Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Pathophysiology
3. Associations of GD with other...
4. Current therapeutics
5. Advances in clinics
6. mRNA/saRNA approach for...
7. Strategy of using mRNA/saRNA...
8. Challenges
9. Concluding remarks and future...
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(6):2111-2129. doi:10.7150/ijbs.87741 This issue Cite
Review
A review on Gaucher disease: therapeutic potential of β-glucocerebrosidase-targeted mRNA/saRNA approach
Shunping Feng1,#, Nino Rcheulishvili1,2,#, Xiaoming Jiang3, Pan Zhu3, Xuehua Pan4, Meilan Wei1, Peng George Wang1, ![]() , Yang Ji1,
, Yang Ji1, ![]() , Dimitri Papukashvili1,2,
, Dimitri Papukashvili1,2, ![]()
1. Department of Pharmacology, School of Medicine, Southern University of Science and Technology, Shenzhen 518000, China.
2. Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China.
3. Cheerland Biomedicine, Shenzhen, China.
4. Shenzhen Pengbo Biotech Co. Ltd, Shenzhen, China.
# Contributed equally.
Received 2023-7-3; Accepted 2024-3-7; Published 2024-3-17
Abstract

Gaucher disease (GD), a rare hereditary lysosomal storage disorder, occurs due to a deficiency in the enzyme β-glucocerebrosidase (GCase). This deficiency leads to the buildup of substrate glucosylceramide (GlcCer) in macrophages, eventually resulting in various complications. Among its three types, GD2 is particularly severe with neurological involvements. Current treatments, such as enzyme replacement therapy (ERT), are not effective for GD2 and GD3 due to their inability to cross the blood-brain barrier (BBB). Other treatment approaches, such as gene or chaperone therapies are still in experimental stages. Additionally, GD treatments are costly and can have certain side effects. The successful use of messenger RNA (mRNA)-based vaccines for COVID-19 in 2020 has sparked interest in nucleic acid-based therapies. Remarkably, mRNA technology also offers a novel approach for protein replacement purposes. Additionally, self-amplifying RNA (saRNA) technology shows promise, potentially producing more protein at lower doses. This review aims to explore the potential of a cost-effective mRNA/saRNA-based approach for GD therapy. The use of GCase-mRNA/saRNA as a protein replacement therapy could offer a new and promising direction for improving the quality of life and extending the lifespan of individuals with GD.
Keywords: Gaucher disease, mRNA, saRNA, GlcCer, GCase, lysosomal storage disease, ERT, SRT, protein replacement therapy
1. Introduction
Gaucher disease (GD) is a rare, autosomal recessive lysosomal storage disease (LSD) (1) and is one of the most common sphingolipidoses and LSDs (2-6). It was first described in 1882 by French physician Philippe Gaucher, in a 32-year-old female patient with an enlarged spleen (7), and initially was thought to be splenetic cancer (8). GD was better understood in 1965, well after its initial discovery (9). GD is caused by mutations in the GBA1 gene, encoding the lysosomal enzyme β-glucocerebrosidase (GCase) (1). In contrast, GBA2 encodes a different, extra-lysosomal enzyme, the so-called second GCase (10,11). It also hydrolyzes GlcCer to glucose and ceramide but operates in a different cellular compartment and is not associated with the lysosomal membrane (12). Unlike GCase, the so-called second GCase is also involved in the metabolism of bile acid 3-O-glucosides (12). The GBA1 gene, located on chromosome 1 (1q22), is composed of 12 exons (NM_001005742.3) and encodes the enzyme GCase, which consists of 497 amino acids. To date, more than 700 mutations have been detected in this gene in individuals diagnosed with GD (13,14) (https://www.hgmd.cf.ac.uk/ac/gene.php?gene=GBA; http://www.gnomad-sg.org/gene/ENSG00000177628?dataset=gnomad_r3). GBA1 mutation leads to a markedly decreased activity of the lysosomal enzyme GCase, whose main function is to cleave its substrate glucosylceramide (GlcCer), also called glucocerebroside, into glucose and ceramide, leading to a significant accumulation of GlcCer in macrophages (15). The accumulation occurs in the mononuclear phagocyte system, mainly in spleen histiocytes, bone marrow, lymph nodes, Kupffer cells in the liver, osteoclasts in bone, microglia in the central nervous system (CNS), alveolar macrophages in lungs, etc. (16). Macrophages engorged with unprocessed GlcCer are called Gaucher cells. The infiltration of Gaucher cells in the spleen, liver, and bone marrow is a hallmark of GD, leading to the characteristic manifestations of the disease. These include splenomegaly, hepatomegaly, cytopenia, and bone lesions (7). GD is categorized into three types: GD1, GD2, and GD3. GD1 is the most prevalent (90%) and typically does not involve neurological impairments. In contrast, GD2 and GD3 are associated with neurological damage (15).
The frequency of GD in the general population is approximately between 1 in 40,000 and 1 in 60,000 individuals while it can reach 1 in 800 in the Ashkenazi Jewish population (15,17,18). Once diagnosed, GD typically requires lifetime treatment (19). Enzyme replacement therapy (ERT) and substrate reduction therapy (SRT) stand as the primary treatment modalities for GD1 and partially GD3. Ideally, the patients should receive treatment before the onset of complications (15). Nucleic acid vaccines, especially those based on messenger RNA (mRNA), have recently garnered significant attention within the scientific community. These innovative vaccines represent a potent, cost-effective, time-saving, and safe alternative to conventional immunization strategies for prophylaxis of infectious diseases (20-24). Moreover, preclinical testing of mRNA technology has yielded positive results, and there are presently active clinical trials testing its utility for protein replacement therapy. Yet, there has been a greater emphasis on the development of prevention strategies for infectious diseases. The approval of mRNA vaccines for coronavirus disease 2019 (COVID-19) represents a landmark in the development of this approach, paving the way for new opportunities in managing various health conditions using this technology. Indeed, except for infectious diseases, it has also been widely tested in plentiful directions, including cancer prevention (25). Remarkably, there is an alternative technology to conventional mRNA vaccines which is called self-amplifying RNA (saRNA). Evidently, the saRNA approach represents a cutting-edge advancement in RNA therapy. It is proposed to offer potential improvements over the conventional mRNA technique, particularly due to its lower dosage requirements (26). This may correlate with relatively fewer side effects making it a promising option. Besides, it possesses long-lasting effects which might be a more suitable option for protein replacement therapy (27). Nonetheless, in order to achieve successful saRNA-based drug development and its approval in clinics, some challenges, particularly with its large size and delivery formulations, that exist in this direction need to be overcome (25). In the context of protein replacement therapy, saRNA has the potential to function similarly to the conventional mRNA, yet possibly with enhanced efficiency. A number of advantages observed in the mRNA-based approach (28) also point to a promising future of saRNA-based protein replacement therapy (25).
The current review addresses the strategy for developing a potentially cost-effective mRNA/saRNA-based approach for GD treatment via protein replacement therapy. This will be one step forward to alleviating the disease and improving both life expectancy and quality of life for GD patients. Moreover, the pathophysiology of the disease, its associations with other health disorders, and currently available therapeutics, along with challenges and perspectives will be discussed, providing a holistic view of the current state and potential improvements in GD therapy.
2. Pathophysiology
In LSDs, enzyme deficiencies cause substrate accumulation in lysosomes (overload disease) (3). GD is a genetic disorder caused by an enzyme GCase deficiency which is a result of mutations in the GBA1 gene (13). More than 700 documented gene mutations (13) cause the deficiency of GCase out of which, the most popular mutations are located in N370S, L444P (29), V394L, D409H, K198T, E326K, and R496H (30,31). Additionally, two mutations, E326K and T369M, are strongly related to Parkinson's disease (PD). Interestingly, they are not known to cause GD in homozygous carriers. However, these mutations may still influence the activity of GCase and subsequently alter the clinical presentation of GD (32). GCase which is activated by saposin C (33,34), is responsible for the lysosomal degradation of GlcCer. GCase maturation occurs in the Golgi apparatus from where it is delivered to lysosomes with the assistance of lysosomal integral membrane protein-2 (LIMP-2) molecule (35). After delivering to the lysosome, the molecular bond is broken down via the acidic pH (36). GCase deficiency leads to GlcCer accumulation. The build-up of GlcCer causes the formation of fibrillar aggregates that accumulate in macrophages, giving the cytoplasm the appearance of “crumpled tissue paper” (35). These Gaucher cells infiltrate various organs such as bone marrow, spleen, and liver, and cause the symptoms of the disease (15,17,18). GlcCer is also a substrate of an alternative metabolic pathway where it can be metabolized by acid ceramidase into glucosylsphingosine (GlcSph) which is less hydrophobic and can diffuse into fluids (37). This pathway becomes prominent in the case of GCase deficiency. GlcSph is metabolized by a cytoplasmic enzyme second GCase which is encoded by the GBA2 gene and functions at neutral pH (15). The so-called second GCase converts GlcSph into sphingosine, which is then phosphorylated, and sphingosine-1-phosphate (S1P) is produced (15). GlcSph, serving as the potential source of S1P, can affect the differentiation, migration, and survival of various cell types, including lymphocytes and macrophages, which may ultimately influence the immune system's function (38). High levels of sphingosine can be toxic to bone (15) while the accumulation of GlcSph can cause neuronal dysfunction and death (15,39). Normally, GlcSph can be detected in the brains of patients with GD-related neurological lesions regardless of the presence of Gaucher cells, while it is absent in the brains of healthy individuals (15) which makes GlcSph a sensitive biomarker (10,40). GlcSph measurements in brain samples of GD patients demonstrate that GlcSph level is remarkably increased in neuronopathic GD, showing the highest levels in GD2 (41).
Saposin C, the activator of GCase, is derived by the cleavage of prosaposin- precursor protein into four homologous proteins (saposins A-D) (42). These mature saposins A-D support the activity of lysosomal hydrolases in the process of sphingolipid degradation (42). Saposin C was discovered in 1971, isolated from the spleen of a 12-year-old female patient with GD3. Later research showed its ability to increase GCase activity in vitro (42). Apparently, except for the normal functioning of GCase, saposin C also plays a crucial role as the mutation of its gene induces lipid accumulation in lysosomes (43). Those patients carrying saposin C mutation develop biochemical phenotypes mimicking GD (44).
Interestingly, mutations in the GBA1 gene were also correlated with an enhanced incidence of PD in GD patients and asymptomatic carriers (45). Indeed, the intricate pathophysiology of GD can result in several related conditions, including skeletal abnormalities and PD (7). The skeletal involvement in GD follows three basic processes: focal disease (irreversible lesions such as osteonecrosis and osteosclerosis), local disease (reversible abnormalities adjacent to heavily involved marrow), and generalized osteopenia (46). In the general population, the likelihood of developing PD after age 60 is about 2-4%. However, this risk is slightly increased in individuals with GD or those carrying the GBA mutation (45,47). These conditions may be a consequence of oxidative stress and inflammatory reactions caused by the interconnection of factors including endoplasmic reticulum (ER) stress, substrate accumulation, defective autophagy, mitochondrial dysfunction, etc. (48).
Infiltration of the spleen by Gaucher cells that exhibit dysregulated expression of surface markers, abnormal release of inflammatory cytokines, and sequestration of iron, leads to splenomegaly. The weight of the spleen in GD patients sometimes reaches several kilograms (49). Indeed, a number of case reports have shown that patients with splenomegaly are often diagnosed with GD and splenomegaly is usually present in most GD patients (50,51). Splenic enlargement which is also reported in patients with chronic kidney disease (52), presents a diagnostic challenge. This similarity of symptoms raises the potential for GD to be misdiagnosed (53). In an in vitro model of GD, the deficiency of GCase significantly hampers the process of human bone marrow hematopoiesis (54). In GD patients, an increased occurrence of erythrophagocytosis is observed compared to that in healthy erythrocytes. This phenomenon is associated with an accumulation of sphingolipids within erythrocytes and a corresponding decrease in their deformability (55). Additionally, the impairment of GCase function hampers the degradation of α-Synuclein (α-Syn) in lysosomes, leading to the accumulation of oligomers in the brain- substantia nigra and inducing neurotoxicity (56). On the other hand, the accumulation of GlcCer due to the functional loss of GCase plays a crucial role in the pathogenesis of PD. It modulates the amyloid formation of α-Syn via stabilizing soluble oligomers. Subsequently, these oligomers aggregate and contribute to the formation of Lewy bodies in nerve cells, the hallmark of PD. This process reveals the molecular link between GD and PD (56). Additionally, Moraitou et al. have demonstrated that α-Syn oligomerization takes place in the red blood cell membranes not only in individuals with GD but also in carriers of GD, even in the absence of PD (57). However, in GD patients, α-Syn oligomerization was correlated with lipid abnormalities, while no lipid abnormalities were detected in carriers of GD (57). Thrombocytopenia, characterized by a deficiency of platelets that are crucial for blood clotting following injury, also occurs, leading to coagulation problems in GD patients (53). Gaucher cells also infiltrate the liver in GD patients, frequently leading to hepatomegaly (58). Common manifestations of GD-related liver involvement include hepatomegaly, non-hepatocellular carcinoma (non-HCC) focal liver lesions, as well as fibrosis (59). In more severe cases, GD patients may develop cirrhosis, portal hypertension, and HCC (59).
The abundance of bioactive glycosphingolipids impacts hematopoiesis and disrupts the equilibrium between osteoblasts and osteoclasts in terms of their numbers and activity (60). An imbalance between osteoblasts, the cells responsible for the formation of new bone tissue, and osteoclasts, which break down and resorb bone tissue, contributes to bone thinning, fragility, and the development of osteolytic lesions (60). In general, bone formation and normal functionality are mediated either by hormone receptors which are present on osteoblasts and osteoclasts, or via an indirect way which implies the presence of cytokines including interleukin (IL)-1, IL-6, and tumor necrosis factor-α (TNF-α) (61). Hence, the cytokines are also responsible for the regulation of osteoclasts and osteoblasts (61). Apparently, the changes in cytokine release in GD are associated with different conditions. For example, IL-10 activity, the release of which is elevated in GD, may lead to the inhibition of osteoblast activity while elevated levels of IL-6 in GD are thought to enhance osteoclast activation and formation (61). Additionally, evidence suggests that systematically increased levels of S1P due to the activity of sphingosine kinase (SphK) 1 or SphK2, in the bloodstream may increase the risk of fractures associated with osteoporosis (62). Moreover, elevated S1P may be considered a biomarker in bone disease (62).
The lung involvement in GD is associated with the infiltration of Gaucher cells into the lung leading to an interstitial disease process that has the potential to progress to pulmonary fibrosis, pulmonary arterial hypertension, or reduction of lung volume due to hepatosplenomegaly (63,64).
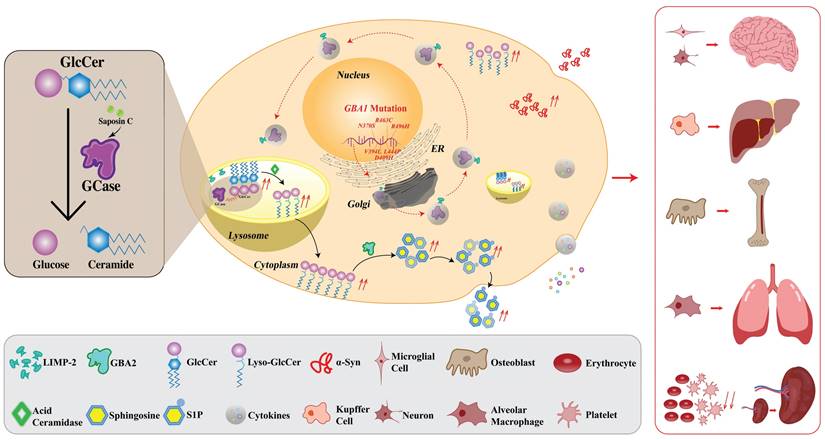
Apparently, except for the GCase deficiency, the anomalies of other implicated members of this molecular cascade such as saposin C or LIMP-2, may also induce GD phenotypes. However, the deficiency of GCase remains the most prevalent and fundamental leading to the manifestation of GD. A schematic representation of the pathophysiology of GD is provided in Fig. 1.
Pathophysiology of GD in macrophage. Normally, GBA1 is transcribed into mRNA which is subsequently exported from the nucleus to the ER where GCase is synthesized. GCase binds to LIMP-2 which is essential for the regulation of GCase transport from ER through the Golgi apparatus (where the GCase protein undergoes maturation) to the lysosomes. The binding occurs at a neutral pH and dissociation takes place within the lysosomal environment, which is characterized by an acidic pH. In the lysosome, with the support of its essential co-factor saposin C, GCase catalyzes the hydrolysis of GlcCer into glucose and ceramide under an acidic environment. In the GD condition, due to the GBA1 mutation, the mutant GCase is produced which is a target for ERAD and proteasomal breakdown. Mutant GCase is unable to reach the lysosome, leading to an accumulation of GlcCer within the lysosomal compartment. The upregulated levels of intracellular GlcCer promote the formation of toxic soluble α-Syn assemblies, exacerbating a pathogenic cycle by impairing the maturation of lysosomes and inhibiting the activity of functional GCase. This impairment results in further GlcCer accumulation and augmented α-Syn formation due to its hampered degradation, leading to the release of inflammatory cytokines. The enzyme known as GBA2 (so-called second GCase) transforms GlcSph (also known as lyso-GlcCer) into sphingosine, which subsequently undergoes phosphorylation, resulting in the production of S1P. Enhanced inflammatory response, along with the associated pathophysiology, contributes to bone fragility, lowering the erythrocyte and platelet levels, reducing lung function, and may also lead to neurodegeneration. Black arrows represent reactions that proceed normally. The red broken arrows indicate the reactions that are typically expected to occur but do not occur. The red solid arrows denote the reactions that take place, but they are harmful. GD, Gaucher disease; GCase, glucocerebrosidase; LIMP-2, lysosomal integral membrane protein 2; ER, endoplasmic reticulum; ERAD, endoplasmic reticulum-associated degradation; GlcCer, glucosylceramide; α-Syn, α-Synuclein; GBA2, β-glucocerebrosidase 2; S1P, sphingosine-1-phosphate; lyso-GlcCer, lyso-glucosylceramide.
The GD has been classified into three forms according to the absence (GD1) or presence and severity (GD2 or GD3) of neurological involvement (53). These three types can be somewhat but not absolutely distinguished by their severity, symptoms, and age of onset. There are also perinatal-lethal and cardiovascular forms of GD (53). The symptoms vary from a few or asymptomatic forms of the disease to chronic and severe complications (53). Visceral involvement and bone disease are commonly observed to some extent across all forms of GD with the potential for symptomatic overlap among the different types (65). In specific circumstances, the distinctions between the three forms of GD may become unclear. Although GD1 is clinically non-neuronopathic, in some cases, patients develop neurological symptoms (66). Besides, some patients exhibit an intermediate phenotype that falls between GD2 and GD3 with a survival range of 3 to 8 years (67). The classification and characterization of GD are given in Table 1.
GD classification and characteristics. GD, Gaucher disease; PD, Parkinson's disease; N/A, not applicable.
| Clinical features | Non-neuronopathic | Neuronopathic | |
|---|---|---|---|
| Acute | Chronic | ||
| Type 1 | Type 2 | Type 3 | |
| Incidence | General population- 1 in 40,000-60,000 | Less than 1 in 100,000 | Less than 1 in 100,000 |
| Ashkenazi Jews- 1 in 800 | |||
| Symptom onset | Any age | Infant | Childhood |
| Symptoms | Distended abdominal | Dermatological abnormalities | Distended abdominal |
| Splenomegaly | Splenomegaly | ||
| Hepatomegaly | Hepatomegaly | ||
| Reduced bone density | Seizures | Seizures | |
| Bone crises | Oculomotor apraxia | ||
| Reduced lung function | Neck rigidity | Thrombocytopenia | |
| Reduced platelet count | Cognitive problems | ||
| Thrombocytopenia | Oculomotor paralysis | Blood disorders | |
| Anemia/cytopenia | Respiratory disorders | ||
| Nosebleeds/bruising | Swallowing disorders | Anemia | |
| Abdominal discomfort | Bone disease | ||
| Life expectancy | Childhood/adulthood | Before 2 years | Childhood/early adulthood |
| Disease course | Progressive | Rapidly progressive | Progressive |
| Associations with other diseases | Osteonecrosis | N/A | Osteonecrosis |
| Osteoporosis | Osteoporosis | ||
| PD | PD | ||
| Neoplasia | Neoplasia | ||
| Cirrhosis | Cirrhosis | ||
| Treatment | ERT/SRT | None/hematopoietic stem cell transplantation | ERT/none |
| Common GBA1 mutations | N370S | Diverse | L444P |
2.1. GD1
Each type of GD has distinct clinical features, severity levels, as well as prognoses, with GD1 being the most common and milder form as it commonly does not affect the CNS. GD1 usually does not involve the nervous system due to the presence of functional GCase in the brain (15,68). It accounts for 90% of all types of GD cases and is generally characterized by a slower progression (69). The mutation in N370S of GBA1 is responsible for approximately 70% of GD1 cases in Ashkenazi Jews (70). GD2 and GD3, especially GD2, are less prevalent as they are characterized by higher rates of mortality at an early age (68). Even though there is a higher occurrence of GD1 among Ashkenazi Jews, the majority of patients with this type of disease are non-Jewish individuals, considering the relatively small size of the Ashkenazi Jewish population on a global scale (65). The clinical phenotype of GD1 is widely acknowledged for its exceptional variability (10). GD1 primarily affects the organs such as the liver and spleen (15). Patients with GD1 generally have a normal life expectancy compared with the other two types of GD (71). The clinical presentation varies widely among individuals and mostly includes organomegaly, bone marrow expansion, osteopenia resulting in bone degeneration and deformities, thrombocytopenia, as well as anemia due to the low levels of red blood cells (72). While CNS involvement is atypical in GD1, a minority of GD1 patients may develop peripheral neuropathy (73). Besides, GD1 patients are at risk for developing Lewy body-associated parkinsonism (48).
2.2. GD2
GD2, an acute neuronopathic form, is the most severe and rapidly progressive type of the disease, typically manifesting in infancy, in the first year of life, with fatal outcomes (71). It affects the brain and nervous system, causing developmental delays, seizures, and muscle rigidity (74). Most children with GD2 do not survive beyond early childhood (71,75). In GD2, Gaucher cells are not only found in the perivascular spaces of the brain (76), which are pial-lined, fluid-filled structures (76), but are also present as free parenchymal Gaucher cells within the cerebral cortex (77). Notably, there is a particular prevalence of these cells in the occipital lobes (77). Farfel-Becker et al. used a mouse model of GD2 and observed that when the continuous neuronal accumulation of GlcCer reaches a certain level, a rapid cascade of neuroinflammation and neurodegeneration is induced in particular regions of the brain and may lead to neuronal cell death (78,79). The symptoms of GD2 include progressive cognitive decline, muscle stiffness, loss of motor skills, difficulty in coordination, respiratory difficulties, and swallowing disorders along with the systemic manifestation of GD (80,81). There are several phenotypes associated with GD2: hydrops fetalis- a condition when an excessive buildup of fluid in various parts of the body occurs; hepatosplenomegaly; skin abnormalities including congenital ichthyosis and abnormal, cellophane-like skin; dysmorphology of face features (82); neurological involvements such as joint contractures, decreased body movements, etc.; thrombocytopenia and anemia (80). Lal et al., have analyzed data from 23 GD2 patients (3 living, 20 deceased) (83). The span of ages at the time of death varied from 3 to 55 months, with an average age of 19.2 months. Despite the aggressive therapeutic interventions, the condition of the fourteen patients receiving ERT, the two treated with SRT, and the three who underwent bone marrow transplantations, worsened along with the disease progression. Apparently, while the current therapeutics can aid in prolonged life, they are unable to repair neurological complications (83). Thus, GD2 remains a profoundly severe and progressive disorder leading to early mortality, underscoring the urgent need for innovative and effective therapies.
2.3. GD3
GD3, also called chronic neuronopathic GD, falls between GD1 and GD2 in terms of severity and usually begins in childhood during the first decade of life. It affects both the visceral organs and the CNS, and the severity of symptoms varies widely (84). GD3 includes patients with neurological manifestations that do not align with the criteria for GD2, representing a highly heterogeneous and phenotypically diverse subgroup (35). Symptoms of GD3 may include enlargement of the spleen and liver, bone abnormalities, eye movement problems, progressive neurological deterioration, and cardiovascular calcification (65). The clinical manifestation of GD3 can vary widely; some affected individuals may only survive their teenage years or early 20s, while others live for much longer. As the condition progresses and symptoms intensify, individuals may require assistance with daily tasks. GD3 has a higher incidence compared to GD2 due to the significantly longer lifespan of individuals affected by the disease (85). While GD is found among different ethnicities, specific clusters of the disease have been extensively studied in Northern Europe, Egypt, and Eastern Asia (65). Because of the common symptoms across different types of GD, a misdiagnosis may take place. For instance, a child with GD3 may not exhibit any neurological symptoms until reaching adolescence, leading to an initial misdiagnosis as GD1 (65).
3. Associations of GD with other diseases
Apart from its primary manifestations, GD is associated with the increased risk and incidence of other health issues. These conditions include PD (86,87), bone complications (46,60), blood disorders (88), and multiple myeloma (89). Additionally, individuals with GD may experience increased susceptibility to infections and cancer.
3.1. Dementia with Lewy bodies
Dementia with Lewy bodies (DLB) is a condition characterized by a gradual deterioration of cognitive functions. The clinical data suggest that there is a strong association between the GBA1 mutation and the DLB (90). These data indicate that the patients carrying GBA1 mutations exhibit a more severe phenotype across disorders with Lewy bodies. These individuals tend to experience symptoms at an earlier age, display intensified motor and cognitive impairments, and have a higher incidence of visual hallucinations as well as rapid eye movement (REM) sleep disorders (90). Mutations of the GBA1 gene are indeed associated with the increased risk of synucleinopathy and lead to DLB via the accumulation of intracellular α-Syn oligomers in the neurons (91,92).
3.2. Parkinson's disease
PD is a slowly advancing neurological disorder that primarily impacts the dopamine-producing neurons responsible for controlling body movements. Although GD1 is considered a non-neuronopathic form of the disease, having it increases the risk of developing PD (93). The association of GD1 with PD has posed a challenge to the established classification paradigm (94). About 5-10% of PD patients have GBA1 mutations (86,87) (https://pdgenetics.shinyapps.io/gba1browser/). The accumulation of α-Syn deposits- Lewy bodies in the brain, in the substantia nigra region that is responsible to produce dopamine, may contribute to the development of PD (95). In addition, mutations in the GBA1 gene, which are associated with GD, have been identified as a risk factor for PD in the general population (96). Studies have also suggested that individuals with GD who develop PD may have a more aggressive form of the disease compared with individuals without GD (95). Alcalay et al. studied the GCase activity in blood samples obtained from 392 PD patients and 175 healthy controls (97). There was a significant correlation between the GBA1 status and GCase activity. Particularly, GCase activity was decreased in GBA1 p.E326K PD carriers compared to the control carriers (97). Remarkably, patients with PD without GBA1 mutations also have lowered levels of GCase (98). Along with the E326K, T369M has been also associated with an increased risk for PD, suggesting that while it may not directly cause GD, it could contribute to the risk of developing PD (99).
α-Syn is a protein that is encoded in humans by the gene SNCA (100). The mutation of the SNCA gene is involved in GD as well as the increased risk of developing PD (100). When the GlcCer accumulation takes place in GD, it elevates levels of the α-Syn protein that contributes to the neurodegeneration and development of symptoms resembling PD (101). Hence, mutations in the SNCA gene are potentially associated with both GD and PD. Nevertheless, further research is essential to fully understand the connection between these conditions (102-104). Current evidence suggests a relationship between GCase deficiency and the accumulation of α-Syn, a protein critically involved in the pathogenesis of PD (105).
3.3. Multiple myeloma
Multiple myeloma is a type of blood cancer when abnormal growth of monoclonal plasma cells in the bone marrow takes place (106). The clinical manifestations include cytopenia and bone lesions that are common for GD as well (89). Individuals with GD have an increased risk of developing multiple myeloma, especially when they have severe bone involvement (107). The exact mechanism of this association is not well understood, but it is thought to be related to immune dysfunction and chronic inflammation associated with GD (107). Indeed, Taddei et al., have evaluated the risk of cancer in 403 patients and found that within the entire cohort, there was an unparallel rise in the lifetime risk of multiple myeloma, primarily observed among N370S homozygous patients (108).
3.4. Gammopathy
Noteworthily, gammopathies which imply conditions with abnormal antibodies in the blood are also associated with GD (88). Multiple myeloma is associated with the wide occurrence of monoclonal gammopathy of undetermined significance (MGUS) which is a condition characterized by the presence of monoclonal gammopathy with uncertain implications (109). A multicenter study investigating 2123 GD1 patients in terms of the cancer risk and gammopathies demonstrated that multiple myeloma after MGUS diagnosis was 7.9% which is similar to that observed in the general population (88). Noteworthily, GD1 patients had a higher risk of developing solid cancers of the liver (2.9 times), melanoma (2.5 times), breast (1.4 times), and kidney (2.8 times) (88).
3.5. Bone manifestations
Osteoporosis is a condition characterized by bone loss and fragility as a result of bone demineralization. Individuals with GD are at increased risk of developing osteoporosis due to the accumulation of GlcCer in the bone marrow (60). The accumulated GlcCer interferes with the normal function of bone cells, leading to bone loss and an increased risk of fractures (46,60). Fortunately, the treatment with ERT has been shown to improve bone density in GD patients, reducing the risk of osteoporosis (46).
4. Current therapeutics
The main therapeutics for GD include ERT and SRT. ERT has been the primary treatment for GD for many years and has been shown to improve bone density, liver and spleen size, and hematological parameters. SRT, on the other hand, is a newer therapeutic option that works by inhibiting the synthesis of the lipids that accumulate in GD (110). The main challenges associated with both therapies include the high cost of ERT and the limited effectiveness of SRT in certain patient populations (53). ERT remains the standard of care for GD, however, new treatment options such as gene therapy and pharmacological chaperones may provide additional benefits in the future (111). Throughout the course of the 15-year retrospective study, a total of 60 patients with various forms of GD were observed where the median age of diagnosis was 2 years. The ERT and SRT could increase the survival of GD patients (110). The Food and Drug Administration (FDA)-approved therapies for GD are given in Table 2.
FDA-approved drugs for treating GD. FDA, Food and Drug Administration; GD, Gaucher disease; ERT, enzyme replacement therapy; SRT, substrate reduction therapy; I.V., intravenous.
| Therapy class | ERT | SRT | |||
|---|---|---|---|---|---|
| Brand name | Cerezyme | Elelyso | VPRIV | Zavesca | Cerdelga |
| Manufacturer | Genzyme | Protalix, Pfizer | Takeda (Shire plc) | Actelion Pharmaceuticals | Sanofi Genzyme |
| Drug name | Imiglucerase | Taliglucerase alfa | Velaglucerase alfa | Miglustat | Eliglustat |
| Molecular formula | C2532H3854N672O711S16 | C2580H3918N680O727S17 | C2532H3850N672O711S16 | C10H21NO4 | C23H36N2O4 |
| Manufacture | CHO cells | Carrot cells | Human fibrosarcoma cells | Chemical synthesis | |
| Dosing (average) | Should be individualized: 2.5-60 U/kg | 60 U/kg | 60 U/kg | 100 mg | 84 mg |
| Administration interval (average) | Once every other week | Once every other week | Once every other week | Once a day | Once/twice a day |
| Administration route | I.V. injection | I.V. injection | I.V. injection | Oral | Oral |
| Mechanism of action | Supplies the mutated GCase activity by increasing the concentration of exogenous GCase | Reduces GlcCer by inhibiting GlcCer synthase | |||
| Main side effects | Abdominal discomfort, nausea, vomiting, diarrhea | Chest tightness, dizziness, feeling of warmth, hives or welts, itching, skin rash | Headache, arthralgia, cough, difficulty with breathing, fever, dizziness, abdominal pain | Diarrhea, stomach pain/bloating, weight loss, upset stomach, vomiting | Fatigue, headache, nausea, diarrhea, back pain, and muscle ache |
| Average annual cost (USD) | 384,814 to 705,493 | 185,940 to 354,977 | 152,909 to 560,666 | ~98,000 | 253,675 to 507,350 |
| FDA approval date | May 23, 1994 | May 1, 2012 | February 26, 2010 | July 31, 2003 | August 19, 2014 |
4.1. ERT
ERT is a well-established treatment for GD which involves intravenous (I.V.) administration of a recombinant form of the deficient enzyme GCase. In general, ERT seems to be effective for the treatment of several LSDs, including GD, Fabry disease, and Pompe disease (112). In GD, the administered enzyme is taken up by the lysosome in the affected cell and helps to break down the accumulated substrate GlcCer (111). Indeed, ERT has been shown to ameliorate the condition of various clinical manifestations of GD, including organomegaly, hematological abnormalities, and bone disease (113). Besides, it has demonstrated improvements in quality of life and survival in GD patients (113). In 1991, ERT was introduced as a treatment for GD, which realized the vision initially proposed by Roscoe Brady in 1966 (114,115). Currently, several ERT products are approved by the FDA and are commercially available for GD treatment (Table 2). These ERTs are imiglucerase (Cerezyme, Sanofi-Genzyme), velaglucerase alfa (VPRIV, Takeda), and taliglucerase alfa (Elelyso, Pfizer) produced via Chinese hamster ovary cells, human fibroblasts, and carrot cells, respectively (111). The results of these three ERT products are promising. Indeed, treatment with imiglucerase demonstrated exceptional and long-lasting efficacy in GD1 patients while minimal toxicity was observed (116). Other two ERTs- velaglucerase and taliglucerase have also demonstrated remarkable efficacy, safety, and long-lasting effects (117,118). Despite the effectiveness of ERT, the treatment does have some limitations. These include the requirement of lifelong I.V. administration and the production of exogenous recombinant GCase antibodies which can decrease the bioavailability of the enzyme and impact clinical outcomes. Besides, the treatment faces challenges due to the short half-life of the enzyme in vivo, the high cost of the treatment, and the insufficient efficacy for all manifestations of the disease (119,120). Notably, ERT is less effective in treating severe neurological symptoms due to the inability to cross the blood-brain barrier (BBB) and reach the CNS (121). Hence, for GD2, a rare and severe form of the disease, ERT has been found to be ineffective. Therefore, alternative treatment approaches are necessary for GD, such as substrate reduction therapy or gene therapy.
4.2. SRT
The pathogenesis of LSDs at a cellular level is complex and not fully understood yet. While some LSDs can be treated with ERT, there are also small molecule therapies such as SRT and chaperone therapy. In addition, gene therapy and genome editing are in advanced preclinical stages and have already made their way to clinical trials for a few disorders (122).
ERT aids the body in recycling more waste material, specifically GlcCer, whereas SRT helps the body generate less of this waste material. SRT does not rely on enzyme replacement but instead reduces the accumulation of glycolipids by balancing the residual activity of the deficient enzyme with lowered substrate levels (123). Recent studies have shown that SRT- oral miglustat, which also can cross BBB (124), may be more effective than ERTs in reducing the size of the spleen and liver, improving bone density, and reducing levels of the biomarker chitotriosidase (123). SRT venglustat with the potential of crossing BBB can alleviate the symptoms and prevent disease progression (125). On the other hand, miglustat has low effectiveness and can cause several side effects (125). For this reason, it has been approved for use in a limited population of adults with GD1 for whom ERT is not feasible (126). As to another SRT eliglustat, the study demonstrated that after a one-year treatment, it is not inferior to ERT (127). Furthermore, the long-term study showed that individuals treated with eliglustat maintained stability for up to four years (128).
4.3. Chaperone therapy
A molecular chaperone is a type of protein that assists in the folding, unfolding, assembling, and disassembling of other proteins in the cell (129). Pharmacological chaperone therapy is an approach that employs small-molecule ligands to selectively stabilize mutant enzymes, elevate their levels within cells, and enhance their activity and lysosomal trafficking (129). The stabilized enzyme is then able to function properly and break down the GlcCer that accumulates in the body of GD patients. It is noteworthy that both miglustat and eliglustat, apart from their inhibitory effect on the synthesis of GlcCer, have also been shown to increase GCase activity by serving as chaperones. Consequently, they help in reducing the levels of GlcCer (130). Pharmacological chaperones have been also developed as an alternative therapy for GD-associated PD (131). N-Octyl-b-valienamine is the first chaperone used for GD. It has been found to enhance the activity of F213I mutant GCase in cultured cells (132). Another chaperone for GD- ambroxol has been evidenced to have a therapeutic effect on bone and hematological symptoms in child with GD1 (133). Moreover, its high dose and long-term application demonstrated promising results in GD1 patient with hepatic fibrosis (134). There are three clinical studies focused on chaperone therapy (NCT01463215, NCT03950050, NCT04388969). Although, chaperone therapy exhibits great promise for GD treatment, its mutation-specific nature limits its applicability. Indeed, Ivanova et al., have shown that the ambroxol increased the GCase activity in cells of patients with L444P/L444P; RecΔ55, RecNciI, and L444P/D409H GBA1 mutations while it could not have positive effects on GCase activity in cells with L444P/L444P, D409H, A456P mutations (130).
4.4. Gene therapy
Gene therapy is a sophisticated modality for treating genetic disorders such as GD, involving the strategic introduction of a functional copy of the defective gene to compensate for the genetic aberration (135). This technique encompasses two primary methodologies: in vivo and ex vivo gene therapy. In vivo gene therapy entails the systemic delivery of a therapeutic gene directly into the patient (136), leveraging vectors such as adeno-associated viruses (AAVs) which are notable for their capability to transverse the BBB- a critical consideration for neurological manifestations of GD (137). The choice of vector is paramount, especially in targeting the central nervous system, where the BBB presents a formidable barrier to many therapeutic agents. AAV vectors, in particular, are engineered for their neurotropic characteristics, enabling them to infiltrate the BBB and deliver therapeutic gene to the affected neuronal cells (138). Conversely, ex vivo gene therapy involves transfecting patient-derived cells with the corrective gene exogenously, followed by the transplantation back into the patient (136). The vectors of choice for this method are typically retroviral (139) or lentiviral vectors (140), each with unique integration properties and long-term expression profiles.
There is certain progress in developing gene therapies for GD1. Currently, there is no approved gene therapy for GD2 (111). Gene editing is one type of gene therapy that implies making precise alterations to the human genome allowing the one-time treatment for genetic disorders including GD (141). Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated protein9 (Cas9) (CRISPR/Cas9) system, zinc finger nucleases (ZFNs), as well as transcription activator-like effector nucleases (TALENs) are the most popular and versatile editing platforms (141,142). CRISPR/Cas9 has been specifically employed to create cell models of GD by editing the GBA1 gene in certain cell lines, providing platforms for pathophysiology studies and drug screening (143). ZFNs have been used in genome editing to increase the levels of lysosomal enzymes. ZFNs are designed to target specific DNA sequences, allowing for precise modifications of the genome. They work by creating double-strand breaks at specific locations, which the cell then repairs, potentially correcting gene mutations (111). Similarly, in case of TALENs, they are engineered to bind to specific DNA sequences, allowing for targeted gene editing. They create breaks in the GBA1 gene that are then repaired, ideally restoring normal function (111). Although these platforms have been utilized for developing gene editing therapies for GD, none have advanced beyond preliminary studies (111). Noteworthily, CRISPR/Cas9 has been successfully used for developing GD cellular models- human monocytic cell line (THP-1) with GBA1 mutation as well as glioblastoma cell line (U87) with GBA1 mutation (143). Additionally, cytosine base editors (CBEs) are enzymes that function as gene editors and can introduce base pair changes (C·G-to-T·A) in genomic DNA. These gene editing enzymes can induce this chemical conversion via enzyme-mediated hydrolytic deamination of cytosine to uracil which is interpreted as thymine by DNA polymerases (144). Generally, these enzymes include the modified CRISPR/Cas9 enzyme which is a naturally occurring cytidine deaminase and an inhibitor of uracil repair. CBEs can be used for gene reversion, thus restoring gene function (144,145).
4.5. Allogeneic hematopoietic stem cell transplantation
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) therapy for GD involves transplanting healthy hematopoietic stem cells from a compatible donor into the patient's bone marrow. The transplanted stem cells produce new, healthy blood cells that can break down the accumulated GlcCer in the patient's body. This therapy can potentially halt the progression of the disease and improve the quality of life for GD patients. However, it is a complex and risky procedure that requires careful consideration of the individual situation (146). Besides, the initial achievements of allo-HSCT therapy were quickly overshadowed by the exceptional effectiveness and low toxicity of ERT which remains the most widespread therapy for GD (147).
5. Advances in clinics
There are ongoing clinical trials for GD using different treatments. Most of the clinical studies focus on ERT, SRT, and chaperone therapy, or already discussed possible curative treatment methods for GD. Clinical trials have shown promising results for ERT. For example, in the clinical trial (NCT05529992) the side effects of velaglucerase alfa were observed in individuals with GD1 with newly diagnosed disease or currently under treatment with ERT. In another clinical study (NCT04656600) efficacy and safety of treatment with imiglucerase were evaluated in Chinese GD3 patients with neurological manifestations. A clinical trial (NCT03485677) studies the efficacy and safety of eliglustat with or without imiglucerase in pediatric patients with GD1 and GD3. Gene therapy approaches have also offered a potential long-term treatment option for GD. In a clinical study (NCT05324943) gene therapy with FLT201, which is a replication-incompetent single-stranded recombinant AAV vector, is investigated. The study is conducted on GD1 patients for enhancing residual GCase expression and activity thus increasing its potential for the improvement of the clinical phenotype and prevention of cellular GlcCer accumulation. Detailed information on recent clinical trials of GD therapies is given in Supplementary Table 1.
6. mRNA/saRNA approach for protein replacement purposes
Along with the global COVID-19 pandemic, a new era of next-generation vaccines, particularly, mRNA vaccines has commenced, offering numerous benefits (20,22,148). mRNA is a molecule that plays a crucial role in protein production. In 1987, Robert Malone successfully transfected the NIH 3T3 mouse cells with the mixture of mRNA and synthetic cationic lipid incorporated into liposome that resulted in the successful expression of the protein (149). Since 1997, Katalin Kariko and Drew Weissman worked together and received the Nobel Prize, recently. They successfully performed the nucleoside modification of mRNA allowing the mRNA to escape innate immune response after delivering into the mice as well as the enhanced translation of the target protein (150). Their approach is successfully utilized in the currently available vaccines of acute respiratory syndrome coronavirus 2 (SARS-CoV-2)- BNT162b2 (Pfizer/BioNTech) and mRNA-1273 (Moderna) (151).
In recent years, researchers have been exploring the use of mRNA as a potential tool for protein replacement therapy. This approach involves the introduction of synthetic mRNA into the patient's cells to produce therapeutic proteins that are missing or defective due to genetic mutations or other factors (25). Once the mRNA enters the cells, it is used as a template to direct the synthesis of a therapeutic protein. The cells then secrete the protein into the bloodstream, where it can travel to the target tissues and exert its therapeutic effects (152,153). mRNA-based protein replacement therapy can be used to treat a wide range of genetic disorders. mRNA-based treatments are also making their way into the field of monoclonal antibody therapies (154). Most importantly, unlike the conventional protein replacement therapies that require the production and purification of large quantities of therapeutic proteins, mRNA-based therapy is much more time-saving. mRNA technology has been studied for protein replacement purposes for certain diseases including heart (155) and alpha 1-antitrypsin deficiency (AATD) (156). There are several clinical trials that use mRNA technology for protein replacement in various diseases including heart failure (NCT03370887), propionic acidemia (NCT04159103), cystic fibrosis (NCT03375047), and ulcers associated with type 2 diabetes mellitus (NCT02935712). This corroborates the idea of mRNA/saRNA technology application for protein replacement therapy for GD. The combination of certain chemical modifications and delivery systems indicates the presence of additional possibilities for optimization (157,158). E.g., N1-methyl-pseudouridine (m1Ψ) nucleoside substitution of uridine in mRNA molecule reduces the detection by the innate immune system, particularly, Toll-like receptors (TLRs) for approximately 100-fold, allowing enhanced protein expression (150,157,159,160).
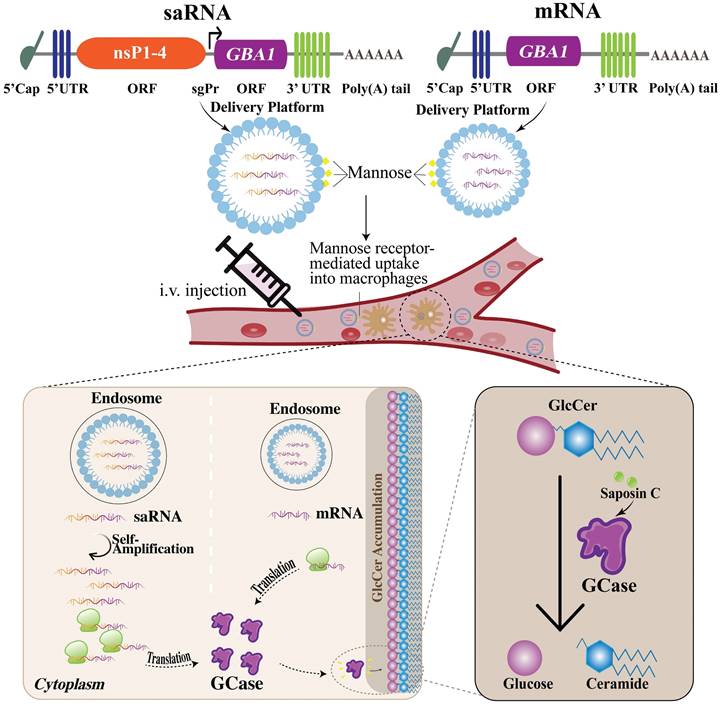
The current standard of care for GD is ERT, which involves the administration of recombinant exogenous GCase. However, the limitation of this approach is the need for frequent infusions and the potential for immune reactions to the exogenous protein. The disadvantages of ERT such as invasiveness and inconvenience of infusion along with the short half-time (119,120) might be overcome by the application of mRNA encoding GCase for protein replacement purposes offering better flexibility (161). Another type of RNA molecule- saRNA has recently garnered the attention of researchers as it has the potential to open a new platform in medicine holding significant potential for creating drugs through a simple and cost-effective approach. There are clinical studies ongoing investigating the application of saRNA as a vaccine for severe acute SARS-CoV-2 (162,163). saRNA can replicate itself, allowing for the sustained production of therapeutic proteins within the cells. saRNA contains not only the genetic instructions for the production of the protein of interest but also the machinery that is necessary for self-amplification, allowing for the continuous production of therapeutic protein within cells. Particularly, saRNA like mRNA is a positive-strand RNA but unlike non-replicating mRNA, except for the gene of interest (GOI), it contains genes encoding viral replicase. Three types of alphaviruses are usually used in the saRNA approach- Venezuelan Equine Encephalitis, Sindbis, and Semliki Forest Viruses (25). In the saRNA molecule, viral structural proteins are deleted, disabling the production of the infectious virus. In the 5'-3' direction, the saRNA molecule comprises 5'cap, 5'untranslated region (UTR), non-structural proteins (nsPs) 1-4, sub-genomic promoter (sgPr), GOI, 3'UTR, and poly(A) tail. When saRNA is delivered into the cell, nsPs form a replicase, also called an RNA-dependent RNA polymerase (RDRP) which then replicates the whole RNA molecule as well as sub-genomic RNA (sgRNA) resulting in the higher and long-lasting protein expression compared to the conventional mRNA outcome (25). saRNA holds remarkable promise as a potential new tool for protein replacement therapy for GD. Nevertheless, before saRNA application launches in clinics, conventional mRNA should not be forgotten since the more research is done on it, the more is known as well. Therefore, thanks to the rapid development of this strategy, here we propose both- conventional mRNA and relatively new saRNA approaches for GD therapy. The schematic illustration of mRNA/saRNA application for GD treatment is given in Fig. 2.
Application of saRNA and mRNA for GD treatment as protein replacement therapy. (Left) when the saRNA is injected and delivered into the cells, the translation of replicase takes place. The replicase then uses the saRNA as a template and makes a negative saRNA strand. Then the negative strand saRNA is used as a template by the replicase and self-amplification occurs. Additionally, replicase recognizes sgPr in the negative strand, and sgRNA is synthesized. Consequently, high levels of GCase are produced. (Right) when the mRNA is injected and delivered into the cells, it is translated into GCase protein. Ultimately, in either case, the produced GCase breaks down the GlcCer into glucose and ceramide leading to the normal condition. saRNA, self-amplifying RNA; mRNA, messenger RNA; sgPr, sub-genomic promoter; GD, Gaucher disease; GCase, glucocerebrosidase; GlcCer, glucosylceramide.
7. Strategy of using mRNA/saRNA approach for GD
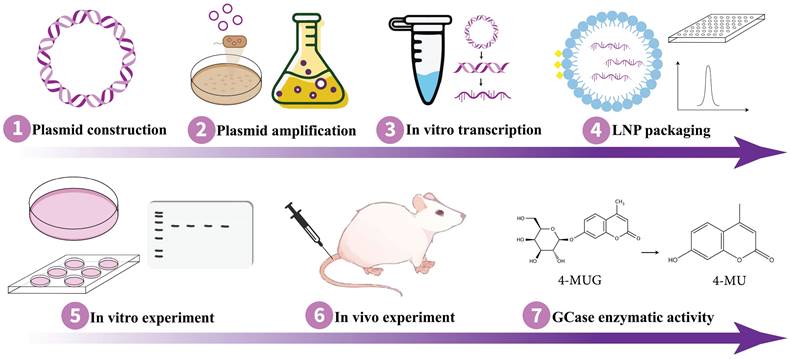
Implementing the appropriate strategy is essential for achieving the intended results. As mRNA and saRNA represent promising avenues for protein replacement therapies, they hold the potential to expedite and enhance the development of GD treatment for therapy. Remarkably, Moderna's mRNA-3927 has been demonstrated to be generally well-tolerated in phase 1/2 clinical trial with promising outcomes for the other inherited disorder propionic acidemia (NCT04159103) where certain parts of fats and proteins cannot be processed properly (164). In order to proceed pre-clinical study on the application of mRNA/saRNA techniques for protein replacement purposes in GD therapy, it is essential to design an expression plasmid containing the sequence of normal GCase and, in the context of saRNA technology, sgPr. Following the synthesis of the expression plasmid, it needs to be transformed into competent cells, such as DH5α, for amplification. Subsequently, the plasmid DNA should be extracted and purified for further use. To verify GCase expression, the plasmid should be transfected into mammalian cells, such as HEK293T, followed by monitoring for the expression of the target protein. Once the optimal protein expression is confirmed, in vitro transcription can be performed to synthesize the mRNA/saRNA. Then the RNA molecule needs to be encapsulated with lipid nanoparticles (LNPs) (165) or another effective delivery system (152,153). Thereafter, validation of GCase protein expression in suitable cells is imperative, which can be followed by the in vivo experiment on mouse models. Fig. 3 outlines a workflow for conducting in vitro and in vivo experiments on GD animal models using mRNA/saRNA technology, delineating the sequential steps from molecular design to therapeutic evaluation.
Schematic of the experimental workflow for studying mRNA/saRNA-based protein replacement therapy in GD. mRNA, messenger RNA; saRNA, self-amplifying RNA; GD, Gaucher disease.
8. Challenges
The primary hurdle in the development of gene therapy for GD has been the absence of an appropriate animal model (166). Fortunately, researchers have successfully developed potential animal models that mimic the neuropathology seen in GD allowing to conduct more studies. Most of the studies use mouse models. For example, Enquist et al., have developed a GD model with Mx1/Cre-loxP system to enable the deletion of GCase exons after birth not to disrupt the GCase activity through the development and skin barrier formation. These mice successfully developed remarkably reduced activity of GCase and significantly elevated GlcCer. Additionally, mice developed infiltration of Gaucher cells in bone marrow, spleen, and liver (167). Mistry et al., have deleted the GBA1 gene in mouse conditionally using an Mx1 promoter that resembled the human GD almost entirely (37). Other GD mouse models such as K14-lnl/lnl (168), Tie2-Cre-LoxP (169) also successfully manifested GD symptoms. Besides mouse models, other animal models have been also studied, e.g., Uemura et al., have found that GBA1-/- medaka (a fish Oryzias latipes) can be used as a neuronopathic GD model (170). Cabaso et al., have demonstrated the relevance of using GBA1bm/m mutant flies for GD studies as they also exhibit remarkably decreased activity of GCase and the accumulation of its substrate (171).
The challenges associated with mRNA-based protein replacement therapy include the necessity of the optimization of mRNA stability, delivery system, translation efficiency, as well as avoidance of innate immune response (172). However, the researchers have made significant progress in the optimization of mRNA sequence and delivery to overcome these challenges (25). LNPs are considered one of the most effective delivery systems which are also employed in FDA-approved mRNA vaccines (173). Furthermore, a small interfering RNA (siRNA) which is the other RNA-based therapeutic (patisiran) approved by FDA also uses LNP as a carrier system (174). Upon entering the host cells, saRNA and its self-replication byproducts induce an innate immune response being recognized as foreign molecules (175). This triggers the release of interferon (IFN) which can interfere with the successful translation of saRNA (176). Besides, the activation of TLRs in the endosomes might be prompted through the endosomal detection mechanisms (20). This will lead to the production of type I IFN along with the other cytokines, resulting in the maturation of dendritic cells (DCs), stimulation of T helper cells and T cell-dependent B cells (176,177). To circumvent the detection of in vitro transcribed mRNA and evade the unwanted immune reaction, nucleoside modifications have been applied, a technique refined by Kariko and colleagues. They demonstrated that the inclusion of pseudouridine (Ψ), a modified nucleoside that occurs naturally, into mRNA achieves two outcomes: it diminishes the immune response triggered by the RNA in vitro as well as in vivo, and it also improves the translation (150). However, this approach is infeasible for saRNA due to its reliance on host cell factors for replication, which would negate the modifications after the initial amplification cycle. Notably, to address this challenge, Blakney et al. have tested saRNA constructs that contained open reading frames (ORFs) of innate inhibiting proteins (IIPs) to see how they affected protein production and immune response. They found that certain proteins, particularly from parainfluenza virus 5 (PIV-5) and Middle East respiratory syndrome coronavirus (MERS-CoV), could significantly increase protein expression and allow the saRNA to evade immune detection (150). Additionally, the fastidious design of mRNA or saRNA constructs necessitates the careful selection of appropriate 5' and 3' UTRs, as well as a signal peptide, along with the addition of the poly-A tail. These elements are crucial for the accurate translation, subsequent secretion of the target protein, as well as stabilization of mRNA construct before translation (178,179).
In order to enhance the effectiveness of LNPs, the incorporation of mannose residues, a process known as mannosylation is also an option. LNPs modified with mannose can improve the uptake ability of macrophages by mannose receptor. Indeed, Goswami et al. claim that mannosylation of LNPs has improved potency for saRNA vaccines (180,181).
A primary obstacle in developing GD therapies utilizing mRNA/saRNA technology lies in the delivery of these molecules to the brain, given the challenges associated with crossing the BBB. BBB is a highly selective and nearly impenetrable protective barrier that separates the blood vessels in the brain from the brain tissue, thereby restricting the entry of various substances including toxins and certain pharmaceuticals, into the brain (182,183). The delivery of drugs across the BBB poses a significant obstacle that hinders the future development of novel therapeutics targeting the brain (184). Nonetheless, some of the large molecules may access the brain via receptor-mediated transport (185). In general, mRNA vaccines, like those developed by Pfizer/BioNTech and Moderna for COVID-19 are not expected to cross the BBB as they are designed to be taken up by cells in the muscle and adjacent lymph nodes at the site of intramuscular (I.M.) injection, with minimal entry into the systemic circulation. However, there is some evidence indicating that small amounts of the mRNA-LNP, particularly, formulated by Moderna for COVID-19 may have the capacity to cross the BBB (186). Gene therapy for GD requires the design of mRNA/saRNA molecules with LNPs to be able to cross BBB and be delivered in the brain, which is challenging. Remarkably, LNPs have been innovatively modified by conjugating monoclonal antibodies (mAbs) to their surface. This modification facilitates the transportation of mRNA into the brain, potentially overcoming the delivery challenges. mAbs ease the vesicular transport of mRNA from one side of a cell to the other side which is called transcytosis. These mAbs are called molecular Trojan horses (187). mAb attaches to a receptor on the BBB, e.g., insulin receptor or transferrin receptor, functioning as a molecular Trojan horse. This facilitates receptor-mediated transcytosis of LNP through the BBB and into the brain extracellular space (187). Notably, multifunctional nanopolymers (MNPs) have been also used for the therapy of CNS owing to the capacity to cross the BBB (188). Remarkably, Sun et al. have studied the application of a CNS-selective delivery system, employing saposin C-dioleoylphosphatidylserine (DOPS) nanovesicles for neuronopathic GD therapy and revealed that the administration of a saposin C-DOPS-GCase successfully delivers functional GCase across a range of tissues, with a pronounced affinity for the brain (189). This method addresses GCase deficiencies within the CNS cells and tissues, demonstrating its effectiveness in ameliorating CNS inflammation and neurological symptoms in a murine model of neuronopathic GD. The research identifies a novel CNS targeting mechanism via a specific phosphatidylserine receptor and the lymphatic system, positioning saposin C-DOPS nanovesicles as a potential new therapeutic avenue for neuronopathic GD (189).
mRNA size usually varies between ~2000-5000 nt while the size of saRNA is much larger- ~15,000 nt (25) and complex that complicates the cellular uptake. This challenge can be surmounted via the application of trans-amplifying RNA (taRNA) which implies the synthesis of saRNA as two separate molecules- mRNA encoding nsPs and mRNA encoding GOI (190,191). Remarkably, taRNA can be as effective as saRNA itself (192). Another obstacle to saRNA approach is the limited clinical data of interactions between nsPs and host factors, the possibility of elevated inflammation due to self-amplification (193), and the generation of innate host immune responses (194,195).
It is also notable that invasive administration of mRNA/saRNA to the brain refers to methods that directly deliver these molecules into the brain tissue or cerebrospinal fluid (CSF), bypassing the BBB. Such methods are considered invasive because they require surgical techniques or other forms of medical intervention that penetrate the protective barriers of the body. Examples of invasive administration routes to the brain include intrathecal injection- delivering substances into the CSF via a lumbar puncture; intracerebroventricular (ICV) injection- injecting directly into the ventricles of the brain; direct parenchymal injection- inserting a needle or catheter directly into the brain tissue, etc. (196). However, these techniques are invasive and painful for patients. Consideration should also be given to potential safety concerns, such as the risk of infection and the possibility of traumatic injuries (197). Besides, the risk of immune response activation in case of both mRNA and saRNA exists that might provoke unintended immune reactions. Moreover, saRNA replication leads to the production of double-stranded RNA intermediates which are known to be highly inflammatory and negatively impact translation efficiency (25). The risk of inducing innate immunity potentially limits repeated administration of saRNA (198). Nonetheless, the benefits of saRNA seem to outweigh the challenges. More research is needed to eliminate these risks to the greatest extent.
The administration routes for mRNA/saRNA therapies include I.V., I.M., as well as intranasal route (199). However, in the context of protein replacement therapy using mRNA or saRNA for GD, the preferred route is I.V. I.V. administration is a common method for systemic delivery, facilitating widespread distribution throughout the body (200). Systemic delivery is important for GD treatment as it targets different organs (199) which are key sites affected by the disease. To circumvent an undesirable immune reaction to administered mRNA, substituting uridines with Ψ is effective (150,159,201). Yet, when multiple injections are required, the development of adaptive immunity and immune memory presents a challenge. When designing mRNA/saRNA constructs, the selection of the appropriate signal peptide is crucial because it significantly influences the final location of the protein once it is translated (179).
9. Concluding remarks and future perspectives
In summary, there are two approaches to specifically treat the abnormal accumulation of GlcCer for GD patients, recovering the enzyme activity and reducing the abnormal levels of GlcCer in the lysosome. There are several remedies for enzyme recovery, ERT, gene therapy, allo-HSCT, and chaperone therapy; for reducing the abnormal levels of the substrate, SRT is available.
To date, out of all the mentioned therapies only ERT and SRT are approved by the FDA. Gene editing is supposed to be the future therapy for GD patients. On the other hand, the approval of mRNA vaccines for COVID-19 marks a significant advancement, opening up new opportunities for managing diverse health conditions. This includes its application in cancer prevention and treating genetic diseases. Besides, saRNA technology, with its lower dosage, reduced side effects, and prolonged efficacy thanks to RDRP, is emerging as a superior alternative for protein replacement therapies. Thus, saRNA appears to become a “game changer” in protein deficiency disorders and it fuels great hope for the GD research community. However, the GD research based on the nucleic acid-based technique may be initiated with a conventional mRNA strategy due to its established sophistication and reliability. Then, it may get advanced with saRNA technology leveraging its potential for increased efficacy. Remarkably, GD2 is a rare and severe form of GD and ERT is not sufficiently effective due to the disability to cross the BBB and reach the CNS where the disease primarily affects. This necessitates alternative treatment approaches such as SRT or gene therapy for GD2. Hence, the proposed GCase-mRNA/saRNA technology as a protein replacement therapy for GD patients is intended to be a step forward to aid in therapy development that will be beneficial in terms of prolonging the lifespan and improving the overall quality of life of the GD population. However, it is important to note that certain obstacles, including the right delivery formulations, therapy crossing the BBB, etc., exist in this direction and need to be surpassed. Taken together, mRNA/saRNA with its feasibility, simplicity, and rapid and cost-effective manufacturing technique may lead to the successful development of protein replacement therapeutics for GD.
Supplementary Material
Supplementary table.
Acknowledgements
Funding
This work was supported by the National Natural Science Foundation of China (General Program, No. 82273841).
Author contributions
S.F. and D.P. conceived the idea and designed the manuscript. S.F. drew the figures. N.R. and D.P. prepared the original draft of the manuscript. S.F., N.R., X.J., Z.P., X.P., M.W., Y.J., and D.P. revised the manuscript. D.P., Y.J., and P.G.W. supervised and gave the final approval of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Institute of Medical Genetics in Cardiff. The Human Gene Mutation Database. The Human Gene Mutation Database. Available from: https://www.hgmd.cf.ac.uk/ac/all.php.
2. National Human Genome Research Institute. Gaucher Disease Clinical Studies at NIH [Internet]. Available from: https://www.genome.gov/Current-NHGRI-Clinical-Studies/Gaucher-Disease#:~:text=URL%3A https%3A%2F%2Fwww.genome.gov%2FCurrent.
3. Rajkumar V, Dumpa V. Lysosomal storage disease. NCBI Books. Lysosomal storage disease. StatPearls Publishing, Treasure Island (FL). 2023
4. Sun A. Lysosomal storage disease overview. Ann Transl Med. 2018;6(24):476
5. Mehta A. Epidemiology and natural history of Gaucher's disease. Eur J Intern Med. 2006;17:S2-5
6. Mehta A, Beck M, Linhart A, Sunder-Plassmann G, Widmer U. History of lysosomal storage diseases: an overview. In: Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis. 2006
7. Stone WL, Basit H, Mukkamalla SKR, Master SR. Gaucher Disease. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 [cited. 2024 Jan 31]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK448080/
8. Gauchers Association. About Gaucher [Internet]. 2024 [cited. 2024 Feb 1]. Available from: https://www.gaucher.org.uk/
9. Mistry PK, Belmatoug N, vom Dahl S, Giugliani R. Understanding the natural history of Gaucher disease. Am J Hematol. 2015Jul;90(Suppl 1):S6-11
10. Mistry PK, Liu J, Sun L, Chuang WL, Yuen T, Yang R. et al. Glucocerebrosidase 2 gene deletion rescues type 1 Gaucher disease. Proc Natl Acad Sci U S A. 2014;111(13):4934-9
11. Harzer K, Yildiz Y. High β-glucosidase (GBA) activity not attributable to GBA1 and GBA2 in live normal and enzyme-deficient fibroblasts may emphasise the role of additional GBAs. Biol Chem. 2015;396(11):1241-6
12. Woeste MA, Wachten D. The enigmatic role of GBA2 in controlling locomotor function. Front Mol Neurosci. 2017;10:386
13. Horowitz M, Braunstein H, Zimran A, Revel-Vilk S, Goker-Alpan O. Lysosomal functions and dysfunctions: Molecular and cellular mechanisms underlying Gaucher disease and its association with Parkinson disease. Adv Drug Deliv Rev. 2022Aug;187:114402
14. Davidson BA, Hassan S, Garcia EJ, Tayebi N, Sidransky E. Exploring genetic modifiers of Gaucher disease: The next horizon. Hum Mutat. 2018;39(12):1739-51
15. Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C. et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci. 2017;18(2):1-30
16. Chen M, Wang J. Gaucher's disease. Arch Pathol Lab Med. 2008;132(5):851-3
17. Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher's disease. The Lancet. 2008;372(9645):1263-71
18. Stirnemann J, Vigan M, Hamroun D, Heraoui D, Rossi-Semerano L, Berger MG. et al. The French Gaucher's disease registry: Clinical characteristics, complications and treatment of 562 patients. Orphanet J Rare Dis. 2012;7:77
19. Gary SE, Ryan E, Steward AM, Sidransky E. Recent advances in the diagnosis and management of Gaucher disease. Expert Rev Endocrinol Metab. 2018Mar;13(2):107-18
20. Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines-a new era in vaccinology. Nat Rev Drug Discov. 2018;17(4):261-79
21. Rcheulishvili N, Papukashvili D, Liu C, Ji Y, He Y, Wang PG. Promising strategy for developing mRNA-based universal influenza virus vaccine for human population, poultry, and pigs- focus on the bigger picture. Front Immunol. 2022;13:1025884
22. Kowalzik F, Schreiner D, Jensen C, Teschner D, Gehring S, Zepp F. mRNA-Based Vaccines. Vaccines Basel. 2021;9(4):390
23. Liu C, Rcheulishvili N, Shen Z, Papukashvili D, Xie F, Wang Z. et al. Development of an LNP-Encapsulated mRNA-RBD Vaccine against SARS-CoV-2 and Its Variants. Pharmaceutics. 2022;14(5):1101
24. Wang X, Liu C, Rcheulishvili N, Papukashvili D, Xie F, Zhao J. et al. Strong immune responses and protection of PcrV and OprF-I mRNA vaccine candidates against Pseudomonas aeruginosa. Npj Vaccines. 2023;8(1):76
25. Papukashvili D, Rcheulishvili N, Liu C, Ji Y, He Y, Wang PG. Self-Amplifying RNA Approach for Protein Replacement Therapy. Int J Mol Sci. 2022;23(21):12884
26. Tregoning JS, Stirling DC, Wang Z, Flight KE, Brown JC, Blakney AK. et al. Formulation, inflammation, and RNA sensing impact the immunogenicity of self-amplifying RNA vaccines. Mol Ther - Nucleic Acids. 2023Mar14;31:29-42
27. Bloom K, van den Berg F, Arbuthnot P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2021Apr;28(3-4):117-29
28. Wadhwa A, Aljabbari A, Lokras A, Foged C, Thakur A. Opportunities and challenges in the delivery of mRNA-based vaccines. Pharmaceutics. 2020;12(2):102
29. Dimitriou E, Moraitou M, Cozar M, Serra-vinardell J, Vilageliu L, Grinberg D. et al. Gaucher disease : Biochemical and molecular findings in 141 patients diagnosed in Greece. Mol Genet Metab Rep. 2020;24:100614
30. Ankleshwaria C, Mistri M, Bavdekar A, Muranjan M, Dave U, Tamhankar P. et al. Novel mutations in the glucocerebrosidase gene of Indian patients with Gaucher disease. J Hum Genet. 2014;59(4):223-8
31. Gatto EM, Da Prat G, Etcheverry JL, Drelichman G, Cesarini M. Parkinsonisms and glucocerebrosidase deficiency: A comprehensive review for molecular and cellular mechanism of glucocerebrosidase deficiency. Brain Sci. 2019;9(2):30
32. Mallett V, Ross JP, Alcalay RN, Ambalavanan A, Sidransky E, Dion PA. et al. GBA P.T369M substitution in Parkinson disease: Polymorphism or association?. A meta-analysis. Neurol Genet. 2016;2:e104
33. Premkumar L, Silman I, Sussman JL, Futerman AH. The X-ray structure of human acid- β-glucosidase: Implications for second-generation enzyme replacement therapy. Futerman AH, editor. Gaucher Disease. Taylor & Francis Group, LLC. 2007:85-96 p
34. Gruschus JM, Jiang Z, Yap TL, Hill SA, Grishaev A, Piszczek G. et al. Dissociation of glucocerebrosidase dimer in solution by its co-factor, saposin C. Biochem Biophys Res Commun. 2015;457(4):561-6
35. Do J, Mckinney C, Sharma P, Sidransky E. Glucocerebrosidase and its relevance to Parkinson disease. Mol Neurodegener. 2019;14:36
36. Gonzalez A, Valeiras M, Sidransky E, Tayebi N. Lysosomal integral membrane protein-2: A new player in lysosome-related pathology. Mol Genet Metab. 2014;111(2):84-91
37. Mistry PK, Liu J, Yang M, Nottoli T, McGrath J, Jain D. et al. Erratum: Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage (Proceedings of the National Academy of Sciences (2010) 107, 45 (19473-19478) DOI: 10.1073/pnas.1003308107. Proc Natl Acad Sci U S A. 2012;109(23):9220
38. Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V. et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355-60
39. Srikanth MP, Jones JW, Kane M, Awad O, Park TS, Zambidis ET. et al. Elevated Glucosylsphingosine in Gaucher Disease induced Pluripotent Stem Cell Neurons Deregulates Lysosomal Compartment through Mammalian Target of Rapamycin Complex 1. Stem Cells Transl Med. 2021Jul1;10(7):1081-94
40. Dekker N, Van Dussen L, Hollak CEM, Overkleeft H, Scheij S, Ghauharali K. et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood. 2011;118(16):118-28
41. Orvisky E, Park JK, LaMarca ME, Ginns EI, Martin BM, Tayebi N. et al. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: Correlation with phenotype and genotype. Mol Genet Metab. 2002;76(4):262-70
42. Tamargo RJ, Velayati A, Goldin E, Sidransky E. The role of saposin C in Gaucher disease. Mol Genet Metab. 2012;106(3):257-63
43. Vaccaro AM, Motta M, Tatti M, Scarpa S, Masuelli L, Bhat M. et al. Saposin C mutations in Gaucher disease patients resulting in lysosomal lipid accumulation, saposin C deficiency, but normal prosaposin processing and sorting. Hum Mol Genet. 2010;19(15):2987-97
44. Rafael J. Tamargo, Velayati A, Goldin E, Sidransky E. The role of saposin C in Gaucher disease Rafael. Occup Env Med. 2012;106(3):257-63
45. Riboldi GM, Di Fonzo AB. GBA, Gaucher disease, and parkinson's disease: From genetic to clinic to new therapeutic approaches. Cells. 2019 8(4)
46. Wenstrup RJ, Roca-Espiau M, Weinreb NJ, Bembi B. Skeletal aspects of Gaucher disease: A review. Br J Radiol. 2002;75(SUPPL. 1):A2-12
47. Zhou Y, Wang Y, Wan J, Zhao Y, Pan H, Zeng Q. et al. Mutational spectrum and clinical features of GBA1 variants in a Chinese cohort with Parkinson's disease. Npj Park Dis. 2023;9:129
48. Roh J, Subramanian S, Weinreb NJ, Kartha R V. Gaucher disease - more than just a rare lipid storage disease. J Mol Med. 2022;100(4):499-518
49. Simpson WL, Hermann G, Balwani M. Imaging of Gaucher disease. World J Radiol. 2014 28;6(9):657-68
50. Garça M, Correia S, Goulart A, Ávila P. Gaucher Disease: One of the Few Causes of Massive Splenomegaly. Eur J Case Rep Intern Med. 2022 9(12)
51. Merra G, Dal Lago A, Ricci R, Antuzzi D, Gasbarrini G, Gasbarrini A. et al. Splenomegaly as a Primary Manifestation of Gaucher Disease in a Young Adult Woman. Case Rep Gastroenterol. 2008;2(3):474-8
52. Platts MM, Anastassiades E, Sheriff S, Smith S, Bartolo DC. Spleen size in chronic renal failure. Br Med J. 1984;289(6456):1415-8
53. Hughes DA, Pastores GM. Gaucher Disease. 2000 Jul 27 [Updated. 2023 Dec 7]. In: Adam MP, Feldman J, Mirzaa GM, et al, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1269/
54. Berger J, Lecourt S, Vanneaux V, Rapatel C, Boisgard S, Caillaud C. et al. Glucocerebrosidase deficiency dramatically impairs human bone marrow haematopoiesis in an in vitro model of Gaucher disease: Research paper. Br J Haematol. 2010;150(1):93-101
55. Dupuis L, Chauvet M, Bourdelier E, Dussiot M, Belmatoug N, Le Van Kim C. et al. Phagocytosis of Erythrocytes from Gaucher Patients Induces Phenotypic Modifications in Macrophages, Driving Them toward Gaucher Cells. Int J Mol Sci. 2022 23(14)
56. Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA. et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146(1):37-52
57. Moraitou M, Sotiroudis G, Papagiannakis N, Ferraz MMJ, Xenakis A, Aerts JMFG. et al. a-Synuclein and lipids in erythrocytes of Gaucher disease carriers and patients before and after enzyme replacement therapy. PLoS ONE. 2023;18(2 February):1-16
58. Starosta RT, Vairo FP e, Dornelles AD, Basgalupp SP, Siebert M, Pedroso MLA. et al. Liver involvement in patients with Gaucher disease types I and III. Mol Genet Metab Rep. 2020;22(September 2019):100564
59. Adar T, Ilan Y, Elstein D, Zimran A. Liver involvement in Gaucher disease - Review and clinical approach. Blood Cells Mol Dis. 2018;68:66-73
60. Hughes D, Mikosch P, Belmatoug N, Carubbi F, Cox TM, Goker-Alpan O. et al. Gaucher Disease in Bone: From Pathophysiology to Practice. J Bone Miner Res. 2019;34(6):996-1013
61. Mikosch P, Hughes D. An overview on bone manifestations in Gaucher disease. Wien Med Wochenschr. 2010;160(23-24):609-24
62. Grewe JM, Knapstein PR, Donat A, Jiang S, Smit DJ, Xie W. et al. The role of sphingosine-1-phosphate in bone remodeling and osteoporosis. Bone Res. 2022;10:34
63. Mistry PK, Sirrs S, Chan A, Pritzker MR, Duffy TP, Grace ME. et al. Pulmonary hypertension in type 1 Gaucher's disease: Genetic and epigenetic determinants of phenotype and response to therapy. Mol Genet Metab. 2002;77(1-2):91-8
64. Farias L de PG de, Padilha IG, Santos CJJ dos, Maranhão CP de M, Miranda CMNR de. Pulmonary involvement in Gaucher disease. Radiol Bras. 2017;50(1):408-9
65. Rosenbloom BE, Weinreb NJ. Gaucher disease: A comprehensive review. In: Critical Reviews in Oncogenesis. 2013 p. 163-75
66. Furderer ML, Hertz E, Lopez GJ, Sidransky E. Neuropathological Features of Gaucher Disease and Gaucher Disease with Parkinsonism. Int J Mol Sci. 2022May23;23(10):5842
67. Goker-Alpan O, Schiffmann R, Park JK, Stubblefield BK, Tayebi N, Sidransky E. Phenotypic continuum in neuronopathic Gaucher disease: An intermediate phenotype between type 2 and type 3. J Pediatr. 2003;143(2):273-6
68. Sidransky E. Gaucher disease: insights from a rare Mendelian disorder. Discov Med. 2012;14(77):273-81
69. Giraldo P, Andrade-Campos M, Morales M, the SEGA (SEguimiento del paciente de GAucher G patient follow up) G. Recommendations on the follow-up of patients with Gaucher disease in Spain: Results from a Delphi survey. JIMD Rep. 2023;64(1):90-103
70. Colombo R. Age estimate of the N370S mutation causing Gaucher disease in Ashkenazi Jews and European populations: A reappraisal of haplotype data. Am J Hum Genet. 2000;66(2):692-7
71. Bossù G, Pedretti L, Bertolini L, Esposito S. Pediatric Gaucher Disease Presenting with Massive Splenomegaly and Hepatic Gaucheroma. Children. 2023May12;10(5):869
72. Grabowski GA. Gaucher disease and other storage disorders. Hematol Am Soc Hematol Educ Program. 2012;2012:13-8
73. Mullin S, Hughes D, Mehta A, Schapira AHV. Neurological effects of glucocerebrosidase gene mutations. Eur J Neurol. 2019;26(3):388-e29
74. Roshan Lal T, Sidransky E. The Spectrum of Neurological Manifestations Associated with Gaucher Disease. Dis Basel Switz. 2017Mar2;5(1):10
75. Mignot C, Doummar D, Maire I, De Villemeur TB, Afenjar A, Bost M. et al. Type 2 Gaucher disease: 15 New cases and review of the literature. Brain Dev. 2006;28(1):39-48
76. Nilsson O, Svennerholm L. Accumulation of Glucosylceramide and Glucosylsphingosine (Psychosine) in Cerebrum and Cerebellum in Infantile and Juvenile Gaucher Disease. J Neurochem. 1982;39(3):709-18
77. Wong K, Sidransky E, Verma A, Mixon T, Sandberg GD, Wakefield LK. et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab. 2004;82(3):192-207
78. Farfel-Becker T, Vitner EB, Pressey SNR, Eilam R, Cooper JD, Futerman AH. Spatial and temporal correlation between neuron loss and neuroinflammation in a mouse model of neuronopathic Gaucher disease. Hum Mol Genet. 2011;20(7):1375-86
79. Farfel-Becker T, Vitner EB, Kelly SL, Bame JR, Duan J, Shinder V. et al. Neuronal accumulation of glucosylceramide in a mouse model of neuronopathic Gaucher disease leads to neurodegeneration. Hum Mol Genet. 2014;23(4):843-54
80. Weiss K, Gonzalez A, Lopez G, Pedoeim L, Groden C, Sidransky E. The Clinical Management of Type 2 Gaucher Disease Karin. Mol Genet Metab. 2015;114(2):110-22
81. Arévalo NB, Lamaizon CM, Cavieres VA, Burgos P V, Álvarez AR, Yañez MJ. et al. Neuronopathic Gaucher disease: Beyond lysosomal dysfunction. Front Mol Neurosci. 2022;15:934820
82. Gupta N, Oppenheim IM, Kauvar EF, Tayebi N, Sidransky E. Type 2 Gaucher disease: Phenotypic variation and genotypic heterogeneity. Blood Cells Mol Dis. 2011;46(1):75-84
83. Roshan Lal T, Seehra GK, Steward AM, Poffenberger CN, Ryan E, Tayebi N. et al. The natural history of type 2 Gaucher disease in the 21st century: A retrospective study. Neurology. 2020;95(15):e2119-30
84. Bennett LL, Fellner C. Pharmacotherapy of Gaucher Disease: Current and Future Options. Pharm Ther. 2018May;43(5):274-309
85. Ramaswami U, Mengel E, Berrah A, AlSayed M, Broomfield A, Donald A. et al. Throwing a spotlight on under-recognized manifestations of Gaucher disease: Pulmonary involvement, lymphadenopathy and Gaucheroma. Mol Genet Metab. 2021Aug1;133(4):335-44
86. Milenkovic I, Blumenreich S, Futerman AH. disease. Curr Opin Neurobiol. 2022;72:148-54
87. Beavan MS, Schapira AHV. Glucocerebrosidase mutations and the pathogenesis of Parkinson disease. Vol. 45, Annals of Medicine. 2013 p. 511-21
88. Rosenbloom BE, Cappellini MD, Weinreb NJ, Dragosky M, Revel-Vilk S, Batista JL. et al. Cancer risk and gammopathies in 2123 adults with Gaucher disease type 1 in the International Gaucher Group Gaucher Registry. Am J Hematol. 2022;97(10):1337-47
89. Costello R, O'Callaghan T, Sébahoun G. Gaucher disease and multiple myeloma. Leuk Lymphoma. 2006;47(7):1365-8
90. Gaubert S, Hourregue C, Mouton-Liger F, Millot P, Franco M, Amar-Bouaziz E. et al. Exploring the link between GBA1 mutations and Dementia with Lewy bodies, A mini-review. Neurosci Biobehav Rev. 2022;141:104856
91. Cullen V, Sardi SP, Ng J, Xu YH, Sun Y, Tomlinson JJ. et al. Acid β-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter α-synuclein processing. Ann Neurol. 2011;69(6):940-53
92. Koss DJ, Erskine D, Porter A, Palmoski P, Menon H, Todd OGJ. et al. Nuclear alpha-synuclein is present in the human brain and is modified in dementia with Lewy bodies. Acta Neuropathol Commun. 2022;10(1):98
93. Kouli A, Torsney KM, Kuan WL. Parkinson's Disease: Etiology, Neuropathology, and Pathogenesis. In: Stoker TB, Greenland JC, editors. Parkinson's Disease: Pathogenesis and Clinical Aspects [Internet]. Brisbane (AU): Codon Publications; 2018 [cited. 2024 Feb 3]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK536722/
94. Bultron G, Kacena K, Pearson D, Boxer M, Yang R, Sathe S. et al. The risk of Parkinson's disease in type 1 Gaucher disease. J Inherit Metab Dis. 2010;33(2):167-73
95. Goldin E. Gaucher disease and parkinsonism, a molecular link theory. Mol Genet Metab. 2010;101(4):307-10
96. Stoker TB, Torsney KM, Barker RA. Pathological Mechanisms and Clinical Aspects of GBA1 Mutation-Associated Parkinson's Disease. In: Stoker TB, Greenland JC, editors. Parkinson's Disease: Pathogenesis and Clinical Aspects [Internet]. Brisbane (AU): Codon Publications; 2018 [cited. 2024 Feb 4]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK536716/
97. Alcalay RN, Wolf P, Chiang MSR, Helesicova K, Zhang XK, Merchant K. et al. Longitudinal Measurements of Glucocerebrosidase activity in Parkinson's patients. Ann Clin Transl Neurol. 2020;7(10):1816-30
98. Murphy KE, Gysbers AM, Abbott SK, Tayebi N, Kim WS, Sidransky E. et al. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson's disease. Brain. 2014;137(3):834-48
99. Ghatti S, Yoon E, Lopez G, Ehrlich D, Horovitz SG. Imaging and genetics in Parkinson's disease: assessment of the GBA1 mutation. J Neurol. 2022;269(10):5347-55
100. Srinivasan E, Chandrasekhar G, Chandrasekar P, Anbarasu K, Vickram AS, Karunakaran R. et al. Alpha-Synuclein Aggregation in Parkinson's Disease. Front Med. 2021Oct18;8:736978
101. Li Y, Li P, Liang H, Zhao Z, Hashimoto M, Wei J. Gaucher-Associated Parkinsonism. Cell Mol Neurobiol. 2015;35(6):755-61
102. Burré J. The synaptic function of α-synuclein. J Park Dis. 2015;5(4):699-713
103. Blandini F, Cilia R, Cerri S, Pezzoli G, Schapira AHV, Mullin S. et al. Glucocerebrosidase mutations and synucleinopathies: Toward a model of precision medicine. Mov Disord. 2019;34(1):9-21
104. Gegg ME, Schapira AHV. The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J. 2018;285(19):3591-603
105. Manning-Boǧ AB, Schüle B, Langston JW. Alpha-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: A biological link between Gaucher disease and parkinsonism. NeuroToxicology. 2009;30(6):1127-32
106. Albagoush SA, Shumway C, Azevedo AM. Multiple Myeloma. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 [cited. 2024 Feb 4]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK534764/
107. Chadha J, Sahai T, Schwimmer J, Shani D. A 30-Year-Old Carrier of Gaucher Disease with Multiple Myeloma. Case Rep Oncol Med. 2019;2019:6469196
108. Taddei TH, Kacena KA, Yang M, Yang R, Malhotra A, Boxer M. et al. The underrecognized progressive nature of N370S Gaucher disease and assessment of cancer risk in 403 patients. Am J Hematol. 2009;84(4):208-14
109. Arends M, Van Dussen L, Biegstraaten M, Hollak CEM. Malignancies and monoclonal gammopathy in Gaucher disease; a systematic review of the literature. Br J Haematol. 2013;161(6):832-42
110. Barney AM, Danda S, Abraham A, Fouzia NA, Gowdra A, Abraham SSC. et al. Clinicogenetic Profile, Treatment Modalities, and Mortality Predictors of Gaucher Disease: A 15-Year Retrospective Study. Public Health Genomics. 2021;24(3-4):139-48
111. Kong W, Lu C, Ding Y, Meng Y. Update of treatment for Gaucher disease. Eur J Pharmacol. 2022;926(February):175023
112. Katsigianni EI, Petrou P. A systematic review of the economic evaluations of Enzyme Replacement Therapy in Lysosomal Storage Diseases. Cost Eff Resour Alloc. 2022Sep19;20(1):51
113. El-Beshlawy A, Abdel-Azim K, Abdel-Salam A, Gebril NA, Selim YMM, Said F. Clinical Characteristics, Molecular Background, and Survival of Egyptian Patients With Gaucher Disease Over a 20-Year Follow-up. J Pediatr Hematol Oncol. 2022;44(5):243-8
114. Brady RO, Kanfer JN, Bradley RM, Shapiro D. Demonstration of a deficiency of glucocerebroside-cleaving enzyme in Gaucher's disease. J Clin Invest. 1966;45(7):1112-5
115. Barton NW, Brady RO, Dambrosia JM, Bisceglie AM Di, S.H.Doppelt, Hill SC. et al. Replacement therapy for inherited enzyme deficiency-macrophage-targeted glucocerebrosidase for Gaucher's disease. 1999.
116. Weinreb NJ, Camelo JS, Charrow J, McClain MR, Mistry P, Belmatoug N. Gaucher disease type 1 patients from the ICGG Gaucher Registry sustain initial clinical improvements during twenty years of imiglucerase treatment. Mol Genet Metab. 2021;132(2):100-11
117. Zimran A, Durán G, Giraldo P, Rosenbaum H, Giona F, Petakov M. et al. Long-term efficacy and safety results of taliglucerase alfa through 5 years in adult treatment-naïve patients with Gaucher disease. Blood Cells Mol Dis. 2019;78:14-21
118. Titievsky L, Schuster T, Wang R, Younus M, Palladino A, Quazi K. et al. Safety and effectiveness of taliglucerase alfa in patients with Gaucher disease: an interim analysis of real-world data from a multinational drug registry (TALIAS). Orphanet J Rare Dis. 2022;17(1):1-11
119. Concolino D, Deodato F, Parini R. Enzyme replacement therapy: Efficacy and limitations. Ital J Pediatr. 2018 44(Suppl 2)
120. Silva AKA, Sagné C, Gazeau F, Abasolo I. Enzyme replacement therapy: current challenges and drug delivery prospects via extracellular vesicles. Rare Dis Orphan Drugs J. 2022;1(3):13
121. Revel-Vilk S, Szer J, Mehta A, Zimran A. How we manage Gaucher Disease in the era of choices. Br J Haematol. 2018;182(4):467-80
122. Platt FM, D'Azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nat Rev Dis Primer. 2018;4(1):27
123. Giuffrida G, Lombardo R, Di Francesco E, Parrinello L, Di Raimondo F, Fiumara A. Successful switch from enzyme replacement therapy to miglustat in an adult patient with type 1 Gaucher disease: a case report. J Med Case Reports. 2016;10:315
124. Ceravolo F, Grisolia M, Sestito S, Falvo F, Moricca MT, Concolino D. Combination therapy in a patient with chronic neuronopathic Gaucher disease: a case report. J Med Case Reports. 2017;11(1):19
125. Sam R, Ryan E, Daykin E, Sidransky E. Current and emerging pharmacotherapy for Gaucher disease in pediatric populations. Expert Opin Pharmacother. 2021Aug;22(11):1489-503
126. Kleytman N, Ruan J, Ruan A, Zhang B, Murugesan V, Lin H. et al. Incremental biomarker and clinical outcomes after switch from enzyme therapy to eliglustat substrate reduction therapy in Gaucher disease. Mol Genet Metab Rep. 2021;29:100798
127. Cox TM, Drelichman G, Cravo R, Balwani M, Burrow TA, Martins AM. et al. Eliglustat compared with imiglucerase in patients with Gaucher's disease type 1 stabilised on enzyme replacement therapy: A phase 3, randomised, open-label, non-inferiority trial. The Lancet. 2015;385(9985):2355-62
128. Cox TM, Drelichman G, Cravo R, Balwani M, Burrow TA, Martins AM. et al. Eliglustat maintains long-term clinical stability in patients with Gaucher disease type 1 stabilized on enzyme therapy. Blood. 2017;129(17):2375-83
129. Parenti G, Andria G, Valenzano KJ. Pharmacological chaperone therapy: Preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Mol Ther. 2015;23(7):1138-48
130. Ivanova MM, Dao J, Kasaci N, Adewale B, Nazari S, Noll L. et al. Cellular and biochemical response to chaperone versus substrate reduction therapies in neuropathic Gaucher disease. PLoS ONE. 2021;16(10 October):1-22
131. Han TU, Sam R, Sidransky E. Small Molecule Chaperones for the Treatment of Gaucher Disease and GBA1-Associated Parkinson Disease. Front Cell Dev Biol. 2020 8(May)
132. Lin H, Sugimoto Y, Ohsaki Y, Ninomiya H, Oka A, Taniguchi M. et al. N-Octyl-β-valienamine up-regulates activity of F213I mutant β-glucosidase in cultured cells: A potential chemical chaperone therapy for Gaucher disease. Biochim Biophys Acta - Mol Basis Dis. 2004;1689(3):219-28
133. Jiang W, Yi M, Maegawa GHB, Zhang H. Ambroxol improves skeletal and hematological manifestations on a child with Gaucher disease. J Hum Genet. 2020;65(3):345-9
134. Zhang P, Zheng MF, Cui SY, Zhang W, Gao RP. Ambroxol Chaperone Therapy for Gaucher Disease Type I-Associated Liver Cirrhosis and Portal Hypertension: A Case Report. Endocr Metab Immune Disord - Drug Targets. 2022;22(6):658-62
135. Tang R, Xu Z. Gene therapy: a double-edged sword with great powers. Mol Cell Biochem. 2020;474(1-2):73-81
136. Anguela XM, High KA. Entering the modern era of gene therapy. Annu Rev Med. 2019;70:273-88
137. Zhang H, Yang B, Mu X, Ahmed SS, Su Q, He R. et al. Several rAAV vectors efficiently cross the blood-brain barrier and transduce neurons and astrocytes in the neonatal mouse central nervous system. Mol Ther. 2011;19(8):1440-8
138. Peters CW, Maguire CA, Hanlon KS. Delivering AAV to the central nervous and sensory systems. Trends Pharmacol Sci. 2021Jun;42(6):461-74
139. Maetzig T, Galla M, Baum C, Schambach A. Gammaretroviral vectors: Biology, technology and application. Viruses. 2011;3(6):677-713
140. Schambach A, Zychlinski D, Ehrnstroem B, Baum C. Biosafety features of lentiviral vectors. Hum Gene Ther. 2013;24(2):132-42
141. Bak RO, Gomez-Ospina N, Porteus MH. Gene Editing on Center Stage. Trends Genet. 2018;34(8):P600-611
142. Zhang D, Hussain A, Manghwar H, Xie K, Xie S, Zhao S. et al. Genome editing with the CRISPR-Cas system: an art, ethics and global regulatory perspective. Plant Biotechnol J. 2020;18(8):1651-69
143. Pavan E, Ormazabal M, Peruzzo P, Vaena E, Rozenfeld P, Dardis A. Crispr/cas9 editing for gaucher disease modelling. Int J Mol Sci. 2020;21(9):3268
144. Lam DK, Feliciano PR, Arif A, Bohnuud T, Fernandez TP, Gehrke JM. et al. Improved cytosine base editors generated from TadA variants. Nat Biotechnol. 2023;41:686-97
145. Li A, Mitsunobu H, Yoshioka S, Suzuki T, Kondo A, Nishida K. Cytosine base editing systems with minimized off-target effect and molecular size. Nat Commun. 2022;13:4531
146. Naphade S, Sharma J, Gaide Chevronnay HP, Shook MA, Yeagy BA, Rocca CJ. et al. Brief reports: Lysosomal cross-correction by hematopoietic stem cell-derived macrophages via tunneling nanotubes. Stem Cells. 2015;33(1):301-9
147. Ito S, Barrett AJ. Gauchers disease-A reappraisal of hematopoietic stem cell transplantation. Pediatr Hematol Oncol. 2013;30(2):61-70
148. Damase TR, Sukhovershin R, Boada C, Taraballi F, Pettigrew RI, Cooke JP. The Limitless Future of RNA Therapeutics. Front Bioeng Biotechnol. 2021;9:628137
149. Malone RW, Felgner PL, Verma IM. Cationic liposome-mediated RNA transfection. Proc Natl Acad Sci U S A. 1989;86:6077-81
150. Karikó K, Muramatsu H, Welsh FA, Ludwig J, Kato H, Akira S. et al. Incorporation of Pseudouridine Into mRNA Yields Superior Nonimmunogenic Vector With Increased Translational Capacity and Biological Stability. Am Soc Gene Ther. 2008;16(11):1833-40
151. Nance KD, Meier JL. Modifications in an Emergency: The Role of N1- Methylpseudouridine in COVID-19 Vaccines. ACS Cent Sci. 2021;7(5):748-56
152. Kowalski PS, Rudra A, Miao L, Anderson DG. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol Ther. 2019;27(4):710-28
153. Yang J, Jia C, Yang J. Designing Nanoparticle-based Drug Delivery Systems for Precision Medicine. Int J Med Sci. 2021;18(13):2943-9
154. Van Hoecke L, Roose K. How mRNA therapeutics are entering the monoclonal antibody field. J Transl Med. 2019;17(1):1-14
155. Magadum A, Kaur K, Zangi L. mRNA-Based Protein Replacement Therapy for the Heart. Mol Ther. 2019;27(4):785-93
156. Karadagi A, Cavedon AG, Zemack H, Nowak G, Eybye ME, Zhu X. et al. Systemic modified messenger RNA for replacement therapy in alpha 1-antitrypsin deficiency. Sci Rep. 2020;10(1):7052
157. Rohner E, Yang R, Foo KS, Goedel A, Chien KR. Unlocking the promise of mRNA therapeutics. Nat Biotechnol. 2022;40(11):1586-600
158. Vaidyanathan S, Azizian KT, Haque AKMA, Henderson JM, Hendel A, Shore S. et al. Uridine Depletion and Chemical Modification Increase Cas9 mRNA Activity and Reduce Immunogenicity without HPLC Purification. Mol Ther - Nucleic Acids. 2018;12(September):530-42
159. Anderson BR, Muramatsu H, Nallagatla SR, Bevilacqua PC, Sansing LH, Weissman D. et al. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res. 2010;38(17):5884-92
160. Andries O, Mc Cafferty S, De Smedt SC, Weiss R, Sanders NN, Kitada T. N1-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J Controlled Release. 2015;217:337-44
161. Vavilis T, Stamoula E, Ainatzoglou A, Sachinidis A, Lamprinou M, Dardalas I. et al. mRNA in the Context of Protein Replacement Therapy. Pharmaceutics. 2023;15(1):166
162. de Alwis R, Gan ES, Chen S, Leong YS, Tan HC, Zhang SL. et al. A single dose of self-transcribing and replicating RNA-based SARS-CoV-2 vaccine produces protective adaptive immunity in mice. Mol Ther. 2021;29(6):1970-83
163. Fenton C, Lamb YN. COVID-19: State of the Vaccination. Drugs Ther Perspect. 2021;37(11):508-18
164. Clinical Trials. Open-Label Study of mRNA-3927 in Participants With Propionic Acidemia. NCT0415910: November 12. 2019
165. Mitchell MJ, Billingsley MM, Haley RM, Langer R, Wechsler ME, Peppas NA. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov. 2021;20:101-24
166. Mistry PK, Lopez G, Schiffmann R, Barton NW, Weinreb NJ, Sidransky E. Gaucher disease: Progress and ongoing challenges. Mol Genet Metab. 2017Jan1;120(1):8-21
167. Enquist IB, Nilsson E, Ooka A, Månsson JE, Olsson K, Ehinger M. et al. Effective cell and gene therapy in a murine model of Gaucher disease. Proc Natl Acad Sci U S A. 2006;103(37):13819-24
168. Enquist IB, Bianco C Lo, Ooka A, Nilsson E, Månsson JE, Ehinger M. et al. Murine models of acute neuronopathic Gaucher disease. Proc Natl Acad Sci U S A. 2007;104(44):17483-8
169. Sinclair GB, Jevon G, Colobong KE, Randall DR, Choy FYM, Clarke LA. Generation of a conditional knockout of murine glucocerebrosidase: Utility for the study of Gaucher disease. Mol Genet Metab. 2007;90(2):148-56
170. Uemura N, Koike M, Ansai S, Kinoshita M, Ishikawa-Fujiwara T, Matsui H. et al. Viable Neuronopathic Gaucher Disease Model in Medaka (Oryzias latipes) Displays Axonal Accumulation of Alpha-Synuclein. PLoS Genet. 2015;11(4):1-23
171. Cabasso O, Paul S, Dorot O, Maor G, Krivoruk O, Pasmanik-Chor M. et al. Drosophila melanogaster mutated in its GBA1b ortholog recapitulates neuronopathic Gaucher disease. J Clin Med. 2019;8(9):1-23
172. Rohner E, Yang R, Foo KS, Goedel A, Chien KR. Unlocking the promise of mRNA therapeutics. Nat Biotechnol. 2022Nov;40(11):1586-600
173. Tenchov R, Bird R, Curtze AE, Zhou Q. Lipid Nanoparticles─From Liposomes to mRNA Vaccine Delivery, a Landscape of Research Diversity and Advancement. ACS Nano. 2021Nov23;15(11):16982-7015
174. Kristen A V, Ajroud-Driss S, Conceição I, Gorevic P, Kyriakides T, Obici L. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener Dis Manag. 2019;9(1):5-23
175. Kim DY, Atasheva S, McAuley AJ, Plante JA, Frolova EI, Beasley DWC. et al. Enhancement of protein expression by alphavirus replicons by designing self-replicating subgenomic RNAs. Proc Natl Acad Sci U S A. 2014;111(29):10708-13
176. Pepini T, Pulichino AM, Carsillo T, Carlson AL, Sari-Sarraf F, Ramsauer K. et al. Induction of an IFN-Mediated Antiviral Response by a Self-Amplifying RNA Vaccine: Implications for Vaccine Design. J Immunol. 2017;198(10):4012-24
177. Minnaert A Katrien, Vanluchene H, Verbeke R, Lentacker I, Smedt SC De, Raemdonck K. et al. Strategies for controlling the innate immune activity of conventional and self-amplifying mRNA therapeutics: Getting the message across. Adv Drug Deliv Rev. 2021;176:113900
178. Qin S, Tang X, Chen Y, Chen K, Fan N, Xiao W. et al. mRNA-based therapeutics: powerful and versatile tools to combat diseases. Signal Transduct Target Ther. 2022;7(1):166
179. Owji H, Nezafat N, Negahdaripour M, Hajiebrahimi A, Ghasemi Y. A comprehensive review of signal peptides: Structure, roles, and applications. Eur J Cell Biol. 2018;97(6):422-41
180. Goswami R, Chatzikleanthous D, Lou G, Giusti F, Bonci A, Taccone M. et al. Mannosylation of LNP Results in Improved Potency for Self-Amplifying RNA (SAM) Vaccines. ACS Infect Dis. 2019;5(9):1546-58
181. Goswami R, O'hagan DT, Adamo R, Baudner BC. Conjugation of mannans to enhance the potency of liposome nanoparticles for the delivery of RNA vaccines. Pharmaceutics. 2021;13(2):240
182. Pardridge WM. A historical review of brain drug delivery. Pharmaceutics. 2022;14(6):1283
183. Hauck ES, Hecker JG. Non-Viral Delivery of RNA Gene Therapy to the Central Nervous System. Pharmaceutics. 2022;14(1):165
184. Pardridge WM. Blood-Brain Barrier and Delivery of Protein and Gene Therapeutics to Brain. Front Aging Neurosci. 2020;11:373
185. Pardridge WM. Targeted Delivery of Protein and Gene Medicines Through the Blood-Brain Barrier. Clin Pharmacol Ther. 2015;97(4):347-61
186. European Medicines Agency. Moderna Assessment Report COVID-19 Vaccine Moderna.:EMA/15689/2021 Corr.1*1. 2021
187. Pardridge WM. Brain gene therapy with Trojan horse lipid nanoparticles. Trends Mol Med. 2023 xx(xx):1-11
188. Patil R, Sun T, Rashid MH, Israel LL, Ramesh A, Davani S. et al. Multifunctional nanopolymers for blood-brain barrier delivery and inhibition of glioblastoma growth through egfr/egfrviii, c-myc, and pd-1. Nanomaterials. 2021;11:2892
189. Sun Y, Liou B, Chu Z, Fannin V, Blackwood R, Peng Y. et al. Systemic enzyme delivery by blood-brain barrier-penetrating SapC-DOPS nanovesicles for treatment of neuronopathic Gaucher disease. EBioMedicine. 2020;55:102735
190. Kim J, Eygeris Y, Gupta M, Sahay G. Self-assembled mRNA vaccines. Adv Drug Deliv Rev. 2021;170:83-112
191. Blakney AK, McKay PF, Shattock RJ. Structural Components for Amplification of Positive and Negative Strand VEEV Splitzicons. Front Mol Biosci. 2018;5:71
192. Beissert T, Perkovic M, Vogel A, Erbar S, Walzer KC, Hempel T. et al. A Trans-amplifying RNA Vaccine Strategy for Induction of Potent Protective Immunity. Mol Ther. 2020;28(1):119-28
193. Maruggi G, Zhang C, Li J, Ulmer JB, Yu D. mRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol Ther. 2019;27(4):757-72
194. Ballesteros-Briones MC, Silva-Pilipich N, Herrador-Canete G, Vanrell L, Smerdou C. A new generation of vaccines based on alphavirus self-amplifying RNA. Curr Opin Virol. 2020;44:145-53
195. Ly HH, Daniel S, Soriano SK V, Blakney AK. Optimization of Lipid Nanoparticles for saRNA Expression and Cellular Activation Using a Design-of-Experiment Approach. Mol Pharm. 2022;19(6):1892-905
196. Atkinson AJ. Intracerebroventricular drug administration. Transl Clin Pharmacol. 2017Sep;25(3):117-24
197. Li MX, Weng JW, Ho E, Chow S, Tsang C. Brain delivering RNA-based therapeutic strategies by targeting mTOR pathway for axon regeneration after central nervous system injury. Neural Regen Res. 2022;17(10):2157-65
198. Schmidt C, Schnierle BS. Self-Amplifying RNA Vaccine Candidates: Alternative Platforms for mRNA Vaccine Development. Pathogens. 2023 13;12(1):138
199. Yuan M, Han Z, Liang Y, Sun Y, He B, Chen W. et al. mRNA nanodelivery systems: targeting strategies and administration routes. Biomater Res. 2023;27:90
200. Kim J, De Jesus O. Medication Routes of Administration. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 [cited. 2024 Feb 5]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK568677/
201. Pardi N, Parkhouse K, Kirkpatrick E, McMahon M, Zost SJ, Mui BL. et al. Nucleoside-modified mRNA immunization elicits influenza virus hemagglutinin stalk-specific antibodies. Nat Commun. 2018;9(1):3361
Author contact
![]() Corresponding authors: Dimitri Papukashvili: papukashvili.dimitriac.cn, Yang Ji: jiyedu.cn, and Peng George Wang: wangp6edu.cn.
Corresponding authors: Dimitri Papukashvili: papukashvili.dimitriac.cn, Yang Ji: jiyedu.cn, and Peng George Wang: wangp6edu.cn.