Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(3):1093-1109. doi:10.7150/ijbs.85443 This issue Cite
Research Paper
Promotion of ROS-mediated apoptosis, G2/M arrest, and autophagy by naringenin in non-small cell lung cancer
Tsung-Ming Chang1,2, Miao-Ching Chi3,4,5,6, Yao-Chang Chiang3,7, Chieh-Mo Lin3,5,8, Mei-Ling Fang9,10, Chiang-Wen Lee3,4,6,11 ![]() , Ju-Fang Liu12,13,14
, Ju-Fang Liu12,13,14 ![]() , Yu Ru Kou2,15
, Yu Ru Kou2,15 ![]()
1. School of Dental Technology, College of Oral Medicine, Taipei Medical University, Taipei 11031, Taiwan.
2. Department and Institute of Physiology, College of Medicine, National Yang-Ming Chiao Tung University, Taipei 11221, Taiwan.
3. Department of Nursing, Division of Basic Medical Sciences, and Chronic Diseases and Health Promotion Research Center, Chang Gung University of Science and Technology, Chiayi 61363, Taiwan.
4. Department of Safety Health and Environmental Engineering, Ming Chi University of Technology, New Taipei City 24301, Taiwan.
5. Division of Pulmonary and Critical Care Medicine, Chang Gung Memorial Hospital, Chiayi 61363, Taiwan.
6. Department of Respiratory Care, Chang Gung University of Science and Technology, Chiayi 61363, Taiwan.
7. Research Center for Industry of Human Ecology and Research Center for Chinese Herbal Medicine, Chang Gung University of Science and Technology, Taoyuan 33303, Taiwan.
8. Graduate Institute of Clinical Medical Sciences, College of Medicine, Chang Gung University, Taoyuan 33302, Taiwan.
9. Center for Environmental Toxin and Emerging-Contaminant Research, Cheng Shiu University, Kaohsiung 83347, Taiwan.
10. Super Micro Research and Technology Center, Cheng Shiu University, Kaohsiung 83347, Taiwan.
11. Department of Orthopaedic Surgery, Chang Gung Memorial Hospital, Chiayi 61363, Taiwan.
12. School of Oral Hygiene, College of Oral Medicine, Taipei Medical University, Taipei 11031, Taiwan.
13. Translational Medicine Center, Shin-Kong Wu Ho-Su Memorial Hospital, Taipei 11101, Taiwan.
14. Department of Medical Research, China Medical University Hospital, China Medical University, Taichung 40402, Taiwan.
15. Department of Medical Research, Hualien Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, Hualien 97002, Taiwan.
Received 2023-4-19; Accepted 2023-12-17; Published 2024-1-21
Abstract

Background: As lung cancer is the leading cause of cancer death worldwide, the development of new medicines is a crucial endeavor. Naringenin, a flavanone derivative, possesses anti-cancer and anti-inflammatory properties and has been reported to have cytotoxic effects on various cancer cells. The current study investigated the underlying molecular mechanism by which naringenin induces cell death in lung cancer.
Methods: The expression of apoptosis, cell cycle arrest, and autophagy markers in H1299 and A459 lung cancer cells was evaluated using a terminal deoxynucleotidyl transferase dUTP nick end labeling assay (TUNEL), Western blot, Annexin V/PI stain, PI stain, acridine orange staining, and transmission electron microscopy (TEM). Using fluorescence microscopy, DALGreen was used to observe the degradation of p62, a GFP-LC3 plasmid was used to evaluate puncta formation, and a pcDNA3-GFP-LC3-RFP-LC3ΔG plasmid was used to evaluate autophagy flux. Furthermore, the anti-cancer effect of naringenin was evaluated in a subcutaneous H1299 cell xenograft model.
Results: Naringenin treatment of lung cancer cells (H1299 and A459) reduced cell viability and induced cell cycle arrest. Pretreatment of cells with ROS scavengers (N-acetylcysteine or catalase) suppressed the naringenin-induced cleavage of apoptotic protein and restored cyclin-dependent kinase activity. Naringenin also triggered autophagy by mediating ROS generation, thereby activating AMP-activated protein kinase (AMPK) signaling. ROS inhibition not only inhibited naringenin-induced autophagic puncta formation but also decreased the ratio of microtubule-associated proteins 1A/1B light chain 3 II (LC3II)/LC3I and activity of the AMPK signaling pathway. Furthermore, naringenin suppressed tumor growth and promoted apoptosis in the xenograft mouse model.
Conclusion: This study demonstrated the potent anti-cancer effects of naringenin on lung cancer cells, thereby providing valuable insights for developing small-molecule drugs that can induce cell cycle arrest, apoptosis, and autophagic cell death.
Keywords: human lung cancer, naringenin, apoptosis, ROS, autophagy
Introduction
Lung cancer is the most common form of cancer [1, 2]. The two types of lung cancer, namely small cell (SCLC, 20%) and non-small cell (NSCLC, 80%), differ in terms of symptoms and treatments [3]. Treatment options for NSCLC largely depend on cancer stage, cancer cell subtype, and patient's physical condition [4]. Whether adjuvant or neoadjuvant therapy is administered, chemotherapy remains a standard treatment for almost all NSCLC patients. However, the cytotoxicity of chemotherapy medicines on normal cells reduces their clinical beneficial effects [5, 6]. Thus, there is a pressing need to develop new anti-cancer medication.
Small molecule compounds targeting programmed cell death in human cancers have rapidly progressed [7]. Programmed cell death is a process in which cells following stimulation regulate spontaneous as well as programmed death through a series of signaling pathways to maintain homeostasis, such as autophagy-dependent cell death and apoptosis [8-11]. Autophagy involves the formation of autolysosomes to maintain cellular homeostasis in response to stress. However, in times of excessive damage, cells initiate programmed autophagic cell death [12, 13]. In cancer progression, autophagy plays a dual role: conferring tolerance to adverse growth and enhancing chemotherapeutic resistance [14, 15]. In contrast, various natural extracts have shown anti-cancer effects by inducing autophagic death in cancer cells [16-18]. These effects of which in inducing apoptosis and autophagy are more pronounced in oral and liver cancer cells [19-21]. Therefore, it is crucial to determine whether candidate anti-cancer compounds that induce apoptosis also simultaneously trigger autophagic death.
Naringenin is a citrus flavonoid with anti-inflammatory and anti-viral properties. Previous studies have demonstrated its potential in addressing several lung diseases, including chronic obstructive pulmonary disease, asthma, and coronavirus disease 2019 [22-24]. Beyond its impact on respiratory conditions, naringenin exhibits remarkable anti-cancer effects, marked by inhibiting cancer cell proliferation and promoting cellular apoptosis [25-27]. Naringenin has been shown to suppress the progression of gastric cancer cells by upregulating apoptosis-related proteins, Bax and cleaved caspase-3, while reducing Bcl-2 [27]. In breast cancer cells, naringenin arrests the cell cycle in the G2/M phase, thereby inhibiting cancer cell proliferation and promoting apoptosis [28]. Importantly, studies have elucidated the potential of naringenin in lung cancer, including reducing cellular oxidation and decreasing the initiation and metastasis of lung cancer cells [29-32]. Moreover, naringenin can exert its anti-cancer potential by increasing oxidative stress. In pancreatic cancer and placental choriocarcinoma, naringenin induces apoptosis by generating reactive oxygen species (ROS) [27, 33-35]. Naringenin also promotes ROS-induced cell cycle arrest in epidermoid carcinoma cells [36]. In addition, naringenin protects against palmitate-induced umbilical vein endothelial cell damage and β-amyloid-induced neurotoxicity by activating autophagy [37, 38]. Despite numerous studies reporting the anti-cancer activity of naringenin, the relationship between naringenin-induced autophagy and apoptosis in lung cancer has yet to be elucidated.
Our findings in the current study indicate that naringenin promoted cell cycle arrest and apoptosis in lung cancer cells. It also increased acidic organelles, autolysosomes, and autophagic cell death through the AMP-activated protein kinase (AMPK)/mammalian target on the rapamycin (mTOR) signaling pathway. Finally, we confirmed that ROS-mediated naringenin-induced apoptosis and autophagy in NSCLC.
Materials and Methods
Chemicals
Primary antibodies specific to mTOR were purchased from Abcam (Cambridge, MA, USA). Akt, phospho-mTOR (Ser2448), phospho-Akt (Ser473), phospho-AMPKα (Thr172), AMPKα, cleaved caspase 3, and cleaved caspase 9 (Cell Signaling; Danvers, MA, USA); Bcl-XL, Bcl-2, Bax, Bak, poly ADP-ribose polymerase (PARP), p62, microtubule-associated proteins 1A/1B light chain 3B (LC3B), and GAPDH (Genetex; Irvine, CA, USA); and cyclin-dependent kinase 1 (CDK1) and cyclin B1 (Merck Millipore; Burlington, MA, USA). Anti-rabbit polyclonal and anti-mouse monoclonal antibodies were purchased from Santa Cruz Biotechnology (Dallas, Texas, USA). ROS scavengers N-acetylcysteine (NAC) and H2O2-scavenging enzyme catalase were purchased from Merck Millipore (Burlington, MA, USA). Pan-caspase inhibitor z-VAD-FMK was purchased from R&D Systems (Minneapolis, MN, USA). Naringenin (N5893) and all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
Cell lines used in this study were as follows: H1299 p53-null human NSCLC cells (American Type Culture Collection; Manassas, VA, USA), A549 p53 wildtype human NSCLC cells, and MRC-5 human fetal normal lung fibroblasts (Bioresource Collection and Research Center; Hsinchu, Taiwan). The cells were maintained according to the recommendations from the suppliers: H1299 (Roswell Park Memorial Institute 1640 medium), A549 (Ham's F-12 nutrient mixture), and MRC-5 (Eagle's minimum essential medium). All cell lines were supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin and incubated at 37° C under 5% CO2 in air.
Cell viability assay
Cell viability was assessed using a cell counting kit-8 (CCK-8; Sigma-Aldrich, St. Louis, MO, USA). Cells (1×104) were seeded in 48-well plates and treated with naringenin (25-500 μM) or pretreated with ROS scavengers (catalase, 50 U/mL; NAC, 1 mM), autophagy inhibitors (bafilomycin A1, 100 nM; chloroquine, 50 μM; 3-methyladenine, 100 μM), or a pan-caspase inhibitor (z-VAD-FMK, 20 μM) for 1 h, and then treated with naringenin (250 μM) for another 24 h. Subsequently, the cells were incubated in media with CCK-8 solution (10 μL/well) at 37°C for 4 h. The optical density was measured using a spectrophotometer at 450 nm (BioTek, Winooski, VT, USA).
Colony formation assay
Cells (1.5×105) were seeded in 6-well plates with a medium containing naringenin (25-500 μM). After 6 h, cells (1×103) were transferred to a new 6-well plate to undergo culturing in naringenin-free medium containing 10% FBS. The culture medium was changed at intervals of three days for 14 days, after which the cells were fixed and stained with 0.05% crystal violet solution. Colony-forming cells were then imaged. Subsequently, an acetic acid solution (33% v/v) was added to the plates, and the absorbance was measured at 550 nm using a microplate reader.
Apoptosis assay
An Annexin V/PI (Sigma-Aldrich, St. Louis, MO, USA) assay was used to identify apoptotic and necrotic cells following incubation with naringenin (100-500 μM) for 24 h. Following the manufacturer's instructions, live cells were harvested and stained (1 μg/mL PI and 0.025 μg/mL FITC-conjugated annexin V) for 15 min in the dark at room temperature, before analysis using a flow cytometer (Accuri C5, BD, East Rutherford, NJ, USA). In the resulting dot plots, the X-axis represents the intensity of green fluorescence (Annexin V) and the Y-axis represents the intensity of red fluorescence (PI). Cell population distribution analysis was divided into four quadrants: cells that were Annexin V-/PI-, Annexin V+/PI-, Annexin V-/PI+, and Annexin V+/PI+, respectively, represented normal state, early apoptosis, necrosis, and late apoptosis.
DNA fragmentation and chromatin condensation analysis
DNA damage and chromatin condensation were monitored using 4,6-diamidino-2-phenylindol staining (DAPI; Merck Millipore, Burlington, MA, USA) and terminal deoxynucleotidyl transferase dUTP nick end labeling assays (TUNEL; BD Biosciences Clontech, Palo Alto, CA, USA). Cells were incubated with naringenin (25-500 μM) for 24 h. After fixation and permeabilization, cells were incubated with a TUNEL reaction mixture, and DNA strand breaks were observed using a Nikon Eclipse Ti fluorescence microscope (Nikon, Tokyo, Japan). In chromatin condensation analysis, fixed cells were incubated with DAPI solution (1 μg/ml) for 5 min, and the nucleus morphology was observed using a fluorescence microscope.
ROS production analysis
Intracellular ROS production was detected via flow cytometry using H2DCFDA (Thermo Fisher, Waltham, MA, USA) after treating cells (5×105) to naringenin at various concentrations with H2DCFDA at a concentration of 1 μM at 37°C for 30 min. And then, live cells were harvested and removed residual media. In the resulting histogram plots, the X-axis represents the intensity of green fluorescence (2', 7'-DCF) and the Y-axis represents cell counts. The area under the curve is the mean green fluorescence value representing intracellular ROS production of the cells.
Mitochondria membrane potential
Mitochondrial membrane potential (MMP) and permeability transition, both of which are related to apoptosis, were monitored using JC-1 dye (Thermo Fisher, Waltham, MA, USA). Cells treated with naringenin (25-500 μM) for 24 h were incubated with media containing JC-1 (5 μg/mL) for 30 min. Images were acquired using a fluorescence microscope. The red/green fluorescence ratio was measured respectively at Ex/Em 585/590 nm and Ex/Em 514/529 nm using a microplate reader (Varioskan LUX; Thermo Fisher, Waltham, MA, USA).
Immunoblotting analysis
Proteins were resolved using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to immobilon polyvinyldifluoride membranes (Merck Millipore, Burlington, MA, USA). The membranes were blocked and probed using primary antibodies (1:1000) at 4°C overnight. The blots were then incubated with anti-rabbit peroxidase-conjugated secondary antibodies (1:10000) at room temperature for 1 h, after which signals were detected using enhanced chemiluminescence and visualized with UVP chemiluminescence detection system (Analytik Jena US, CA, USA).
Cell cycle analysis
Cells (5×105) were incubated in 6-well plates with naringenin (25-500 μM) for 24 h. Cells were harvested and fixed in ethanol at -20 °C for 2 h. After removing ethanol, cells were stained using a propidium iodide (PI) solution (100 μg/mL; Sigma-Aldrich, St. Louis, MO, USA), followed by flow cytometry. In the resulting histogram, the X-axis represents PI fluorescence intensity and the Y-axis represents the number of cells.
Detection of vesicle formation
Cells (5×105) treated with naringenin (25-500 μM) for 24 h were imaged, and the number of cells with vesicles was counted. Data is presented as a percentage of the number of cells containing vesicles and the total number of cells.
Detection of acidic vesicular organelles
The formation of acidic vacuoles (a hallmark of autophagy) in NSCLC cells was detected using acridine orange staining (1 µg/mL; Sigma-Aldrich, St. Louis, MO, USA). AO emits red fluorescence when excited in an acidic environment and is used to detect the formation of acidic vesicles. Following treatment with naringenin, viable cells were stained with AO for 30 min and the development of acidic organelles was examined using a fluorescence microscope.
Transmission electron microscopy
H1299 cells (5×105) treated with naringenin (250 μM) for 6 h were first washed to remove the medium and trypsin, after which the suspended cells were immediately fixed in 70% Karnovsky fixative at 4°C until embedding. The cells were then observed under a JEOL JEM-1400 transmission electron microscope (Tokyo, Japan).
DALGreen autophagy detection
Cells (4×104) were cultured in a 6-well plate and incubated with DALGreen solution (1 μM; Dojindo, Kumamoto, JP) for 30 min, washed twice with culture medium, and then cultured in medium with or without naringenin (250 μM) for 6 h. DALGreen binding to autolysosomes was observed using a fluorescence microscope.
Autophagy sensor p62-GFP assay
Adherent cells (4×104) were incubated in 6-well plates with 12 μL of p62-GFP reagent (1×108 PremoTM particles/mL; Thermo Fisher, Waltham, MA, USA) for at least 16 h. The cells were then incubated in medium with or without 250 μM naringenin for 6 h. p62-GFP was detected using a fluorescent microscope.
GFP-LC3 puncta formation and autophagic flux assay
Autophagic activity was assessed in terms of GFP-LC3 fluorescence intensity, GFP-LC3 quench, and GFP-LC3 puncta formation. To measure autophagic flux via GFP-LC3 fluorescence intensity and quench, cells were transfected with GFP-LC3 plasmid or pcDNA3-GFP-LC3-RFP-LC3ΔG (Addgene, MA, USA) using Lipofectamine 3000 transfection reagent (Thermo Fisher, Waltham, MA, USA) for 24 h. Transfected cells (5×105) were then pretreated with or without the autophagy inhibitor bafilomycin A1, followed by incubation in media containing naringenin (250 μM). Fluorescence intensity was evaluated using flow cytometry and a fluorescence microscope. The fluorescence intensity of GFP-LC3 was normalized to RFP-LC3ΔG, and the percentage of each group relative to the control group was quantified. To observe puncta formation, transfected cells (5×105) were pretreated with ROS inhibitors prior to incubation in medium with or without naringenin (250 μM). Fluorescence intensity was evaluated using a fluorescence microscope. Puncta formation is presented as the percentage of the number of cells with puncta and the total number of cells.
Xenograft assay
All animal experiments were performed following a protocol approved by the Institutional Animal Care and Use Committees of Shin‐Kong Wu Ho‐Su Memorial Hospital (Taipei, Taiwan; IACUC Approval No: 111MOST007). Male 4-week-old Nu/Nu mice were purchased from Lasco (BioLASCO Co., Ltd., Taipei, Taiwan) and housed under pathogen‐free conditions. H1299 cells (2×106 cells in 100 μL) were subcutaneously injected into the dorsal region of the mice. Tumors were allowed to develop until they reached approximately 100 mm3 before naringenin treatment was initiated. Mice were randomly assigned to groups of 10. We referred to studies by Qin et al. and Zhang et al. on the anti-cancer effect of naringenin in our selection of doses of 100 mg/kg to 200 mg/kg in vivo for experiments [39, 40]. In our study, mice were administered a vehicle or naringenin (100 mg/kg) daily for 21 days (10 mice/group) to evaluate the inhibitory effects on tumor growth [39, 40]. The volume of the implanted tumor on the dorsal side of the mice was measured every three days using a caliper based on the following formula: V = (L × W2)/2, where V is the volume (mm3), L is the largest diameter (mm), and W is the smallest diameter (mm). After 21 days, the mice were sacrificed via CO2 inhalation.
Immunohistochemistry analysis
Tissue sections (3 μm) were prepared from paraffin-embedded tissues, deparaffinized in xylene, rehydrated in a series of graded alcohols, and washed in deionized water. After performing antigen retrieval, the NovolinkTM polymer detection system (Leica, Wetzlar, Germany) was used to conduct IHC assay according to the manufacturer's instructions. Tissue sections were incubated overnight at 4°C with a primary antibody specific for cleaved caspase 3 (1:200) and then washed with PBST. Novolink Polymer was applied at room temperature for 1 h. The stained sections were detected using 3,3′-diaminobenzidine tetrahydrochloride, counterstained with hematoxylin, and observed under a light microscope. The H score was calculated as the sum of the signal intensity (from 0 to 3) multiplied by the percentage of the total area and multiplied by 100. H scores were independently interpreted by three investigators.
Statistical analysis
SigmaStat 3.5 statistical software was used for processing experimental data. Values were reported as mean ± standard deviation (SD). Statistical comparisons between two samples were performed using the two-tailed test. In contrast, comparisons among more than two groups were performed using a one-way ANOVA with Fisher-LSD post-hoc test. In all cases, p ≤ 0.05 was considered significant.
Results
Naringenin suppressed cancer cell proliferation and induced cell apoptosis
We first examined the cytotoxic effects of naringenin (Figure 1A) in normal lung fibroblasts (MRC-5) and lung cancer cells (H1299 and A549) using cell viability assay and cell proliferation assay. We found that naringenin significantly decreased the survival rates of H1299 and A549 cells in a dose-dependent manner, with respective IC50 values of 330.7 μM and 400.4 μM (Figure 1B); note that the viability of MRC-5 cells was not affected. In colony formation assays, naringenin significantly suppressed cancer cell proliferation in both NSCLC cell lines, but it did not in MRC-5 cells (Figure 1C-D and S1). These results suggest that naringenin possessed anti-cancer effects on lung cancer cells without noticeable adverse effects on normal cells.
Naringenin suppressed proliferation and induced apoptosis in non-small cell lung cancer cells. (A) Chemical structure of naringenin. (B) MRC-5, H1299, and A549 cells were treated with naringenin (100-500 μM) for 24 h, after which cell viability was assessed by CCK-8 assay (n=4). (C) H1299 and (D) A549 cells were treated with naringenin (25-500 μM) for 6 h and then incubated in naringenin-free medium for a further 14 days, after which cell reproductive viability was assessed by colony formation assay (n=4). (E) H1299 and (F) A549 cells were treated with naringenin (100-500 μM) for 24 h and then stained with Annexin V/PI; cell death was analyzed by flow cytometry (n=4). (G) H1299 and (H) A549 cells were treated with naringenin (25-500 μM) for 24 h, and then visualized by optical microscopy. DNA fragmentation and chromatin condensation were respectively examined by TUNEL assay and DAPI staining via fluorescence microscopy. Untreated cells were used as control. Scale bar = 20 μm. Results are shown as means ± SD. *p < 0.05 compared with untreated control.
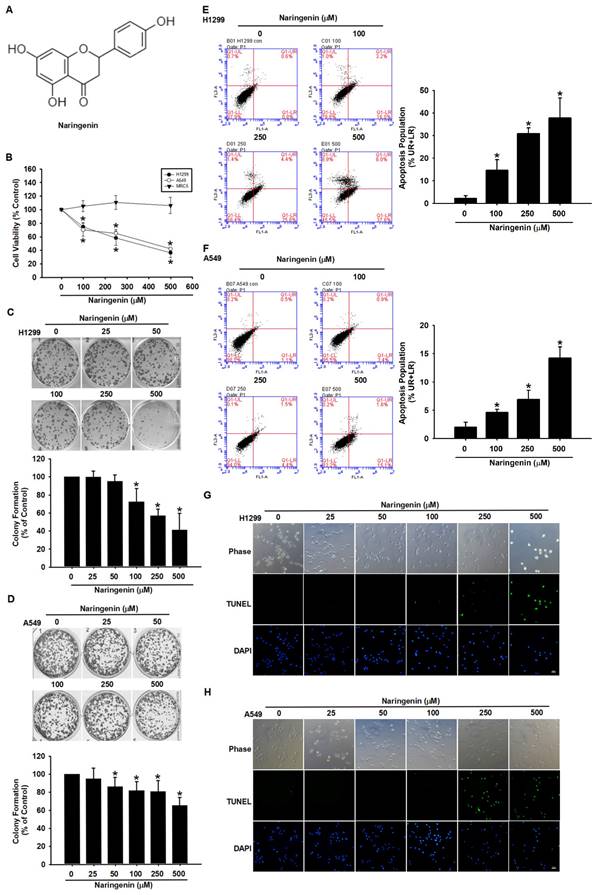
The mechanism underlying naringenin-induced cell death was explored using Annexin V/PI double staining. Annexin V binds to phosphatidylserine, which is exposed on the outer membrane during early apoptosis, whereas PI stains the DNA of dead cells. The percentage of apoptotic H1299 and A549 cells significantly increased after incubation with naringenin for 24 h (H1299: 100 μM, 14.6±4.71%; 250 μM, 30.9±2.60%; 500 μM, 37.8±8.81%; and A549: 100 μM, 4.7±0.54%; 250 μM, 7.0±1.59%; 500 μM, 14.2±1.99%) (Figure 1E-F). An examination of the modes of apoptotic cell death using DAPI and TUNEL staining revealed that nuclei fragmentation and chromatin condensation dramatically increased following naringenin treatment (Figure 1G-H). Taken together, these results indicate that naringenin caused non-small cell lung cancer cell death by promoting apoptosis and inhibiting cell proliferation.
Naringenin promoted apoptosis through ROS production and MMP perturbation
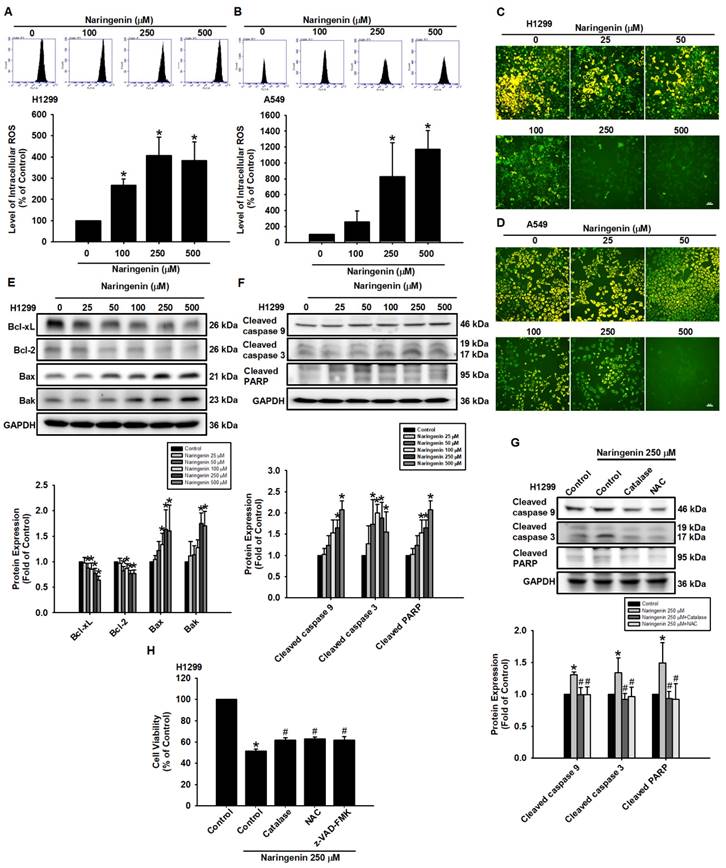
The relationship between ROS and apoptosis has been addressed in previous studies [41, 42]. In the present study, we sought to further investigate the anti-cancer effects of naringenin by examining ROS production and the mechanism of naringenin-induced cell death in NSCLC cells. Treatment of A549 and H1299 cells with naringenin significantly increased intracellular ROS production levels in a dose-dependent manner (Figure 2A-B) while reducing MMP (Figure 2C-D and S2). The contribution of MMP depolarization on intrinsic apoptosis was explored by examining the expression of the apoptosis-related Bcl-2 family proteins Bcl-2 and Bcl-xL, which are responsible for stabilizing the integrity of the mitochondrial outer membrane [43]. Stimulating NSCLC cells with naringenin decreased the expression of Bcl-xL and Bcl-2 while promoting the expression of Bax and Bak proteins, which are known to perforate the mitochondrial outer membrane to initiate apoptosis (Figure 2E and S5A). The loss of MMP led to a corresponding cleavage of caspase 9, caspase 3, and PARP (Figure 2F and S5B). Pretreatment of cells with ROS scavengers NAC and catalase significantly reduced naringenin-induced cleavage of caspase 9, caspase 3, and PARP while slightly but significantly restoring naringenin-mediated cell death (Figure 2G-H). Taken together, these results indicate that naringenin promoted apoptosis in lung cancer cells by inducing ROS.
Naringenin induced intracellular ROS and promoted apoptosis by altering MMP in non-small cell lung cancer cells. (A) H1299 and (B) A549 cells were treated with naringenin (100-500 μM) and then inculcated with H2DCFDA for 30 min, after which ROS production was evaluated by flow cytometry (n=4). (C) H1299 and (D) A549 cells were treated with naringenin (25-500 μM) for 24 h and then incubated with JC-1 for 30 min. MMP was observed by fluorescence microscopy (n=4). Scale bar = 20 μm. (E-F) H1299 cells were treated with naringenin (25-500 μM) for 8 h; by Western blot, expression levels of (E) Bcl-xL, Bcl-2, Bak, and Bax and (F) Cleaved caspase 3, cleaved caspase 9, and cleaved PARP were examined (n=4). (G) H1299 cells were pretreated with ROS scavengers, catalase (H2O2-scavenging enzyme, 50 U/mL) or N-acetylcysteine (NAC, 1 mM), for 1 h and then treated with naringenin (250 μM) for 8 h; protein expression levels of cleaved caspase 3, cleaved caspase 9, and cleaved PARP were examined by Western blot (n=4). (H) H1299 cells were pretreated with catalase (50 U/mL), NAC (1 mM), or pan-caspase inhibitor z-VAD-FMK (20 μM) for 1 h and then treated with naringenin (250 μM) for 24 h; cell viability was assessed using a CCK-8 assay (n=4). Untreated cells were used as control. Results are shown as means ± SD. *p < 0.05 compared with untreated control. #p < 0.05 compared with group treated with naringenin.
Naringenin induced cell cycle arrest via ROS production in NSCLC cells
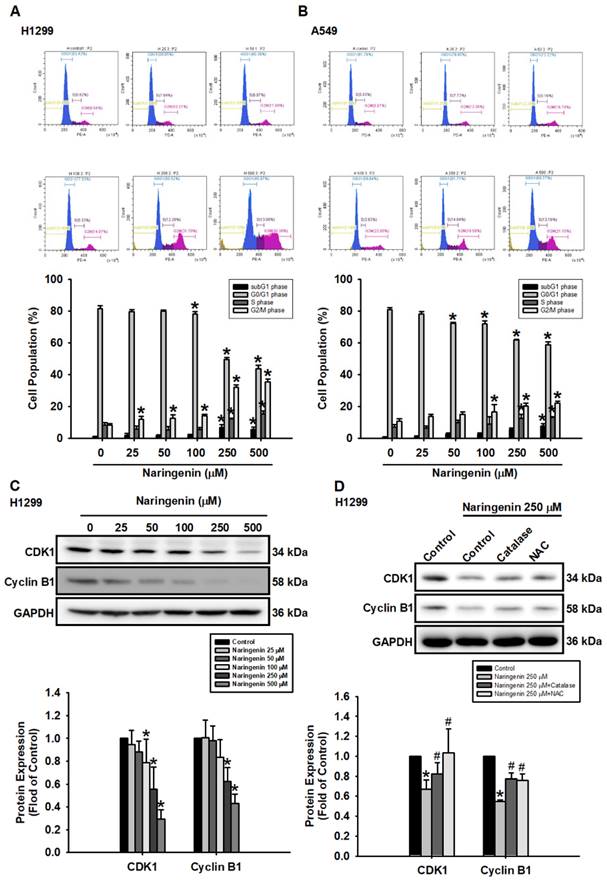
Treating H1299 and A549 cells with naringenin at various concentrations (25-500 μM) for 24 h significantly shifted the distribution of cells from the G0/G1 phase to the subG1 and G2/M phase (Figure 3A and 3B). G2/M phase arrest was associated with the downregulation of CDK1 and cyclin B1 (Figure 3C and S6A), the effects of which were reversed following treatment with ROS scavengers (Figure 3D and S6B). Taken together, these results confirm that the effects of naringenin in reducing cell viability were accompanied by the arrest of the G2/M cell cycle and the downregulated expression of CDK1 and cyclin B1 via ROS production.
Naringenin induced cell cycle arrest via ROS production in non-small cell lung cancer cells. (A) H1299 and (B) A549 cells were treated with naringenin (25-500 μM) for 24 h and then stained with PI for 30 min; cell cycle was evaluated by flow cytometry (n=4). (C) H1299 cells were treated with naringenin (25-500 μM) for 6 h; CDK1 and cyclin B1 protein expression was examined by Western blot (n=4). (D) H1299 cells were pretreated with ROS scavengers, catalase (50 U/mL) or NAC (1 mM), for 1 h and then treated with naringenin (250 μM) for 6 h; protein expression levels of CDK1 and cyclin B1 were examined by Western blot (n=4). Untreated cells were used as control. Results are shown as means ± SD. *p < 0.05 compared with untreated control. #p < 0.05 compared with group treated with naringenin.
Naringenin promoted autophagy through ROS-induced activation of AMPK in NSCLC cells
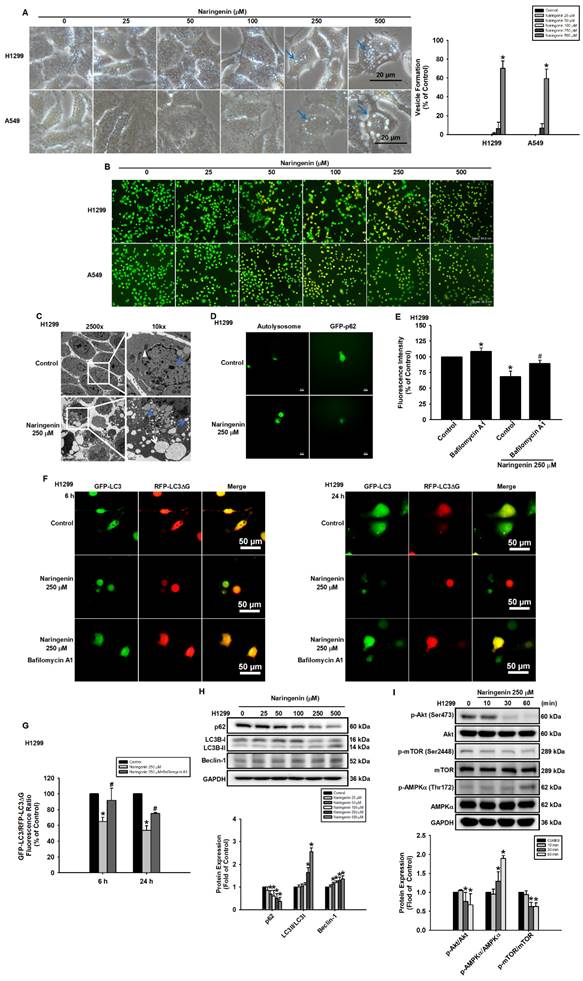
Previous studies have reported that naringenin induces autophagy in neural cells and mice [44, 45]. This prompted us to examine whether mechanisms associated with autophagy were altered in lung cancer cells in response to naringenin. Microscopy image analysis revealed the presence of vesicles, which are hallmarks of autophagy, following naringenin treatment in H1299 and A549 cells (Figure 4A). Acridine orange staining further revealed the formation of acidic autophagic vesicles, as evidenced by bright red staining (Figure 4B). Following the confirmation of autolysosomes in naringenin-treated H1299 cells by TEM (Figure 4C), fluorescence microscopy image analysis was used to clarify changes in autophagy molecular markers. We found that naringenin induced autolysosome expression and decreased p62 expression (Figure 4D), which indicated substantial autophagy activity. Pretreatment with an autophagy inhibitor (bafilomycin A1) blocked naringenin-induced autolysosome formation and reduced the degradation of GFP-LC3 (Figure 4E and S3). In order to examine autophagy flux, a pcDNA3-GFP-LC3-RFP-LC3ΔG plasmid was used as a fluorescent probe. The cleavage of the C-terminus of LC3 by autophagy-related 4 cysteine peptidase leads to the cleavage of the GFP-LC3-RFP-LC3ΔG tandem protein into GFP-LC3 and RFP-LC3ΔG. GFP-LC3 can then conjugate to phosphatidylethanolamine and is subsequently degraded by autolysosomes, while RFP-LC3ΔG cannot conjugate to phosphatidylethanolamine and remains in the cytosol. This served as an internal control to evaluate autophagic flux by observing a decrease in GFP fluorescence compared to RFP fluorescence. The quenching of green fluorescence following 6 h of naringenin treatment, indicating the degradation of GFP-LC3. This degradation persisted until at least 24 h (Figure 4F). However, pretreatment of H1299 cells with bafilomycin A1 blocked naringenin-induced green fluorescence quenching, providing further evidence that naringenin increased autophagy flux via autolysosome formation (Figure 4F and 4G). Western blot analysis revealed that naringenin reduced the expression of p62 as well as increased the expression of Beclin-1 and the LC3II/LC3I ratio (Figure 4H and S7A). As researchers have speculated that AMPK/Akt/mTOR signaling plays an essential role in autophagy by regulating autolysosome formation [46], we investigated whether AMPK/Akt/mTOR signaling was involved in naringenin-induced autophagy. Compared to the control group, naringenin significantly decreased Akt and mTOR phosphorylation and promoted AMPKα phosphorylation in NSCLC cells (Figure 4I and S7B), indicating that naringenin altered the expression of autophagy-related proteins by activating AMPKα and inactivating the Akt and mTOR signaling pathways. This could explain the production of autolysosomes and the promotion of autophagic activity in NSCLC cells.
Naringenin promoted autophagy in non-small cell lung cancer cells. (A) Left: H1299 and A549 cells were treated with naringenin (25-500 μM) for 24 h, after which cell morphology (blue arrows: vesicles) was observed by optical microscopy. Scale bar = 20 μm. Right: Quantitative analysis of vesicle formation (n=4). (B) H1299 and A549 cells were treated with naringenin (25-500 μM) for 24 h and then stained with acridine orange for 30 min; acidic organelles were visualized by fluorescence microscopy. Scale bar = 50 μm. (C) H1299 cells were treated with naringenin (250 μM) for 6 h, after which autolysosomes (white arrow) were observed by TEM; white triangle indicates mitochondria, blue triangles indicate lysosomes, the blue arrows indicate vacuoles, and the white arrows indicate autolysosomes. 2,500×: Scale bar = 5 μm. 10,000×: Scale bar = 1 μm. (D) H1299 cells were incubated with DALGreen solution or p62-GFP reagent and then treated with naringenin for 6 h, after which autolysosomes and GFP-p62 were observed by fluorescence microscopy. Scale bar = 20 μm. (E) H1299 cells transfected with GFP-LC3 were pretreated with autophagy inhibitor bafilomycin A1 (100 nM) for 1 h and then treated with naringenin (250 μM) for 6 h; autophagic activity was assessed by flow cytometry (n=4). (F) H1299 cells transfected with pcDNA3-GFP-LC3-RFP-LC3ΔG were pretreated with autophagy inhibitor bafilomycin A1 (100 nM) for 1 h and then treated with naringenin (250 μM) for 6 h and 24 h, after which autophagic flux was observed by fluorescence microscopy (n=4). Scale bar = 50 μm. (G) Quantitative analysis of GFP-LC3/RFP-LC3△G fluorescence ratio (n=4). (H) H1299 cells were treated with naringenin (25-500 μM) for 6 h; protein expression of p62 and Beclin-1 as well as the LC3II/LC3I ratio were examined by Western blot (n=4). (I) H1299 cells were treated with naringenin (250 μM) for the indicated times, after which the phosphorylation of Akt, mTOR, and AMPKα was examined by Western blot (n=4). Untreated cells were used as control. Results are shown as means ± SD. *p < 0.05 compared with untreated control. #p < 0.05 compared with control treated with naringenin.
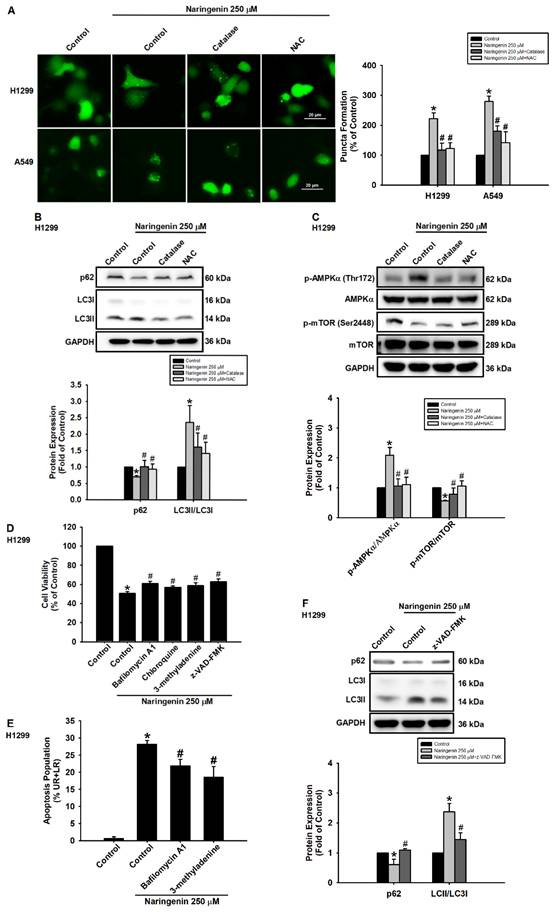
The role of ROS in naringenin-induced autophagy via the formation of puncta was explored by transiently transfecting H1299 and A549 cells with a plasmid, GFP-LC3 plasmid and then pretreating the cells with or without ROS inhibitors before naringenin treatment. Pretreatment with ROS scavengers, catalase and NAC, suppressed the naringenin-induced formation of GFP-LC3 puncta (Figure 5A). In NSCLC cells, the ROS inhibitors also induced naringenin-reduced p62 expression, decreased the LC3II/LC3I ratio (Figure 5B and S8A), and reversed naringenin-induced AMPKα phosphorylation and naringenin-suppressed mTOR phosphorylation (Figure 5C and S8B). We next sought to confirm the biological role of naringenin in the regulation of autophagic flux in lung cancer cells. Pretreatment of H1299 cells with bafilomycin A1 (an inhibitor of autophagosome-lysosome fusion), chloroquine (CQ, an inhibitor of autophagosomes/lysosomes), and 3-methyladenine (3-MA, an inhibitor of the PI3 complex and autophagosomes) prior to naringenin treatment was shown to modulate naringenin-induced cell death and improve cell viability (Figure 5D). In addition, 3-methyladenine and bafilomycin A1 significantly inhibited naringenin-induced apoptosis in H1299 cells (Figure 5E), and z-VAD-FMK (a pan-caspase inhibitor) reversed the naringenin-induced changes in autophagy proteins (Figure 5F). These results suggest that naringenin-induced autophagy and apoptosis worked synergistically to regulate cell death and that naringenin triggered ROS production to induce autophagy via AMPK/Akt/mTOR signaling, leading to cell death.
Naringenin promoted cell death via ROS-induced autophagy. (A) Left: H1299 and A549 cells transfected with GFP-LC3 were pretreated with ROS scavengers, catalase (50 U/mL) or NAC (1 mM), for 1 h and then treated with naringenin (250 μM) for 6 h; puncta formation was observed by fluorescence microscopy. Scale bar = 20 μm. Right: Quantitative analysis of puncta formation (n=4). (B) H1299 cells were pretreated with ROS scavengers for 1 h and then treated with naringenin (250 μM) for 6 h; expression of p62 protein and LC3II/LC3I ratio were measured by Western blot (n=4). (C) H1299 cells were pretreated with ROS scavengers for 1 h and then treated with naringenin (250 μM) for 1 h; phosphorylation of AMPKα and mTOR was examined by Western blot (n=4). (D) H1299 cells were pretreated with autophagy inhibitors bafilomycin A1 (100 nM), chloroquine (50 μM), or 3-methyladenine (100 μM) as well as pan-caspase inhibitor z-VAD-FMK (20 μM) for 1 h and then treated with naringenin (250 μM) for 24 h; cell viability was assessed by CCK-8 assay (n=4). (E) H1299 cells were pretreated with bafilomycin A1 (100 nM) and 3-methyladenine (100 μM) for 1 h and then treated with naringenin (250 μM) for 24 h; cell apoptosis was assessed by Annexin V/PI stain (n=4). (F) H1299 cells were pretreated with z-VAD-FMK (20 μM) for 1 h and then treated with naringenin (250 μM) for 6 h; expression of p62 and the LC3II/LC3I ratio was measured by Western blot (n=4). Results are shown as means ± SD. *p < 0.05 compared with untreated control. #p < 0.05 compared with group treated with naringenin.
Naringenin suppressed lung cancer growth in a mouse xenograft model
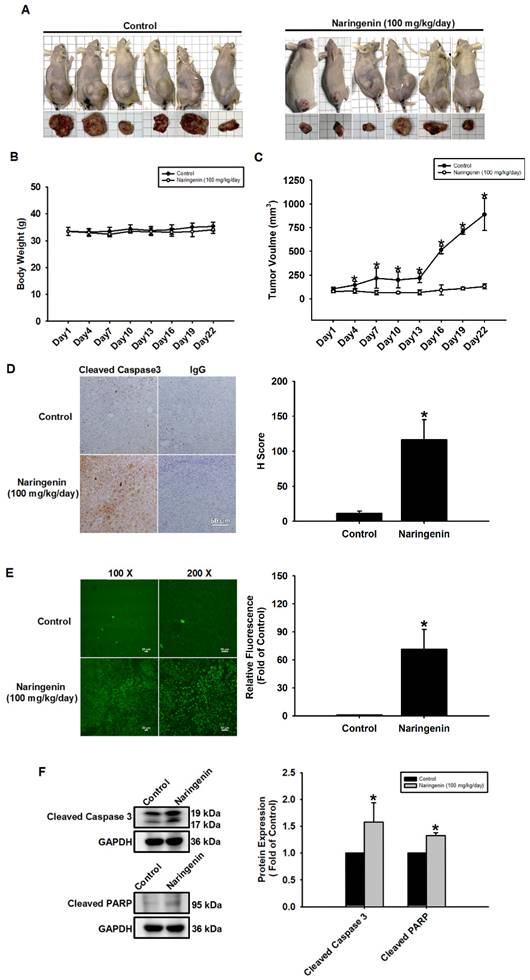
A subcutaneous xenograft model was used to investigate the therapeutic effects of naringenin on lung cancer and tumor growth. Mice were treated daily with a vehicle or naringenin (100 mg/kg) from the day that tumor volume reached approximately 100 mm3. Tumor volume was measured at 3-day intervals using a caliper. Compared to the vehicle-treated control group, naringenin significantly inhibited tumor growth from day 4 (Figure 6A and 6C) with no effects on body weight (Figure 6B). IHC and TUNEL assays of tumor sections revealed that naringenin can promote lung cancer cell apoptosis (Figure 6D and 6E). Western blot analysis of the tumors revealed that the expression of cleaved caspase 3 and cleaved PARP was significantly higher with naringenin treatment (Figure 6F). Taken together, these findings demonstrate the efficacy of naringenin in suppressing tumor growth by promoting apoptosis in H1299 cells in vivo.
Naringenin suppressed tumor growth and induced apoptosis in vivo. (A) Representative images of mice and tumors. (B) Body weight and (C) tumor volume of control and naringenin-treated groups (n=6). (D) Left: Expression of cleaved caspase 3 in control and naringenin-treated tumor tissues was analyzed by IHC. Scale bar = 20 μm. Right: Quantitative analysis of H score of cleaved caspase3. (E) Left: Apoptosis in control and naringenin-treated tumor tissues was analyzed by TUNEL assay. Scale bar = 50 μm. Right: Quantitative analysis of relative fluorescence of TUNEL-positive cells in control and naringenin-treated groups. (F) Expression levels of cleaved caspase 3 and cleaved PARP in control and naringenin-treated tumor tissues were examined by Western blot (n=4). Results are shown as means ± SD. *p < 0.05 compared with untreated control.
Discussion
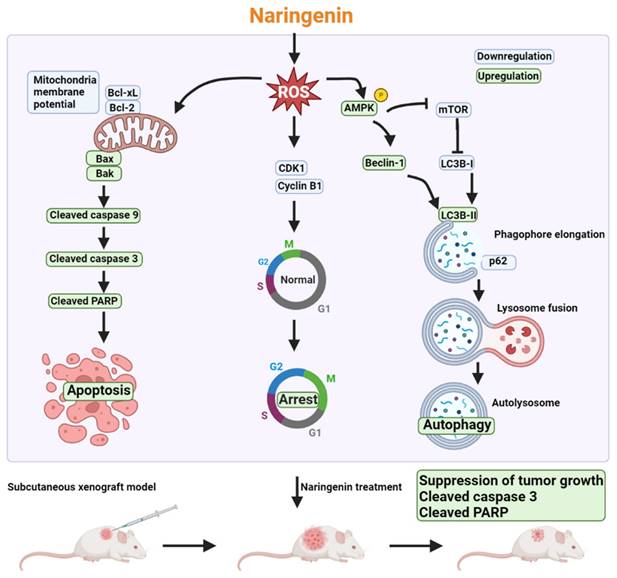
Despite the numerous treatment strategies available for various stages of NSCLC, lung cancer remains the leading cause of cancer death worldwide [2]. Chemotherapy is a standard treatment prescribed to most lung cancer patients; however, the low specificity of chemotherapy drugs lowers their clinical applicability [4]. Various natural products have been shown to exhibit anti-cancer activity by promoting apoptosis or autophagy [47, 48]. Naringenin has demonstrated anti-cancer or adjunctive effects when combined with chemotherapy, such as the inhibition of cell proliferation and the promotion of apoptosis [49-52]. Nonetheless, the mechanisms underlying the anti-cancer effects of naringenin in lung cancer remain unclear. Our results revealed that the potent effects of naringenin on lung cancer can be attributed to promote cell cycle arrest, apoptosis, and autophagy, all of which leads to cell death (Figure 7).
Schematic mechanism of naringenin promoting apoptosis, cell cycle arrest, and autophagy in lung cancer. Triggering of intracellular ROS by naringenin reduces mitochondrial membrane potential (MMP) as well as CDK1 and cyclin B1 expression and activates AMPK. Loss of MMP results in a reduction of anti-apoptotic proteins Bcl-xL and Bcl-2 but an increase in pro-apoptotic proteins Bax and Bak to subsequently initiate the caspase 9/3 cascade. Suppression of CDK1 and cyclin B1 promotes cell cycle arrest in the G2/M phase. Naringenin not only induces AMPK activation but also inhibits mTOR phosphorylation to subsequently trigger autophagic cell death by inducing the expression of Beclin-1 and LC3B-II, degradation of p62, and formation of autolysosome. The anti-cancer effect of naringenin was further confirmed in a subcutaneous xenograft lung cancer model.
Intracellular ROS production has been shown to induce mitochondrial dysfunction and thus apoptosis [53, 54]. The well-documented roles of ROS in promoting cell cycle arrest in many cancers have led to the inclusion of ROS in the design of many anti-cancer drugs [55, 56]. In addition, several chemotherapeutic agents, including gemcitabine, trichostatin A, epigallocate-3-gallate, capsaicin, and benzyl isothiocyanate, aim to increase cellular ROS levels in a bid to enhance the apoptosis of tumor cells [57-62]. Flavonoids such as naringenin act as antioxidants under normal conditions but as potent pro-oxidants in cancer cells that modulate apoptotic pathways [63], such as the inhibition of glutathione reductase activity [64]. In a previous study on human epidermoid cancer cells, naringenin was shown to trigger ROS production and promote cancer cell apoptosis and cell cycle arrest [36]. Similar to Liu et al. [4], we observed a remarkable increase in ROS production following naringenin treatment, resulting in the activation of caspase cascades and the inhibition of cell proliferation (Figure 2G-H). ROS scavengers were also shown to reverse naringenin-induced AMPK signaling and autophagic activity (Figure 5). Our results support previous findings indicating that the effects of naringenin in promoting cell cycle arrest, apoptosis, and autophagy in cancer cells are associated with intracellular ROS production.
During chemotherapy treatment, autophagy acts counterintuitively by maintaining cell homeostasis and promoting cancer cell survival. However, researchers have developed several anti-cancer drugs capable of inducing apoptosis via hyperactive autophagy [65, 66]. In many cancers, flavonoids induce autophagy and apoptosis, as well as inhibit cell progression by modulating ROS [63]. In the current study, naringenin promoted autophagy by inducing ROS production in NSCLC cells. Pretreatment with autophagy inhibitors restored naringenin-suppressed viability in H1299 cells (Figure 5D). The phosphatidylinositol 3-kinase/Akt/mTOR and AMPK pathways are crucial regulator of autophagy, proliferation, and survival of cancer cells [67, 68]. Indeed, we found that naringenin inhibited mTOR phosphorylation and promoted AMPK activation, while pretreatment with ROS scavengers reversed these effects (Figure 5C and S8B). These results suggest that naringenin causes autophagy via the ROS-mediated phosphatidylinositol 3-kinase/Akt/mTOR and AMPK signaling pathway in human NSCLC cells. In Figure 5D, the results further indicated that naringenin causes autophagic cell death.
Interestingly, autophagy inhibitors only slightly but significantly reversed naringenin-induced cell death. The result led us to investigate whether the mechanism of naringenin-induced cell death is related to the interaction between apoptosis and autophagy. Autophagy and apoptosis are inter-connected in the anti-cancer process, exerting synergistic, promotional, and antagonistic effects [69]. In addition, autophagic cell death highly depends on the cell type, the source of signals, and the duration of stimulation [70]. We observed that autophagy inhibitors and apoptosis inhibitors respectively reduced the number of apoptotic cells and autophagy proteins induced by naringenin, thus confirming that naringenin-induced apoptosis and autophagic cell death synergistically (Figure 5E and 5F). Our findings reveal that naringenin promotes cell death in lung cancer cells due to the lethal synergistic effects of ROS stimulating apoptosis and autophagy.
A growing body of evidence suggests that naringenin suppresses tumor growth in mice with breast cancer, glioblastoma, and skin cancer [39, 71, 72]. Previous studies have found that ingesting 600 to 900 mg of naringenin had no significant adverse effects on heart rate or blood pressure. Similarly, no significant adverse effects were observed following the administration of naringenin (up to 100 mM) on rat intestinal epithelial cells, indicating a comparable safety profile in vivo and in vitro studies [73, 74]. In the current study, naringenin significantly decreased tumor volume in vivo and promoted tumor apoptosis. The fact that there was no indication of naringenin cytotoxicity in healthy lung cells and no adverse effects on body weight further supports naringenin as a potent candidate compound in the development of novel cancer treatments (Figure 1B, 6B, and S1).
The reason for this result may be due to the mechanisms of naringenin activity involved the downregulation of CDKs, cyclins, and anti-apoptotic proteins with a corresponding activation of pro-apoptotic proteins in cancer cells. Note that the levels of CDKs and cyclins in normal cells remained at basal levels, i.e., naringenin did not affect cell cycle activity at basal levels. Our findings suggest that in NSCLC cells, naringenin suppressed the expression of CDK1, cyclin B1, and anti-apoptotic proteins as well as upregulated pro-apoptotic pathways. This evidence underscores the pivotal role of naringenin as a promising and versatile candidate in the development of innovative therapeutic strategies for non-small cell lung cancer.
Conclusion
This study confirms the mechanisms of naringenin in inhibiting the proliferation of NSCLC cells and inducing cell death both in vitro and in vivo. Our results have further elucidated the potent anti-cancer effects of naringenin in non-small cell lung cancer and provide strategies for developing small-molecule compounds that induce cell cycle arrest, apoptosis, and autophagy through enhanced ROS production.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by grants from the National Science and Technology Council (NSTC112-2320-B-038-029), the Chang Gung University of Science Foundation grants (ZRRPF6N0011), and Chang Gung Medical Research Program Foundation Grants CMRPF6K0083.
Ethics statement
All animal experiments were carried out in accordance with the protocols approved by the Institutional Ethics Committee of Shin Kung Wu Ho Su Memorial Hospital (IACUC Approval No: 111MOST007).
Data availability
The datasets generated for this study can be accessed upon request to the corresponding author.
Author contributions
Chiang-Wen Lee, Ju-Fang Liu, and Yu Ru Kou: Conceptualization; Investigation. Tsung-Ming Chang, Miao-Ching Chi, and Yao-Chang Chiang: Data curation; Investigation; Methodology. Chieh-Mo Lin, Mei-Ling Fang, Chiang-Wen Lee, and Ju-Fang Liu: Funding acquisition; Supervision. Tsung-Ming Chang, Chiang-Wen Lee, Ju-Fang Liu, and Yu Ru Kou: Writing - original draft; Writing - review and editing.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wu F, Wang L, Zhou C. Lung cancer in China: current and prospect. Curr Opin Oncol. 2021;33:40-6
2. Sung H, Ferlay J, Siegel RL. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209-49
3. Oser MG, Niederst MJ, Sequist LV. et al. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 2015;16:e165-72
4. Liu X, Zhao T, Shi Z. et al. Synergism Antiproliferative Effects of Apigenin and Naringenin in NSCLC Cells. Molecules. 2023 28
5. Chen P, Liu Y, Wen Y. et al. Non-small cell lung cancer in China. Cancer Commun (Lond). 2022;42:937-70
6. de Scordilli M, Michelotti A, Bertoli E. et al. Targeted Therapy and Immunotherapy in Early-Stage Non-Small Cell Lung Cancer: Current Evidence and Ongoing Trials. Int J Mol Sci. 2022 23
7. Peng F, Liao M, Qin R. et al. Regulated cell death (RCD) in cancer: key pathways and targeted therapies. Signal Transduct Target Ther. 2022;7:286
8. Galluzzi L, Senovilla L, Vitale I. et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869-83
9. Zhu H, Luo H, Zhang W. et al. Molecular mechanisms of cisplatin resistance in cervical cancer. Drug Des Devel Ther. 2016;10:1885-95
10. Florea AM, Busselberg D. Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancers (Basel). 2011;3:1351-71
11. Siddik ZH. Biochemical and molecular mechanisms of cisplatin resistance. Cancer Treat Res. 2002;112:263-84
12. Martinez J, Verbist K, Wang R. et al. The relationship between metabolism and the autophagy machinery during the innate immune response. Cell Metab. 2013;17:895-900
13. Rashid HO, Yadav RK, Kim HR. et al. ER stress: Autophagy induction, inhibition and selection. Autophagy. 2015;11:1956-77
14. Wang J, Dong Z, Sheng Z. et al. Hypoxia-induced PVT1 promotes lung cancer chemoresistance to cisplatin by autophagy via PVT1/miR-140-3p/ATG5 axis. Cell Death Discov. 2022;8:104
15. Chen YY, Chen SY, Li TJ. et al. 4-Acetylantroquinonol B enhances cell death and inhibits autophagy by downregulating the PI3K/Akt/MDR1 pathway in gemcitabine- resistant pancreatic cancer cells. Oncol Lett. 2022;23:128
16. Shao X, Chen X, Wang Z. et al. Diprenylated flavonoids from licorice induce death of SW480 colorectal cancer cells by promoting autophagy: Activities of lupalbigenin and 6,8-diprenylgenistein. J Ethnopharmacol. 2022;296:115488
17. Wang R, Ha KY, Dhandapani S. et al. Biologically synthesized black ginger- selenium nanoparticle induces apoptosis and autophagy of AGS gastric cancer cells by suppressing the PI3K/Akt/mTOR signaling pathway. J Nanobiotechnology. 2022;20:441
18. Tao Y, Liu Y, Dong Z. et al. Cellular Hypoxia Mitigation by Dandelion-like Nanoparticles for Synergistic Photodynamic Therapy of Oral Squamous Cell Carcinoma. ACS Appl Mater Interfaces. 2022;14:44039-53
19. Eisenberg-Lerner A, Bialik S, Simon HU. et al. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16:966-75
20. Yin S, Jin W, Qiu Y. et al. Solamargine induces hepatocellular carcinoma cell apoptosis and autophagy via inhibiting LIF/miR-192-5p/CYR61/Akt signaling pathways and eliciting immunostimulatory tumor microenvironment. J Hematol Oncol. 2022;15:32
21. Ji Y, Hu W, Jin Y. et al. Liquiritigenin exerts the anti-cancer role in oral cancer via inducing autophagy-related apoptosis through PI3K/AKT/mTOR pathway inhibition in vitro and in vivo. Bioengineered. 2021;12:6070-82
22. Zhou W, Dong M, Wu H. et al. Common mechanism of Citrus Grandis Exocarpium in treatment of chronic obstructive pulmonary disease and lung cancer. Chin Herb Med. 2021;13:525-33
23. Jasemi SV, Khazaei H, Fakhri S. et al. Naringenin Improves Ovalbumin-Induced Allergic Asthma in Rats through Antioxidant and Anti-Inflammatory Effects. Evid Based Complement Alternat Med. 2022;2022:9110798
24. Wu WY, Jiao X, Song WX. et al. Network pharmacology and bioinformatics analysis identifies potential therapeutic targets of Naringenin against COVID-19/LUSC. Front Endocrinol (Lausanne). 2023;14:1187882
25. El-Wafaey DI, Nafea OE, Faruk EM. Naringenin alleviates hepatic injury in zinc oxide nanoparticles exposed rats: impact on oxido-inflammatory stress and apoptotic cell death. Toxicol Mech Methods. 2022;32:58-66
26. Sahin A, Sakat MS, Kilic K. et al. The protective effect of Naringenin against ovalbumin-induced allergic rhinitis in rats. Eur Arch Otorhinolaryngol. 2021;278:4839-46
27. Bao L, Liu F, Guo HB. et al. Naringenin inhibits proliferation, migration, and invasion as well as induces apoptosis of gastric cancer SGC7901 cell line by downregulation of AKT pathway. Tumour Biol. 2016;37:11365-74
28. Qi Z, Kong S, Zhao S. et al. Naringenin inhibits human breast cancer cells (MDA- MB-231) by inducing programmed cell death, caspase stimulation, G2/M phase cell cycle arrest and suppresses cancer metastasis. Cell Mol Biol (Noisy-le-grand). 2021;67:8-13
29. Bodduluru LN, Kasala ER, Madhana RM. et al. Naringenin ameliorates inflammation and cell proliferation in benzo(a)pyrene induced pulmonary carcinogenesis by modulating CYP1A1, NFkappaB and PCNA expression. Int Immunopharmacol. 2016;30:102-10
30. Zhang L, Xu X, Jiang T. et al. Citrus aurantium Naringenin Prevents Osteosarcoma Progression and Recurrence in the Patients Who Underwent Osteosarcoma Surgery by Improving Antioxidant Capability. Oxid Med Cell Longev. 2018;2018:8713263
31. Lian GY, Wang QM, Mak TS. et al. Inhibition of tumor invasion and metastasis by targeting TGF-beta-Smad-MMP2 pathway with Asiatic acid and Naringenin. Mol Ther Oncolytics. 2021;20:277-89
32. Shi X, Luo X, Chen T. et al. Naringenin inhibits migration, invasion, induces apoptosis in human lung cancer cells and arrests tumour progression in vitro. J Cell Mol Med. 2021;25:2563-71
33. Zhao Z, Jin G, Ge Y. et al. Naringenin inhibits migration of breast cancer cells via inflammatory and apoptosis cell signaling pathways. Inflammopharmacology. 2019;27:1021-36
34. Park HJ, Choi YJ, Lee JH. et al. Naringenin causes ASK1-induced apoptosis via reactive oxygen species in human pancreatic cancer cells. Food Chem Toxicol. 2017;99:1-8
35. Park S, Lim W, Bazer FW. et al. Naringenin suppresses growth of human placental choriocarcinoma via reactive oxygen species-mediated P38 and JNK MAPK pathways. Phytomedicine. 2018;50:238-46
36. Ahamad MS, Siddiqui S, Jafri A. et al. Induction of apoptosis and antiproliferative activity of naringenin in human epidermoid carcinoma cell through ROS generation and cell cycle arrest. PLoS One. 2014;9:e110003
37. Zhao Q, Yang H, Liu F. et al. Naringenin Exerts Cardiovascular Protective Effect in a Palmitate-Induced Human Umbilical Vein Endothelial Cell Injury Model via Autophagy Flux Improvement. Mol Nutr Food Res. 2019;63:e1900601
38. Ahsan AU, Sharma VL, Wani A. et al. Naringenin Upregulates AMPK-Mediated Autophagy to Rescue Neuronal Cells From beta-Amyloid (1-42) Evoked Neurotoxicity. Mol Neurobiol. 2020;57:3589-602
39. Qin L, Jin L, Lu L. et al. Naringenin reduces lung metastasis in a breast cancer resection model. Protein Cell. 2011;2:507-16
40. Zhang YS, Li Y, Wang Y. et al. Naringin, a natural dietary compound, prevents intestinal tumorigenesis in Apc (Min/+) mouse model. J Cancer Res Clin Oncol. 2016;142:913-25
41. Cui Q, Wang JQ, Assaraf YG. et al. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist Updat. 2018;41:1-25
42. Gao L, Loveless J, Shay C. et al. Targeting ROS-Mediated Crosstalk Between Autophagy and Apoptosis in Cancer. Adv Exp Med Biol. 2020;1260:1-12
43. Do BH, Nguyen TPT, Ho NQC. et al. Mitochondria-mediated Caspase-dependent and Caspase-independent apoptosis induced by aqueous extract from Moringa oleifera leaves in human melanoma cells. Mol Biol Rep. 2020;47:3675-89
44. Ahsan AU, Sharma VL, Wani A. et al. Naringenin Upregulates AMPK-Mediated Autophagy to Rescue Neuronal Cells From beta-Amyloid (1-42) Evoked Neurotoxicity. Mol. Neurobiol. 2020;57:3589-602
45. Zhao R, Xiao H, Jin T. et al. Naringenin promotes cell autophagy to improve high- fat-diet-induced atherosclerosis in ApoE-/- mice. Braz. J. Med. Biol. Res. 2021;54:e9764
46. Liang P, Jiang B, Li Y. et al. Autophagy promotes angiogenesis via AMPK/Akt/mTOR signaling during the recovery of heat-denatured endothelial cells. Cell Death Dis. 2018;9:1152
47. Efferth T. From ancient herb to modern drug: Artemisia annua and artemisinin for cancer therapy. Semin Cancer Biol. 2017;46:65-83
48. Jendzelovska Z, Jendzelovsky R, Kucharova B. et al. Hypericin in the Light and in the Dark: Two Sides of the Same Coin. Front Plant Sci. 2016;7:560
49. Shi X, Luo X, Chen T. et al. Naringenin inhibits migration, invasion, induces apoptosis in human lung cancer cells and arrests tumour progression in vitro. J Cell Mol. Med. 2021;25:2563-71
50. Askar MA, El Shawi OE, Abou Zaid OAR. et al. Breast cancer suppression by curcumin-naringenin-magnetic-nano-particles: In vitro and in vivo studies. Tumour Biol. 2021;43:225-47
51. Qi Z, Kong S, Zhao S. et al. Naringenin inhibits human breast cancer cells (MDA- MB-231) by inducing programmed cell death, caspase stimulation, G2/M phase cell cycle arrest and suppresses cancer metastasis. Cell Mol. Biol. (Noisy-le-grand). 2021;67:8-13
52. Arul D, Subramanian P. Naringenin (citrus flavonone) induces growth inhibition, cell cycle arrest and apoptosis in human hepatocellular carcinoma cells. Pathol. Oncol. Res. 2013;19:763-70
53. Alnahdi A, John A, Raza H. Augmentation of Glucotoxicity, Oxidative Stress, Apoptosis and Mitochondrial Dysfunction in HepG2 Cells by Palmitic Acid. Nutrients. 2019 11
54. Rizwan H, Pal S, Sabnam S. et al. High glucose augments ROS generation regulates mitochondrial dysfunction and apoptosis via stress signalling cascades in keratinocytes. Life Sci. 2020;241:117148
55. Li B, Zhou P, Xu K. et al. Metformin induces cell cycle arrest, apoptosis and autophagy through ROS/JNK signaling pathway in human osteosarcoma. Int. J. Biol. Sci. 2020;16:74-84
56. Wang S, Li Z, Liu W. et al. Neohesperidin Induces Cell Cycle Arrest, Apoptosis, and Autophagy via the ROS/JNK Signaling Pathway in Human Osteosarcoma Cells. Am. J. Chin. Med. 2021;49:1251-74
57. Zhang R, Humphreys I, Sahu RP. et al. In vitro and in vivo induction of apoptosis by capsaicin in pancreatic cancer cells is mediated through ROS generation and mitochondrial death pathway. Apoptosis. 2008;13:1465-78
58. Gahr S, Ocker M, Ganslmayer M. et al. The combination of the histone- deacetylase inhibitor trichostatin A and gemcitabine induces inhibition of proliferation and increased apoptosis in pancreatic carcinoma cells. Int J Oncol. 2007;31:567-76
59. Lee KH, Lotterman C, Karikari C. et al. Epigenetic silencing of MicroRNA miR- 107 regulates cyclin-dependent kinase 6 expression in pancreatic cancer. Pancreatology. 2009;9:293-301
60. Shankar S, Ganapathy S, Hingorani SR. et al. EGCG inhibits growth, invasion, angiogenesis and metastasis of pancreatic cancer. Front Biosci. 2008;13:440-52
61. Shankar S, Suthakar G, Srivastava RK. Epigallocatechin-3-gallate inhibits cell cycle and induces apoptosis in pancreatic cancer. Front Biosci. 2007;12:5039-51
62. Srivastava SK, Singh SV. Cell cycle arrest, apoptosis induction and inhibition of nuclear factor kappa B activation in anti-proliferative activity of benzyl isothiocyanate against human pancreatic cancer cells. Carcinogenesis. 2004;25:1701-9
63. Kopustinskiene DM, Jakstas V, Savickas A. et al. Flavonoids as Anticancer Agents. Nutrients. 2020 12
64. Yen GC, Duh PD, Tsai HL. et al. Pro-oxidative properties of flavonoids in human lymphocytes. Biosci Biotechnol Biochem. 2003;67:1215-22
65. Peng J, Drobish JK, Liang G. et al. Anesthetic preconditioning inhibits isoflurane- mediated apoptosis in the developing rat brain. Anesth. Analg. 2014;119:939-46
66. Li C, Zhang Y, Liu J. et al. Mitochondrial DNA stress triggers autophagy- dependent ferroptotic death. Autophagy. 2021;17:948-60
67. Ma RF, Liu H, Zhao XC. et al. Meroterpenoids from Daphne genkwa shows promising in vitro antitumor activity via inhibiting PI3K/Akt/mTOR signaling pathway in A549 cells. Bioorg Chem. 2023;140:106803
68. Peng Y, Wang Y, Zhou C. et al. PI3K/Akt/mTOR Pathway and Its Role in Cancer Therapeutics: Are We Making Headway? Front Oncol. 2022;12:819128
69. Xie Q, Liu Y, Li X. The interaction mechanism between autophagy and apoptosis in colon cancer. Transl Oncol. 2020;13:100871
70. Athamneh K, Alneyadi A, Alsamri H. et al. Origanum majorana Essential Oil Triggers p38 MAPK-Mediated Protective Autophagy, Apoptosis, and Caspase- Dependent Cleavage of P70S6K in Colorectal Cancer Cells. Biomolecules. 2020 10
71. Aroui S, Fetoui H, Kenani A. Natural dietary compound naringin inhibits glioblastoma cancer neoangiogenesis. BMC Pharmacol. Toxicol. 2020;21:46
72. Kumar R, Bhan Tiku A. Naringenin Suppresses Chemically Induced Skin Cancer in Two-Stage Skin Carcinogenesis Mouse Model. Nutr. Cancer. 2020;72:976-83
73. Rebello CJ, Beyl RA, Lertora JJL. et al. Safety and pharmacokinetics of naringenin: A randomized, controlled, single-ascending-dose clinical trial. Diabetes Obes Metab. 2020;22:91-8
74. Surampalli G, Nanjwade BK, Patil PA. Safety evaluation of naringenin upon experimental exposure on rat gastrointestinal epithelium for novel optimal drug delivery. Drug Deliv. 2016;23:512-24
Author contact
![]() Corresponding authors: Chiang-Wen Lee, Department of Respiratory Care, Chang Gung University of Science and Technology, Chiayi 61363, Taiwan; Tel: (886) 5-3628800 ext. 2620; E-mail: cwleecgust.edu.tw. Or Ju-Fang Liu, School of Oral Hygiene, College of Oral Medicine, Taipei Medical University, Taipei 11031, Taiwan; Tel: (886) 2-28332211 ext. 9420; E-mail: jufangliuedu.tw. Or Yu Ru Kou, Department of Medical Research, Hualien Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, Hualien 97002, Taiwan; Tel: (886) 3-8561825 ext. 15933; E-mail: yrkouedu.tw.
Corresponding authors: Chiang-Wen Lee, Department of Respiratory Care, Chang Gung University of Science and Technology, Chiayi 61363, Taiwan; Tel: (886) 5-3628800 ext. 2620; E-mail: cwleecgust.edu.tw. Or Ju-Fang Liu, School of Oral Hygiene, College of Oral Medicine, Taipei Medical University, Taipei 11031, Taiwan; Tel: (886) 2-28332211 ext. 9420; E-mail: jufangliuedu.tw. Or Yu Ru Kou, Department of Medical Research, Hualien Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, Hualien 97002, Taiwan; Tel: (886) 3-8561825 ext. 15933; E-mail: yrkouedu.tw.