Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(5):1796-1814. doi:10.7150/ijbs.91396 This issue Cite
Research Paper
Carbonic Anhydrase 3 is required for cardiac repair post myocardial infarction via Smad7-Smad2/3 signaling pathway
Yuanyuan Su1#, Dongmei Shi1#, Guofang Xia1, Yujia Liu2, Longwei Xu3, Ling Dao3, Xia Lu1, Chengxing Shen1 ![]() , Congfeng Xu1,4
, Congfeng Xu1,4 ![]()
1. Department of Cardiology, Shanghai Sixth People's Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai 200233, China.
2. Department of Neurology, Tongren Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai 200042, China.
3. Department of Cardiology, the First Affiliated Hospital of Zhengzhou University, Zhengzhou 450000, China.
4. Shanghai Key Laboratory of Sleep Disordered Breathing, Shanghai 200233, China.
#These authors contributed equally to this work.
Received 2023-10-22; Accepted 2024-2-12; Published 2024-2-25
Abstract

Appropriate fibrosis is required to prevent subsequent adverse remodeling and heart failure post myocardial infarction (MI), and cardiac fibroblasts (CFs) play a critical role during the process. Carbonic anhydrase 3 (CAR3) is an important mediator in multiple biological processes besides its CO2 hydration activity; however, the role and underlying mechanism of CAR3 on cardiac repair post MI injury remains unknown. Here, we found that CAR3 expression was up-regulated in cardiac tissue in infarct area at the reparative phase of MI, with a peak at 7 days post MI. The upregulation was detected mainly on fibroblast instead of cardiomyocyte, and primary cardiac fibroblasts treated with TGF-β1 recaptured our observation. While CAR3 deficiency leads to weakened collagen density, enlarged infarct size and aggravated cardiac dysfunction post-MI. In fibroblast, we observed that CAR3 deficiency restrains collagen synthesis, cell migration and gel contraction of cardiac fibroblasts, whereas overexpression of CAR3 in CFs improves wound healing and cardiac fibroblast activation. Mechanistically, CAR3 stabilizes Smad7 protein via modulating its acetylation, which dampens phosphorylation of Smad2 and Smad3, thus inhibiting fibroblast transformation. In contrast, inhibition of Smad7 acetylation with C646 blunts CAR3 deficiency induced suppression of fibroblast activation and impaired cardiac healing. Our data demonstrate a protective role of CAR3 in cardiac wound repair post MI via promoting fibroblasts activation through Smad7‐TGF-β/Smad2/3 signaling pathway.
Keywords: cardiac repair, myocardial infarction, fibroblast activation, CAR3, Smad7 acetylation
Introduction
Coronary heart disease (CHD) remains the leading cause of death worldwide, of which myocardial infarction (MI) is the largest contributor to cardiovascular morbidity and mortality1-3. Though substantial improvements have been made in decreasing acute mortality of MI via timely percutaneous coronary intervention over past decades, the risk in survived patients to progressive development of left ventricular (LV) remodeling and heart failure remains unacceptably high4-6. Cardiac wound healing post-MI undergoes a finely orchestrated series of events including immune cell recruitment and formation of stable infarct scars composed of extracellular matrix (ECM)5,6. Appropriate healing process is essential for preserving cardiac structure and function, whereas inadequate healing may lead to infarct expansion, severe dilatation of LV, heart failure and even cardiac rupture5,7,8.
During repair processes, timely and well controlled deposition of ECM is critical for stable collagen-based scar formation in the infarct area9. And fibroblasts are the dominant cells during the proliferative phase for its crucial role during synthesis and secretion of structural ECM10-12. In response to stimuli, quiescent fibroblasts can be activated and transformed into myofibroblasts with featured expression of α-smooth muscle actin (⍺-SMA), and enhanced migratory, proliferative, contractile and secretory properties7,11,13. Of all the well-established factors, TGF-β and its downstream signaling have been highlighted to play a central role in myofibroblast differentiation post-MI12,14-17. Considering the complicated and diverse biology of myofibroblast differentiation in early infarct repair and subsequent cardiac remodeling, it is of greatly value to further understand the regulatory factors involved in TGF-β signaling for development of therapeutics.
Carbonic anhydrases (CAs) are a family of zinc-containing metalloenzymes that catalyze the reversible hydration of CO2 (CO2 + H2O ⟷ HCO3- + H+), allowing these enzymes to regulate intra- and extra-cellular concentrations of CO2, and pH value18,19. Among sixteen different members identified so far, there are eight cytosolic proteins (CA1, CA2, CA3, CA7, CA8, CA10, CA11, CA13), three transmembrane proteins (CA9, CA12, CA14), two mitochondrial matrix proteins (CA5A, CA5B), two glycosylphosphatidylinositol (GPI)-anchored proteins (CA4 and CA15) and one secreted protein (CA6). Extensive evidence has underscored the participation of CAs in numerous biological processes, such as adipogenesis, gluconeogenesis, acid-base balance, ion transport and calcification20,21. Among the family, carbonic anhydrase 3 (CA3) is special because of its negligent catalytic capability (about only 3 % of that of CA2)22,23. Mainly expressed in skeletal muscle, liver and adipose tissues, CA3 has been verified to involve in various biological processes including alleviation of oxidative stress24-27, regulation of lipogenesis28-30, promotion of cell migration31,32. Recently, CA3 has been detected in heart tissue22, and may involve in multiple cardiovascular diseases including dilated cardiomyopathy (DCM) with HF33, hypertrophic cardiomyopathy (HCM)34, doxorubicin-induced cardiomyopathy35, and ischemia-reperfusion Injury36, suggesting a potential role of CA3 in cardiovascular system. However, the exact biological function of CA3 remains unrevealed. In this study, we found that CAR3 (the mouse homolog for human CA3) regulates fibroblast activation through TGF-β/Smad2/3 signaling via mediating acetylation of Smad7. Our study provides evidence that CAR3 mediates fibroblast transformation to promote wound healing post myocardial infarction, and identifies a potential therapeutic mediator for cardiac repair and heart failure post-MI.
Materials and Methods
Animals and myocardial infarction model
The animal experimental protocols were reviewed and approved by the Animal Ethics Committee of Shanghai Jiao Tong University. Car3‐deficient staggerer (sg/sg) mice were obtained from Jackson Laboratory (Bar Harbor, ME) as previously described37. Car3‐deficient mice and their wild‐type (WT) littermates aged 8 to 10-week-old were housed at 24°C ± 2°C, humidity of 40% ± 5%, under a constant 12 h light/dark cycle with available water and standard rodent chow ad libitum.
The previously described surgical procedures were followed to induce mouse MI model38. Briefly, 8-week-old male mice were anesthetized with a mixture of isoflurane (1.5%) and oxygen (0.5 L/min) inhalation and were placed on a temperature-controlled surgical table before the surgical procedure but without ventilation. An incision was performed in the fourth intercostal space of the left chest to expose the heart. The left anterior descending (LAD) coronary artery was permanently ligated 2 mm from the first branch with a 6-0 silk suture after the heart smoothly and gently externalized. Afterwards, the heart is immediately placed back into the intrathoracic space followed by closure of the skin incision. Sham‐operated mice underwent the abovementioned procedure without LAD occlusion. The blind method was used for the operation and subsequent echocardiographic evaluation in this study. Mice were euthanized with isoflurane/oxygen and sacrificed on the corresponding days after MI surgery to obtain their heart samples for corresponding analyses. To inhibit the acetyltransferase activity of p300 in vivo, mice were injected intraperitoneally with C646 (15 mg/kg; MCE, USA) or DMSO once a day for consecutive 14 days39.
Echocardiography
In vivo cardiac function of mice was evaluated by echocardiography with a high-resolution imaging system (Vevo2100, Visual Sonic Inc., CA) 7 days or 4 weeks after MI, as previously described40. The mice were anesthetized using a mixture of isoflurane and oxygen followed by removal of chest hair and placed in the supine position. The two‐dimensional and M-mode echocardiographic views of the mid‐ventricular short axis were acquired at the level of the papillary muscle tips below the mitral valve. Variables including left ventricular internal diameter at the end of diastolic (LVIDd) and systolic (LVIDs) were measured on M‐mode echocardiography. Left ventricular ejection fraction (LVEF), fractional shortening (LVFS) and left ventricular end-diastolic volume (LVEDV) and end-systolic volume (LVESV) were calculated as reported previously38,40. During echocardiography assessment, the heart rate of mice was maintained between 450 and 550 beats per minute.
Triphenyltetrazolium chloride (TTC) staining
To assess the infarct size, the TTC assays were performed as previously described in detail41. In brief, the heart was harvested and frozen at -80°C, then quickly sliced into five pieces from the apex to base perpendicular to the long axis of the heart. The sections were subsequently incubated in 1% TTC solution (Sigma Aldrich) dissolved in PBS at 37°C for 20 minutes. After stopping the staining process with PBS, the slices were photographed. The infarct size was calculated as the percentage of infarct over the ventricular area. ImageJ software (1.52 V) was used to determine the infarcted area.
Isolation, culture and treatment of primary cells
Neonatal rat cardiac fibroblasts (NRCFs) and neonatal rat cardiac myocytes (NRCMs) were isolated from 1 to 2-day-old neonatal Sprague-Dawley rats. The hearts were removed from the chest after euthanasia and immediately cut into about 1 mm3 cardiac tissues in ice-cold phosphate buffered saline (PBS). The small pieces were then collected into a cell culture dish and digested with 0.25% trypsin (Gibco) at 4°C for 8 hours. After digestion, the supernatants were passed through 70 μm nylon mesh filter, and plated for 2 hours to allow fast-adherent cells (mainly NRCFs) to attach. Afterwards, the medium was replaced with fresh, prewarmed Dulbecco's Modified Eagle Medium (DMEM, Invitrogen Corporation, USA) with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. The collected supernatant (mainly NRCMs) was centrifuged and resuspended in DMEM with 10% FBS and BrdU (0.1 mM, inhibiting the proliferation of fibroblasts, Sigma).
Adult mouse primary cardiac fibroblasts (ACFs) were isolated from 8-10 weeks old WT mice or Car3-deficient mice. The hearts were minced into small pieces after removing atriums and connective tissues. Subsequently, the pieces were digested with 0.1% type II collagenase (Worthington Biochemical) for 1 hour at 37°C with shaking. Digested cell suspension was resuspended in DMEM with 10% FBS and then passed through 70 μm filter, followed by seeding into culture dish for further experiments.
All kinds of cells were cultured at 37°C in a humidified incubator containing 5% CO2. CFs of the 2nd-4th passages cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin were used for subsequent experiments. To induce fibroblast activation, CFs were put on serum‐free DMEM for 24 hours prior to stimulation with 10 ng/ml TGF-β1 (PeproTech) for appropriate time. To inhibit the acetyltransferase activity of P300, CFs were exposed to serum‐free DMEM for 24 hours prior followed by pretreatment with compound C64642 (3 μM, MCE, USA) or DMSO followed by the presence or absence of TGF-β1 (10 ng/ml). After purification by differential preplating, NRCMs were seeded at a density of 1 × 106 cells/ml into corresponding culture dishes and cultured with DMEM with 10% FBS. To simulate ischemic injury, the NRCMs were cultured in serum-free and glucose deprived DMEM under hypoxia for 6 hours after starvation with serum‐free DMEM for 12 hours, which was induced by a hypoxia chamber (Thermo, HERA cell 150i) with 5% CO2, 1% O2, and 94% N2. The same protocols were also used for NRCFs/ACFs. Human Umbilical Vein Endothelial Cells (HUVECs) from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin.
Construction and transduction of recombinant adenovirus
Recombinant adenovirus (Adv) packaged by Genechem (Shanghai, China) were used to manipulate the expression of CAR3 in cultured fibroblasts. Gene sequence of Car3 was referred to Genbank (NM_007606). To overexpress CAR3, CFs were subsequently infected with Adv-GFP (MOI = 30) or Adv-Car3 (MOI = 30) for 4 hours, and then the medium containing the adenovirus was removed and replaced by DMEM supplemented with 10% FBS. The cells were cultured in serum-free DMEM for 24 hours and subsequently treated with TGF-β1 or not.
Western blot analysis
Homogenates of cardiac tissue or cultured primary CFs were prepared using RIPA buffer with complete protease and phosphatase inhibitors (Roche). Lysates protein concentrations were quantified using Pierce BCA Protein Assay Kit (Thermo Fisher). Equal amount of protein lysates (20‐40 μg/lane) were used for SDS‐PAGE Gels. The SDS/PAGE Gels proteins were electro‐transferred onto Polyvinylidene difluoride (PVDF) membranes and then incubated overnight at 4°C with primary antibodies. Followed were all the commercially available antibodies used by Western blotting analysis: anti-CAR3 (sc-50715, Santa Cruz, 1:1000), anti-CAR2 (sc-133111, Santa Cruz, 1:1000), anti-α-SMA (ab7817, abcam,1:2000), anti-Collagen1 (72026, CST, 1:1000), anti-Collagen3 (184993, abcam, 1:1000), anti-TGFβR1 (310328, zen-bio, 1:1000), anti-TGFβR2 (346596, zen-bio, 1:1000), anti-Smad7 (sc-365846, Santa Cruz, 1:1000), anti-p-Smad2 (188334, abcam, 1:1000), anti-p-Smad3 (52903, abcam, 1:1000), anti-Smad2/3 (202445, abcam, 1:1000), anti-Ac-lysine (sc-32268, Santa Cruz, 1:1000), anti-P300 (AF6795, Beyotime 1:1000), anti-HSP90 (4877, CST 1:1000), anti-α-tubulin (52866, abcam, 1:1000). α-tubulin and HSP90 served as the loading control. After incubating with the corresponding peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch, 1:20000) for 1 hour at room temperature, the immunoblot bands were detected with the enhanced-chemiluminescent (ECL) system (Tanon, China) and calculated with ImageJ software.
Real‐time quantitative polymerase chain reaction (RT q‐PCR)
Total RNA was extracted from cardiac tissues or cells using TRIzol Reagent (Sigma-Aldrich, USA), according to the manufacturer's instructions. cDNA was synthesized using HiScript® II Q RT SuperMix for qPCR (+gDNA wiper) (Vazyme, China), and then amplified by AceQ® Universal SYBR qPCR Master Mix (Vazyme, China) in the LightCycler® 96 Real‐Time PCR System (Roche, USA). β-actin was used as the housekeeping gene. Results from LightCycler® 96 analysis software are presented as Ct values and calculated with standard comparative Ct method. The primer sequences utilized for real‐time PCR in this study are presented in Table S1.
Immunofluorescence and immunohistochemistry staining
For immunofluorescence (IF) staining, paraffin-embedded sections (5-6 μm thick) of heart tissue chips were dewaxed and rehydrated, followed by antigen retrieval in 10 mM citric acid sodium buffer. In addition, cultured cells were washed with PBS and then fixed with 4% paraformaldehyde. The heart sections and fixed cells were subsequently permeabilized with 0.1% Triton X-100 and blocked in PBS containing with 10% goat serum albumin for 1 hour at room temperature. Next, the samples were incubated with indicated primary antibodies overnight at 4°C. The corresponding primary antibodies used were as follows: anti-CAR3 (196835, abcam, 1:100), anti-α-SMA (7817, abcam, 1:100), anti-Vimentin (8978, abcam,1:100), anti-Collagen1 (72026, CST, 1:1000), anti-Collgen3 (184993, abcam, 1:100), anti-cTnT (8295, abcam, 1:100). After washing with PBS, the slices were incubated with appropriate fluorescent-labeled secondary antibodies for 1 hour at room temperature. Nuclei were counterstained with 4′,6-diamidino- 2-phenylindol (DAPI, Invitrogen, USA). Fluorescent images were obtained by fluorescent confocal microscopy (Leica TCS SP8; Leica, Germany) and fluorescent intensity of cells was quantified using ImageJ software.
The myocardial tissue chips were dewaxed and rehydrated for immunohistochemistry. After antigen retrieval with citric acid sodium solution, the washed tissue slices were permeabilized, blocked and stained with primary antibodies against CAR3 (196835, abcam, 1:100; DAPI, Invitrogen) overnight at 4°C. After incubating with the biotin-conjugated secondary antibodies, staining signals were estimated with an Avidin-Biotin Complex peroxidase system. A 3,3′- diaminobenzidine (DAB) peroxidase substrate kit (K5007, DAKO) was used to visualize antibody binding. Bright-field images were obtained by a fluorescence microscope (Leica, Germany).
Histological analysis
Hearts were harvested from each group under anesthesia and perfused with PBS followed by fixed with 4% paraformaldehyde. After embedding in paraffin, the heart tissue samples were cut into 5μm thick sections and prepared for hematoxylin and eosin (HE) staining to observe general morphology, Masson's trichrome staining and Sirius Red staining to assess collagen content. Infarct size was determined by infarct area/total LV area from each section and expressed as a percentage. In addition, the ventricular wall thickness of the scar was also calculated using ImageJ software.
Immunoprecipitation/Co-immunoprecipitation
Prepared heart tissue and cell samples were solubilized in immunoprecipitation lysis buffer (20 mM Tris pH7.5, 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 5% glycerol, protease inhibitor cocktail) and precipitated by centrifugation. The prepared tissue and cell supernatants (800 μg) were incubated with anti-CAR3 (Santa Cruz, 1μg per 250 μg of total protein) antibody overnight at 4°C followed by incubation with 30 μL of protein G Agarose beads (Santa Cruz) for 4 hours at 4°C. After being washed three times with cold wash buffer and once with lysis buffer, the immunoprecipitations were resuspended in 30 μL of loading buffer and subjected to western blot for examination of Smad7. Following consecutive washes, the immunoprecipitations containing the CAR3-bound or IgG control-bound proteins were resuspended in 30 μL of loading buffer and subjected to western blot for examination of Smad7. Whole lysate/input sample was used as a control of the immunoprecipitation enrichment. Then, tissue and cell lysates (800 μg) were prepared and carried out by incubation with anti-Smad7 ((Santa Cruz, 1 μg per 250 μg of total protein) antibody overnight at 4°C followed by addition of 30 μL of protein G Agarose beads. The eluted samples were further analyzed via western blotting to detect the Ac-lysine modification of Smad7.
Cell migration assay
For the wound healing assay, the cardiac fibroblasts isolated from WT or Car3-deficient adult mice were plated in 6-well plates. The confluent monolayer cells were then wounded by creating a linear wound area with a 200 μL pipette tip to evaluate the migratory ability of cardiac fibroblasts. The scratched plate was then gently washed to remove detached cells followed by treatment with TGF-β1 or not for 24 hours. Cells were photographed at time of injury and 24 hours after injury. The migratory capacity of fibroblasts was briefly calculated by measuring the wound recovered area using the ImageJ software.
For the transwell assay, a system (Corning Costar, USA) with upper chamber polycarbonate filters of 8 mm pore size was used to carry out migratory assay. ACFs were harvested and seeded at a density of 3 ×104 cells per well to the top chamber in serum-free medium whereas the bottom well was added with DMEM with 10% FBS. After incubation for 24 hours at 37°C, cells on the upper surface of the membrane were wiped off with a cotton swab, the cells on the lower chamber were fixed with 4% paraformaldehyde, followed by staining with 0.01% Crystal Violet (Sigma, USA) and then counted five fields randomly.
Collagen gel contraction assay
Isolated ACFs were cultured in DMEM with 10% FBS at 37°C in a 5% CO2 incubator for further experiments. Digested fibroblasts were suspended in complete media to prepare a mixture with a bovine collagen solution as the kit protocols described (Cell Biolabs, USA) for a final collagen concentration of 1.5 mg/mL collagen and 2 ×105 cells/mL. 250 μL of solution was seeded in a 48-well plate within a suspension cell culture plate and then incubated at 37°C for 1.5 hours for the gel to coagulate. Subsequently, the collagen gel was detached from the edges of the plate with a pipette tip followed by the correspondent stimuli. Pictures of the surface area of each matrix were photographed immediately after release as well as 12, 24, 48, and 72 hours following. Gel area was measured using ImageJ software and normalized to original gel area.
Statistical analysis
All quantitative values were presented as mean ± SEM. Data distribution normality was determined by Shapiro-Wilk test. When comparing between two groups, the equality of variances was evaluated by the Levene test. Student's t-test (equal variances) or unequal variance t-test (unequal variances) was used for normally distributed variables whereas the Mann-Whitney U test was used for variables not normally distributed. For comparison of more than two groups, the Brown-Forsythe test was used to evaluate the homogeneity of variance. One-way or two-way analysis of variance (ANOVA) followed by the Bonferroni post hoc test was used if equal variances were assumed in multiple‐group comparisons, otherwise the Tamhane post hoc test was selected if equal variances were not assumed. Statistical analysis was carried out using SPSS (version 26.0) or GraphPad Prism (ver. 9). p < 0.05 was regarded as statistically significant.
Results
Increased expression of CAR3 in cardiac tissues at the infarction site
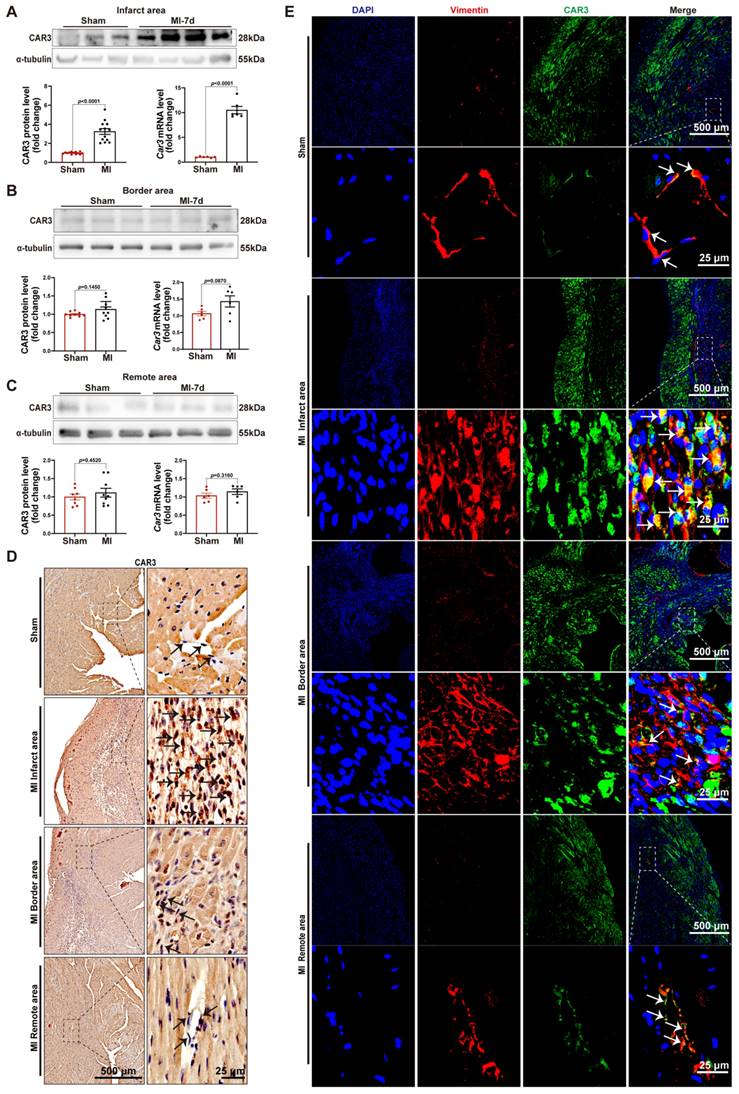
In order to investigate the role of CAR3 during myocardial infarction, we established animal model by ligation of LAD coronary artery with mice. As expected, the expression of CAR3 in myocardial tissue from sham mice was hardly observed, instead it could be detected easily using immunoblotting assay (Figure S1A). While the infarct region displayed increased expression post MI, with a peak at 7 days post MI and rapidly back to base level at day 14 post-MI (Figure 1A; Figure S1B, C). The expression pattern of Car3 mRNA was similar to protein (Figure 1A; Figure S1C). We subsequently detected the expression of Carbonic anhydrase 2 (CAR2), which shares similar sequence identity and structure with CAR3 but is well known for its efficient and specific activity of CO2 hydration-dehydration reaction. The data found that CAR2 expression was considerably up-regulated at day 1 and persisted up to day 3 post myocardial infarction (Figure S1B, C), different from expression trend of CAR3. However, we observed no obvious expression change of CAR3 in the border and remote areas (Figure 1B, C; Figure S1D, E). Also, immunohistochemistry and immunofluorescence staining showed clearly higher CAR3 protein expression in the infarct myocardium than in sham controls, but no change for CAR3 levels in the border and remote regions of hearts post-MI (Figure 1D, E). Our data indicate a dramatical elevation of CAR3 in the infarct area of heart after MI, foretelling a potential role of CAR3 for this disease.
CAR3 expression is increased at the infarct area in response to myocardial infarction. A, Expression of CAR3 was measured using Western blot analysis (n = 12-13 mice per group) and RT q‐PCR (n = 6 mice per group) in the infarct area of WT mice at day 7 post-MI as well as in sham control. Results were normalized against α-tubulin and converted to fold induction relative to sham‐operated mice. B, CAR3 level was determined by Western blot analysis (n = 9 mice per group) and RT q‐PCR (n = 6 mice per group) in the border area of MI-operated hearts and their sham controls. Corresponding statistic of CAR3 was shown. C, CAR3 level was determined by Western blot analysis (n = 8-9 mice per group) and RT q‐PCR (n = 6 mice per group) in the remote area of MI-operated hearts and their sham controls. Corresponding statistic of CAR3 was shown. D, Immunohistochemistry for cardiac fibroblast CAR3 was indicated by black arrows in the sham or MI-operated heart tissue (n = 4 samples per group, the left scale bar =500 μm, the right scale bar =25 μm). E, Immunofluorescence co-staining for vimentin with CAR3 and DAPI in cardiac fibroblasts was shown by white arrows in the heart of WT mice at day 7 post-MI (n = 4 samples per group, the upper scale bar =500 μm, the lower scale bar =25 μm). CAR3 was labeled in green. vimentin was labeled in red. Nuclei stained with DAPI were blue. The data are expressed as mean ± SEM. The data shown in A-C were analyzed by Student's t-test. CAR3, Carbonic anhydrase 3; RT q‐PCR, real‐time quantitative polymerase chain reaction; WT, wild type; MI, myocardial infarction; DAPI, 4'6-diamidino-2-phenylindole; MI-7d, 7 days post myocardial infarction.
Upregulation of CAR3 in CFs upon MI operation and TGF-β1 treatment in vitro
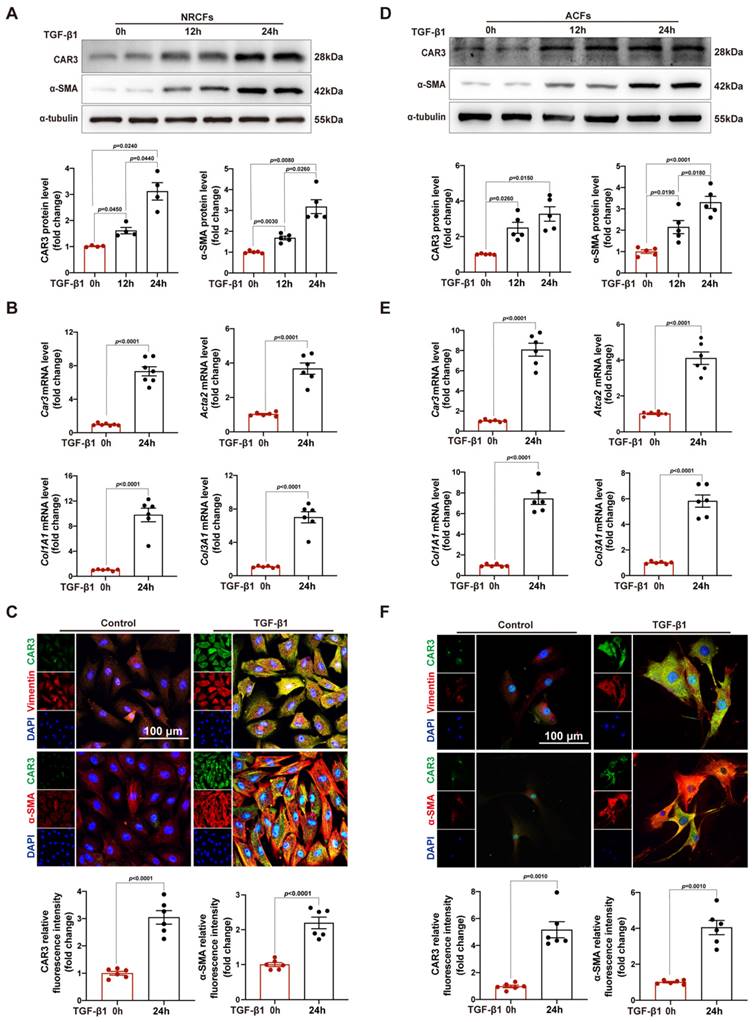
After increase of CAR3 in infarcted heart was identified, we further identify the cellular specificity of CAR3 expression change in cardiac tissue after MI. IF staining showed that the CAR3 in infarct area was colocalized with vimentin, a specific marker of cardiac fibroblasts, while there was only sporadic colocalization in the border and remote areas post-MI or sham-operated controls (Figure 1E). We then detected the expression of CAR3 in cultured NRCMs, NRCFs isolated from neonatal rat hearts and HUVEC cell line, and Western blot analysis demonstrated that NRCMs and NRCFs expressed CAR3 moderately, while no CAR3 was observed in cultured HUVECs (Figure S2A). To simulate ischemia condition, we cultured the cells by deprivation of glucose and oxygen, and observed no significant alteration of CAR3 and cTNT expression in NRCMs by either Western bolt analysis, RT-qPCR study, or IF staining (Figure S2B-D). We also observed no distinct expression difference for CAR3 and α-SMA in NRCFs and ACFs regardless exposed to hypoxia treatment or not (Figure S2E, F). While the expression of CAR3 markedly increased in NRCFs treated with TGF-β1, accompanied by increased expression of α-SMA (Figure 2A, B), and collagen-related genes such as Col1A1 and Col3A1 as well (Figure 2B). IF analysis further confirmed the colocalization of CAR3 with vimentin or α-SMA in NRCFs (Figure 2C). In addition, ACFs from WT mice displayed similar results by Western blotting analysis, RT-qPCR study and immunofluorescence staining (Figure 2D-F). While CAR2 protein levels in NRCFs and ACFs showed no dramatic changes between TGF-β1-treated groups and their controls (Figure S2G, H). These results confirmed specific CAR3 upregulation in cardiac fibroblasts but not cardiomyocytes in infarct area post MI.
CAR3 level is up-regulated in TGF-β1-treated cardiac fibroblasts. A, Expression of CAR3 was determined using Western blot (n = 4-5 independent experiments per group) in TGF-β1‐treated NRCFs. Results were normalized against α-tubulin and converted to fold induction relative to control‐treated group. B, NRCFs were administrated by TGF-β1 or not for 24h. Relative mRNA expressions of Car3, Acta2, Col1A1and Col3A1 were examined by RT q‐PCR (n = 6 independent experiments per group). Corresponding statistics were shown. C, Representative CAR3 immunofluorescence images of NRCFs treated with TGF-β1 or Control. CAR3 was labeled in green. Fibroblast markers α-SMA and vimentin were labeled in red. Nuclei stained with DAPI were blue. Fluorescence intensity of CAR3 and α-SMA staining was determined and expressed as fold change relative to control group (n = 6 independent experiments per group, scale bar = 100 μm). D, CAR3 and α-SMA Western blot bands (n = 5 per group) were shown in ACFs after stimulation with TGF-β1 or control. Corresponding statistics of CAR3 and α-SMA were shown. E, Relative mRNA expressions of Car3, Acta2, Col1A1and Col3A1 were examined by RT q‐PCR (n = 6 per group) in ACFs treated by TGF-β1 for 24h. F, IF co-staining for α-SMA or vimentin with CAR3 and DAPI in TGF-β1-treated ACFs. Fluorescence intensity was measured and the results were presented as fold change against the corresponding controls (n = 6 independent experiments per group, scale bar = 100 μm). Data are expressed as mean ± SEM. The data shown in A and B were analyzed by one-way ANOVA followed by Bonferroni post hoc test. Data shown in B, C, E, and F were analyzed by Student's t-test. TGF-β1, transforming growth factor-β1; NRCFs, neonatal rat cardiac fibroblasts; α-SMA, α-smooth muscle actin; Acta2, actin alpha 2, smooth muscle, aorta; Col1A1, collagen type I alpha 1 chain; Col3A1, collagen type III alpha 1 chain; ACFs, adult mouse cardiac fibroblasts.
CAR3 deficiency impaired cardiac repair and aggravated cardiac dysfunction 7d post-MI
The altered expression of CAR3 in myocardial tissue and TGF-β1-treated fibroblasts suggested its underlying role in the regulation of cardiac repair post-MI. Then, we subjected Car3‐deficient mice and their WT littermates to MI model. Echocardiographic parameters indicated a similar cardiac phenotype of Car3‐deficient mice and WT littermates at baseline (Table S2). However, triphenyltetrazolium chloride (TTC) staining revealed that CAR3 deficiency considerably enlarged left ventricular infarct area 7 days after MI surgery (Figure 3B). Similarly, cardiac function assessed by ultrasound showed a significantly worsen deterioration of left ventricular ejection fraction (LVEF) and fractional shortening (LVFS) in Car3‐deficient mice subjected to LAD ligation, and other parameters such as LV internal diameter at end diastole (LVIDd) and at end systole (LVIDs), LV end-diastolic volume (LVEDV) and end-systolic volume (LVESV), also suggested aggravated LV dilatation (Figure 3C, D).
CAR3 deficiency impaired cardiac wounding healing and exacerbated cardiac dysfunction 7d after MI. A, Schematic protocol of the experimental approach. B, The infarct size of WT and Car3‐deficient mouse heart at 7 days after MI operation was determined by TTC staining and expressed as the percentage of infarct over ventricular area (n = 5 mice per group). C, Representative serial echocardiography via short axis acquired at 7d post-MI and relative quantifications (n = 8 mice per group). D, Cardiac function parameters, namely LVEF, LVFS, LVIDs, LVIDd, LVEDV, and LVESV were measured by echocardiography in indicated groups (n = 8 mice per group). E, Representative Western blot analysis and quantitative results of Col1, Col3, and α-SMA. Results were normalized against α-tubulin and converted to fold induction relative to their respective controls (n = 4-5 mice per group). F, Transcription levels of Col1A1, Col3A1, postn, and Acta2 were measured by RT q‐PCR in the cardiac infarcted tissues of the indicated groups (n = 6 mice per group). G, Representative micrographs of Col1 expression by immunofluorescence staining in the infarct size at day 7 post-MI (n = 4 mice per group, the upper scale bar =100 μm, the lower scale bar =20 μm). H, Representative images of Col3 expression by IF in the infarct area 7 days post-MI (n = 4 mice per group, the upper scale bar =100 μm, the lower scale bar =20 μm). Data are presented as mean ± SEM, by one-way ANOVA followed by Bonferroni post hoc test. TTC, Triphenyltetrazolium chloride staining; LVEF, left ventricular ejection fraction; LVFS, left ventricular fractional shortening; LVIDs, left ventricular end‐systolic diameter; LVIDd, left ventricular end‐diastolic diameter; LVEDV, left ventricular end-diastolic volume; LVESV, left ventricular end-systolic volume.
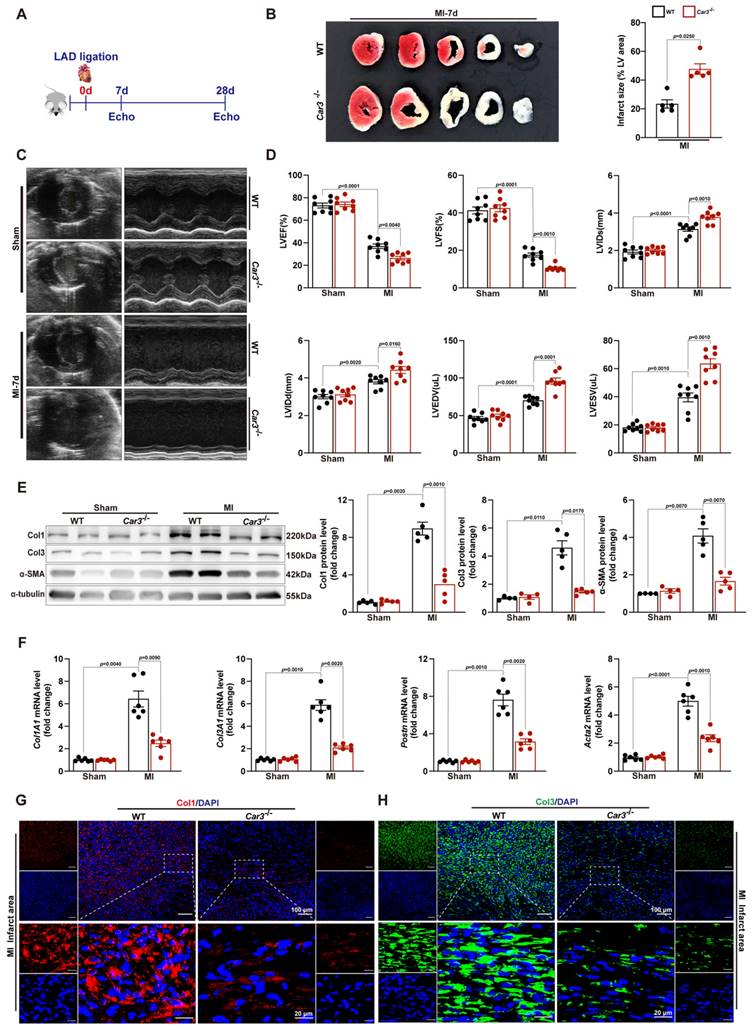
To explore the effects of CAR3 on cardiac healing post-MI, the dynamic change and composition of the ECM in infarcted myocardium were investigated. We found that on day 7 post MI, α-SMA, Col1 and Col3 were upregulated in infarct zone, whereas CAR3 deficiency largely obviated the up-regulated expression (Figure 3E). In addition, CAR3 deficiency dampened the mRNA levels of ECM-related genes Col1A1, Col3A1, Postn and Acta2 after myocardial infarction (Figure 3F). Consistently, IF staining also presented remarkable accumulation of Col1 and Col3 in the infarcted hearts of WT mice, but dramatic restrain of Col1 and Col3 increase in Car3-deficient mice (Figure 3G, H). Collectively, these results reveal that CAR3 deficiency worsened MI-induced cardiac dysfunction and infarct size, possibly by impairing deposition of collagen proteins, supporting a beneficial role for CAR3 in the proliferative phase of infarct healing.
CAR3 deficiency contributed infarct size enlargement and heart failure
Perturbed deposition of new structural matrix because of inadequate wound healing post-MI may lead to lethal cardiac rupture, enlarged infarct area and severe late-onset heart failure7. To test the effects of CAR3 deficiency on long-term cardiac function, we monitored cardiac function and pathological change 28 days after LAD ligation. Heart images and HE staining demonstrated that left ventricular scar formation in Car3-deficient mice was dramatically increased compared with WT mice (Figure 4A, B).
CAR3 deficiency enlarged infarct size and aggravated heart failure 28d post-MI. A, Representative pictures of the whole heart and HE staining of WT and Car3‐deficient mice after Sham or MI operation for 4 weeks (n = 6-7 mice per group). B, Infarct size was measured as the percentage of infarct over ventricular areas. C, Representative echocardiograms via short axis acquired from WT and Car3-knockout mice at 28 days after MI or sham operation and relative quantifications (n = 8 mice per group). D, Echocardiographic parameters of cardiac function, namely LVEF, LVFS, LVIDs, LVIDd, LVEDV, and LVESV, were measured in the indicated groups 4 weeks after MI (n = 8 mice per group). E, Representative photographs of Masson's trichrome staining in sections of hearts obtained from WT and Car3-deficient mice at day 28 after LAD ligation (n = 6-7 mice per group, the upper scale bar =2.5 mm, the lower scale bar =100 μm). F, Picrosirius red staining was performed to examine collagen density in the scar of different groups 4 weeks post-MI (n = 6-7 mice per group). G, Quantitative assessment of the ventricular wall thickness as well as the volume of collagen in the infarct area at 28d after MI (n=6-7 mice per group). H, Quantification of fibrotic areas in the border zone and remote zone at 28d post-MI in indicated groups (n=6-7 mice per group). The data are presented as mean ± SEM. B and D were analyzed by one-way ANOVA followed by Bonferroni post hoc test; G and H were analyzed by Student's t test.
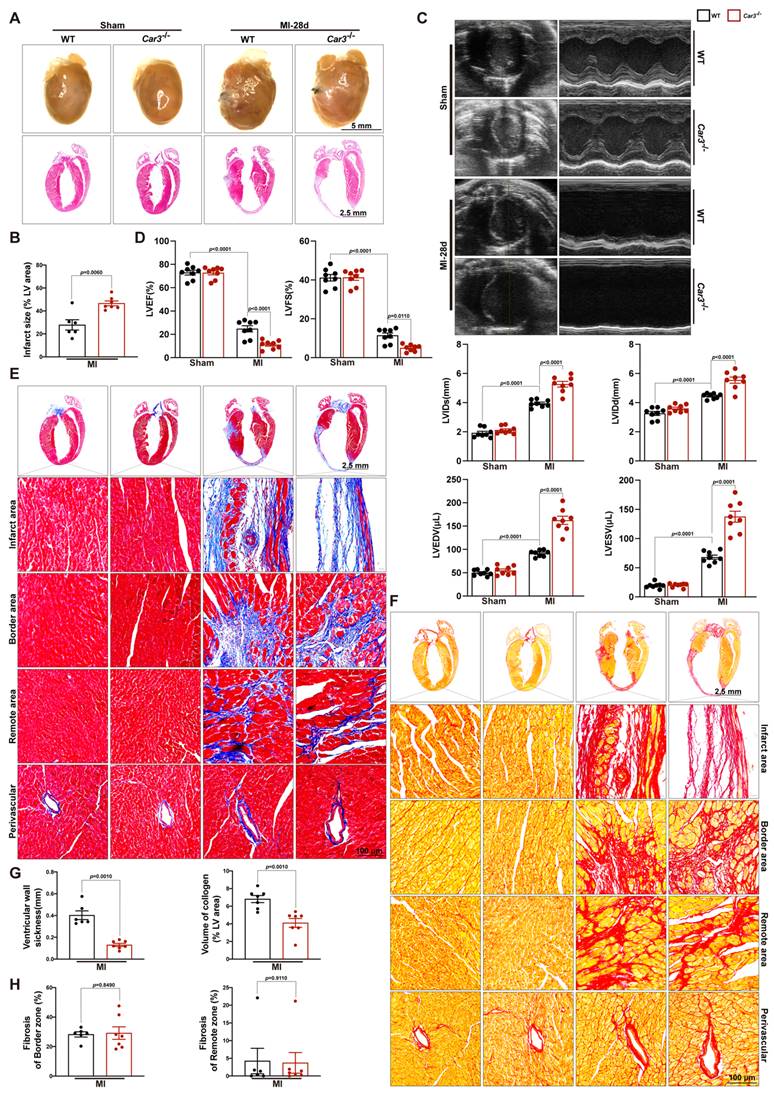
Echocardiographic parameters showed that permanent ischemia injury induced more deteriorating heart failure in Car3-knockout mice, testified by notably diminished LVEF and LVES and remarkably expanded LVIDs, LVIDd, LVESV and LVEDV (Figure 4C, D). Moreover, Masson trichrome and picrosirius red staining revealed that CAR3 deficiency strikingly inhibited the accumulation of collagen in infarct area 28 days after myocardial infarction, accompanied by distinctly thinner ventricular wall compared with WT mice (Figure 4E-H). Impressively, no significant difference of cardiac fibrosis in border zone, interstitial fibrosis in the remote zone or perivascular fibrosis was detected between two groups (Figure 4E-H). Thus, our studies indicate that CAR3 deficiency aggravated cardiac infarct size expansion and heart failure post-myocardial infarction.
CAR3 deficiency restrained wound healing and cardiac fibroblasts activation
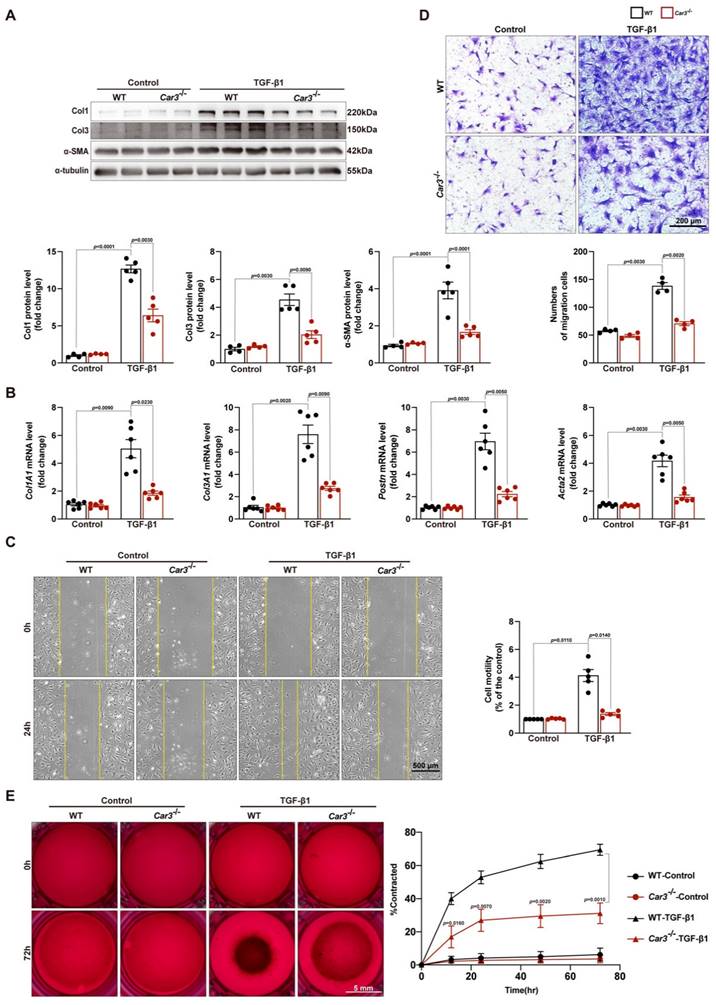
Cardiac fibroblasts are key cells for replacing damaged and lost cardiomyocytes and prominent for contributing to cardiac repair after the acute injury period post-MI10,43. The decreased ECM deposition in the infarct area of Car3-dificient mice indicated fibroblasts activation disorder. To further investigate the effects of CAR3 on fibroblast activation, ACFs isolated from mice were cultured and treated with TGF-β1 (10ng/ml). TGF-β1 stimulation could clearly activate fibroblasts from WT mice based on up-regulation of protein levels of α-SMA, Col1 and Col3 as well as mRNA levels of Col1A1, Col3A1, Postn and Acta2 (Figure 5A, B). In contrast, CAR3 deficiency obviously obviated the increase of α-SMA and collagen proteins (Figure 5A, B). Similarly, enhanced migratory capacity of fibroblasts from WT mice upon TGF-β1 stimulation were abolished by CAR3 deficiency based on transwell and wound-healing assays (Figure 5C, D). Gel contraction in fibroblasts lacking CAR3 was also dramatically hindered compared to activated myofibroblasts from WT mice (Figure 5E). On the whole, these results suggest that CAR3 deficiency significantly blunted TGF-β1 induced transformation of quiescent cardiac fibroblasts to myofibroblast based on collagen synthesis, cell migration and gel contraction.
CAR3 deficiency weakened wound healing and cardiac fibroblasts activation in vitro. A, Western blot analysis and quantitative results of Col1, Col3, and α-SMA in ACFs treated with TGF-β1 for 24h isolated from WT and Car3-dificient mice. Results were normalized against α-tubulin and converted to fold induction relative to their respective controls (n = 5 independent experiments per group). B, RT q‐PCR was used to determine the transcription levels of Col1A1, Col3A1, postn, and Acta2 in the indicated groups (n = 6 independent experiments per group). C, The migratory capacity of fibroblasts from WT and Car3-dificient mice respectively was assessed by the covering area of the scratch after administration with TGF-β1 for 24 h (n = 4 independent experiments per group, scale bar = 500 μm). D, Representative images of transwell assay after stimulation with TGF-β1 for 24h (n = 5 independent experiments per group, scale bar = 200 μm). E, Representative experiments of collagen gel contraction assay were investigated at different time points (n = 6 independent experiments per group, scale bar = 5 mm). Data are presented as mean ± SEM. The data shown in A-D were analyzed by one-way ANOVA followed by Bonferroni post hoc test. E was analyzed by two-way ANOVA followed by Bonferroni post hoc test.
CAR3 mediated cardiac fibroblasts activation by stabilizing inhibitory Smad7
Overwhelming evidence indicated that TGF-β and its downstream Smad-dependent signaling is a key inducer of fibroblast-to-myofibroblast conversion and ECM production17,44. To explore the molecular mechanism underlying CAR3-dependent fibroblast activation, we first investigated the expression of TGFβR1 and TGFβR2. Although TGFβR1 and TGFβR2 were increased in the infarct area of hearts after MI and in TGF-β1 stimulated CFs, CAR3 deficiency exerted no effects on their expressions (Figure S5A-D). We then explored the effects of CAR3 on TGF-β-Smad2/3 signaling. Up-regulation of phosphorylation of Smad2 and Smad3 were observed in ACFs treated with TGF-β1 as well as in infarcted cardiac tissues of WT mice 7d after MI, and CAR3 knockout strikingly inhibited the activation signal of Smad2 and Smad3 (Figure 6A, B). Smad6 and Smad7 (I-Smads) are well-studied negative signal regulators in TGF-β pathways, including phosphorylation of Smad2/345,46. As Smad6 preferentially inhibits bone morphogenetic protein (BMP)-induced Smad signaling47 while Smad7 restrains both BMP- and TGF-β-induced Smad signaling48, we subsequently detected the role of CAR3 on Smad7 expression change. After exposed to TGF-β1 treatment for 6 hours, Smad7 protein level was notably decreased in CFs from WT mice, which could be reversed when lacking CAR3 (Figure 6C). In parallel, CAR3 deficiency also dramatically weakened the reduction of Smad7 protein expression in infarcted cardiac tissue (Figure 6D). However, no significant expression change of Smad7 transcription level were observed both in vitro and in vivo (Figure 6C, D), which prompted us to explore how CAR3 affected Smad7 stability or turnover.
CAR3 deficiency inhibited the activation of TGF-β-Smad2/3 signaling pathway via mediating the stability of Smad7. A, Western blot analysis and statistical results of the levels of phosphorylated Smad2 and Smad3 in ACFs treated with TGF-β1 for 6h were examined and shown (n = 4 per group). B, Protein levels of phosphorylated Smad2 and Smad3 as well as Col1, Col3 and α-SMA were determined by Western blot (n = 4‐5 mice per group) in the infarcted heart of MI‐treated mice. C, Expression of Smad7 was measured using Western blot and RT q‐PCR (n = 4‐5 independent experiments per group) in cultured ACFs from WT and Car3-deficient mice 6h after TGF-β1 treatment. Results were normalized against α-tubulin and converted to fold induction relative to control‐treated group. D, Representative Western blots and statistical results of Smad7 in the infarct zone 7d post-MI (n = 5 mice per group). mRNA level of Smad7 was detected by RT q‐PCR in the indicated groups (n = 5 mice per group). E, Interaction of CAR3 with Smad7 in cultured ACFs of different groups was determined by immunoprecipitation with anti-CAR3 antibody followed by immunoblot with anti-Smad7 antibody. Following immunoprecipitation of Smad7, the acetylation of Smad7 was detected with anti-acetylated lysine antibody (Acy-K). IgG as a negative control (n = 3). F, Co-immunoprecipitation and Western blots of interaction of CAR3 with Smad7 and the change of Smad7 acetylation in infarcted heart tissues. IgG as a negative control (n = 3). The data are shown as the means ± SEM. The data shown in A, B, C, and D were analyzed by one-way ANOVA followed by Bonferroni post hoc test. Acy-K, acetylated lysine.
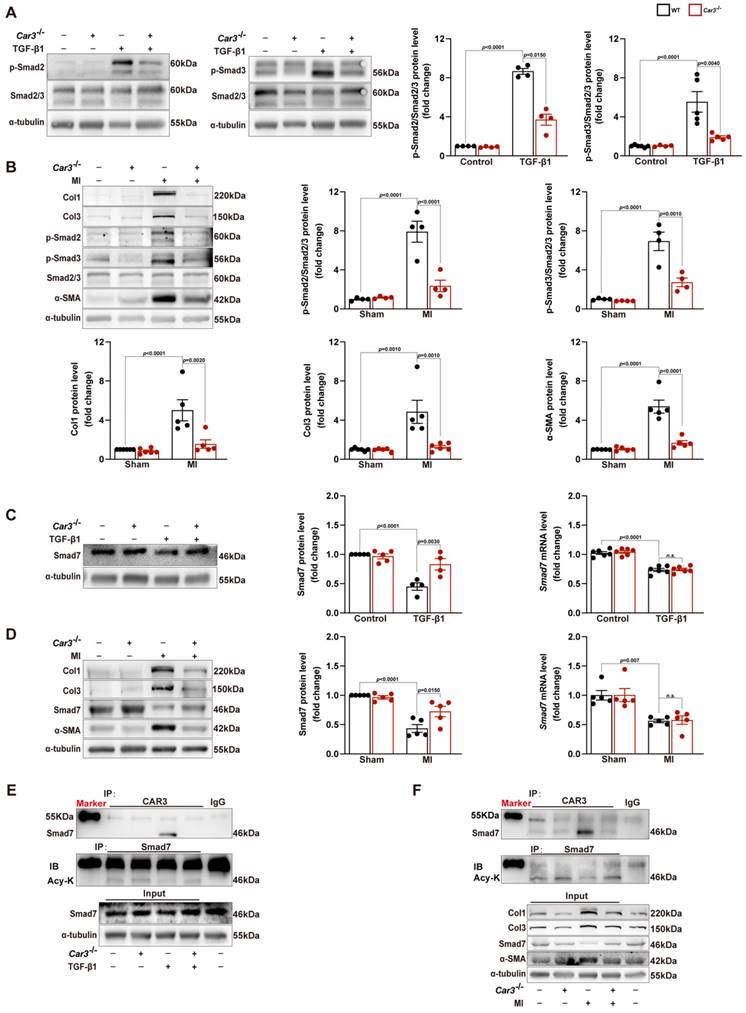
So we first evaluated the interaction between CAR3 and Smad7 in cardiac fibroblasts lysates by co-immunoprecipitation assays, and an increased association of CAR3 and Smad7 was observed in ACFs if treated with TGF-β1 (Figure 6E). As acetylation modification promotes Smad7 protein stability while TGF-β signaling inhibits its acetylation49,50, we then explored the function of CAR3 on Smad7 acetylation.
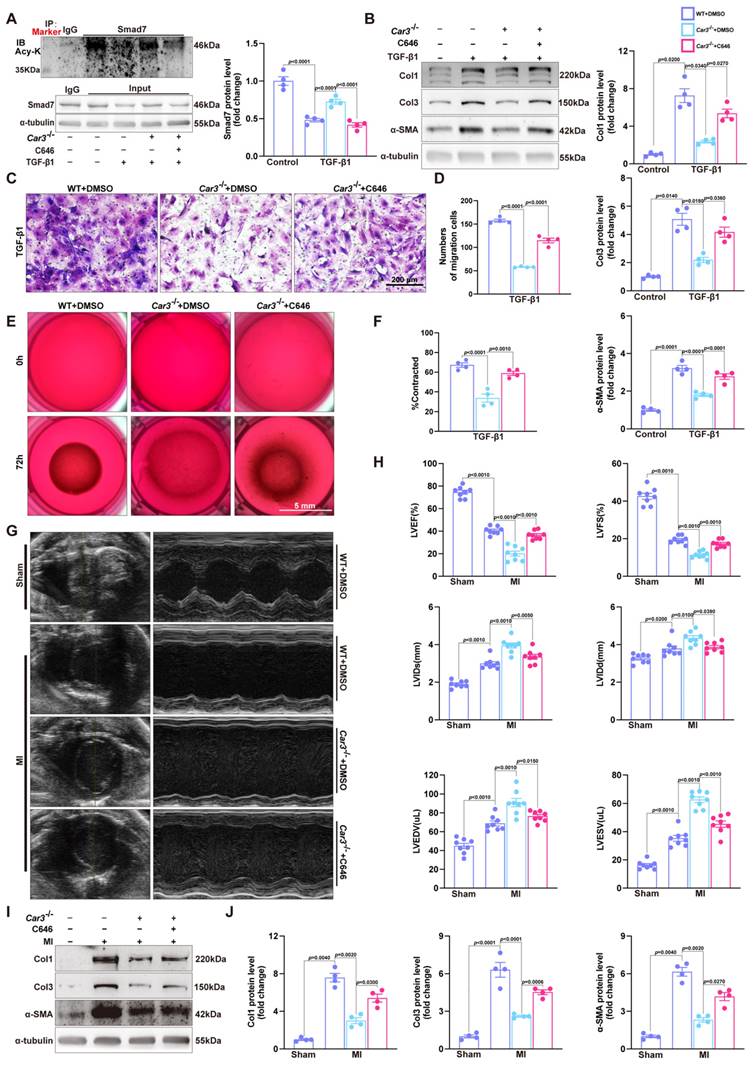
Co-immunoprecipitation and Western blot analysis demonstrated that TGF-β1 treatment caused decrease of Smad7 acetylation in CFs from WT mice, while CAR3 deficiency increased the acetylation of Smad7 (Figure 6E). Unsurprisingly, in vivo investigations showed that the enhanced association of CAR3 and Smad7 induced in MI was revoked by CAR3 deficiency, so do reduction of Smad7 acetylation (Figure 6F). To further assessed whether CAR3 mediated Smad7 protein stability through regulating acetylation, compound C646 experiments were performed to inhibit p30042, which is the acetyltransferase responsible for Smad7 acetylation. C646 treatment of fibroblast cells restrained the increased acetylation of Smad7, thus inhibiting the upregulation of Smad7 protein level induced by CAR3 deficiency (Figure 7A; S6A, B). And accordingly, inhibition of fibroblast activation was released in CAR3 deficiency condition (Figure 7B-F). This function was mirrored by alleviated CAR3 deficiency-exerted impaired cardiac healing, and improved cardiac function accompanied with increased collagen synthesis in the infarcted hearts 7d post-MI (Figure 7G-J). These data demonstrate that CAR3 plays a role in regulating Smad7 protein stability by modifying p300-dependent acetylation.
C646-pretreatment reversed CAR3 deficiency induced suppression of fibroblasts activation in vitro and impairment of cardiac healing and cardiac dysfunction in vivo. A, WT, Car3-knockout, or C646-preincubated Car3-knockout ACFs were stimulated with TGF-β1 for 6 h. Expressions of Smad7 and its acetylation were determined using Western blot (n = 4 independent experiments per group). B, ACFs were preincubated with DMSO or C646 followed by treatment with TGF-β1 for 24 h. Protein levels of Col1, Col3, and α-SMA were detected by Western blot (n = 4 per group). C, Representative images of transwell assay in indicated groups (n = 4 independent experiments per group, scale bar = 200 μm). D, Quantitative results for the migration capacity were measured. E, Representative experiments of collagen gel contraction assay in ACFs preincubated with DMSO or C646 followed by TGF-β1 treatment (n = 4 independent experiments per group, scale bar = 5 mm). F, Quantitative results for collagen gel contraction ability in indicated groups. G, WT, Car3-knockout, or C646-pretreated Car3-knockout mice were followed to induce mouse MI model. Representative serial echocardiography acquired by short axis at 7d post-MI and relative quantifications (n = 8 mice per group). H, Cardiac function parameters (LVEF, LVFS, LVIDs, LVIDd, LVEDV, and LVESV) were measured by echocardiography in indicated groups (n = 8 mice per group). I, Representative Western blot analysis (n = 4 mice per group) and quantitative results of Col1, Col3, and α-SMA. J, Statistical results of Col1, Col3, and α-SMA in the infarct zone 7d post-MI. Data are presented as mean ± SEM, by one-way ANOVA followed by Bonferroni post hoc test. DMSO, Dimethyl sulfoxide.
CAR3 overexpression promoted wound healing and cardiac fibroblasts activation in vitro
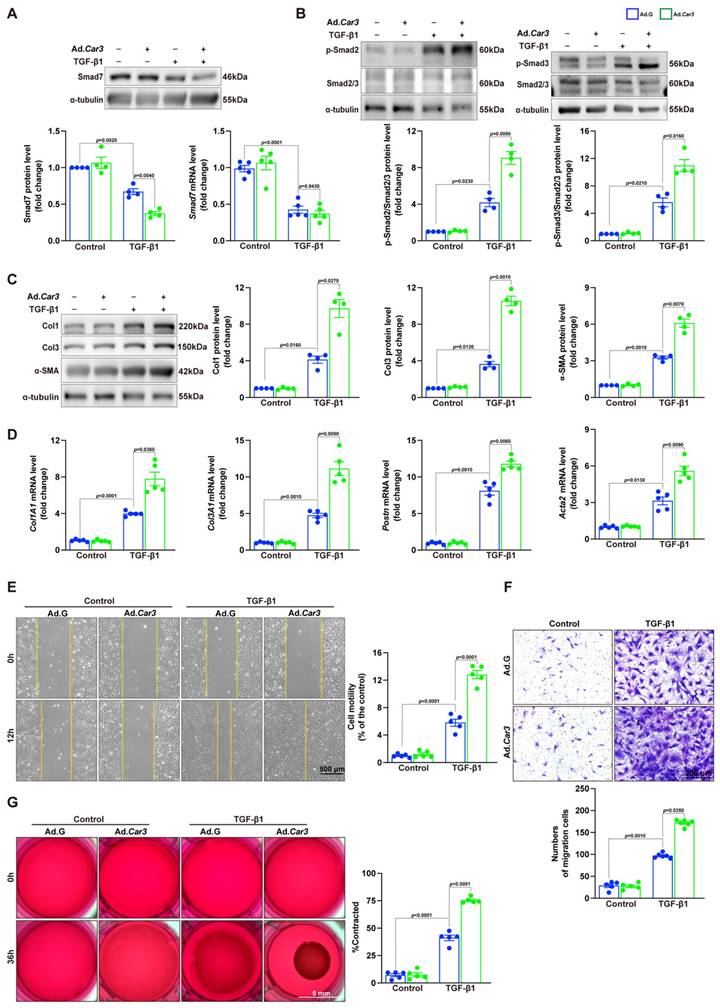
To further confirm the effects of CAR3 on fibroblast activation, we conducted experiments with Adv-GFP or Adv-Car3 to overexpress CAR3 or not (Figure S7A-C). Western blotting revealed that CAR3 overexpress in fibroblasts exerted a further reduction of Smad7 protein level under TGF-β1 treatment, unsurprisingly, no significant difference of Smad7 mRNA level was detected (Figure 8A). We then observed that fibroblasts infected with Adv-Car3 signally increased the expression of phosphorylation of Smad2 and Smad3 compared to Adv-GFP group after TGF-β1 treatment for 6 hours (Figure 8B). Accordingly, the expression of α-SMA and related collagen proteins were promoted further in CAR3 overexpression group upon TGF-β1 stimulation (Figure 8C, D). Moreover, overexpression of CAR3 promoted both cellular migration and contraction of fibroblasts (Figure 8E-G). Overall, these data confirm that CAR3 plays a key role in activation of cardiac fibroblasts and synthesis of ECM.
CAR3 overexpression promoted cardiac fibroblasts activation and wound healing in vitro. A, ACFs isolated from WT mice were infected with Adv-Car3 (Ad.Car3) or Adv-GFP (Ad.G) for 48 h before the cells were treated with TGF-β1 for 6 h. Expression of Smad7 was determined using Western blot and RT q‐PCR (n = 4‐5 independent experiments per group). B, Expression level of phosphorylated Smad2 and Smad3 in ACFs were examined by Western blot. Corresponding statistics were shown (n = 4 per group). C, ACFs were infected with Adv-Car3 or Adv-GFP for 48 h followed by treatment with TGF-β1 for 24 h. Protein levels of Col1, Col3, and α-SMA and statistical results were detected by Western blot (n = 4 per group). D, The transcription levels of Col1A1, Col3A1, postn, and Acta2 were determined by RT q‐PCR in the indicated groups (n = 4 per group). E, Quantitative results for the migration ability of ACFs in indicated groups was measured by the covering area of the scratch (n = 5 independent experiments per group, scale bar = 500 μm). F, Representative images of transwell assay in ACFs transduced with Adv-Car3 or Adv-GFP for 48 hours followed by TGF-β1 treatment (n = 5-6 independent experiments per group, scale bar = 200 μm). G, Collagen gel contraction assay was carried out to test the contraction capacity of ACFs after 36h administration with TGF-β1 in indicated groups (n = 6 independent experiments per group, scale bar = 5 mm). Data are presented as mean ± SEM, by one-way ANOVA followed by Bonferroni post hoc test. GFP, green fluorescent protein.
Discussion
In the present work, we report a cardioprotective role of CAR3 in cardiac repair post-MI. The up-regulation of CAR3, mainly in fibroblasts, is responsive upon myocardial injury, and is beneficial for cardiac healing, while CAR3 deficiency in mice significantly impaired cardiac wounding healing, leading to deteriorated cardiac function and increased infarct size. Mechanistically, CAR3 involved in conversion of fibroblasts to myofibroblasts via activation of TGF-β-Smad2/3 signaling pathway through regulating acetylation of Smad7. Thus our current study provides evidence that CAR3 promotes CF activation and potentially anticipates a promising therapeutic target for cardiac repair post-MI.
Substantial evidence has verified the effects of CAR3 in regulating not only physiological processes (fatty acid metabolism and oxidative stress response) but pathological conditions (muscle dysfunction, cancer progression, and autoimmune diseases)19,22,51-53. Dramatic elevation of CAR3 was observed in multiple cardiovascular biological processes, suggesting the potential involvement of CAR3 in the pathogenesis of cardiovascular diseases33-36,54. However, these studies failed to explore whether and how CAR3 works during myocardial ischemia injury. In this study, we find that CAR3 plays a critical role on cardiac fibroblasts during cardiac wound healing post-MI. CAR3 deficiency dramatically hampers transformation of cardiac fibroblast to myofibroblast, while CAR3 overexpression facilitates it.
As a key source of ECM, cardiac fibroblasts involve in controlling the homeostasis of microenvironment in mammalian heart and maintaining cardiac architecture and performance post-MI14,55. Upon exposure to MI injury, timely and sufficient transformation of fibroblasts to myofibroblasts is vital to promote cardiac repair, prevent cardiac rupture, and alleviate later myocardial remodeling and cardiac dysfunction10,43,56,57. During ischemic myocardial injury, TGF-β and its downstream signaling pathway is one of the most potent regulatory cascades for fibroblast differentiation44,58,59. TGF-β binds a heterodimeric receptor in the plasma membrane consisting of the TGF-β receptor type 1 (TGFβR1) and 2 (TGFβR2), which subsequently activates canonical Smad or non-canonical signaling cascades16.
Schematic diagram depicting the role of CAR3 in cardiac repair post MI. CAR3 deficiency aggravated infarct size enlargement and cardiac dysfunction after myocardial infarction through inhibition of cardiac fibroblasts transformation. Mechanistically, CAR3 promotes fibroblast transformation by TGF-β-Smad2/3 signaling via repressing acetylation of inhibitory Smad7, which ultimately improves timely cardiac repair post MI.
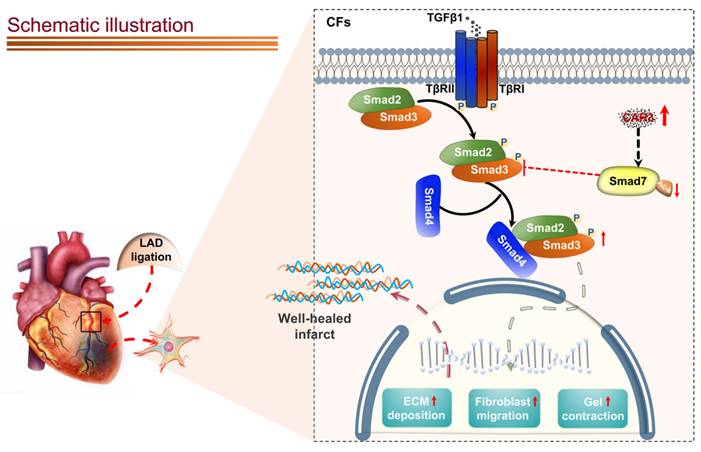
Canonical TGF-β-Smad2/3 signaling is well-documented for dominating myofibroblast activation and matrix protein synthesis60-64. Our data show that CAR3 deficiency notably suppresses activation of Smad2/3 signaling pathway in the infarct size of cardiac tissue post-MI and in cardiac fibroblasts under TGF-β1 treatment.
A variety of positive and negative regulators involves in regulating TGF-β-Smad2/3 signaling16. Among them, Smad7 has aroused great interest as a pleiotropic endogenous inhibitor that represses intracellular signaling through interactions with activated type I receptors (TGFβRs) or R-Smads46. Our current study showed that Smad7 protein level was down-regulated in infarcted heart tissue post MI, while CAR3 deficiency reversed the decrease of Smad7 protein expression with no observable change of Smad7 mRNA expression. We did observe an increased interaction of CAR3 and Smad7 both in vivo and in vitro suggesting a potential role of CAR3 on posttranslational modification of Smad7 protein. Modifications of Smad7 are associated with its stability and interaction with other proteins, which in turn regulates its functions46. Lys64 and Lys70 in the N terminus of Smad7 can be acetylated by acetyltransferase p300 and thus stabilizes Smad7, preventing it from subsequent ubiquitination degradation46,49. Our data showed that LAD-ligation and TGF-β1 treatment strikingly reduced Smad7 acetylation, while CAR3 deficiency restored the acetylation of Smad7. Meanwhile, C646, a specific inhibitor for p300 acetyltransferase, blunt CAR3 deficiency-exerted increase of Smad7 acetylation and freed restrain of fibroblast activation, and improved impaired cardiac repair and collagen synthesis. These results provide evidence supporting the hypothesis that CAR3 regulates the acetylation of Smad7 mediated by acetyltransferase p300. To our knowledge, this is the first study to identify CAR3 as a crucial regulator of cardiac fibroblasts activation via the TGF-β-Smad2/3 pathway and Smad7 protein as a target of CAR3.
However, some limitations exist in the current study, and need further research. The first one is what causes the increase of CAR3 expression in infarcted myocardial tissue. The second question is that we may need a cell-specific, like fibroblast-specific Car3 deficient mice, to exclude the effects of CAR3 on cardiomyocytes during MI. The module of how CAR3 affects p300-mediated Smad7 acetylation is also not clear. Other interesting issue also include the individual role of CAR3 in other phases, which are necessary to further investigations about therapeutic time window for MI. Elucidating these issues may provide translational application for CAR3 in intervening cardiovascular disease.
Abbreviations
MI: myocardial infarction; CF: cardiac fibroblast; CAR3: carbonic anhydrase 3; ACF: adult mouse primary cardiac fibroblast; CHD: Coronary heart disease; ECM: extracellular matrix; ⍺-SMA: α-smooth muscle actin; DCM: dilated cardiomyopathy; HCM: hypertrophic cardiomyopathy; WT: wild‐type; LAD: left anterior descending artery; LVID: left ventricular internal diameter; LVEF: left ventricular ejection fraction; LVFS: left ventricular fractional shortening; LVEDV: left ventricular end-diastolic volume; LVESV: left ventricular end-systolic volume; TTC: triphenyltetrazolium chloride; NRCF: neonatal rat cardiac fibroblasts; NRCM: neonatal rat cardiac myocyte; PBS: phosphate buffered saline; FBS: fetal bovine serum; HUVEC: human umbilical vein endothelial cells; Adv: recombinant adenovirus; PVDF: polyvinylidene difluoride membranes; ECL: enhanced-chemiluminescent system; RT q‐PCR: real‐time quantitative polymerase chain reaction; IF: immunofluorescence; DAPI: 4′,6-diamidino-2-phenylindol; HE: hematoxylin and eosin; ANOVA: analysis of variance; BMP: bone morphogenetic protein; TGFβR: TGF-β receptor.
Supplementary Material
Supplementary materials and methods, figures and tables.
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (82171812, 31870885, and 82000262).
Author contributions
C.X. obtained the fund support, conceived and supervised the study. Y.S. designed the study, performed the experiments, analyzed the data and drafted the manuscript. D.S. conducted animal models and experiments. G.X., Y.L, L.X, L.D, X.L. assisted study design and performed a series of experiments. C.S. and C.X. revised the paper. All authors have read and approved the article.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS. et al. Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association. Circulation. 2022;145:e153-e639
2. Vaduganathan M, Mensah GA, Turco JV, Fuster V, Roth GA. The Global Burden of Cardiovascular Diseases and Risk: A Compass for Future Health. J Am Coll Cardiol. 2022;80:2361-2371
3. Mensah GA, Roth GA, Fuster V. The Global Burden of Cardiovascular Diseases and Risk Factors: 2020 and Beyond. J Am Coll Cardiol. 2019;74:2529-2532
4. Niccoli G, Montone RA, Ibanez B, Thiele H, Crea F, Heusch G. et al. Optimized Treatment of ST-Elevation Myocardial Infarction. Circ Res. 2019;125:245-258
5. Prabhu SD, Frangogiannis NG. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ Res. 2016;119:91-112
6. Frangogiannis NG. Pathophysiology of Myocardial Infarction. Compr Physiol. 2015;5:1841-75
7. van den Borne SW, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol. 2010;7:30-7
8. Frangogiannis NG. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ Res. 2019;125:117-146
9. Frangogiannis NG. The extracellular matrix in myocardial injury, repair, and remodeling. J Clin Invest. 2017;127:1600-1612
10. Daseke MJ 2nd, Tenkorang MAA, Chalise U, Konfrst SR, Lindsey ML. Cardiac fibroblast activation during myocardial infarction wound healing: Fibroblast polarization after MI. Matrix Biol. 2020;91-92:109-116
11. Ma Y, Iyer RP, Jung M, Czubryt MP, Lindsey ML. Cardiac Fibroblast Activation Post-Myocardial Infarction: Current Knowledge Gaps. Trends Pharmacol Sci. 2017;38:448-458
12. Chen W, Frangogiannis NG. Fibroblasts in post-infarction inflammation and cardiac repair. Biochim Biophys Acta. 2013;1833:945-53
13. Talman V, Ruskoaho H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016;365:563-81
14. Humeres C, Frangogiannis NG. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl Sci. 2019;4:449-467
15. Kong P, Shinde AV, Su Y, Russo I, Chen B, Saxena A. et al. Opposing Actions of Fibroblast and Cardiomyocyte Smad3 Signaling in the Infarcted Myocardium. Circulation. 2018;137:707-724
16. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577-84
17. Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184-95
18. Susan C. Frost and Robert McKenna. Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications. Subcellular biochemistry. 2014
19. Aspatwar A, Tolvanen MEE, Barker H, Syrjänen L, Valanne S, Purmonen S. et al. Carbonic anhydrases in metazoan model organisms: molecules, mechanisms, and physiology. Physiol Rev. 2022;102:1327-1383
20. Herbert JD, Coulson RA. A role for carbonic anhydrase in de novo fatty acid synthesis in liver. Ann N Y Acad Sci. 1984;429:525-7
21. Sly WS, Hu PY. Human carbonic anhydrases and carbonic anhydrase deficiencies. Annu Rev Biochem. 1995;64:375-401
22. Harju AK, Bootorabi F, Kuuslahti M, Supuran CT, Parkkila S. Carbonic anhydrase III: a neglected isozyme is stepping into the limelight. J Enzyme Inhib Med Chem. 2013;28:231-9
23. Supuran CT. Carbonic anhydrases-an overview. Curr Pharm Des. 2008;14:603-14
24. Silagi ES, Batista P, Shapiro IM, Risbud MV. Expression of Carbonic Anhydrase III, a Nucleus Pulposus Phenotypic Marker, is Hypoxia-responsive and Confers Protection from Oxidative Stress-induced Cell Death. Sci Rep. 2018;8:4856
25. Shi C, Uda Y, Dedic C, Azab E, Sun N, Hussein AI. et al. Carbonic anhydrase III protects osteocytes from oxidative stress. FASEB J. 2018;32:440-452
26. Zimmerman UJ, Wang P, Zhang X, Bogdanovich S, Forster R. Anti-oxidative response of carbonic anhydrase III in skeletal muscle. IUBMB Life. 2004;56:343-7
27. Räisänen SR, Lehenkari P, Tasanen M, Rahkila P, Härkönen PL, Väänänen HK. Carbonic anhydrase III protects cells from hydrogen peroxide-induced apoptosis. Faseb j. 1999;13:513-22
28. Liu D, Wong CC, Zhou Y, Li C, Chen H, Ji F. et al. Squalene Epoxidase Induces Nonalcoholic Steatohepatitis Via Binding to Carbonic Anhydrase III and is a Therapeutic Target. Gastroenterology. 2021;160:2467-2482 e3
29. Mitterberger MC, Kim G, Rostek U, Levine RL, Zwerschke W. Carbonic anhydrase III regulates peroxisome proliferator-activated receptor-gamma2. Exp Cell Res. 2012;318:877-86
30. Yamamoto H, Uramaru N, Kawashima A, Higuchi T. Carbonic anhydrase 3 increases during liver adipogenesis even in pre-obesity, and its inhibitors reduce liver adipose accumulation. FEBS Open Bio. 2022;12:827-834
31. Chu YH, Su CW, Hsieh YS, Chen PN, Lin CW, Yang SF. Carbonic Anhydrase III Promotes Cell Migration and Epithelial-Mesenchymal Transition in Oral Squamous Cell Carcinoma. Cells. 2020 9
32. Dai HY, Hong CC, Liang SC, Yan MD, Lai GM, Cheng AL. et al. Carbonic anhydrase III promotes transformation and invasion capability in hepatoma cells through FAK signaling pathway. Mol Carcinog. 2008;47:956-63
33. Su H, Hu K, Liu Z, Chen K, Xu J. Carbonic anhydrase 2 and 3 as risk biomarkers for dilated cardiomyopathy associated heart failure. Ann Palliat Med. 2021;10:12554-12565
34. Coats CJ, Heywood WE, Virasami A, Ashrafi N, Syrris P, Dos Remedios C. et al. Proteomic Analysis of the Myocardium in Hypertrophic Obstructive Cardiomyopathy. Circ Genom Precis Med. 2018;11:e001974
35. Shuai Y, Guo J, Dong Y, Zhong W, Xiao P, Zhou T. et al. Global gene expression profiles of MT knockout and wild-type mice in the condition of doxorubicin-induced cardiomyopathy. Toxicol Lett. 2011;200:77-87
36. Veltman D, Wu M, Pokreisz P, Claus P, Gillijns H, Caluwé E. et al. Clec4e-Receptor Signaling in Myocardial Repair After Ischemia-Reperfusion Injury. JACC Basic Transl Sci. 2021;6:631-646
37. Kim G, Lee TH, Wetzel P, Geers C, Robinson MA, Myers TG. et al. Carbonic anhydrase III is not required in the mouse for normal growth, development, and life span. Mol Cell Biol. 2004;24:9942-7
38. Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M. et al. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res. 2010;107:1445-53
39. Ono H, Kato T, Murase Y, Nakamura Y, Ishikawa Y, Watanabe S. et al. C646 inhibits G2/M cell cycle-related proteins and potentiates anti-tumor effects in pancreatic cancer. Sci Rep. 2021;11(1):10078
40. Pistner A, Belmonte S, Coulthard T, Blaxall B. Murine echocardiography and ultrasound imaging. J Vis Exp. 2010
41. Huang S, Li X, Zheng H, Si X, Li B, Wei G. et al. Loss of Super-Enhancer-Regulated circRNA Nfix Induces Cardiac Regeneration After Myocardial Infarction in Adult Mice. Circulation. 2019;139:2857-2876
42. Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA. et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol. 2010;17:471-82
43. Jugdutt BI. Ventricular remodeling after infarction and the extracellular collagen matrix: when is enough enough? Circulation. 2003;108:1395-403
44. Stempien-Otero A, Kim DH, Davis J. Molecular networks underlying myofibroblast fate and fibrosis. J Mol Cell Cardiol. 2016;97:153-61
45. Li X, Guo L, Liu Y, Su Y, Xie Y, Du J. et al. MicroRNA-21 promotes wound healing via the Smad7-Smad2/3-Elastin pathway. Exp Cell Res. 2018;362:245-251
46. Miyazawa K, Miyazono K. Regulation of TGF-beta Family Signaling by Inhibitory Smads. Cold Spring Harb Perspect Biol. 2017 9
47. Goto K, Kamiya Y, Imamura T, Miyazono K, Miyazawa K. Selective inhibitory effects of Smad6 on bone morphogenetic protein type I receptors. J Biol Chem. 2007;282:20603-11
48. Hanyu A, Ishidou Y, Ebisawa T, Shimanuki T, Imamura T, Miyazono K. The N domain of Smad7 is essential for specific inhibition of transforming growth factor-beta signaling. J Cell Biol. 2001;155:1017-27
49. Grönroos E, Hellman U, Heldin CH, Ericsson J. Control of Smad7 stability by competition between acetylation and ubiquitination. Mol Cell. 2002;10:483-93
50. Simonsson M, Heldin CH, Ericsson J, Grönroos E. The balance between acetylation and deacetylation controls Smad7 stability. J Biol Chem. 2005;280:21797-803
51. Okada K, Ikemoto M. Carbonic Anhydrase III Has Potential as a Biomarker for Experimental Colitis and Functions as an Immune Regulator by Inhibiting Inflammatory Cytokine Secretion. Biology (Basel). 2022 11
52. Du A, Huang S, Zhao X, Feng K, Zhang S, Huang J. et al. Suppression of CHRN endocytosis by carbonic anhydrase CAR3 in the pathogenesis of myasthenia gravis. Autophagy. 2017;13:1981-1994
53. Du AL, Ren HM, Lu CZ, Tu JL, Xu CF, Sun YA. Carbonic anhydrase III is insufficient in muscles of myasthenia gravis patients. Autoimmunity. 2009;42:209-15
54. Zhang XM, Tao YH, Zhou XL, Shang XL, Gong XB, Liu YC. et al. The role of carbonic anhydrase III and autophagy in type 2 diabetes with cardio-cerebrovascular disease. Metab Brain Dis. 2021;36:2329-2341
55. Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255-78
56. Li YL, Hao WJ, Chen BY, Chen J, Li GQ. Cardiac Fibroblast-Specific Activating Transcription Factor 3 Promotes Myocardial Repair after Myocardial Infarction. Chin Med J (Engl). 2018;131:2302-2309
57. Ruiz-Villalba A, Romero JP, Hernández SC, Vilas-Zornoza A, Fortelny N, Castro-Labrador L. et al. Single-Cell RNA Sequencing Analysis Reveals a Crucial Role for CTHRC1 (Collagen Triple Helix Repeat Containing 1) Cardiac Fibroblasts After Myocardial Infarction. Circulation. 2020;142:1831-1847
58. Plikus MV, Wang X, Sinha S, Forte E, Thompson SM, Herzog EL. et al. Fibroblasts: Origins, definitions, and functions in health and disease. Cell. 2021;184:3852-3872
59. Frangogiannis NG. Transforming growth factor-β in myocardial disease. Nat Rev Cardiol. 2022;19:435-455
60. Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T. et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. 2017;127:3770-3783
61. Huang S, Chen B, Humeres C, Alex L, Hanna A, Frangogiannis NG. The role of Smad2 and Smad3 in regulating homeostatic functions of fibroblasts in vitro and in adult mice. Biochim Biophys Acta Mol Cell Res. 2020;1867:118703
62. Huang S, Chen B, Su Y, Alex L, Humeres C, Shinde AV. et al. Distinct roles of myofibroblast-specific Smad2 and Smad3 signaling in repair and remodeling of the infarcted heart. J Mol Cell Cardiol. 2019;132:84-97
63. Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G. et al. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. 2007;116:2127-38
64. Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF. et al. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res. 2010;107:418-28
Author contact
![]() Corresponding authors: Congfeng Xu: Email: cxuedu.cn, Tel./Fax: +86-21-24058721; Chengxing Shen: Email: shencxedu.cn, Tel./Fax: +86-21-24058441.
Corresponding authors: Congfeng Xu: Email: cxuedu.cn, Tel./Fax: +86-21-24058721; Chengxing Shen: Email: shencxedu.cn, Tel./Fax: +86-21-24058441.