14]. ABL-dependent pathways and non-dependent pathways may work in synergy, leading to the accumulation of key events at the DNA, RNA, and protein levels, causing abnormal cell cycle control, differentiation, apoptosis failure, and eventually drug resistance.
ABL-dependent mechanism
BCR-ABL kinase domain mutations
Point mutations in the BCR-ABL fusion gene are the most common cause of TKI resistance, with about 31%-63% of imatinib (IM) resistance attributed to point mutations[19-21]. According to the published literature, more than 100 mutations have been found, distributed in the ABL amino acid range of 220-500 (Figure 2)[19]. Four main point mutation sites leading to IM resistance are: a). Gatekeepers (IM-binding site): F317I/L and T315I; b). ATP-binding loop (P-loop): E255K, Q252H, Y253F; c). catalytic-loop (C-loop): M351T; and d). activation loop (A-loop): H396P. The T315I mutation was the most common mutation (about 13%-16%). E255K and M351T mutations accounted for 9%-14% of mutations. Y253F, Q252H, F317L and other mutations accounted for 3%-6% of mutations, while V299L mutations were rare[6, 16, 22]. It is worth noting that E255K, Y253F, and Q252H, the mutations with higher mutation rates, are located in the p-loop, followed by the mutations at IM-binding sites[23, 24].
The location of hotspot mutations in the kinase domain.
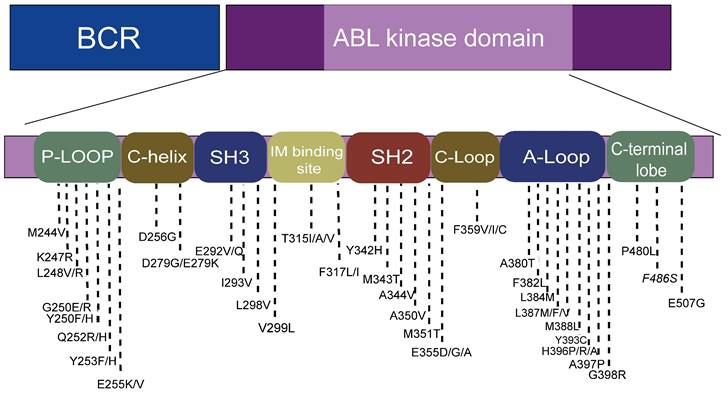
Compound mutations (variants containing ≥ 2 mutations within the same BCR-ABL1 allele that presumably arise sequentially or simultaneously) exhibit different spectra of resistance from single mutations[10]. For example, while ponatinib is effective in treating a CML with a single T315I mutation, most compound mutations containing T315I are resistant to ponatinib[25, 26]. In addition to T315I, there are also E255V mutations that contain second mutations that exhibit different resistance to ponatinib. Meanwhile, compound mutations are mostly located in the p-loop in patients with CML, yet more likely to contain T315I[27]. Patients with compound mutations are more likely to progress during follow-up [28, 29].
Both ELN and NCCN recommend screening for BCR-ABL kinase domain mutations and providing the best drug regimen for a subset of well-documented mutation types, but there may not be definite TKI options for mutations listed in official guidelines.
Heat maps are widely used in the literature, which use the same reds, greens, and yellows as traffic lights to indicate the sensitivity of mutations to different TKI, allowing the selection of appropriate TKI for treatment[6]. It should be noted that heat maps are mutant models based on mouse cell lines and in vitro experiments. The sensitivity of in vitro generated data to TKI may vary in vivo. Similarly, there may be genetic differences between humans and mice. In addition, the choice of using heat maps to predict TKI is based on resistance caused by KD mutations as a prerequisite. If the resistance is driven by other cells or other pathways and is not related to KD mutations, the use of heat maps to predict TKI response is ineffective[15]. Even so, heat maps can still be used as a reference, but at the same time in vivo responses should be monitored to assess the biological behavior of subclonals.
Historically, Sanger sequencing (SS) has been used in most clinical practice to identify BCR-ABL1 mutations associated with TKI resistance. However, SS is unable to detect mutations present in 10% to 20% cells and does not allow direct detection of compound mutations, although their presence can be inferred in certain cases (when ≥ 2 mutations are detected at a combined frequency > 100%)[29-31]. Next‐generation sequencin (NGS) also enables direct detection of BCR-ABL1 compound mutations. However, it has recently been shown that polymerase chain reaction (PCR) mediated recombination may cause compound mutation frequencies to be overestimated when PCR amplicons are used for NGS[32]. Thus, the ability of NGS to reliably detect compound mutations, and therefore their prevalence in CML, remains to be established. NGS can be used to better monitor the size of the subclonal KD mutant and adjust the treatment regimen in time. Although SS cannot distinguish between polyclonal and compound mutations, unless the combined mutant allele burden clearly exceeds 100%, NGS identifies compound mutations as long as the average read length exceeds the distance between the 2 single nucleotide variations[30, 33].
BCR-ABL overexpression
BCR-ABL overexpression is another mechanism of ABL-dependent resistance that causes IM resistance, but its clinical significance for resistance is far less than that of ABL mutation. BCR-ABL overexpression refers to any abnormality of the regulatory mechanism of ABL genes. Differential regulation or gene amplification can lead to increased expression of BCR-ABL[34]. Therefore, higher level of BCR-ABL proteins can still cause disease progression despite the administration of TKI. Compared with CML cells with low BCR-ABL expression levels in the chronic phase, high-expression cells are less sensitive to IM. The high expression of BCR-ABL is more pronounced in the accelerated phase (AP). It may be the reason why patients in accelerated phase treated with IM respond less well than patients in chronic phase.
DNA damage repair and Gene instability
Genomic instability leads to the accumulation of mutations at the ABL KD and other molecular or chromosomal aberrations. Vice versa, overexpression of BCR-ABL fusion proteins can also lead to genomic instability in CML cells[20]. Partial deletions of RUNX1 and PMRD16, expression of RUNX1/PMRD16, and mutations in GATA2 activation are also associated with CML progression and their presence may be detected in CML[14, 35].
Abnormally active tyrosine kinase activity causes the accumulation of reactive oxygen species (ROS). Increased ROS can damage DNA, leading to alkaline oxidative damage, DNA double-strand breaks (DSBs), and mismatch repair. Ultimately, ROS-induced genomic instability and subsequent genetic events, such as mutations, chromosomal translocations, and deletions, can lead to drug resistance[24, 36]. ROS involvement in genomic instability and CML progression has been widely evaluated. BCR-ABL kinase activity has been found to increase intracellular ROS levels, which is significantly more pronounced in CML-BP cells, exhibiting higher BCR-ABL levels than in CP CML cells[19]. Elevated ROS levels and exogenous factors, such as radiation or genotoxic compounds, may enhance oxidative DNA damage. On the other hand, DNA repair mechanisms are dysregulated due to the loss or acquisition of BCR-ABL-positive cell function. In human cells, DSBs are preferentially repaired by homologous recombination (HR) or non-homologous termination (NHEJ), but sometimes highly unfaithful single-strand annealing (SSA) mechanisms may occur[37]. Novicki et al. have demonstrated that HR and NHEJ are enhanced in ROS-mediated DSB repair in BCR-ABL cells, where these mechanisms lead to mutations and a large number of deletions. In fact, BCR-ABL (non-mutant and T315I mutants) has been shown to bind and phosphorylate RAD51 and its paralog RAD51B, promoting unfaithful homologous HR in a dose-dependent manner[20].
ABL-independent mechanism
Alternative pathways
TKI inhibits the BCR-ABL kinase activity through competitive binding, but does not eliminate CML cells. This means that CML stem cells can survive through other signaling pathways, such as SRC, JAK/STAT, RAS/MAPK, and PI3K/AKT[38]. Activation of alternative pathways may reveal why the disease still recurs after patients who achieve an excellent response stop treatment[14, 39, 40].
Overexpression of SRC family kinase proteins, such as LYN and HCK, is essential for cell proliferation, survival, and adhesion[19, 20]. SRC proteins lead to AKT activation and promote survival and STAT5 activation to stimulate proliferation. Overexpression of SRC proteins in CML is a rationale for the development and use of dual SRC/ABL inhibitors, such as dasatinib and bosutinib[35].
In addition, STAT can activate the JAK2 protein by responding to cytokines released by cancer cells and bone marrow niche cells. JAK2 is activated and subsequently phosphorylated by one of the 7 STAT members. STAT3 and STAT5 have been identified as the most relevant STAT proteins in cancer[36, 41]. After STAT phosphorylation, this protein migrates to the nucleus, where it regulates transcription of various target genes, such as c-MYC.
GAB2 is a member of the GAB family of docking proteins that play a key role in CML by amplifying BCR-ABL signaling. Dysregulation of this protein leads to increased proliferation, decreased demand for growth factors, and increased cell viability[20]. In addition, continuous phosphorylation of GAB2 leads to activation of substrates, such as the stabilization of RAS proteins in active form after GAB2 activation. An increase in protein kinase C (PKC) expression has also been observed in TKI-resistant CML cells[39].
After PI3K is activated, AKT is subsequently phosphorylated, affecting a variety of downstream proteins[42]. BAD is one of the AKT targets that reduces the signal of apoptosis. After phosphorylation, BAD becomes inactive and therefore does not inhibit anti-apoptotic proteins, such as BCL-2 and BCL-XL. Another AKT target is the FOXO transcription factor, which regulates cell cycle arrest and apoptosis under normal conditions. AKT-induced FOXO phosphorylation blocks its activity, and avoids apoptosis and promotes cell cycle progression. In addition, mTOR is a serine/threonine kinase that is activated by AKT and regulates mRNA translation, controlling cell growth and proliferation. Similarly, NF-kB is also indirectly activated by AKT, promoting gene transcription. AKT targets IKK, a natural inhibitor of NF-kB, and releases this inhibitory signal from NF-kB[34, 38, 43]
Quiescent CML stem cells
CML stem cells account for about 0.5% of the CD34+ population, and they do not require BCR-ABL kinase activity to survive[42]. CML stem cells are resistant to TKI, and resistance/recurrence is presumed to come primarily from these cells. In vitro studies have shown that "quiescent" leukemia stem cells are highly resistant to IM. Even if complete molecular biological remission is obtained, leukemia stem cells in some patients can still survive for a long time[34, 43]. This could explain why TKI cannot kill all leukemia cells even in patients with optimal response. So far, combining TKI with another drug to remove residual stem cells and identifying the underlying signaling pathways of CML stem cells seems to be the most promising way to overcome treatment failure[44].
Epigenetic alterations
There is now ample evidence that mutations in epigenetic regulatory genes, such as DNMT3A, TET2, EZH2, and ASXL1, are relatively rare in chronic-phase CML[22, 45], but the incidence of these mutations increases during disease progression and accelerates leukemia stem cell production, maintenance, and progression of CML[35, 46, 47].
The most common mechanisms of epigenetic modification include methylation, acetylation, and phosphorylation[35]. Their role is to regulate chromatin structure and remodeling, providing a site for the recruitment of other transcription factors, followed by altering the cell cycle, apoptosis, and expression of tumor suppressor genes. DNA hypermethylation is a common carcinogenic process in many solid and hematological tumors[48]. It has been reported in detail in patients with CML, especially in patients with AP and BC. Although ABL hypermethylation has been demonstrated, its role in the pathophysiology of disease progression is unclear.
Bioavailability and blood concentration
As an oral drug, IM is first affected by patient compliance, followed by IM absorption through the gastrointestinal tract, and is influenced by first-pass metabolism. About 95% of IM binds to plasma proteins (mainly albumin) and α-1 acid glycoprotein (AGP, a hepatic acute phase protein)[49]. It has been proposed that the combination of AGP with IM in plasma can reduce the accessibility[34, 35, 50].
TKIs (imatinib, nilotinib, dasatinib) are metabolized primarily by the cytochrome P450 system, mainly involving the isoenzyme CYP3A4. The activity of this isoenzyme varies from individual to individual and is affected by concomitant drugs, which may also lead to differences in IM concentrations.
Drug influx/efflux pump
Resistance is related to the expression level and function of solute carrier (SLC) transporters. According to the direction of transportation, it is divided into influx type and efflux type transporter. Influx transporters include organic cation transporter 1 (OCT1 or SLC22A1), organic anion-transporting polypeptide 1A2 (SCL01A2 or OATP1A2), OCTN2 and MATE1[46, 51]. OCT1 is the main transporter responsible for TKI uptake, and its expression or activity affects the level of drug response[7, 13, 27]. Other transporters have been identified as intermediaries in TKI transportation.
Efflux transporters include ATP-binding cassette subfamily B member 1 (ABCB1) also known as P-glycoprotein or MDR1, and ATP-binding cassette G subfamily member (ABCG2) also known as breast cancer resistance protein (BCRP)[52]. All TKIs approved for CML therapy are recognized P-gp substrates, and high levels of ABCB1 expression (genes encoding P-gp) are associated with poorer long-term outcomes and advanced disease. Another basic transporter of TKI drug resistance is breast cancer resistance protein (BCRP), encoded by the ABCG2 gene[35]. This protein is found in stem cells and its function is particularly relevant to leukemia stem cells (LSCs), protecting them from TKI action.
Reduced ABCG2 and increased SLC22A1 mRNA expression are associated with imatinib response in chronic myeloid leukemia. High expression of ABCB1 is more likely to be observed in patients with acute phase CML than in patients with CML in the chronic phase[47]. In patients undergoing IM therapy, higher OCT-1 activity was associated with increased MMR, EFS, and OS, while cell uptake of second-generation TKIs (dasatinib and nilotinib) appeared to be independent of OCT expression, thus supporting the theory that dasatinib or nilotinib might be superior[19, 39, 50].
Innovative strategies
Currently approved TKIs mainly target the ATP binding site of BCR-ABL1. Asciminib (ABL001) is a potent, specific, orally bioavailable BCR-ABL1 inhibitor that is distinct from approved ABL1 kinase inhibitors in that it does not bind to the ATP-binding site of the kinase[53]. In contrast, asciminib acts as an allosteric inhibitor and engages a vacant pocket at a site of the kinase domain normally occupied by the myristoylated N-terminal of ABL1— a motif that serves as an allosteric negative regulatory element lost on fusion of ABL1 to BCR[2]. By binding the myristoyl site, asciminib mimics myristate and restores inhibition of kinase activity. Owing to the distinct conformation of the myristoyl pocket, asciminib has high selectivity for ABL1 ABL kinase mutations, including T315I[11, 15]. Asciminib targets both native and mutated BCR-ABL1, including the gatekeeper T315I mutant.
Conclusion
The prognosis of CML has been significantly improved since the discovery of molecularly targeted therapies, but due to the heterogeneity of the mechanisms of resistance, the focus is shifting to find an inhibitor with broad utility that is conducive to overcoming drug resistance. Therefore, the management of patients with drug-resistant CML, including next-generation treatment options and higher sensitivity monitoring techniques, remains a challenge.
Acknowledgements
This work was supported by the National Natural Science Foundation of China grant (82371872, 31430021, 81272294, 32000431), the National Key R&D Program of China (2018YFA0106902, 2020YFA0707704), the Innovative Program of National Natural Science Foundation of China (82050003); Bethune project of Jilin technology project (20210101327JC); and California Institute of Regenerative Medicine (CIRM)grant (RT2-01942).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hochhaus A, Baccarani M, Silver RT, Schiffer C, Apperley JF, Cervantes F. et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020;34:966-84
2. Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2022 update on diagnosis, therapy, and monitoring. Am J Hematol. 2022;97:1236-56
3. Hehlmann R, Lauseker M, Saussele S, Pfirrmann M, Krause S, Kolb HJ. et al. Assessment of imatinib as first-line treatment of chronic myeloid leukemia: 10-year survival results of the randomized CML study IV and impact of non-CML determinants. Leukemia. 2017;31:2398-406
4. Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP. et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N Engl J Med. 2017;376:917-27
5. Hehlmann R. Chronic Myeloid Leukemia in 2020. Hemasphere. 2020;4:e468
6. Benjamin C, Murugan S, Hoosen S, Rapiti N. Chronic myeloid leukemia kinase domain mutations: A retrospective descriptive study on the therapeutic and prognostic significance in patients at King Edward VIII Hospital, KwaZulu-Natal, South Africa. Health Sci Rep. 2023;6:e1376
7. Branford S, Apperley JF. Measurable residual disease in chronic myeloid leukemia. Haematologica. 2022;107:2794-809
8. Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF. et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122:872-84
9. Deininger MW, Shah NP, Altman JK, Berman E, Bhatia R, Bhatnagar B. et al. Chronic Myeloid Leukemia, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2020;18:1385-415
10. Shahrin NH, Wadham C, Branford S. Defining Higher-Risk Chronic Myeloid Leukemia: Risk Scores, Genomic Landscape, and Prognostication. Curr Hematol Malig Rep. 2022;17:171-80
11. Senapati J, Jabbour E, Kantarjian H, Short NJ. Pathogenesis and management of accelerated and blast phases of chronic myeloid leukemia. Leukemia. 2023;37:5-17
12. Rinaldi I, Winston K. Chronic Myeloid Leukemia, from Pathophysiology to Treatment-Free Remission: A Narrative Literature Review. J Blood Med. 2023;14:261-77
13. Brown G. Hematopoietic and Chronic Myeloid Leukemia Stem Cells: Multi-Stability versus Lineage Restriction. Int J Mol Sci. 2022 23
14. Bavaro L, Martelli M, Cavo M, Soverini S. Mechanisms of Disease Progression and Resistance to Tyrosine Kinase Inhibitor Therapy in Chronic Myeloid Leukemia: An Update. Int J Mol Sci. 2019 20
15. Patel AB, O'Hare T, Deininger MW. Mechanisms of Resistance to ABL Kinase Inhibition in Chronic Myeloid Leukemia and the Development of Next Generation ABL Kinase Inhibitors. Hematol Oncol Clin North Am. 2017;31:589-612
16. Iriyama N. Chronic myeloid leukemia: the cutting-edge evidence and things we should know. Int J Hematol. 2023;117:1-2
17. Yanhong C, Xiucai X. Reserch progress of chronic myeloidleukemia drug resistance J. Practical Medical Research. 2018;34(11):1804-7
18. Soverini S, Branford S, Nicolini FE, Talpaz M, Deininger MW, Martinelli G. et al. Implications of BCR-ABL1 kinase domain-mediated resistance in chronic myeloid leukemia. Leuk Res. 2014;38:10-20
19. Poudel G, Tolland MG, Hughes TP, Pagani IS. Mechanisms of Resistance and Implications for Treatment Strategies in Chronic Myeloid Leukaemia. Cancers (Basel). 2022 14
20. Kaehler M, Cascorbi I. Molecular Mechanisms of Tyrosine Kinase Inhibitor Resistance in Chronic Myeloid Leukemia. Handb Exp Pharmacol. 2023;280:65-83
21. Copland M. Treatment of blast phase chronic myeloid leukaemia: A rare and challenging entity. Br J Haematol. 2022;199:665-78
22. Amarante-Mendes GP, Rana A, Datoguia TS, Hamerschlak N, Brumatti G. BCR-ABL1 Tyrosine Kinase Complex Signaling Transduction: Challenges to Overcome Resistance in Chronic Myeloid Leukemia. Pharmaceutics. 2022 14
23. Kantarjian H M, Jabbour E, Deininger M. et al. Ponatinib after failure of second-generation tyrosine kinase inhibitor in resistant chronic-phase chronic myeloid leukemia. Am J Hematol. 2022;97(11):1419-26
24. Shi DY, Qin YZ, Lai YY, Shi HX, Huang XJ, Jiang Q. Variables associated with BCR-ABL kinase domain mutation in TKI-resistant patients with chronic myeloid leukemia. Zhonghua xue ye xue za zhi = Zhonghua xueyexue zazhi. 2020;41:469-76
25. Iqbal Z, Absar M, Mahmood A, Aleem A, Iqbal M, Jameel A. et al. Discovery and Protein Modeling Studies of Novel Compound Mutations Causing Resistance to Multiple Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia. Asian Pac J Cancer Prev. 2020;21:3517-26
26. Radich J. Structure, function, and resistance in chronic myeloid leukemia. Cancer Cell. 2014;26:305-6
27. Ansari S, Verma M. Control of Ph(+) and additional chromosomal abnormalities in chronic myeloid leukemia by tyrosine kinase inhibitors. Med Oncol. 2023;40:237
28. Deininger MW, Hodgson JG, Shah NP, Cortes JE, Kim DW, Nicolini FE. et al. Compound mutations in BCR-ABL1 are not major drivers of primary or secondary resistance to ponatinib in CP-CML patients. Blood. 2016;127:703-12
29. Kang KH, Kim SH, Choi SY, Yoo HL, Lee MY, Song HY. et al. Compound mutations involving T315I and P-loop mutations are the major components of multiple mutations detected in tyrosine kinase inhibitor resistant chronic myeloid leukemia. Leuk Res. 2019;76:87-93
30. Soverini S, Abruzzese E, Bocchia M, Bonifacio M, Galimberti S, Gozzini A. et al. Next-generation sequencing for BCR-ABL1 kinase domain mutation testing in patients with chronic myeloid leukemia: a position paper. J Hematol Oncol. 2019;12:131
31. Marin AM, Wosniaki DK, Sanchuki HBS, Munhoz EC, Nardin JM, Soares GS. et al. Molecular BCR::ABL1 Quantification and ABL1 Mutation Detection as Essential Tools for the Clinical Management of Chronic Myeloid Leukemia Patients: Results from a Brazilian Single-Center Study. Int J Mol Sci. 2023 24
32. Soverini S, Bavaro L, De Benedittis C, Martelli M, Iurlo A, Orofino N. et al. Prospective assessment of NGS-detectable mutations in CML patients with nonoptimal response: the NEXT-in-CML study. Blood. 2020;135:534-41
33. Shokeen Y, Sharma NR, Vats A, Taneja V, Minhas S, Jauhri M. et al. Identification of Prognostic and Susceptibility Markers in Chronic Myeloid Leukemia Using Next Generation Sequencing. Ethiop J Health Sci. 2018;28:135-46
34. Yaghmaie M, Yeung CC. Molecular Mechanisms of Resistance to Tyrosine Kinase Inhibitors. Curr Hematol Malig Rep. 2019;14:395-404
35. Alves R, Goncalves AC, Rutella S, Almeida AM, De Las Rivas J, Trougakos IP. et al. Resistance to Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia-From Molecular Mechanisms to Clinical Relevance. Cancers (Basel). 2021 13
36. Warsch W, Grundschober E, Berger A, Gille L, Cerny-Reiterer S, Tigan AS. et al. STAT5 triggers BCR-ABL1 mutation by mediating ROS production in chronic myeloid leukaemia. Oncotarget. 2012;3:1669-87
37. Xu X, Yin S, Ren Y, Hu C, Zhang A, Lin Y. Proteomics analysis reveals the correlation of programmed ROS-autophagy loop and dysregulated G1/S checkpoint with imatinib resistance in chronic myeloid leukemia cells. Proteomics. 2022;22:e2100094
38. Braun TP, Eide CA, Druker BJ. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell. 2020;37:530-42
39. Minciacchi VR, Kumar R, Krause DS. Chronic Myeloid Leukemia: A Model Disease of the Past, Present and Future. Cells. 2021 10
40. Singh P, Kumar V, Gupta SK, Kumari G, Verma M. Combating TKI resistance in CML by inhibiting the PI3K/Akt/mTOR pathway in combination with TKIs: a review. Med Oncol. 2021;38:10
41. Putri A, Rinaldi I, Louisa M, Koesnoe S. The Role of STAT5 in Tyrosine Kinase Inhibitor (IMATINIB) Resistance in CML Patients. Acta Med Indones. 2019;51:348-52
42. Al Hamad M. Contribution of BCR-ABL molecular variants and leukemic stem cells in response and resistance to tyrosine kinase inhibitors: a review. F1000Res. 2021;10:1288
43. Houshmand M, Simonetti G, Circosta P, Gaidano V, Cignetti A, Martinelli G. et al. Chronic myeloid leukemia stem cells. Leukemia. 2019;33:1543-56
44. Eldesouki RE, Wu C, Saleh FM, Mohammed EA, Younes S, Hassan NE. et al. Identification and Targeting of Thomsen-Friedenreich and IL1RAP Antigens on Chronic Myeloid Leukemia Stem Cells Using Bi-Specific Antibodies. Onco Targets Ther. 2021;14:609-21
45. Rinke J, Hochhaus A, Ernst T. CML - Not only BCR-ABL1 matters. Best Pract Res Clin Haematol. 2020;33:101194
46. Zhou T, Medeiros LJ, Hu S. Chronic Myeloid Leukemia: Beyond BCR-ABL1. Curr Hematol Malig Rep. 2018;13:435-45
47. Meenakshi Sundaram DN, Jiang X, Brandwein JM, Valencia-Serna J, Remant KC, Uludag H. Current outlook on drug resistance in chronic myeloid leukemia (CML) and potential therapeutic options. Drug Discov Today. 2019;24:1355-69
48. Yang J, Surapaneni M, Schiffer CA. An evaluation of ponatinib as a therapy in adult patients with resistant/intolerant chronic-phase chronic myeloid leukemia. Expert Rev Hematol. 2022;15:393-402
49. Rudich A, Garzon R, Dorrance A. Non-Coding RNAs Are Implicit in Chronic Myeloid Leukemia Therapy Resistance. Int J Mol Sci. 2022 23
50. Talati C, Pinilla-Ibarz J. Resistance in chronic myeloid leukemia: definitions and novel therapeutic agents. Curr Opin Hematol. 2018;25:154-61
51. Kaehler M, Cascorbi I. Pharmacogenomics of Impaired Tyrosine Kinase Inhibitor Response: Lessons Learned From Chronic Myelogenous Leukemia. Front Pharmacol. 2021;12:696960
52. Ammar M, Louati N, Frikha I, Medhaffar M, Ghozzi H, Elloumi M. et al. Overexpression of P-glycoprotein and resistance to Imatinib in chronic myeloid leukemia patients. J Clin Lab Anal. 2020;34:e23374
53. Hughes TP, Mauro MJ, Cortes JE, Minami H, Rea D, DeAngelo DJ. et al. Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibitor Failure. N Engl J Med. 2019;381:2315-26
Author contact
![]() Corresponding authors: Ji-Fan Hu, Palo Alto Veterans Institute for Research, Palo Alto, CA 94304, USA, Tel: 650-852-3275, Fax: 650-849-1213, e-mail: jifanedu or hujifanedu.cn; or Sujun Gao, Hematology Department, First hospital of Jilin University, Changchun, Jilin, 130021, P.R. China, e-mail: sjgaoedu.cn; or Yehui Tan, Hematology Department, First hospital of Jilin University, Changchun, Jilin, 130021, P.R. China, e-mail: yhtanedu.cn.
Corresponding authors: Ji-Fan Hu, Palo Alto Veterans Institute for Research, Palo Alto, CA 94304, USA, Tel: 650-852-3275, Fax: 650-849-1213, e-mail: jifanedu or hujifanedu.cn; or Sujun Gao, Hematology Department, First hospital of Jilin University, Changchun, Jilin, 130021, P.R. China, e-mail: sjgaoedu.cn; or Yehui Tan, Hematology Department, First hospital of Jilin University, Changchun, Jilin, 130021, P.R. China, e-mail: yhtanedu.cn.