Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(5):1547-1562. doi:10.7150/ijbs.83040 This issue Cite
Research Paper
Inhibition of BRD4 Attenuates ER Stress-induced Renal Ischemic-Reperfusion Injury
Paula Diaz-Bulnes1,2, Ramon M Rodríguez1,3, Elisenda Banon-Maneus2,4, María Laura Saiz1,2, Cristian Ruiz Bernet1,2, Viviana Corte-Iglesias1,2, Maria Jose Ramirez-Bajo2,4, Marta Lazo-Rodriguez4, Isaac Tamargo-Gómez5, Raúl R. Rodrigues-Diez1,2, Ana B Sanz6,2, Carmen Diaz-Corte1,2,7, Marta Ruiz-Ortega8,2, Fritz Diekmann2,4,9, Ana M Aransay10,11, Carlos Lopez-Larrea1,2, ![]() , Beatriz Suarez-Alvarez1,2,
, Beatriz Suarez-Alvarez1,2, ![]()
1. Translational Immunology, Instituto de Investigación Sanitaria del Principado de Asturias (ISPA), 33011 Oviedo, Spain.
2. RICORS2040 (Kidney Disease), Instituto de Salud Carlos III, 28029 Madrid, Spain.
3. Lipids in Human Pathology, Health Research Institute of the Balearic Islands (IdISBa), Research Unit, University Hospital Son Espases, 07120 Palma, Spain.
4. Laboratori Experimental de Nefrologia i Trasplantament (LENIT), Institut d'Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona, Spain.
5. Autophagy and Metabolism Laboratory, Instituto de Investigación Sanitaria del Principado de Asturias (ISPA), Oviedo, Spain.
6. Laboratorio de Nefrología Experimental, Instituto de Investigación Sanitaria-Fundación Jiménez Díaz, Universidad Autónoma de Madrid, Madrid, Spain.
7. Servicio de Nefrología, Hospital Universitario Central de Asturias, Oviedo, Asturias, Spain.
8. Cellular Biology in Renal Diseases Laboratory, IIS-Fundación Jiménez Díaz, Universidad Autónoma Madrid, 28040 Madrid, Spain.
9. Departmento de Nefrologia y Transplante Renal, Institut Clínic de Nefrologia i Urologia, Hospital Clínic Barcelona, Barcelona, Spain
10. Genome Analysis Platform, CIC bioGUNE-BRTA, Derio, Spain.
11. Centro de Investigación Biomédica en Red en el Área Temática de Enfermedades Hepáticas (CIBERehd), Madrid, Spain.
Received 2023-1-30; Accepted 2024-1-17; Published 2024-2-11
Abstract

Renal ischemia-reperfusion injury (IRI) leads to endoplasmic reticulum (ER) stress, thereby initiating the unfolded protein response (UPR). When sustained, this response may trigger the inflammation and tubular cell death that acts to aggravate the damage. Here, we show that knockdown of the BET epigenetic reader BRD4 reduces the expression of ATF4 and XBP1 transcription factors under ER stress activation. BRD4 is recruited to the promoter of these highly acetylated genes, initiating gene transcription. Administration of the BET protein inhibitor, JQ1, one hour after renal damage induced by bilateral IRI, reveals reduced expression of ATF4 and XBP1 genes, low KIM-1 and NGAL levels and recovery of the serum creatinine and blood urea nitrogen levels. To determine the molecular pathways regulated by ATF4 and XBP1, we performed stable knockout of both transcription factors using CRISPR-Cas9 and RNA sequencing. The pathways triggered under ER stress were mainly XBP1-dependent, associated with an adaptive UPR, and partially regulated by JQ1. Meanwhile, treatment with JQ1 downmodulated most of the pathways regulated by ATF4 and related to the pathological processes during exacerbated UPR activation. Thus, BRD4 inhibition could be useful for curbing the maladaptive UPR activation mechanisms, thereby ameliorating the progression of renal disease.
Keywords: BRD4, ER stress, UPR, renal damage, Hypoxia
Introduction
Tubular epithelial cells (TECs) are especially vulnerable to a variety of insults, such as hypoxia, drugs, toxins and proteinuria. They respond to injury not only by triggering mechanisms and pathways to restore cellular homeostasis, but also by inducing the production of inflammatory and pro-fibrotic factors that determine the progression of chronic kidney disease (CKD) [1]. Protein homeostasis is disturbed by renal ischemia-reperfusion injury (IRI), inducing the accumulation of unfolded proteins within the endoplasmic reticulum (ER) of tubular cells [2]. When proteostasis is disrupted and unfolded proteins accumulate, ER stress initiates the unfolded protein response (UPR) that attenuates protein synthesis, eliminates misfolded proteins and enhances the ability of the ER to fold proteins, allowing cell survival [3]. However, under sustained ER stress, the adaptive ability of UPR is lost, leading to cell death by apoptosis. Targeting prolonged UPR activation might offer protection from renal damage and its progression [4,5]. Pharmacological inhibition of ER stress with the chemical chaperones TUDCA (tauroursodeoxycholic acid) and 4-PBA (4-phenylbutyric acid) reduces inflammation and tubular cell death, alleviating damage and preventing renal failure [6, 7].
Under ER stress, the resident chaperone BiP/GRP78 (78-kDa glucose-related protein) engages with unfolded proteins to release three major sensors: IRE1α, PERK and ATF6. IRE1α allows processing of XBP1 to a splicing form (XBP1s) that is functionally active as a transcription factor and that induces JNK, NF-κB and NLRP3-inflammasome activation [8-10]. The PERK branch phosphorylates and activates eukaryotic initiation factor 2 alpha (eIF2α), enabling the expression of ATF4. This transcription factor helps regulate autophagy and promote the expression of the pro-apoptotic factor CHOP (C/EBP-homologous protein), and is a metabolic effector of mTORC1 [11, 12]. The third sensor, ATF6, is transported to the Golgi, where it is cleaved and moves to the nucleus, where it induces expression of XBP1 and CHOP, among others [13, 14]. Therefore, each branch of the UPR pathway facilitates the transcription of many genes, some of which restore ER homeostasis, while others aggravate the damage.
BRD4, the best characterized member of the bromodomain and extra-terminal (BET) protein family, recognizes and binds to acetylated lysine on histones and other non-histone proteins and recruits transcriptional complexes to gene promoters to initiate this transcription [15]. Therefore, pharmacological targeting of BRD4 has been proposed as an alternative therapy for treating a wide range of human diseases, including those of the kidney [16, 17]. We and other groups have reported that BRD4 regulates the expression of pro-inflammatory genes, blocks the NF-κB pathway and NLRP3-inflammasome activation, and modulates the neutrophil activation associated with renal injury [18-20]. Blockage of BRD4 has also been shown to be a potent antifibrotic therapy in renal cells and in a mouse model of UUO-induced renal damage [21, 22]. However, its role in modulating UPR genes and the consequent pathological processes associated with renal damage has not yet been explored.
Here, we set out to determine the role of BRD4 in regulating UPR genes and the underlying mechanisms generated as a consequence of renal ischemic damage. We explore the outcome of BRD4 targeting using the JQ1 inhibitor in an in vivo IRI model, and show that BRD4 is a potential target for downregulating ATF4 and XBP1 expression, and for alleviating the ischemic renal damage produced by sustained ER stress.
Methods
Cell culture and treatments
Human proximal tubular epithelial cells (TECs) (HK-2 cell line; CRL-2190; ATCC, VA) were maintained in RPMI 1640 medium (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, 1% insulin transferrin selenite (Gibco) and 5 ng/ml hydrocortisone (Sigma-Aldrich, St. Louis, MO, USA). Cells were cultured at 0.25x106 cells/ml in serum-free medium for 24 h before the assay and further stimulated with thapsigargin (Tg; 4 μM; 24 h; Sigma-Aldrich). Inhibitors used were: JQ1(+) or its inactive enantiomer JQ1(-) (100-500 nM; 24 h; Selleckchem, Houston, TX, USA); I-BET 726 (400-1000 nM; 24 h; Cayman Chemical, Ann Arbor, MI, USA) and 5,6-dichlorobenzimidazole 1-β-D-ribofuranoside DRB (20-60 μM; 6 h; Sigma-Aldrich). For hypoxic conditions, cells were cultured in medium without nutrients for 12 h in a hypoxic incubator (1% O2, 94% N2 and 5% CO2, 37°C). In some conditions, HK-2 cells were followed by reoxygenation at different times (2, 4, and 6 h) with fresh complete medium, and in 5% CO2 and 95% air at 37°C.
IR injury animal model and animal studies
All animal procedures were performed in accordance with the guidelines for animal research in the European Community and with the approval of the local government authorities at IDIBAPS, Hospital Clinic, Barcelona. Briefly, bilateral ischemia reperfusion injury (IRI) was induced in 9-12-week-old male C57BL/6 mice (Charles River Laboratories, Wilmington, MA, USA) by clamping both renal pedicles for 45 min at 37°C, adopting a retroperitoneal approach, under isofluorane anesthesia. After removing the clamps, reperfusion was verified by visual inspection of the kidney. Following surgery, mice were randomly assigned to three groups (n=5/group): Sham, IRI and IRI + JQ1. Sham group mice underwent the same procedure except the renal pedicles were not clamped. The BET bromodomain inhibitor, JQ1(+), supplied by James Bradner's laboratory (Dana-Farber Cancer Institute, Boston, MA, USA), was dissolved in 10% hydroxypropyl β-cyclodextrin (Sigma Aldrich) and administered by intraperitoneal injection of a single dose of 100 mg/kg 1 h after surgery. Mice were euthanized at different times after reperfusion, and the kidneys and blood collected for further analysis.
Assessment of renal injury
Kidney sections (2-5 μm) were stained with periodic acid-Schiff (PAS) reagent to assess renal injury and quantify the percentage of necrotic tubules by an experienced pathologist and an observer blinded to the experimental conditions. Images were acquired with a Leica DFC700T optical microscope (Leica Biosystems, Wetzlar, Germany). Blood samples were collected in heparin tubes and serum was obtained from whole blood after centrifugation. Serum creatinine and urea were determined using a creatinine assay kit (Abcam, Cambridge, UK) and a blood urea nitrogen (BUN) colorimetric detection kit (Arbor Assays, Ann Arbor, MI, USA), respectively, and according to the manufacturer's instructions.
Gene expression studies
Total RNA from HK-2 cells or frozen kidneys was isolated using a GeneMATRIX Universal RNA purification kit (EURx, Gdansk, Poland) following the manufacturer's instructions. Purified RNA (1 µg) was reversed-transcribed to cDNA using a high-capacity cDNA reverse-transcription kit (Applied Biosystems, Foster City, CA, USA). Quantification was performed by the reverse-transcription polymerase chain reaction (RT-PCR) using TB Green Premix Ex TaqII (Takara Bio Inc., Kusatsu, Japan) or TaqMan Gene Expression Master Mix (Applied Biosystems), and analyzed with a StepOnePlusTM Real-Time PCR System (Applied Biosystems). Expression of each gene of interest was normalized to the housekeeping gene Gapdh and data were calculated by the ΔCt method. The primers and TaqMan assays used are listed in Table S1.
CRISPR-Cas9
HK-2 cells were transfected with a single guide RNA (sgRNA) specific to ATF4 and XBP1 and designed using the https://cctop.cos.uni-heidelberg.de website. Plasmids expressing Cas9 and sgRNA were generated by inserting custom sgRNA into lentiCRISPRv2 (#52961; Addgene, Watertown, MA, USA) following the manufacturer's instructions. Briefly, lentiviruses were packaged in HEK 293T cells by transfection with specific sgRNA/Cas9-expressing lentiCRISPRv2 and the packaging plasmid psPAX-2 (#12260; Addgene) and psPMD2g (#12259; Addgene) at 1:1:1 molar ratio using the Lipofectamine 3000 Reagent (Invitrogen). Medium was replaced after 6 h and virus samples collected at 24, 48 and 72 h. HK-2 cells were infected with lentivirus in the presence of polybrene (8 μg/ml) and further selected with 1 μg/ml of puromycin to obtain single clones. sgRNAs and primers are listed in Table S2.
Gene silencing
HK-2 cells were transfected with variable quantities of ON-TARGETplus siRNAs specific to human BRD4 (L-004937-00-0005) or of the non-targeting control pool as a negative control (D-001810-10-05) using DharmaFECT1 Transfection Reagent. All reagents were supplied by GE Healthcare Dharmacon, Lafayette, CO, USA.
Protein analysis
Proteins were extracted using radioimmunoprecipitation assay (RIPA) lysis buffer supplemented with a protease and phosphatase inhibitor cocktail (Merck Millipore, Burlington, MA, USA) for 30 min on ice, quantified using the Bradford protein assay (Bio-Rad), and separated on 10-12% polyacrylamide-SDS gels. Membranes were incubated overnight at 4°C with the following primary antibodies: XBP1 spliced form (NBP2-20917, 1:1000; Novus Biologicals, Centennial, CO, USA), ATF4 (11815S, 1:1000; Cell Signaling Technology, Danvers, MA, USA), ATF6 (MAB71527-SP, 1:2000; R&D Systems, Minneapolis, MI, USA), BRD4 (A301-985A; Bethyl Laboratories, Montgomery, TX, USA), KIM-1 (AF1817, 1:1000; R&D Systems), NGAL (Sc-50350, 1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA), and β-actin (4967S, 1:2000; Cell Signaling Technology); followed by incubation with HRP-conjugated IgG secondary antibody for 1 h. Immunostained bands were visualized using a chemiluminescence kit (Luminata Forte Western HRP Substrate; Merck Millipore), detected on an ImageLab system (Bio-Rad, Hercules, CA, USA), and quantified with ImageJ version 1.53c software (NIH Image).
Chromatin immunoprecipitation assay
HK-2 cells (107) were fixed with 1% formaldehyde (Sigma-Aldrich) in RPMI medium for 30 min at 4°C, followed by quenching with 0.125 M glycine for 10 min at 4°C. Further, chromatin was shared on a BioRuptor (Diagenode, Liège, Belgium) into DNA fragments between 500-1000 bp long. 100 μg of chromatin was diluted into ChIP dilution buffer (0.01% SDS; 1.1% Triton X100; 1.2 mM EDTA; 16.7 mM Tris-HCL, pH 8.1 and 167 mM NaCl) containing protease and phosphatase inhibitor (Thermo Scientific) and incubated with specific antibodies to BRD4 (A301-985A; Bethyl Laboratories), anti-phospho RNA pol II (Ser 2) (MABE953; Merck), acetylated H3 (06-599; Merck), acetylated H4 (06-598; Merck) or normal rabbit IgG (A300-109A; Bethyl Laboratories) overnight at 4°C. Antibody-chromatin complexes were recovered with Salmon Sperm DNA/Protein A-Agarose beads (Merck) for 1 h at 4°C, washed and eluted from the beads with elution buffer (1% SDS, 0.1 M NaHCO3). After reverse-crosslinking and proteinase K treatment, DNA was extracted and analyzed by quantitative RT-PCR using specific primers (Table S1). Chromatin obtained before immunoprecipitation was used as the input control. For ChIP assay in kidney samples, kidneys were immediately fixed with 1% paraformaldehyde after extraction, quenched with 0.125 M glycine and stored at -80°C until use. The High Cell ChIP Kit (Diagenode) was used in accordance with the manufacturer's instructions, employing specific primers (Table S1). In both assays, enrichment was calculated using the formula: ΔCt=Ct (bound) - [Ct (input) - log2 (Input dilution factor)] and data are expressed as the fold enrichment (FE) of each specific antibody relative to the negative control, where FE=2 exp - [ΔCt (specific antibody) - ΔCt (normal IgG)].
RNA sequencing and gene expression analysis
RNA from HK-2 cells was isolated with a PureLink™ RNA Mini Kit (Invitrogen, Carlsbad, CA, USA). The integrity of the RNA was analyzed in RNA 6000 Nano Chips with a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA) and quantified using a fluorometric Qubit RNA HS assay kit (Invitrogen, Carlsbad, CA, USA). Briefly, starting from 1 µg of total RNA, sequencing libraries were prepared following the TruSeq Stranded mRNA Sample Preparation Guide (Part #15031058 Rev. E), using the TruSeq Stranded mRNA Library Prep kit (Illumina Inc., Cat. #20020294) and TruSeq RNA Single Indexes (Illumina Inc., Cat. #20020492 and 20020493). Sequencing was carried out using a HiSeq 2500 platform (Illumina Inc.). Files of data generated by the sequencing were aligned against the human genome (Homo_sapiens.GRCh38.94.gtf) using the STAR program [23], and the genes and transcripts were analyzed with the RSEM program [24] using GENCODE v26 [25]. Expression data were analyzed using R package, the DEQseq2 method of the Bioconductor project (www.bioconductor.org), and lumi packages. To detect genes differentially expressed between groups, the moderated t-test was used. Genes with a >2-fold change and an adjusted p<0.05 were considered significant. Functional interaction network data were obtained from STRING v10 [26], generated using the Fruchterman-Reingold clustering algorithm, and then built using Cytoscape software [27]. The resulting network was exported to Gephi (http://gephi.org). Gene functional analysis was carried out using the DAVID Go web-based tool [28]. Raw mRNAseq data have been deposited in the NCBI Gene Expression Omnibus under accession number GEO:GSE217103.
Statistical analysis
Data were summarized as the mean ± standard error of the mean (SEM) of at least two or three independent experiments. Characteristics of groups were compared using Fisher´s exact test for categorical variables, and the Wilcoxon paired-samples test, or the Mann-Whitney U test for unpaired samples. Statistical analyses were carried out with IBM SPSS Statistics, version 27.0 (IBM SPSS, Armonk, NY, USA) and GraphPad-Prism v7 (San Diego, CA, USA). Statistical significance was concluded for values of p<0.05. Statistical details of the experiments and significance are noted in the respective figures and legends.
Results
Blockage of BET proteins attenuates UPR activation triggered in human TECs
To evaluate the role of BET proteins in UPR pathway activation in tubular cells, we used HK-2 cells treated with thapsigargin (Tg, 4 μM), an inhibitor of the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) that impairs calcium access to the ER, inducing the three branches of the UPR pathway. After Tg stimulation, increased expression of the three downstream transcription factors (ATF4, XBP1 and ATF6) was detected (Figure 1A). For XBP1 only the active XBP1s form was analyzed (and referenced as XBP1 from now on). The induction of these genes declined significantly and dose-dependently when JQ1(+) was added (Figure 1A and Figure S1A), except for ATF6, whose expression levels remained unchanged. Similar results were observed at the protein level (Figure 1B) and with other BET protein inhibitors, such as I-BET762 (Figure S1B). Additionally, specific BRD4 silencing significantly decreased the induced ATF4 and XBP1 expression, but not ATF6, after sustained UPR activation with Tg (Figure S1C and S1D).
Treatment with JQ1 ameliorates overexpression of UPR genes induced by thapsigargin and hypoxia in HK-2 cells. Renal TECs (HK-2 cell line) were treated with DMSO (Ctrl) or Tg (4 μM, 24 h) (A,B) or cultured under hypoxic conditions (1% O2, 5% CO2) at different times (t0, normoxia; t12, 12 h) (C,D). The inhibitor of BET proteins, JQ1(+), or its inactive enantiomer, JQ1(-), were co-cultured at the same indicated times under both cultured conditions. Gene expression levels of ATF4, XBP1 and ATF6 were analyzed by RT-PCR (A,C) and protein levels were analyzed by western blot (B,D). Data are expressed as the mean ± SEM of at least three independent experiments. GADPH and β-actin were used as housekeeping markers of RT-PCR and western blot, respectively. Statistical analyses involved use of the two-tailed Student's paired t-test and the Wilcoxon test. *p<0.05 vs. control (DMSO) or t0 (normoxia) and # vs. cells treated with JQ1(-) + Tg or cells in hypoxia (t12) treated with JQ1(-).
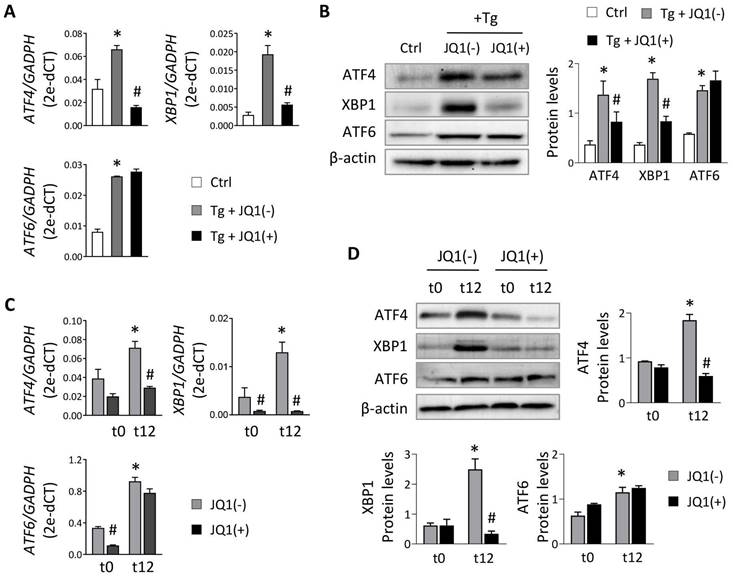
To verify these data under physiological conditions, HK-2 cells were subjected to different periods of hypoxia and the highest expression of ATF4, XBP1 and ATF6 was detected at 12 h (t12) (Figure S2). At this time, when cells were treated with the JQ1(+) inhibitor, the expression of ATF4 and XBP1 was not induced, suggesting that their regulation was dependent on BRD4 (Figure 1C and 1D). These results were corroborated using specific siRNA silencing of BRD4 (Figure 2A), where the increased ATF4 and XBP1 expression observed under hypoxic conditions (t12) was again reduced after BRD4 blockage reaching similar levels to that detected in normoxia (t0) (Figure 2B and 2C). Similarly to what was observed when induced with Tg, the ATF6 protein levels remained unchanged (Figure 2C).
Specific BRD4 silencing impairs expression of UPR genes induced under hypoxia. HK-2 cells were transfected with a specific siRNA against BRD4 (siRNA BRD4) or control siRNA (siRNA Ctrl, 40 nM, 48 h) before exposure to conditions of normoxia (t0) or hypoxia (t12). (A) Transcriptional and protein levels of BDR4 after specific silencing. Transcriptional levels of ATF4, XBP1 and ATF6 were determined by RT-PCR (B) and protein levels (C) assayed by western blot. Data are expressed as mean ± SEM of three independent experiments. Statistical analyses involved use of the two-tailed Student's paired t-test and the Wilcoxon test. *p<0.05 vs. t0 (normoxia), # vs. siRNA Ctrl-treated cells.

The cellular response to hypoxia and reoxygenation (H/R) is a complex and tightly regulated process. We aimed to determine whether the downregulation of ATF4 and XBP1 by JQ1(+) is maintained after reperfusion. Our findings demonstrate an increase of the transcription levels of ATF4 and XBP1 after 2 h of reoxygenation, that are subsequently sustained (Figure S3A). These results were confirmed at the protein level, although ATF4 reverts to hypoxic levels 6 h after reperfusion (Figure S3B). Once again, the treatment with JQ1(+) effectively prevents the upregulation of both ATF4 and XBP1 during reoxygenation (Figure S3A and S3B).
In order to evaluate whether the activation of the UPR pathway under these conditions induces changes in BRD4 expression, we evaluated the transcriptional and protein levels of BRD4 expression in HK-2 cells treated with Tg (Figure S4A) or cultured under hypoxic and H/R conditions. (Figure S4B and S4C). Across all conditions, BRD4 levels remained unaltered compared to the basal conditions, suggesting that its role in the cellular response to ER stress may not be primarily governed by changes in its abundance but rather by its binding to specific regulatory regions of these genes.
BRD4 is recruited to promoter regions of UPR genes to induce their expression
BRD4 recognizes and binds to acetylated lysine of regulatory regions, thereby allowing the recruitment of the P-TEFb complex that phosphorylates RNA polymerase II (RNA pol II), and initiating gene transcription. To explore this, we performed ChIP assays with an anti-BRD4 antibody in HK-2 cells cultured under normoxia (t0) or hypoxia (t12) in the presence of the JQ1(+) inhibitor or its enantiomer, JQ1(-). Data show that in the presence of the nonactive control, JQ1(-), BRD4 is highly enriched in the promoter regions of the ATF4 and XBP1 genes after hypoxia (Figure 3). However, this recruitment was significantly reduced after treatment with JQ1(+). Similarly, the enrichment of the phosphorylated RNA pol II (Ser 2) observed under hypoxia in these genes was significantly reduced after blockage of the BRD4 binding by JQ1(+) (Figure 3). We also investigated whether enhancement of the levels of acetylated histones is the initial point for the recruitment of BRD4 to these genes. Acetylation levels in histone H3 (AcH3) and H4 (AcH4) were highly enriched in the promoter region of ATF4 and XBP1 genes under hypoxia, and thereby associated with their enhanced expression (Figure 3). Treatment with JQ1(+) reduced these acetylation levels at similar levels to observe under normoxia. Similar results were observed under Tg induction (Figure S5A).
JQ1 inhibits direct binding of BRD4 protein to UPR genes induced by hypoxia in HK-2 cells. HK-2 cells were pretreated with or without JQ1 (+) or its enantiomer JQ1 (-) (500 nM, 24h), and subsequently cultured under conditions of normoxia (t0) and hypoxia (t12) at the times indicated. ChIP assays were performed with specific antibodies against BRD4, RNA POL II, AcH3 and AcH4, and the region of interest of ATF4 (A) and XBP1 (B) genes was amplified using specific primers (dashed arrows) by RT-PCR. Results are represented as the relative enrichment of each antibody relative to the IgG control. Data are expressed as the mean ± SEM of three independent experiments; *p<0.05 vs. t0 (normoxia) and # vs. cells treated with JQ1 (-).
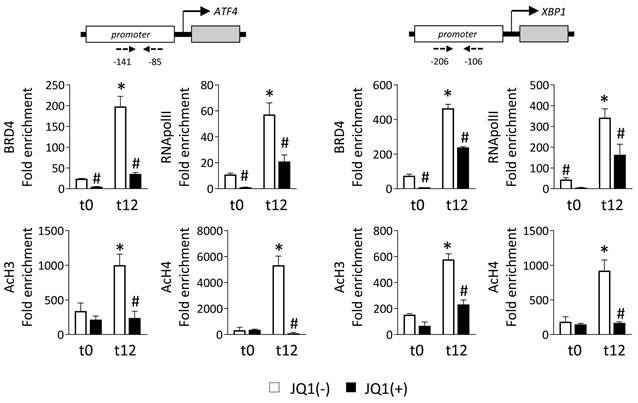
Additionally, HK-2 cells induced with Tg were treated with DRB, a specific inhibitor of the component of the P-TEFb complex CDK9 (cyclin-dependent kinase 9). Results showed a dose-dependent decrease in ATF4 and XBP1 transcription levels (Figure S5B). Thus, we can suggest that recruitment of BRD4 and its binding to acetylated histones facilitate activation of the P-TEFb complex and, consequently, the phosphorylation of RNA pol II, which triggers the rapid expression of these UPR genes.
Moreover, consistent with our prior findings, BRD4 inhibition does not influence ATF6 expression. To corroborate this, the recruitment of BRD4 was analyzed along the ATF6 promoter region, spanning approximately 900 bp and using H3 histone as a positive control. We observed that the recruitment of BRD4 was not enhanced in the promoter region of ATF6 under hypoxic conditions, and consequently the treatment with the JQ1(+) inhibitor did not show any effect, reinforcing that BRD4 is not directly involved in the increased ATF6 expression after UPR activation (Figure S6).
JQ1 reverts renal ischemia-mediated ER stress, ameliorating kidney function
Next, the effect of JQ1 was evaluated in an in vivo model of renal damage induced by IRI. JQ1 (100 mg/kg) was administered intraperitoneally 1 h after injury to evaluate its therapeutic potential. Phenotypic and functional changes were analyzed at 3 h (t3) and 24 h (t24) after reperfusion (Figure 4A). The expression of ATF4 and XBP1 genes increased soon after damage remaining elevated even after 24 h (Figure 4B). Administering JQ1 impaired the overexpression of these genes, restoring similar levels to those in the sham group (Figure 4B and 4C).
Administration of JQ1 after IRI blocks BRD4 recruitment to UPR genes, decreasing their expression. (A) C57BL/6 mice were treated with JQ1 (100 mg/kg) or vehicle (10% cyclodextran) 1 h after IRI. Sham mice were used as the control group. Samples were obtained at 3 h (t3) and 24 h (t24) post-ischemic damage. Groups: Sham, IRI (with vehicle) and IRI+JQ1 (administration of JQ1). (B) Gene expression levels were analyzed by RT-PCR at 3 h and 24 h. Results are means ± SEM of five animals per group. *p<0.05 vs. control and # vs. IRI group. (C) ATF4 and XBP1 protein levels were detected by western blot assay 3 h after IRI. (D) A ChIP assay was carried out in renal samples from the Sham, IRI and IRI+JQ1 groups using specific antibodies for BRD4, AcH3 and AcH4. Rabbit IgG was used as a negative control. Enrichment of binding regions in the atf4 and xbp1 promoters was quantified by RT-PCR using specific primers (dashed arrows). Data are summarized as the mean ± SEM of three independent experiments and expressed as the enrichment relative to the IgG negative control. Statistical analyses involved use of the two-tailed Student's unpaired t-test and the U-Mann Whitney test. *p<0.05 vs. Sham group; # p<0.05 vs. IRI group.
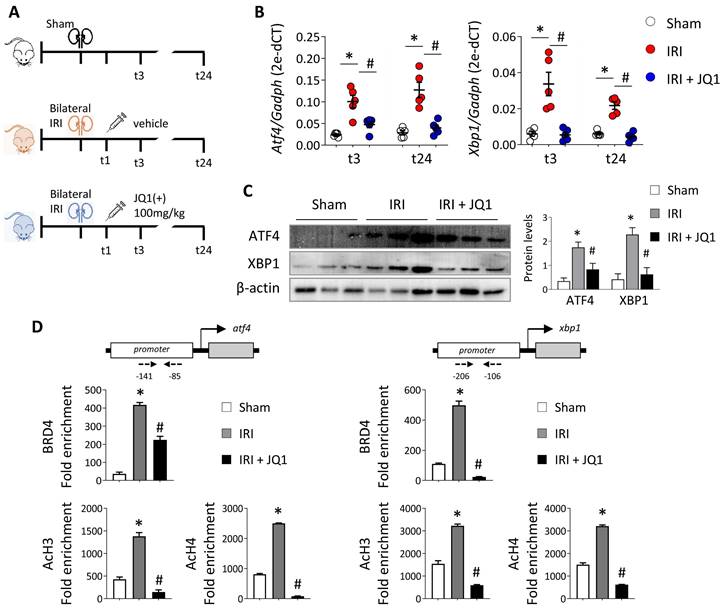
We corroborated the recruitment of BRD4 to the regulatory region of these genes in the kidneys during IRI, associated with elevated levels of AcH3 and AcH4 (Figure 4D). However, the recruitment of BRD4 was significantly reverted in the kidneys of JQ1-treated mice and so the acetylated histone levels decreased at lower levels to that observed in the sham group. These results indicate that JQ1 acts to impair binding of BRD4 at the promoter region of these genes and, with it, the recruitment of the transcriptional machinery, while preventing their overexpression even in the presence of renal damage.
In line with our in vitro models, analysis of BRD4 levels showed a slight increase in BRD4 mRNA levels at 3h post-reperfusion, returning to control (sham) levels afterwards (Figure S7). Nevertheless, despite the substantial recruitment of BRD4 to the regulatory regions of ATF4 and XBP1, there were no significant changes in BRD4 protein levels.
Additionally to these changes, JQ1 administration is associated to a reduced expression of the kidney injury markers, KIM-1 and NGAL (Figure 5A). PAS staining in the corticomedullary junction, the kidney region most susceptible to renal ischemic injury, shows that mice in the IRI group have tubular damage with loss of the tubular structure and epithelial cell connections as well as urinary cast within the tubule compared with the sham group (Figure 5B). However, mice treated with JQ1 had significantly fewer damaged tubules and, although some intratubular deposits were observed, the integrity and structure of the TECs was preserved (Figure 5B). Moreover, the significant enhancement of BUN and creatinine levels observed at 24 h in IRI mice was partially restored after JQ1 administration (Figure 5C). Thus, pharmacological blockage of ATF4 and XBP1 expression by JQ1 not only reduced UPR activation but also partially contributes to reduce the associated renal damage.
Inhibition of BET proteins with JQ1 restores renal damage induced by in vivo IRI. C57BL/6 mice were treated with JQ1 (100 mg/kg) or vehicle (10% cyclodextran) 1 h after IRI. Sham mice were used as the control group. Samples were obtained at 3 (t3) and 24 (t24) h post-ischemic damage. Groups: Sham, IRI (with vehicle) and IRI+JQ1 (administration of JQ1). (A) Renal KIM-1 and NGAL protein levels were analyzed by western blot 24 h post-damage. Data are summarized as the mean ± SEM of five animals per group. The western blot image shows the expression levels of three representative animals per group. Statistical analysis involved use of the two-tailed Student's unpaired t-test and the Mann-Whitney U test. *p<0.05 vs. Sham, and # vs. IRI group. (B) Representative PAS-stained sections for Sham, IRI and IRI+JQ1 groups. Scale bar= 100 µm and arrows indicate presence of tubular casts. C) Serum creatinine (mg/dl) and blood urea nitrogen (BUN, mg/dl) in mice from the three groups at 3 h (t3) and 24 h (t24) post-ischemic damage. The Mann-Whitney test was used. *p<0.05 vs. Sham and # vs. IRI group.

BET proteins differentially regulate molecular pathways triggered by ATF4 and XBP1 under ER stress
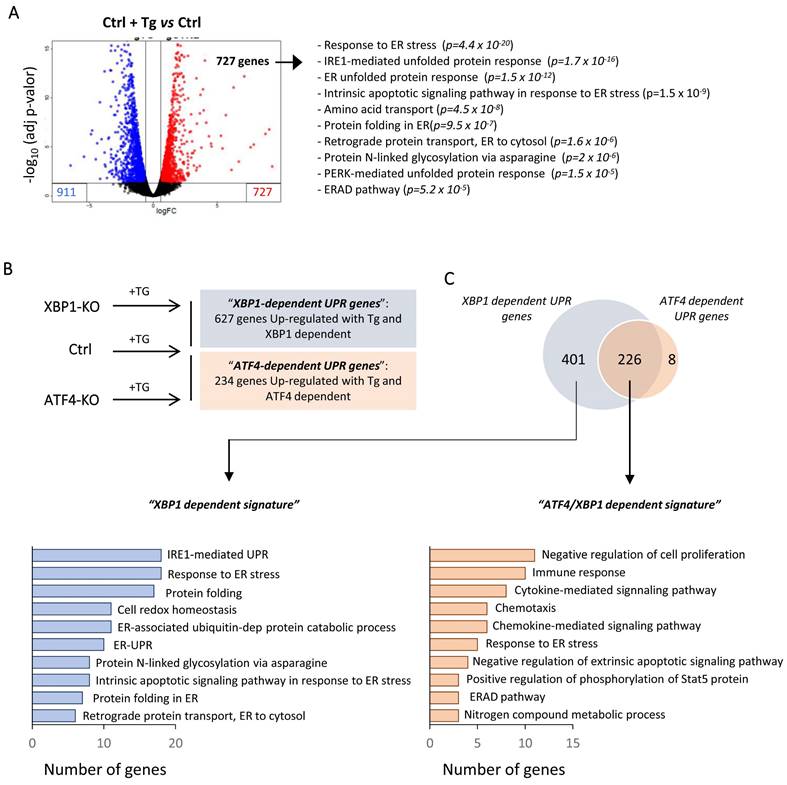
Furthermore, we examined whether the administration of JQ1 could modulate the ATF4 and XBP1 downstream pathways in a similar way. For that, we generated stable knockout of ATF4 and XBP1 in HK-2 cells using CRISPR-Cas9 technology followed by transcriptomic analysis through RNA-seq. The complete absence of ATF4 and XBP1 before and after Tg-mediated ER stress induction was confirmed by sequencing (Figure S8A) and expression studies (Figure S8B and S8C).
Differential gene expression analysis revealed that under Tg induction of HK-2 cells, 727 and 911 genes were upregulated and downregulated, respectively (Figure 6A and Table S3). As expected, upregulated genes showed enrichment in biological processes mainly associated with “response to ER stress”, “protein folding in ER” or downstream pathways such as the “ERAD pathway” or “cell death” (Table S3). To elucidate the upregulated genes by ATF4 and/or XBP1 under ER stress, we adopted a similar approach (UPR induction by Tg) using ATF4 (ATF4-KO) and XBP1 (XBP1-KO) knockout cells and making further comparison of upregulated genes under both conditions with control cells treated with Tg (Figure 6B). We identified 627 genes regulated by XBP1 and 234 ATF4-dependent genes. Surprisingly, we found that 96.6% (226/234) of the ATF4-dependent genes were also regulated by XBP1 and only eight genes (SLC18B1, EFCAB6, CLDN11, MST1, AHRR, BBUF1, GPT2, TMEM184A) were exclusively regulated by ATF4. Gene ontology (GO) analysis showed that the genes regulated only by XBP1 were associated with early processes induced under UPR activation, such as “protein folding”, “response to ER stress”, “IRE1-mediated UPR” and “protein folding”, among others (Figure 6C and Table S4). By contrast, ATF4 in collaboration with XBP1 (ATF4/XBP1) regulated mainly biological pathways related to “negative regulation of cell proliferation”, “immune response” or “cytokine and chemokine signaling pathways” (Figure 6C and Table S4). Thus, XBP1 mainly activates processes that can restore ER homeostasis, but also, in conjunction with ATF4, regulates pathological processes that perpetuate the damage.
XBP1 and ATF4-dependent transcriptional pathways following UPR activation in HK-2 cells. Control HK-2 cells, and XBP1 (XBP1-KO) and ATF4 (ATF4-KO)-deficient cells were untreated or treated with Tg (4 μM, 24 h) before mRNA isolation and RNA sequencing analysis. (A) Volcano plot of genes differentially expressed between HK-2 cells treated with Tg relative to untreated cells. Upregulated and downregulated genes are shown in red and in blue, respectively. The ten most significant gene ontology (GO) pathways of upregulated genes after Tg treatment are shown, with the probability of each category in parentheses. (B) Scheme of the different HK-2 cell types (Ctrl, XBP1-KO, and ATF4-KO) treated with Tg and comparisons between them. “XBP1-dependent UPR genes” (blue) are derived from the genes upregulated in control (Ctrl) cells but not in XBP1 KO cells. “ATF4-dependent UPR genes” (red) are derived from the genes upregulated in control (Ctrl) cells but not in ATF4-KO cells. (C) Venn diagram of the comparison between “XBP1-dependent UPR genes” and “ATF4-dependent UPR genes” and GO analysis of the ten most significant categories of each one according to the number of genes.
To confirm the transcriptional signature induced under ER stress and modulated by the BETs inhibitor, HK-2 cells treated with Tg and JQ1(+) were analyzed by RNA-seq. We found 2515 genes differentially expressed relative to those in the control cells (979 upregulated, 1536 downregulated genes) (data not shown). Additionally, upregulated genes were compared with those in HK-2 cells treated only with Tg to identify the genes downmodulated by JQ1 under UPR activation (Figure 7A). We identified 407 genes whose expression, in the presence of JQ1(+), was not increased by Tg, and which were therefore regulated by BET proteins (Table S5). Using Venn diagrams and the previously determined XBP1- and ATF4/XBP1-dependent signatures, we found that 51.3% (206/401) of the XBP1-regulated genes and 66.3% (150/226) of the XBP1/ATF4-regulated genes were downmodulated by JQ1 (Table S5). Most significant pathways regulated exclusively by XBP1 and downmodulated by JQ1(+) were associated with “protein glycosylation” (OSCT, ALG5), the “defense response to virus” (RPN1), or “positive regulation of IFN-γ and IL-6 production” (IRF1, TLR3, IL6, XBP1) (Figure 7B and Table S5). However, pathways related to the immediate response mechanisms and involved in the degradation process of misfolding proteins aimed to restore the ER stress, such as “ERAD pathway” (DERL3, OS9,), “protein folding in ER” (HSP90/GPR94 and PDIA3), “response to ER stress” (ERN1/IRE1A) or “response to unfolded proteins” MANF) were not regulated by JQ1(+) (Figure S9 and Table S5). These data were further confirmed in the in vitro and in vivo studies previously performed (Figure S10A and S10B).
JQ1 reverts expression of molecular pathways regulated by XBP1 and ATF4 under UPR activation. (A) Scheme showing the comparative of HK-2 cells treated with Tg or the combination of Tg plus JQ1 to determine the signature of genes upregulated by Tg and modulated by JQ1, “JQ1-dependent signature”. (B) Venn diagrams showing comparisons between “XBP1 dependent signature” or “ATF4/ XBP1 dependent signature” and “JQ1 dependent signature” to select the 206 genes regulated by XBP1 and downmodulated by JQ1, and the 150 genes regulated by ATF4/XBP1 and downmodulated by JQ1. GO analysis of the ten most significant categories of genes whose expression was reduced by JQ1 under UPR activation and functional interaction networks of genes downmodulated by JQ1 and regulated by XBP1 or ATF4/XBP1. Network centrality is indicated by the color scale and node size.

Additionally, ATF4/XBP1-regulated and BET-dependent genes were associated with immune and inflammatory responses (IL1A, CD274, CSF3, VNN1), chemotaxis and cytokine signaling pathways (CXCL8, CXCL3, CX3CL1), or the response to unfolded proteins (TRAM1, DERL1) (Figure 7B and Table S5). Some of these genes associated with the inflammatory response (IL6, VNN1), immune defense (IL23, TLR3) and cell death (TRIB3, NUPR1, MST1), and regulated by XBP1 (IL6, IL23A, TLR3, TRIB3, NUPR1), ATF4 (MST1) or both (VNN1, CX3CL1), were significantly reduced after JQ1(+) treatment in the in vitro and in vivo models previously established (Figure 8A and 8B). These results demonstrate that inhibition of BET proteins impairs the transcriptional programs associated with the renal damage triggered by persistent UPR activation, without affecting the initial mechanisms of the ER stress response, thus enabling early repair.
Downregulation of immune, inflammatory and apoptotic genes as consequence of blockage of BET proteins under UPR activation. (A) HK-2 cells were untreated or treated with Tg (4 μM, 24 h) in the presence of JQ1 (+) or its enantiomer, JQ1 (-). (B) Kidney samples were obtained from the sham, IRI and IRI+JQ1 mouse groups (n=5 per group) at 3 (t3) and 24 (t24) h post-reperfusion. Expression of XBP1-dependent genes (IL6, IL23A, TLR3, TRIB3, NURP1), XBP1/ATF4-dependent genes (CXCL3, VNN1) and ATF4-dependent genes (MST1) was measured by RT-PCR analysis; GAPDH was used as a housekeeping gene. Data are summarized as the mean ± SEM of at least three independent experiments. Statistical analyses involved use of the two-tailed Student's paired t-test and Mann-Whitney U test. *p<0.05 vs. control (Ctrl, DMSO) or sham group, and # vs. cells treated with Tg + JQ1 (-) or IRI group.

Discussion
BET proteins are involved in many inflammatory and pro-fibrotic processes triggered during chronic kidney damage. However, their role in the initial stages after acute kidney injury is poorly understood and some apparently contradictory observations have been reported. Here, we demonstrate that pharmacological inhibition of BET proteins blocks the ATF4 and XBP1-mediated maladaptive UPR pathway that is activated under sustained ER stress and is triggered by ischemic renal injury.
Numerous studies had previously suggested that disturbance of ER proteostasis and thereby activation of the UPR pathway is involved in several kidney-associated pathologies [29-31]. Therefore, modulation of the balance between adaptive and maladaptive UPR activation represents a crucial therapeutic strategy for preventing or attenuating the progression of renal damage [32]. However, the involvement of epigenetic mechanisms in regulating and modulating the UPR pathway has not been thoroughly explored. Our group recently reported that the methylation dynamics of H3K9 and H3K27 histones are key to modulating expression of the ATF4 and XBP1 transcription factors under ER stress conditions in tubular cells [33].
BRD4 belongs to a family of transcriptional mediators, the BET proteins, that recognize and bind to acetylated residues to further recruit the p-TEFb elongation complex and activate RNA pol II, leading to gene transcription [34]. Our results reveal that in tubular cells exposed to hypoxia, BRD4 is highly recruited to the promoter region of ATF4 and XBP1 genes, a process abolished in the presence of JQ1. The reduction in the BRD4 recruitment is correlated with an impaired RNA pol II phosphorylation, leading to the inhibited transcription of these genes. BRD4 also binds to enhancer regions, regulating the expression of genes requiring a quick response, such as those mediated by NF-κB or the proto-oncogene c-MYC [35, 36]. The activation of UPR after an ischemic insult needs to be immediately triggered in the cell if the damage is to be repaired and if ER homeostasis is to recover. Wilflingseder et al. [37] recently demonstrated that, after ischemic renal damage, BRD4 is located next to the acetylation of H3K27 residues in “enhancer” and "super-enhancer" regions, allowing rapid enhancement of gene transcription in response to the initial stimulus. These data reinforce our results by revealing the key role of BET proteins in the rapid and efficient response following to an ischemic damage. Moreover, these authors demonstrated that the BRD4 expression in the renal tissue is not modified after ischemia, in accordance with our results where the BRD4 protein levels remained unchanged, both in vitro and in vivo, following UPR pathway activation. Thus, the changes induced by BRD4 after an ischemic insult are mainly mediated by the recruitment of this epigenetic remodeler to the regulatory regions of the ATF4 and XBP1 genes. However, our results differ from those published by Liu et al. [38] who reported a significant increase in BRD4 protein levels during hypoxia and subsequent reoxygenation which were then reduced in the presence of JQ1. Their study concluded that the contribution of JQ1 to alleviate the ischemic renal damage is attributed to the modulation of BRD4 expression, but in any case, they demonstrated the changes in the recruitment of BRD4 to the analyzed genes. Moreover, their animal model of IRI, involving right kidney nephrectomy and 30 min of ischemia in the left kidney, differ from us, so these results should be analyzed with caution.
In our in vitro cellular context, ATF6 expression was induced after UPR activation, which was not attenuated by JQ1 or specific BRD4 blockage. Our data indicate that BRD4 is recruited to the regulatory region of determined genes contributing to modulate their expression. So, the treatment with JQ1 will affect only to the expression of specific genes without global alterations, as evidenced by the unchanged ATF6 protein levels that were detected. The contribution of the three different pathways of the UPR to the renal damage is a complex process, because of their interplay [14]. In fact, we repot that in absence of ATF4, the expression of XBP1 is reduced and both are modulating jointly the expression of several genes. However, neither ATF4 nor XBP1 modulates the expression of ATF6. Thus, it will be the final balance between the adaptive and maladaptive pathways that ultimately dictates the progression of the damage.
Until now, some studies have proposed administration of JQ1 as a possible treatment against AKI induced by IRI, but results remain contradictory. Liu et al. [38] showed that 7 days pre-treatment with JQ1 in an I/R mouse model prevents apoptosis mediated by ROS production, leading to the maintenance of tubular morphology, which favors survival. Likewise, 7 days treatment with MS417, another specific BRD4 inhibitor, followed by unilateral IRI, could attenuate neutrophil infiltration in the kidney, which is associated with less tubular damage and the reduction of KIM-1 and NGAL in the face of early damage [18]. Consistent with these reports, we found that administering JQ1 reduced expression of the neutrophil chemoattractants, CXCL8 (IL8) and CXCL2 (MIP2b), which could be associated with the observed in the lower levels of KIM-1 and NGAL and, eventually, functional recovery. Nevertheless, it has been reported that administering JQ1 at the time of ischemic damage not only fails to alter renal function but also produces higher mortality [37]. However, the progressive delay of JQ1 administration after the ischemia event, has different molecular effects and reduces systemic toxicity. In our experience, administering JQ1 only 1 h after the damage does not affect survival one day after the ischemic insult. Consistent with our results, Wilflingseder et al. [37] reported that surviving mice in the JQ1-treated group had the lowest creatinine value on day 1 post-surgery, indicating recovery of renal function in 20% of mice treated at the time of surgery. It is important to note that the effect of the treatment may depend on the intensity of the damage because the ischemia we performed was more severe and the molecular mechanisms of damage response activated could have differed. In another pathological context, administration of BRD4 inhibitors, 2 or 4 h post-ischemic stroke, protected against brain damage by reducing inflammation, oxidative stress and the activation of NLRP3 inflammasome [39, 40]. More studies are needed to determine the optimal timing for administering BET inhibitors after IR kidney injury.
The role of XBP1 differs with the time since injury and the cellular context. In initial stages, XBP1 is essential for cell adaptation and restoring ER homeostasis, but sustained and aggravated UPR activation triggers oxidative stress and an inflammatory response, aggravating the loss of renal function [41-43]. However, overexpression of ATF4 during renal pathology is mainly associated with development and progression of the disease [44-47]. Accordingly, our data revealed that 96.6% of ATF4-dependent genes are also regulated by XBP1. It is known that ATF6 and XBP1 form heterodimers to induce the expression of the main components of the ERAD response [48, 49], but it is the first time, to our knowledge, that ATF4 is proposed to regulate a coordinated response with XBP1.
Apoptosis and inflammation are essential to the pathogenesis of a wide variety of renal diseases and to the AKI-to-CKD transition [50, 51]. Among UPR-induced genes and JQ1-downregulated genes, TRIB3 and NUPR1 have been shown to reduce apoptosis, favoring autophagy and cell survival [52-54]. Additionally, blockage of IL6 and TLR3 signaling reduces inflammation, and inhibition of the IL23-Th17 axis might reduce the infiltration of T lymphocytes [55, 56]. Others, such as CXCL2, CX3CL1, IL-1A and VNN1, had already been proposed as targets for alleviating renal damage [57-59]. We also observed downregulation of the DDOST gene by JQ1, which is involved in oxidative stress and chronic inflammation in the kidney [60].
It is worth emphasizing that JQ1 does not inhibit the adaptive pathways mediated by ATF4 and XBP1, aimed to restore ER homeostasis. Chaperones involved in degrading misfolding proteins and cell survival pathways (GRP94), molecules key to promote the re-folding of misfolded proteins (PDIA3) and genes such as DERL3 and OS9 associated to ERAD protective mechanism remain unmodified. Likewise, the expression of genes such as MANF, which is a cytoprotective gene with anti-inflammatory and anti-oxidative properties in kidney damage, is not modulated by JQ1 [61-64]. It has been previously reported that genes such as HSPA5 or PDIA3 are only partially regulated by XBP1, and ATF6 (not modulated by JQ1) could be regulating them [65, 66]. Likewise, we cannot obvious that JQ1 could be involved in the regulation of numerous genes that in addition to UPR pathway contribute to the renal damage as it has been previously reported by our group among others [19-22, 37]. Additional studies using ATF4 and XBP1 knockout mice in tubular cells will be required to analyze the specific involvement of these genes in AKI.
In conclusion, our findings identify BRD4 as a key regulator of overexpression of the ATF4 and XBP1 UPR genes after acute kidney injury induced by IRI. Administration of the potent BET protein inhibitor, JQ1, soon after damage might tip the balance between the adaptive and maladaptive UPR pathways. JQ1 could block the molecular pathways related to renal damage such as inflammation, oxidative stress and cell death, while preserve the mechanisms that restores ER homeostasis.
Abbreviations
BET: bromodomain and extra-terminal; CKD: chronic kidney disease; ER: endoplasmic reticulum; IRI: Ischemia reperfusion injury; KO: knockout; TECs: tubular epithelial cells; Tg: Thapsigargin; UPR: Unfolded protein response.
Supplementary Material
Supplementary figures and tables 1-2, table legends for tables 3-5.
Supplementary table 3.
Supplementary table 4.
Supplementary table 5.
Acknowledgements
This study was funded by the Instituto de Salud Carlos III (ISCIII) through the projects “PI19/00184”, “PI20/00639” and “PI22/00738, co-funded by the European Union; Red de Investigación Renal RICORS2040 KIDNEY DISEASE (ISCIII) (RD21/0005/0017, RD21/0005/0001, RD21/0005/0002, and RD21/0005/0003) also founded by the European Union - NextGeneration EU which finance the actions of the Recovery and Resilience Mechanism; “RD16/0009/0020”, co-funded by European Union ERDF, “A way to make Europe”. Gobierno del Principado de Asturias, PCTI-Plan de Ciencia, Tecnología e Innovación 2021-2023 (Grant number IDI/2021/000032). Instituto de Salud Carlos III (ISCIII); CD20/00042 (Sara Borrell 'program) to RRR-D, CM20/00115 (Rio Hortega´ program) to VC-I, co-funded by the European Union, CP18/00106 (Miguel Servet I´ program) to BS-A, co-funded by ESF, “Investing in your future”, and Scientific Foundation of the Spanish Association Against Cancer, INVES222995RODR to RMR. We want to thank Aurora Astudillo (Biobank, Instituto de Investigación Sanitaria del Principado de Asturias, Asturias, Spain) for her help with the immunohistochemical analysis, and James Bradner (Dana-Farber Cancer Institute, Boston, MA, USA) for providing us with the BET bromodomain inhibitor, JQ1.
Author contributions
PD-B, RMR and MLS conducted the in vitro and in vivo experiments and analyzed the data, and helped supervise the production of the manuscript. EB-M, MJR and ML-R and RRR-D performed the in vivo experiments, designed the mouse model and immunohistochemical techniques. ABS provides material, IT-L and VC-I contributed to the in vitro experiments. CRB performed the BRD4 analysis. RMR and AMA helped design, perform and analyze the of RNA sequencing experiments and the histological studies. CD-C, MR-O and FD helped to revise the manuscript. CL-L and BS-A designed the experiments, interpreted the data, wrote the manuscript and provided the resources. All authors approved the final version of the manuscript for publication.
Data statement
Sequencing RNA data have been deposited in the Gene Expression Omnibus under accession code GSE217103.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Takaori K, Nakamura J, Yamamoto S, Nakata H, Sato Y, Takase M. et al. Severity and Frequency of Proximal Tubule Injury Determines Renal Prognosis. Journal of the American Society of Nephrology: JASN. 2016;27:2393-406
2. Maekawa H, Inagi R. Stress Signal Network between Hypoxia and ER Stress in Chronic Kidney Disease. Frontiers in physiology. 2017;8:74
3. Almanza A, Carlesso A, Chintha C, Creedican S, Doultsinos D, Leuzzi B. et al. Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications. The FEBS journal. 2019;286:241-78
4. Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nature reviews Nephrology. 2017;13:681-96
5. Shu S, Zhu J, Liu Z, Tang C, Cai J, Dong Z. Endoplasmic reticulum stress is activated in post-ischemic kidneys to promote chronic kidney disease. EBioMedicine. 2018;37:269-80
6. Gao X, Fu L, Xiao M, Xu C, Sun L, Zhang T. et al. The nephroprotective effect of tauroursodeoxycholic acid on ischaemia/reperfusion-induced acute kidney injury by inhibiting endoplasmic reticulum stress. Basic & clinical pharmacology & toxicology. 2012;111:14-23
7. Liu SH, Yang CC, Chan DC, Wu CT, Chen LP, Huang JW. et al. Chemical chaperon 4-phenylbutyrate protects against the endoplasmic reticulum stress-mediated renal fibrosis in vivo and in vitro. Oncotarget. 2016;7:22116-27
8. Kaneko M, Niinuma Y, Nomura Y. Activation signal of nuclear factor-kappa B in response to endoplasmic reticulum stress is transduced via IRE1 and tumor necrosis factor receptor-associated factor 2. Biological & pharmaceutical bulletin. 2003;26:931-5
9. Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A. et al. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell metabolism. 2012;16:250-64
10. Talty A, Deegan S, Ljujic M, Mnich K, Naicker SD, Quandt D. et al. Inhibition of IRE1alpha RNase activity reduces NLRP3 inflammasome assembly and processing of pro-IL1beta. Cell death & disease. 2019;10:622
11. B'Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y. et al. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic acids research. 2013;41:7683-99
12. Torrence ME, MacArthur MR, Hosios AM, Valvezan AJ, Asara JM, Mitchell JR. et al. The mTORC1-mediated activation of ATF4 promotes protein and glutathione synthesis downstream of growth signals. eLife. 2021 10
13. Yang H, Niemeijer M, van de Water B, Beltman JB. ATF6 Is a Critical Determinant of CHOP Dynamics during the Unfolded Protein Response. iScience. 2020;23:100860
14. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881-91
15. Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nature reviews Drug discovery. 2014;13:337-56
16. Morgado-Pascual JL, Rayego-Mateos S, Tejedor L, Suarez-Alvarez B, Ruiz-Ortega M. Bromodomain and Extraterminal Proteins as Novel Epigenetic Targets for Renal Diseases. Frontiers in pharmacology. 2019;10:1315
17. Tejedor-Santamaria L, Morgado-Pascual JL, Marquez-Exposito L, Suarez-Alvarez B, Rodrigues-Diez RR, Tejera-Munoz A. et al. Epigenetic Modulation of Gremlin-1/NOTCH Pathway in Experimental Crescentic Immune-Mediated Glomerulonephritis. Pharmaceuticals. 2022 15
18. Reid S, Fine N, Bhosle VK, Zhou J, John R, Glogauer M. et al. Inhibition of BRD4 Reduces Neutrophil Activation and Adhesion to the Vascular Endothelium Following Ischemia Reperfusion Injury. International journal of molecular sciences. 2020;21:9620
19. Suarez-Alvarez B, Morgado-Pascual JL, Rayego-Mateos S, Rodriguez RM, Rodrigues-Diez R, Cannata-Ortiz P. et al. Inhibition of Bromodomain and Extraterminal Domain Family Proteins Ameliorates Experimental Renal Damage. Journal of the American Society of Nephrology: JASN. 2017;28:504-19
20. Tan YF, Wang M, Chen ZY, Wang L, Liu XH. Inhibition of BRD4 prevents proliferation and epithelial-mesenchymal transition in renal cell carcinoma via NLRP3 inflammasome-induced pyroptosis. Cell death & disease. 2020;11:239
21. Xiong C, Masucci MV, Zhou X, Liu N, Zang X, Tolbert E. et al. Pharmacological targeting of BET proteins inhibits renal fibroblast activation and alleviates renal fibrosis. Oncotarget. 2016;7:69291-308
22. Zhou B, Mu J, Gong Y, Lu C, Zhao Y, He T. et al. Brd4 inhibition attenuates unilateral ureteral obstruction-induced fibrosis by blocking TGF-beta-mediated Nox4 expression. Redox biology. 2017;11:390-402
23. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15-21
24. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics. 2011;12:323
25. Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome research. 2012;22:1760-74
26. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic acids research. 2015;43:D447-52
27. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research. 2003;13:2498-504
28. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44-57
29. Fan Y, Lee K, Wang N, He JC. The Role of Endoplasmic Reticulum Stress in Diabetic Nephropathy. Current diabetes reports. 2017;17:17
30. Mohammed-Ali Z, Cruz GL, Dickhout JG. Crosstalk between the unfolded protein response and NF-kappaB-mediated inflammation in the progression of chronic kidney disease. Journal of immunology research. 2015;2015:428508
31. Noh MR, Kim JI, Han SJ, Lee TJ, Park KM. C/EBP homologous protein (CHOP) gene deficiency attenuates renal ischemia/reperfusion injury in mice. Biochimica et biophysica acta. 2015;1852:1895-901
32. Cao AL, Wang L, Chen X, Wang YM, Guo HJ, Chu S. et al. Ursodeoxycholic acid and 4-phenylbutyrate prevent endoplasmic reticulum stress-induced podocyte apoptosis in diabetic nephropathy. Laboratory investigation; a journal of technical methods and pathology. 2016;96:610-22
33. Diaz-Bulnes P, Saiz ML, Corte-Iglesias V, Rodrigues-Diez RR, Bernardo Florez A, Ruiz Bernet C. et al. Demethylation of H3K9 and H3K27 Contributes to the Tubular Renal Damage Triggered by Endoplasmic Reticulum Stress. Antioxidants. 2022;11:1355
34. Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. The Journal of biological chemistry. 2007;282:13141-5
35. Brown JD, Lin CY, Duan Q, Griffin G, Federation A, Paranal RM. et al. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Molecular cell. 2014;56:219-31
36. Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR. et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320-34
37. Wilflingseder J, Willi M, Lee HK, Olauson H, Jankowski J, Ichimura T. et al. Enhancer and super-enhancer dynamics in repair after ischemic acute kidney injury. Nature communications. 2020;11:3383
38. Liu H, Wang L, Weng X, Chen H, Du Y, Diao C. et al. Inhibition of Brd4 alleviates renal ischemia/reperfusion injury-induced apoptosis and endoplasmic reticulum stress by blocking FoxO4-mediated oxidative stress. Redox biology. 2019;24:101195
39. Liu L, Yang C, Lavayen BP, Tishko RJ, Larochelle J, Candelario-Jalil E. Targeted BRD4 protein degradation by dBET1 ameliorates acute ischemic brain injury and improves functional outcomes associated with reduced neuroinflammation and oxidative stress and preservation of blood-brain barrier integrity. Journal of neuroinflammation. 2022;19:168
40. Zhou Y, Gu Y, Liu J. BRD4 suppression alleviates cerebral ischemia-induced brain injury by blocking glial activation via the inhibition of inflammatory response and pyroptosis. Biochemical and biophysical research communications. 2019;519:481-8
41. Ferre S, Deng Y, Huen SC, Lu CY, Scherer PE, Igarashi P. et al. Renal tubular cell spliced X-box binding protein 1 (Xbp1s) has a unique role in sepsis-induced acute kidney injury and inflammation. Kidney international. 2019;96:1359-73
42. Madhusudhan T, Wang H, Dong W, Ghosh S, Bock F, Thangapandi VR. et al. Defective podocyte insulin signalling through p85-XBP1 promotes ATF6-dependent maladaptive ER-stress response in diabetic nephropathy. Nature communications. 2015;6:6496
43. Zhang J, Zhang J, Ni H, Wang Y, Katwal G, Zhao Y. et al. Downregulation of XBP1 protects kidney against ischemia-reperfusion injury via suppressing HRD1-mediated NRF2 ubiquitylation. Cell death discovery. 2021;7:44
44. Liang Q, Liu T, Guo T, Tao W, Chen X, Chen W. et al. ATF4 promotes renal tubulointerstitial fibrosis by suppressing autophagy in diabetic nephropathy. Life sciences. 2021;264:118686
45. Liu J, Huang K, Cai GY, Chen XM, Yang JR, Lin LR. et al. Receptor for advanced glycation end-products promotes premature senescence of proximal tubular epithelial cells via activation of endoplasmic reticulum stress-dependent p21 signaling. Cellular signalling. 2014;26:110-21
46. Luo B, Lin Y, Jiang S, Huang L, Yao H, Zhuang Q. et al. Endoplasmic reticulum stress eIF2alpha-ATF4 pathway-mediated cyclooxygenase-2 induction regulates cadmium-induced autophagy in kidney. Cell death & disease. 2016;7:e2251
47. Wang MG, Fan RF, Li WH, Zhang D, Yang DB, Wang ZY. et al. Activation of PERK-eIF2alpha-ATF4-CHOP axis triggered by excessive ER stress contributes to lead-induced nephrotoxicity. Biochimica et biophysica acta Molecular cell research. 2019;1866:713-26
48. Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C. et al. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell reports. 2013;3:1279-92
49. Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H. et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Developmental cell. 2007;13:365-76
50. Priante G, Gianesello L, Ceol M, Del Prete D, Anglani F. Cell Death in the Kidney. International journal of molecular sciences. 2019;20:3598
51. Sato Y, Yanagita M. Immune cells and inflammation in AKI to CKD progression. American journal of physiology Renal physiology. 2018;315:F1501-F12
52. Ding WY, Li WB, Ti Y, Bi XP, Sun H, Wang ZH. et al. Protection from renal fibrosis, putative role of TRIB3 gene silencing. Experimental and molecular pathology. 2014;96:80-4
53. Wang W, Cheng J, Sun A, Lv S, Liu H, Liu X. et al. TRB3 mediates renal tubular cell apoptosis associated with proteinuria. Clinical and experimental medicine. 2015;15:167-77
54. Zhou R, Liao J, Cai D, Tian Q, Huang E, Lu T. et al. Nupr1 mediates renal fibrosis via activating fibroblast and promoting epithelial-mesenchymal transition. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2021;35:e21381
55. Hu X, Chen L, Li T, Zhao M. TLR3 is involved in paraquat-induced acute renal injury. Life sciences. 2019;223:102-9
56. Paust HJ, Turner JE, Steinmetz OM, Peters A, Heymann F, Holscher C. et al. The IL-23/Th17 axis contributes to renal injury in experimental glomerulonephritis. Journal of the American Society of Nephrology: JASN. 2009;20:969-79
57. Chen J, Lu H, Wang X, Yang J, Luo J, Wang L. et al. VNN1 contributes to the acute kidney injury-chronic kidney disease transition by promoting cellular senescence via affecting RB1 expression. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2022;36:e22472
58. Furuichi K, Gao JL, Murphy PM. Chemokine receptor CX3CR1 regulates renal interstitial fibrosis after ischemia-reperfusion injury. The American journal of pathology. 2006;169:372-87
59. Schunk SJ, Triem S, Schmit D, Zewinger S, Sarakpi T, Becker E. et al. Interleukin-1alpha Is a Central Regulator of Leukocyte-Endothelial Adhesion in Myocardial Infarction and in Chronic Kidney Disease. Circulation. 2021;144:893-908
60. Wu XQ, Zhang DD, Wang YN, Tan YQ, Yu XY, Zhao YY. AGE/RAGE in diabetic kidney disease and ageing kidney. Free radical biology & medicine. 2021;171:260-71
61. Hou C, Mei Q, Song X, Bao Q, Li X, Wang D. et al. Mono-macrophage-Derived MANF Protects Against Lipopolysaccharide-Induced Acute Kidney Injury via Inhibiting Inflammation and Renal M1 Macrophages. Inflammation. 2021;44:693-703
62. Kim Y, Lee H, Manson SR, Lindahl M, Evans B, Miner JH. et al. Mesencephalic Astrocyte-Derived Neurotrophic Factor as a Urine Biomarker for Endoplasmic Reticulum Stress-Related Kidney Diseases. Journal of the American Society of Nephrology: JASN. 2016;27:2974-82
63. Li X, Zou Y, Fu YY, Xing J, Wang KY, Wan PZ. et al. A-Lipoic Acid Alleviates Folic Acid-Induced Renal Damage Through Inhibition of Ferroptosis. Frontiers in physiology. 2021;12:680544
64. Zhu J, Fu M, Gao J, Dai G, Guan Q, Du C. Upregulation of Thioredoxin Reductase 1 Expression by Flavan-3-Ols Protects Human Kidney Proximal Tubular Cells from Hypoxia-Induced Cell Death. Antioxidants. 2022;11:1399
65. Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Molecular and cellular biology. 2003;23:7448-59
66. Vidal RL, Figueroa A, Court FA, Thielen P, Molina C, Wirth C. et al. Targeting the UPR transcription factor XBP1 protects against Huntington's disease through the regulation of FoxO1 and autophagy. Human molecular genetics. 2012;21:2245-62
Author contact
![]() Corresponding author: Carlos Lopez-Larrea, inmunoes; Beatriz Suarez-Alvarez beatriz.suarezes.
Corresponding author: Carlos Lopez-Larrea, inmunoes; Beatriz Suarez-Alvarez beatriz.suarezes.