Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Pathophysiology of CRC
KRAS signaling pathway in CRC
Mutated KRAS and the CRC TME
Treatments targeting KRAS and...
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(5):1947-1964. doi:10.7150/ijbs.88779 This issue Cite
Review
Impact of KRAS mutation on the tumor microenvironment in colorectal cancer
Yiru Zhou1, Yeye Kuang1, Chan Wang1, Yijian Yu2,3, Lijuan Pan4,5, Xiaotong Hu1,2 ![]()
1. Biomedical Research Center and Key Laboratory of Biotherapy of Zhejiang Province, Sir Run Run Shaw Hospital, Zhejiang University, Hangzhou, Zhejiang, China.
2. Department of Pathology, Sir Run Run Shaw Hospital, Zhejiang University, Hangzhou, Zhejiang, China.
3. Taizhou Hospital of Zhejiang Province affiliated to Wenzhou Medical University, Taizhou, Zhejiang, China.
4. School of Pharmacy, Hangzhou Normal University, Hangzhou, Zhejiang, China.
5. Key Laboratory of Elemene Class Anti-Cancer Chinese Medicines; Engineering Laboratory of Development and Application of Traditional Chinese Medicines; Collaborative Innovation Center of Traditional Chinese Medicines of Zhejiang Province, Hangzhou Normal University, Hangzhou, Zhejiang, China.
Received 2023-8-3; Accepted 2024-1-21; Published 2024-3-3
Abstract

Kirsten rat sarcoma viral oncogene homolog (KRAS) is an oncogene implicated in the pathophysiology of many cancers. Increasing evidence shows that KRAS mutation is correlated with poor prognosis in numerous cancers, including colorectal cancer (CRC), breast cancer, and melanoma. KRAS also participates in regulating the CRC microenvironment. However, the direct and indirect therapeutic targets of KRAS in CRC have not been identified; thus, elucidating the mechanisms and interactions between KRAS and the tumor microenvironment (TME) in-depth is paramount. Herein, we present some of the major roles KRAS plays in shaping the heterogeneity of the TME and propose a potential strategy for targeting the downstream components of the KRAS signaling pathway and the TME in CRC.
Keywords: Colorectal cancer, KRAS mutation, Tumor microenvironment, Immunotherapy
Introduction
KRAS is one of the most frequently mutated oncogenes across malignancies. KRAS mutations are present in approximately 40% of colorectal cancer (CRC) patients [1]. Phosphorylated and dephosphorylated oncogenic KRAS is essential for maintaining the epithelial structure of CRC cells in 3D culture and assisting in the hypodermic growth of tumors in mice [2]. Patients with metastatic CRC (mCRC) with KRAS and BRAF mutations have worse progression-free survival (PFS) and overall survival (OS) outcomes than those without these mutations [3]. CRC patients with KRAS mutations and liver metastases have more micrometastases, potentially resulting in recurrence after surgery [4]. KRAS mutation, which is more common in right-sided colon cancer (RCC) than in left-sided colon cancer (LCC), is correlated with a negative prognosis in LCC patients but not in RCC patients [5]. In addition, KRAS mutation is positively correlated with the TMEM16A protein, a calcium-activated chloride channel associated with tumorigenesis and progression in various cancers [6]. However, patients harboring multiple KRAS mutations have better prognoses than those harboring a single mutation [7]. Nevertheless, enriched KRAS signaling gene sets have been linked to inflammation, a favorable tumor immune microenvironment, and improved survival in triple-negative breast cancer [8].
The rates of KRAS mutation in codons 12, 13, and 61 are 73.3% (11/15), 20% (3/15), and 6.67% (1/15), respectively [9]. KRAS G12C is the most prevalent driver mutation and is found in a sizable proportion of CRC patients [10]. KRAS G12D is associated with poor survival outcomes and is a marker of poor prognosis in Chinese patients [11]. Fortunately, effective and potent G12D inhibitors have recently been developed [12]. The difference between G12D and G13D mutations lies in the activation of different signaling molecules [13].
In this review, we first present the pathophysiology and treatment of CRC. The role of KRAS mutation in the development and progression of CRC is then discussed. We also address the impact of KRAS mutation on the TME, including alterations in the immune response, metabolism, and angiogenesis.
Pathophysiology of CRC
CRC is one of the deadliest diseases worldwide and has serious implications for families and society. An estimated 1.2 million people are diagnosed with CRC annually, and the incidence rate is higher in men than in women [14]. The 5-year survival rate has remained at 64% over the past 10 years, and the overall cure rate of CRC has not increased [15]. Diet is an important factor affecting the incidence rate; those with a diet containing fiber, milk, calcium, or whole grain have a lower risk of CRC than those with a diet containing processed and red meat [16]. A person's lifestyle can be improved by following Mediterranean diet principles to help prevent cancer and other gut-related disorders [17]. Polyunsaturated fatty acids may be important in the development of G>A transitions in the KRAS oncogene, potentially contributing to the etiology of CRC [18]. In addition, smokers are prone to CpG island methylator phenotype (CIMP)-positive (several CpG island sites simultaneously hypermethylated in CRC) and high-MSI (deficient in mismatch repair system) CRC subtypes [19]. A particular cholesterol metabolic abnormality, a potential therapeutic target, was recently discovered in the liver metastases of CRC patients [20]. Typically, there are four consensus molecular subtypes (CMSs) of CRC: (1) CMS1 features immune infiltration and mismatch repair mutations. (2) CMS2 cells display the hallmarks WNT and MYC. (3) CMS3 is characterized by KRAS mutation and alterations in metabolism. (4) CMS4 is enriched in the stroma and is associated with the worst prognosis [21]. The TME in obese patients promotes the transition of CMS2 CRC epithelial cells toward the CMS4 mesenchymal subtype at the transcriptomic expression level [22].
Multiple KRAS-mutant alleles contribute to the heterogeneous landscape of the TME in KRAS-mutant CRC. Herein, we summarize several features identified by multiomics studies. Based on integrated multiomics data, Chong et al. reported that the KRAS-M1 (KM1) subtype was related to the CMS4 subtype, which, as opposed to the KRAS-M2 (KM2) subtype, is associated with a poor prognosis and increased angiogenesis, EMT, and TGF-β pathways [23]. Liu et al. integrated drug sensitivity information and the phosphoproteome and reported that the combination of SHP2 and DOT1L inhibitors is an effective treatment for patients with subset 2 KRAS-mutant cancers [24]. Using liquid chromatography-mass spectrometry (LC‒MS/MS) and enzyme-linked immunosorbent assay (ELISA), common methods for proteomic analysis, Lim et al. found that APOPA1 continually increased in the serum of CRC patients compared with that in healthy controls; thus, APOPA1 could serve as a potential biomarker for CRC patients [25]. A study of the proteome of the intermediate-stage colorectal cancer cell line Caco2 revealed that the mutant KRAS (V12) contributed to an increased H-Ras protein level, indicating that HRAS might be a pivotal factor involved in the effects of KRAS mutation [26].
At present, the first-line treatments for CRC are surgery, chemotherapy, immunotherapy, and targeted therapy. Current approaches, such as anti-epidermal growth factor receptor (EGFR)-targeted therapies and nonspecific cytotoxic chemotherapeutic regimens, have shown little benefit. Immune checkpoint blockade can be used to treat cancer effectively since glycolysis in cancer cells can be stimulated by interactions between programmed death ligand 1 (PD-L1) on murine CRC cells and PD1 on T cells, increasing effector T-cell access to glucose [27]. Regarding targeted therapy, it is extremely difficult to inhibit KRAS directly due to the limited number of available KRAS protein pockets [28]. The efficacy of indirect KRAS inhibition is also limited by other cancer-related cellular processes, such as cell Warburg metabolism, a process in which cancer cells preferentially use glycolysis to consume glucose and generate lactate despite the presence of oxygen; this process can be triggered by the mutant KRAS protein and maintain tumor growth [29]. The direct inhibitory effects of KRAS mainly include covalent binding to a residue of the protein and blocking the interaction between Ras and its ligands. When KRAS G12C is GDP-bound, these substances covalently attach to the mutant cysteine residue and occupy a novel allosteric pocket [30]. Strategies targeting KRAS indirectly include 1) inhibiting the nucleotide exchange cycle, 2) disrupting membrane localization and KRAS processing, and 3) inhibiting downstream signaling pathways. Here, we list several clinical trials to evaluate approaches that directly or indirectly target KRAS. MCLA-158, an LGR5 EGFR bispecific antibody, has therapeutic efficacy in preclinical models of various epithelial cancer types by inhibiting the growth of KRAS-mutant colorectal tumors, blocking metastasis initiation, and suppressing tumor expansion [31]. The novel therapies available for KRAS-mutant CRC include sotorasib [32], beta-elemene, cetuximab [33], and adagrasib (MRTX849) [34]. Target-specific therapeutic alternatives are needed because most of these clinically approved conventional regimens are frequently accompanied by dose-related toxicity, drug resistance, and unfavorable physiological side effects [35]. Another obstacle in CRC treatment is the disease's resistance to epidermal growth factor receptor (EGFR) inhibition conferred by KRAS mutation. The EGFR signaling pathway is the main cause of resistance to KRAS G12C inhibitors; consequently, EGFR and KRAS G12C should be inhibited together [4]. Thus, it is imperative to elucidate the role of KRAS mutation in CRC.
KRAS signaling pathway in CRC
KRAS activation and downstream effectors
The KRAS signaling pathway is critical for the regulation of cellular growth, division, and survival. The pathway is activated by KRAS, a small GTP-binding protein. KRAS activation leads to the recruitment and activation of downstream effectors, such as RAF, MEK, and ERK, ultimately resulting in the activation of transcription factors, such as ELK1, leading to the expression of genes that regulate cell proliferation, survival, and angiogenesis.
KRAS mutations and cancer development
The KRAS signaling pathway is tightly regulated in normal cells but is often hyperactivated in cancer cells due to KRAS gene mutations. This hyperactivation leads to uncontrolled cell proliferation and increased cell survival, ultimately leading to the development and progression of cancer. KRAS signaling pathway inhibition has been a major focus of research in cancer therapy, and drugs targeting different pathway components are currently being evaluated in clinical trials.
Canonical pathways downstream of KRAS mutations
Mutations can permanently activate KRAS, leading to uncontrolled cell growth and proliferation. The downstream signaling pathway of mutated KRAS involves a complex network of proteins and pathways that promote cell proliferation and survival. The main downstream effectors of KRAS are the RAF/MEK/ERK and PI3K/AKT/mTOR pathways. KRAS interacts with multiple signaling pathways, such as the phosphoinositide 3-kinase (PI3K)-protein kinase B (AKT)-mammalian target of rapamycin (mTOR) signaling pathway and the rapidly accelerated fibrosarcoma (RAF)-mitogen-activated protein kinase (MEK)-extracellular regulated protein kinase (ERK) signaling pathway [36]. KRAS with the G12D mutation modulates translation via the MNK/eIF4E pathway and results in sustained expression of c-MYC, a common proto-oncogene involved in many cancers [21]. KRAS activity is increased by sequence-specific RNA splicing modifications resulting from p53 mutations in pancreatic cancer [37]. In addition to the RAF/MEK/ERK and PI3K/AKT/mTOR pathways, KRAS mutations can activate other downstream signaling pathways that promote cell growth and proliferation. The pathways directly activated by KRAS are listed in Table 1 and Figure 1.
Canonical pathways downstream of KRAS mutations
| Pathway | Directly activated by KRAS? | Downstream/upstream effectors of the pathway | Main role | References |
|---|---|---|---|---|
| RAS-RAF-MEK-ERK1/2 | Yes | AP-1, ELK1, p90RSK, MAPKAPK2, CREB | Regulating cell proliferation, differentiation, and survival. | [38-41] |
| RAS-RAF-MEK-ERK3 | Yes | Cell proliferation, differentiation, migration, and invasion | [42] | |
| RAS-RAF-MEK-ERK5 | Yes | Survival, migration, proliferation, and differentiation | [43, 44] | |
| PI3K/Akt | Yes | p70S6K (ribosomal protein S6 kinase) and 4E-BP1 (eukaryotic translation initiation factor 4E-binding protein 1) | Cell growth, survival, and metabolism | [45-47] |
| TGF-β | Yes | Cell growth, differentiation, and immune responses | SMAD transcription factors. | [48, 49] |
| RalGDS | Yes | RalA and RalB | Cytoskeletal dynamics, cell migration and invasion, and endocytosis. | [50-53] |
| Notch | Yes | Jagged1 | Regulation of the activity of the transcription factor CSL (CBF1/RBPJk in mammals), a key component of the Notch pathway. Regulation of cell fate determination and differentiation | [54-56] |
| p38 MAPK | No | ATF2, CREB (cAMP response element-binding protein), | Stress responses, inflammation, cell survival. | [57-61] |
| JNK pathway | No | c-Jun, ATF2 | Apoptosis, proliferation, and migration, | [62-64] |
| Hippo | No | YAP (Yes-associated protein) | Organ size, tissue regeneration, and cell fate determination, | [65, 66] |
| Hedgehog | No | A downstream effector of RAF/MEK/MAPK | Cell fate determination and tissue patterning during development. | [67-69] |
| NF-κB | No | IL-6 | Regulation of immune responses, inflammation, and cell survival. | [70, 71] |
Canonical pathways downstream of KRAS mutations. KRAS mutation leads to permanent activation of the KRAS protein, activating several downstream pathways both directly (represented by components in green) and indirectly (represented by components in yellow) regulated by KRAS. Five pathways are directly under the control of KRAS: 1) the RAF-MEK-ERK pathway, 2) the RalGDS pathway, 3) the TGF pathway, 4) the PI3K-AKT-mTOR pathway, and 5) the JNK pathway. The JNK pathway is named after the final kinases (ERK, JNK, and p38) of the RAF-MEK-ERK pathway. The KRAS:1 indirectly regulates five pathways: 1) the p38-MAPK pathway, 2) the NF-kB pathway, 3) the Hippo-YAP pathway, 4) the Notch pathway, and 5) the Hedgehog pathway. (→direct regulated; --> indirectly regulated).
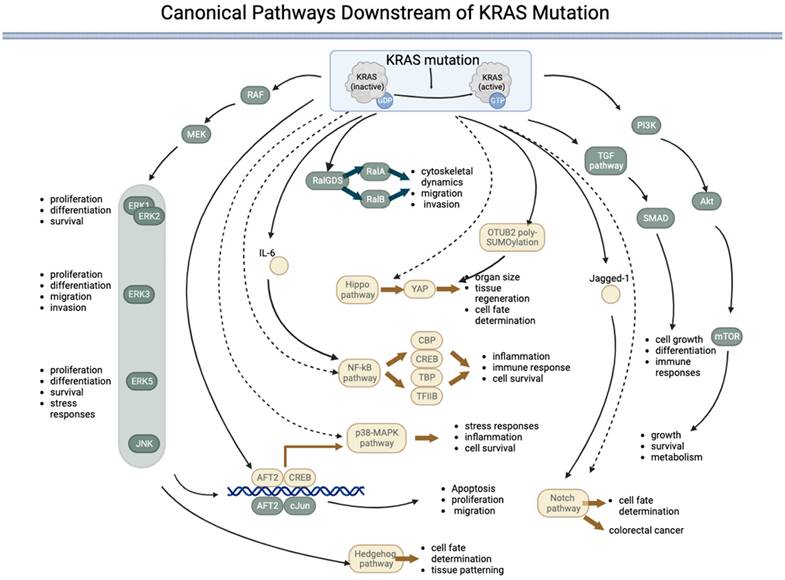
The RAS-RAF-MEK-ERK signaling pathway is a highly conserved signaling pathway that regulates various cellular processes, including cell proliferation, differentiation, survival, and stress responses [38]. The pathway comprises a cascade of kinases sequentially activated downstream of RAS, including RAF, MEK, and ERK proteins (ERK1/2, ERK3, ERK4, and ERK5). Although the core elements of the pathway are similar, there are several key differences between the different ERKs. ERK1 and ERK2, often referred to as canonical ERKs, are the most thoroughly studied members of the ERK family and are ubiquitously expressed. These ERKS regulate numerous cellular processes, including cell proliferation, differentiation, and survival. ERK3, which is less well studied than ERK1 and ERK2, has been implicated in regulating cell proliferation, differentiation, migration, and invasion [42]. ERK5 is the most divergent member of the ERK family and is activated by a distinct signaling pathway involving the MEK5 kinase. ERK5 regulates cell survival, migration, and the stress response [43, 44].
The PI3K-AKT-mTOR pathway is activated when KRAS binds to PI3K, leading to the activation of AKT and, subsequently, mTOR. This pathway regulates cell growth, survival, and metabolism [45]. The TGF-β pathway regulates cell growth, differentiation, and immune responses. The RAS gene interacts with and regulates TGF-β receptor activity, activating downstream effectors, such as SMAD transcription factors [48, 49]. The RalGDS pathway is activated when KRAS binds to RalGDS, activating RalA and RalB [50, 51]. This pathway regulates cytoskeletal dynamics, cell migration, and invasion [52, 53]. The downstream effectors of KRAS are not always activated in a linear, sequential manner. Instead, there is significant crosstalk between the different pathways, and the activation of one pathway can influence the activity of others. RalGDS is the key component that connects the KRAS pathway to other pathways.
KRAS indirectly activates some pathways, i.e., KRAS interacts with these pathways via an effector. The Notch pathway regulates cell fate determination and differentiation. KRAS can upregulate the expression of Jagged1, a ligand of the Notch pathway, and promote Notch signaling in colorectal cancer cells [54]. KRAS's activation of the p38 MAPK pathway can activate transcription factors, such as ATF2 and cAMP response element-binding protein (CREB), and regulate cellular processes, such as stress responses, inflammation, and cell survival [57-61]. The p38 MAPK pathway has also been implicated in the development of cancer and other diseases and is a potential therapeutic target. Activation of the JNK pathway by KRAS can lead to the phosphorylation of transcription factors, such as c-Jun and ATF2, which can regulate gene expression and cellular processes, such as apoptosis, proliferation, and migration [62-64]. The Hippo pathway regulates organ size, tissue regeneration, and cell fate determination [65]. KRAS has been shown to indirectly activate Yes-associated protein (YAP), the downstream effector of the Hippo pathway, by inducing OTUB2 poly-SUMOylation, which can regulate gene expression and cellular proliferation [66]. The Hedgehog pathway regulates cell fate determination and tissue patterning during development [67]. KRAS has been shown to activate the downstream effector RAF/MEK/MAPK pathway rather than the PI3K-AKT pathway, which can activate the Hedgehog pathway [68]. In addition, KRAS has been implicated in regulating the expression of Gli proteins in the KRAS-androgen axis in prostate cancer [69]. The NF-κB pathway regulates immune responses, inflammation, and cell survival. Controlled by IL-6, KRAS has been shown to activate the NF-κB pathway during EMT [70], which subsequently regulates downstream effectors, such as transcription factors such as TFIIB, TATA-binding protein (TBP), and CREB binding protein (CBP) [71].
For unknown reasons, each RAS mutation has unique, tissue-specific oncogenic properties. KRAS is an isoform of the Ras family that is ubiquitously expressed in mammals. KRAS mutations are primarily found in pancreatic ductal and colorectal adenocarcinomas [72], whereas NRAS at codon 61 mutations are primarily found in cutaneous malignant melanoma [73]. In the GTP hydrolysis cycle, KRAS is in either a GTP-bound active state or a GDP-bound inactive state. KRAS is one of the most common oncogenes in numerous cancers. The three cancers with the highest KRAS mutation rates are pancreatic ductal adenocarcinoma (PDA; > 95%), non-small cell lung cancer (NSCLC), and colorectal carcinoma (CRC; 40%) [74]. KRAS gene somatic point mutations account for 30-50% of all CRC cases [36, 75].
The common mechanism of action of the KRAS mutation is glycine substitution with aspartic acid. The wild-type (wt) KRAS protein is momentarily activated after receptor tyrosine kinase (RTK) signal transduction; however, a mutated KRAS protein forces the constitutive activation of downstream signaling pathways, thereby driving a toxic cellular program that is tumorigenic and frequently associated with targeted therapy resistance [76, 77]. This mutation allows constitutive KRAS protein activation, thus driving pro-growth and anti-apoptotic signaling pathways without stimulation. Among all patients with positive KRAS mutations, 73.3% (11/15), 20% (3/15), and 6.67% (1/15) of the mutations are in codons 12, 13, and 61, respectively [9]. The KRAS G12C mutation is the most prevalent driver mutation and is present in a considerable proportion of CRC patients [10]. Patients with KRAS G12D mutations have poor survival outcomes, and this mutation is a marker of poor prognosis in Chinese patients [11]. Fortunately, effective and potent G12D inhibitors have been developed in recent years [78]. The G12D and G13D mutations activate different signaling molecules [13]. Due to the intriguing link between KRAS and other signaling pathways, the efficacy of RAF, MEK, or ERK inhibitors alone is generally unsatisfactory; therefore, combining these inhibitors with other MAPK pathway inhibitors is worthy of investigation [1]. The regression of cancers resistant to anti-EGFR therapy requires the concurrent inhibition of PI3K, HER2, and MEK [79]. KRAS siRNA and p38α siRNA can be used as a combination treatment to suppress genes involved in CRC [80].
Mutated KRAS and the CRC TME
Components of the TME
The TME comprises intestinal microbiota, immune cells (such as neutrophils, T lymphocytes, dendritic cells, and macrophages), stromal cells (such as cancer-associated endothelial cells and fibroblasts), and noncellular components (such as the extracellular matrix (ECM)). The TME is essential for regulating or promoting tumor growth. Innate and adaptive immune cells interact with tumor cells to form the TME (Figure 2).
Construction of the tumor microenvironment. The tumor microenvironment (TME) mainly consists of interstitial cells, infiltrating immune cells, the extracellular matrix (ECM), and microbes. Immune cells, such as Tregs, TAMs, and MDSCs, are at the core of the establishment of an immune-suppressive microenvironment and can aid tumor cells in escaping the surveillance of the immune system. In addition, interstitial cells, such as CAFs, assist in the growth and metastasis of tumor cells. The extracellular matrix and microbes together influence the dynamics of the TME.
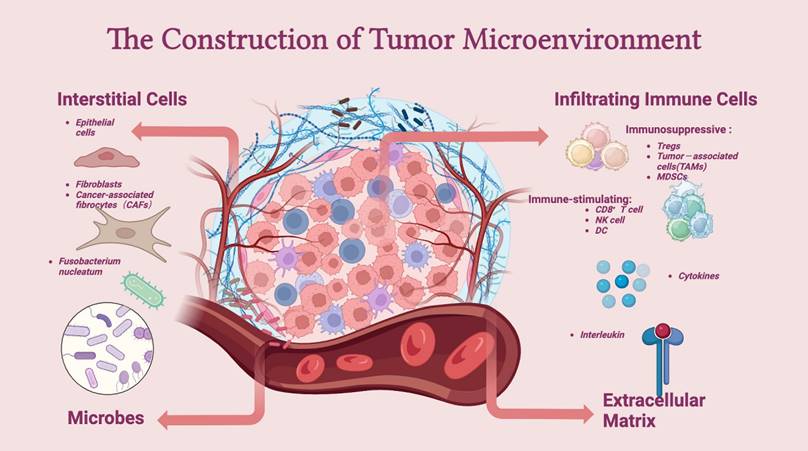
The role of T cells in the TME is well established. The ECM supports and connects tissues under normal circumstances and preserves physiologically normal functions [81]. An abnormal ECM affects the epithelial-mesenchymal transition (EMT), directly promoting cell transformation and metastasis [82]. An important mechanism for the occurrence and growth of CRC is angiogenesis, for which vascular endothelial growth factor (VEGF) is the most important growth factor. Furthermore, CRC cells express three VEGF receptors. Tumor grade, Dukes stage, and lymph node involvement are associated with VEGFR-1, and lymph node involvement is associated with VEGFR-2; however, no clinicopathological variables are associated with VEGFR-3 [83]. Cancer-associated fibroblasts have been shown to directly secrete exosomes, promoting chemical resistance and CRC metastasis by increasing CRC cell stemness and promoting epithelial-mesenchymal transformation [84]. In solid tumor tissues, tumor-associated macrophages (TAMs) are considered the most prevalent immune cell population [85]. Through EMT remodeling, TAMs encourage the growth and invasion of colon cancer cells [86]. Following infection, the body releases chemokines, causing neutrophils to migrate and detect pathogens [87]. An increase in neutrophils in tumors has been linked to a malignant phenotype and can indicate a poor prognosis in CRC patients [88]. Tumor-infiltrating lymphocytes (TILs) also play an important role in the TME. Lymphocytes, which include T, B, NK, and NKT cells, are the primary immune cells in tumors. Among these factors, Tregs play an interesting role in the prognosis of cancer. Tregs are a subtype of CD4+ T cells that participate in allergic disease overactivity, immunosuppression, and inflammation [89]. However, infiltrating Tregs play different roles in some cancers. In a model of mismatch repair deficiency (dMMR)-CRC, a high level of Foxp3+ Treg infiltration was associated with a higher survival rate [90]. However, in a model of lung cancer, Tregs induced metastasis [91]. Btla is a coinhibitory receptor that is expressed mainly on B and T cells and is responsible for limiting innate and adaptive immune responses. Btla expressed on Tregs plays a pivotal role in microbial homeostasis. The loss of Btla on lymphocytes is associated with the disruption of microbial homeostasis and elevations in pathogenic and commensal bacteria [92]. Decreased CD8+ T-cell effector function in the presence of antigens is related to a dysfunctional immune response in the TME [93]. B cells also have antitumor properties as they preferentially localize to the TME [94]. However, the role of B cells in the TME is complex. Most studies have shown that the presence of B cells is associated with improved outcomes in cancer patients. However, B cells can generate immunosuppressive cytokines in both animal models and humans, resulting in impaired antitumor immunity and poor clinical outcomes [95].
Many TME cells can produce proinflammatory cytokines (such as TNF-a, IL-1, and IL-6) and certain chemokines, such as CXCL8/IL-8, and the roles of these cytokines and chemokines are related to CRC-associated cachexia [96]. Exosomes secreted by tumor and interstitial cells are key components of the TME. The exosome contents include proteins, DNA, miRNA, mRNA, long noncoding RNA, and even virus/prion genetic material [97]. SATB2-AS1, a long noncoding RNA, is specifically expressed in colorectal tissues and downregulated in CRC. Survival analyses indicate that decreased SATB2-AS1 expression is associated with poor survival outcomes [98]. Functional experiments and bioinformatics analyses revealed that SATB2-AS1 inhibits CRC cell metastasis and regulates TH1-type chemokine expression and immune cell density in CRC [99].
The gut microbial flora plays an important role in the CRC microenvironment. The role of bacteria is complex. Some bacterial families contribute to antitumor immunity, while others promote oncogenesis and progression [100]. Microbial dysbiosis can promote colon tumor susceptibility by hyperstimulating CD8+ T cells, leading to early T-cell exhaustion and chronic inflammation, which can jeopardize antitumor immunity [101]. The miscellaneous microbiota in neoplasms is also associated with colorectal carcinogenesis [102]. One of the potential risk factors for the development and progression of CRC is Fusobacterium nucleatum (F. nucleatum). The most crucial processes of F. nucleatum in CRC are predominantly related to virulence factors, such as FadA and Fap2; microRNAs, such as miR-21, bacteria metabolism, and immunological regulatory components, including myeloid-derived suppressor cells and inhibitory receptors of natural killer cells [103]. Toll-like receptors, microRNAs, and autophagy are coordinated by F. nucleatum to therapeutically, physiologically, and mechanistically control CRC chemoresistance [104]. CRC heterogeneity can be attributed in part to differences in the variety and composition of microbes and their interactions in humans [105]. Oral bacterial cancer therapy (BCT) directly affects the tumor epithelium and tumor stem cells [106].
Pathways involved in the immune system of the TME
Numerous axes or pathways affecting immune cell interactions are engaged in the tumor environment (Figure 3).
Signaling pathways in the TME. PD-1 is an important receptor expressed on the surface of T cells and exerts immunosuppressive effects by prohibiting T cells from reacting excessively. Accordingly, tumor cells produce the corresponding ligand (PD-L1) on their surfaces to prevent T cells from attacking them. In this process, the interaction between PD-1 and PD-L1 leads to immunosuppression in the TME. The Notch signaling pathway mainly affects the integrity of vascular endothelial cells, therefore promoting metastasis. The (CXCL)3-CXCR2 axis promotes hepatic metastasis by attracting immunosuppressive neutrophils to the TME, while the Wnt signaling pathway mainly infiltrates granulocytes. The MondoA-TXNIP axis affects the identity of Treg cells. In addition, HH and the Gas6/Axl pathway participate in the formation of an immunosuppressive TME.
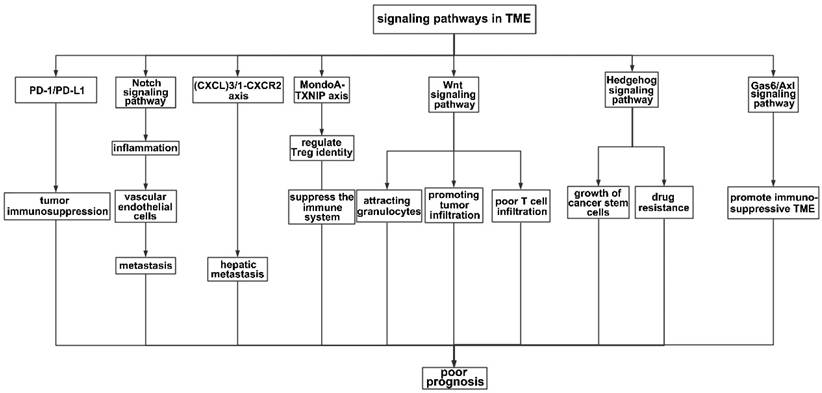
The KRAS signaling pathway plays a crucial role in regulating the immune system, from the activation and differentiation of immune cells to the regulation of immune cell apoptosis (i.e., the programmed death of immune cells). The downregulation of the immune system ensures that the immune response is regulated appropriately, preventing excessive inflammation. One of the most important signaling pathways in the TME is the programmed death ligand-1/programmed death-1 (PD-L1/PD-1) signaling pathway, which plays a key role in tumor immunosuppression. However, therapies targeting this pathway remain ineffective.
Inflammatory factors in the TME may be the underlying reason for the failure of anti- PD-L1/PD-1 therapies because inflammation may induce the production of PD-L1, interfering with the efficiency of PD-L1/PD-1 blockade [107]. In KRAS G12D-driven serrated cancer, activation of NOTCH1 signaling in the murine intestinal epithelium results in highly penetrant metastasis by creating a TME similar to that of human CRC subtypes with poor prognoses (CMS4 and CRIS-B) [108]. Notch activation contributes to metastasis, possibly by influencing vascular endothelial cells. Wieland et al. reported that Notch activation leads to endothelial cell senescence, inducing inflammation and increasing metastasis [109]. The chemokine (C-X-C motif) ligand (CXCL) family plays a key role in inflammation, and the level of CXCL is associated with tumor prognosis. Low levels of CXCR1 and CXCR3 are associated with a decreased tumor volume, decreased alpha fetoprotein levels, and a decreased TNM tumor stage [110]. The TGF-beta-chemokine (C-X-C motif) ligand (CXCL)3/1-CXCR2 axis can be controlled by KIAA1199, facilitating the infiltration of immunosuppressive neutrophils and contributing to hepatic metastasis [111]. The MondoA-TXNIP axis is a crucial metabolic regulator of Treg identity and activity in the microenvironment of CRC and a possible target for cancer therapy [112]. In CRC liver metastasis, activation of the Wnt signaling pathway may be crucial for attracting granulocytes and promoting tumor infiltration [113]. Poor spontaneous T-cell infiltration is frequently correlated with WNT/β-catenin signaling activation across most human cancers; thus, combination cancer therapy should block Wnt/β-catenin signaling to increase T-cell infiltration [114]. The Hedgehog (HH) signaling pathway participates in inflammation, tumor immune tolerance, tumor growth, and drug resistance in the TME. Both the growth of cancer stem cells (CSCs) and the drug resistance of gastrointestinal tumors can be facilitated by aberrant HH signaling activation [115]. The Gas6/Axl signaling pathway has been implicated in promoting the immunosuppressive TME [116]. In addition, the KRAS pathway has been implicated in the development of various autoimmune diseases, such as rheumatoid arthritis, systemic lupus erythematosus, and multiple sclerosis. In these diseases, an overactive KRAS signaling pathway leads to a hyperactive immune response and chronic inflammation.
KRAS mutation participates in metabolic interactions
KRAS mutation can influence the TME, thus further affecting the prognosis of CRC patients. The oncogenic mutant KRAS controls divergent and convergent metabolic networks in the colon (Figure 4).
KRAS mutation-induced metabolic alterations KRAS mutation can induce oncogenesis by regulating metabolism, including lactate accumulation, hypoxia, PPINs, glycolysis, and lipid metabolism. These alterations influence immune cells and create an immunosuppressive environment, which is necessary for cancer cells to evade immune surveillance. The common patterns for the interaction between metabolism and the immune microenvironment are as follows: exclusion of immune cells with antitumor ability, enhancement of immunosuppressive cells, deprivation of oxygen to immune cells, and inflammation in the microenvironment. Therefore, given its polyfunctionality, mutant KRAS may be a critical target for treating CRC. (→promote; --| inhibit)
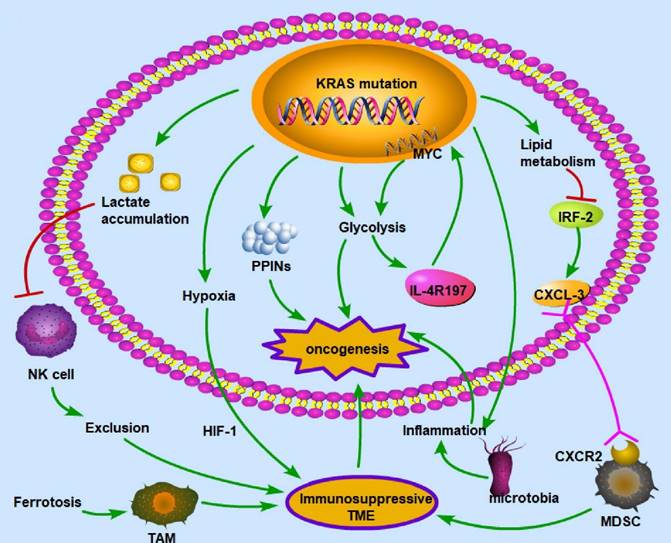
In the TME, cancer and noncancer cells participate in metabolic interactions by limiting nutrient availability and the activity of the immune system [117]. The KRAS signaling pathway in the TME of CRC primarily regulates hypoxia and inflammation, glucose competition, lactate accumulation, amino acid levels, and lipid metabolism [118]. For example, the mutated KRAS signaling pathway cooperates with the microbiota in the colon to produce an inflammatory environment conducive to activating oncogenic signaling [119]. There is a vicious cycle between KRAS mutation and inflammation in the process of lung tumorigenesis: KRAS mutation can promote inflammation, while enhanced inflammation may also increase the KRAS mutation rate [120]. KRAS mutation promotes the autonomous expression of type I cytokine receptor complexes (IL2r-IL4r and IL2r-IL13r1) in cancer cells, which can then bind to and take up cytokine growth signals (IL4 or IL13) released by Th2 cells in the microenvironment [121]. Hypoxia is also a key factor in CRC development. In a KRAS-driven mouse model of pancreatic ductal adenocarcinoma, hypoxia results in B-cell exclusion, and it is speculated that the underlying mechanism is that in a hypoxic environment, KRAS mutation results in the exclusion of immune cells via hypoxia-inducible factor-1 [122]. The hypoxic TME promotes CRC cell stemness and resistance to chemotherapy; thus, this environment could be targeted to mitigate chemoresistance [123]. Moreover, glycolysis plays an important role in the immune system. In a mouse model of PDA, KRAS mutation increased the surface expression of IL-4R197, increasing the vulnerability of cancer cells to cytokines produced by TH2 cells [121]. This effect increased MYC activity through JAK-STAT signaling and elevated glycolytic gene expression and flux in cancer cells, facilitating tumor growth [121]. Furthermore, lactate is a metabolic byproduct of KRAS-mutant metabolism in cancer cells and can create an immunosuppressive TME. Due to a decrease in the intracellular pH and the formation of mitochondrial ROS, lactate in the TME of colon liver metastases causes the apoptosis of infiltrating natural killer cells [124]. In the mouse intestinal epithelium, simultaneous mutation of APC and KRAS strongly dysregulates metabolism, enhancing glutamine consumption; thus, the glutamine antiporter SLC7A5 is essential for the development of colorectal tumors [125]. KRAS mutation can rewire protein-protein interaction networks (PPINs), altering the cellular phenotype, signal flow, transcriptional regulation, and protein complexes [126].
KRAS mutation can downregulate miR-139-5p and derepress the epithelial-to-mesenchymal transition and oncogenic signaling pathways by disrupting the reciprocal negative feedback mechanism of miR-139-5p/Wnt signaling [127]. The KRAS mutation status might be implicated in the development of an oxidative phenotype [128].
KRAS regulates immune and nonimmune cells (Figure 5)
Immune cells play an important role in the TME. Obesity caused by a high-fat diet inhibits CD8+ T-cell activity in the mouse TME, hastening tumor progression [129]. Racial discrepancies in survival outcomes are mediated by the lymphocyte response in the tumor [130]. Moreover, nonimmune cells impact the tumor immune microenvironment in CRC [131]. For example, chimeric extracellular vesicles are created when tumor cells come into contact with platelets; these vesicles inhibit the growth of the main tumor by activating tumor-eliminating macrophages while promoting metastasis through EMT and endothelial activation [132]. Hepatocytes produce various substances that promote cancer cell spreading, attracting or activating stromal cells and immune cells to the liver and creating a favorable premetastatic niche and an immunosuppressive liver milieu conducive to tumor cell colonization and propagation [133]. Colon fibroblasts, not tumor cells, are the main cell type that directs infiltrating monocytes toward a particular macrophage population that exhibits high levels of CD163 expression and CCL2 production [134]. KRAS can also impact the TME by regulating immune cells, thus affecting the host immune system. KRAS mutations increase the immunogenicity of tumors by increasing the mutational load and hampering DNA repair, thus enhancing neoantigen production [135]. KRAS-mediated repression of IRF2 leads to high expression of CXCL3, which binds to CXCR2 on myeloid-derived suppressor cells, thus promoting their migration to the TME [136]. Similarly, Treg cells, which play immunosuppressive roles, can also be regulated by KRAS. Mechanistically, mutant KRAS transforms conventional T cells into Treg cells by stimulating the secretion of TGF-beta1 and IL-10 [137]. The KRAS G12D mutation contributes to regulatory T-cell conversion through activation of the MEK/ERK pathway in pancreatic cancer [138]. In a previous study, Tregs were shown to be associated with CD8+ T cell dysfunction and ILC2 upregulation, contributing to KRAS-associated chemoresistance and lymph node metastasis [139]. CD8+ T cells, T helper cells, and dendritic cells play crucial roles in the antitumor immune response. Lal et al. analyzed The Cancer Genome Atlas (TGCA) CRC datasets and reported decreased infiltration of cytotoxic cells and a reduced Th1-mediated immune response in patients with KRAS-mutant tumors [140]. Moreover, statin-mediated ICD enhances the cross-priming ability of dendritic cells, thereby stimulating CD8+ T-cell immune responses against KRAS mutant tumors [141]. The antitumor immune response is also jeopardized through CD47, an antiphagocytosis signal that desensitizes tumor cells to phagocytosis [142]. Ferroptosis, which is dependent on autophagy, promotes the polarization of TAMs through the release and uptake of the oncogenic KRAS protein [143]. Programmed death ligand 1 translation is suppressed in KRAS G12D tumors, resulting in evasion of immune attack [144]. Studies have revealed a greater level of PD-L1 protein expression in KRAS- or TP53-mutant NSCLC than in nonmutant tumors, suggesting that KRAS may participate in immune system inhibition via PD-L1 [145]. Higher PD-L1 expression enables patients with NSCLC to benefit from immune checkpoint blockade therapy [146]. However, another subtype of KRAS mutation (KRAS G12D) was found to be associated with a lower mutation burden in lung adenocarcinoma, resulting in a suppressed immune response [147]. In addition, patients with KRAS mutations can also produce a series of cytokines that establish an immunosuppressive microenvironment. In a model of pancreatic cancer, C-C motif chemokine ligand (CCL)15 can be upregulated by KRAS and increase migration and invasion [148]. Another C-C motif chemokine ligand (CCL)28 can also be driven by KRAS, which subsequently promotes the upregulation of Fos-like antigen 2 (FOSL2), a novel transcription factor [149]. In a KRAS-mutant lung cancer model, the inhibition of CCL5 and interleukin (IL)-6 could hamper cancer growth, suggesting that KRAS mutation may be involved in tumor progression via the production of cytokines [150].
KRAS mutations affect the tumor immune microenvironment KRAS mutations play a dual role in the tumor immune microenvironment. The main role of mutant KRAS is destroying the antitumor immune response. KRAS mutation can dampen the function of CD8+ T cells and T helper cells and promote phagocytosis while also promoting immunosuppressive Treg cells (→promote; --| inhibit).
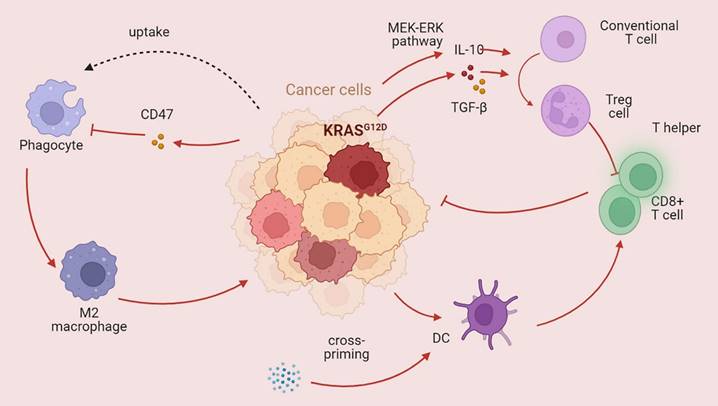
The efficacy of immunotherapy is largely dictated by PD-L1 dysregulation. Zhou et al. reported that dysregulation of PD-L1 induced by UFMylation could result in tumor immune evasion; thus, UFMylation is a potential therapeutic target [151]. High PD-L1 or PD-L2 expression is strongly related to poor outcomes, and this dysregulation strongly impacts signaling pathways such as the mTOR, HIF-1, and ERBB pathways [152]. T cell-tumor interactions and responses to PD-L1/PD-1 blockade are influenced by molecules expressed on tumor cells, such as CD58 and CMTM6 [153]. Cytotoxic T cells can be inactivated by PD-L1, leading to restricted immunosurveillance of the TME [154]. Therefore, we infer that PD-L1 expression must be fully considered when administering immunotherapy.
Promotion of angiogenesis by KRAS mutations
Downstream signaling pathways are overactivated when the KRAS gene is mutated, stimulating the production of angiogenic factors and promoting the growth of new blood vessels. This increased angiogenesis is a hallmark of many cancers and contributes to tumor progression, metastasis, and chemotherapy resistance. It is speculated that oncogenes, which are hallmarks of the coagulopathies that occur in many cancers, may also play crucial roles in hemostasis and angiogenesis [155]. Activation of the MAPK pathway is associated with upregulated expression of VEGF and improved cell growth and survival in melanoma cells [156]. The underlying mechanism of increased VEGF expression might be regulated by activating transcription factors, such as AP-1 (activator protein 1) [157]. Endothelial cells outside vascular beds can be stimulated by Ras activation through molecules such as VEGF and COX-derived prostaglandins. In addition, Ras activation can increase uPA/MMP expression and decrease TSP expression, resulting in extracellular matrix remodeling [158]. ZNF322A transcriptional activation can also promote neoangiogenesis in lung cancer when KRAS is activated [159].
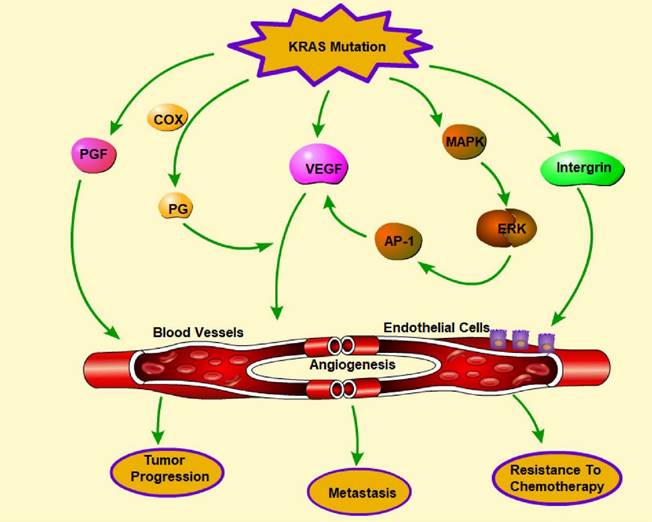
Phenformin, commonly used to treat diabetes, targets extracellular regulated protein kinase (ERK), which is a downstream effector of the KRAS pathway and leads to the inhibition of the expression of proangiogenic molecules [160]. ASP13 mutation, which is less harmful than KRAS mutation, can induce a significant VEGF-A-associated vascular network via the RAF-RAS-ERK pathway by activating the VEGF-A promoter when the HIF-1 level is low [161]. KRAS mutations can lead to increased fibroblast growth factor expression and activity, resulting in enhanced angiogenesis. KRAS can interact with proteins involved in angiogenesis, such as integrin β4, to promote blood vessel formation, resulting in increased invasion and metastasis [162]. Overall, the presence of KRAS mutations in cancer cells can promote angiogenesis, which can lead to aggressive tumor growth and progression (Figure 6). Targeting the KRAS signaling pathway and angiogenesis may be a promising strategy for developing new cancer treatments.
Regulation of angiogenesis by KRAS mutations. Angiogenesis plays a pivotal role in tumor progression, metastasis, and chemotherapy resistance. KRAS mutation promotes angiogenesis through the release of multiple molecules, including VEGF, PGF, PG, and integrin, which can influence endothelial cells and the formation of blood vessels. In addition, activation of the MARK-ERK-AP-1 pathway indirectly facilitates angiogenesis by promoting VEGF. (→promote; --| inhibit)
Treatments targeting KRAS and the TME
Ongoing clinical trials or recently approved drugs targeting the TME
Since KRAS mutations are known to cause many cancers at the genetic level, we now focus on some ongoing clinical trials and recently approved drugs targeting KRAS. Inhibitors can target mutations at different sites. Sotorasib, an inhibitor selectively and irreversibly targeting KRAS (G12C), is used to treat lung cancers caused by the KRAS p.G12C mutation [163]. Sotorasib exhibited safe and strong anticancer effects among patients with the KRAS p.G12C mutation who had previously received treatment [164]. Adagrasib demonstrates encouraging clinical efficacy and is well tolerated in patients pretreated with KRASG12C-mutated solid tumors [165]. Targeting the constitutively active KRAS G12C oncogenic driver with sotorasib and adagrasib has shown promising results, highlighting the need for additional studies to enhance the therapeutic use of these agents in this high-risk population [166]. Although sotorasib and adagrasib are currently approved for use in patients with advanced/metastatic NSCLC, regular biomarker testing of KRAS G12C mutations should include testing and reporting before first-line therapy since these tests may aid in clinical decision-making in patients with KRAS G12C-mutated advanced NSCLC [167]. In addition to inhibitors that target the G12C mutation (sotorasib and adagrasib), drugs targeting G12D have been developed, such as MRTX1133. Unfortunately, drugs targeting other mutations remain scarce [168].
Inhibitors and immunotherapies targeting KRAS and its downstream pathways and the TME
A greater comprehension of the effector pathways downstream of KRAS that are either directly or indirectly regulated by KRAS could lead to improved therapeutic interventions. Here, we describe several cutting-edge therapeutic approaches that endeavor to block or hinder the activation of downstream pathways. In KRAS-mutant GC cells, increased expression of DNMT1 was induced by KRAS knockdown, and the combination of a MEK/ERK inhibitor with DNA methyltransferases (DNMTs) might be a promising strategy for GC [169]. Dactolisib, a dual PI3K/mTOR inhibitor, can result in greater therapeutic benefits for those with KRAS-mutated lung cancer when combined with Lys05, a dimeric chloroquine [170]. Exosomes engineered to promote M1 polarization and inhibit the IL-4 receptor can reprogram TAMs from the M2-like to M1-like phenotype [171]. However, macrophage-mediated immune reprogramming and macrophage-mediated drug delivery approaches require substantial optimization [172]. Since PI3K-mTOR is the downstream pathway of KRAS, alpelisib, a PI3K inhibitor, was investigated in KRASG12C mutant cancer cells (pancreatic ductal adenocarcinoma [173], ovarian cancer [174], and thyroid cancer [175]) and demonstrated synergistic effects with sotorasib, reducing cell viability [176]. The development of immunotherapy has improved treatments for several cancers, and it has been shown that the immune system can be trained to identify and eliminate cancer cells. Recently, the use of an enhancer RNA-based subtyping system has shown great potential in prognosis prediction and immunotherapy management in stage II/III CRC patients [177].
Immune checkpoint inhibitors (ICIs) against PD-1 and its ligand PD-L1 effectively treat many cancer patients. However, it is unclear whether KRAS oncogene substitutions impact ICI efficacy. Responses to ICIs that block PD-1/PD-L1 may be highly dependent on concurrent mutations [178]. According to several studies, although ICIs can effectively treat patients with gastrointestinal cancers or NSCLC harboring a KRAS mutation, primary or acquired resistance to ICIs may occur [179]. Targeted next-generation sequencing of lung adenocarcinoma revealed that KRAS mutation with TP53 and MET mutations is strongly linked to increased PD-L1 expression [180]. In KRAS-mutant lung adenocarcinoma, a low TMB and high copy number alteration are potential biomarkers predicting a worse response to ICIs [181]. Liu et al. reported that in patients with NSCLC, the KRAS-G12D mutation promoted ICI resistance and immunosuppression, while the combination of chemotherapy and ICI therapy had better efficacy than either treatment alone [182]. KRAS mutations cooccurring with TP53 mutations resulted in a notable therapeutic benefit after PD-1 inhibitor administration; therefore, mutations in KRAS and TP53 could be used as predictors for ICI efficacy [119]. The mechanism by which KRAS mutation promotes resistance to ICIs may involve the production of immune-suppressive cytokines, such as vascular endothelial growth factor [183], and the suppressive effect on T-cell recruitment [184].
Role of the microbiome in the treatment of CRC
Microbiomes have been applied in CRC treatment. For example, Tfh-associated antitumor immunity in the colon can be boosted by introducing immunogenic intestinal bacteria, such as Helicobacter hepaticus [185]. Aryl hydrocarbon receptor activation in tumor-associated macrophages by tryptophan-derived microbial metabolites inhibits antitumor immunity [186]. Fecal microbiota transplantation, in which feces from a specific donor are given to a recipient, is a popular therapeutic strategy in cancer treatment. Fecal microbiota transplantation has been used to treat several clinical conditions, including ulcerative colitis, Clostridium difficile infection, and other digestive diseases [187]. Furthermore, patients who were previously resistant to immune checkpoint inhibitors may respond to therapy after receiving a fecal microbiota transplant [188].
Beta-elemene, a natural product, can induce ferroptosis and prevent epithelial-mesenchymal transformation and can be combined with cetuximab to treat KRAS-mutant CRC [33]. Bile acids can also be used to modulate immune cells. Through the activation and recruitment of immune cells that can kill tumors, such as natural killer T cells, bile acids can help promote antitumor immune responses [189].
The butyrophilin molecules BTN2A1 and BTN3A1 can be manipulated to activate Vγ9Vδ2+ T cells, a component of the innate immune system [190]. Icariside I improves the microbiota community structure, alleviates inflammation, promotes the restoration of the intestinal barrier, and has significant antitumor effects [191]. Characterized by antitumor and anticachexia properties, a ketogenic diet can be used as an adjuvant therapy by reprogramming the epigenome, cell metabolism, and gut microbiome [192].
Atractylenolide I, a component extracted from Rhizoma Atractylodis macrocephalae, may inhibit dysbacteriosis by regulating TLR4/MyD88/NF-κB signaling [193]. Alpha-galactosylceramide, produced by bacteria such as Bacteroides fragilis, Bacteroides vulgatus, and Prevotella copri, exerts antitumor effects by activating nonconventional T cells, such as invariant natural killer T (iNKT) cells and γδ T cells [194]. AGPs exert chemopreventive effects by restoring dysbiosis and maintaining enteric homeostasis [195].
Limitations of current targeted therapies
Targeting the interactions between mutated KRAS and the TME is an active area of novel therapeutic research aimed at overcoming KRAS-driven resistance to therapy and improving patient outcomes (Figure 7).
Targeted treatment of CRC KRAS mutation is the cause of resistance to anti-EGFR treatment, as it leads to constitutive activation of the MAPK signaling pathway and activation of Warburg metabolism. (→promote; --| inhibit)
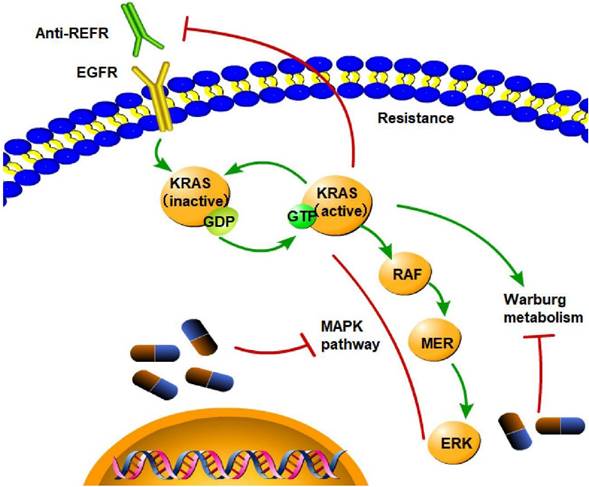
Since KRAS mutation is related to poor outcomes in CRC patients, KRAS mutation can serve as a target for treating CRC. In-depth research has been conducted using structural and biochemical methods to create KRAS inhibitors. Unfortunately, no specific treatments for KRAS-mutant malignancies have been authorized to date despite the success of small-molecule inhibitor development for other cancer drivers, such as BCR-ABL, C-kit, EGFR, BRAF, and ALK. However, KRAS-mutant CRC is resistant to EGFR inhibitors, and the mechanism is complex. The only reliable biomarkers for determining which patients will benefit from anti-EGFR therapy are skin toxicity, KRAS status, and the European Organization for Research and Treatment of Cancer Classification [196]. KRAS mutations are not the primary mechanism of resistance to chemotherapy or anti-EGFR mAbs [197]; however, the KRAS G13D mutation is an exception. Cetuximab, an EGFR-blocking antibody, is beneficial for treating KRAS G13D CRC patients [198]. In KRAS G12C-mutant NSCLC, epithelial-to-mesenchymal transition causes intrinsic and acquired resistance to KRAS G12C inhibitors [199]. The KRAS gene interacts with the KEAP1 gene. Loss of KEAP1 increases NRF2 activity and facilitates KRAS-driven lung adenocarcinoma in mice [200]. Thus, it can be inferred that inhibiting the KEAP1 gene might be a potential mechanism for treating CRC. In numerous pancreatic cancer mouse models, the administration of iExosomes that can target oncogenic KRAS-suppressed cancer markedly improves overall survival outcomes [201].
Conclusion
KRAS mutation plays a crucial role in the development and progression of CRC; therefore, therapies targeting the KRAS gene have become a research focus. Targeted therapeutic drugs that specifically target (directly or indirectly) the KRAS gene, such as sotorasib, adagrasib, and MRTX1133, have been developed and applied in clinical settings. This review summarized current understanding of the mechanism of KRAS mutation and its relationship with the TME. In addition, we promote the potential of targeting KRAS in immune and targeted therapy for CRC by discussing the mechanism underlying the aberrant activation of KRAS signaling coincident with the loss of the antitumor immune responses, including molecular changes in the TME, interactions with the immune system, alterations in metabolism and angiogenesis. Investigating these mechanisms will help develop new therapies targeting these signaling pathways and the TME, improve the prognosis of CRC patients, and identify novel CRC biomarkers.
However, specific and precise targeted therapies are still lacking, and drug resistance and unfavorable physiological side effects remain unresolved issues. In addition, it is difficult to determine which patients are suitable for targeted therapy, and this process requires the use of biomarkers. Consequently, further research should focus on the tumor immune microenvironment and take advantage of the antitumor response to achieve better treatment strategies and reduce resistance to therapy.
Abbreviations
AP-1: Activator protein 1; BCT: Bacterial cancer therapy; CIMP: CpG island methylator phenotype; CMS: Consensus molecular subtypes; CRC: Colorectal cancer; CREB: cAMP response element-binding; CREBP: cAMP response element-binding protein; CSCs: Cancer stem cells; dMMR: mismatch repair deficiency; ECM: Extracellular matrix; EGFR: Epidermal growth factor receptor; EMT: Epithelial-mesenchymal transition; ERK: Extracellular regulated protein kinases; F. nucleatum: Fusobacterium nucleatum; HH: Hedgehog; ICIs: Immunocheckpoint inhibitors; IKK: IκB kinase; iNKT: Invariant natural killer T; KRAS: Kirsten rat sarcoma viral oncogene homolog; LCC: Left-sided colon cancer; MAPK: Mitogen-activated protein kinase; mCRC: Metastatic colorectal cancer; MEK: Mitogen-activated protein kinase; MSI: Microsatellite instability; mTOR: Mammalian target of rapamycin; NSCLC: Non-small cell lung cancer; OS: Overall survival; p70S6K: Ribosomal protein S6 kinase; PDA: Pancreatic ductal adenocarcinoma; PD-L1: Programmed death ligand 1; PFS: Progression-free survival; PI3K: Phosphoinositide 3-kinase; PPINs: Protein-protein interaction networks; RAF: Rapidly accelerated fibrosarcoma; RCC: Right-sided colon cancer; RTK: Receptor tyrosine kinase; TAMs: Tumor-associated macrophages; TILs: Tumor-infiltrating lymphocytes; TME: Tumor microenvironment; VEGF: Vascular endothelial growth factor; wt: wild-type; YAP: Yes-associated protein; 4E-BP1: Eukaryotic translation initiation factor 4E-binding protein 1.
Acknowledgements
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Funding
This work was supported by the National Natural Science Foundation of China [grant number 81972716].
Availability of data and materials
All the generated data are included in this published article.
Author contributions
YZ drafted the manuscript and prepared the figures. YK, CW, YY, and LP helped collect the related literature and participated in the discussion. XH designed the review and revised the manuscript. All the authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhu G, Pei L, Xia H, Tang Q, Bi F. Role of oncogenic KRAS in the prognosis, diagnosis and treatment of colorectal cancer. Mol Cancer. 2021;20:143
2. Cabot D, Brun S, Paco N, Ginesta MM, Gendrau-Sanclemente N, Abuasaker B. et al. KRAS phosphorylation regulates cell polarization and tumorigenic properties in colorectal cancer. Oncogene. 2021;40:5730-40
3. Modest DP, Ricard I, Heinemann V, Hegewisch-Becker S, Schmiegel W, Porschen R. et al. Outcome according to KRAS-, NRAS- and BRAF-mutation as well as KRAS mutation variants: pooled analysis of five randomized trials in metastatic colorectal cancer by the AIO colorectal cancer study group. Ann Oncol. 2016;27:1746-53
4. Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A. et al. EGFR Blockade Reverts Resistance to KRAS(G12C) Inhibition in Colorectal Cancer. Cancer Discov. 2020;10:1129-39
5. Xie MZ, Li JL, Cai ZM, Li KZ, Hu BL. Impact of primary colorectal Cancer location on the KRAS status and its prognostic value. BMC Gastroenterol. 2019;19:46
6. Li H, Yang Q, Huo S, Du Z, Wu F, Zhao H. et al. Expression of TMEM16A in Colorectal Cancer and Its Correlation With Clinical and Pathological Parameters. Front Oncol. 2021;11:652262
7. Ucar G, Ergun Y, Akturk Esen S, Acikgoz Y, Dirikoc M, Esen I. et al. Prognostic and predictive value of KRAS mutation number in metastatic colorectal cancer. Medicine (Baltimore). 2020;99:e22407
8. Tokumaru Y, Oshi M, Katsuta E, Yan L, Satyananda V, Matsuhashi N. et al. KRAS signaling enriched triple negative breast cancer is associated with favorable tumor immune microenvironment and better survival. Am J Cancer Res. 2020;10:897-907
9. Gorukmez O, Yakut T, Gorukmez O, Sag SO, Karkucak M, Kanat O. Distribution of KRAS and BRAF Mutations in Metastatic Colorectal Cancers in Turkish Patients. Asian Pac J Cancer Prev. 2016;17:1175-9
10. Araujo LH, Souza BM, Leite LR, Parma SAF, Lopes NP, Malta FSV. et al. Molecular profile of KRAS G12C-mutant colorectal and non-small-cell lung cancer. BMC Cancer. 2021;21:193
11. Yuan Y, Liu Y, Wu Y, Zhang J, Shen C, Zhang F. et al. Clinical characteristics and prognostic value of the KRAS mutation in Chinese colorectal cancer patients. Int J Biol Markers. 2021;36:33-9
12. Moore AR, Rosenberg SC, McCormick F, Malek S. Author Correction: RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discov. 2020;19:902
13. Tahir R, Renuse S, Udainiya S, Madugundu AK, Cutler JA, Nirujogi RS. et al. Mutation-Specific and Common Phosphotyrosine Signatures of KRAS G12D and G13D Alleles. J Proteome Res. 2021;20:670-83
14. Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383:1490-502
15. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7-34
16. Song M, Garrett WS, Chan AT. Nutrients, foods, and colorectal cancer prevention. Gastroenterology. 2015;148:1244-60 e16
17. Illescas O, Rodriguez-Sosa M, Gariboldi M. Mediterranean Diet to Prevent the Development of Colon Diseases: A Meta-Analysis of Gut Microbiota Studies. Nutrients. 2021 13
18. El Asri A, Ouldim K, Bouguenouch L, Sekal M, Moufid FZ, Kampman E. et al. Dietary Fat Intake and KRAS Mutations in Colorectal Cancer in a Moroccan Population. Nutrients. 2022 14
19. Wang X, Amitay E, Harrison TA, Banbury BL, Berndt SI, Brenner H. et al. Association Between Smoking and Molecular Subtypes of Colorectal Cancer. JNCI Cancer Spectr. 2021 5
20. Zhang KL, Zhu WW, Wang SH, Gao C, Pan JJ, Du ZG. et al. Organ-specific cholesterol metabolic aberration fuels liver metastasis of colorectal cancer. Theranostics. 2021;11:6560-72
21. Knight JRP, Alexandrou C, Skalka GL, Vlahov N, Pennel K, Officer L. et al. MNK Inhibition Sensitizes KRAS-Mutant Colorectal Cancer to mTORC1 Inhibition by Reducing eIF4E Phosphorylation and c-MYC Expression. Cancer Discov. 2021;11:1228-47
22. Di Franco S, Bianca P, Sardina DS, Turdo A, Gaggianesi M, Veschi V. et al. Adipose stem cell niche reprograms the colorectal cancer stem cell metastatic machinery. Nat Commun. 2021;12:5006
23. Chong W, Zhu X, Ren H, Ye C, Xu K, Wang Z. et al. Integrated multi-omics characterization of KRAS mutant colorectal cancer. Theranostics. 2022;12:5138-54
24. Liu Z, Liu Y, Qian L, Jiang S, Gai X, Ye S. et al. A proteomic and phosphoproteomic landscape of KRAS mutant cancers identifies combination therapies. Mol Cell. 2021;81:4076-90 e8
25. Lim LC, Looi ML, Zakaria SZ, Sagap I, Rose IM, Chin SF. et al. Identification of Differentially Expressed Proteins in the Serum of Colorectal Cancer Patients Using 2D-DIGE Proteomics Analysis. Pathol Oncol Res. 2016;22:169-77
26. Ikonomou G, Kostourou V, Shirasawa S, Sasazuki T, Samiotaki M, Panayotou G. Interplay between oncogenic K-Ras and wild-type H-Ras in Caco2 cell transformation. J Proteomics. 2012;75:5356-69
27. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD. et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015;162:1229-41
28. Papke B, Der CJ. Drugging RAS: Know the enemy. Science. 2017;355:1158-63
29. Serna-Blasco R, Sanz-Alvarez M, Aguilera O, Garcia-Foncillas J. Targeting the RAS-dependent chemoresistance: The Warburg connection. Semin Cancer Biol. 2019;54:80-90
30. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548-51
31. Herpers B, Eppink B, James MI, Cortina C, Canellas-Socias A, Boj SF. et al. Functional patient-derived organoid screenings identify MCLA-158 as a therapeutic EGFR x LGR5 bispecific antibody with efficacy in epithelial tumors. Nat Cancer. 2022;3:418-36
32. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI. et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med. 2020;383:1207-17
33. Chen P, Li X, Zhang R, Liu S, Xiang Y, Zhang M. et al. Combinative treatment of beta-elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial-mesenchymal transformation. Theranostics. 2020;10:5107-19
34. Ou SI, Janne PA, Leal TA, Rybkin II, Sabari JK, Barve MA. et al. First-in-Human Phase I/IB Dose-Finding Study of Adagrasib (MRTX849) in Patients With Advanced KRAS(G12C) Solid Tumors (KRYSTAL-1). J Clin Oncol. 2022;40:2530-8
35. Teo MYM, Fong JY, Lim WM, In LLA. Current Advances and Trends in KRAS Targeted Therapies for Colorectal Cancer. Mol Cancer Res. 2022;20:30-44
36. Uprety D, Adjei AA. KRAS: From undruggable to a druggable Cancer Target. Cancer Treat Rev. 2020;89:102070
37. Escobar-Hoyos LF, Penson A, Kannan R, Cho H, Pan CH, Singh RK. et al. Altered RNA Splicing by Mutant p53 Activates Oncogenic RAS Signaling in Pancreatic Cancer. Cancer Cell. 2020;38:198-211 e8
38. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11-22
39. Khan NM, Ahmad I, Haqqi TM. Nrf2/ARE pathway attenuates oxidative and apoptotic response in human osteoarthritis chondrocytes by activating ERK1/2/ELK1-P70S6K-P90RSK signaling axis. Free Radic Biol Med. 2018;116:159-71
40. Miguel SM, Namdar-Attar M, Noh T, Frenkel B, Bab I. ERK1/2-activated de novo Mapkapk2 synthesis is essential for osteogenic growth peptide mitogenic signaling in osteoblastic cells. J Biol Chem. 2005;280:37495-502
41. Lenz B, Klafki HW, Hillemacher T, Frieling H, Clepce M, Gossler A. et al. ERK1/2 protein and mRNA levels in human blood are linked to smoking behavior. Addict Biol. 2012;17:1026-35
42. An HJ, Lee CJ, Lee GE, Choi Y, Jeung D, Chen W. et al. FBXW7-mediated ERK3 degradation regulates the proliferation of lung cancer cells. Exp Mol Med. 2022;54:35-46
43. Stecca B, Rovida E. Impact of ERK5 on the Hallmarks of Cancer. Int J Mol Sci. 2019 20
44. Gomez N, Erazo T, Lizcano JM. ERK5 and Cell Proliferation: Nuclear Localization Is What Matters. Front Cell Dev Biol. 2016;4:105
45. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261-74
46. Zhao XF, Gartenhaus RB. Phospho-p70S6K and cdc2/cdk1 as therapeutic targets for diffuse large B-cell lymphoma. Expert Opin Ther Targets. 2009;13:1085-93
47. Quan Z, Yang Y, Zheng H, Zhan Y, Luo J, Ning Y. et al. Clinical implications of the interaction between PD-1/PD-L1 and PI3K/AKT/mTOR pathway in progression and treatment of non-small cell lung cancer. J Cancer. 2022;13:3434-43
48. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577-84
49. Lee SH, Kim O, Kim HJ, Hwangbo C, Lee JH. Epigenetic regulation of TGF-beta-induced EMT by JMJD3/KDM6B histone H3K27 demethylase. Oncogenesis. 2021;10:17
50. Lim KH, Baines AT, Fiordalisi JJ, Shipitsin M, Feig LA, Cox AD. et al. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell. 2005;7:533-45
51. Zhu Z, Xiao S, Hao H, Hou Q, Fu X. Kirsten Rat Sarcoma Viral Oncogene Homologue (KRAS) Mutations in the Occurrence and Treatment of Pancreatic Cancer. Curr Top Med Chem. 2019;19:2176-86
52. Yoshizawa R, Umeki N, Yanagawa M, Murata M, Sako Y. Single-molecule fluorescence imaging of RalGDS on cell surfaces during signal transduction from Ras to Ral. Biophys Physicobiol. 2017;14:75-84
53. Gentry LR, Martin TD, Reiner DJ, Der CJ. Ral small GTPase signaling and oncogenesis: More than just 15minutes of fame. Biochim Biophys Acta. 2014;1843:2976-88
54. Pelullo M, Nardozza F, Zema S, Quaranta R, Nicoletti C, Besharat ZM. et al. Kras/ADAM17-Dependent Jag1-ICD Reverse Signaling Sustains Colorectal Cancer Progression and Chemoresistance. Cancer Res. 2019;79:5575-86
55. Zeng C, Xing R, Liu J, Xing F. Role of CSL-dependent and independent Notch signaling pathways in cell apoptosis. Apoptosis. 2016;21:1-12
56. Ohashi S, Natsuizaka M, Yashiro-Ohtani Y, Kalman RA, Nakagawa M, Wu L. et al. NOTCH1 and NOTCH3 coordinate esophageal squamous differentiation through a CSL-dependent transcriptional network. Gastroenterology. 2010;139:2113-23
57. Park MT, Kim MJ, Suh Y, Kim RK, Kim H, Lim EJ. et al. Novel signaling axis for ROS generation during K-Ras-induced cellular transformation. Cell Death Differ. 2014;21:1185-97
58. Li H, Wicks WD. Retinoblastoma protein interacts with ATF2 and JNK/p38 in stimulating the transforming growth factor-beta2 promoter. Arch Biochem Biophys. 2001;394:1-12
59. Hwang YS, Oh SW, Park SH, Lee J, Yoo JA, Kwon K. et al. Melanogenic Effects of Maclurin Are Mediated through the Activation of cAMP/PKA/CREB and p38 MAPK/CREB Signaling Pathways. Oxid Med Cell Longev. 2019;2019:9827519
60. Hotamisligil GS, Davis RJ. Cell Signaling and Stress Responses. Cold Spring Harb Perspect Biol. 2016 8
61. Yong HY, Koh MS, Moon A. The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin Investig Drugs. 2009;18:1893-905
62. Roman M, Lopez I, Guruceaga E, Baraibar I, Ecay M, Collantes M. et al. Inhibitor of Differentiation-1 Sustains Mutant KRAS-Driven Progression, Maintenance, and Metastasis of Lung Adenocarcinoma via Regulation of a FOSL1 Network. Cancer Res. 2019;79:625-38
63. Yang F, Li M, Xu D, Jiang Z, Jiang H, Xiao Y. et al. Inhibition of JNK/c-Jun-ATF2 Overcomes Cisplatin Resistance in Liver Cancer through down-Regulating Galectin-1. Int J Biol Sci. 2023;19:2366-81
64. Pinal N, Calleja M, Morata G. Pro-apoptotic and pro-proliferation functions of the JNK pathway of Drosophila: roles in cell competition, tumorigenesis and regeneration. Open Biol. 2019;9:180256
65. Akrida I, Bravou V, Papadaki H. The deadly cross-talk between Hippo pathway and epithelial-mesenchymal transition (EMT) in cancer. Mol Biol Rep. 2022;49:10065-76
66. Zhang Z, Du J, Wang S, Shao L, Jin K, Li F. et al. OTUB2 Promotes Cancer Metastasis via Hippo-Independent Activation of YAP and TAZ. Mol Cell. 2019;73:7-21 e7
67. Echevarria-Andino ML, Franks NE, Schrader HE, Hong M, Krauss RS, Allen BL. CDON contributes to Hedgehog-dependent patterning and growth of the developing limb. Dev Biol. 2023;493:1-11
68. Ji Z, Mei FC, Xie J, Cheng X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J Biol Chem. 2007;282:14048-55
69. Wu M, Ingram L, Tolosa EJ, Vera RE, Li Q, Kim S. et al. Gli Transcription Factors Mediate the Oncogenic Transformation of Prostate Basal Cells Induced by a Kras-Androgen Receptor Axis. J Biol Chem. 2016;291:25749-60
70. Siddiqui I, Erreni M, Kamal MA, Porta C, Marchesi F, Pesce S. et al. Differential role of Interleukin-1 and Interleukin-6 in K-Ras-driven pancreatic carcinoma undergoing mesenchymal transition. Oncoimmunology. 2018;7:e1388485
71. Perkins ND. The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer. 2012;12:121-32
72. Cheng D, Tuveson D. Kras in Organoids. Cold Spring Harb Perspect Med. 2018 8
73. Teixido C, Castillo P, Martinez-Vila C, Arance A, Alos L. Molecular Markers and Targets in Melanoma. Cells. 2021 10
74. Prior IA, Hood FE, Hartley JL. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020;80:2969-74
75. Timar J, Kashofer K. Molecular epidemiology and diagnostics of KRAS mutations in human cancer. Cancer Metastasis Rev. 2020;39:1029-38
76. Asati V, Mahapatra DK, Bharti SK. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur J Med Chem. 2016;109:314-41
77. Yaeger R, Chatila WK, Lipsyc MD, Hechtman JF, Cercek A, Sanchez-Vega F. et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell. 2018;33:125-36 e3
78. Moore AR, Rosenberg SC, McCormick F, Malek S. RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discov. 2020;19:533-52
79. Mangiapane LR, Nicotra A, Turdo A, Gaggianesi M, Bianca P, Di Franco S. et al. PI3K-driven HER2 expression is a potential therapeutic target in colorectal cancer stem cells. Gut. 2022;71:119-28
80. Kamran S, Seyedrezazadeh E, Shanehbandi D, Asadi M, Zafari V, Shekari N. et al. Combination Therapy with KRAS and P38alpha siRNA Suppresses Colorectal Cancer Growth and Development in SW480 Cell Line. J Gastrointest Cancer. 2022;53:597-604
81. Cox TR. The matrix in cancer. Nat Rev Cancer. 2021;21:217-38
82. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
83. Rmali KA, Puntis MC, Jiang WG. Tumour-associated angiogenesis in human colorectal cancer. Colorectal Dis. 2007;9:3-14
84. Hu JL, Wang W, Lan XL, Zeng ZC, Liang YS, Yan YR. et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol Cancer. 2019;18:91
85. Cassetta L, Pollard JW. Tumor-associated macrophages. Curr Biol. 2020;30:R246-R8
86. Fu XT, Dai Z, Song K, Zhang ZJ, Zhou ZJ, Zhou SL. et al. Macrophage-secreted IL-8 induces epithelial-mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int J Oncol. 2015;46:587-96
87. Irimia D, Balazsi G, Agrawal N, Toner M. Adaptive-control model for neutrophil orientation in the direction of chemical gradients. Biophys J. 2009;96:3897-916
88. Rao HL, Chen JW, Li M, Xiao YB, Fu J, Zeng YX. et al. Increased intratumoral neutrophil in colorectal carcinomas correlates closely with malignant phenotype and predicts patients' adverse prognosis. PLoS One. 2012;7:e30806
89. Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol. 2018;19:665-73
90. Frey DM, Droeser RA, Viehl CT, Zlobec I, Lugli A, Zingg U. et al. High frequency of tumor-infiltrating FOXP3(+) regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. Int J Cancer. 2010;126:2635-43
91. Marshall EA, Ng KW, Kung SH, Conway EM, Martinez VD, Halvorsen EC. et al. Emerging roles of T helper 17 and regulatory T cells in lung cancer progression and metastasis. Mol Cancer. 2016;15:67
92. Stienne C, Virgen-Slane R, Elmen L, Veny M, Huang S, Nguyen J. et al. Btla signaling in conventional and regulatory lymphocytes coordinately tempers humoral immunity in the intestinal mucosa. Cell Rep. 2022;38:110553
93. Hashimoto M, Kamphorst AO, Im SJ, Kissick HT, Pillai RN, Ramalingam SS. et al. CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Annu Rev Med. 2018;69:301-18
94. Schrama D, thor Straten P, Fischer WH, McLellan AD, Brocker EB, Reisfeld RA. et al. Targeting of lymphotoxin-alpha to the tumor elicits an efficient immune response associated with induction of peripheral lymphoid-like tissue. Immunity. 2001;14:111-21
95. Downs-Canner SM, Meier J, Vincent BG, Serody JS. B Cell Function in the Tumor Microenvironment. Annu Rev Immunol. 2022;40:169-93
96. Kasprzak A. The Role of Tumor Microenvironment Cells in Colorectal Cancer (CRC) Cachexia. Int J Mol Sci. 2021 22
97. Silva J, Garcia V, Rodriguez M, Compte M, Cisneros E, Veguillas P. et al. Analysis of exosome release and its prognostic value in human colorectal cancer. Genes Chromosomes Cancer. 2012;51:409-18
98. Li C. New Functions of Long Noncoding RNAs during EMT and Tumor Progression. Cancer Res. 2019;79:3536-8
99. Xu M, Xu X, Pan B, Chen X, Lin K, Zeng K. et al. LncRNA SATB2-AS1 inhibits tumor metastasis and affects the tumor immune cell microenvironment in colorectal cancer by regulating SATB2. Mol Cancer. 2019;18:135
100. Shelkey E, Oommen D, Stirling ER, Soto-Pantoja DR, Cook KL, Lu Y. et al. Immuno-reactive cancer organoid model to assess effects of the microbiome on cancer immunotherapy. Sci Rep. 2022;12:9983
101. Yu AI, Zhao L, Eaton KA, Ho S, Chen J, Poe S. et al. Gut Microbiota Modulate CD8 T Cell Responses to Influence Colitis-Associated Tumorigenesis. Cell Rep. 2020;31:107471
102. Liu W, Zhang X, Xu H, Li S, Lau HC, Chen Q. et al. Microbial Community Heterogeneity Within Colorectal Neoplasia and its Correlation With Colorectal Carcinogenesis. Gastroenterology. 2021;160:2395-408
103. Hashemi Goradel N, Heidarzadeh S, Jahangiri S, Farhood B, Mortezaee K, Khanlarkhani N. et al. Fusobacterium nucleatum and colorectal cancer: A mechanistic overview. J Cell Physiol. 2019;234:2337-44
104. Yu T, Guo F, Yu Y, Sun T, Ma D, Han J. et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell. 2017;170:548-63 e16
105. Zhao L, Cho WC, Nicolls MR. Colorectal Cancer-Associated Microbiome Patterns and Signatures. Front Genet. 2021;12:787176
106. Mackie GM, Copland A, Takahashi M, Nakanishi Y, Everard I, Kato T. et al. Bacterial cancer therapy in autochthonous colorectal cancer affects tumor growth and metabolic landscape. JCI Insight. 2021 6
107. Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B. et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. 2019;18:10
108. Jackstadt R, van Hooff SR, Leach JD, Cortes-Lavaud X, Lohuis JO, Ridgway RA. et al. Epithelial NOTCH Signaling Rewires the Tumor Microenvironment of Colorectal Cancer to Drive Poor-Prognosis Subtypes and Metastasis. Cancer Cell. 2019;36:319-36 e7
109. Wieland E, Rodriguez-Vita J, Liebler SS, Mogler C, Moll I, Herberich SE. et al. Endothelial Notch1 Activity Facilitates Metastasis. Cancer Cell. 2017;31:355-67
110. Chen X, Chen R, Jin R, Huang Z. The role of CXCL chemokine family in the development and progression of gastric cancer. Int J Clin Exp Pathol. 2020;13:484-92
111. Wang H, Zhang B, Li R, Chen J, Xu G, Zhu Y. et al. KIAA1199 drives immune suppression to promote colorectal cancer liver metastasis by modulating neutrophil infiltration. Hepatology. 2022;76:967-81
112. Lu Y, Li Y, Liu Q, Tian N, Du P, Zhu F. et al. MondoA-Thioredoxin-Interacting Protein Axis Maintains Regulatory T-Cell Identity and Function in Colorectal Cancer Microenvironment. Gastroenterology. 2021;161:575-91 e16
113. Zhang Y, Song J, Zhao Z, Yang M, Chen M, Liu C. et al. Single-cell transcriptome analysis reveals tumor immune microenvironment heterogenicity and granulocytes enrichment in colorectal cancer liver metastases. Cancer Lett. 2020;470:84-94
114. Li X, Xiang Y, Li F, Yin C, Li B, Ke X. WNT/beta-Catenin Signaling Pathway Regulating T Cell-Inflammation in the Tumor Microenvironment. Front Immunol. 2019;10:2293
115. Zhang J, Fan J, Zeng X, Nie M, Luan J, Wang Y. et al. Hedgehog signaling in gastrointestinal carcinogenesis and the gastrointestinal tumor microenvironment. Acta Pharm Sin B. 2021;11:609-20
116. Tanaka M, Siemann DW. Gas6/Axl Signaling Pathway in the Tumor Immune Microenvironment. Cancers (Basel). 2020 12
117. Andrejeva G, Rathmell JC. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017;26:49-70
118. Kerk SA, Papagiannakopoulos T, Shah YM, Lyssiotis CA. Metabolic networks in mutant KRAS-driven tumours: tissue specificities and the microenvironment. Nat Rev Cancer. 2021;21:510-25
119. Ternet C, Kiel C. Signaling pathways in intestinal homeostasis and colorectal cancer: KRAS at centre stage. Cell Commun Signal. 2021;19:31
120. Keohavong P, Peter Di Y. Pulmonary Inflammation and KRAS Mutation in Lung Cancer. Adv Exp Med Biol. 2021;1303:71-87
121. Dey P, Li J, Zhang J, Chaurasiya S, Strom A, Wang H. et al. Oncogenic KRAS-Driven Metabolic Reprogramming in Pancreatic Cancer Cells Utilizes Cytokines from the Tumor Microenvironment. Cancer Discov. 2020;10:608-25
122. Lee KE, Spata M, Bayne LJ, Buza EL, Durham AC, Allman D. et al. Hif1a Deletion Reveals Pro-Neoplastic Function of B Cells in Pancreatic Neoplasia. Cancer Discov. 2016;6:256-69
123. Tang YA, Chen YF, Bao Y, Mahara S, Yatim S, Oguz G. et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1alpha and TGF-beta2 to promote chemoresistance in colorectal cancer. Proc Natl Acad Sci U S A. 2018;115:E5990-E9
124. Harmon C, Robinson MW, Hand F, Almuaili D, Mentor K, Houlihan DD. et al. Lactate-Mediated Acidification of Tumor Microenvironment Induces Apoptosis of Liver-Resident NK Cells in Colorectal Liver Metastasis. Cancer Immunol Res. 2019;7:335-46
125. Najumudeen AK, Ceteci F, Fey SK, Hamm G, Steven RT, Hall H. et al. The amino acid transporter SLC7A5 is required for efficient growth of KRAS-mutant colorectal cancer. Nat Genet. 2021;53:16-26
126. Kennedy SA, Jarboui MA, Srihari S, Raso C, Bryan K, Dernayka L. et al. Extensive rewiring of the EGFR network in colorectal cancer cells expressing transforming levels of KRAS(G13D). Nat Commun. 2020;11:499
127. Du F, Cao T, Xie H, Li T, Sun L, Liu H. et al. KRAS Mutation-Responsive miR-139-5p inhibits Colorectal Cancer Progression and is repressed by Wnt Signaling. Theranostics. 2020;10:7335-50
128. Rebane-Klemm E, Truu L, Reinsalu L, Puurand M, Shevchuk I, Chekulayev V. et al. Mitochondrial Respiration in KRAS and BRAF Mutated Colorectal Tumors and Polyps. Cancers (Basel). 2020 12
129. Ringel AE, Drijvers JM, Baker GJ, Catozzi A, Garcia-Canaveras JC, Gassaway BM. et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell. 2020;183:1848-66 e26
130. Wallace K, Lewin DN, Sun S, Spiceland CM, Rockey DC, Alekseyenko AV. et al. Tumor-Infiltrating Lymphocytes and Colorectal Cancer Survival in African American and Caucasian Patients. Cancer Epidemiol Biomarkers Prev. 2018;27:755-61
131. Mei Y, Xiao W, Hu H, Lu G, Chen L, Sun Z. et al. Single-cell analyses reveal suppressive tumor microenvironment of human colorectal cancer. Clin Transl Med. 2021;11:e422
132. Plantureux L, Mege D, Crescence L, Carminita E, Robert S, Cointe S. et al. The Interaction of Platelets with Colorectal Cancer Cells Inhibits Tumor Growth but Promotes Metastasis. Cancer Res. 2020;80:291-303
133. Zeng X, Ward SE, Zhou J, Cheng ASL. Liver Immune Microenvironment and Metastasis from Colorectal Cancer-Pathogenesis and Therapeutic Perspectives. Cancers (Basel). 2021 13
134. Stadler M, Pudelko K, Biermeier A, Walterskirchen N, Gaigneaux A, Weindorfer C. et al. Stromal fibroblasts shape the myeloid phenotype in normal colon and colorectal cancer and induce CD163 and CCL2 expression in macrophages. Cancer Lett. 2021;520:184-200
135. Dong ZY, Zhong WZ, Zhang XC, Su J, Xie Z, Liu SY. et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin Cancer Res. 2017;23:3012-24
136. Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P. et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell. 2019;35:559-72 e7
137. Zdanov S, Mandapathil M, Abu Eid R, Adamson-Fadeyi S, Wilson W, Qian J. et al. Mutant KRAS Conversion of Conventional T Cells into Regulatory T Cells. Cancer Immunol Res. 2016;4:354-65
138. Cheng H, Fan K, Luo G, Fan Z, Yang C, Huang Q. et al. Kras(G12D) mutation contributes to regulatory T cell conversion through activation of the MEK/ERK pathway in pancreatic cancer. Cancer Lett. 2019;446:103-11
139. Domvri K, Petanidis S, Zarogoulidis P, Anestakis D, Tsavlis D, Bai C. et al. Treg-dependent immunosuppression triggers effector T cell dysfunction via the STING/ILC2 axis. Clin Immunol. 2021;222:108620
140. Lal N, White BS, Goussous G, Pickles O, Mason MJ, Beggs AD. et al. KRAS Mutation and Consensus Molecular Subtypes 2 and 3 Are Independently Associated with Reduced Immune Infiltration and Reactivity in Colorectal Cancer. Clin Cancer Res. 2018;24:224-33
141. Nam GH, Kwon M, Jung H, Ko E, Kim SA, Choi Y. et al. Statin-mediated inhibition of RAS prenylation activates ER stress to enhance the immunogenicity of KRAS mutant cancer. J Immunother Cancer. 2021 9
142. Hu H, Cheng R, Wang Y, Wang X, Wu J, Kong Y. et al. Oncogenic KRAS signaling drives evasion of innate immune surveillance in lung adenocarcinoma by activating CD47. J Clin Invest. 2023 133
143. Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ. et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;16:2069-83
144. Xu Y, Poggio M, Jin HY, Shi Z, Forester CM, Wang Y. et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat Med. 2019;25:301-11
145. Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y. et al. PDL1 Regulation by p53 via miR-34. J Natl Cancer Inst. 2016 108
146. Liu C, Zheng S, Jin R, Wang X, Wang F, Zang R. et al. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020;470:95-105
147. Gao G, Liao W, Ma Q, Zhang B, Chen Y, Wang Y. KRAS G12D mutation predicts lower TMB and drives immune suppression in lung adenocarcinoma. Lung Cancer. 2020;149:41-5
148. Messex JK, Adams KLA, Hawkins WG, DeNardo D, Bardeesy N, Billadeau DD. et al. Oncogenic Kras-Mediated Cytokine CCL15 Regulates Pancreatic Cancer Cell Migration and Invasion through ROS. Cancers (Basel). 2022 14
149. Zhang S, Li P, Li J, Gao J, Qi Q, Dong G. et al. Chromatin accessibility uncovers KRAS-driven FOSL2 promoting pancreatic ductal adenocarcinoma progression through up-regulation of CCL28. Br J Cancer. 2023
150. Zhu Z, Aref AR, Cohoon TJ, Barbie TU, Imamura Y, Yang S. et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014;4:452-65
151. Zhou J, Ma X, He X, Chen B, Yuan J, Jin Z. et al. Dysregulation of PD-L1 by UFMylation imparts tumor immune evasion and identified as a potential therapeutic target. Proc Natl Acad Sci U S A. 2023;120:e2215732120
152. Zhao Q, Guo J, Zhao Y, Shen J, Kaboli PJ, Xiang S. et al. Comprehensive assessment of PD-L1 and PD-L2 dysregulation in gastrointestinal cancers. Epigenomics. 2020;12:2155-71
153. Miao B, Hu Z, Mezzadra R, Hoeijmakers L, Fauster A, Du S. et al. CMTM6 shapes antitumor T cell response through modulating protein expression of CD58 and PD-L1. Cancer Cell. 2023;41:1817-28 e9
154. Gulley JL, Schlom J, Barcellos-Hoff MH, Wang XJ, Seoane J, Audhuy F. et al. Dual inhibition of TGF-beta and PD-L1: a novel approach to cancer treatment. Mol Oncol. 2022;16:2117-34
155. Rak J, Klement G. Impact of oncogenes and tumor suppressor genes on deregulation of hemostasis and angiogenesis in cancer. Cancer Metastasis Rev. 2000;19:93-6
156. Graells J, Vinyals A, Figueras A, Llorens A, Moreno A, Marcoval J. et al. Overproduction of VEGF concomitantly expressed with its receptors promotes growth and survival of melanoma cells through MAPK and PI3K signaling. J Invest Dermatol. 2004;123:1151-61
157. Kleiner J, Hollborn M, Wiedemann P, Bringmann A. Activator protein-1 contributes to the NaCl-induced expression of VEGF and PlGF in RPE cells. Mol Vis. 2018;24:647-66
158. Kranenburg O, Gebbink MF, Voest EE. Stimulation of angiogenesis by Ras proteins. Biochim Biophys Acta. 2004;1654:23-37
159. Lin CC, Kuo IY, Wu LT, Kuan WH, Liao SY, Jen J. et al. Dysregulated Kras/YY1/ZNF322A/Shh transcriptional axis enhances neo-angiogenesis to promote lung cancer progression. Theranostics. 2020;10:10001-15
160. Wang ZD, Wei SQ, Wang QY. Targeting oncogenic KRAS in non-small cell lung cancer cells by phenformin inhibits growth and angiogenesis. Am J Cancer Res. 2015;5:3339-49
161. Figueras A, Arbos MA, Quiles MT, Vinals F, Germa JR, Capella G. The impact of KRAS mutations on VEGF-A production and tumour vascular network. BMC Cancer. 2013;13:125
162. Choi SH, Kim JK, Chen CT, Wu C, Marco MR, Barriga FM. et al. KRAS Mutants Upregulate Integrin beta4 to Promote Invasion and Metastasis in Colorectal Cancer. Mol Cancer Res. 2022;20:1305-19
163. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J. et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N Engl J Med. 2021;384:2371-81
164. Strickler JH, Satake H, George TJ, Yaeger R, Hollebecque A, Garrido-Laguna I. et al. Sotorasib in KRAS p.G12C-Mutated Advanced Pancreatic Cancer. N Engl J Med. 2023;388:33-43
165. Bekaii-Saab TS, Yaeger R, Spira AI, Pelster MS, Sabari JK, Hafez N. et al. Adagrasib in Advanced Solid Tumors Harboring a KRAS(G12C) Mutation. J Clin Oncol. 2023;41:4097-106
166. Mausey N, Halford Z. Targeted Therapies for Previously "Undruggable" KRAS-Mutated Non-Small Cell Lung Cancer: A Review of Sotorasib and Adagrasib. Ann Pharmacother. 2023: 10600280231197459.
167. Lim TKH, Skoulidis F, Kerr KM, Ahn MJ, Kapp JR, Soares FA. et al. KRAS G12C in advanced NSCLC: Prevalence, co-mutations, and testing. Lung Cancer. 2023;184:107293
168. Khan S, Budamagunta V, Zhou D. Targeting KRAS in pancreatic cancer: Emerging therapeutic strategies. Adv Cancer Res. 2023;159:145-84
169. Chen Z, Zhang L, Yang Y, Liu H, Kang X, Nie Y. et al. DNMT1 expression partially dictates 5-Azacytidine sensitivity and correlates with RAS/MEK/ERK activity in gastric cancer cells. Epigenetics. 2023;18:2254976
170. Abdelwahab M, Saeed H, Elnikhely N, Nematalla H. Synergistic effect of Dactolisib/Lys05 combination on autophagy in A549 cells. Acta Biochim Pol. 2023
171. Gunassekaran GR, Poongkavithai Vadevoo SM, Baek MC, Lee B. M1 macrophage exosomes engineered to foster M1 polarization and target the IL-4 receptor inhibit tumor growth by reprogramming tumor-associated macrophages into M1-like macrophages. Biomaterials. 2021;278:121137
172. Xia Y, Rao L, Yao H, Wang Z, Ning P, Chen X. Engineering Macrophages for Cancer Immunotherapy and Drug Delivery. Adv Mater. 2020;32:e2002054
173. Ma Y, Schulz B, Trakooljul N, Al Ammar M, Sekora A, Sender S. et al. Inhibition of KRAS, MEK and PI3K Demonstrate Synergistic Anti-Tumor Effects in Pancreatic Ductal Adenocarcinoma Cell Lines. Cancers (Basel). 2022 14
174. Kim MJ, Lee SJ, Ryu JH, Kim SH, Kwon IC, Roberts TM. Combination of KRAS gene silencing and PI3K inhibition for ovarian cancer treatment. J Control Release. 2020;318:98-108
175. Jin N, Jiang T, Rosen DM, Nelkin BD, Ball DW. Synergistic action of a RAF inhibitor and a dual PI3K/mTOR inhibitor in thyroid cancer. Clin Cancer Res. 2011;17:6482-9
176. Chan CH, Chiou LW, Lee TY, Liu YR, Hsieh TH, Yang CY. et al. PAK and PI3K pathway activation confers resistance to KRAS(G12C) inhibitor sotorasib. Br J Cancer. 2023;128:148-59
177. Bu X, Liu S, Zhang Z, Wu J, Pan S, Hu Y. Comprehensive profiling of enhancer RNA in stage II/III colorectal cancer defines two prognostic subtypes with implications for immunotherapy. Clin Transl Oncol. 2023
178. Cascetta P, Marinello A, Lazzari C, Gregorc V, Planchard D, Bianco R. et al. KRAS in NSCLC: State of the Art and Future Perspectives. Cancers (Basel). 2022 14
179. Kato S, Fujiwara Y, Hong DS. Targeting KRAS: Crossroads of Signaling and Immune Inhibition. J Immunother Precis Oncol. 2022;5:68-78
180. Schoenfeld AJ, Rizvi H, Bandlamudi C, Sauter JL, Travis WD, Rekhtman N. et al. Clinical and molecular correlates of PD-L1 expression in patients with lung adenocarcinomas. Ann Oncol. 2020;31:599-608
181. Xiang L, Fu X, Wang X, Li W, Zheng X, Nan K. et al. A Potential Biomarker of Combination of Tumor Mutation Burden and Copy Number Alteration for Efficacy of Immunotherapy in KRAS-Mutant Advanced Lung Adenocarcinoma. Front Oncol. 2020;10:559896
182. Liu C, Zheng S, Wang Z, Wang S, Wang X, Yang L. et al. KRAS-G12D mutation drives immune suppression and the primary resistance of anti-PD-1/PD-L1 immunotherapy in non-small cell lung cancer. Cancer Commun (Lond). 2022;42:828-47
183. Okada F, Rak JW, Croix BS, Lieubeau B, Kaya M, Roncari L. et al. Impact of oncogenes in tumor angiogenesis: mutant K-ras up-regulation of vascular endothelial growth factor/vascular permeability factor is necessary, but not sufficient for tumorigenicity of human colorectal carcinoma cells. Proc Natl Acad Sci U S A. 1998;95:3609-14
184. Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P. et al. Correction: RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin Cancer Res. 2019;25:1437
185. Overacre-Delgoffe AE, Bumgarner HJ, Cillo AR, Burr AHP, Tometich JT, Bhattacharjee A. et al. Microbiota-specific T follicular helper cells drive tertiary lymphoid structures and anti-tumor immunity against colorectal cancer. Immunity. 2021;54:2812-24 e4
186. Hezaveh K, Shinde RS, Klotgen A, Halaby MJ, Lamorte S, Ciudad MT. et al. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity. 2022;55:324-40 e8
187. Matson V, Chervin CS, Gajewski TF. Cancer and the Microbiome-Influence of the Commensal Microbiota on Cancer, Immune Responses, and Immunotherapy. Gastroenterology. 2021;160:600-13
188. Hayase E, Jenq RR. Role of the intestinal microbiome and microbial-derived metabolites in immune checkpoint blockade immunotherapy of cancer. Genome Med. 2021;13:107
189. Sipe LM, Chaib M, Pingili AK, Pierre JF, Makowski L. Microbiome, bile acids, and obesity: How microbially modified metabolites shape anti-tumor immunity. Immunol Rev. 2020;295:220-39
190. Rigau M, Uldrich AP, Behren A. Targeting butyrophilins for cancer immunotherapy. Trends Immunol. 2021;42:670-80
191. Chen G, Cao Z, Shi Z, Lei H, Chen C, Yuan P. et al. Microbiome analysis combined with targeted metabolomics reveal immunological anti-tumor activity of icariside I in a melanoma mouse model. Biomed Pharmacother. 2021;140:111542
192. Cortez NE, Mackenzie GG. Ketogenic Diets in Pancreatic Cancer and Associated Cachexia: Cellular Mechanisms and Clinical Perspectives. Nutrients. 2021 13
193. Liu P, Zhao G, Zhang L, Gong Y, Gu Y. Atractylenolide I inhibits antibiotic-induced dysbiosis of the intestinal microbiome. Ann Transl Med. 2021;9:1539
194. Ustjanzew A, Sencio V, Trottein F, Faber J, Sandhoff R, Paret C. Interaction between Bacteria and the Immune System for Cancer Immunotherapy: The alpha-GalCer Alliance. Int J Mol Sci. 2022 23
195. Wang CZ, Huang WH, Zhang CF, Wan JY, Wang Y, Yu C. et al. Role of intestinal microbiome in American ginseng-mediated colon cancer prevention in high fat diet-fed AOM/DSS mice [corrected]. Clin Transl Oncol. 2018;20:302-12
196. Paez D, Pare L, Espinosa I, Salazar J, del Rio E, Barnadas A. et al. Immunoglobulin G fragment C receptor polymorphisms and KRAS mutations: are they useful biomarkers of clinical outcome in advanced colorectal cancer treated with anti-EGFR-based therapy? Cancer Sci. 2010;101:2048-53
197. Tougeron D, Cortes U, Ferru A, Villalva C, Silvain C, Tourani JM. et al. Epidermal growth factor receptor (EGFR) and KRAS mutations during chemotherapy plus anti-EGFR monoclonal antibody treatment in metastatic colorectal cancer. Cancer Chemother Pharmacol. 2013;72:397-403
198. McFall T, Diedrich JK, Mengistu M, Littlechild SL, Paskvan KV, Sisk-Hackworth L. et al. A systems mechanism for KRAS mutant allele-specific responses to targeted therapy. Sci Signal. 2019 12
199. Adachi Y, Ito K, Hayashi Y, Kimura R, Tan TZ, Yamaguchi R. et al. Epithelial-to-Mesenchymal Transition is a Cause of Both Intrinsic and Acquired Resistance to KRAS G12C Inhibitor in KRAS G12C-Mutant Non-Small Cell Lung Cancer. Clin Cancer Res. 2020;26:5962-73
200. Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE. et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med. 2017;23:1362-8
201. Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA. et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546:498-503
Author contact
![]() Corresponding author: Xiaotong Hu, Department of Pathology, Sir Run Run Shaw Hospital, Zhejiang University, Hangzhou, Zhejiang, China. Telephone: +86 13588708170; Email address: hxt_hzedu.cn.
Corresponding author: Xiaotong Hu, Department of Pathology, Sir Run Run Shaw Hospital, Zhejiang University, Hangzhou, Zhejiang, China. Telephone: +86 13588708170; Email address: hxt_hzedu.cn.