Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
1. Tumor immunotherapy...
2. Microbiota impact on cancer...
3 Impact of...
4. Conclusion and prospects
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(6):2264-2294. doi:10.7150/ijbs.91771 This issue Cite
Review
Influence of Microbiota on Tumor Immunotherapy
Xin Yu1,†, Wenge Li2,†, Zhi Li3,†, Qi Wu4,1 ![]() , Shengrong Sun1
, Shengrong Sun1 ![]()
1. Department of Breast and Thyroid Surgery, Renmin Hospital of Wuhan University, Wuhan, Hubei, P. R. China.
2. Department of Oncology, Shanghai Artemed Hospital, Shanghai, P. R. China.
3. Department of Orthopedics, Affiliated Provincial Hospital of Anhui Medical University, Hefei, Anhui, P. R. China.
4. Tongji University Cancer Center, Shanghai Tenth People's Hospital, School of Medicine, Tongji University, Shanghai, P. R. China.
†These authors have contributed equally to this work and share first authorship.
Received 2023-11-1; Accepted 2024-3-26; Published 2024-3-31
Abstract

The role of the microbiome in immunotherapy has recently garnered substantial attention, with molecular studies and clinical trials providing emerging evidence on the pivotal influence of the microbiota in enhancing therapeutic outcomes via immune response modulation. However, the impact of microbial communities can considerably vary across individuals and different immunotherapeutic approaches, posing prominent challenges in harnessing their potential. In this comprehensive review, we outline the current research applications in tumor immunotherapy and delve into the possible mechanisms through which immune function is influenced by microbial communities in various body sites, encompassing those in the gut, extraintestinal barrier, and intratumoral environment. Furthermore, we discuss the effects of diverse microbiome-based strategies, including probiotics, prebiotics, fecal microbiota transplantation, and the targeted modulation of specific microbial taxa, and antibiotic treatments on cancer immunotherapy. All these strategies potentially have a profound impact on immunotherapy and pave the way for personalized therapeutic approaches and predictive biomarkers.
Keywords: microbiota, immunotherapy, fecal microbiota transplantation, antibiotic
Introduction
Immunotherapy has ushered in a transformative era in oncological interventions, achieving significant advancements in this discipline [1]. Approaches such as anti-programmed cell death-1 (PD-1) antibodies [2] and chimeric antigen receptor (CAR)-T cell therapy[3] can rejuvenate the immune milieu and hinder the proliferation and dissemination of malignant cells. These therapies have been found to greatly enhance the survival rate and overall well-being of a substantial cohort of patients with cancer. However, patients undergoing immunotherapy often exhibit variable responses to such therapies, which can be partly attributed to the intricate interactions within the tumor immune microenvironment [1-3].
Researchers are increasingly acknowledging the pivotal role of the microbiota in regulating the immune system [4]. The human microbiota is a diverse ecosystem comprising varied microorganisms, including bacteria, fungi, and viruses, which predominantly inhabit body sites such as the gastrointestinal tract, skin, and oral cavity. These microbiota engage in complex and nuanced interactions with the immune system to potentially influence the nature and magnitude of immune responses, thereby leading to a profound impact on the effectiveness of tumor immunotherapy [4].
Moreover, this relationship between the microbiota and the immune system has prompted comprehensive exploration into microbiome-based strategies for optimizing the efficacy of tumor immunotherapy [5]. Various interventions, such as probiotics, prebiotics, fecal microbiota transplantation (FMT), and antibiotics, have been identified as potential tools for directly modulating the microbiome and its influence on immune responses [6-9]. Therefore, altering human microbiota composition via these interventions is a promising approach for regulating the complicated interactions within the tumor immune microenvironment and improving immunotherapy effectiveness.
This comprehensive review aims to elaborate on the numerous immunotherapy applications in oncology and explore the complex interplay between microbiota and tumor immunotherapy. Moreover, we attempt to examine the influence of microbiota on the immune system and discuss the potential associations of microbiome-based therapies and antibiotics with tumor immunotherapy. Lastly, based on our extensive investigations into these aspects, we hope to provide further insights to facilitate the development of personalized strategies for tumor immunotherapy, ultimately enhancing treatment outcomes for patients with cancer.
1. Tumor immunotherapy development and historical findings on the impact of bacteria on immunotherapy
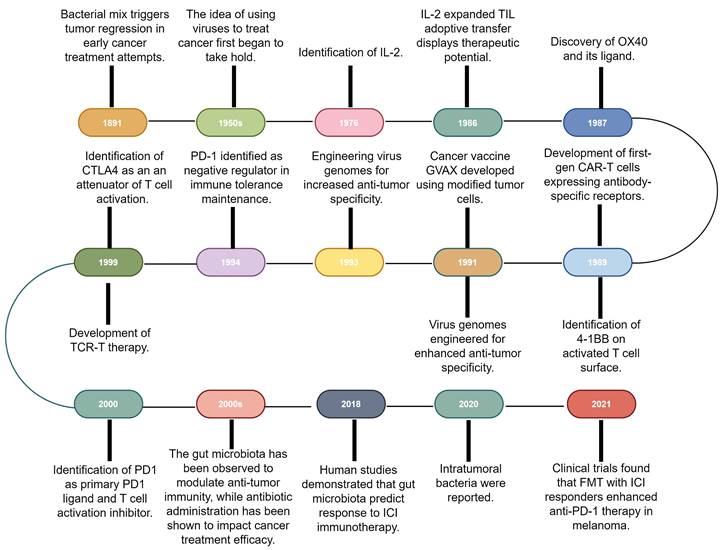
Immunotherapy, which primarily aims to enhance the innate immune mechanisms against malignant cells, is a groundbreaking advancement in cancer therapeutics and catalyzes a paradigm shift in oncology. Although the concept of leveraging the host immune system for eradicating cancer has existed for a century [10], remarkable progress and transformative developments in fundamental scientific investigations and clinical research endeavors have only been recently achieved [1]. This field has genuinely succeeded in only recent years via significant advancements across a spectrum of therapeutic techniques, including oncolytic viruses, cancer vaccines, cytokine therapy, adoptive cell therapy (ACT), immune checkpoint inhibitors (ICIs), and immune-agonists [11-15]. One intriguing aspect contributing to the progress in tumor immunotherapy is the historical findings related to the impact of bacteria on this therapeutic approach [16]. Recent research has shed light on the intricate interplay between bacteria and the immune system, highlighting their potential roles in modulating immunotherapy response [17,18]. These observations have added a new dimension to our understanding of immunotherapy, resulting in the influence of bacteria becoming increasingly recognized and integrated into the broader landscape of cancer treatment strategies (Fig. 1).
Research history of immunotherapy and the role of microbiota in immunotherapy. The timeline of immunotherapy advancements spans over a century, starting with William Coley's work in 1891. Milestones include IL-2 identification in 1976, OX40 receptor characterization in 1987, and first-gen CAR-T cells in the late 1980s. Other breakthroughs include virus genome engineering, MAGE-encoded antigen identification, CTLA4 discovery in the late 1990s, and PD-1/PD-L1 recognition as immune regulators. Gut microbiota's impact on anti-tumor immunity and antibiotic influence emerged, with 2018 studies linking microbiota diversity to ICI therapy response. In 2020, intratumoral bacteria presence was discovered, deepening knowledge of microbiota's role in cancer immunotherapy. 2021 trials showcased FMT's potential with anti-PD-1 therapy in melanoma patients. IL-2: Interleukin-2; CAR-T: Chimeric antigen receptor T-cell; CTLA4: Cytotoxic T-lymphocyte-associated antigen 4; FMT: Fecal microbiota transplantation; GVAX: GM-CSF gene transduced autologous tumor vaccine; ICI: Immune checkpoint inhibitor; PD-1: Programmed cell death-1; TCR-T: T cell receptor-engineered T cell; TIL: Tumor-infiltrating lymphocyte.
1.1 Oncolytic virus therapy
A cornerstone of the historical development of immunotherapy can be traced back to the pioneering observations of Virchow in 1863, who identified white blood cells of the immune system in tumor tissues, hinting at the intricate connection between inflammation and tumorigenesis [10]. Subsequently, the visionary William Coley initiated a remarkable research direction in 1891 by experimenting with a mixture of live and inactivated cultures of pyogenic streptococci and Bacillus [19]. This groundbreaking approach suggested the potential cancer-combating role of the immune system; however, the unknown mechanisms and inherent infection risks hampered the progress of this research perspective.
In the ensuing decades, oncolytic virus therapy has emerged as a beacon of hope. In this radical endeavor, a novel strategy harnessing the latent capabilities of meticulously designed genetically modified viruses that specifically targeted neoplastic cells was employed. This precise intervention initiates a pro-inflammatory microenvironment, which amplifies the systemic anti-tumor immune responses [20]. Recent advancements in genetic engineering and viral manipulation methodologies have further thrust oncolytic virus therapy into the forefront of cancer treatment research. One such example of this advancement is the application of talimogene laherparepvec, commonly referred to as T-VEC or Imlygic, an innovatively engineered herpes simplex virus [21]. T-VEC has demonstrated remarkable clinical efficacy, particularly in patients with advanced melanoma [21,22]. In 2015, the United States Food and Drug Administration (FDA) approved the use of T-VEC, representing a significant milestone in oncolytic virotherapy. This approval, which was provided to T-VEC for managing the challenging clinical condition of metastatic melanoma, was a pivotal moment. Moreover, T-VEC is currently the only oncolytic virus immunotherapy sanctioned by the FDA. This revolutionary therapy is a second-generation approach that utilizes the herpes simplex virus type 1 (HSV-1) for treating metastatic melanoma. T-VEC strategically includes granulocyte-macrophage colony-stimulating factor (GM-CSF), a cytokine that is critical in immune system modulation.
The other oncolytic virus candidates in clinical trials cover a broad spectrum of solid tumors [23,24]. Among them, coxsackievirus A21 is a naturally occurring virus that targets intercellular adhesion molecule-1. It exhibited well-tolerated characteristics when administered in conjunction with pembrolizumab in a phase Ib clinical trial. Furthermore, this combination treatment has been found to partially upregulate the expression of programmed cell death ligand-1 (PD-L1) on tumor cells [24]. Another virus candidate, DNX-2401, also known as Delta-24-RGD, is an engineered adenovirus tailored for preferential replication within cells exhibiting Rb defects. A phase I trial showed promising clinical responses in 20% of patients with recurrent malignant glioma following the intratumoral injection of DNX-2401[25]. The viruses predominantly utilized in current clinical trials for oncolytic virus-mediated tumor therapy encompass adenoviruses, HSV-1, and poxviruses, reflecting the growing knowledge of deoxyribonucleic acid (DNA)-based viral agents [26]. Additionally, a type 3 deattenuated strain of the orthoreovirus, which is from the family of non-enveloped double-stranded ribonucleic acid (RNA) viruses known as reoviruses, has been extensively investigated as an oncolytic agent [27].
Previous studies have also demonstrated that combining oncolytic viruses with conventional clinical anti-cancer therapies can enhance therapeutic efficacy. For example, integrating the measles virus with conventional chemotherapeutic agents, such as gemcitabine, has been reported to facilitate the lysis of senescent cancer cells across diverse tumor types [28,29]. A phase III clinical trial involving the synergistic administration of PD-1/PD-L1 blockade and TG4010 (an engineered vaccinia virus Ankara) revealed markedly enhanced treatment efficacy among patients with advanced cancer [30,31]. A study by Nishio et al. demonstrated that CAR-T cells armed with adenovirus expressing chemokine (C-C motif) ligand 5 (CCL5, better known as RANTES) and cytokine interleukin (IL)-15, specifically augmented CAR-T cell migration and proliferation in a xenotransplant human neuroblastoma murine model, ultimately leading to enhanced tumor-bearing mice survival [32].
1.2 Tumor vaccines
The elucidation of the immune system evasion mechanisms of cancer cells has resulted in the multifaceted advancement of cancer immunotherapy. This progress entails a broad spectrum of resources, including antibodies, peptides, proteins, nucleic acids, and immunocompetent cells, collectively contributing to the ongoing development of cancer immunotherapy [33]. In the case of cancer vaccines, vaccination strategies can be grouped into three major categories according to their format and composition, i.e., protein-based vaccines, cell-based vaccines (encompassing immune or tumor cells), and nucleic acid-based vaccines.
The domain of cancer vaccines has heralded a paradigm change by utilizing tumor-specific antigens to provoke T cell-mediated immune responses that target tumors. One such pioneering research has identified the potential application of MZ2-E and MZ2-D, derivatives of melanoma-associated antigen gene family-encoded antigens with specific relevance to melanoma. These antigens have been shown to trigger cytotoxic T cells, inciting robust anti-tumor immune responses [11,12]. Furthermore, the melanoma antigen gp100 has emerged as a pivotal factor in directing in vivo tumor rejection responses via immune reactions facilitated by tumor-infiltrating lymphocytes (TILs) in individuals with melanoma [34]. All these findings have laid the groundwork for establishing tumor antigens as the cornerstone of cancer immunotherapy.
Autologous tumor cell vaccines using cells are one of the vaccine strategies currently under evaluation. Of these approaches, dendritic cell (DC)-based vaccination has emerged as a potent and promising strategy. DCs are often considered the quintessential antigen-presenting cells (APCs) with a central role in the anti-tumor immune responses. After activation by tumor antigens, DCs swiftly engulf, process, and present the resulting epitopes to T cells, eliciting cytotoxic T lymphocyte (CTL) responses. The development of DC-based vaccines involves the isolation of DCs, pulsing them with tumor antigens or tumor cell lysates, and ex vivo stimulation using precisely defined maturation cocktails [35]. A prominent example of this approach is sipuleucel-T, a DC-based immunotherapy that has received approval for the treatment of advanced prostate cancer [36]. Furthermore, using whole tumor cells as a therapeutic modality has been gaining momentum. The introduction of GM-CSF gene transduced autologous tumor vaccine (GVAX), an immunotherapeutic vaccine designed from autologous tumor cells that have been genetically modified to produce GM-CSF, has demonstrated significant potential in enhancing tumor-specific immune responses in a diverse array of cancer malignancies [37-39]. All these advancements underscore the profound impact of tumor vaccines on the clinical landscape of cancer therapy.
DNA vaccines have also displayed promise in several initial studies [40,41]. Among them, VGX3100, a DNA vaccine for cervical cancer, is currently undergoing phase 3 clinical trials [42]. A total of 53 (49.5%) of 107 patients administered VGX-3100 exhibited histopathological regression. This outcome highlights the potential of VGX-3100 as a non-surgical therapeutic alternative for cervical intraepithelial neoplasia 2/3 (CIN2/3), potentially reshaping the treatment field for this prevalent ailment. In contrast to DNA vaccines, RNA vaccines do not integrate into the genome, mitigating concerns of carcinogenicity [43]. Additionally, RNA vaccines function within the cytoplasm, thereby bypassing the need for nuclear entry [44]. Consequently, the clearance of RNA vaccines is rapid, leading to minimal side effects. Although RNA is inherently less stable than DNA, various modifications, including formulations with liposomes or stabilizing adjuvants, can help enhance its stability [45-47]. Other techniques have also been devised to fortify the RNA molecule, such as incorporating a 5′ cap structure, untranslated regions, and optimized codon usage within translated regions [48]. Thus, continued advancement in nucleic acid delivery methods is a promising step toward the transformative development of the field of nucleic acid vaccines.
1.3 Cytokine therapy
Cytokines are versatile messengers within the complex immune network and are pivotal in modulating immune responses [49]. Of these, IL-2 is a critical cytokine, originally regarded as a T-cell growth factor [50]. IL-2 possesses a remarkable capacity for T-cell expansion in vitro and in vivo, manifesting potent immunostimulatory properties [51]. Furthermore, the clinical administration of high IL-2 doses has demonstrated compelling evidence of cancer regression in patients with metastatic malignancies [52,53].
Another prominent therapeutic cytokine in cancer treatment is interferon-alpha (IFN-α) [54]. This multifaceted type I IFN has a dual role in tumor control. The first role consists of the direct elimination of tumor cells via the induction of senescence and apoptosis, whereas the second one includes enhancing the effectiveness of anti-tumor immune responses by stimulating DC maturation and augmenting T-cell cytotoxicity [55]. Clinical investigations have also underscored the therapeutic efficacy of high-dose IFN-α in conditions such as chronic myeloid leukemia and melanoma [56,57].
Moreover, chemokine networks are often dysregulated in cancer, with chemokines being significantly involved in the neovascularization processes. Malignant cells also regularly exploit the chemotactic activity of chemokines [58]. Consequently, targeting specific chemokines or their tumor receptors has a solid preclinical rationale [49]. C-X-C chemokine receptor type 4 (CXCR4), a chemokine receptor overexpressed in >75% of cancers, is crucial for tumor cell proliferation, dissemination, and angiogenesis [59]. CXCR4 antagonists have demonstrated efficacy in restricting tumor growth in various experimental murine models. Plerixafor is one of the most common CXCR4 antagonists used in clinical applications. It has received approval for mobilizing hematopoietic stem cells, particularly in patients with non-Hodgkin lymphoma or multiple myeloma [60].
Although cytokine therapy has potential clinical value, its practical adoption as a standalone treatment has been impeded by challenges concerning its tolerability and severe toxicity [49]. Nevertheless, cytokines are still helpful when employed in conjunction with other immunotherapeutic approaches, particularly with ACT, effectively alleviating these obstacles.
1.4 ACT
ACT is a groundbreaking strategy that utilizes autologous immune cells, primarily T cells, natural killer (NK) cells, and macrophages. In this therapy, such cells are isolated, genetically modified, expanded ex vivo, and reintroduced into the patients. The primary objective of ACT is eliminating cancer cells and accomplishing sustained clinical effectiveness [13]. The employment of highly selective, tumor-reactive T cells has emerged as a transformative strategy, particularly in ACT for patients with metastatic melanoma presenting with the characteristic overexpression of endogenous differentiation antigens [61]. This approach has yielded sustained clonal expansion of T cells in patients with cancer. Moreover, the innovative use of genetically engineered and custom-designed T cells targeting novel antigens has gained considerable traction. Subsequently, two distinct categories of transgenic T cells with substantial contributions to the treatment of malignant tumors have been developed, namely CAR-T cells and T-cell receptor (TCR)-engineered T cells [62].
CAR-T cell therapy, which utilizes antibody fragments to precisely target the surface antigens of cancer cells, has undergone considerable development. The transition from first-generation CAR-T cells containing immunoglobulin-TCR chimeric receptors [63] to second-generation CARs incorporating co-stimulatory molecules such as the cluster of differentiation (CD) 28 marks a pivotal milestone in this progress [64]. This transformation has enabled modified T cells to exhibit prolonged in vivo persistence. Furthermore, researchers have explored the effectiveness of combining other molecules within CARs, demonstrating success in the gene modification of autologous T cells to express an anti-CD4 antibody connected to CD19-zeta and 19-3BB signaling domains. This combination strategy in CARs has effectively induced immune responses in patients with chronic lymphocytic leukemia [65,66]. All these findings stress the anti-tumor potential of CAR-T therapy across a range of human cancers.
T cells interact with peptides derived by means of a cell-surface receptor, the T-cell receptor (TCR), which is a disulfide-bonded heterodimeric protein composed of α and β chains. This receptor complex is augmented in functionality by associations with the CD3ε, γ, δ, and ζ subunits [67]. Upon encountering peptides that have been processed and displayed on major histocompatibility complex (MHC) molecules, TCRs initiate signaling events that lead to T-cell activation. In humans, these antigen-presenting MHC molecules are categorized into human leukocyte antigen (HLA) class I and HLA class II, with the former typically presenting peptides from cytoplasmic sources and the latter from extracellular compartments [68]. The engagement of the TCR with MHC molecules is facilitated by coreceptors CD8 and CD4, respectively, which are integral for enhancing the sensitivity of TCR-mediated antigen recognition [69]. Upon TCR binding to its cognate MHC, a cascade of intracellular events is triggered, including the phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) within the CD3 subunits. This phosphorylation is instrumental in activating the T cell, leading to a suite of effector responses, such as cell proliferation, cytokine production, and the execution of cytotoxic functions mediated by perforin and granzyme release [70]. The development of TCR-engineered T cells, commonly known as TCR-T cell therapy, can be attributed to the groundbreaking work by Clay et al. [71]. This pioneering approach involved the transfer of TCR genes into peripheral blood lymphocytes obtained from patients with melanoma, ultimately leading to the ex vivo generation of tumor-reactive effector T cells. Subsequently, clinical validation was quickly attained, wherein patients with metastatic melanoma experienced tumor regression following TCR-T cell therapy [72]. Moreover, transgenic TCR-T cells targeting the cancer-testis antigen NY-ESO-1 demonstrated sustained antigen-specific anti-tumor effects [73], eventually resulting in tumor regression. Hence, CAR-T cell and TCR-T cell therapies have made pronounced strides in cancer treatment, offering encouraging clinical outcomes [74,75]. CAR T-cell therapy requires the incorporation of autologous T cells to address concerns related to allogeneic activity and graft-versus-host disease. However, many patients exhibit diminished peripheral blood T-cell counts due to extended pre-CAR-T treatment regimens. This situation substantially delays therapy and can render CAR-T manufacturing impractical. Additionally, the CAR-T manufacturing process is protracted and intricate, leading to an increasing number of patients becoming ineligible or experiencing disease progression post-treatment. A prior study revealed that 22.5% (16/71) of patients failed to achieve the targeted CD3+ T-cell yield for CAR-T production via autologous lymphapheresis [76].
Considering that NK cells can activate autonomously (regardless of the MHC pathway) and exhibit diminished alloreactivity risks, the generation of CAR-NK cells does not require autologous NK cells. The available resources, such as NK92 cell lines, umbilical cord blood, and induced pluripotent stem cells (iPSCs), can be utilized for this purpose [77]. To date, most CARs employed in CAR-NK cell research have applied CAR architectures identical to those employed in CAR-T cells. These CAR structures typically contain either the same CD3ζ intracellular domain observed in first-generation CAR-T cells [78] or the CD3ξ co-stimulatory domain used in second-generation CAR-T cells, such as 4-1BB[79]. The inclusion of the 4-1BB co-stimulatory domain has been found to significantly enhance the activation and cytotoxic potential of NK cells as well as the secretion of key cytokines, including IFN-γ and GM-CSF. Li et al. conducted a comprehensive comparative analysis between a CAR-T cell construct and various CAR-NK cell variants to further refine the design of CAR-NK cells. Their results showed that CAR-NK cells had unique transmembrane and intracellular signaling domains that effectively targeted a specific antigen. Furthermore, CAR-NK cells with the NKG4D transmembrane domain, 2B4 co-stimulatory domain, and CD3ζ signaling domain demonstrated remarkable cytotoxicity that was robust and antigen-specific. CAR-NK cells derived from iPSCs harboring this specific construct also exhibited a characteristic NK cell phenotype, showing substantial anti-tumor activity and prolonged in vivo persistence [80].
More recently, CAR-macrophages have emerged as a highly promising and innovative therapeutic approach for cancer treatment [81]. Compared to CAR-T and CAR-NK cells, CAR-macrophages offer unique and distinct advantages for addressing two critical challenges of managing solid tumors. These problems pertain to the trafficking and infiltration of immune cells into the tumor microenvironment (TME) and the amelioration of the immunosuppressive conditions within the TME [82]. According to their functional properties and major activation states, macrophages can be classified into the following two groups: pro-inflammatory M1 and anti-inflammatory M2 macrophages. Among these, tumor-associated macrophages, particularly those of the M2 subtype, are widely acknowledged as a pivotal group of immunosuppressive cells within the TME [83]. Despite their immunosuppressive effects on other immune cells, M2 macrophages retain their phagocytic capabilities, exhibiting a higher phagocytic activity than M1 macrophages. Moreover, macrophages possess a significant degree of phenotypic plasticity, enabling them to adapt to environmental cues and modify their phenotypes [84]. CAR constructs employed in CAR-engineered macrophages display structural components similar to those observed in CAR-T cells. These structures consist of an extracellular antigen-binding domain, hinge region, transmembrane domain, and intracellular domain. Nevertheless, one distinctive feature of CAR-engineered macrophages is the composition of their intracellular signaling domains. Similar to CAR-T cells, CAR-macrophages utilize the CD3ζ intracellular domain, which incorporates ITAMs derived from immune receptor tyrosine kinases of the Src family [85-87]. In CAR-T cells, the ITAMs undergo phosphorylation after CAR engagement by Src family kinases and subsequently interact with the tSH70 domain in zeta-chain-associated protein kinase 2 (ZAP2), culminating in CAR-T cell activation for cytotoxic function. In contrast, macrophages lack ZAP2 expression. Thus, they express the kinase Syk, which houses a tSH3 domain that can bind to CD100ζ, ultimately mediating phagocytic signaling in the macrophages [88]. Currently, only one phase I clinical trial of CAR-macrophages has been officially registered (ClinicalTrials.gov identifier: NCT04660929). This clinical trial involves the CAR-macrophages originally developed by Klichinsky et al. In that approach, the researchers modified CAR-macrophages by integrating a hybrid adenovirus vector Ad5f35 and a specific targeting mechanism utilizing single-chain variable fragments against human epidermal growth factor receptor 2. The adenoviral infection triggers macrophage polarization toward a pro-inflammatory M1-like phenotype. However, no outcomes or findings of this clinical trial have been released since its initiation in 2021. Simultaneously, a second study involving CAR macrophages has also been registered (ClinicalTrials.gov identifier: NCT05007379). This observational study is designed to evaluate the anti-tumor activity of CAR-macrophages derived from the organotypic cultures of patients.
1.5 ICIs
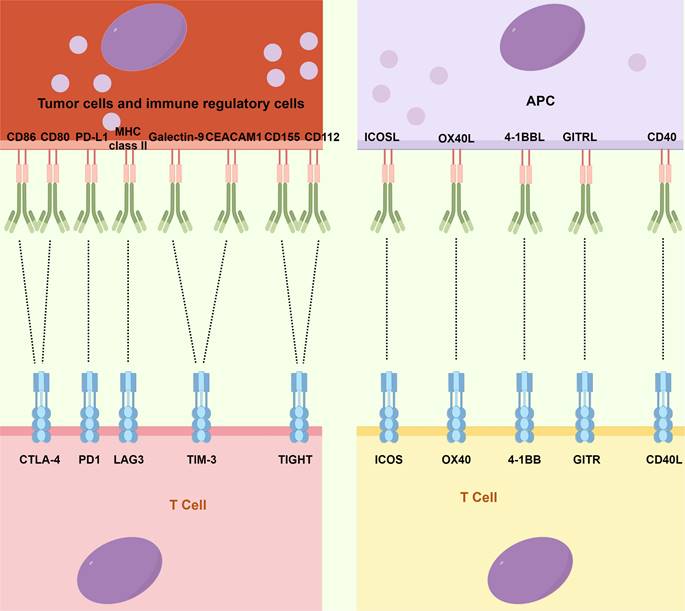
Although ACT has made significant advancements, the emergence of ICIs, a novel category of monoclonal antibodies, has taken center stage in the field of immunotherapy and has assumed a pivotal role in therapeutic interventions [14]. Immune checkpoints, which constitute molecules participating in co-inhibitory signaling pathways, are critical in maintaining immune tolerance. However, these checkpoints are frequently co-opted by cancer cells to evade immune surveillance (Fig. 2). ICIs are specifically engineered to reinvigorate anti-tumor immune responses by disrupting these co-inhibitory signaling pathways, eventually facilitating the immune-mediated elimination of malignant cells [89,90]. The primary targets of ICIs include cytotoxic T-lymphocyte-associated antigen 4 (CTLA4), PD-1, PD-L1, lymphocyte-activation gene 3 (LAG3), T-cell immunoglobulin and mucin domain-containing protein 3 (TIM3), and T-cell immunoglobulin and ITIM domain (TIGIT).
The primary immune checkpoints and immune-agonists site on cancer cells and immune cells. CEACAM1: Carcinoembryonic antigen cell adhesion molecule 1; CTLA4: Cytotoxic T-lymphocyte-associated antigen 4; GITR: Glucocorticoid-induced TNF receptor; ICOS: Inducible T-cell costimulator; LAG3: Lymphocyte-activation gene 3; MHC: Major histocompatibility complex; PD-1: Programmed cell death-1; PD-L1: Programmed cell death ligand-1; TIGHT: T-cell immunoglobulin and ITIM domain; TIM3: T-cell immunoglobulin and mucin domain-containing protein 3.
CTLA4 is an inhibitory molecule expressed on T cells and is crucial in modulating T-cell activation following the interaction of the MHC with the TCR [91,92]. The primary function of CTLA4 is to preserve immune homeostasis by negatively regulating T-cell activation [93]. This inhibitory activity of CTLA4 is attributed to its ability to outcompete the co-stimulatory receptor CD28 for binding to the B7 co-stimulatory ligands, CD80 and CD86, via its relatively higher binding affinity and avidity [94]. Consequently, CTLA4 effectively dampens T-cell activation. Additionally, CTLA4 can reduce the surface expression of CD80 and CD86 on APCs via trans-endocytosis, thereby curtailing cytotoxic T-cell activation [95]. The TME of various cancers has been shown to accumulate CD4+CD25+Foxp3+ regulatory T cells (Tregs), which also express elevated levels of CTLA4 [96]. The stabilization of CTLA4 expression by Foxp3, in turn, amplifies the attenuation of T-cell response in the context of cancer [97]. Seminal research has further demonstrated that the antibody blocking of CTLA4 can evoke robust immune responses, leading to tumor regression [98]. Subsequent clinical trials and efficacy assessments resulted in ipilimumab, a CTLA4-targeting monoclonal antibody, receiving approval as the first ICI for cancer treatment [99,100].
PD-1, originally linked to programmed cell death, is an immune checkpoint expressed on the T-cell surface [101]. The mechanisms underlying PD-1 regulation remained unclear until the discovery of its ligand, PD-L1. This ligand is not only expressed in normal tissues but also exploited by tumor cells to evade immune surveillance [102]. Moreover, the classical two-signal hypothesis has underscored the necessity of proper T-cell activation, which requires TCR and MHC engagement and ensuing co-stimulatory signals mediated by CD28 via the B7 ligands [103]. Previous research has demonstrated that blocking the PD-1/PD-L1 pathway can rejuvenate the cytotoxic potential of T cells and elicit tumor regression, thus establishing PD-1 and PD-L1 as promising therapeutic targets [104,105]. After the groundbreaking approval of pembrolizumab for advanced melanoma treatment in 2014, the clinical utility of PD-1/PD-L1 inhibitors has expanded to various cancer types, including head and neck squamous cell carcinoma [106-108], non-small cell lung cancer (NSCLC) [109], and renal cell carcinoma (RCC) [110]. The widespread use of ICIs has also uncovered a range of independent immune-related adverse events. Some patients experience severe immune-related side effects, leading to complications such as pneumonia, myocarditis, or hepatitis [111,112]. Additionally, a substantial proportion of patients exhibit primary resistance that arises from diverse mechanisms, including the inherent resistance of tumor cells to T-cell engagement, limited tumor immunogenicity, impaired CD8+ T-cell migration to the tumor site, and immune inhibitory factors, such as myeloid-derived suppressor cells (MDSCs) and Tregs, within the TME [2,111].
The LAG3 gene is expressed by T and NK cells after the ligation of MHC class II molecules [113]. Although the precise mechanism remains elusive, LAG3 is suggested to be pivotal in negatively regulating T-cell function, thus protecting against tissue damage and autoimmunity. LAG3 is often co-expressed and upregulated on TILs along with PD-1, contributing to immune exhaustion and facilitating tumor growth [114]. Consequently, LAG3 blockade was found to enhance anti-tumor immunity and complement other immunotherapeutic approaches via a distinct mechanism involving the inhibition of cell cycle progression [115]. Although the concurrent administration of anti-LAG3 and anti-PD-1 therapies are considered synergistic, the effectiveness of combining anti-LAG3 therapy with other ICIs is still uncertain [116]. Further, the clinical benefits of such combination therapies are potentially accompanied by an increased occurrence of autoimmune toxicities [117].
TIM3 is a crucial direct negative regulator of T cells and is also present in NK cells and macrophages [118]. This receptor influences the immunosuppressive milieu indirectly, chiefly by promoting MDSC expansion [119]. The heightened TIM3 levels are closely correlated with T-cell dysfunction and exhaustion, emphasizing its pivotal role in various malignancies. In particular, the presence of TIM3-expressing T cells in NSCLC and follicular lymphoma is closely associated with disease severity and an unfavorable prognosis [120]. Conversely, diminished TIM3 levels have been linked to autoimmune conditions, such as diabetes and multiple sclerosis [119]. The intervention of TIM3 blockade via monoclonal antibodies may enhance T-cell proliferation and cytokine production, thereby indicating its potential anti-tumor efficacy and simultaneous risk of exacerbating autoimmune diseases. Therefore, the application of these antibodies is concerning because certain acute infections, including Listeria infections, may be aggravated by TIM3-mediated CD8+ T-cell enhancement [121]. Additionally, several ligands, such as galectin-9, phosphatidylserine, and carcinoembryonic antigen cell adhesion molecule 1, have been reported to modulate the TIM3 pathway [120]. These ligands are vital for carcinogenesis, tumor survival, and the progression of various malignancies, including melanoma, gastrointestinal cancer, and lung cancer [122-124]. In contrast to other immune inhibitory pathways that interfere with cellular function, TIM3 mainly modulates cell apoptosis. This feature may explain its augmented effects when combined with other ICIs [118]. However, the optimal molecule that can be combined with TIM3 is yet to be identified.
TIGIT is a CD28 family-like receptor expressed on NK and T cells. This receptor directly suppresses immune responses and indirectly regulates the release of immunoregulatory cytokines, including IL-10. TIGIT also diminishes IFN-γ and IL-17 production and impedes DC maturation [125]. The two agonists of TIGIT, CD155 (also known as poliovirus receptor [PVR]) and CD112 (alternatively termed as PVRL2 or nectin-2), are widely expressed by immune, non-immune, and tumor cells, including melanoma cells [126]. TILs often co-express high levels of TIGIT as well as PD-1, TIM3, and LAG3, indicating a dysfunctional phenotype [127]. Preliminary studies have shown that simultaneously blocking TIGIT and either PD-1 or TIM3 promotes immune cell proliferation, cytokine release, and degranulation and reverses T-cell exhaustion, which in turn results in tumor rejection and protective memory responses [120,128]. Moreover, TIGIT expression is more prominent in the TME than in peripheral cells, thereby allowing more precise and less toxic therapy. Additionally, TIGIT mainly alters cytokine production and CD8 T-cell function, suggesting its complementary role with other ICIs [129].
1.6 Immune agonists
Checkpoint inhibitors have led to notable success in cancer treatment. However, over 80% of patients do not achieve a favorable response after ICI treatment or develop resistance over time. Consequently, research efforts have noticeably shifted toward enhancing anti-tumor activity by manipulating signaling pathways with agonistic antibodies targeting specific molecules to augment anti-tumor T-cell responses [15]. The comprehensive activation of T cells requires a trio of signals, including TCR signaling, co-stimulatory signaling, and cytokine support [130]. TCR signaling primarily revolves around the recognition of neoantigens, which exhibit distinct expression patterns exclusive to tumor cells. These neoantigens originate from genetic mutations within tumor cells, resulting in peptide epitopes distinct from those originating from the standard human genome [131]. The neoantigen peptide-MHCs are then conspicuously displayed on the surfaces of tumor cells and APCs, providing a strong target for the TCRs on antigen-specific T cells [132]. Furthermore, interventions focused on regulating TCR signaling, such as the pioneering approach of CAR-T therapy, have already proven their worth in clinical applications [133].
Various co-stimulatory pathways are involved in T-cell activation [134]. One key co-stimulatory cascade that significantly contributes to T-cell activation and cytokine release is the CD80/CD86-CD28 signaling pathway [135]. Conversely, T-cell suppression occurs due to the relatively higher binding affinity of CTLA4 for CD80/CD86 [136]. Another inducible co-stimulatory receptor expressed on activated T cells is the inducible T-cell co-stimulator (ICOS), which interacts with the ICOS ligand [137]. Apart from these co-stimulatory receptors, receptors such as OX40, 4-1BB, glucocorticoid-induced tumor necrosis factor (TNF) receptor (GITR), and other TNF superfamily members can synergize with TCR signaling, ultimately enhancing T-cell responses and survival. Additionally, alternative mechanisms for boosting the anti-tumor responses of T cells involve the co-stimulatory receptors, such as CD40, on APCs [138].
ICOS, a member of the CD28 superfamily, undergoes rapid induction following T-cell activation to deliver secondary co-stimulatory signals [139,140]. Moreover, its ligand, known as the ICOS ligand, is predominantly expressed on B cells, macrophages, and DCs. The ICOS ligand-ICOS signaling pathway moderately facilitates T-cell proliferation, augments the production of cytokines (including IL-4, IL-5, IL-10, IFN-γ, and TNF-α), initiates Foxp3 transcription, and represses Tregs [141]. Although the humanized ICOS agonist JTX2011 has delivered encouraging results in murine cancer models, it has shown limited efficacy when administered as monotherapy in clinical trials [142,143]. Nevertheless, its use in conjunction with other checkpoint inhibitors has yielded slightly more promising results.
OX40 (also known as CD134) is a member of the TNFRSF4 family and is predominantly expressed on activated CD4/CD8+ T cells and Foxp3CD4 Tregs, especially within the intratumoral Treg population. Moreover, OX40 expression is transient, peaking at 24-48 h post-activation and persisting for 3-4 days [144]. The ligand of OX40, CD252 (also designated as OX40 ligand [OX40L]), is found on activated APCs, including DCs, B cells, and macrophages [144,145]. The OX40-OX40L signaling pathway enhances effector T-cell expansion and survival, eventually promoting memory T cells and inhibiting Tregs [146]. Prior clinical trials have demonstrated that combining OX40 agonists with PD-1 blockade improves treatment outcomes in murine models; however, the results in patients were less favorable [147].
The co-stimulatory receptor 4-1BB is expressed on CD4+ and CD8+ T cells in murine models, whereas its expression in humans is predominantly in activated CD8+ T cells, NK cells, NKT cells, Tregs, DCs, and various myeloid cells. The cognate ligand, 4-1BBL, shows inducible expression patterns on activated APCs, myeloid progenitors, and hematopoietic stem cells. After its activation, 4-1BB engages with CD137L to initiate the activation of multiple signaling pathways, including nuclear factor κappa-B (NF-κB), extracellular signal-regulated protein kinases, c-Jun N-terminal kinase, and mitogen-activated protein kinase pathways, culminating in increased cytotoxic T-cell activity [148]. Nevertheless, the role of 4-1BB in the regulation of Tregs is complex and context-dependent. Multiple 4-1BB-targeting agonists have been developed, which have the potential to enhance T-cell persistence, functionality, and expansion [149]. Clinical trials have also explored the combination of 4-1BB agonists with immunotherapy or chemotherapy and yielded diverse outcomes, encompassing heightened anti-tumor efficacy and improved survival rates in select cases [150-152].
GITR (also termed CD357) is a member of the TNF receptor superfamily 18 and exhibits robust expression on Tregs, thus wielding a significant influence on their expansion and differentiation [153]. GITR is expressed at lower levels on naive and memory T cells [154], while its ligand, the GITR ligand, has limited expression in APCs, including DCs, macrophages, and B cells. GITR is also pivotal in enhancing T-lymphocyte activity via the upregulation of IL-2 and IFN-γ and the concurrent inhibition of TCR activation-induced apoptosis, thereby aiding T-cell survival [155-157]. Preclinical investigations involving GITR agonists in murine models have showcased their effectiveness in promoting the activation of CD4+ and CD8+ T cells and the suppression of intratumoral Tregs [158]. Early-phase clinical trials centered on GITR agonists have also reported favorable safety profiles, with certain patients displaying sustained disease stability [159].
CD40, a constituent of the TNFR superfamily 5, is expressed on various immune cell types, including DCs, B cells, monocytes, and vascular/epithelial cells. Additionally, its ligand, CD40L (also known as CD154), is transitorily expressed on activated T cells [160]. CD40 signaling in DCs is crucial in promoting the release of cytokines, upregulation of co-stimulatory molecules, cross-presentation of antigens, and modulation of pro- and anti-apoptotic gene expression [161]. CD40 ligation can also induce apoptosis in some malignancies [162], while CD40-activated macrophages have been shown to exhibit tumoricidal activity, deplete tumor stroma, and induce tumor regression, all independently of T cells [163]. Clinical trials investigating CD40 agonists have demonstrated good safety and objective responses in some patients, with combination therapies incorporating CD40 agonists also showing promise in overcoming immune refractoriness [164-166].
1.7 Bacterial insights in immunotherapy history
In the first decade of the 21st century, a notable advancement was achieved in understanding the modulation of CD8 T-cell activity by the gut microbiota and the consequential stimulation of anti-tumor immune responses [167]. Furthermore, the efficacy of cancer treatments was reported to be compromised in the presence of antibiotics or germ-free mouse models, with these observations being contingent upon the specific species within the gut microbial community. In a seminal investigation in 2015, a groundbreaking correlation was established between the gut microbiota and ICI responses in murine models [168,169]. The research underscored the impact of gut microbiota composition on anti-PD-L1 therapy response and showed that FMT could mitigate the response discrepancies. Moreover, the oral administration of Bifidobacterium was revealed to facilitate DC maturation, thereby enhancing the initiation and accumulation of CD8 T cells in the TME. Conversely, this restoration of anti-tumor efficacy was impaired by PD-L1 blockade [168].
Parallel studies of anti-CTLA4 therapy have indicated that antibiotics suppress the anti-tumor effects produced by ICIs. In contrast, supplementation with susceptible Bacteroides fragilis in germ-free or antibiotic-treated melanoma mice accentuated the therapeutic effects of anti-CTLA4. Furthermore, the microbiota-dependent anti-tumor effects were found to be intricately linked to the induction of Th1 cell activation in tumor-draining lymph nodes (LNs) and the maturation of intratumoral DCs [169]. A series of human studies published in 2018 collectively proposed that the composition and diversity of the gut microbiota might serve as predictive indicators of ICI immunotherapy responses [170,171].
In line with this notion, FMT from ICI-responsive patients to germ-free or antibiotic-treated mice ameliorated tumor control and heightened ICI responses, whereas FMT from non-responders failed to yield these beneficial effects [163,164]. Patients with NSCLC or RCC who exhibited elevated gut bacterial diversity also demonstrated heightened sensitivity to anti-PD-1 therapy [170]. Similarly, an investigation involving patients with metastatic melanoma detected elevated baseline levels of Bifidobacterium longum, Collinsella aerofaciens, and Faecalibacterium prausnitzii in the feces of responder patients [164]. Prospective studies conducted between 2019 and 2020 have further confirmed the noteworthy correlation between the gut microbiota of patients with NSCLC, hepatocellular carcinoma, or RCC and ICI outcomes [172-175]. In 2020, two extensive studies examined the diverse intratumoral microbiota across more than 30 cancer types. Poore et al. researched the varied microbial communities within tumors and proposed a novel diagnostic tool based on the cancer-associated microbiota [176]. Subsequently, Ravid et al. comprehensively analyzed seven tumor microbiome profiles and presented imaging evidence of the spatial distribution and intracellular localization of these microbial communities within tumors [177]. A surprising finding from a 2021 clinical trial indicated that FMT from ICI responders coupled with anti-PD-1 therapy enabled patients with melanoma to overcome their resistance to PD-1 blockade therapy [8]. Therefore, a growing body of evidence in the current landscape of tumor immunotherapy research underscores the significance of the complex and diverse gut microbiota in modulating immune-based treatment outcomes.
2. Microbiota impact on cancer immunity
The human microbial community primarily resides within the gastrointestinal tract, followed by other physiological interfaces between the human body and the external environment. These microbial communities predominantly orchestrate diverse regulatory effects on various host functions via immunomodulation [178]. Additionally, recent research findings underline the substantial contribution of intratumoral microbiota to cancer immunodynamics [179]. Microbes residing at distinct anatomical locations can also have varying impacts on systemic immunity and the immune milieu within tumors. Thus, a comprehensive exploration of this complex interplay between the microbial community and immune responses continues to be a focal point of contemporary research.
2.1 The intricate relationship between gut microbiota and tumors
2.1.1 Influence of gut microbiota on adaptive immune responses
The emerging body of evidence has emphasized the complicated connections between anti-cancer therapies and specific commensal-driven immune responses, thereby contributing to a deeper understanding of their mechanistic foundations (Fig. 3, Table 1) [180,181]. Some notable associations include the promotion of Enterococcus gallinarum translocation following cyclophosphamide treatment, which results in pathogenic T helper cell (Th)17 responses and the production of IFN-producing CD8 T effector cells. This coordinated immune response effectively restrains tumor growth in sarcoma and lung adenocarcinoma models [182,183]. Another important connection is the increased presence of B. fragilis and Bifidobacterium species in the fecal samples of patients with melanoma who received CTLA4 blockade. Subsequently, this enrichment activates toll-like receptor 4 (TLR4) and IL-12-dependent Th1 responses, contributing to therapeutic effectiveness [169]. Studies have also detected a prominent relationship, wherein the effectiveness of PD-L1 inhibition in eliciting T-cell responses against melanoma is significantly enhanced in hosts harboring Bifidobacterium species within their microbiota [168,171]. Lastly, oxaliplatin-induced ileal enterocyte death has been demonstrated to be a pivotal factor regulating the balance between anti-tumor follicular T helper (TFH) cells and potentially deleterious Th17 cells in colon cancer. This balance is finely modulated by the ratio of immunogenic Enterococcaceae to tolerogenic Lactobacillaceae [184].
Mechanistic insights into the impact of gut microbiota on tumor immunity. Gut microbiota play a pivotal role in modulating adaptive immune responses through DCs present in the gut-associated lymphoid tissues, spleen, or tumor-draining lymph nodes. Additionally, metabolites produced by gut microbiota, such as butyrate and propionate, exert effects on regulating immune cell activation and resistance to certain immunotherapies. Furthermore, the gut microbial community impacts the systemic immune response by influencing bone marrow cellularity and thymic cellularity. Moreover, the intricate effects of the gut microbiome extend to its influence on the local and distant tumor microenvironment, orchestrating a complex interplay. DC: Dendritic cell; GPR109A: G-coupled receptor-109a; NK: Natural killer; OMV: Outer membrane vesicles.
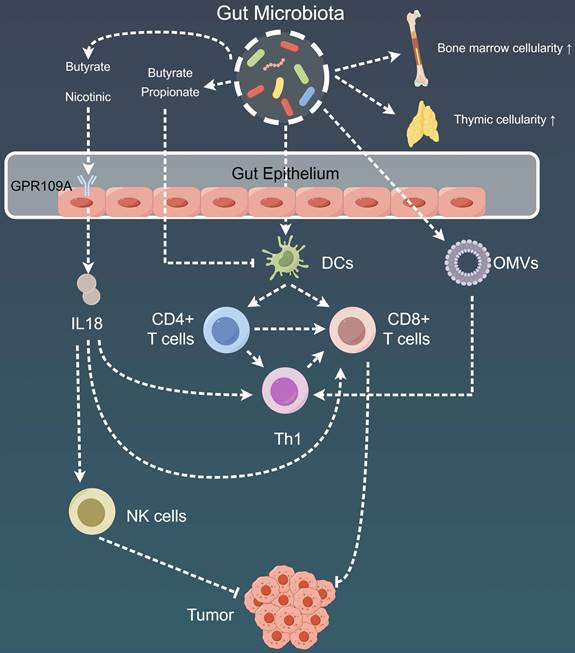
The intricate relationship between gut microbiota and tumors.
| Gut Microbiota | Model | Mechanism |
|---|---|---|
| Enterococcus hirae; Bacteroides intestinihominis | C57BL / 6J mice, MCA205 sarcoma cell, Ret melanoma cell | Enterococcus hirae increased the intratumoral CD8/Treg ratio and Bacteroides intestinihominis promoted the infiltration of IFN-γ-producing γδT cells in cancer lesions [183]. |
| Bacteroides thetaiotaomicron; Bacteroides fragilis | C57BL/6J and BALB/c mice, mouse fibrosarcoma MCA205 cell, murine colon carcinoma MC38 cell, RET melanoma cell, mouse colon carcinoma CT26 cell | The efficacy of CTLA4 blockade is influenced by the microbiota composition. The microbiota composition affects IL-12-dependent TH1 immune responses [169]. |
| Bifidobacterium spp. | C57BL/6 mice, melanoma cell line B16 F10, MB49 bladder cancer cell | Commensal Bifidobacterium-derived signals modulated the activation of DCs in the steady state, which in turn supports improved effector function of tumor-specific CD8+ T cells [168]. |
| Bifidobacterium breve | C57BL/6 mice, 2C TCR transgenic mice, melanoma cell line B16 F10, murine T-cell lymphoma RMA-S | Commensal bacteria can stimulate antitumor immune responses via cross-reactivity and bacterial antigens affect the T cell landscape [193]. |
| Bacteroides fragilis; Erysipelotrichaceae | C57BL/6J and BALB/c mice, mouse colon cancer cell MC38, mouse fibrosarcoma cell line MCA205, mouse colon cancer cell CT26, mouse breast cancer cell 4T1 cell | Ileal microbiota dictates tolerogenic versus immunogenic cell death of ileal intestinal epithelial cells (IECs) and the accumulation of TFH cells in patients [184]. |
| A consortium of 11 bacterial strains | C57BL/6, BALB/c and IQI mice, MC38 adenocarcinoma cell | 11 healthy human-associated bacterial strains act together to induce IFN-γ CD8 T cells, confer resistance to the intracellular pathogen Listeria, and are effective at inhibiting tumour growth in conjunction with ICIs [194]. |
| Bacteroides pseudolongum | C57BL/6J, B6(Cg), Zbtb46tm1(HBEGF)Mnz/J, 129S-Adora2atm1Jfc/J mice, MC38 adenocarcinoma cell, MB49 bladder cancer cell | Intestinal Bacteroides pseudolongum activated antitumor T cells through production of the metabolite inosine [212]. |
| Bacteroides spp. | C57BL/6 mice, murine colon carcinoma MC38 cell, melanoma cell YUMM1.5 | Changes in gut microbiota resulted in enhanced expression of select chemokines and cytokines, which have known roles in the activation of DCs and T cells [199]. |
| Bifidobacterium | C57BL/6 and Balb/c mice, murine colon adenocarcinoma cell MC38, murine T cell lymphoma cell EG7. | Bifidobacterium potently stimulates STING signaling and increases cross-priming of dendritic cells after anti-CD47 treatment [186]. |
Moreover, in most of these models, DCs derived from gut-associated lymphoid tissues (GALTs), spleen, or tumor-draining LNs played a critical role as intermediaries, with the DCs actively sensing various commensal microorganisms, including Bacteroides, B. fragilis, Mucispirillum, Ruminococcaceae, and Rodentibacter. These interactions further triggered immune responses via the IFN-I and IL-12-mediated pathways [168,169,185-187]. In addition to their role as DC adjuvants, the gut microbes are a rich source of antigens capable of eliciting commensal-specific T-cell responses on a systemic level. These responses can either exert detrimental or protective effects on the host, depending on the specific peptides involved.
A study by Gil-Cruz et al. elucidated how shared structural features between a β-galactosidase enzyme derived from Bacteroides thetaiotaomicron and host cardiac myosin heavy chain-6 could instigate a devastating autoimmune myocarditis condition [188]. In contrast, Nanjundappa et al. indicated that cross-reactivity between an integrase enzyme obtained from Prevotella and the host islet-specific glucose-6-phosphatase catalytic subunit-related protein could redirect autoreactive CD8 T cells, thus ameliorating colitis [189].
Recent investigations have improved our understanding of cross-reactive homologs and enabled the inclusion of exogenous dietary antigens, particularly in the case of human leukocyte antigen-DQ2.5-mediated celiac disease [190]. Molecular simulation studies have offered insights into the interplay between cancer and microbial antigens [191,192], wherein the concept of molecular mimicry between cancer and microbial antigens has been explored in-depth. Specifically, immune responses mediated by H-2Kb-restricted T cells against a spectrum of enterococcal bacteriophages exhibited cross-reactivity with an oncogenic driver factor, the proteasome 20S subunit beta 4 (PSMB4). Oral administration of bacteria carrying these bacteriophages led to enhanced bacteriophage-specific T-cell responses during cyclophosphamide or anti-PD4 antibody treatment, effectively countering extraintestinal tumors overexpressing PSMB4[192]. Correspondingly, T cells targeting a specific peptide expressed in the commensal bacterium Bifidobacterium showed a cross-reaction with a model neoantigen expressed in mouse melanoma B16-SIY [193]. Furthermore, some human T cells with specificity for naturally processed melanoma epitopes showed recognition of microbial peptides, implying its clinical relevance [192].
Nevertheless, mechanisms other than molecular mimicry may also be involved in enhancing anti-tumor immunity. For instance, Tanoue et al. revealed an interesting consortium of 11 bacteria that could promote tumor antigen-specific CD8 IFN-γ T-cell responses during ICI. Moreover, these responses demonstrated no cross-reactivity with microbial antigens and did not originate from the colon. All these findings collectively highlight the intricate interplay between the gut microbiota and host immune responses in the context of cancer, thereby providing novel perspectives on potential avenues for therapeutic intervention [194].
Tumor irradiation has been proven to be significantly boosted in the presence of vancomycin, which eradicates the immunosuppressive metabolites, especially butyrate and propionate, originating from Clostridia [185,195]. This elimination process is suggested to function via the elevated presentation of DC antigen and the concurrent priming of CD8 T cells. Conversely, metabolites derived from the gut microbiota, such as propionate, and those associated with the tryptophan pathway (+1H-indole-3-aldehyde and quinolinic acid), have been shown to confer long-term protection against radiation in vivo [196].
Moreover, increased butyrate and propionate concentrations have been linked to CTLA4 blockade resistance in murine models and patients with melanoma. This resistance is accompanied by an increased proportion of Tregs, diminished activation of DCs and effector T cells, and weakened responses to IL-2[197]. These metabolites have also been found to correlate with extended progression-free survival (PFS) after anti-PD-1 therapy [198].
Prebiotic inulin-type fructans have also been found to act as potent modulators of the gut microbiota, promoting the in vitro growth of Bacteroides species. This modulation mechanism exerts a restraining effect on the growth kinetics of the gut microbiota, effectively curbing T cell-dependent invasive melanoma [199]. The action of prebiotic inulin occurs via distinct mechanisms, including facilitating the dominance of Bacteroides species in the gut microbiota, improving CTL function within the spleen, and successfully combating melanoma resistance to mitogen-activated extracellular signal-regulated kinase inhibitors [199]. This sophisticated interplay between the gut microbiota, immunosuppressive metabolites, and therapeutic interventions underlines the intricate landscape of tumor immune responses and opens up promising avenues for therapeutic exploration.
2.1.2 Significance of gut microbiota in systemic immunity
The establishment of a resilient immune system is of paramount significance in the context of allogeneic hematopoietic stem cell transplantation (HSCT), exhibiting a critical role in relapse management and the reduction of transplant-associated mortality in patients [200,201]. Recent extensive clinical trials conducted across various centers and countries have shed light on a strong relationship between the richness of the gut microbiota and decreased mortality rates in individuals who have undergone allogeneic HSCT [202]. Another comprehensive longitudinal investigation analyzed over 10,000 fecal specimens from patients who underwent allogeneic HSCT and meticulously tracked daily fluctuations in the differential blood cell counts of those patients. The study results implied a close-knit interplay between the dynamics of immune reconstitution and the complex composition of the gut microbiota [203].
This notable connection between the gut microbiota and the various facets of post-transplant biology has been corroborated via experimental findings in murine models, which have further established that the influence of this association extends to nutritional factors, post-transplant bone marrow integrity, thymic cell function, lymphocyte homeostasis, and the intricate processes of hematopoiesis [204]. This heterogeneous influence is partly attributable to the endogenous ligands for retinoic acid-inducible gene I, including 3pRNA and RNA from various sources such as viruses, phages, or bacteria. These ligands were found to induce a protective fibronectin I signaling pathway and facilitate intestinal barrier repair in the intestinal cells [205]. The generation of lymphocytes in the post-transplant phase is also intrinsically linked to the efficient extraction of energy from dietary sources, which may hinge upon the genomic repertoire of the carbohydrate-active enzymes in the gut microbiota [204].
2.1.3 Role of gut microbiota in TME regulation
The complicated impact of the gut microbiome extends to local and distant tumors, coordinating a complex interplay involving the modulation of immune responses, bone marrow and lymphocyte trafficking, inflammation, and metabolic patterns. In this intricate network, secretions from the gut microbiota are the primary players. For example, commensal microorganisms release outer membrane vesicles that have the remarkable capability to reprogram the TME toward a pro-Th1 profile, leading to the upregulation of cytokines such as CXCL10 and IFN-γ [206]. Moreover, metabolites, including butyrate and nicotinic acid, from these microorganisms serve as mediators of G-coupled receptor-109a-dependent induction of IL-18 in the colonic epithelial cells, along with the dampening of colitis and colitis-associated tumorigenesis [207]. Additionally, the nicotinamide adenine dinucleotide phosphate oxidase 2-mediated decrease in the production of myeloid cell reactive oxygen species in the context of tumor-associated oxidative stress after antibiotic treatment or in germ-free conditions was reported to compromise the effectiveness of tumor therapies, thus highlighting the cooperative role of commensal microorganisms in cancer development [208]. The mono-association of gnotobiotic mice with specific bacteria in colonized gut-draining LNs induces CTLs to produce TNFα [209]. Furthermore, endogenous gut bacterial translocation in Tet2-deficient mice driven by Tet2 intrinsic factors was found to be crucial in instigating IL-6-dependent pre-leukemic myeloproliferation. Moreover, this phenomenon was amendable via antibiotic intervention and completely abrogated in germ-free mice, indicating its potential application in clinical management [210,211]. However, subsequent findings indicated that a complete gut microbiota was essential in preventing leukemia progression in genetically predisposed mice.
Non-hematopoietic constituents of the gut mucosa also have a crucial association with the TME. Genetic deficiencies in mice and bone marrow chimeras have unveiled the integral role of ring-finger protein 5 (RNF5), an E3 ubiquitin ligase, in melanoma immune surveillance. Specifically, the absence of RNF5 in mice resulted in the diminished secretion of antimicrobial peptides and heightened epithelial cell apoptosis in cryptopatches, ultimately altering the gut microbiota composition. This mucosal damage, in turn, increased DC mobilization toward melanoma-draining LNs, resulting in the enhanced intratumoral infiltration of IFN-γ-generating T lymphocytes. Furthermore, cohousing Rnf5-/- and wild-type mice or administering antibiotics confirmed the microbiota-mediated effects and ultimately restored tumor invasiveness [187]. Another study observed a correlation between the overrepresentation of immunogenic bacteria (particularly TLR2 agonists) and oxaliplatin-induced crypt cell apoptosis in the ileal mucosa [184]. This finding corresponded to the priming of TFH cells in LNs, ultimately leading to B cell activation, Ig production, and TIL infiltration in patients and mice with colon cancer. Moreover, the disruption of the intestinal barrier function by anti-CTLA4 treatment was found to be vital for the systemic translocation of adenosine derived from Bifidobacterium, consequently promoting Th1 activation and anti-tumor immunity via T cell-specific A2AR signaling [212]. All these results strongly suggest that gut barrier dysfunction or the translocation of microbiota metabolites is closely tied to the composition of local microbial communities, which in turn mobilizes DCs within and outside GALTs and significantly contributes to T-cell infiltration in the TME.
Additionally, the immune components within tumors not only encompass stromal, tumor, and endothelial cells and hematopoietic progenitors but also comprise a compact network of intricate connections to adrenergic nerve fibers [213-216]. Further, the neuron subsets within the gut nervous system are responsive to the gut microbiota and function in a region-dependent manner to independently influence metabolic control beyond the regulation by the central nervous system [217]. All these findings imply a close relationship between mucosal or tumor-associated commensal microorganisms and the nerve fibers innervating the tumors, thereby warranting further exploration in this aspect.
2.2 Microbiota at extraintestinal barriers and its influence on cancer immunity
Considering that the intestinal barrier is the most expansive interface between the host organism and its microbiota and has the highest microbial diversity, research endeavors have predominantly investigated the impact of the gut microbiota on cancer development and prognosis [179]. These investigations can potentially uncover the causal relationships between alterations in gut microbiota composition and the impairment of tumor immunosurveillance. More importantly, these effects may extend beyond intestinal malignancies and include extraintestinal cancers. However, extraintestinal cancers have often been shown to originate in tissues harboring their distinct microbiota, indicating that these microbial ecosystems may be pivotal in tumor progression [218-220].
For example, the lung, which has a substantial surface area of approximately 1 m2/kg of body weight, does harbor microorganisms [221]. In the case of oncogene-driven native lung cancer, studies involving murine models have suggested that local symbiotic relationships may be disrupted during carcinogenesis. These changes initiate complex interactions between alveolar macrophages and lung-residing γδ T cells, ultimately resulting in lung tumor advancement [222]. In line with this finding, Le Noci et al. demonstrated that the reduction in bacterial biomass owing to the administration of aerosolized antibiotics was associated with enhanced anti-tumor immune responses, possibly involving the activation of T and NK cells, as well as a decrease in immunosuppressive Tregs. Furthermore, the use of probiotic Lactobacillus rhamnosus GG could mitigate immunosuppression, inhibit lung tumor engraftment, and decrease tumor metastasis under antibiotic and probiotic conditions [223]. The clinical relevance of these observations has recently become prominent in a study involving patients with lung cancer [224]. In that study, Tsay et al. reported that the microaspiration of upper airway symbionts in patients with lung cancer substantially influences therapy response and overall survival (OS). This result may be closely linked to the aggravation of Th17-mediated inflammation, an expected consequence of immune checkpoint inhibition [224]. Another research by Greathouse et al. has proposed a connection between TP53 and changes in the lung microbiota. In particular, they detected an abundance of the Acidovorax genus in lung biopsy specimens from individuals with squamous cell carcinoma, along with the further enrichment of a comparable taxon in lung biopsies of patients with squamous cell carcinoma and TP53 mutations [225].
The skin, which is the largest and outermost organ of the human body, plays a pivotal role in preserving host homeostasis by actively communicating with its resident microbiota, keratinocytes, and an array of skin immune components [221]. This intricate interplay occurs through a nexus of metabolic, innate, and homologous immune responses. Previous studies have demonstrated that alterations in the skin microbiota composition can significantly affect the onset of non-melanoma skin carcinogenesis [226]. In support of this notion, a cell culture study incorporating Staphylococcus epidermidis, a specific strain of skin commensal bacterium, exhibited a potent protective effect against skin cancer, emphasizing the profound impact of the commensal microorganisms on the skin. These S. epidermidis strains were found to produce 6-N-hydroxyaminopurine, a DNA polymerase activity inhibitor capable of suppressing the proliferation of tumor cell lines in culture [227]. Hoste et al. employed a mouse model of wound-induced skin cancer to delve deeper into the mechanisms underlying the promotion of inflammation and tumorigenesis by skin microbiota. Their investigation revealed that skin microbiota was indispensable for promoting inflammation and tumorigenesis. Furthermore, eliminating skin microbiota prevented tumor development, primarily by dismantling several key innate immune sensors, including TLR5, with inflammation as a pivotal correlate of tumorigenesis. Lastly, they observed that antibiotic treatment effectively inhibited tumor formation in a TLR5-dependent manner [228].
Correspondingly, cervical cancers arising from persistent high-risk human papillomavirus infections are frequently linked to an imbalance in cervical microbial communities [229,230]. These intricate interactions emerging between microbial symbionts and virus-related cancers, along with their potential synergistic effects on tumorigenesis, necessitate thorough investigation. Therefore, the cancer-microbiota interactions at extraintestinal barriers beyond the gut barrier should be comprehensively explored.
2.3 Impact of intratumoral microbiota on the TME
In-depth studies on the mechanistic underpinnings of intratumoral microbiota are notably scarce. Nevertheless, previous investigations have demonstrated the multifaceted effects of this microbiota on the TME, highlighting their potential complex modulations of local anti-tumor immunity [231-233] (Fig. 4). Current research indicates that the intratumoral microbial community predominantly colonizes within tumors through three main routes. The first route involves mucosal barrier origins, encompassing gastrointestinal tumors such as colorectal and pancreatic cancers, as well as pulmonary and cervical cancers. Given that these organs possess externalized cavities, the microbes colonizing the mucosal surfaces may infiltrate tumors via mucosal disruptions occurring during tumorigenesis [231,234]. The second route is via neighboring normal tissues. Prior studies have detected certain bacteria within organs that were initially considered sterile, with the microbial composition in the tumor tissue closely resembling that in the adjacent normal cellular tissue. Furthermore, the immunosuppressive milieu and hypoxic microenvironment of tumors have been found to facilitate microbial colonization [177]. However, the source of microbiota in normal tissues remains unclear and may have spread from tumor sites; thus, additional research is required to substantiate this notion. Finally, the third possible route of intratumoral microbial colonization is through hematogenous dissemination, wherein microbiota originating from the oral cavity, intestines, and other potential sites may be transported to tumor locations via the bloodstream and gain entry into tumors via compromised vascular permeation [235].
Mechanistic insights into the dual role of intratumoral microbiota in cancer. Intratumoral microbiota play a dual role in the TME. On the one hand, intra-tumoral bacteria contribute to tumorigenesis, progression, drug resistance, and metastasis by releasing genotoxins, promoting the WNT/β-catenin signaling pathway, inducing host complement C3, fostering cancer cell autophagy, and secreting metalloproteinases. On the other hand, these intra-tumoral bacteria facilitate anti-tumor immunity through mechanisms involving immune cell recruitment, activation of CD8+ T cells, induction of chemotactic factors, and stimulation of anti-tumor immune responses via bacterial antigen elicitation. TME: Tumor microenvironment; DC: Dendritic cell; STING: Stimulator of interferon genes.
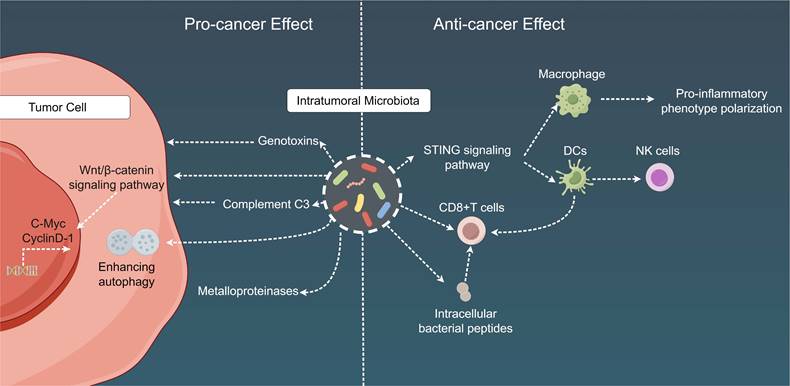
Additionally, intratumoral microbiota can exert the following cancer-specific effects: (1) influence carcinogenesis in the gastrointestinal and urogenital tracts, mainly through the secretion of genotoxins, such as colibactin toxin from pks+ Escherichia coli and toxins from B. fragilis[236-239]; (2) affect chemotherapy resistance either directly via microbial metabolism (e.g., gemcitabine degradation by cytidine deaminase in pancreatic cancer) or indirectly by augmenting cancer cell autophagy in colon cancer[240,241]; (3) promote tumor proliferation in pancreatic cancer through the fungal activation of the host C3 complement cascade[242]; (4) escalate breast and lung cancer metastasis via the upregulation of tumor stromal metalloproteinases or the attenuation of tumor immunosurveillance[243,244]; and (5) engage with host oncogenic pathways through microbial products within the TME, potentially resulting in the upregulation or activation of these oncogenic pathways, with a particular impact on specific pathways such as the Wnt/β-catenin signaling pathway. Disruptions in β-catenin signal transduction can stimulate the transcription of key cancer-associated genes, including cellular myelocytomatosis oncogene and cyclin D-1, thereby contributing to the advancement of carcinogenesis and tumor progression [245,246]. In terms of immunological ramifications, intratumoral microbial populations frequently establish tolerogenic programming via their interactions with pattern recognition receptors, which can result in diminished proportions of TILs (such as CD8 T cells) and an increased number of Tregs. These findings have been documented in lung, breast, colorectal, and pancreatic cancers [243,244,247-250]. Moreover, multiple studies have implied that imbalances within local bacterial communities can elicit a persistent proinflammatory immune response, thus fostering cancer progression. For instance, this phenomenon may develop from the microbial activation of NF-κB, a pivotal regulatory factor in cancer-associated inflammation [246,251,252].
Recent research has also determined that tumor-related microbiota can bolster anti-tumor immunity through multiple mechanisms. For example, bacteria, such as Bifidobacterium, migrate to colorectal cancer (CRC) sites, where they establish residence and subsequently activate DCs via the stimulator of interferon genes (STING) signaling pathway [186]. Additionally, STING agonists derived from Akkermansia muciniphila have been shown to stimulate IFN-I production by intra-tumoral monocytes, thus promoting macrophage reprogramming and communication between NK cells and DCs. Ultimately, these changes were found to enhance the effectiveness of ICI in patients with melanoma [253]. Another mechanism by which the tumor microbiome can shape anti-tumor immunity is encouraging the recruitment and activation of CD8+ T cells. For example, Saccharopolyspora, Pseudoxanthomonas, and Streptomyces in pancreatic ductal adenocarcinoma (PDAC) tissues were demonstrated to favor the activation of CD8 T cells, resulting in anti-tumor immune responses [254]. Furthermore, the tumor microbiome, including lactobacilli, Epstein-Barr virus, hepatitis B virus, and Merkel cell polyomavirus, can induce chemokine production, thereby influencing the infiltration of CD8 T cells into tumor tissues and potentially enhancing the survival rates of patients with cutaneous melanoma [255-259]. Lastly, antigens originating from the tumor-associated microbiome can also provoke anti-tumor immune responses. One study revealed an increase in IFN-γ-secreting and melanoma-infiltrating lymphocytes after exposure to various bacterial peptides compared to control cells not loaded with these peptides. This finding indicates that intracellular bacterial peptides presented by tumor cells can trigger T-cell immune responses, possibly serving as viable targets for combating tumor cells [260]. Furthermore, other studies have revealed that the intratumoral administration of bacteria or their antigens can paradoxically induce immune stimulatory effects, as observed in notable research based on Coley's toxins and bacterial cancer therapies [19,261]. Experiments involving intratumoral Bacteroides in the context of breast cancer have underscored the significance of lymphoid lineage cells as intermediaries influencing the impact of intratumoral microbial communities on tumor immune surveillance [243]. However, the precise mechanistic basis of these associations remains unknown.
3 Impact of microbiota-associated therapy and antibiotics on cancer immunotherapy
3.1 Impact of microbiota on cancer immunotherapy response
Mounting evidence suggests that microbiota members could serve as prognostic biomarkers for predicting patient responses to immune checkpoint blockade (ICB) therapy. Previous researchers have identified substantial differences in the microbiome composition of patients with varying ICB prognoses (Table 2) [180,262]. For instance, a pioneering global prospective study analyzed the fecal samples of 39 patients with melanoma who underwent combined immunotherapy with anti-CTLA4 (ipilimumab) and anti-PD-1 (nivolumab) or anti-PD-1 treatment alone [263]. The results showed that Holdemania filiformis, B. thetaiotaomicron, and F. prausnitzii were enriched in patients responding favorably to the combination therapy, whereas Dorea formicigenerans was abundant in those responding to anti-PD-1 treatment [263]. Additionally, 16S ribosomal RNA sequencing of archived fecal samples unveiled increased proportions of Burkholderiales and Bacteroidales in the anti-CTLA4 responders, highlighting the role of CTLA4 blockade-induced mucosal damage in regulating the microbiota [169]. In the case of non-melanoma cancers, a relatively larger cohort of patients with advanced epithelial tumors demonstrated a correlation between a higher abundance of muciniphila bacteria in their feces and more favorable responses to anti-PD-1 therapy [170]. Moreover, FMT from responders (R-FMT) and non-responders (NR-FMT) to germ-free mice provided compelling evidence for the modulatory role of microbiota in anti-tumor immune responses [169,170]. The NR-FMT mice displayed enhanced tumor growth rates and diminished responsiveness to anti-PD-1 therapy compared to their R-FMT counterparts, thereby reinforcing the notion that microbiota has a crucial regulatory function in ICI therapy.
Microbiota impact on cancer immunotherapy response.
| Immunotherapy | Disease | Microbiota | Mechanism |
|---|---|---|---|
| Anti-PD-1 treatment | Melanoma | Dorea formicigenerans | The gut bacteria induced maturation of anti-melanoma DCs and T cells, and increased T-cell interferon γ production [263]. |
| Advanced epithelial tumors | Akkermansia muciniphilic | Akkermansia muciniphila increased the recruitment of CCR9 +CXCR3 +CD4 + T lymphocytes into mouse tumor beds [168]. | |
| Non-small cell lung cancer | Enterococcus hirae strain 13144 | Enterococcus hirae strain 13144 enhanced peripheral CD8 and CD4 T-cell responses, amplified production of IFN-γ, and an extension of PFS [170]. | |
| Metastatic melanoma, Non-small cell lung cancer, and Renal cell carcinoma | Akkermansia and Prevotella copri | Akkermansia and Prevotella copri promoted the recruitment of CCR9+CXCR3+CD4+T lymphocytes to the tumor bed and appeared to be related to type 1 immunity [264,265]. | |
| Anti-PD-L1 treatment | Melanoma | Live Bifidobacterium spp. | Live Bifidobacterium spp. inhibited melanoma growth and enhanced tumor-specific CD8 T-cell responses, similar to anti-PD-L1 therapy [168]. |
| Anti-CTLA4 treatment | Melanoma | Burkholderiales and Bacteroidales | The microbiota composition affects IL-12-dependent TH1 immune responses [170]. |
| Melanoma | Bacteroides and theta/thetaaaomicron | Bacteroides and theta/thetaaaomicron enhanced LN and intratumoral DC maturation, leading to heightened TH1 immune responses and a restoration of anti-CTLA4 efficacy [169]. | |
| Anti-CTLA4 and anti-PD-1 treatment | Melanoma | Holdemania filiformis, Bacteroides thetaiotaomicron, and Faecalibacterium prausnitzii | The gut bacteria induced maturation of anti-melanoma DCs and T cells, and increased T-cell interferon γ production [263]. |
Although the link between the microbiota and ICB therapy response is evident, the exact fundamental mechanisms are still ambiguous. Most researchers have concentrated on the adaptive immune responses influenced by the microbiota during ICB therapy. One plausible mechanism posits that the microbiota augments CD8 T-cell responses against tumors, with Bacteroides abundance being linked to CD8 T-cell activity in early mouse experiments. Furthermore, a probiotic mixture containing live Bifidobacterium spp. was found to inhibit melanoma growth and enhance tumor-specific CD8 T-cell responses, similar to that observed in anti-PD-L1 therapy [168]. In a clinical trial of patients with NSCLC, responders showed increased levels of the Enterococcus hirae strain 13144. These increased proportions of E. hirae corresponded to heightened peripheral CD8 and CD4 T-cell responses, amplified IFN-γ production, and an extended PFS [170]. Another study of healthy donor feces identified 11 bacterial strains capable of promoting the accumulation and recruitment of intestinal IFN-γ CD103 T cells. This ability of the bacterial strains was independent of innate immune regulation and instead relied on resident lamina propria DCs and MHC Ia class molecules [194].
An additional potential mechanism pertains to the impact of microbiota on Th1 immune responses and its subsequent influence on the TME. The immunogenicity of certain Bacteroides or Thauomyces spp. has been associated with IL-12-dependent Th1 immune responses, which in turn have implications for the effectiveness of anti-CTLA4 therapy [169]. In a rearranged during transfection (RET) melanoma mouse model, a 2-week broad-spectrum antibiotic regimen in germ-free and specific pathogen-free mice led to reduced anti-CTLA4 effects. However, orally supplementing the mice with a mixture of B. fragilis and Burkholderia cepacia resulted in enhanced LN and intratumoral DC maturation, culminating in heightened Th1 immune responses and restored anti-CTLA4 efficacy [169].
Microbiota may also affect the TME via its influence on Th17 cells. Earlier investigations have explored whether metastatic PDAC harbors a microbiota composition similar to that of the gut. After oral antibiotic administration, the unique TME of PDAC, characterized by IL-17A CD4 Th17 cells that inhibit the differentiation of anti-cancer IFN-γ CD4 Th1 cells, exhibited a significant reduction in tumor burden and a shift toward immunogenicity, with the shift being particularly evident in the context of adaptive immune responses [248].
Moreover, A. muciniphila has been identified as a microbiota member influencing ICB responses through innate immunity modulation. Although the specific mechanisms are yet to be deciphered, Akkermansia has emerged as a possible candidate for predicting or enhancing ICB responses owing to its reported mucosal healing capabilities. For example, the oral administration of Akkermansia in NR-FMT mice preserved the efficacy of anti-PD-1 therapy, with IL-12 promoting the recruitment of CCR9+CXCR3+CD4+T lymphocytes to the tumor bed [264]. Another study found that the concurrent administration of Akkermansia and Prevotella copri in germ-free mice significantly potentiated anti-PD-1 therapy effectiveness against metastatic melanoma, NSCLC, and RCC [170]. Furthermore, a previous investigation highlighted the role of Akkermansia in stimulating T follicular helper cell-dependent IgG1 responses in mice. However, the precise processes underlying these effects of Akkermansia require further elucidation, with its association with type 1 immunity being proposed as a potential mechanism [265].
Initial evidence on the association between microbiota and ACT effectiveness was observed in a murine model with a deficiency in CD14 and TLR4 receptors [167]. This association manifested after combining ACT with total body irradiation (TBI), a form of lymphodepletion. The modulation of the microbiota via antibiotic treatment or inhibiting lipopolysaccharide (LPS) signaling constituents ultimately resulted in the impaired functionality of the infused CD8+ T cells and a decrease in activated DCs. Consequently, the therapeutic efficacy of ACT was compromised. In contrast, introducing LPS to TBI-treated, microbiota-depleted mice substantially amplified the proliferation and functionality of reinfused T cells and even induced sustained remission in mice with sizable tumors. In terms of the underlying mechanism, the TBI procedure is suggested to trigger microbial translocation, particularly that of gram-negative bacteria proficient in LPS production, into the mesenteric LNs. These translocated microorganisms, in turn, activate the TLR4 pathway by expressing varied TLR4 agonists, including LPS and peptidoglycan. This alteration subsequently heightens DC activation and increases the secretion of pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α, within the gastrointestinal tract. LPS administration has also been found to bolster the anti-cancer response mediated by the transferred CD8+ T cells in TBI-untreated mice [167].
Uribe-Herranz et al. investigated the administration of ACT for treating tumors in C57BL/6 mice procured from two distinct sources: the Jackson Laboratory (JAX) and Harvard (HAR) [266]. The results unveiled a marked contrast in the tumor growth between these two sets of mice. The HAR mice exhibited nearly complete suppression of tumor growth, whereas the JAX mice did not show any such response. Subsequent analysis of 16S rRNA sequencing data demonstrated obvious differences in the fecal microbiota composition between these two mice cohorts. The HAR mice displayed a diverse array of Bacteroidetes taxa, while the JAX mice were predominantly characterized by Bacteroidales S24-7. This difference hinted at a potential correlation between ACT efficacy and specific genera of Bacteroidetes, specifically Bacteroides and Parabacteroides. Subsequently, the JAX and HAR mice were administered vancomycin, an antibiotic targeting the gram-negative Bacteroidetes phylum, to further assess this correlation. The vancomycin intervention had no significant effect on the HAR mice, whereas it remarkably enhanced tumor regression in the JAX mice, effectively aligning their response with that of the HAR mice without vancomycin treatment. This improved response in the JAX mice was ascribed to a bolstered Th-1-mediated immune response and an increased accumulation of peripheral DCs, all of which led to the heightened expansion and activity of the transferred T cells [266]. Moreover, the antibiotic treatments employing neomycin and metronidazole failed to induce any discernible phenotypic changes, highlighting the specific role of certain bacterial species in orchestrating the host response to ACT.
However, the constrained approval timeline has caused a scarcity of available data on the association of ACT effectiveness and tolerance with the microbiota. Hence, controlled investigations are imperative to evaluate the fecal bacterial communities in these patient cohorts. The diversity of prior treatments and sample profile variability also pose significant challenges to attaining satisfactory outcomes. Nevertheless, the current recommendation still advocates broad-spectrum antibiotics for patients undergoing CAR-T cell therapy and autologous transplantation [267].
3.2 Impact of microbiota transplantation and dietary regulation on cancer immunotherapy efficacy
FMT has been granted FDA approval for treating recurrent and refractory Clostridioides difficile infections [268]. Preliminary research by Wang et al. has additionally highlighted the transformative potential of FMT in immunotherapy by detailing the successful treatment of immunotherapy-induced colitis using FMT enriched with beneficial Bifidobacterium species [269]. However, this initial study warrants future clinical trials to validate the therapeutic potential of FMT in immunotherapy. Moreover, recent proof-of-concept clinical trials have established the safety and efficacy of FMT in augmenting anti-PD-1 monoclonal antibody responses in patients with refractory melanoma. These studies indicated that combining microbial depletion via antibiotic treatment with subsequent FMT and reinitiation of anti-PD-1 monoclonal antibody treatment resulted in partial or complete remission in select patients. This outcome underscores the therapeutic capability of FMT in strengthening immunotherapy responses, with its characteristic sustained impact on the gut microbiota and the requirement for fewer frequent interventions than other modulation methods [8]. Currently, a series of clinical trials investigating the use of microbiota transplantation in boosting immunotherapy efficacy is underway (Table 3). Nonetheless, future research undertakings should incorporate ICI biomarkers, including PD-L1 expression and tumor mutational burden, to assess the indispensable role of pre-existing adaptive immunity in facilitating effective FMT [270,271].
Clinical trials of microbiota-based therapy modulate the efficacy and AEs of ICl.
| NCT number | Cancer types | Interventions | Outcome | Stage |
|---|---|---|---|---|
| NCT03341143 | Melanoma | FMT + Pembrolizumab | Objective Response Rate | Phase 2 |
| NCT03353402 | Melanoma | FMT + Anti-PD-1 therapy | Incidence of FMT-related Adverse Events; Proper implant engraftment | Phase 1 |
| NCT03353402 | Melanoma | FMT + ICI | Incidence of FMT-related Adverse Events | Phase 1 |
| NCT03686202 | Solid Tumors | MET-4 + ICI | Cumulative relative abundance of immunotherapy-responsiveness associated species at day 12 of MET-4; Changes in relative abundance of immunotherapy-responsiveness associated MET-4 strains between baseline and approximately day 12; Number of participants with treatment-related adverse events assessed by CTCAE v.5.0 | Phase 2-3 |
| NCT03772899 | Melanoma | FMT + Pembrolizumab/Nivolumab | To evaluate the safety of combining FMT using intestinal bacteria existing in the stool of healthy donors with immunotherapy in melanoma patients. | Phase 1 |
| NCT03819296 | Solid tumors | FMT + Infliximab/Vedolizumab | Difference in stool microbiome pattern; Incidence of adverse events of FMT | Phase 1-2 |
| NCT03891979 | Pancreatic cancer | Pembrolizumab + Ciprofloxacin + Metronidazole | Change in immune activation in pancreatic tumor tissue following treatment with antibiotics and pembrolizumab measured by activation of HLA-DR | Phase 4 |
| NCT04038619 | Renal Cell Carcinoma | Loperamide + FMT + ICI | Incidence of FMT-related adverse events; Clinical response/remission of immune-related diarrhea/colitis. | Phase 1 |
| NCT04056026 | Mesothelioma | FMT + Keytruda | Progression free survival | Phase 1 |
| NCT04116775 | Prostate | FMT + Enzalutamide + Pembrolizumab | Anticancer effect of fecal microbiota transplant from responders to pembrolizumab to non-responders. | Phase 2 |
| NCT04130763 | Gastrointestinal | FMT + Anti-PD-1 therapy | Objective Response Rate;Rate of abnormal vital signs and laboratory test results;The number of adverse events | Phase 1 |
| NCT04163289 | Renal Cell Carcinoma | FMT + Ipilimumab/Nivolumab | Occurence of immune-related colitis associated with ipilimumab/nivolumab treatment | Phase 1 |
| NCT04163289 | Renal Cell Carcinoma | FMT + Nivolumab /lpilimumab | Occurence of immune-related colitis associated with ipilimumab/nivolumab treatment | Phase 1 |
| NCT04264975 | Solid Tumors | FMT + ICI | Overall Response Rate | Not Applicable |
| NCT04521075 | Melanoma,Non-small Cell Lung Cancer | FMT + Nivolumab | Incidence of FMT-related Adverse Events; Overall Response Rate | Phase 1-2 |
| NCT04577729 | Melanoma | FMT + ICI | Progression free survival | Not Applicable |
| NCT04699721 | Non-small Cell Lung Cancer | Bifidobacterium trifidum live powder + Nivolumab + Paclitaxel + Carboplatin AUC5 | Adverse effects;Surgical complications; Non-R0 surgical events | Phase 1 |
| NCT04729322 | Colorectal cancer | FMT + Pembrolizumab/Nivolumab | Objective response rate | Phase 1 |
| NCT04758507 | Renal Cell Carcinoma | FMT + ICI | Number of participants who will be free from tumor progression, as assessed by RECIST criteria v. 1.1. | Phase 1-2 |
| NCT04883762 | Solid tumors | FMT + ICI | Incidence of FMT-related adverse events | Phase 1 |
| NCT04924374 | Advanced Lung Cancer | FMT + Anti-PD-1 therapy | Measure of safety | Not Applicable |
| NCT04988841 | Melanoma | FMT + Pembrolizumab /Nivolumab | To assess whether the safety of a 23-week treatment with MaaT013, combined with ipilimumab+nivolumab, is different from that of ipilimumab+nivolumab+placebo in patients with melanoma naïve to Ipilimumab and anti-PD-1 | Phase 2 |
| NCT05008861 | Non-Small Cell Lung Cancer | FMT+Anti-PD-1/PD-L1 therapy+Platinum based chemotherapy | Incidence of FMT-related Adverse Events;Incidence of anti-PD-1/PD-L1-related Adverse Events | Phase 1 |
| NCT05220124 | Bladder Urothelial Carcinoma | Bifidobacterium Lactobacillus and Enterococcus Capsules+ICI | Progression-free survival | Phase 4 |
| NCT05251389 | Melanoma Stage IIIMelanoma Stage IV | FMT+ICI | Efficacy, defined as clinical benefit (SD, PR, CR) | Phase 1Phase 2 |
| NCT05279677 | Colorectal Cancer | FMT + Sintilimab + Fruquintinib | Overall Response Rate | Phase 2 |
| NCT05286294 | Melanoma; Head and Neck Squamous Cell Carcinoma; Cutaneous Squamous Cell Carcinoma; MSI-High; Clear Cell Renal Cell Carcinoma; Non-small Cell Lung Cancer | FMT+ICI | Safety evaluation of FMT in advanced cancer patients;Tumor response evaluation | Phase 2 |
| NCT05462496 | Pancreatic cancer | FOLFIRINOX + Ciprofloxacin + Metronidazole + Pembrolizumab | Achievement of overall immune response | Phase 2 |
| NCT05690048 | Hepatocellular carcinoma | FMT/Vancomycin + Atezolizumab + Bevacizumab | Differential tumoral CD8 T-cell infiltration; Adverse event documentation of FMT in advanced HCC | Phase 2 |
| NCT05750030 | Hepatocellular carcinoma | FMT + Atezolizumab + Bevacizumab | Safety of atezolizumab/bevacizumab in combination with FMT, measured by incidence and severity of treatment-related adverse events, determined according to National Cancer Institute CTCAE v.5.0. | Phase 2 |
Abbreviations: FMT: Fecal microbiota transplantation; MET: Microbial ecosystem therapeutics; SD: stable disease, PR: partial response, CR: complete response; PD-1: programmed cell death protein 1; PD-L1: Programmed cell death 1 ligand 1; ICI: Immune checkpoint inhibitor.
Another approach to enhance ICI responses involves the application of prebiotics and dietary interventions. Common prebiotics, including inulin and oligofructose, are capable of enriching beneficial bacterial species such as Bifidobacterium, Lactobacillus, and Faecalibacterium in the human gut [272,273], yielding improved anti-cancer immune responses [168,169,194]. However, additional research is essential to elucidate the direct impact of prebiotics on ICIs. Dietary modulation has also exhibited efficacy in improving treatment outcomes. For instance, high-fiber diets have been associated with the enrichment of Bifidobacterium species and improved clinical outcomes in patients with metastatic NSCLC and melanoma undergoing ICI [274]. Mechanistic studies support these findings, illustrating that high-fiber diets elevate TIL levels in ICI-treated mice and that fiber supplementation leads to the enrichment of the Ruminococcaceae family that in turn facilitates T-cell activation and tumor infiltration [7]. Additionally, ketogenic diets and the resulting ketone bodies have proven instrumental in augmenting ICI efficacy in mice by promoting CD8+ T-cell proliferation and suppressing PD-L1 expression, thereby sustaining T-cell activation to exert anti-cancer effects [275]. Furthermore, the rapid responses of microbiota to dietary changes are noteworthy and suggest the transient nature of dietary modulation [276,277]. Therefore, comprehensive research should focus on strategies to prolong and sustain dietary effects for achieving optimal ICI responses.
A parallel strategy to modulate immunotherapeutic responses entails the utilization of probiotics. Commercial probiotics containing microbiota species, such as B. longum and L. rhamnosus GG, linked to enhanced immunotherapy responses have shown promise in preclinical investigations [168,278]. These probiotics have demonstrated potential in enhancing anti-cancer immunity by reducing Treg levels, promoting CD8+ T-cell activation and CD4+ T-cell differentiation, and facilitating intratumoral NK cell infiltration [279,280]. Moreover, a meticulously curated consortium of 11 bacterial strains, comprising seven Bacteroides and four non-Bacteroides species, has been reported to induce IFN-γ-producing CD8+ T cells mediated by CD103+ DCs, thereby enhancing ICI efficacy in syngeneic tumor-bearing mice. All these findings stress the potential of probiotics as supplementary agents to amplify immunotherapy effectiveness. Previous clinical trials have observed that certain probiotic strains, especially Bifidobacterium lactis Bl-04 and Lactobacillus acidophilus NCFM, cause an increase in butyrate-producing species, including Faecalibacterium and Clostridiales, within the microbiota of patients with CRC. This proliferation coincides with the improved immunotherapy responses [281].
However, current clinical research primarily addresses the impact of probiotics on microbiota composition and does not identify any direct causative links with immunotherapy outcomes. Additionally, recent findings suggest that individuals using probiotics may experience a potential reduction in microbial diversity, a characteristic commonly associated with non-responsiveness to immunotherapy [282]. This phenomenon has been substantiated by preclinical mechanistic studies that have shown that mice treated with probiotics including B. longum or L. rhamnosus GG displayed attenuated responses to anti-PD-L1 monoclonal antibody treatment and reduced levels of IFN-γ+ CD8+ T cells within the TME, which contrasted prior research findings [7]. Consequently, the indiscriminate use of over-the-counter probiotics in patients undergoing immunotherapy should be discouraged due to the existing knowledge gaps.
3.3 Impact of antibiotics on cancer immunotherapy efficacy
Antibiotics are a standard intervention in the prophylactic and therapeutic care of patients with cancer, owing to their infection susceptibility. However, recent research suggests that the timing and duration of antibiotic use may significantly influence immunotherapy effectiveness [170,175,283-291] (Table 4). Thus, clinical practitioners must exercise caution when considering antibiotic application in this patient population.
The Impact of antibiotics on cancer immunotherapy efficacy.
| Disease | Therapy | Antibiotic intervention | Outcome |
|---|---|---|---|
| Advancer non-small cell lung cancer, renal cell carcinoma and urothelial carcinoma | Anti-PD-1 and anti-PD-L1 treatment | Retrospective analysis of the antibiotic utilization of patients within a window of 60 days before and 30 days after the start of treatment ICI initiation. | Patients who have undergone antibiotic treatment experience a poorer prognosis [170]. |
| Metastatic renal cell carcinoma | Systemic therapy | Oral or intravenous systemic antibiotic treatment | Antibiotic use was associated with worse outcomes in patients treated with either contemporary PD-1/PD-L1-based ICIs or cytokines [284]. |
| Metastatic renal cell carcinoma | Anti-CTLA4 and anti-PD-1 treatment | Retrospective analysis of the antibiotic utilization of patients within a window of 30 days before and 30 days after the start of treatment ICI initiation. | Use of antibiotic before ICIs treatment is a predictor of poor ICIs response in metastatic renal cell carcinoma [285]. |
| Non-small cell lung cancer | Anti-PD-L1 treatment | Retrospective analysis of the antibiotic utilization of patients in the 1 month prior to ICI initiation. | Antibiotics use in patients with metastatic non-small cell lung cancer is associated with poor outcome and may influence the efficacy of ICI [286]. |
| Multiple advanced cancers | Anti-PD-1 and anti-PD-L1 treatment | Retrospective analysis of the antibiotic utilization of patients within a window of 14 days before and 14 days after the start of treatment ICI initiation. | Patients who have undergone antibiotic treatment experience a poorer prognosis [288]. |
| Advanced non-small cell lung cancer | Anti-PD-1 and anti-PD-L1 treatment | Retrospective analysis the antibiotic utilization of patients in the 1 month prior to ICI initiation. | Patients who have undergone antibiotic treatment exhibit lower alpha diversity in the gut microbiota and experience a poorer prognosis [290]. |
| Renal cell carcinoma and non-small-cell lung cancer | Anti-PD-1, anti-PD-L1 and anti-CTLA4 treatment | Retrospective analysis of the antibiotic utilization of patients in the 2 months prior to ICI initiation. | Patients who have received antibiotic treatment exhibit a poorer prognosis, and those who received antibiotics within the 30 days preceding ICIs therapy have a worse prognosis compared to patients who received antibiotics within the 60 days preceding ICI therapy[291]. |
| Renal Cell Carcinoma | Anti-PD-1 treatment | Retrospective analysis of the antibiotic utilization of patients in the 2 months prior to ICI initiation. | Patients who have undergone antibiotic treatment experience a poorer prognosis [175]. |
| Multiple cancers | ICIs | Prospective analysis of the antibiotic utilization of patients up to 30 days prior to or concurrent with ICI therapy. | The administration of antibiotics up to 30 days prior to ICIs therapy is associated with diminished efficacy of ICI treatment, and this phenomenon is observed independent of the specific tumor site [299]. |
Broad-spectrum antibiotics, even when administered for extended periods in cases of evident or latent infections, have been implicated in disrupting the delicate microbiota balance and impairing immune cell responses [292,293]. Moreover, patients who received antibiotics immediately before or after anti-PD-1 treatment experienced an approximately 50% reduction in median survival compared to those not administered antibiotics [170]. Similarly, patients with late-stage cancer concurrently using antibiotics with ICI therapy demonstrated diminished response rates and shorter OS or PFS [288,294]. Prolonged antibiotic exposure is also positively associated with the risk of various cancers [284,285,295-297]. In line with these results, murine models of NSCLC and melanoma revealed that the co-administration of broad-spectrum antibiotics, such as vancomycin, ampicillin, metronidazole, and neomycin, suppressed the protective IL-17-producing γδT17 cell response, ultimately promoting metastasis [298]. All these findings underscore the adverse effects of antibiotics on tumor progression.
Nonetheless, the existing body of evidence on antibiotic use and cancer treatments primarily consists of animal experiments and retrospective investigations, making it challenging to confirm the direct negative impact of antibiotics on anti-PD-1 efficacy. Hence, prospective research is necessary to validate these research results. A multicenter, prospective cohort study involving 196 patients with NSCLC, melanoma, RCC, or head and neck cancer reported that those who received antibiotics prior to PD-1/PD-L1 antibody treatment had poorer responses and OS [299]. Furthermore, patients who were administered a single dose of broad-spectrum antibiotics 1 month before ICB treatment fared worse clinically than those undergoing antibiotic therapy simultaneously with ICB treatment [299]. Additionally, ICB treatment following antibiotic administration appeared less favorable than no initial treatment. Another clinical trial assessed the outcomes of patients with late-stage RCC or NSCLC who received anti-PD-L1 monoclonal antibodies alone or in combination with antibiotics (quinolones or β-lactams) within 4 days of treatment initiation and observation that the addition of antibiotics reduced OS in NSCLC and PFS in those with RCC [291]. The study also found that patients receiving antibiotics within the first 30 days before ICB treatment experienced poorer clinical outcomes than those receiving antibiotics within the first 60 days [291]. Overall, these findings emphasize the critical significance of antibiotic administration timing in immunotherapy.
Moreover, specific antibiotics, including ampicillin, vancomycin, and lincomycin as well as imipenem, are linked with microbiota alterations, which in turn affect anti-CTLA4 therapy and weaken anti-tumor effects [169]. Studies involving K/BxN mice have consistently proven the potent inhibitory effects of antibiotics, such as vancomycin and ampicillin, on the progression of rheumatoid arthritis. However, a broader microbiota-directed antibiotic regimen incorporating vancomycin to target gram-negative bacteria, metronidazole and neomycin for anaerobic bacteria, and ampicillin for gram-positive bacteria led to a reduction in Th17 cell populations, with each antibiotic acting via distinct mechanisms [300,301]. Conversely, microbiota restoration was able to achieve Th17 cell recovery, suggesting that microbiota regulation can mitigate antibiotic-induced immune dysfunction and influence the occurrence and severity of gut lesions caused by antibiotic therapy, ultimately enhancing therapeutic efficacy.
Some researchers have reported contrasting findings, wherein antibiotics were synergistically utilized with other drugs to elicit anti-cancer effects. For example, a lipid delivery system co-loading curcumin and doxorubicin has been developed to exploit their synergistic anti-cancer effects, resulting in tumor attenuated and efficient therapeutic outcomes and consequently enhancing tumor control [302]. The antibiotic tigecycline has emerged as an effective agent for highly malignant double-hit lymphoma, with characteristic activation of MYC and B-cell lymphoma-2 cancer genes. The combination of tigecycline with B-cell lymphoma-2 inhibitors such as venetoclax exhibited significant anti-cancer effects, presenting a promising front-line treatment strategy for lymphomas [303]. The addition of antibiotics offers advantages in modulating the effects of antibiotic-induced imbalance of a single microbial community [304]. Given the propensity for gut tumors to co-occur with microbial dysbiosis, polysaccharide regulation may be a potent approach to maintaining a balanced gut microbiota, with studies revealing its ability to counteract gut microbiota dysbiosis caused by paclitaxel chemotherapy in a 4T1 breast cancer mouse model [305].
Furthermore, combining antibiotics with radiation therapy has yielded improved treatment outcomes. For example, the addition of vancomycin to radiation therapy was superior to individual drug utilization in model animals, leading to modification of the TME and enhancement of local antigen presentation, as well as tumor infiltration by IFN-γ- and CD8-dependent cytotoxic T cells [306]. A positive correlation has also been reported between antibiotic use and tumor immunogenicity. For instance, erythromycin, a clinically effective macrolide-type anti-tumor antibiotic, was found to enhance tumor-infiltrating T-cell populations. Fusobacterium nucleatum is commonly observed in CRC tissues, and metronidazole treatment of colon cancer xenograft mice was shown to reduce these bacterial loads in the TME and slow tumor cell proliferation [185]. Additionally, short-chain fatty acids (SCFAs) generated by the gut microbiota, particularly clostridia, exert inhibitory effects on APC function and diminish the radiotherapy-boosted anti-tumor efficacy of vancomycin. Moreover, vancomycin has been demonstrated to specifically modulate gram-positive bacterial populations, such as clostridia, leading to reduced SCFA and C4 concentrations within fecal and tissue samples. These findings underscore the benefits of precision antibiotic therapy aimed at cancer-associated microbial communities, thereby laying the groundwork for the development of potential therapeutic strategies for treating individuals with cancer [185]. Antibiotic interventions in the gut microbiota positively affect tumor immunity. In some patients with pancreatic cancer, the gut microbiota was found to promote immunosuppression and Treg cell proliferation via Foxp3 induction [307]. A judicious application of antibiotics can be used to regulate the advancement of premalignant lesions and cancer progression. Butyrate has been shown to induce alterations in T-cell behavior via epigenetic modifications in the Foxp3 gene, indicating that antibiotics can influence the gut microbiota to alter the generation of these critical metabolic byproducts. Antibiotic cocktails (ABX) can disrupt microbiota, ultimately suppressing PDAC infiltration and inducing immunogenic reprogramming of the TME after its dissolution. ABX administration is also associated with increased M1 macrophage and Th1 CD4+ T-cell differentiation and CD8+ T-cell activation, as well as significantly upregulates PD-1 expression on effector T cells and promotes checkpoint-targeted immunotherapy outcomes. All these results imply that antibiotics can assist in enhancing cancer immunotherapy by establishing an equilibrium within microbial communities, influencing tumor-associated microbiota, and augmenting anti-tumor advantages [9]. Therefore, formulating oral antibiotics to target the microbiota is a promising approach for further improving immunotherapy outcomes.
In light of these findings, the use of antibiotics and probiotics to restore a healthy gut microenvironment is especially relevant. Correspondingly, previous studies have observed that replenishing the gut microbiota in mice effectively reversed the intratumoral immunogenic alterations in their tumors, which were initially ascribed to the depletion of bacteria by antibiotic treatment. This reversal also translated into the increased expression of genes linked to T cell-mediated immune activation in the tumors of mice that had received antibiotic treatment. These results indicated that the reconstituted microbiota could more effectively regulate immune activation. Furthermore, antibiotics aid in preventing gut bacterial translocation, thus maintaining a stable distribution of bacteria in the body. For example, ABX administration efficiently mitigated bacterial translocation to the liver and gut, wherein the disruption of the gut vascular barrier was responsible for the systemic dissemination of gut bacteria and the subsequent colonization of the liver in CRC [248].
As mentioned above, prior studies have unveiled a compelling association between antibiotic utilization and lower PFS, OS, and response rates, with the timing of antibiotic administration being critical. A comprehensive meta-analysis has noted that patients without antibiotic exposure within 42 days before commencing ICI therapy had 3.43 times longer OS than those who received antibiotics within 60 days preceding ICI initiation [308]. These findings are congruent with research reporting the restoration of microbial composition to nearly baseline levels within 42 days after administering healthy individuals with an ABX (meropenem, gentamicin, and vancomycin) for 4 days [309].
Apart from epidemiological observations, emerging studies have delineated distinct microbial signatures in antibiotics-exposed patients, with a characteristic reduction in microbial diversity and enrichment of Clostridium hathewayi and features associated with diminished survival rates. Consequently, a prudent approach should be followed, wherein antibiotic use is avoided before initiating ICI therapy [175,290]. Alternatively, FMT or probiotics may be a viable option to rectify antibiotic-induced dysbiosis preceding ICI therapy. However, treating PDAC may require a different strategy because preclinical study findings indicate that antibiotics targeting intratumoral bacteria can potentiate immunotherapy efficacy in PDAC [248].
Based on all these results, although antibiotic use may hinder anti-cancer responses to some extent by influencing the microbial composition, the potential to expand the richness and diversity of the gut microbiota via the synergistic interactions between different antibiotic drugs should not be underestimated. Additionally, meticulously assessing the influence of antibiotics and microbiota across diverse cancer types is imperative, considering the varied factors including host immune status, tumor genetic factors, TME, and microbiota modulation. Lastly, remarkable improvements in tumor control can be achieved by developing antibiotic combinations to suppress tumor development, regulating the antibiotic-induced ecological imbalance that can lead to tumor progression, and enhancing the efficacy of antibiotic detoxification within the TME.
4. Conclusion and prospects
Previous studies have made substantial progress in comprehending the role of the host microbiota in normal physiology and disease, as well as shed light on the potential therapeutic strategy of targeting microbial communities within the gastrointestinal tract and other ecological niches for treating diseases and promoting holistic well-being. However, this field is still at a nascent stage, displaying valuable perspectives for further unraveling the mechanisms by which these microorganisms affect various physiological and pathological processes. Moreover, identifying the most effective approach, such as dietary interventions and other modalities, for targeting these microorganisms is vital.
Further, the increasing recognition of the microbiome as a pivotal determinant of health and disease has raised the prospect of incorporating the assessment and regulation of microbial communities in the gut and other ecological habitats into precision oncology care. This convergent application can potentially foster the evolution of comprehensive precision health paradigms. Currently, personalized cancer care involves the histopathological characterization of precancerous or cancerous tissues via targeted gene profiling or next-generation sequencing methods [310,311]. Other processes employed include the analysis of genomic or proteomic alterations, as well as the limited assessment of immune cells (utilizing PD-L1, CD8, and other biomarkers) at baseline and during treatment to guide therapy and determine treatment responses [312-314].
Recent developments have introduced more comprehensive strategies for cancer prevention and treatment [195,277], with the potential to improve health using multifaceted monitoring, feedback, and early interventions. These advances enable a more holistic approach to cancer care and involve the assessment of somatic and lineage mutations in tissue/tumors and blood, as well as the feature evaluation of microbiota derived from tissue and blood samples. Thus, in-depth examinations of systemic and tissue/tumor-based immunity beyond the current capabilities of conventional biomarkers are warranted. Such assessments will provide a foundation to explore innovative immune mechanisms and can revolutionize cancer prevention and treatment approaches by enhancing immune surveillance [315].
The analysis of microbial communities in the gastrointestinal tract and other ecological niches has indeed shown promise in the field of oncology, particularly in understanding the complex interplay between the host immune system and cancer development. In addition to investigating systemic inflammation markers and lifestyle factors [316], recent research focused on the role of immune-modulating metabolites or compounds within the microbiome that could potentially influence cancer progression and treatment response [317]. Immunostimulatory microbial metabolites, such as SCFAs, polysaccharides, and LPS, have been shown to modulate the immune response of host cells [195]. Moreover, the manipulation of the microbiome through methods such as FMT or the use of narrow-spectrum antibiotics can be tailored based on individual patient data monitoring. By identifying specific microbial signatures associated with favorable or adverse cancer outcomes, healthcare providers can design personalized dietary interventions and microbial community manipulations to modulate the levels of these immunostimulatory metabolites. Fusing multi-omics data using artificial intelligence also facilitates the implementation of mathematical modeling and other methodologies in this field, thus bolstering strategies for the treatment, interception, and prevention of cancer. This process entails a dynamic cycle of iteration and refinement of current techniques [318,319].
However, current research in this budding field is accompanied by certain limitations. The complexity of the microbiome and its interaction with the host immune system poses challenges in differentiating causation from correlation. Moreover, identifying optimal strategies for manipulating microbial communities continues to be a hurdle due to the variations in individual responses to interventions. Additionally, ethical considerations, including the long-term effects of the interventions and potential unintended consequences, require careful examination.
Furthermore, the possible impact of microbiome-focused strategies on tumor immunotherapy necessitates thorough investigation. Although these strategies may be valuable in enhancing immune surveillance, the chances for unforeseen outcomes, such as exacerbating inflammation or compromising the efficacy of existing immunotherapies, must be carefully evaluated. Thus, the future direction not only involves expanding our understanding of the microbiome but also encompasses critically assessing the translational implications of such interventions in cancer immunotherapy.
In conclusion, integrating microbiome research into precision oncology may be a promising avenue with transformative potential. However, these developments should be considered with caution and a clear recognition of the existing limitations. Thus, future research addressing these challenges can help pave the way for innovative and personalized approaches to cancer prevention and treatment, thereby providing profound implications for the field of tumor immunotherapy.
Abbreviations
ACT: Adoptive cell therapy; APCs: Antigen-presenting cells; CAR-T: Chimeric antigen receptor T-cell; CCL5, better known as RANTES: Chemokine (C-C motif) ligand 5; CD: Cluster of differentiation; CEACAM1: Carcinoembryonic antigen cell adhesion molecule 1; CIN2/3: cervical intraepithelial neoplasia 2/3; c-Myc: Cellular myelocytomatosis; CRC: Colorectal cancer; CTL: Cytotoxic T lymphocyte; CTLA4: Cytotoxic T-lymphocyte-associated antigen 4; CXCR4: C-X-C chemokine receptor type 4; DC: Dendritic cell; DNA: Deoxyribonucleic acid; DOXO: Doxorubicin; EBV: Epstein-Barr virus; ERK: Extracellular regulated protein kinases; FDA: Food and drug administration; FMT: Fecal microbiota transplantation; FN-I: Fibronectin I; GALT: gut-associated lymphoid tissues; GF: Germ-free; GITR: glucocorticoid-induced TNF receptor; GM-CSF: Granulocyte-macrophage colony-stimulating factor; Gpr109a: G-coupled receptor-109a; GVAX: GM-CSF gene transduced autologous tumor vaccine; GVHD: Graft-versus-host disease; HBV: Hepatitis B virus; HCC: Hepatocellular carcinoma; HER2: Human epidermal growth factor receptor 2; HLA: Human leukocyte antigen; HSCT: Hematopoietic stem cell transplantation; HSV-1: Herpes simplex virus type 1; ICAM-1: Intercellular adhesion molecule-1; ICI: Immune checkpoint inhibitor; ICOS: Inducible T-cell costimulator; IFN: Interferon; IGRP: Islet-specific glucose-6-phosphatase catalytic subunit-related protein; IL-2: Interleukin-2; iPSC: induced pluripotent stem cell; irAEs: Immune-related adverse events; ITAM: Immunoreceptor tyrosine-based activation motifs; JNK: Jun N-terminal kinase; LAG3: Lymphocyte-activation gene 3; LN: lymph node; LPS: lipopolysaccharide; MAGE: Melanoma-associated antigen; MAPK: Mitogen-activated protein kinase; MCPyV: Merkel cell polyomavirus; MDSC: Myeloid-derived suppressor cells; MEK: Mitogen-activated extracellular signal-regulated kinase; MeV: Measles virus; MHC: Major histocompatibility complex; NF-κB: Nuclear factor κappa-B; NK: Natural killer; NOX2: Nicotinamide adenine dinucleotide phosphate oxidase 2; NSCLC: Non-small cell lung cancer; OMV: Outer membrane vesicles; ORR : Objective response rate; OS: Overall survival; PBL: Peripheral blood lymphocyte; PD-1: Programmed cell death-1; PDAC: pancreatic ductal adenocarcinoma; PD-L1: Programmed cell death ligand-1; PFS: Progression-free survival; PMP: Precursor myeloproliferation; PSMB4: Proteasome 20S subunit beta 4; PTX: Paclitaxel; PVR: Poliovirus receptor; RCC: Renal cell carcinoma; RET: Rarranged during transfection; RIG-I: Retinoic acid-inducible gene I; RNA: Ribonucleic acid; RNF5: Ring finger protein 5; ROS: Reactive Oxygen Species; SCFAs: Short-chain fatty acids; scFv: Single-chain variable fragments; SPF: Specific pathogen-free; STING: Stimulator of interferon genes; TAM: Tumor-associated macrophage; TBI: total body irradiation; TCR: T cell receptor; TFH: Follicular helper T; TIGIT: T-cell immunoglobulin and ITIM domain; TILs: Tumor-infiltrating lymphocytes; TIM3: T-cell immunoglobulin and mucin domain-containing protein 3; TLR4: Toll-like receptor 4; TME: Tumor microenvironment; TNF: tumor necrosis factor; Tregs: Regulatory T cells; T-VEC: Talimogene laherparepvec; ZAP2: Zeta-chain-associated protein kinase.
Acknowledgements
We thank Figdraw (www.figdraw.com) for expert assistance in the pattern drawing. The authors thank professional English editor (Bullet Edits) for assistance in improving the quality of language.
Funding
This work was also supported by a Research Foundation of Shanghai Artemed Hospital (Grant NO: ATM2022YJ01) to Dr. Wenge Li, a National Natural Science Foundation of China (NSFC) (Grant NO: 82203629) and a Shanghai Pujiang Program (Grant NO: 22PJD054) to Dr. Qi Wu.
Author contributions
QW and SS performed and conceived the review. XY, WL and ZL wrote the manuscript. XY drew all the figures. QW and SS assisted in improving the quality of language and supplying financial support. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. 2020;17:807-21
2. Yi M, Zheng X, Niu M, Zhu S, Ge H, Wu K. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer. 2022;21:28
3. Amini L, Silbert SK, Maude SL. et al. Preparing for CAR T cell therapy: patient selection, bridging therapies and lymphodepletion. Nat Rev Clin Oncol. 2022;19:342-55
4. Matson V, Chervin CS, Gajewski TF. Cancer and the Microbiome-Influence of the Commensal Microbiota on Cancer, Immune Responses, and Immunotherapy. Gastroenterology. 2021;160:600-13
5. El Tekle G, Garrett WS. Bacteria in cancer initiation, promotion and progression. Nat Rev Cancer. 2023;23:600-18
6. Samanta S. Potential Impacts of Prebiotics and Probiotics on Cancer Prevention. Anticancer Agents Med Chem. 2022;22:605-28
7. Spencer CN, McQuade JL, Gopalakrishnan V. et al. Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science. 2021;374:1632-40
8. Baruch EN, Youngster I, Ben-Betzalel G. et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science. 2021;371:602-9
9. Jing Y, Chen X, Li K. et al. Association of antibiotic treatment with immune-related adverse events in patients with cancer receiving immunotherapy. J Immunother Cancer. 2022;10:e003779
10. Virchow R. Cellular pathology. As based upon physiological and pathological histology. Lecture XVI-Atheromatous affection of arteries. 1858. Nutr Rev. 1989;47:23-5
11. van der Bruggen P, Traversari C, Chomez P. et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643-7
12. Gaugler B, Van den Eynde B, van der Bruggen P. et al. Human gene MAGE-3 codes for an antigen recognized on a melanoma by autologous cytolytic T lymphocytes. J Exp Med. 1994;179:921-30
13. Pan K, Farrukh H, Chittepu VCSR, Xu H, Pan C-X, Zhu Z. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J Exp Clin Cancer Res. 2022;41:119
14. Naimi A, Mohammed RN, Raji A. et al. Tumor immunotherapies by immune checkpoint inhibitors (ICIs); the pros and cons. Cell Commun Signal. 2022;20:44
15. Choi Y, Shi Y, Haymaker CL, Naing A, Ciliberto G, Hajjar J. T-cell agonists in cancer immunotherapy. J Immunother Cancer. 2020;8:e000966
16. Zitvogel L, Ma Y, Raoult D, Kroemer G, Gajewski TF. The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science. 2018;359:1366-70
17. Ting NL-N, Lau HC-H, Yu J. Cancer pharmacomicrobiomics: targeting microbiota to optimise cancer therapy outcomes. Gut. 2022;71:1412-25
18. Hayase E, Jenq RR. Role of the intestinal microbiome and microbial-derived metabolites in immune checkpoint blockade immunotherapy of cancer. Genome Med. 2021;13:107
19. Starnes CO. Coley's toxins in perspective. Nature. 1992;357:11-2
20. Ma R, Li Z, Chiocca EA, Caligiuri MA, Yu J. The emerging field of oncolytic virus-based cancer immunotherapy. Trends Cancer. 2023;9:122-39
21. Ferrucci PF, Pala L, Conforti F, Cocorocchio E. Talimogene Laherparepvec (T-VEC): An Intralesional Cancer Immunotherapy for Advanced Melanoma. Cancers (Basel). 2021;13:1383
22. Dummer R, Gyorki DE, Hyngstrom J. et al. Neoadjuvant talimogene laherparepvec plus surgery versus surgery alone for resectable stage IIIB-IVM1a melanoma: a randomized, open-label, phase 2 trial. Nat Med. 2021;27:1789-96
23. Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016;107:1373-9
24. Boyero L, Sánchez-Gastaldo A, Alonso M, Noguera-Uclés JF, Molina-Pinelo S, Bernabé-Caro R. Primary and Acquired Resistance to Immunotherapy in Lung Cancer: Unveiling the Mechanisms Underlying of Immune Checkpoint Blockade Therapy. Cancers (Basel). 2020;12:3729
25. Lang FF, Conrad C, Gomez-Manzano C. et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J Clin Oncol. 2018;36:1419-27
26. Macedo N, Miller DM, Haq R, Kaufman HL. Clinical landscape of oncolytic virus research in 2020. J Immunother Cancer. 2020;8:e001486
27. Gong J, Sachdev E, Mita AC, Mita MM. Clinical development of reovirus for cancer therapy: An oncolytic virus with immune-mediated antitumor activity. World J Methodol. 2016;6:25-42
28. May V, Berchtold S, Berger A. et al. Chemovirotherapy for pancreatic cancer: Gemcitabine plus oncolytic measles vaccine virus. Oncol Lett. 2019;18:5534-42
29. Weiland T, Lampe J, Essmann F. et al. Enhanced killing of therapy-induced senescent tumor cells by oncolytic measles vaccine viruses. Int J Cancer. 2014;134:235-43
30. Saab S, Zalzale H, Rahal Z, Khalifeh Y, Sinjab A, Kadara H. Insights Into Lung Cancer Immune-Based Biology, Prevention, and Treatment. Front Immunol. 2020;11:159
31. Quoix E, Lena H, Losonczy G. et al. TG4010 immunotherapy and first-line chemotherapy for advanced non-small-cell lung cancer (TIME): results from the phase 2b part of a randomised, double-blind, placebo-controlled, phase 2b/3 trial. Lancet Oncol. 2016;17:212-23
32. Nishio N, Diaconu I, Liu H. et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014;74:5195-205
33. Igarashi Y, Sasada T. Cancer Vaccines: Toward the Next Breakthrough in Cancer Immunotherapy. J Immunol Res. 2020;2020:5825401
34. Kawakami Y, Eliyahu S, Delgado CH. et al. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci U S A. 1994;91:6458-62
35. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245-52
36. Kantoff PW, Higano CS, Shore ND. et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411-22
37. Salgia R, Lynch T, Skarin A. et al. Vaccination with irradiated autologous tumor cells engineered to secrete granulocyte-macrophage colony-stimulating factor augments antitumor immunity in some patients with metastatic non-small-cell lung carcinoma. J Clin Oncol. 2003;21:624-30
38. Jaffee EM, Hruban RH, Biedrzycki B. et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J Clin Oncol. 2001;19:145-56
39. Dranoff G, Jaffee E, Lazenby A. et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A. 1993;90:3539-43
40. Ferraro B, Morrow MP, Hutnick NA, Shin TH, Lucke CE, Weiner DB. Clinical applications of DNA vaccines: current progress. Clin Infect Dis. 2011;53:296-302
41. Sardesai NY, Weiner DB. Electroporation delivery of DNA vaccines: prospects for success. Curr Opin Immunol. 2011;23:421-9
42. Trimble CL, Morrow MP, Kraynyak KA. et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: a randomised, double-blind, placebo-controlled phase 2b trial. Lancet. 2015;386:2078-88
43. Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines - a new era in vaccinology. Nat Rev Drug Discov. 2018;17:261-79
44. Gote V, Bolla PK, Kommineni N. et al. A Comprehensive Review of mRNA Vaccines. Int J Mol Sci. 2023;24:2700
45. Espuelas S, Roth A, Thumann C, Frisch B, Schuber F. Effect of synthetic lipopeptides formulated in liposomes on the maturation of human dendritic cells. Mol Immunol. 2005;42:721-9
46. Fotin-Mleczek M, Zanzinger K, Heidenreich R. et al. Highly potent mRNA based cancer vaccines represent an attractive platform for combination therapies supporting an improved therapeutic effect. J Gene Med. 2012;14:428-39
47. Qiu P, Ziegelhoffer P, Sun J, Yang NS. Gene gun delivery of mRNA in situ results in efficient transgene expression and genetic immunization. Gene Ther. 1996;3:262-8
48. Lorentzen CL, Haanen JB, Met Ö, Svane IM. Clinical advances and ongoing trials on mRNA vaccines for cancer treatment. Lancet Oncol. 2022;23:e450-8
49. Propper DJ, Balkwill FR. Harnessing cytokines and chemokines for cancer therapy. Nat Rev Clin Oncol. 2022;19:237-53
50. Morgan DA, Ruscetti FW, Gallo R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science. 1976;193:1007-8
51. Hernandez R, Põder J, LaPorte KM, Malek TR. Engineering IL-2 for immunotherapy of autoimmunity and cancer. Nat Rev Immunol. 2022;22:614-28
52. Majidpoor J, Mortezaee K. Interleukin-2 therapy of cancer-clinical perspectives. Int Immunopharmacol. 2021;98:107836
53. Wrangle JM, Patterson A, Johnson CB. et al. IL-2 and Beyond in Cancer Immunotherapy. J Interferon Cytokine Res. 2018;38:45-68
54. Yu R, Zhu B, Chen D. Type I interferon-mediated tumor immunity and its role in immunotherapy. Cell Mol Life Sci. 2022;79:191
55. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15:405-14
56. Magenau JM, Peltier D, Riwes M. et al. Type 1 interferon to prevent leukemia relapse after allogeneic transplantation. Blood Adv. 2021;5:5047-56
57. Eigentler TK, Gutzmer R, Hauschild A. et al. Adjuvant treatment with pegylated interferon α-2a versus low-dose interferon α-2a in patients with high-risk melanoma: a randomized phase III DeCOG trial. Ann Oncol. 2016;27:1625-32
58. Graham GJ, Handel TM, Proudfoot AEI. Leukocyte Adhesion: Reconceptualizing Chemokine Presentation by Glycosaminoglycans. Trends Immunol. 2019;40:472-81
59. Domanska UM, Kruizinga RC, Nagengast WB. et al. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer. 2013;49:219-30
60. Lee EQ, Duda DG, Muzikansky A. et al. Phase I and Biomarker Study of Plerixafor and Bevacizumab in Recurrent High-Grade Glioma. Clin Cancer Res. 2018;24:4643-9
61. Dudley ME, Wunderlich JR, Robbins PF. et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850-4
62. Wachsmann TLA, Wouters AK, Remst DFG. et al. Comparing CAR and TCR engineered T cell performance as a function of tumor cell exposure. Oncoimmunology. 2022;11:2033528
63. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86:10024-8
64. Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol. 2002;20:70-5
65. Brentjens RJ, Davila ML, Riviere I. et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38
66. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725-33
67. Baulu E, Gardet C, Chuvin N, Depil S. TCR-engineered T cell therapy in solid tumors: State of the art and perspectives. Sci Adv. 2023;9:eadf3700
68. Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419-66
69. Mørch AM, Bálint Š, Santos AM, Davis SJ, Dustin ML. Coreceptors and TCR Signaling - the Strong and the Weak of It. Front Cell Dev Biol. 2020;8:597627
70. Klebanoff CA, Chandran SS, Baker BM, Quezada SA, Ribas A. T cell receptor therapeutics: immunological targeting of the intracellular cancer proteome. Nat Rev Drug Discov. 2023;22:996-1017
71. Clay TM, Custer MC, Sachs J, Hwu P, Rosenberg SA, Nishimura MI. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol. 1999;163:507-13
72. Morgan RA, Dudley ME, Wunderlich JR. et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126-9
73. Jungbluth AA, Antonescu CR, Busam KJ. et al. Monophasic and biphasic synovial sarcomas abundantly express cancer/testis antigen NY-ESO-1 but not MAGE-A1 or CT7. Int J Cancer. 2001;94:252-6
74. Rapoport AP, Stadtmauer EA, Binder-Scholl GK. et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med. 2015;21:914-21
75. Robbins PF, Morgan RA, Feldman SA. et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917-24
76. Allen ES, Stroncek DF, Ren J. et al. Autologous lymphapheresis for the production of chimeric antigen receptor T cells. Transfusion. 2017;57:1133-41
77. Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine. 2020;59:102975
78. Kruschinski A, Moosmann A, Poschke I. et al. Engineering antigen-specific primary human NK cells against HER-2 positive carcinomas. Proc Natl Acad Sci U S A. 2008;105:17481-6
79. Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106:376-83
80. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell. 2018;23:181-192.e5
81. Sloas C, Gill S, Klichinsky M. Engineered CAR-Macrophages as Adoptive Immunotherapies for Solid Tumors. Front Immunol. 2021;12:783305
82. Ritchie D, Mileshkin L, Wall D. et al. In vivo tracking of macrophage activated killer cells to sites of metastatic ovarian carcinoma. Cancer Immunol Immunother. 2007;56:155-63
83. Wu K, Lin K, Li X. et al. Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front Immunol. 2020;11:1731
84. Liu M, Liu J, Liang Z. et al. CAR-Macrophages and CAR-T Cells Synergistically Kill Tumor Cells In Vitro. Cells. 2022;11:3692
85. Morrissey MA, Williamson AP, Steinbach AM. et al. Chimeric antigen receptors that trigger phagocytosis. Elife. 2018;7:e36688
86. Klichinsky M, Ruella M, Shestova O. et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. 2020;38:947-53
87. Niu Z, Chen G, Chang W. et al. Chimeric antigen receptor-modified macrophages trigger systemic anti-tumour immunity. J Pathol. 2021;253:247-57
88. Bu JY, Shaw AS, Chan AC. Analysis of the interaction of ZAP-70 and syk protein-tyrosine kinases with the T-cell antigen receptor by plasmon resonance. Proc Natl Acad Sci U S A. 1995;92:5106-10
89. Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell. 2021;184:5309-37
90. Galluzzi L, Humeau J, Buqué A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol. 2020;17:725-41
91. Brunet JF, Denizot F, Luciani MF. et al. A new member of the immunoglobulin superfamily-CTLA-4. Nature. 1987;328:267-70
92. Walunas TL, Lenschow DJ, Bakker CY. et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405-13
93. Wing K, Onishi Y, Prieto-Martin P. et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271-5
94. Kraehenbuehl L, Weng CH, Eghbali S, Wolchok JD, Merghoub T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nature reviews Clinical oncology. 2021/09/29 ed. 2022;19:37-50
95. Oladejo M, Paulishak W, Wood L. Synergistic potential of immune checkpoint inhibitors and therapeutic cancer vaccines. Semin Cancer Biol. 2023;88:81-95
96. Matoba T, Imai M, Ohkura N. et al. Regulatory T cells expressing abundant CTLA-4 on the cell surface with a proliferative gene profile are key features of human head and neck cancer. Int J Cancer. 2019;144:2811-22
97. Zhao Y, Yang W, Huang Y, Cui R, Li X, Li B. Evolving Roles for Targeting CTLA-4 in Cancer Immunotherapy. Cell Physiol Biochem. 2018;47:721-34
98. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734-6
99. Hodi FS, O'Day SJ, McDermott DF. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-23
100. Ledford H. Melanoma drug wins US approval. Nature. 2011;471:561
101. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887-95
102. Doroshow DB, Bhalla S, Beasley MB. et al. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat Rev Clin Oncol. 2021;18:345-62
103. Sun C, Mezzadra R, Schumacher TN. Regulation and Function of the PD-L1 Checkpoint. Immunity. 2018;48:434-52
104. Hirano F, Kaneko K, Tamura H. et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089-96
105. Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol. 2005;17:133-44
106. Kwok G, Yau TCC, Chiu JW, Tse E, Kwong Y-L. Pembrolizumab (Keytruda). Hum Vaccin Immunother. 2016;12:2777-89
107. Al Hadidi SA, Lee HJ. Pembrolizumab for the treatment of Hodgkin Lymphoma. Expert Opin Biol Ther. 2020;20:1275-82
108. de Sousa LG, Ferrarotto R. Pembrolizumab in the first-line treatment of advanced head and neck cancer. Expert Rev Anticancer Ther. 2021;21:1321-31
109. Qu J, Wang L, Jiang M. et al. A Review About Pembrolizumab in First-Line Treatment of Advanced NSCLC: Focus on KEYNOTE Studies. Cancer Manag Res. 2020;12:6493-509
110. Chau V, Bilusic M. Pembrolizumab in Combination with Axitinib as First-Line Treatment for Patients with Renal Cell Carcinoma (RCC): Evidence to Date. Cancer Manag Res. 2020;12:7321-30
111. Wang Q, Wu X. Primary and acquired resistance to PD-1/PD-L1 blockade in cancer treatment. Int Immunopharmacol. 2017;46:210-9
112. Johnson DB, Nebhan CA, Moslehi JJ, Balko JM. Immune-checkpoint inhibitors: long-term implications of toxicity. Nat Rev Clin Oncol. 2022;19:254-67
113. Hu S, Liu X, Li T, Li Z, Hu F. LAG3 (CD223) and autoimmunity: Emerging evidence. J Autoimmun. 2020;112:102504
114. Shi A-P, Tang X-Y, Xiong Y-L. et al. Immune Checkpoint LAG3 and Its Ligand FGL1 in Cancer. Front Immunol. 2021;12:785091
115. He Y, Rivard CJ, Rozeboom L. et al. Lymphocyte-activation gene-3, an important immune checkpoint in cancer. Cancer Sci. 2016;107:1193-7
116. Andrews LP, Marciscano AE, Drake CG, Vignali DAA. LAG3 (CD223) as a cancer immunotherapy target. Immunol Rev. 2017;276:80-96
117. Carlino MS, Larkin J, Long GV. Immune checkpoint inhibitors in melanoma. Lancet. 2021;398:1002-14
118. Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol. 2020;20:173-85
119. Du W, Yang M, Turner A. et al. TIM-3 as a Target for Cancer Immunotherapy and Mechanisms of Action. Int J Mol Sci. 2017;18:645
120. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity. 2016;44:989-1004
121. Gorman JV, Starbeck-Miller G, Pham N-LL. et al. Tim-3 directly enhances CD8 T cell responses to acute Listeria monocytogenes infection. J Immunol. 2014;192:3133-42
122. Fiori V, Magnani M, Cianfriglia M. The expression and modulation of CEACAM1 and tumor cell transformation. Ann Ist Super Sanita. 2012;48:161-71
123. Ebrahim AH, Alalawi Z, Mirandola L. et al. Galectins in cancer: carcinogenesis, diagnosis and therapy. Ann Transl Med. 2014;2:88
124. Ohue Y, Kurose K, Nishio Y, Isobe M, Oka M, Nakayama E. Abstract A101: Role of TIM-3/Galectin-9 pathway in lung cancer. Cancer Immunology Research. 2016;4:A101-A101
125. Lian J, Yue Y, Yu W, Zhang Y. Immunosenescence: a key player in cancer development. J Hematol Oncol. 2020;13:151
126. Casado JG, Pawelec G, Morgado S. et al. Expression of adhesion molecules and ligands for activating and costimulatory receptors involved in cell-mediated cytotoxicity in a large panel of human melanoma cell lines. Cancer Immunol Immunother. 2009;58:1517-26
127. Johnston RJ, Yu X, Grogan JL. The checkpoint inhibitor TIGIT limits antitumor and antiviral CD8+ T cell responses. Oncoimmunology. 2015;4:e1036214
128. Chauvin J-M, Pagliano O, Fourcade J. et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J Clin Invest. 2015;125:2046-58
129. Zhu C, Anderson AC, Schubart A. et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245-52
130. Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591-619
131. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69-74
132. Neefjes J, Jongsma MLM, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. 2011;11:823-36
133. Mchayleh W, Bedi P, Sehgal R, Solh M. Chimeric Antigen Receptor T-Cells: The Future is Now. J Clin Med. 2019;8:207
134. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227-42
135. Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 Costimulation: From Mechanism to Therapy. Immunity. 2016;44:973-88
136. Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol. 2001;1:220-8
137. Dong C, Juedes AE, Temann UA. et al. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature. 2001;409:97-101
138. Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229:152-72
139. Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2:116-26
140. Wikenheiser DJ, Stumhofer JS. ICOS Co-Stimulation: Friend or Foe? Front Immunol. 2016;7:304
141. Swallow MM, Wallin JJ, Sha WC. B7h, a novel costimulatory homolog of B7.1 and B7.2, is induced by TNFalpha. Immunity. 1999;11:423-32
142. Chen Q, Mo L, Cai X. et al. ICOS signal facilitates Foxp3 transcription to favor suppressive function of regulatory T cells. Int J Med Sci. 2018;15:666-73
143. Tu J-F, Ding Y-H, Ying X-H. et al. Regulatory T cells, especially ICOS+ FOXP3+ regulatory T cells, are increased in the hepatocellular carcinoma microenvironment and predict reduced survival. Sci Rep. 2016;6:35056
144. Aspeslagh S, Postel-Vinay S, Rusakiewicz S, Soria J-C, Zitvogel L, Marabelle A. Rationale for anti-OX40 cancer immunotherapy. Eur J Cancer. 2016;52:50-66
145. Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol Rev. 2009;229:173-91
146. Croft M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu Rev Immunol. 2010;28:57-78
147. Messenheimer DJ, Jensen SM, Afentoulis ME. et al. Timing of PD-1 Blockade Is Critical to Effective Combination Immunotherapy with Anti-OX40. Clin Cancer Res. 2017;23:6165-77
148. Martinez-Forero I, Azpilikueta A, Bolaños-Mateo E. et al. T cell costimulation with anti-CD137 monoclonal antibodies is mediated by K63-polyubiquitin-dependent signals from endosomes. J Immunol. 2013;190:6694-706
149. Kim AMJ, Nemeth MR, Lim S-O. 4-1BB: A promising target for cancer immunotherapy. Front Oncol. 2022;12:968360
150. Ying Z, He T, Wang X. et al. Parallel Comparison of 4-1BB or CD28 Co-stimulated CD19-Targeted CAR-T Cells for B Cell Non-Hodgkin's Lymphoma. Mol Ther Oncolytics. 2019;15:60-8
151. Maude SL, Laetsch TW, Buechner J. et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med. 2018;378:439-48
152. Massarelli E, Segal NH, Ribrag V. et al. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Journal for ImmunoTherapy of Cancer. 2016;4:82
153. Ephrem A, Epstein AL, Stephens GL, Thornton AM, Glass D, Shevach EM. Modulation of Treg cells/T effector function by GITR signaling is context-dependent. Eur J Immunol. 2013;43:2421-9
154. Knee DA, Hewes B, Brogdon JL. Rationale for anti-GITR cancer immunotherapy. Eur J Cancer. 2016;67:1-10
155. Riccardi C, Ronchetti S, Nocentini G. Glucocorticoid-induced TNFR-related gene (GITR) as a therapeutic target for immunotherapy. Expert Opin Ther Targets. 2018;22:783-97
156. Schaer DA, Murphy JT, Wolchok JD. Modulation of GITR for cancer immunotherapy. Curr Opin Immunol. 2012;24:217-24
157. Nocentini G, Giunchi L, Ronchetti S. et al. A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor-induced apoptosis. Proc Natl Acad Sci U S A. 1997;94:6216-21
158. Cohen AD, Schaer DA, Liu C. et al. Agonist anti-GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra-tumor accumulation. PLoS One. 2010;5:e10436
159. Denlinger CS, Infante JR, Aljumaily R. et al. A phase I study of MEDI1873, a novel GITR agonist, in advanced solid tumors. Annals of Oncology. 2018;29:viii411
160. Tang T, Cheng X, Truong B, Sun L, Yang X, Wang H. Molecular basis and therapeutic implications of CD40/CD40L immune checkpoint. Pharmacol Ther. 2021;219:107709
161. Muik A, Adams 3rd HC, Gieseke F. et al. DuoBody-CD40x4-1BB induces dendritic-cell maturation and enhances T-cell activation through conditional CD40 and 4-1BB agonist activity. J Immunother Cancer. 2022;10:e004322
162. Laman JD, Claassen E, Noelle RJ. Functions of CD40 and Its Ligand, gp39 (CD40L). Crit Rev Immunol. 2017;37:371-420
163. Vonderheide RH. Prospect of targeting the CD40 pathway for cancer therapy. Clin Cancer Res. 2007;13:1083-8
164. Beatty GL, Torigian DA, Chiorean EG. et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;19:6286-95
165. Vonderheide RH, Burg JM, Mick R. et al. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology. 2013;2:e23033
166. Nowak AK, Cook AM, McDonnell AM. et al. A phase 1b clinical trial of the CD40-activating antibody CP-870,893 in combination with cisplatin and pemetrexed in malignant pleural mesothelioma. Ann Oncol. 2015;26:2483-90
167. Paulos CM, Wrzesinski C, Kaiser A. et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest. 2007;117:2197-204
168. Sivan A, Corrales L, Hubert N. et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350:1084-9
169. Vetizou M, Pitt JM, Daillere R. et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079-84
170. Routy B, Le Chatelier E, Derosa L. et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91-7
171. Matson V, Fessler J, Bao R. et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359:104-8
172. Song P, Yang D, Wang H. et al. Relationship between intestinal flora structure and metabolite analysis and immunotherapy efficacy in Chinese NSCLC patients. Thorac Cancer. 2020;11:1621-32
173. Zheng Y, Wang T, Tu X. et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J Immunother Cancer. 2019;7:193
174. Jin Y, Dong H, Xia L. et al. The Diversity of Gut Microbiome is Associated With Favorable Responses to Anti-Programmed Death 1 Immunotherapy in Chinese Patients With NSCLC. J Thorac Oncol. 2019;14:1378-89
175. Derosa L, Routy B, Fidelle M. et al. Gut Bacteria Composition Drives Primary Resistance to Cancer Immunotherapy in Renal Cell Carcinoma Patients. European urology. 2020/05/08 ed. 2020;78:195-206
176. Poore GD, Kopylova E, Zhu Q. et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature. 2020;579:567-74
177. Nejman D, Livyatan I, Fuks G. et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science. 2020;368:973-80
178. Cullin N, Azevedo Antunes C, Straussman R, Stein-Thoeringer CK, Elinav E. Microbiome and cancer. Cancer Cell. 2021;39:1317-41
179. Sepich-Poore GD, Zitvogel L, Straussman R, Hasty J, Wargo JA, Knight R. The microbiome and human cancer. Science. 2021;371:eabc4552
180. Derosa L, Routy B, Desilets A. et al. Microbiota-Centered Interventions: The Next Breakthrough in Immuno-Oncology? Cancer Discov. 2021;11:2396-412
181. Conti G, D'Amico F, Fabbrini M, Brigidi P, Barone M, Turroni S. Pharmacomicrobiomics in Anticancer Therapies: Why the Gut Microbiota Should Be Pointed Out. Genes (Basel). 2022;14:55
182. Viaud S, Saccheri F, Mignot G. et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342:971-6
183. Daillère R, Vétizou M, Waldschmitt N. et al. Enterococcus hirae and Barnesiella intestinihominis Facilitate Cyclophosphamide-Induced Therapeutic Immunomodulatory Effects. Immunity. 2016;45:931-43
184. Roberti MP, Yonekura S, Duong CPM. et al. Chemotherapy-induced ileal crypt apoptosis and the ileal microbiome shape immunosurveillance and prognosis of proximal colon cancer. Nature medicine. 2020;26:919-31
185. Uribe-Herranz M, Rafail S, Beghi S. et al. Gut microbiota modulate dendritic cell antigen presentation and radiotherapy-induced antitumor immune response. The Journal of clinical investigation [Internet]. 2019 Available at: http://www.ncbi.nlm.nih.gov/pubmed/31815742 https://dm5migu4zj3pb.cloudfront.net/manuscripts/124000/124332/JCI124332.v1.pdf
186. Shi Y, Zheng W, Yang K. et al. Intratumoral accumulation of gut microbiota facilitates CD47-based immunotherapy via STING signaling. J Exp Med. 2020;217:e20192282
187. Li Y, Tinoco R, Elmén L. et al. Gut microbiota dependent anti-tumor immunity restricts melanoma growth in Rnf5-/- mice. Nat Commun. 2019;10:1492
188. Gil-Cruz C, Perez-Shibayama C, De Martin A. et al. Microbiota-derived peptide mimics drive lethal inflammatory cardiomyopathy. Science. 2019;366:881-6
189. Hebbandi Nanjundappa R, Ronchi F, Wang J. et al. A Gut Microbial Mimic that Hijacks Diabetogenic Autoreactivity to Suppress Colitis. Cell. 2017;171:655-667.e17
190. Petersen J, Ciacchi L, Tran MT. et al. T cell receptor cross-reactivity between gliadin and bacterial peptides in celiac disease. Nat Struct Mol Biol. 2020;27:49-61
191. Snyder A, Makarov V, Merghoub T. et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189-99
192. Fluckiger A, Daillère R, Sassi M. et al. Cross-reactivity between tumor MHC class I-restricted antigens and an enterococcal bacteriophage. Science. 2020;369:936-42
193. Bessell CA, Isser A, Havel JJ. et al. Commensal bacteria stimulate antitumor responses via T cell cross-reactivity. JCI Insight. 2020;5:e135597 135597
194. Tanoue T, Morita S, Plichta DR. et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature. 2019;565:600-5
195. Wu Q, Yu X, Li J, Sun S, Tu Y. Metabolic regulation in the immune response to cancer. Cancer Commun (Lond). 2021;41:661-94
196. Guo H, Chou W-C, Lai Y. et al. Multi-omics analyses of radiation survivors identify radioprotective microbes and metabolites. Science. 2020;370:eaay9097
197. Coutzac C, Jouniaux J-M, Paci A. et al. Systemic short chain fatty acids limit antitumor effect of CTLA-4 blockade in hosts with cancer. Nat Commun. 2020;11:2168
198. Nomura M, Nagatomo R, Doi K. et al. Association of Short-Chain Fatty Acids in the Gut Microbiome With Clinical Response to Treatment With Nivolumab or Pembrolizumab in Patients With Solid Cancer Tumors. JAMA Netw Open. 2020;3:e202895
199. Li Y, Elmén L, Segota I. et al. Prebiotic-Induced Anti-tumor Immunity Attenuates Tumor Growth. Cell Rep. 2020;30:1753-1766.e6
200. Gournay V, Vallet N, Peux V. et al. Immune landscape after allo-HSCT: TIGIT- and CD161-expressing CD4 T cells are associated with subsequent leukemia relapse. Blood. 2022;140:1305-21
201. Sendker S, Reinhardt D, Niktoreh N. Redirecting the Immune Microenvironment in Acute Myeloid Leukemia. Cancers (Basel). 2021;13:1423
202. Peled JU, Gomes ALC, Devlin SM. et al. Microbiota as Predictor of Mortality in Allogeneic Hematopoietic-Cell Transplantation. N Engl J Med. 2020;382:822-34
203. Schluter J, Peled JU, Taylor BP. et al. The gut microbiota is associated with immune cell dynamics in humans. Nature. 2020/11/27 ed. 2020;588:303-7
204. Staffas A, Burgos da Silva M, Slingerland AE. et al. Nutritional Support from the Intestinal Microbiota Improves Hematopoietic Reconstitution after Bone Marrow Transplantation in Mice. Cell Host Microbe. 2018;23:447-457.e4
205. Fischer JC, Bscheider M, Eisenkolb G. et al. RIG-I/MAVS and STING signaling promote gut integrity during irradiation- and immune-mediated tissue injury. Sci Transl Med. 2017;9:eaag2513
206. Kim OY, Park HT, Dinh NTH. et al. Bacterial outer membrane vesicles suppress tumor by interferon-γ-mediated antitumor response. Nat Commun. 2017;8:626
207. Singh N, Gurav A, Sivaprakasam S. et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40:128-39
208. Iida N, Dzutsev A, Stewart CA. et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342:967-70
209. Wang L, Tang L, Feng Y. et al. A purified membrane protein from Akkermansia muciniphila or the pasteurised bacterium blunts colitis associated tumourigenesis by modulation of CD8+ T cells in mice. Gut. 2020;69:1988-97
210. Meisel M, Hinterleitner R, Pacis A. et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature. 2018;557:580-4
211. Vicente-Dueñas C, Janssen S, Oldenburg M. et al. An intact gut microbiome protects genetically predisposed mice against leukemia. Blood. 2020;136:2003-17
212. Mager LF, Burkhard R, Pett N. et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science. 2020;369:1481-9
213. Amit M, Takahashi H, Dragomir MP. et al. Loss of p53 drives neuron reprogramming in head and neck cancer. Nature. 2020;578:449-54
214. Venkataramani V, Tanev DI, Strahle C. et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature. 2019;573:532-8
215. Zeng Q, Michael IP, Zhang P. et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature. 2019;573:526-31
216. Venkatesh HS, Morishita W, Geraghty AC. et al. Electrical and synaptic integration of glioma into neural circuits. Nature. 2019;573:539-45
217. Muller PA, Matheis F, Schneeberger M, Kerner Z, Jové V, Mucida D. Microbiota-modulated CART+ enteric neurons autonomously regulate blood glucose. Science. 2020;370:314-21
218. Goto T. Microbiota and lung cancer. Semin Cancer Biol. 2022;86:1-10
219. Rizzo A, Santoni M, Mollica V, Fiorentino M, Brandi G, Massari F. Microbiota and prostate cancer. Semin Cancer Biol. 2022;86:1058-65
220. Woo YR, Cho SH, Lee JD, Kim HS. The Human Microbiota and Skin Cancer. Int J Mol Sci. 2022;23:1813
221. O'Dwyer DN, Dickson RP, Moore BB. The Lung Microbiome, Immunity, and the Pathogenesis of Chronic Lung Disease. J Immunol. 2016;196:4839-47
222. Jin C, Lagoudas GK, Zhao C. et al. Commensal Microbiota Promote Lung Cancer Development via γδ T Cells. Cell. 2019;176:998-1013.e16
223. Le Noci V, Guglielmetti S, Arioli S. et al. Modulation of Pulmonary Microbiota by Antibiotic or Probiotic Aerosol Therapy: A Strategy to Promote Immunosurveillance against Lung Metastases. Cell Rep. 2018;24:3528-38
224. Tsay J-CJ, Wu BG, Sulaiman I. et al. Lower Airway Dysbiosis Affects Lung Cancer Progression. Cancer Discov. 2021;11:293-307
225. Greathouse KL, White JR, Vargas AJ. et al. Interaction between the microbiome and TP53 in human lung cancer. Genome Biol. 2018;19:123
226. Squarzanti DF, Zavattaro E, Pizzimenti S, Amoruso A, Savoia P, Azzimonti B. Non-Melanoma Skin Cancer: news from microbiota research. Crit Rev Microbiol. 2020;46:433-49
227. Nakatsuji T, Chen TH, Butcher AM. et al. A commensal strain of Staphylococcus epidermidis protects against skin neoplasia. Sci Adv. 2018;4:eaao4502
228. Hoste E, Arwert EN, Lal R. et al. Innate sensing of microbial products promotes wound-induced skin cancer. Nat Commun. 2015;6:5932
229. Liu J, Luo M, Zhang Y, Cao G, Wang S. Association of high-risk human papillomavirus infection duration and cervical lesions with vaginal microbiota composition. Ann Transl Med. 2020;8:1161
230. So KA, Yang EJ, Kim NR. et al. Changes of vaginal microbiota during cervical carcinogenesis in women with human papillomavirus infection. PLoS One. 2020;15:e0238705
231. Wong-Rolle A, Wei HK, Zhao C, Jin C. Unexpected guests in the tumor microenvironment: microbiome in cancer. Protein & cell. 2020/12/10 ed. 2021;12:426-35
232. Oliva M, Mulet-Margalef N, Ochoa-De-Olza M. et al. Tumor-Associated Microbiome: Where Do We Stand? Int J Mol Sci. 2021;22:1446
233. Chen Y, Wu F-H, Wu P-Q, Xing H-Y, Ma T. The Role of The Tumor Microbiome in Tumor Development and Its Treatment. Front Immunol. 2022;13:935846
234. Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol. 2012;10:575-82
235. Helmink BA, Khan MAW, Hermann A, Gopalakrishnan V, Wargo JA. The microbiome, cancer, and cancer therapy. Nat Med. 2019;25:377-88
236. Barrett M, Hand CK, Shanahan F, Murphy T, O'Toole PW. Mutagenesis by Microbe: the Role of the Microbiota in Shaping the Cancer Genome. Trends Cancer. 2020;6:277-87
237. Boot A, Ng AWT, Chong FT. et al. Characterization of colibactin-associated mutational signature in an Asian oral squamous cell carcinoma and in other mucosal tumor types. Genome Res. 2020;30:803-13
238. Pleguezuelos-Manzano C, Puschhof J, Rosendahl Huber A. et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature. 2020;580:269-73
239. Dejea CM, Fathi P, Craig JM. et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science. 2018;359:592-7
240. Geller LT, Barzily-Rokni M, Danino T. et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science. 2017;357:1156-60
241. Yu T, Guo F, Yu Y. et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell. 2017;170:548-563.e16
242. Aykut B, Pushalkar S, Chen R. et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature. 2019;574:264-7
243. Parhi L, Alon-Maimon T, Sol A. et al. Breast cancer colonization by Fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nat Commun. 2020;11:3259
244. Le Noci V, Guglielmetti S, Arioli S. et al. Modulation of Pulmonary Microbiota by Antibiotic or Probiotic Aerosol Therapy: A Strategy to Promote Immunosurveillance against Lung Metastases. Cell reports. 2018;24:3528-38
245. Shang S, Hua F, Hu Z-W. The regulation of β-catenin activity and function in cancer: therapeutic opportunities. Oncotarget. 2017;8:33972-89
246. Garrett WS. Cancer and the microbiota. Science. 2015;348:80-6
247. Jin C, Lagoudas GK, Zhao C. et al. Commensal Microbiota Promote Lung Cancer Development via γδ T Cells. Cell. 2019/02/05 ed. 2019;176:998-1013.e16
248. Pushalkar S, Hundeyin M, Daley D. et al. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov. 2018;8:403-16
249. Mima K, Sukawa Y, Nishihara R. et al. Fusobacterium nucleatum and T Cells in Colorectal Carcinoma. JAMA Oncol. 2015;1:653-61
250. Hamada T, Zhang X, Mima K. et al. Fusobacterium nucleatum in Colorectal Cancer Relates to Immune Response Differentially by Tumor Microsatellite Instability Status. Cancer Immunol Res. 2018;6:1327-36
251. Kostic AD, Chun E, Robertson L. et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14:207-15
252. Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther. 2020;5:209
253. Lam KC, Araya RE, Huang A. et al. Microbiota triggers STING-type I IFN-dependent monocyte reprogramming of the tumor microenvironment. Cell. 2021;184:5338-5356.e21
254. Riquelme E, Zhang Y, Zhang L. et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell. 2019;178:795-806.e12
255. Zhu G, Su H, Johnson CH, Khan SA, Kluger H, Lu L. Intra-tumor microbiome associated with the infiltration of cytotoxic CD8+ T cells and patient survival in cutaneous melanoma. Eur J Cancer. 2021;151:25-34
256. Wang J, Li R, Cao Y. et al. Intratumoral CXCR5+CD8+T associates with favorable clinical outcomes and immunogenic contexture in gastric cancer. Nat Commun. 2021;12:3080
257. Cheng Y, Gunasegaran B, Singh HD. et al. Non-terminally exhausted tumor-resident memory HBV-specific T cell responses correlate with relapse-free survival in hepatocellular carcinoma. Immunity. 2021;54:1825-1840.e7
258. Miller NJ, Church CD, Dong L. et al. Tumor-Infiltrating Merkel Cell Polyomavirus-Specific T Cells Are Diverse and Associated with Improved Patient Survival. Cancer Immunol Res. 2017;5:137-47
259. Welters MJP, Ma W, Santegoets SJAM. et al. Intratumoral HPV16-Specific T Cells Constitute a Type I-Oriented Tumor Microenvironment to Improve Survival in HPV16-Driven Oropharyngeal Cancer. Clin Cancer Res. 2018;24:634-47
260. Kalaora S, Nagler A, Nejman D. et al. Identification of bacteria-derived HLA-bound peptides in melanoma. Nature. 2021;592:138-43
261. Zhou S, Gravekamp C, Bermudes D, Liu K. Tumour-targeting bacteria engineered to fight cancer. Nat Rev Cancer. 2018;18:727-43
262. Lu Y, Yuan X, Wang M. et al. Gut microbiota influence immunotherapy responses: mechanisms and therapeutic strategies. J Hematol Oncol. 2022;15:47
263. Frankel AE, Coughlin LA, Kim J. et al. Metagenomic Shotgun Sequencing and Unbiased Metabolomic Profiling Identify Specific Human Gut Microbiota and Metabolites Associated with Immune Checkpoint Therapy Efficacy in Melanoma Patients. Neoplasia. 2017;19:848-55
264. Alam A, Leoni G, Quiros M. et al. The microenvironment of injured murine gut elicits a local pro-restitutive microbiota. Nat Microbiol. 2016;1:15021
265. Ansaldo E, Slayden LC, Ching KL. et al. Akkermansia muciniphila induces intestinal adaptive immune responses during homeostasis. Science. 2019;364:1179-84
266. Uribe-Herranz M, Bittinger K, Rafail S. et al. Gut microbiota modulates adoptive cell therapy via CD8α dendritic cells and IL-12. JCI Insight. 2018;3:e94952
267. Yakoub-Agha I, Chabannon C, Bader P. et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Haematologica. 2020;105:297-316
268. Borody TJ, Khoruts A. Fecal microbiota transplantation and emerging applications. Nat Rev Gastroenterol Hepatol. 2011;9:88-96
269. Wang Y, Wiesnoski DH, Helmink BA. et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat Med. 2018;24:1804-8
270. Davar D, Dzutsev AK, McCulloch JA. et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science. 2021;371:595-602
271. Woelk CH, Snyder A. Modulating gut microbiota to treat cancer. Science. 2021;371:573-4
272. Dewulf EM, Cani PD, Claus SP. et al. Insight into the prebiotic concept: lessons from an exploratory, double blind intervention study with inulin-type fructans in obese women. Gut. 2013;62:1112-21
273. Azcarate-Peril MA, Ritter AJ, Savaiano D. et al. Impact of short-chain galactooligosaccharides on the gut microbiome of lactose-intolerant individuals. Proc Natl Acad Sci U S A. 2017;114:E367-75
274. Richard C, Benlaifaoui M, Ouarzadi OE. et al. 679 High fiber diet modifies gut microbiome, propionate production, intratumor immune response and is associated with outcome in patients with lung cancer treated with immune checkpoint inhibitors. J Immunother Cancer [Internet]. 2020 [cited 30 October 2023]; 8. Available at: https://jitc.bmj.com/content/8/Suppl_3/A408.1
275. Ferrere G, Tidjani Alou M, Liu P. et al. Ketogenic diet and ketone bodies enhance the anticancer effects of PD-1 blockade. JCI Insight. 2021;6:e145207 145207
276. David LA, Maurice CF, Carmody RN. et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559-63
277. Wu Q, Gao Z-J, Yu X, Wang P. Dietary regulation in health and disease. Signal Transduct Target Ther. 2022;7:252
278. Si W, Liang H, Bugno J. et al. Lactobacillus rhamnosus GG induces cGAS/STING- dependent type I interferon and improves response to immune checkpoint blockade. Gut. 2022;71:521-33
279. Jacouton E, Chain F, Sokol H, Langella P, Bermúdez-Humarán LG. Probiotic Strain Lactobacillus casei BL23 Prevents Colitis-Associated Colorectal Cancer. Front Immunol. 2017;8:1553
280. Hu J, Wang C, Ye L. et al. Anti-tumour immune effect of oral administration of Lactobacillus plantarum to CT26 tumour-bearing mice. J Biosci. 2015;40:269-79
281. Hibberd AA, Lyra A, Ouwehand AC. et al. Intestinal microbiota is altered in patients with colon cancer and modified by probiotic intervention. BMJ Open Gastroenterol. 2017;4:e000145
282. Spencer CN, Gopalakrishnan V, McQuade J. et al. Abstract 2838: The gut microbiome (GM) and immunotherapy response are influenced by host lifestyle factors. Cancer Research. 2019;79:2838
283. Pinato DJ, Howlett S, Ottaviani D. et al. Association of Prior Antibiotic Treatment With Survival and Response to Immune Checkpoint Inhibitor Therapy in Patients With Cancer. JAMA Oncol. 2019;5:1774-8
284. Lalani A-KA, Xie W, Braun DA. et al. Effect of Antibiotic Use on Outcomes with Systemic Therapies in Metastatic Renal Cell Carcinoma. Eur Urol Oncol. 2020;3:372-81
285. Ueda K, Yonekura S, Ogasawara N. et al. The Impact of Antibiotics on Prognosis of Metastatic Renal Cell Carcinoma in Japanese Patients Treated With Immune Checkpoint Inhibitors. Anticancer Res. 2019;39:6265-71
286. Chalabi M, Cardona A, Nagarkar DR. et al. Efficacy of chemotherapy and atezolizumab in patients with non-small-cell lung cancer receiving antibiotics and proton pump inhibitors: pooled post hoc analyses of the OAK and POPLAR trials. Ann Oncol. 2020;31:525-31
287. Khan U, Ho K, Hwang EK. et al. Impact of Use of Antibiotics on Response to Immune Checkpoint Inhibitors and Tumor Microenvironment. Am J Clin Oncol. 2021;44:247-53
288. Ahmed J, Kumar A, Parikh K. et al. Use of broad-spectrum antibiotics impacts outcome in patients treated with immune checkpoint inhibitors. Oncoimmunology. 2018;7:e1507670
289. Reikvam DH, Erofeev A, Sandvik A. et al. Depletion of murine intestinal microbiota: effects on gut mucosa and epithelial gene expression. PLoS One. 2011;6:e17996
290. Hakozaki T, Richard C, Elkrief A. et al. The Gut Microbiome Associates with Immune Checkpoint Inhibition Outcomes in Patients with Advanced Non-Small Cell Lung Cancer. Cancer Immunol Res. 2020;8:1243-50
291. Derosa L, Hellmann MD, Spaziano M. et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann Oncol. 2018;29:1437-44
292. Yoon MY, Yoon SS. Disruption of the Gut Ecosystem by Antibiotics. Yonsei Med J. 2018;59:4-12
293. Weersma RK, Zhernakova A, Fu J. Interaction between drugs and the gut microbiome. Gut. 2020;69:1510-9
294. Zhao S, Gao G, Li W. et al. Antibiotics are associated with attenuated efficacy of anti-PD-1/PD-L1 therapies in Chinese patients with advanced non-small cell lung cancer. Lung Cancer. 2019;130:10-7
295. Friedman GD, Oestreicher N, Chan J, Quesenberry CP, Udaltsova N, Habel LA. Antibiotics and risk of breast cancer: up to 9 years of follow-up of 2.1 million women. Cancer Epidemiol Biomarkers Prev. 2006;15:2102-6
296. Simin J, Tamimi RM, Engstrand L, Callens S, Brusselaers N. Antibiotic use and the risk of breast cancer: A systematic review and dose-response meta-analysis. Pharmacol Res. 2020;160:105072
297. Simin J, Fornes R, Liu Q. et al. Antibiotic use and risk of colorectal cancer: a systematic review and dose-response meta-analysis. Br J Cancer. 2020;123:1825-32
298. Cheng M, Qian L, Shen G. et al. Microbiota modulate tumoral immune surveillance in lung through a γδT17 immune cell-dependent mechanism. Cancer Res. 2014;74:4030-41
299. Pinato DJ, Howlett S, Ottaviani D. et al. Association of Prior Antibiotic Treatment With Survival and Response to Immune Checkpoint Inhibitor Therapy in Patients With Cancer. JAMA oncology. 2019/09/13 ed. 2019;5:1774-8
300. Wu H-J, Ivanov II, Darce J. et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32:815-27
301. Ivanov II, Frutos R de L, Manel N. et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4:337-49
302. Misra R, Sahoo SK. Coformulation of doxorubicin and curcumin in poly(D,L-lactide-co-glycolide) nanoparticles suppresses the development of multidrug resistance in K562 cells. Mol Pharm. 2011;8:852-66
303. Ravà M, D'Andrea A, Nicoli P. et al. Therapeutic synergy between tigecycline and venetoclax in a preclinical model of MYC/BCL2 double-hit B cell lymphoma. Sci Transl Med. 2018;10:eaan8723
304. Liu L, Li M, Yu M. et al. Natural polysaccharides exhibit anti-tumor activity by targeting gut microbiota. Int J Biol Macromol. 2019;121:743-51
305. Su J, Li D, Chen Q. et al. Anti-breast Cancer Enhancement of a Polysaccharide From Spore of Ganoderma lucidum With Paclitaxel: Suppression on Tumor Metabolism With Gut Microbiota Reshaping. Front Microbiol. 2018;9:3099
306. Uribe-Herranz M, Rafail S, Beghi S. et al. Gut microbiota modulate dendritic cell antigen presentation and radiotherapy-induced antitumor immune response. J Clin Invest. 2020;130:466-79
307. Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res. 2006;12:5423-34
308. Wilson BE, Routy B, Nagrial A, Chin VT. The effect of antibiotics on clinical outcomes in immune-checkpoint blockade: a systematic review and meta-analysis of observational studies. Cancer Immunol Immunother. 2020;69:343-54
309. Palleja A, Mikkelsen KH, Forslund SK. et al. Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat Microbiol. 2018;3:1255-65
310. Overman MJ, Lonardi S, Wong KYM. et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol. 2018;36:773-9
311. Giannakis M, Mu XJ, Shukla SA. et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016;15:857-65
312. Chen P-L, Roh W, Reuben A. et al. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov. 2016;6:827-37
313. Tumeh PC, Harview CL, Yearley JH. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568-71
314. Topalian SL, Hodi FS, Brahmer JR. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-54
315. Lei X, Lei Y, Li J-K. et al. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020;470:126-33
316. Menzel A, Samouda H, Dohet F, Loap S, Ellulu MS, Bohn T. Common and Novel Markers for Measuring Inflammation and Oxidative Stress Ex Vivo in Research and Clinical Practice-Which to Use Regarding Disease Outcomes? Antioxidants (Basel). 2021;10:414
317. Shapiro H, Thaiss CA, Levy M, Elinav E. The cross talk between microbiota and the immune system: metabolites take center stage. Current Opinion in Immunology. 2014;30:54-62
318. Bucci V, Xavier JB. Towards predictive models of the human gut microbiome. J Mol Biol. 2014;426:3907-16
319. Dohlman AB, Arguijo Mendoza D, Ding S. et al. The cancer microbiome atlas: a pan-cancer comparative analysis to distinguish tissue-resident microbiota from contaminants. Cell Host Microbe. 2021;29:281-298.e5
Author contact
![]() Corresponding authors: Dr. Shengrong Sun, Department of Breast and Thyroid Surgery, Renmin Hospital of Wuhan University, 238 Ziyang Road, Wuhan 430060, Hubei Province, P. R. China. E-mail: sun137com; Dr. Qi Wu, Tongji University Cancer Center, Shanghai Tenth People's Hospital, School of Medicine, Tongji University, Shanghai, P. R. China. E-mail: waiwaiedu.cn.
Corresponding authors: Dr. Shengrong Sun, Department of Breast and Thyroid Surgery, Renmin Hospital of Wuhan University, 238 Ziyang Road, Wuhan 430060, Hubei Province, P. R. China. E-mail: sun137com; Dr. Qi Wu, Tongji University Cancer Center, Shanghai Tenth People's Hospital, School of Medicine, Tongji University, Shanghai, P. R. China. E-mail: waiwaiedu.cn.