Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(5):1688-1704. doi:10.7150/ijbs.93120 This issue Cite
Research Paper
Interruption of p38MAPK-MSK1-CREB-MITF-M pathway to prevent hyperpigmentation in the skin
Song-Hee Kim1,*, Jiyeon Lee1,*, Jihye Jung1, Ga Hyun Kim1, Cheong-Yong Yun1, Sang-Hun Jung2, Bang Yeon Hwang1, Jin Tae Hong1, Sang-Bae Han1, Jae-Kyung Jung1, ![]() , Youngsoo Kim1,
, Youngsoo Kim1, ![]()
1. College of Pharmacy, Chungbuk National University, Cheongju 28160, Korea.
2. College of Pharmacy, Chungnam National University, Daejeon 34134, Korea.
*Co-first authorship: S.-H.K. and J.L. equally contributed to this study.
Received 2023-12-11; Accepted 2024-2-5; Published 2024-2-17
Abstract

Background: Melanocortin 1 receptor (MC1R), a receptor of α-melanocyte-stimulating hormone (α-MSH), is exclusively present in melanocytes where α-MSH/MC1R stimulate melanin pigmentation through microphthalmia-associated transcription factor M (MITF-M). Toll-like receptor 4 (TLR4), a receptor of endotoxin lipopolysaccharide (LPS), is distributed in immune and other cell types including melanocytes where LPS/TLR4 activate transcriptional activity of nuclear factor (NF)-κB to express cytokines in innate immunity. LPS/TLR4 also up-regulate MITF-M-target melanogenic genes in melanocytes. Here, we propose a molecular target of antimelanogenic activity through elucidating inhibitory mechanism on α-MSH-induced melanogenic programs by benzimidazole-2-butanol (BI2B), an inhibitor of LPS/TLR4-activated transcriptional activity of NF-κB.
Methods: Ultraviolet B (UV-B)-irradiated skins of HRM-2 hairless mice and α-MSH-activated melanocyte cultures were employed to examine melanogenic programs.
Results: Topical treatment with BI2B ameliorated UV-B-irradiated skin hyperpigmentation in mice. BI2B suppressed the protein or mRNA levels of melanogenic markers, such as tyrosinase (TYR), MITF-M and proopiomelanocortin (POMC), in UV-B-exposed and pigmented skin tissues. Moreover, BI2B inhibited melanin pigmentation in UV-B-irradiated co-cultures of keratinocyte and melanocyte cells and that in α-MSH-activated melanocyte cultures. Mechanistically, BI2B inhibited the activation of cAMP response element-binding protein (CREB) in α-MSH-induced melanogenic programs and suppressed the expression of MITF-M at the promoter level. As a molecular target, BI2B primarily inhibited mitogen-activated protein kinase (MAPK) kinase 3 (MKK3)-catalyzed kinase activity on p38MAPK. Subsequently, BI2B interrupted downstream pathway of p38MAPK-mitogen and stress-activated protein kinase-1 (MSK1)-CREB-MITF-M, and suppressed MITF-M-target melanogenic genes, encoding enzymes TYR, TYR-related protein-1 (TRP-1) and dopachrome tautomerase (DCT) in melanin biosynthesis, and encoding proteins PMEL17 and Rab27A in the transfer of pigmented melanosomes to the overlaying keratinocytes in the skin.
Conclusion: Targeting the MKK3-p38MAPK-MSK1-CREB-MITF-M pathway was suggested as a rationale to inhibit UV-B- or α-MSH-induced facultative melanogenesis and as a strategy to prevent acquired pigmentary disorders in the skin.
Keywords: Melanin pigmentation, UV-B, α-MSH, MKK3, benzimidazole-2-butanol
Introduction
Imbalanced overproduction and patched distribution of heavily pigmented melanosomes can cause acquired pigmentary diseases, such as melasma and freckles [1]. Facultative melanogenesis is inducible and determines the degree of hyperpigmentation in the skin, in which ultraviolet B (UV-B) irradiation and melanogenic hormones are major stimulating factors [2, 3]. Natural product/chemical-based control of facultative melanogenesis has been applied for pharmacological treatment of acquired pigmentary diseases in the skin as well as for cosmetic purpose to obtain a lightener skin appearance [4].
UV-B irradiation from sunlight up-regulates the expression of melanogenic hormones, such as α-melanocyte-stimulating hormone (α-MSH), and stem cell factor at epidermal keratinocytes in the skin [2]. In a paracrine fashion, melanogenic hormones from keratinocytes bind to their specific receptors in melanocytes, which transmit melanocyte-specific signals to express the microphthalmia-associated transcription factor M (MITF-M), thus regulating melanosome pigmentation and the transfer of pigmented melanosomes to keratinocytes at the overlaying epidermis to induce hyperpigmentation in the skin [2, 5]. Moreover, UV-B irradiation onto melanocytes transmits melanogenic signals through mitogen-activated protein kinase (MAPK) family, such as p38MAPK, extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK), in the expression of MITF-M [6, 7].
α-MSH binds to the extracellular domain of melanocortin 1 receptor (MC1R) at epidermal melanocytes in the skin, which initiates intracellular signals to increase cAMP levels via activation of adenylate cyclase [3]. The second messenger cAMP binds to the regulatory subunit (R) of inactive protein kinase A (PKA) holoenzyme (R2C2) and dissociates the catalytic subunit (C), thus gaining the kinase activity of PKA [8]. MITF-M is inducible in response to α-MSH stimulation or UV-B irradiation [5]. The promoter region of MITF-M encodes several cis-acting consensus, including the cAMP response DNA element (CRE) and the lymphoid enhancer binding factor 1 (LEF1)-binding motifs [9-11]. Transcriptional activation of MITF-M at the promoter level is maximally up-regulated by specific interactions between the CRE and the heteromer of CRE-binding protein (CREB) and CREB regulated transcription coactivators (CRTCs) as well as those between the LEF1-binding motifs and the heteromer of LEF1 and β-catenin [9-11]. Nuclear CREB and LEF1 directly interact with corresponding cis-acting consensus in the promoter region of MITF-M [9-11]. After translocation to the nucleus, CRTCs or β-catenin respectively coactivate CREB or LEF1 that already occupy the promoter region of MITF-M [9-11].
Transcriptional activities of CREB, CRTCs and β-catenin are tightly regulated by posttranslational modification of reversible phosphorylation and dephosphorylation [9, 10]. PKA activity in the axis with cAMP directly phosphorylates nuclear CREB at the S133 residue, a co-substrate of the kinase activity of the mitogen- and stress-activated protein kinase-1 (MSK1) in the axis with p38MAPK or that of p90 ribosomal S6 kinase (p90RSK) in the axis with ERK [12-14]. The phosphor (p)-CREB is mostly dephosphorylated in the nucleus by protein phosphatase 2A, which cannot alter the affinity of p-CREB or CREB to the CRE in promoter region of MITF-M [15]. In α-MSH-induced melanogenic programs, PKA directly phosphorylates the salt-inducible kinases (SIKs), the AMP-activated protein kinase (AMPK), and the glycogen synthase kinase-3β (GSK3β) [16-18]. Kinase activities of SIKs and AMPK phosphorylate the conserved Ser residues of CRTCs (S64, S151 and S245 residues in CRTC1; S70, S171 and S275 residues in CRTC2; and S62, S162 and S273 residues in CRTC3) in the cytosol, which are reversibly dephosphorylated by calcineurin, a Ca2+/calmodulin-dependent protein phosphatase [19, 20]. UV-B-induced, cAMP-dependent or anisomycin-activated kinase activity of JNK also phosphorylates the conserved Ser residues of CRTC3, leading to the tethering of p-CRTC3 with 14-3-3 protein and sequestering CRTC3 in the cytosol [21, 22]. Kinase activity of ERK directly phosphorylates CRTC3 at the S391 residues, which facilitates the recruitment of calcineurin, and dephosphorylates p-CRTC3 at the S162 and S273 residues, thus resulting in the dissociation of 14-3-3 protein [20]. In α-MSH-induced melanogenic programs, kinase activity of GSK3β in the axis with PKA phosphorylates β-catenin at the S675 residue in the cytosol, thus stabilizing p-β-catenin from degradation by ubiquitination and proteasome system [18, 23].
Human melanocytes are not simply melanin pigment-producing cells but also implicated in innate immunity through Toll-like receptors (TLRs) [24]. Endotoxin lipopolysaccharide (LPS), an agonistic ligand of TLR4, stimulates nuclear translocation of nuclear factor-κB (NF-κB) after degradation of inhibitor of NF-κB (IκB) to up-regulate NF-κB-dependent expression of cytokines in immune and other cell types including melanocytes [24, 25]. In melanocytes, LPS/TLR4 transmit intracellular signals to regulate MITF-M-dependent expression of melanogenic genes, in addition to NF-κB-associated innate immunity [24, 25].
In our previous work, a derivative of benzimidazole-2-butanol (BI2B, Figure 1A) inhibits LPS/TLR4-activated transcriptional activity of NF-κB in immune cells [26]. Moreover, BI2B inhibited LPS/TLR4-induced melanin production in melanocytes (Figure S1). We then hypothesized that BI2B could negatively regulate intracellular signals of melanin production coupling with those of NF-κB activation. In the current study, we propose a molecular target of antimelanogenic activity by elucidating the inhibitory activity and mechanism of BI2B on facultative melanogenesis via UV-B irradiation or α-MSH stimulation. Topical treatment with BI2B ameliorated hyperpigmentation in UV-B-irradiated skins of mice. Moreover, BI2B inhibited melanin pigmentation in UV-B-irradiated co-cultures of keratinocyte and melanocyte cells and α-MSH-activated melanocytes. As a mechanism, BI2B primarily inhibited MAPK kinase 3 (MKK3)-catalyzed kinase activity on p38MAPK, thus interrupting the downstream pathway of p38MAPK-MSK1-CREB-MITF-M in α-MSH-induced melanogenic programs.
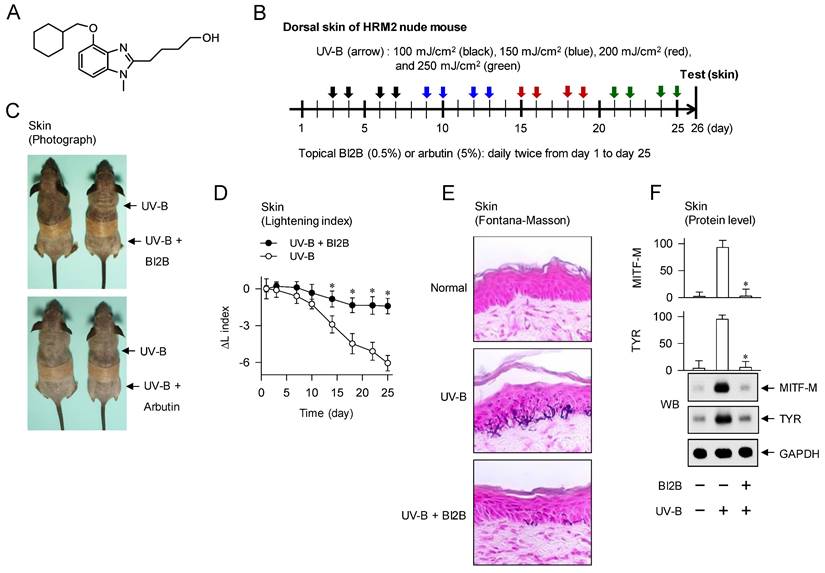
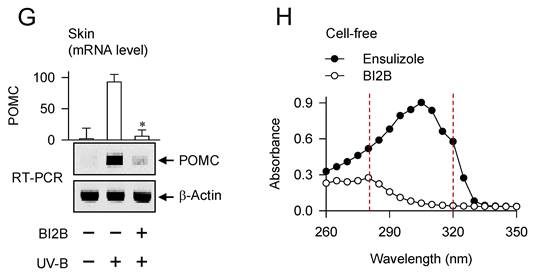
Effect of BI2B on skin hyperpigmentation in vivo. (A) Chemical structure of BI2B. (B) Experimental protocol of skin hyperpigmentation. The dorsal skins of HRM2 hairless mice were topically treated with BI2B (0.5%) or arbutin (5%) in a daily twice regimen for consecutive 25 days, and irradiated with increasing doses (100-250 mJ/cm2) of UV-B at the time points indicated by arrows. Melanogenic markers were examined in alive or biopsied skins at day 26. (C) BI2B inhibited UV-B irradiated visual hyperpigmentation in alive skins, as did arbutin. (D) BI2B restored UV-B-induced change of lightening (ΔL) index in alive skins during 25 days to normal status. (E) BI2B inhibited the deposit of melanin granules in UV-B-exposed and pigmented skin tissues. (F, G) BI2B suppressed the protein levels of MITF-M or TYR and mRNA levels of POMC in UV-B-exposed and pigmented skin tissues. (H) BI2B did not absorb UV-B light, in which ensulizole was employed as a positive control agent. Three independent tests with two mice per group. *P < 0.05 vs. UV-B alone.
Materials and Methods
Drugs and chemicals
BI2B (purity, >95%) was prepared as described previously [26]. Melanogenic stimuli were α-MSH (Sigma-Aldrich, M4125) as hormone; and N6,2'-O-dibutyryl-cAMP (db-cAMP; Sigma-Aldrich, D0627) as cAMP agonist. Pharmacological agents were arbutin (Sigma-Aldrich, A4256) as skin whitener; ensulizole (Sigma-Aldrich, 437166) as ingredient of sun blocker; KT 5720 (Sigma-Aldrich, K3761) as PKA inhibitor; SB2020190 (Sigma-Aldrich, S7067) as p38MAPK inhibitor; SB203580 (Sigma-Aldrich, S8307) as p38MAPK inhibitor; SB-747651A (Tocris, 4630) as MSK1 inhibitor; and RMM-46 (Tocris, 5909) as MSK1 inhibitor.
Skin hyperpigmentation by UV-B irradiation
HRM-2 hairless mice were obtained from Central Lab Animals (Eumsung, Korea), and housed in the facility under relative humidity 55±5%, temperature 22±2°C, and a 12-h light-dark cycle. Experimental protocol of hyperpigmentation in the skin is shown in Figure 1B. BI2B (0.5%) or arbutin (5%) was dissolved in a vehicle of propylene glycol: ethyl alcohol: H2O (5: 3: 2) and topically treated to the dorsal skins of mice in a daily twice regimen for consecutive 25 days. Increasing doses (100-250 mJ/cm2) of UV-B were irradiated at the time points indicated by arrows. Lightening index of UV-B-exposed and pigmented skins in alive mice was measured once per every three days using a chromameter. The UV-B-exposed and pigmented skins were biopsied at day 26. Skin tissues were fixed in p-formaldehyde, embedded in paraffin, sectioned in a 5-μm thickness, and reacted with Fontana-Masson silver nitrate (Scytek, FMS-1) to stain melanin pigments in deposit. Protein extracts or total RNAs were separted from skin tissues, and subjected to Western blot (WB) or RT-PCR analysis. All procedures for animal study were approved as permission number CBNUR-1253-19 by the Animal Experimentation Ethics Committee of CBNU institute.
Cell culture
B16F0 mouse melanoma cells (ATCC, CRL-6322) or HaCaT human keratinocyte cells (AddexBio, T0020001) were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, D2902) containing 10% fetal bovine serum (Coring, 35-01-CV) and antibiotic-antimycotic solution (Gibco, 15240062) at 37°C in a 5% CO2 atmosphere. For co-culture, B16F0 cells were seeded on 6-well plates and HaCaT cells on the trans-well insert containing a pore size of 0.4 μm (Corning, 3412). HaCaT cells on the trans-well inserts were transferred to B16F0 cells in the 6-well plates, and cultured in the same condition as described above. Human epidermal melanocyte (HEM; Thermo Fischer Scientific, C1025C) was cultured in medium 254 (Gibco, M254) supplemented with melanocyte growth supplement (Gibco, S002) at 37°C in a 5% CO2 atmosphere.
Western blot analysis
Protein extracts were prepared from skin tissues, HEM or B16F0 cells, resolved on sodium dodecyl sulfate (SDS)-acrylamide gels by electrophoresis, and transferred to the membranes of PVDF (Merck-Millipore, IPVH00010) using a semidry blotting apparatus. After blocking with 5% non-fat milk (BD Biosciences, 232100) in Tris-buffered saline (TBS) with Tween 20 (Biosesang, TR2007-100-74), blots were reacted with primary antibody overnight at 4°C. After washing with TBS containing Tween 20, blots were reacted with horseradish peroxidase (HRP)-labeled secondary antibody for 1-3 h and visualized the immune complex by chemiluminescence kit (GE Healthcare, RPN2232). Primary antibodies were anti-MITF-M (Abcam, ab12039); anti-TYR (Santa Cruz Biotechnology, sc-7833); anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Cell Signaling Technology, 5174); anti-p-CREB (Cell Signaling Technology, 9198); anti-CREB (Cell Signaling Technology, 9197); anti-p-CRTC1 (Cell Signaling Technology, 3359); anti-CRTC1 (Cell Signaling Technology, 2587); anti-p-β-catenin (Cell Signaling Technology, 9567); anti-β-catenin (Cell Signaling Technology, 9562); anti-SRY-box transcription factor 10 (SOX10; Santa Cruz Biotechnology, sc-365692); anti-lamin A (Santa Cruz Biotechnology, sc-20680); anti-p-p38MAPK (Cell Signaling Technology, 9211); anti-p38MAPK (Cell Signaling Technology, 9212); anti-p-ERK (Cell Signaling Technology, 9101); anti-ERK (Cell Signaling Technology, 9102); anti-p-JNK (Cell Signaling Technology, 9251); anti-JNK (Cell Signaling Technology, 9252); anti-p-MSK1 (Cell Signaling Technology, 9595); anti-MSK1 (Cell Signaling Technology, 3489); anti-p-MKK3/6 (Cell Signaling Technology, 9236); anti-MKK3/6 (Cell Signaling Technology, 8535); anti-tyrosinase (TYR)-related protein-1 (TRP-1; Santa Cruz Biotechnology, sc-10446); and anti-dopachrome tautomerase (DCT; Santa Cruz Biotechnology, sc-10451). Secondary antibodies were HRP-labeled anti-rabbit IgG (Thermo Fisher Scientific, 31460) and HRP-labeled anti-mouse IgG (Thermo Fisher Scientific, 31430).
RT-PCR analysis
Total RNAs were extracted from skin tissues, HEM or B16F0 cells using NucleoZOL kit (Macherey-Nagel, 740404). Complementary DNA (cDNA) was prepared from 1 ug of total RNAs using a reverse transcription kit (Intron, 25087) and subjected to PCR analysis to determine the levels of each transcript, such as POMC, MITF-M, CREB, CRTC1, β-catenin, TYR, TRP-1, DCT, PMEL17, Rab27A or β-catenin. Nucleotide sequences of PCR primers were described in Table S1. RT-PCR condition was as follows: reverse transcription at 95°C for 3 min followed by 25-30 cycles of PCR at 94°C for 30 sec (denaturation), 50-60°C for 1 min (annealing), and 72°C for 1 min (extension). Amplified transcripts were resolved on agarose gels by electrophoresis and visualized by staining with EcoDye (Biofact, ES301).
Quantification of melanin pigments
B16F0 or HEM cells were respectively seeded on 96-well or 6-well plates, and stimulated with 100 nM α-MSH or 3 mM db-cAMP for 72 h. Extracellular melanin pigments were collected from the medium. To extract intracellular melanin pigments, the cells were disrupted using 0.85 N NaOH and 20% dimethyl sulfoxide (DMSO; Sigma-Aldrich, 34869) at 80℃. The extracellular and intracellular melanin pigments were quantified by measuring the absorbance values at wavelength 405 nm.
Cell viability assay
B16F0 or HEM cells were cultured with BI2B for 72 h, and reacted with 0.5 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich, M5655) for another 30 min. Insoluble formazan crystals were dissolved in 50% DMSO and their absorbance values were measured at wavelength 590 nm.
Luciferase reporter assay
B16F0 cells were transfected with plasmid construct encoding firefly luciferase reporter fused to the promoter region of MITF-M (MITF-M-Luc) or that of TYR (TYR-Luc) along with Renilla control vector using lipofectamine kit (Invitrogen, 11668), and incubated for 48 h. The cells harboring each reporter plasmid were stimulated with 100 nM α-MSH in the presence of BI2B for 20 h, and harvested for dual-luciferase assays (Promega, E1910). The promoter activity of MITF-M or TYR was measured by Firefly luciferase activity, and normalized by Renilla luciferase activity, indicating the transfection efficiency.
Small interfering (si) RNA assay
The siRNAs against CREB, CRTC1 or β-catenin were supplied from Bioneer (Cheongju, Korea), and their nucleotide sequences were 5'-GAUUCACAGGAGUCUGUGG-3' (CREB), 5'-UGGACAGAGUAUAUCGUGA-3' (CRTC1), and 5'-CUGUUGGAUUGAUUCGAAA-3' (β-catenin). B16F0 cells were transfected with each siRNA using lipofectamine kit, and incubated for 48 h, and stimulated with α-MSH for 2 h. Total RNAs were subjected to RT-PCR analysis of MITF-M, CREB, CRTC1 and β-catenin as described above.
Confocal microscopy
B16F0 cells were seeded on poly-Lys-coated glass slides, fixed with p-formaldehyde, and permeabilized with PBS containing 0.5% Triton X-100 (Biosesang, PR4007) for 10 min. The cells on glass slides were incubated with PBS containing 1% bovine serum albumin (BSA; Sigma-Aldrich, A9647), and reacted with each antibody against CREB, p-CREB or CRTC1. After washing, the cells on glass slides were stained with Alexa Fluor 488-conjugated secondary antibody (Invitrogen, A-11034) and also with Vecashield antifade medium containing 4',6-diamidino-2-phenylindole (DAPI; Vector Lab, H1200), and examined using confocal microscope (Carl Zeiss LSM 980).
Kinase assay
To determine cellular PKA activity, lysates of B16F0 cells were reacted with kemptide (Promega, V5601) as PKA-specific peptide substrate, and 10 μCi [γ-32P]-ATP (Perkin Elmer, NEG002A) as a probe for 30 min at 30°C. To determine in vitro kinase activity of MKK3 in cell-free conditions, catalytically active rhMKK3 (SignalChem, M04-10G) was reacted with p38αMAPK (SignalChem, M37-14G) as a substrate in presence of 10 μCi [γ-32P]-ATP as a probe for 30 min at 30°C. Similarly, catalytically active TGF-β-activated kinase 1 (rhTAK1)-TAK1 binding protein 1 (TAB1) (Promega, V4088) was reacted with major basic protein (MBP; SignalChem, M42-51) as an exogenic substrate in presence of 10 μCi [γ-32P]-ATP as a probe for 30 min at 30°C. The mixtures of kinase reactions were spotted on a P81 phosphocellulose paper and washed with 0.75% H3PO4 followed by 100% acetone. Radioactivity on the paper was determined as count per min (cpm) using scintillation counting to quantify the kinase activity.
Statistical analysis
Results are expressed as the mean ± SEM of 3 independent experiments (n = 3). Data were statistically analyzed using the analysis of variance (ANOVA) or Student's t-test. P < 0.05 were considered as statistically significant.
Results
Amelioration of UV-B-irradiated hyperpigmentation in the skin of mice by BI2B
To examine antimelanogenic activity of BI2B in vivo, we employed hyperpigmentation in UV-B-irradiated skins of mice. The dorsal skins of HRM-2 hairless mice were topically treated with BI2B (0.5%) or arbutin (5%, a dose approved by Korea FDA) in a daily twice regimen for consecutive 25 days, and stimulated with UV-B irradiation at the time points indicated by arrows (Figure 1B). UV-B-exposed and pigmented skins were biopsied at day 26, and their melanogenic markers were evaluated.
Topical treatment with BI2B decreased visual hyperpigmentation in UV-B-irradiated alive skins at day 26, as did that with arbutin (Figure 1C). Arbutin is a Korea FDA-approved lightening drug, thus employing it as a positive control agent in this study. The percentage change of lightening (ΔL) index, inversely correlating with the degree of hyperpigmentation, was gradually decreased in UV-B-irradiated alive skins during 25 days, which was restored to basal status by treatment with BI2B (Figure 1D). UV-B-irradiated skins were then biopsied at day 26. To examine the deposit of melanin granules, skin tissues were embedded in paraffin, sectioned in a 5-μm thickness, and stained with Fontana-Masson silver nitrate. Treatment with BI2B decreased the content of black-colored melanin granules in UV-B-irradiated skins, more evident in the border between epidermis and dermis where melanocytes produce pigmented melanosomes (Figure 1E). The protein or mRNA levels of melanogenic markers, such as MITF-M, TYR and proopiomelanocortin (POMC) were up-regulated in UV-B-irradiated skins, which were suppressed by treatment with BI2B (Figure 1F and G). However, BI2B did not absorb the UV-B light at wavelengths 280-320 nm, in which ensulizole is a UV-B-protecting ingredient and was employed as a positive control agent (Figure 1H). These results suggest that topical treatment with BI2B could ameliorate hyperpigmentation in UV-B-irradiated skins of mice.
Inhibition of UV-irradiated, α-MSH-induced, or cAMP-dependent melanin production in melanocyte culture by BI2B
α-MSH is secreted from keratinocytes and melanocytes at the epidermis of skin after posttranslational processing of the precursor protein POMC that is up-regulated in response to UV-B [2, 27]. In paracrine and autocrine fashions, α-MSH binds to its receptor MC1R at melanocytes, which stimulates cAMP-dependent melanin production to pigment the melanosomes [3, 8].
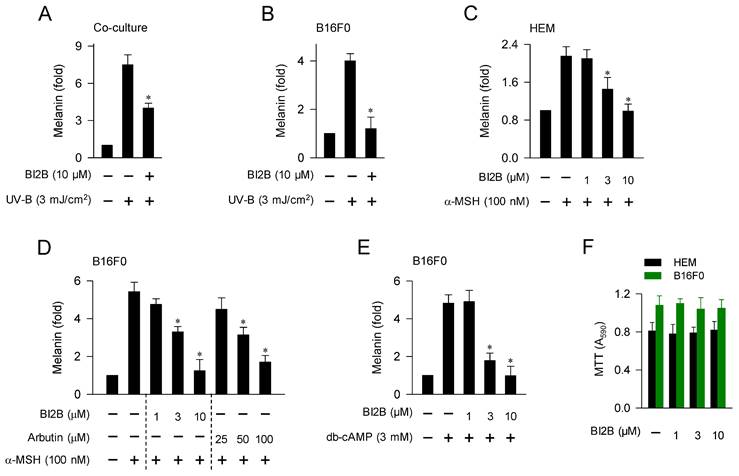
We then examined whether antimelanogenic activity of BI2B in vivo could be translated in the facultative melanogenesis in vitro using cell-culture models. Upon a single irradiation with UV-B, co-cultures of HaCaT keratinocyte and B16F0 melanoma cells stimulated melanin production, which was inhibited by treatment with BI2B (Figure 2A). In this co-culture model, melanogenic hormones including α-MSH might be secreted from HaCaT cells and interacted with their receptors including MC1R in B16F0 cells to stimulate melanin production. Monocultures of HEM or B16F0 cells also stimulated melanin production upon a single irradiation with UV-A or UV-B, which were also inhibited by treatment with BI2B (Figure 2B; Figure S2). In this monoculture model, UV-B might stimulate α-MSH-induced melanin production in an autocrine manner or might activate MAPKs-signaled melanogenic programs in melanocytes. Treatment with BI2B inhibited α-MSH-induced melanin production in HEM or B16F0 cells (Figure 2C and D), suggesting its antimelanogenic activity regardless of the melanocyte origins from human and mouse. BI2B showed more effective activity (approximately 10-fold) than arbutin in the comparison with their IC50 values on α-MSH-induced melanin production (Figure 2D), which was applied to select the dose of BI2B in vivo (Figure 1B). Treatment with BI2B also inhibited db-cAMP-induced melanin production in B16F0 cells (Figure 2E). The db-cAMP was employed as a cAMP agonist. However, BI2B at 1-10 μM did not alter MTT assay-based viability of HEM or B16F0 cells (Figure 2F), excluding from non-specific cytotoxicity. These results suggest that BI2B could inhibit various models of facultative melanogenesis in melanocyte cultures. We then focused on α-MSH-induced melanogenic programs in melanocytes to elucidate the mechanism of BI2B on antimelanogenic activity.
Effect of BI2B on melanin production in cell-culture model. (A) Co-cultures of HaCaT keratinocyte and B16F0 melanoma cells were stimulated with a single irradiation of UV-B and treated with BI2B for 72 h. BI2B inhibited melanin production in UV-B-irradiated co-cultures. (B) Monocultures of B16F0 cells were stimulated with a single irradiation of UV-B and treated with BI2B for 72 h. BI2B inhibited melanin production in UV-B-irradiated monocultures. (C-E) HEM or B16F0 cells were stimulated with α-MSH or db-cAMP in the presence of BI2B for 72 h. BI2B inhibited α-MSH- or db-cAMP-induced melanin production. (F) HEM or B16F0 cells were incubated with BI2B for 72 h. BI2B did not alter MTT assay-based cell viability. *P < 0.05 vs. UV-B alone, α-MSH alone or db-cAMP alone.
Down-regulation of MITF-M expression at the promoter level in α-MSH-induced melanogenic programs by BI2B
MITF-M is inducible in melanocytes and functions as a master transcription factor that regulates the biogenesis, pigmentation and transfer of melanosomes [5, 10, 28]. We evaluated whether BI2B could affect the expression of MITF-M in α-MSH-activated melanocyte cultures, since topical treatment with BI2B suppressed the protein levels of MITF-M in UV-B-irradiated and pigmented skins of mice (Figure 1F, upper). HEM or B16F0 cells increased the protein and mRNA levels of MITF-M in response to α-MSH, which were suppressed by treatment with BI2B (Figure 3A and B). In contrast, treatment with arbutin did not alter the mRNA levels of MITF-M in α-MSH-activated B16F0 cells (Figure 3C), suggesting that BI2B may be different in the antimelanogenic mechanism from arbutin.
Effect of BI2B on α-MSH-induced MITF-M expression. HEM or B16F0 cells were pretreated with BI2B for 2 h and stimulated with α-MSH for another 2 h (mRNA levels at A-C) or 4 h (protein levels at A, B) in the presence of BI2B. (A, B) BI2B suppressed α-MSH-induced expression of MITF-M in melanocytes. (C) Arbutin did not alter α-MSH-induced transcription of MITF-M. (D) B16F0 cells were transfected with MITF-M-Luc reporter, and stimulated with α-MSH in the presence of BI2B for 20 h. BI2B inhibited α-MSH-induced promoter activity of MITF-M. *P < 0.05 vs. α-MSH alone.
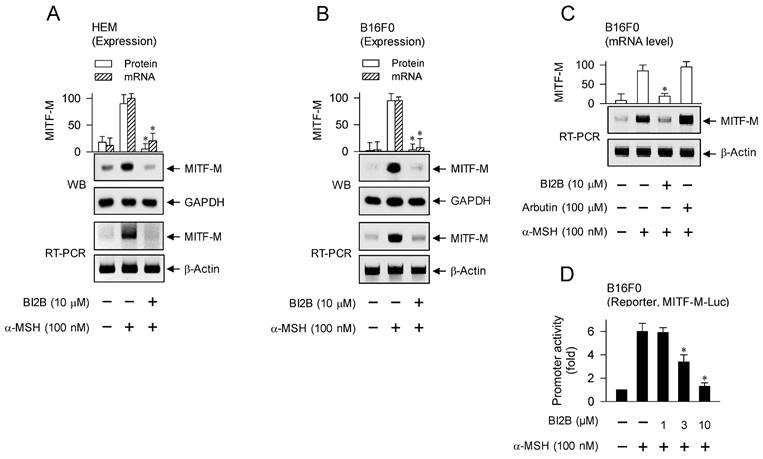
Transcriptional regulation of MITF-M by BI2B was further characterized using luciferase reporter assay-based promoter activity. B16F0 cells were transfected with MITF-M-Luc, a plasmid containing the promoter region (-2200/+95) of MITF-M and the reporter of firefly luciferase. B16F0 cells harboring MITF-M-Luc increased firefly luciferase activity in response to α-MSH, reporting the promoter activity of MITF-M, which was inhibited by treatment with BI2B (Figure 3D). These results suggest that BI2B could regulate the expression of MITF-M at the promoter level.
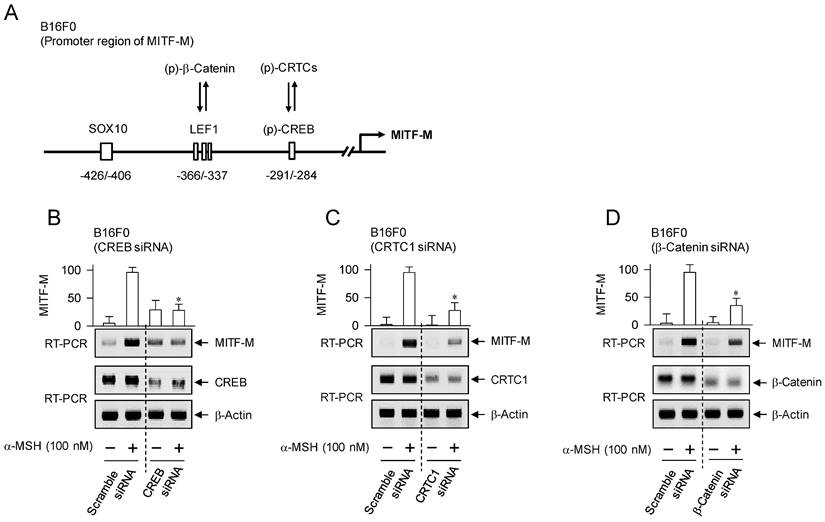
The promoter region of mouse MITF-M encodes several cis-acting elements, such as the CRE at -291/-284, LEF1-binding sites at -366/-337 and SOX10-binding site at -426/-406 [11], as represented in Figure 4A. After translocation from the cytosol to the nucleus, CRTCs and β-catenin coactivate CREB and LEF1 that already occupy the corresponding cis-acting elements on MITF-M promoter [9, 10]. Each CRTC in the nucleus can dimerize with CREB, which makes more tight occupancy of CREB on the promoter [29]. Moreover, another domain of CRTC interacts with basal transcription factor TFIID in the transcription machinery, thus facilitating the recruitment of RNA polymerase II onto the promoter [10, 29]. Nuclear β-catenin dimerizes with LEF1 that is usually surrounded by transcriptional corepressors, which induces conformational changes leading to the dissociation of corepressor complex from LEF1, and alters a heteromer of LEF1/β-catenin to become transcriptional activator of MITF-M promoter [10, 18, 23]. Melanocyte-specific SOX10 cooperates with ubiquitous CREB/CRTCs, which restricts the expression of MITF-M in melanocytes [30]. To examine which transcription factors could be associated with the expression of MITF-M in response to α-MSH, gene-specific siRNA assay was employed. As shown in Figure 4B-D, knockdown of CREB, CRTC1 or β-catenin suppressed the transcription of MITF-M in α-MSH-activated B16F0 cells. These results suggest that CREB/CRTCs and LEF1/β-catenin could be responding to α-MSH-induced melanogenic programs as transcriptional activators of MITF-M promoter.
Transcriptional activators on MITF-M promoter in response to α-MSH. (A) An illustrated structure of MITF-M promoter: transcriptional activators and their cis-acting DNA elements. CREB and LEF1 constitutively interact with the corresponding cis-acting DNA elements. After nuclear translocation, CRTCs and β-catenin coactivate CREB and LEF1, respectively. Transcriptional activities of CREB, CRTCs and catenin are regulated by posttranslational modification of reversible phosphorylation and dephosphorylation. (B-D) B16F0 cells were transfected with each siRNA, incubated for 48 h and stimulated with α-MSH for 2 h. Knockdown of CREB, CRTC1 or β-catenin suppressed α-MSH-induced transcription of MITF-M. *P < 0.05 vs. scramble siRNA plus α-MSH.
Inhibition of CREB phosphorylation but not nuclear translocation of CREB, CRTC1, β-catenin or SOX10 in α-MSH-induced expression of MITF-M by BI2B
Melanogenic programs in response to α-MSH increase the phosphorylation of CREB coupling with the dephosphorylation of CRTCs for transcriptional activation of MITF-M through the complex of CREB/CRTCs [9, 10]. Phosphorylation of CREB at the S133 residue promotes its affinity with CREB-binding protein (CBP) and paralogue p300 (CBP/p300), thus recruiting CBP/p300 onto the promoter region of MITF-M, where CBP/p300 catalyze the acetylation of nucleosomal histones for disassembly, thus becoming as transcriptionally active in chromatin structure [31, 32]. Phosphorylation of CRTCs, including p-CRTC1 at the S151 residue, sequesters them in the cytosol, but dephosphorylation of them is essentially required for nuclear translocation [16, 33]. Phosphorylation of β-catenin at the S675 residue stabilizes its conformation from proteasome-mediated degradation, resulting in its accumulation in the cytosol [18, 23]. The p-β-catenin is then allowed to translocate into the nucleus, and interacts with LEF1 for transcriptional activation of MITF-M promoter [10, 18, 23].
After stimulation with α-MSH, B16F0 cells increased the phosphorylation of CREB at the S133 residue, decreased that of CRTC1 at the S151 residue, and increased that of β-catenin at the S675 residue (Figure 5A). Treatment with BI2B decreased the levels of p-CREB but did not alter those of p-CRTC1 and p-β-catenin in α-MSH-activated B16F0 cells (Figure 5A). HEM cells also increased the phosphorylation of CREB coupling with dephosphorylation of CRTC1 in response to α-MSH (Figure S3), suggesting that phosphorylation circuits on CREB and CRTCs may be commonly operated in human and mouse melanocytes. Treatment with BI2B consistently inhibited the phosphorylation of CREB but bypassed the dephosphorylation of CTRC1 in α-MSH-activated HEM cells (Figure S3). Moreover, B16F0 cells increased the protein and mRNA levels of SOX10 in response to α-MSH, which were unaffected by treatment with BI2B (Figure S4A and B).
Effects of BI2B on the phosphorylation of CREB and the nuclear-cytosolic shuttling of CREB, CRTC1, β-catenin or SOX10 in response to α-MSH. B16F0 cells were pretreated with BI2B for 2 h and stimulated with α-MSH for 30 min (A) or 1 h (B) in the presence of BI2B. (A) BI2B inhibited α-MSH-induced phosphorylation of CREB but not those of CRTC1 and β-catenin. (B) BI2B did not alter α-MSH-induced nuclear translocation of CREB, CRTC1, β-catenin or SOX10. *P < 0.05 vs. α-MSH alone.
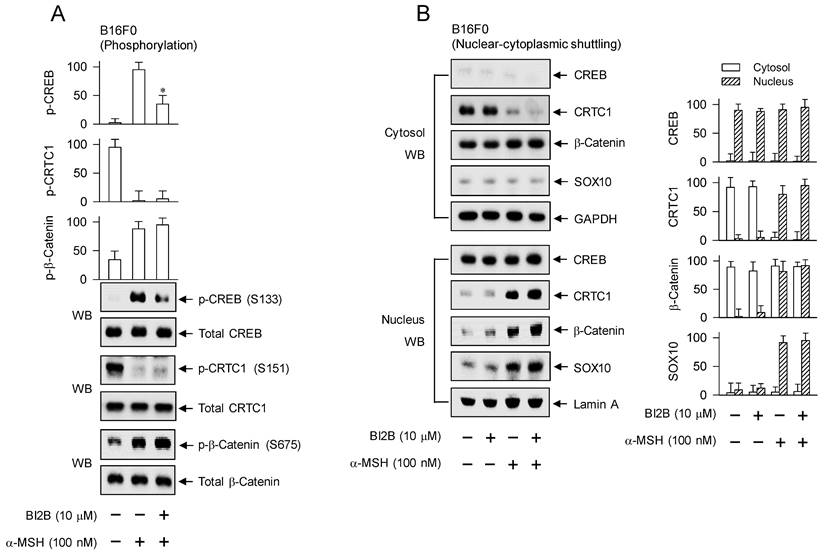
To examine nuclear translocation of the transcription factors in response to α-MSH, cytosolic and nuclear fractions were separated and then subjected to Western blot analysis with anti-CREB, anti-CRTC1, anti-β-catenin or anti-SOX10 antibody (Figure 5B). Anti-CREB antibody could react with dephosphorylated and phosphorylated CREBs, anti-CRTC1 antibody with dephosphorylated and phosphorylated CRTC1s, or anti-β-catenin antibody with dephosphorylated and phosphorylated β-catenins. GAPDH was employed as a marker of the cytosol and lamin A as that of the nucleus. Both dephosphorylated CREB in the presence of medium alone and phosphorylated CREB under stimulation with α-MSH were localized in the nucleus (Figure 5B). Phosphorylated CRTC1 in the presence of medium alone was found in the cytosol but dephosphorylated CRTC1 under stimulation with α-MSH was translocated to the nucleus (Figure 5B). Dephosphorylated β-catenin in the presence of medium alone was present in the cytosol but phosphorylated β-catenin under stimulation with α-MSH was partitioned between the nucleus and the cytosol (Figure 5B). SOX10 was localized in the nucleus after up-regulating its expression in response to α-MSH (Figure 5B; Figure S4A and B). Treatment with BI2B did not alter nuclear-cytosolic shuttling of CREB, CRTC1, β-catenin or SOX10 in α-MSH-activated B16F0 cells (Figure 5B).
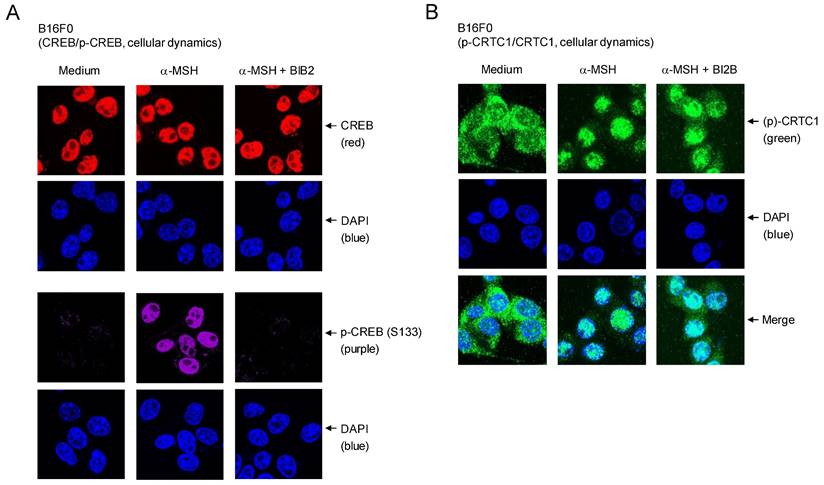
Cellular dynamics of CREB and CRTC1 in response to α-MSH were further characterized using confocal microscopy. Location of dephosphorylated CREB (red) was overlapped with that of DAPI (blue), staining nucleic acids in the nucleus (Figure 6A). B16F0 cells increased nuclear levels of p-CREB (purple) after stimulation with α-MSH (Figure 6A). Treatment with BI2B inhibited the phosphorylation of CREB in the nucleus of α-MSH-activated B16F0 cells but did not alter the localization of CREB or p-CREB in the nucleus (Figure 6A). On the other hand, p-CRTC1 (green) was stained with anti-CRTC1 antibody and mainly found in the cytosol of B16F0 cells in the presence of medium alone, but dephosphorylated CRTC1 (green) under stimulation with α-MSH was translocated to the nucleus (Figure 6B), suggesting that nuclear-cytosolic shuttling of CRTC1 and p-CRTC1 could be regulated by their reversible phosphorylation and dephosphorylation. Treatment with BI2B did not alter nuclear entry of dephosphorylated CRTC1 in α-MSH-activated B16F0 cells (Figure 6B). Overall, BI2B could inhibit the phosphorylation of CREB in α-MSH-induced melanogenic programs through the expression of MITF-M. However, BI2B did not alter the dephosphorylation of CRTC1, the phosphorylation of β-catenin, the up-regulation of SOX10, and the nuclear-cytosolic shuttling of CREB, CRTC1, β-catenin or SOX10 in α-MSH-activated melanocyte cultures.
Effects of BI2B on cellular dynamics of CREB and CRTC1 in response to α-MSH. B16F0 cells were pretreated with BI2B for 2 h and stimulated with α-MSH for 1 h in the presence of BI2B. (A) Both CREB (red) in the presence of medium and p-CREB (purple) under stimulation with α-MSH were overlapped with the location of DAPI (blue), staining nucleic acids in the nucleus. BI2B inhibited α-MSH-induced phosphorylation of CREB but did not alter localization of CREB or p-CREB in the nucleus. (B) p-CRTC1 (green) in the presence of medium alone was stained with anti-CRTC1 antibody and predominantly localized in the cytosol. Dephosphorylated CRTC1 (green) under stimulation with α-MSH was found in the nucleus. BI2B did not alter α-MSH-induced nuclear translocation of CRTC1.
Interruption of p38MAPK-MSK1 axis but not cAMP-PKA axis in α-MSH-induced melanogenic programs by BI2B
α-MSH binds to its receptor MC1R on the surface of melanocytes, which increases intracellular levels of cAMP, and activates PKA as well as MAPKs, thus up-regulating the expression of MITF-M in the melanogenic programs [3]. B16F0 cells increased the kinase activity of PKA after stimulation with α-MSH, which was unaffected by treatment with BI2B but inhibited by that with KT 5720 (Figure 7A). KT 5720 was employed as an inhibitor of PKA-catalyzed kinase activity. From these results, BI2B might bypass cAMP-PKA axis-mediated signals to the expression of MITF-M, such as PKA-catalyzed phosphorylation (activation) of CREB [12], PKA-SIKs axis- or PKA-AMPK axis-mediated phosphorylation (inactivation) of CRTCs [16, 17], and PKA-GSK3β axis-mediated phosphorylation (activation) of β-catenin [18, 23].
Effects of BI2B on cAMP-PKA and p38MAPK-MSK1 axes in α-MSH-induced melanogenic programs. B16F0 cells were pretreated with BI2B for 2 h and stimulated with α-MSH for 30 min in the presence of BI2B. (A) BI2B did not alter α-MSH-induced PKA activity. (B) BI2B inhibited α-MSH-induced phosphorylation of p38MAPK but did not alter those of ERK and JNK. (C) BI2B inhibited α-MSH-induced phosphorylation of MSK1. (D) BI2B did not alter α-MSH-induced phosphorylation of MKK3/6. (E) Catalytically active rhMKK3 was treated with BI2B for 10 min, and its kinase activity was measured in cell-free reactions. BI2B inhibited MKK3-catalyzed kinase activity on p38MAPK. *P < 0.05 vs. α-MSH alone or rhMKK3 alone.
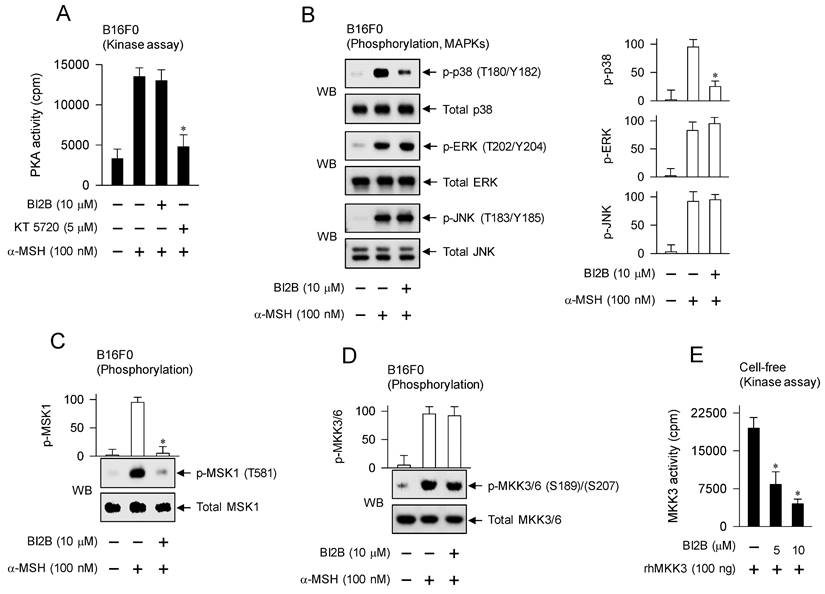
We then evaluated whether BI2B could affect MAPKs-mediated signals to the expression of MITF-M in the melanogenic programs. B16F0 cells increased the phosphorylation of p38MAPK at the T180 and Y182 residues, that of ERK at the T202 and Y204 residues, and that of JNK at the T183 and Y185 residues after stimulation with α-MSH (Figure 7B). Treatment with BI2B decreased the levels of p-p38MAPK but did not alter those of p-ERK and p-JNK in α-MSH-activated B16F0 cells (Figure 7B). From these results, BI2B might bypass ERK- and JNK-dependent expression of MITF-M, such as ERK-MSK1 axis- or ERK-p90RSK axis-mediated phosphorylation (activation) of CREB [14, 34], ERK-calcineurin axis-mediated dephosphorylation (activation) of CRTCs [20], and JNK-catalyzed phosphorylation (inactivation) of CRTCs [21].
BI2B had inhibited not only the activation of CREB through phosphorylation (Figure 5A; Figure 6A) but also the activation of p38MAPK in α-MSH-activated melanocyte cultures (Figure 7B). We then considered MSK1 as a candidate of linker between p38MAPK and CREB, since MSK1 can be activated by p38MAPK-catalyzed phosphorylation [35, 36], and kinase activity of MSK1 phosphorylates CREB [13, 37]. B16F0 cells increased the phosphorylation of MSK1 at the T581 residue after stimulation with α-MSH, which was inhibited by treatment with BI2B (Figure 7C).
To elucidate a primary target by BI2B, we examined the upstream kinases that activate p38MAPK. MKK3/6 can directly phosphorylate p38MAPK [35, 36]. B16F0 cells increased the phosphorylation of MKK3 at the S189 residue and that of MKK6 at the S207 residue after stimulation with α-MSH (Figure 7D). Treatment with BI2B did not alter α-MSH-induced phosphorylation (activation) of MKK3/6 at all (Figure 7D). To elucidate whether BI2B could directly affect catalytic activity of MKK3, we employed in vitro kinase assays. Treatment with BI2B inhibited rhMKK3-catalyzed phosphorylation of GST-p38αMAPK in cell-free conditions (Figure 7E). In another, kinase activity of TAK1, a MAPK kinase kinase (MAPKKK), can phosphorylate MKK3/6 and lead to the activation of p38MAPK activity [35]. Treatment with BI2B did not alter catalytically active rhTAK1-TAB1-catalyzed kinase activity in cell-free reactions (Figure S5). These results suggest that BI2B could primarily inhibit the MKK3-catalyzed kinase activity on p38MAPK, thus interrupting the downstream pathway of p38MAPK-MSK1-CREB-MITF-M in α-MSH-induced melanogenic programs.
Interruption of α-MSH-induced melanogenic programs by specific inhibitors of p38MAPK and MSK1
We confirmed the importance of α-MSH-induced p38MAPK-MSK1-CREB-MITF-M pathway in the melanogenic programs using chemical inhibitors of p38MAPK and MSK1. Treatment with small-molecule inhibitors of p38MAPK or MSK1 decreased the phosphorylation of CREB in α-MSH-activated B16F0 cells but bypassed the dephosphorylation of CRTC1, as did that with BI2B (Figure 8A). Pyridinylimidazole derivatives SB202190 and SB203580 were employed as ATP-competitive inhibitors of p38MAPK-catalyzed kinase activity [38]. Structurally unrelated chemicals SB-747651A and RMM-46 were employed as MSK1 inhibitors, in which SB-747651A targets to the kinase activity of N-terminal kinase domain on protein substrates including CREB, and RMM-46 to C-terminal kinase domain-catalyzed autophosphorylation (activation) of N-terminal kinase domain [37].
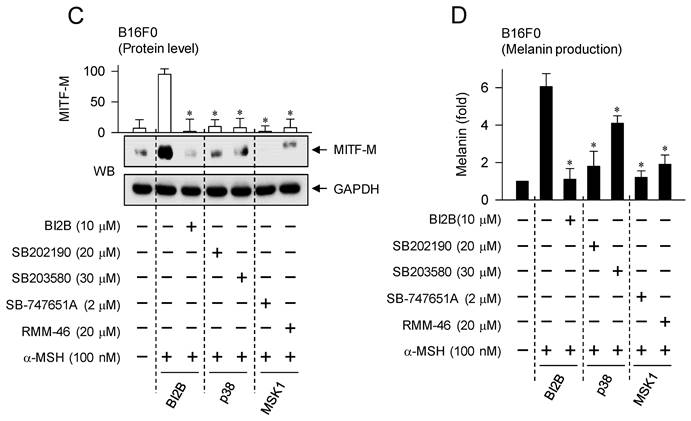
Effects of p38MAPK and MSK1 inhibitors on α-MSH-induced melanogenic programs. SB202190 and SB203580 were employed as p38MAPK inhibitors, SB-747651A and RMM-46 as MSK1 inhibitors, and KT 5720 as a PKA inhibitor. B16F0 cells were pretreated with each kinase inhibitor for 2 h and stimulated with α-MSH for 30 min (A, B), 4 h (C) or 72 h (D) in the presence of each kinase inhibitor. (A) Both p38MAPK and MSK1 inhibitors decreased the phosphorylation of CREB in response to α-MSH but bypassed the dephosphorylation of CRTC1, as did BI2B. (B) PKA inhibitor decreased α-MSH-induced phosphorylation of CREB coupling with dephosphorylation of CRTC1. (C) Both p38MAPK and MSK1 inhibitors suppressed α-MSH-induced protein levels of MITF-M, as did BI2B. (D) Both p38MAPK and MSK1 inhibitors decreased α-MSH-induced melanin production, as did BI2B. *P < 0.05 vs. α-MSH alone.
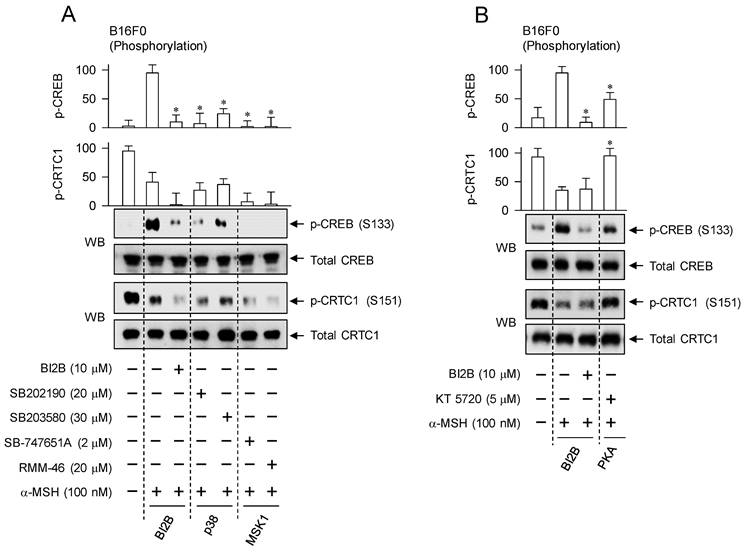
However, treatment with PKA inhibitor (KT 5720) partially inhibited the phosphorylation of CREB but completely restored the dephosphorylation of CRTC1 to basal status in α-MSH-activated B16F0 cells (Figure 8B). KT 5720 suppresses α-MSH-induced expression of MITF-M in melanogenic programs [39], and inhibited melanin production in α-MSH- or db-cAMP-activated B16F0 cells (Figure S6). Besides PKA activity, the exchange protein directly activated by cAMP (EPAC) also transmits intracellular signals to regulate cAMP-dependent gene expression through the phosphorylation of CREB [40]. EPAC inhibitor (ESI-09) decreases α-MSH- or db-cAMP-induced melanin production [41]. These results suggest that signaling pathways through p38MAPK-MSK1 and cAMP-PKA axes could undertake different phosphorylation circuits on CREB and CRTC1 in response to α-MSH.
Treatment with p38MAPK or MSK1 inhibitors consequently decreased cellular levels of MITF-M as well as melanin production in α-MSH-activated B16F0 cells, as did that with BI2B (Figure 8C and D). Moreover, the chemical inhibitors did not alter MTT assay-based viability of B16F0 cells (Figure S7), excluding from non-specific cytotoxicity. Overall, BI2B and chemical inhibitors of p38MAPK or MSK1 could interrupt the same melanogenic signaling pathway in the expression of MITF-M in response to α-MSH.
Suppression of α-MSH-induced expression of MITF-M-target melanogenic genes by BI2B
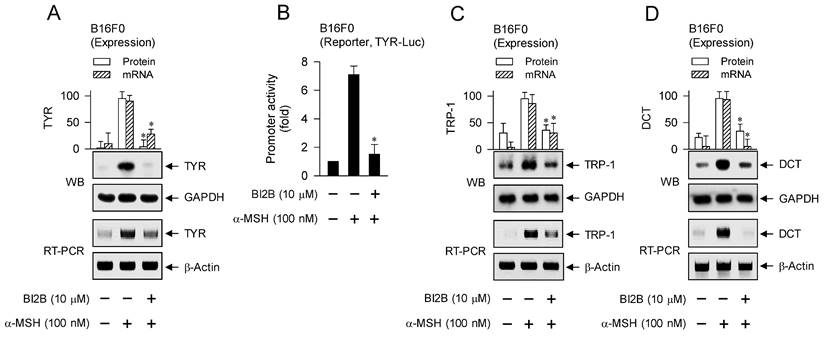
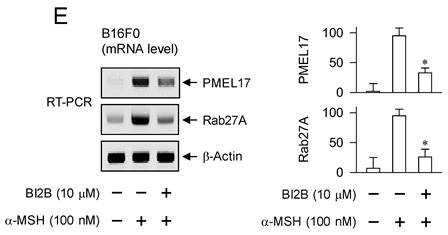
MITF-M up-regulates the expression of melanogenic genes encoding TYR, TRP-1 or DCT in melanin biosynthesis at melanosomes and PMEL17 or Rab27A in the transfer of pigmented melanosomes from melanocytes to overlaying keratinocytes in the skin [10, 28, 42]. α-MSH-induced melanogenic programs up-regulated the expression of MITF-M in melanocyte cultures, which was suppressed by treatment with BI2B (Figure 3A and B). Subsequently, we examined that BI2B could affect MITF-M-dependent expression of melanogenic genes. B16F0 cells up-regulated the expression of TYR upon stimulation with α-MSH, which was suppressed by treatment with BI2B (Figure 9A). To clarify transcriptional regulation of TYR by BI2B, B16F0 cells were transfected with TYR-Luc, a plasmid construct containing the promoter region (-2236/+59) of TYR and the reporter, encoding firefly luciferase. B16F0 cells harboring the reporter plasmid increased firefly luciferase activity upon stimulation with α-MSH, indicating that α-MSH could stimulate the promoter activity of TYR (Figure 9B). Treatment with BI2B inhibited α-MSH-induced promoter activity of TYR (Figure 9B). These results suggest that BI2B could suppress α-MSH-induced expression of TYR at the promoter level. Notably, treatment with BI2B also suppressed the expression of TRP-1 or DCT (Figure 9C and D) as well as the transcription of PMEL17 or Rab27A in α-MSH-activated B16F0 cells (Figure 9E). Overall, treatment with BI2B could suppress the expression of MITF-M-target melanogenic genes.
Effect of BI2B on α-MSH-induced expression of MITF-M-target melanogenic genes. B16F0 cells were stimulated with α-MSH in the presence of BI2B for 24 h (mRNA levels at A, C-E) or 48 h (protein levels at A, C, D). (A) BI2B suppressed α-MSH-induced expression of TYR. (B) B16F0 cells harboring TYR-Luc reporter were stimulated with α-MSH in the presence of BI2B for 20 h. BI2B inhibited α-MSH-induced promoter activity of TYR. (C, D) BI2B suppressed α-MSH-induced expression of TRP-1 and DCT. (E) BI2B suppressed α-MSH-induced transcription of PMEL17 and Rab27A. *P < 0.05 vs. α-MSH alone.
Discussion
In the current study, we propose a molecular target of antimelanogenic efficacy by elucidating the inhibitory mechanism of BI2B on facultative melanogenesis. Topical treatment with BI2B ameliorated skin hyperpigmentation via UV-B-irradiated facultative melanogenesis in mice. BI2B also suppressed the protein or mRNA levels of melanogenic markers, such as MITF-M, TYR and POMC, in UV-B-exposed and pigmented skins. Moreover, BI2B inhibited melanin production in cell-culture models, such as UV-B-irradiated co-cultures of keratinocyte and melanocyte cells and α-MSH-activated melanocyte cultures. Mechanistically, BI2B inhibited the phosphorylation of CREB at the S133 residue in α-MSH-induced melanogenic programs, and suppressed the expression of MITF-M at the promoter. As shown in Figure 10, BI2B directly inhibited MKK3-catalyzed kinase activity on p38MAPK. Consequently, BI2B interrupted the downstream melanogenic pathway of p38MAPK-MSK1-CREB in α-MSH-induced expression of MITF-M. However, BI2B bypassed the axes of CRTC1, β-catenin and SOX10 in α-MSH-induced melanogenic programs.
A proposed mechanism of BI2B on antimelanogenic activity. MC1R, a receptor of α-MSH, is exclusively present in melanocytes. BI2B suppressed the expression of MITF-M via decreasing the phosphorylation of CREB in α-MSH-induced melanogenic programs. As a molecular target, BI2B directly inhibited MKK3-catalyzed kinase activity on p38MAPK, and interrupted the downstream pathway of p38MAPK-MSK1-CREB in the expression of MITF-M through the phosphorylation of CREB. TLR4, a receptor of LPS, is distributed in immune and other cell types including melanocytes. LPS/TLR4 transmit intracellular signals to activate TAK1 activity, which is then branched to either MKK3/6 for MITF-M expression in pigmentation or IKKβ for NF-κB activation in immunity.
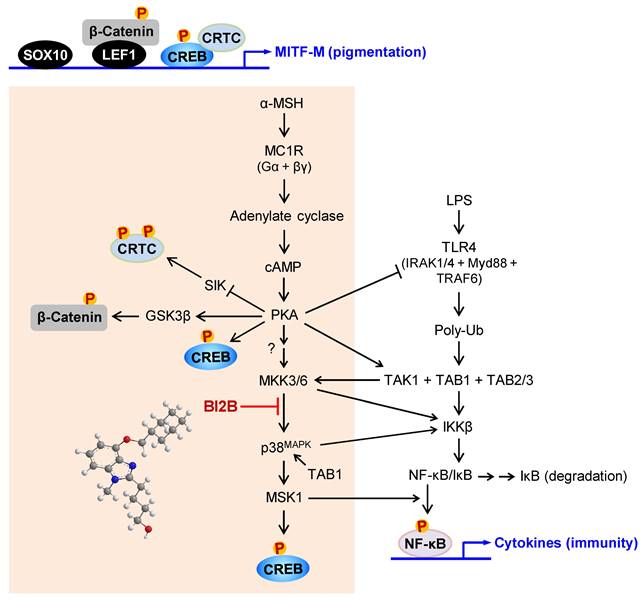
BI2B inhibited the phosphorylation (activation) of p38MAPK but not those of ERK and JNK in α-MSH-activated melanocyte cultures. Mammalian p38MAPK family consists of four isoforms, such as p38α, β, γ, and σ [35]. The p38α and β are ubiquitously expressed, whereas p38γ and σ are more tissue-specific, and cellular p38α are usually at higher levels than other isoforms [35]. The p38MAPK family is activated by almost all environmental stresses, including UV radiation and inflammatory stimuli, and integrates may types of intracellular signals, thus contributing to the biological responses [35]. The p38MAPK activity is tightly regulated, in which upstream kinases MKK3/6 directly phosphorylate p38MAPK for activation [35, 43]. MKK3 preferentially activates p38α and β, and MKK6 can phosphorylate all isoforms [35, 43]. In another, the kinase activity of TAK1 phosphorylates MKK3/6, thus linking to the activation of p38MAPK activity [35, 43]. The p38MAPK can be activated in MKK3/6-independent mechanism by directly interacting with TAB1, thus leading to the autophosphorylation (activation) of MKK3/6 [44]. Chemical inhibitors of p38MAPK activity are clinically applied to the patients with inflammation and autoimmune disorders, such as early Alzheimer disease, multiple myeloma, cardiomyopathy and muscular dystrophy [45]. Here, we propose that chemical inhibition of MKK3 activity in the axis with p38MAPK could prevent acquired hyperpigmentation in the skin.
BI2B inhibits LPS/TLR4-mediated transcriptional activity of NF-κB [26], as well as melanin production in the current study. Melanogenic programs can cross-talk with signaling pathways of NF-κB activation through TLRs [24], as represented in Figure 10. LPS/TLR4 transmit intracellular signals to induce the autophosphorylation TAK1 at the T187 residue, which results in partial gain of TAK1 activity [46]. PKA in melanogenic programs to regulate the expression of MITF-M can phosphorylate TAK1 at the S412 residue, thus fully activating the kinase activity of TAK1 [47]. TAK1 activity is then branched to MKK3/6 or IKKβ [46]. Ten kinds of MAPKKKs are known to activate MKK3/6 [35, 36]. However, it remains to be defined which MAPKKKs like TAK1 are involved in α-MSH-induced melanogenic programs. IKKβ phosphorylates IκB, and p-IκB is degraded by ubiquitination-proteasome system [48]. NF-κB freed from IκB can be translocated into the nucleus, where a heterodimer of NF-κB p50 and p65 binds to the cis-acting κB consensus on promoter regions [48]. Nuclear MSK1 phosphorylates NF-κB p65 at the S276 residue in addition to the phosphorylation of CREB at the S133 residue, which facilitates the recruitment of CBP/p300 [13, 31, 48]. CBP/p300 can acetylate not only nucleosomal histones to become transcriptionally active in chromatin structure but also NF-κB p65 to enhance the transcriptional activity of NF-κB in another mechanism [13, 48].
Conclusions
Taken together, we propose the interruption of MKK3-p38MAPK-MSK1-CREB pathway in the expression of MITF-M as a rationale to ameliorate hyperpigmentation via facultative melanogenesis and a potential strategy to treat acquired pigmentary disorders in the skin.
Supplementary Material
Supplementary figures and table.
Acknowledgements
This work was financially supported by grants (2020R1F1A1072049 & 2023R1A2C2003964) from the National Research Foundation of Korea, and also by Youngsoo Kim's Research Year of Chungbuk National University in 2023. We appreciated prof. Key-Hwan Lim for his help to keep a zero-tolerance plagiarism.
Data Availability Statement
The data obtained during the study are available from the corresponding author (Youngsoo Kim) upon reasonable request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Liu W, Chen Q, Xia Y. New mechanistic insights of melasma. Clin Cosmet Investig Dermatol. 2023;16:429-42
2. Yardman-Frank JM, Fisher DE. Skin pigmentation and its control: from ultraviolet radiation to stem cells. Exp Dermatol. 2021;30:560-71
3. Abdel-Malek ZA, Knittel J, Kadekaro AL, Swope VB, Starner R. The melanocortin 1 receptor and the UV response of human melanocytes: a shift in paradigm. Photochem Photobiol. 2008;84:501-8
4. Rachmin I, Ostrowski SM, Weng QY, Fisher DE. Topical treatment strategies to manipulate human skin pigmentation. Adv Drug Deliv Rev. 2020;153:65-71
5. Nguyen NT, Fisher DE. MITF and UV responses in skin: from pigmentation to addiction. Pigment Cell Melanoma Res. 2019;32:224-36
6. Jinlian L, Yingbin Z, Chunbo W. p38 MAPK in regulating cellular responses to ultraviolet radiation. J Biomed Sci. 2007;14:303-12
7. Tagashira H, Miyamoto A, Kitamura S, Tsubata M, Yamaguchi K, Takagaki K. et al. UVB stimulates the expression of endothelin B receptor in human melanocytes via a sequential activation of the p38/MSK1/CREB/MITF pathway which can be interrupted by a French maritime pine bark extract through a direct inactivation of MSK1. PLoS One. 2015;10:e0128678
8. Rodríguez CI, Setaluri V. Cyclic AMP (cAMP) signaling in melanocytes and melanoma. Arch Biochem Biophys. 2014;563:22-7
9. Wan P, Hu Y, He L. Regulation of melanocyte pivotal transcription factor MITF by some other transcription factors. Mol Cell Biochem. 2011;354:241-6
10. Hartman ML, Czyz M. MITF in melanoma: mechanisms behind its expression and activity. Cell Mol Life Sci. 2015;72:1249-60
11. Shibahara S, Takeda K, Yasumoto K, Udono T, Watanabe K, Saito H. et al. Microphthalmia-associated transcription factor (MITF): multiplicity in structure, function, and regulation. J Investig Dermatol Symp Proc. 2001;6:99-104
12. Torres-Quesada O, Mayrhofer JE, Stefan E. The many faces of compartmentalized PKA signalosomes. Cell Signal. 2017;37:1-11
13. Vermeulen L, Vanden Berghe W, Beck IM, De Bosscher K, Haegeman G. The versatile role of MSKs in transcriptional regulation. Trends Biochem Sci. 2009;34:311-8
14. Ludwik KA, Lannigan DA. Ribosomal S6 kinase (RSK) modulators: a patent review. Expert Opin Ther Pat. 2016;26:1061-78
15. Wang H, Xu J, Lazarovici P, Quirion R, Zheng W. cAMP response element-binding protein (CREB): a possible signaling molecule link in the pathophysiology of schizophrenia. Front Mol Neurosci. 2018;11:255
16. Darling NJ, Cohen P. Nuts and bolts of the salt-inducible kinases (SIKs). Biochem J. 2021;478:1377-97
17. Aslam M, Ladilov Y. Emerging role of cAMP/AMPK signaling. Cells. 2022 11
18. Bellei B, Pitisci A, Catricalà C, Larue L, Picardo M. Wnt/β-catenin signaling is stimulated by α-melanocyte-stimulating hormone in melanoma and melanocyte cells: implication in cell differentiation. Pigment Cell Melanoma Res. 2011;24:309-25
19. Tasoulas J, Rodon L, Kaye FJ, Montminy M, Amelio AL. Adaptive transcriptional responses by CRTC coactivators in cancer. Trends Cancer. 2019;5:111-27
20. Sonntag T, Ostojić J, Vaughan JM, Moresco JJ, Yoon YS, Yates JR 3rd. et al. Mitogenic signals stimulate the CREB coactivator CRTC3 through PP2A recruitment. iScience. 2019;11:134-45
21. Kim JH, Hong AR, Kim YH, Yoo H, Kang SW, Chang SE. et al. JNK suppresses melanogenesis by interfering with CREB-regulated transcription coactivator 3-dependent MITF expression. Theranostics. 2020;10:4017-29
22. Sonntag T, Vaughan JM, Montminy M. 14-3-3 proteins mediate inhibitory effects of cAMP on salt-inducible kinases (SIKs). FEBS J. 2018;285:467-80
23. Valenta T, Hausmann G, Basler K. The many faces and functions of β-catenin. Embo J. 2012;31:2714-36
24. Koike S, Yamasaki K. Melanogenesis connection with innate immunity and Toll-like receptors. Int J Mol Sci. 2020 21
25. Ahn JH, Jin SH, Kang HY. LPS induces melanogenesis through p38 MAPK activation in human melanocytes. Arch Dermatol Res. 2008;300:325-9
26. Boggu P, Venkateswararao E, Manickam M, Kim Y, Jung SH. Exploration of SAR for novel 2-benzylbenzimidazole analogs as inhibitor of transcription factor NF-κB. Arch Pharm Res. 2017;40:469-79
27. Cui R, Widlund HR, Feige E, Lin JY, Wilensky DL, Igras VE. et al. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell. 2007;128:853-64
28. Goding CR, Arnheiter H. MITF-the first 25 years. Genes Dev. 2019;33:983-1007
29. Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. 2011;12:141-51
30. Huber WE, Price ER, Widlund HR, Du J, Davis IJ, Wegner M. et al. A tissue-restricted cAMP transcriptional response: SOX10 modulates α-melanocyte-stimulating hormone-triggered expression of microphthalmia-associated transcription factor in melanocytes. J Biol Chem. 2003;278:45224-30
31. Solt I, Magyar C, Simon I, Tompa P, Fuxreiter M. Phosphorylation-induced transient intrinsic structure in the kinase-inducible domain of CREB facilitates its recognition by the KIX domain of CBP. Proteins. 2006;64:749-57
32. Zhou S, Zeng H, Huang J, Lei L, Tong X, Li S. et al. Epigenetic regulation of melanogenesis. Ageing Res Rev. 2021;69:101349
33. Bittinger MA, McWhinnie E, Meltzer J, Iourgenko V, Latario B, Liu X. et al. Activation of cAMP response element-mediated gene expression by regulated nuclear transport of TORC proteins. Curr Biol. 2004;14:2156-61
34. Yoon Y, Bae S, Kim TJ, An S, Lee JH. Nodakenin Inhibits Melanogenesis Via the ERK/MSK1 Signaling Pathway. Pharmazie. 2023;78:6-12
35. Canovas B, Nebreda AR. Diversity and versatility of p38 kinase signalling in health and disease. Nat Rev Mol Cell Biol. 2021;22:346-66
36. Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92:689-737
37. Sattarifard H, Safaei A, Khazeeva E, Rastegar M, Davie JR. Mitogen- and stress-activated protein kinase (MSK1/2) regulated gene expression in normal and disease states. Biochem Cell Biol. 2023;101:204-19
38. Jackson PF, Bullington JL. Pyridinylimidazole based p38 MAP kinase inhibitors. Curr Top Med Chem. 2002;2:1011-20
39. Kim SH, Na C, Yun CY, Kim JG, Baek ST, An HJ. et al. Targeting phosphorylation circuits on CREB and CRTCs as the strategy to prevent acquired skin hyperpigmentation. Int J Biol Sci. 2024;20:312-30
40. Lee K. Epac: new emerging cAMP-binding protein. BMB Rep. 2021;54:149-56
41. Yun CY, Mi Ko S, Pyo Choi Y, Kim BJ, Lee J, Mun Kim J. et al. α-Viniferin improves facial hyperpigmentation via accelerating feedback termination of cAMP/PKA-signaled phosphorylation circuit in facultative melanogenesis. Theranostics. 2018;8:2031-43
42. Vachtenheim J, Borovanský J. "Transcription physiology" of pigment formation in melanocytes: central role of MITF. Exp Dermatol. 2010;19:617-27
43. Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, Tanaka Y. et al. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003;17:1969-78
44. Burton JC, Antoniades W, Okalova J, Roos MM, Grimsey NJ. Atypical p38 signaling, activation, and implications for disease. Int J Mol Sci. 2021 22
45. Yong HY, Koh MS, Moon A. The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin Investig Drugs. 2009;18:1893-905
46. Xu YR, Lei CQ. TAK1-TABs complex: a central signalosome in inflammatory responses. Front Immunol. 2020;11:608976
47. Ouyang C, Nie L, Gu M, Wu A, Han X, Wang X. et al. Transforming growth factor (TGF)-β-activated kinase 1 (TAK1) activation requires phosphorylation of serine 412 by protein kinase A catalytic subunit α (PKACα) and X-linked protein kinase (PRKX). J Biol Chem. 2014;289:24226-37
48. Mulero MC, Huxford T, Ghosh G. NF-κB, IκB, and IKK: integral components of immune system signaling. Adv Exp Med Biol. 2019;1172:207-26
Author contact
![]() Corresponding authors: Youngsoo Kim, Ph.D. and Professor. E-mail: youngsooac.kr & Jae-Kyung Jung, Ph.D. and Professor. E-mail: orgjkjungac.kr.
Corresponding authors: Youngsoo Kim, Ph.D. and Professor. E-mail: youngsooac.kr & Jae-Kyung Jung, Ph.D. and Professor. E-mail: orgjkjungac.kr.