Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. The PI3K/Akt pathway at a...
3. The PI3K/Akt cascade and...
4. The PI3K/Akt cascade and...
5. Clinical implications
6. Conclusions and future...
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(8):3113-3125. doi:10.7150/ijbs.89942 This issue Cite
Review
The PI3K/Akt Pathway and Glucose Metabolism: A Dangerous Liaison in Cancer
Fabrizio Fontana ![]() , Gaia Giannitti, Sara Marchesi, Patrizia Limonta
, Gaia Giannitti, Sara Marchesi, Patrizia Limonta
Department of Pharmacological and Biomolecular Sciences "Rodolfo Paoletti", Università degli Studi di Milano, Milan, Italy.
Received 2023-9-7; Accepted 2024-4-11; Published 2024-5-27
Abstract

Aberrant activation of the PI3K/Akt pathway commonly occurs in cancers and correlates with multiple aspects of malignant progression. In particular, recent evidence suggests that the PI3K/Akt signaling plays a fundamental role in promoting the so-called aerobic glycolysis or Warburg effect, by phosphorylating different nutrient transporters and metabolic enzymes, such as GLUT1, HK2, PFKB3/4 and PKM2, and by regulating various molecular networks and proteins, including mTORC1, GSK3, FOXO transcription factors, MYC and HIF-1α. This leads to a profound reprogramming of cancer metabolism, also impacting on pentose phosphate pathway, mitochondrial oxidative phosphorylation, de novo lipid synthesis and redox homeostasis and thereby allowing the fulfillment of both the catabolic and anabolic demands of tumor cells. The present review discusses the interactions between the PI3K/Akt cascade and its metabolic targets, focusing on their possible therapeutic implications.
Keywords: PI3K, Akt, aerobic glycolysis, Warburg effect, cancer metabolism
1. Introduction
The phosphoinositide 3-kinase (PI3K)/Akt pathway is one of the most frequently deregulated molecular cascades in human malignancies [1]. In physiological conditions, this signaling is triggered by growth factors, cytokines and hormones, and mediates different metabolic processes, particularly glycolysis, to support cell growth and survival. Oncogenic activation of the PI3K/Akt cascade leads to a wide metabolic reprogramming via upregulation of glucose transporters and glycolytic enzymes, also impacting on pentose phosphate pathway (PPP), mitochondrial oxidative phosphorylation, de novo lipid synthesis and redox homeostasis and thus fulfilling both the catabolic and anabolic needs of tumor cells [2]. Based on these observations, a deeper understanding of the metabolic consequences of PI3K/Akt hyperactivation in cancers may open to the identification of novel therapeutic strategies. Herein, we provide an overview of the role of the PI3K/Akt signaling in the modulation of glucose metabolism and its associated biochemical pathways in malignant cells, with a focus on their potential as new pharmacological targets.
2. The PI3K/Akt pathway at a glance
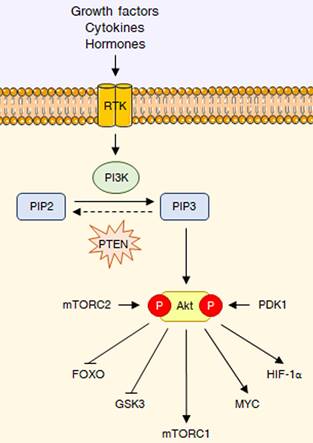
As previously mentioned, the PI3K/Akt signaling is deeply involved in the control of cell growth and proliferation [1]. PI3Ks are plasma membrane-associated lipid kinases, consisting of two regulatory subunits (p85 and p55) and a catalytic subunit (p110) [3]. Depending on their different structures and specific substrates, PI3Ks can be divided into three classes, I, II, and III: among these, PI3K IA represents the most commonly mutated protein in tumors [4]. Under physiological conditions, PI3K is generally activated by a variety of extracellular stimuli, including insulin, growth factors and cytokines [3,5]. Upon stimulation, PI3K mediates the phosphorylation of phosphatidylinositol 4,5-bisphosphate (PIP2) into phosphatidylinositol (3,4,5)-trisphosphate (PIP3), a second messenger able to bind to the pleckstrin-homology (PH) domains of a wide subset of kinases and promote their recruitment to the cell membrane; phosphatase and tensin homologue (PTEN) inhibits the pathway by dephosphorylating PIP3 to PIP2 [3,5]. One of the downstream targets of PIP3 is the serine-threonine kinase Akt (also called protein kinase B or PKB), which becomes fully active via subsequent phosphorylation at T308 by phosphoinositide-dependent protein kinase 1 (PDK1) and at S473 by mechanistic target of rapamycin complex 2 (mTORC2) [6]. Akt governs glucose metabolism either directly, by phosphorylating multiple nutrient transporters and metabolic enzymes, or indirectly, by regulating different signaling networks and transcription factors, including mTORC1, glycogen synthase kinase 3 (GSK3), members of the forkhead box O (FOXO) protein family, MYC and hypoxia-inducible factor 1α (HIF-1α) [2]. It should be noted that the genetic alterations responsible for the constitutive hyperactivation of the PI3K/Akt cascade are well-known cancer drivers. Common tumorigenic changes include: activating mutations in PIK3CA, the p110 catalytic subunit of PI3K1; deletion/loss of function in PTEN; amplification/activation of PI3K upstream regulators, particularly receptor tyrosine kinases (RTKs); amplification/gain of function in Akt [7]. Overall, these modifications result in a plethora of biological effects, such as cell cycle dysregulation, apoptosis escape and epithelial-to-mesenchymal transition (EMT), ultimately culminating in cancer growth, metastasis and drug resistance [8]. The main steps of the PI3K/Akt pathway are represented in Fig. 1.
The PI3K/Akt pathway at a glance. RTK interaction with growth factors, cytokines or hormones leads to PI3K activation, which converts PIP2 into PIP3, that can be dephosphorylated back to PIP2 by PTEN. PIP3 acts as a second messenger to recruit Akt, which it is fully activated through phosphorylation at T308 and S473 by PDK1 and mTORC2, respectively. Akt reprograms glucose metabolism through different key downstream substrates, including FOXO transcription factors, GSK3, mTORC1, MYC and HIF-1α.
3. The PI3K/Akt cascade and glucose metabolism in cancer
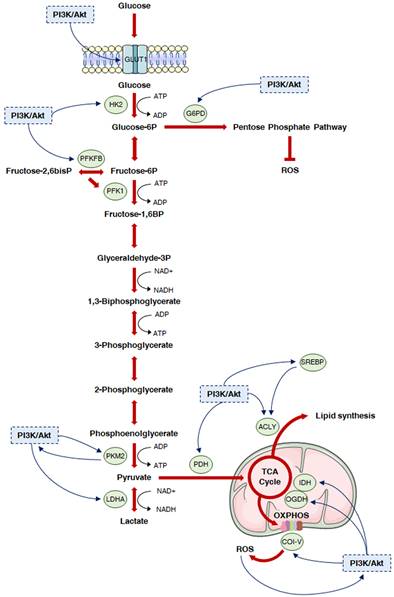
Deregulated glucose metabolism represents a distinctive trait of tumor cells. This peculiar phenotype, characterized by a high rate of glucose fermentation even in the presence of oxygen, was described by Otto Warburg in 1956 and is called aerobic glycolysis or Warburg effect [9,10]. Besides producing ATP, the increase in the glycolytic flux allows glycolytic intermediates to supply subsidiary pathways implicated in the synthesis of various macromolecules, such as nucleotides, proteins and lipids, which are essential for cell growth and survival [9,10]. Moreover, it favors the production of cofactors required for the maintenance of cellular redox state: enhanced glucose uptake allows higher generation of the reducing equivalent NADPH by the oxidative branch of the PPP, and lactic acid fermentation leads to the regeneration of the oxidizing equivalent NAD+ [11]. Finally, it has been recently postulated that the Warburg effect might result from molecular crowding, a common limitation to overall metabolism in rapidly proliferating cells [12]. Despite the enormous genetic heterogeneity of cancers, cell transformation appears to be based on the common induction of the PI3K/Akt signaling to promote the above metabolic rewiring (Fig. 2).
The PI3K/Akt cascade and glucose metabolism in cancer. The PI3K/Akt signaling is known to regulate glycolysis and different subsidiary metabolic pathways in cancer. In particular, it promotes plasma membrane localization of GLUT1, thus increasing glucose uptake. Then, it activates HK2 to facilitate the generation of glucose 6-phosphate, which can fuel both the PPP and the glycolytic flux. In this regard, the PI3K/Akt cascade can trigger nucleotide synthesis by inducing G6PD, while it can further enhance glycolysis by targeting PFKFB3 and 4, which generate the PKF1 activator fructose-2,6-bisphosphate, and PKM2, which is responsible for the production of pyruvate. Pyruvate is either converted into lactate by PI3K/Akt hyperactivation-induced LDHA or can enter the mitochondria; in these organelles, it undergoes a multistep oxidation via the TCA cycle and OXPHOS, that can be boosted by the PI3K/Akt pathway via upregulation of PDH, IDH and respiratory complexes I, III and IV. Moreover, the PI3K/Akt signaling directly stimulates lipid synthesis through direct or SREBP-dependent phosphorylation of ACLY, which generates acetyl-CoA from citrate. Finally, it controls redox homeostasis by replenishing NADPH cytosolic pool and modulating PDH, OGDH and OXPHOS complex I activity.
Glucose serves as a fundamental source of carbon and energy in all organisms. Its uptake into cells is facilitated by the family of glucose transporters (GLUTs), among which GLUT1 plays a pivotal role in tumorigenesis [13]. Interestingly, a positive correlation between GLUT1, PI3K and p-Akt expression has been reported in several cancers, including endometrial and head and neck carcinoma [14-16]. Mechanistically, it has been shown that Akt can promote the translocation of GLUT1 to the plasma membrane by phosphorylating and thus inhibiting thioredoxin-interacting protein (TXNIP), which mediates GLUT1 endocytosis and blocks glucose uptake [13,17]. This results in an increased glycolytic flux, leading to enhanced tumor growth, metastasis and chemoresistance [18]. Almost 40 compounds have been recognized as potential GLUT1 inhibitors. For example, many nutraceuticals, including a variety of alkaloids (i.e. matrine), flavonoids (i.e. apigenin, genistein and quercetin) and non-flavonoid phenolic molecules (i.e. resveratrol and curcumin), can suppress GLUT1 expression either directly or by affecting PI3K activity, while some synthetic drugs, such as BAY-876, STF-31 and WZB117, can interact with the transporter to inhibit glucose transfer [19,20]. Unfortunately, none of these chemicals has been approved by Food and Drug Administration (FDA), due to low activity and poor specificity.
Glycolysis intensity is controlled by the activity of three physiologically irreversible enzymes: hexokinase (HK), phosphofructokinase-1 (PFK-1) and pyruvate kinase (PK). Regarding the first, four different forms exist [21]. HK1, 2 and 3, which are characterized by a high affinity for glucose, consist of two similar 50-kDa N- and C-terminal lobes and can be inhibited by their product, glucose-6-phosphate; only C-terminal domain is functional in HK1 and 3, while both portions are endowed with catalytic activity in HK2. HK4 (also known as glucokinase) has only one 50-kDa lobe with reduced affinity for glucose. The expression of these isozymes varies across different tissues, with HK1 and less abundant HK3 being ubiquitously and stably expressed in all cells, HK2 being finely regulated by various hormonal or metabolic mechanisms in muscles and heart, and HK4 being mainly present in the liver, pancreas, small intestine and brain [21]. Among them, HK2 is a specific downstream target of Akt in cancer [22-30]. In particular, Akt has been observed to mediate the binding of HK2 to the voltage-dependent anion channel (VDAC) on the outer mitochondrial membrane (OMM), thereby enhancing its phosphorylating activity by allowing a direct exploitation of mitochondria-derived ATP [31-33]. Of course, this is also followed by a reduced sensitivity to apoptosis, due to a limited opening of the mitochondrial permeability transition pore (mPTP) [31-33]. The Akt/HK2 cascade is controlled by PH domain leucine-rich repeat protein phosphatase (PHLPP), which can dephosphorylate Akt and promote HK2 translocation from mitochondria to the cytosol [34]. Remarkably, PHLPP is regulated by mTORC1 and in turn inhibits the mTORC1 substrate p70S6K [35], suggesting the existence of a complex regulatory system matching HK2 levels and subcellular localization with the metabolic status of the cell. A further characterization of HK2 functions in tumors has been recently provided by Ciscato et al., who have demonstrated that 80% of this protein lodges in mitochondria-associated membranes (MAMs), small subcellular regions connecting the OMM to the endoplasmic reticulum (ER); there, it can participate to the IP3R-GRP75-VDAC1-mediated control of Ca2+ homeostasis, contributing to the proliferation of cancer cells [36]. Parallelly, Cheung and colleagues have reported the ability of HK2 to translocate to the mitochondrial surface under hypoxia and to bind to TIGAR, a fructose-2,6-bisphosphatase that inhibits glycolysis and promotes PPP; the obtained complex not only stimulates HK2 activity but also lowers mitochondrial ROS levels and protects against cell death [37]. In this intricate scenario, it is worth underlying that HK2 is overexpressed in several human malignancies, such as liver, pancreatic, colon and ovarian carcinoma, and has been associated with tumor initiation and progression in many in vivo models [38-41]. More importantly, it has been recently identified as a negative prognostic marker in glioblastoma multiforme and cervical and hepatocellular cancer, and its induction correlates with resistance to radiation, chemo- and targeted therapy [42-49]. For these reasons, HK2 upregulation has been recently proposed as a metabolic vulnerability for the management of PTEN-deficient and Akt-overexpressing tumors, including prostate cancer [39,50-53]. Indeed, HK2 can be successfully targeted by several drugs, such as the catalytic inhibitors lonidamine and 3-bromopyruvate (3-BrPyr) and the glucose-analogue 2-deoxyglucose (2-DG); however, the use of these compounds has been abandoned after the onset of severe side effects during clinical trials [54]. On the other hand, different natural agents directed against HK2, including berberine, wogonin, epigallocatechin-3-gallate, β-escin and tocotrienols, have shown no toxicity when administered in vivo but little efficacy when given to patients [20]. An alternative anti-HK2 strategy relies on the usage of specific peptides able to remove this specific enzyme from OMM without interfering with the activity of any other isoform. These chemicals contain specific sequences for plasma membrane crossing and can rapidly trigger tumor cell death via Ca2+ flux perturbation [32,36,55,56]. Nonetheless, they lack selectivity for cancer cell entry, and their pharmacological utility still needs to be validated in pre-clinical and clinical settings.
As mentioned above, PFK-1 is another major rate-limiting enzyme of glycolysis, mediating the production of fructose-1,6-bisphosphate from fructose-6-phosphate. Its most potent activator is fructose-2,6-bisphosphate, a product of the reaction catalyzed by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB). In humans, PFKFB is encoded by four genes, with PFKFB1 being found in the liver and skeletal muscle, PFKFB2 predominating in cardiac muscle, PFKFB3 being ubiquitously expressed and PFKFB4 occurring mainly in testes [57,58]. Of note, PFKFB is both a substrate and positive effector of Akt pro-tumor activity. Indeed, isoenzymes 3 and 4 can be directly phosphorylated by this kinase [59]; in turn, PFKB4 can promote Akt action by regulating the expression of histone acetyltransferase GCN5 or by interacting with isoprenylcystein carboxyl methyl transferase (ICMT), a posttranslational modifier of RAS [60,61]. As discussed in the following paragraphs of the present article, PFKFB3 and PFKFB4 affect carcinogenesis in a multidirectional manner, participating in the control of glucose metabolism by enhancing not only glycolysis but also PPP [62]. Unsurprisingly, high levels of these proteins have been found in breast, lung, colon, pancreatic, gastric, ovarian, thyroid and liver cancer, where they correlate with metastases to lymph nodes and advanced tumor staging [63-70]. Thus, inhibitors of PFKFBs have been recently designed to improve therapeutic outcomes. In particular, new small molecules directed against PFKFB3, such as 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO), compound 26, PQP, KAN0438757 and PFK15, have shown promising anti-cancer effects in vitro and in vivo [59]; more importantly, one of them, namely PFK158, has already been enrolled in a Phase I trial for the management of solid malignancies (clinicaltrials.gov #NCT02044861).
As one of the key glycolytic enzymes, PK acts on phosphoenolpyruvate to form pyruvate fueling the tricarboxylic acid (TCA) cycle in mitochondria [71]. It has four different subtypes: PKL is mainly found in the liver, PKR is mainly expressed in red blood cells, PKM1 is distributed in myocardium, skeletal muscle and brain tissue, PKM2 is present in all proliferating cells, especially embryonic cells. PKL and PKR are encoded by the PKLR gene, while PKM1 and PKM2 are transcribed from the PKM gene via alternative splicing; in particular, PKM2 possesses the PKM2-specific exon 10 and lacks the PKM1-specific exon 9. This single exon difference results in important function distinctions: PKM1 constitutively oligomerizes to a highly active tetramer under physiological conditions, while PKM2 may be present as a tetramer or a less active dimer; this implies that the main biological function of PKM1 is the generation of ATP, whereas the tetramer-to-dimer transition of PKM2 supports the formation of glycolytic intermediates for biomass production. It should also be emphasized that several metabolites, such as fructose-1,6-bisphosphate, can act as allosteric activators of PKM2 but not of PKM1 [71]. Intriguingly, a switch from PKM1 to PKM2 has been detected in various cancers, and a reverse isoform shift from PKM2 to PKM1 has been found to inhibit aerobic glycolysis and reduce tumorigenesis in multiple xenograft models [71]. In particular, recent findings point to an Akt-dependent pro-tumor modulation of PKM2 [72-77], which has also emerged as an upstream regulator of the PI3K/Akt pathway itself [78-81]; in particular, it seems to be able to activate mTORC1 by interacting with Akt1 substrate 1 (Akt1S1) [82]. In this context, aberrant PKM2 expression has been widely associated with poor prognosis in patients with solid tumors of the digestive system, such as esophageal, gastric, colorectal and liver carcinoma [83-85]. There are different small molecules able to reduce tumor growth by targeting PKM2 [20,86-88]. These drugs, particularly shikonin and its analogs, bind to the allosteric site of the enzyme, thereby impairing glucose metabolism [20,86-91]; nevertheless, they have only been tested in vitro.
Lactate production largely contributes to malignant progression, not only by replenishing NAD+ for glycolysis itself but also by lowering extracellular pH for invasion and triggering angiogenesis and immune escape [92]. Lactate dehydrogenase A (LDHA) is the main effector of glucose fermentation, converting pyruvate into lactic acid [92]. Increased levels of this enzyme have been reported in numerous malignancies, including pancreatic, nasopharyngeal, gastric, bladder and endometrial cancer, and are commonly linked to PI3K/Akt hyperactivation [92-101]. Considering this, LDHA has been regarded as an intriguing target for tumor prevention and therapy, with pyruvate- and NADH-competitive inhibitors (i.e. oxamate, gossypol, FX11, quinoline 3‐sulfonamides and N‐hydroxyindoles) and free enzyme-binding molecules (i.e. galloflavin) demonstrating great efficacy in a variety of cellular and xenograft cancer models [92]. Yet, according to recent literature, LDH inhibition can disrupt the metabolism of healthy T cells, causing severe immunosuppression [102]; to overcome this issue, lactate oxidase/catalase-displaying nanoparticles have been proposed as useful tools to consume lactate in the tumor microenvironment and effectively suppress cancer growth [103,104].
Besides directly controlling the activity of glucose transporters and glycolytic enzymes, the PI3K/Akt signaling can drive the Warburg effect by modulating the expression of MYC. This transcription factor is one of the most encountered oncogenes in tumors, where it favors aerobic glycolysis by upregulating GLUT1 and most metabolic enzymes, including HK2, PFK-1, enolase 1 (ENO1), pyruvate dehydrogenase kinase 1 (PDK1), PKM2 and LDHA [105]. The PI3K/Akt pathway promotes MYC induction via various molecular mechanisms [105]. For instance, mTORC1 increases MYC translation [106,107], while Akt stabilizes the protein via inactivation of the proteasomal degradation inducer GSK3 [108-110]. Moreover, the PI3K/Akt cascade can inhibit the suppressive effects of FOXO transcription factors on MYC-mediated increase in glycolytic flux [111]. However, it should be emphasized that MYC expression can be finely controlled by a wide range of oncoproteins, including mitogen-activated protein kinases (MAPKs) [112-114]. For this reason, the MYC-related reprogramming of glucose metabolism cannot be exclusively reconducted to the action of Akt.
A distinctive hallmark of several malignancies is that they develop in a hypoxic environment [115]. The metabolic adaptation to hypoxia is orchestrated by HIF-1, a heterodimeric transcription factor composed of two subunits, α and β. In the presence of oxygen, HIF-1α is hydroxylated at pro402 and pro564 by prolyl hydroxylases, resulting in its ubiquitination and proteasomal degradation. Under hypoxic conditions, prolyl hydroxylases are inhibited, and HIF-1α can thus translocate to the nucleus and dimerize with its partner HIF-1β. The HIF-1 dimer then binds to the hypoxia response element site on DNA, inducing the expression of a variety of genes implicated in the hypoxic response; among them, there are numerous enzymes participating in the control of the glycolytic flux, such as HK2, PFK-1, fructose-bisphosphate aldolase A (ALDOA), ENO1, PKM2 and LDHA [116]. Intriguingly, many cancers exhibit constitutive activation of HIF-1α even in normoxic conditions, and Akt is known to participate in this process via mTORC1 upregulation [117-122]. In this context, HIF1α stabilization seems to directly contribute to tumor growth, presumably endowing the tumor mass with the metabolic flexibility necessary to adapt to oxygen fluctuations.
Emerging evidence suggests that MYC and HIF also cooperate to promote the metabolic reprogramming observed in Akt-overexpressing malignancies [123]. Under hypoxia, MYC is inhibited by HIF-1α through disruption of the MYC/MAX complex, resulting in adaptive cellular changes that favor survival in low-oxygen conditions. On the other hand, when MYC is overexpressed, it reduces HIF-1α degradation and increases its activity at the chromatin levels, directly participating in the boost in the glycolytic flux [123]. In particular, it has been recently reported that mTOR-related upregulation of PKM2 in mouse kidney tumors results from HIF-1α-mediated transcription activation and c-Myc-heterogeneous nuclear ribonucleoprotein (hnRNP)-dependent regulation of PKM2 gene splicing [124]. Given the importance of the MYC-HIF interplay in cancer cells, it would be critical to further understand how these proteins precisely interact with each other to jointly regulate the expression of metabolic genes.
Cancer onset and evolution are not only influenced by genetic/epigenetic changes in malignant cells but also by the rearrangement of the components of the tumor microenvironment through a bidirectional and dynamic crosstalk [125]. Among the different cell types that are found in tumor stroma, adipocytes have emerged as critical regulators of cancer metabolism [126]. In particular, periprostatic adipose tissue has been found to induce tumor switch towards the Warburg phenotype through Akt/HIF-1α activation [127]. Likewise, bone marrow adipose cells have been shown to fuel the glycolytic pathway of metastatic prostate carcinoma by promoting Akt phosphorylation [128]. Remarkably, these metabolic alterations seem to be mediated by both soluble and insoluble extracellular factors.
4. The PI3K/Akt cascade and other glycolysis-associated metabolic processes
The above evidence emphasizes the crucial role played by the PI3K/Akt signaling in the induction of aerobic glycolysis in cancer. In addition to accelerating ATP production, this phenotypic switch improves the metabolic flux into various glucose-dependent pathways that are responsible for the synthesis of cellular macromolecules [129]. As discussed in the following paragraphs of this article, these biosynthetic mechanisms are also under control of the PI3K/Akt cascade (Fig. 2).
4.1 Pentose phosphate pathway
Nucleotides serve as monomeric units of DNA and RNA [130]. In tumors, de novo synthesis of these molecules is commonly exacerbated to sustain unlimited cell growth and proliferation [131]. This process is supported by different metabolic mechanisms, including the glucose-dependent PPP. The PPP consists of two reaction sequences, often referred to as the oxidative and non-oxidative branches. In the oxidative arm, glucose 6-phosphate (G6P) is converted to ribulose 5-phosphate (Ru5P) and CO2, with the formation of two molecules of NADPH essential for both anabolic reactions and ROS scavenging; in the non-oxidative phase, three molecules of Ru5P are converted to two molecules of fructose 6-phosphate (F6P) and one molecule of glyceraldehyde 3-phosphate (GAP) [132]. Glucose-6-phosphate dehydrogenase (G6PD) is the rate-controlling enzyme of the PPP, promoting the oxidation of G6P to 6-phosphoglucono-δ-lactone. By boosting the glycolytic flux, the PI3K/Akt signaling not only provides multiple co-factors for nucleotide generation, such as ATP, NADH and NADPH, but also increases the production of G6P [133]; on the other hand, it specifically promotes the induction of G6PD expression [122]. Of note, Akt can also upregulate the PPP by controlling the activity of other enzymes, either directly - as in the case of transketolase (TKT) [134,135] - or indirectly - by stimulating the MYC-related transcription and translation of phosphoribosyl pyrophosphate (PRPP) synthase 2 (PRPS2) [136,137]. Finally, it should be emphasized that Akt-dependent regulation of PFKFB3 and PFKFB4 levels might considerably impact on the crosstalk between glycolysis and PPP [138]. Indeed, it has been shown that inhibition of PFKFB3 reroutes glucose metabolism from glycolytic flux to PP synthesis; conversely, silencing of PFKFB4 causes increases in fructose-2,6-bisphosphate concentrations, which in turn divert more glucose into glycolysis at the expense of PPP and NADPH production. The differences in the function of PFKFB3 and PFKFB4 apparently allow malignant cells to exploit these isozymes to allocate glucose to either the glycolytic process or the PPP, finely regulating the balance between cellular bioenergetics and redox homeostasis [138].
4.2 Oxidative phosphorylation
Contrary to Warburg's original hypothesis that aerobic glycolysis is a consequence of impaired mitochondria, recent investigations have clearly established that mitochondrial function is still intact in many cancers [62,139-142]. In particular, it has been largely demonstrated that oxidative phosphorylation (OXPHOS) substantially contributes to ATP production and redox balance in tumor cells. Despite being at a preliminary stage, recent studies indicate that Akt exerts multiple activities in the control of mitochondrial metabolism, affecting both the TCA cycle and OXPHOS. For example, this oncoprotein has been shown to phosphorylate the catalytic subunit of pyruvate dehydrogenase (PDH), namely PDH-E1α, with great impact on acetyl-CoA generation and overall energy homeostasis [143]. Furthermore, it has been demonstrated to regulate the sterol regulatory element-binding protein (SREBP)-mediated transcription of all three isocitrate dehydrogenase (IDH) isoforms (1, 2 and 3), thus boosting the TCA cycle and linking mitochondrial respiration with lipid synthesis [144]. Finally, improved ATP production as a consequence of active Akt has been associated to mTORC1- and 4E-BP1-related modulation of the respiratory complexes I, III and IV [145]. In the light of this, many efforts are currently being made to develop safe and effective anti-cancer approaches directed against mitochondria, as extensively reviewed in [146,147].
As reported above, the Warburg Effect only partially describes the complexity of cancer metabolism. Recent studies have shown that aerobic glycolysis and OXPHOS are not mutually exclusive, with tumor cells displaying flexible metabolic phenotypes that shift in relation to the specific conditions within the microenvironment [148]. In this context, a newer theory, known as the “reverse Warburg effect”, defines a two-compartment model where cancer-associated fibroblasts (CAFs) are forced by malignant cells to undergo an Akt-dependent increase in the glycolytic flux and then transfer the lactate back to the tumor to fuel mitochondrial OXPHOS [149,150]. These dual interactions allow cancers to quickly respond to changes in nutrient availability to maximize cell proliferation and survival and are primarily mediated by monocarboxylate transporters (MCT): MCT4 promotes the release of lactate from CAFs and is upregulated by HIF-1α and NF-κB; MCT1 is predominantly involved in the uptake of this catabolite by cancer cells and is induced by MYC and TIGAR [149,150]. Of course, there are important clinical implications associated with the expanding knowledge of the tumor metabolic heterogeneity responsible for the reverse Warburg effect. Indeed, both cancer and stroma metabolic parameters might be incorporated into more reliable prognostic models, improving the prediction of disease behavior and outcomes. On the other hand, tumor-stroma metabolic coupling may represent a novel target for anti-cancer strategies.
4.3 De novo lipid synthesis
Whereas non-malignant tissues depend on extracellular fatty acids for their growth and survival, cancer cells increase de novo lipid biosynthesis to rapidly proliferate [144]. Both glycerol-3P from glycolysis and citrate from mitochondrial respiration are essential building blocks for lipid synthesis, evidencing the ability of the PI3K/Akt signaling to indirectly coordinate lipid metabolism [151,152]. On the other hand, Akt has been found to specifically modulate the expression of key enzymes controlling both fatty acid and cholesterol production.
ATP citrate lyase (ACLY) acts as a link between carbohydrate metabolism and fatty acid biosynthesis, by converting citrate into acetyl-CoA. The PI3K/Akt cascade can directly phosphorylate this enzyme and increase its activity, thus favoring tumor growth and affecting histone acetylation [153,154]. ACLY overexpression is commonly observed in a variety of human malignancies, and its targeting reduces cancer cell proliferation both in vitro and in vivo [155]; based on this evidence, ACLY inhibitors, originally designed for metabolic disorders, are now being tested as potential anti-cancer drugs [156].
Akt can also regulate de novo lipid synthesis by inducing SREBPs [157]. When inactive, these transcription factors are located within the endoplasmic reticulum; their activation requires the cleavage into a N-terminal domain which is translocated to the nucleus. Once activated, they bind to specific sterol regulatory element DNA sequences, thereby upregulating the synthesis of several enzymes implicated in fatty acid generation, including ACLY; after that, SREBP mature forms are targeted for ubiquitin-dependent degradation by GSK3-mediated phosphorylation [158]. The PI3K/Akt pathway promotes the above processing via mTORC1 induction and stimulates protein stabilization through GSK3 inhibition [159,160]. Recently, MYC has also been found to contribute to SREBP-mediated lipogenesis in tumors [161]. Of note, SREBP levels are known to be elevated in various malignancies, thus representing intriguing targets for cancer management [162].
4.4 Redox homeostasis
Reactive oxygen species (ROS) are biological byproducts of multiple biochemical processes, including glucose metabolism. Interestingly, moderate ROS generation has been shown to support cancer cell proliferation and survival, whereas higher levels of these molecules are detrimental to intracellular components [163]. In this context, the PI3K/Akt pathway stimulates both ROS-producing and -scavenging mechanisms that vary between malignant settings. For instance, ROS have been reported to promote tumor growth by activating the PI3K/Akt signaling via PTEN inhibition or direct upregulation of Akt protein [164]. Parallelly, the PI3K/Akt cascade has been found to boost the generation of superoxide and H2O2 by abolishing the negative regulation imposed by mitochondrial GSK-3β on PDH and OXPHOS complex I [164]. In addition, oncogenic PIK3CA is known to confer a peculiar metabolic state to cancer cells, characterized by the overexpression of oxoglutarate dehydrogenase (OGDH): this enzyme is not only an important ROS source but also plays a key role in fueling the malate-aspartate shuttle, which is fundamental for the cytoplasmic NAD+ regeneration that supports the rapid glycolytic flux occurring in Akt-overexpressing tumors [165]. On the other hand, Akt is able to increase the cellular reducing power available for antioxidant responses. As illustrated above, it can replenish the cytosolic pool of NADPH by boosting the oxidative branch of PPP. Moreover, it promotes the activity of NAD kinase (NADK), which catalyzes the phosphorylation of NAD+ into NADP+, the rate-limiting substrate for NADPH-producing enzymes [166]. Finally, it contributes to ROS neutralization through stabilization and sustained activation of NRF2 (nuclear factor erythroid 2-related factor 2), a transcription factor that controls the expression of numerous antioxidant genes, including those of the glutathione and thioredoxin systems [167]. Overall, this evidence points to the attractive therapeutic possibility of altering redox homeostasis in cancers exhibiting PI3K/Akt hyperactivation, by either reducing intracellular ROS content or promoting an excessive ROS production [164,168]. In this regard, the apparent cytotoxicity of ROS increase has been recently exploited in the management of PI3K inhibitor-resistant malignancies. Specifically, resistant cells cultured in the absence of PI3K inhibitors (mimicking drug holidays conditions) have been found to develop a proliferative defect due to an mTORC1-dependent accumulation of ROS [169]. Remarkably, these anti-proliferative effects can be counteracted by treatment with ROS scavengers, such as N-acetylcysteine (NAC), therefore highlighting the redox vulnerabilities of the drug-insensitive cell subpopulations emerging through therapies [169]. Of course, despite these promising data, it is important to underline that the pharmacological exploitation of ROS-associated mechanisms in oncology is complex. Indeed, there is still significant debate about the possible systemic consequences of ROS level elevation in cancer patients as well as regarding the actual benefits of antioxidants in cancer therapy [170]. Hence, it is fundamental to deeply characterize tumors at both the genetic and metabolic levels to establish whether targeting their redox homeostasis might represent an appropriate approach for their elimination.
5. Clinical implications
Due to its crucial role as an oncodriver, the PI3K/Akt pathway remains a prime candidate for therapeutic intervention. However, only five PI3K inhibitors (copanlisib, idelalisib, umbralisib, duvelisib and alpelisib) have been approved by FDA so far, while Akt inhibitors, such as MK-2206, AZD5363, GDC-0068 and perifosine, are still being tested in phase I and II clinical trials [171,172]. In particular, most drugs suffer from many adverse effects (i.e. hyperglycemia and consequent hyperinsulinemia) as well as from poor solubility and permeability. Moreover, they have demonstrated limited therapeutic benefit as single agents. Therefore, designing new molecules based on different binding sites could help increase selectivity and reduce toxicity. On the other hand, focusing on other pathways by exploiting targeted and endocrine therapies as a combinatorial approach could enhance the efficacy of available agents [171,172]. As discussed above, pharmacological targeting of specific metabolic transporters and enzymes downstream of the PI3K/Akt signaling might offer interesting alternative anti-cancer strategies to PI3K and Akt inhibitors (Table 1).
Main drugs targeting glycolysis in cancer.
| Drug | Target | Development phase | Main results obtained so far | Ref. |
|---|---|---|---|---|
| Matrine, apigenin, genistein, quercetin, resveratrol and curcumin | GLUT-1 downregulation | In vitro and vivo studies, Phase I and II trials | High activity in vitro, no toxicity but modest efficacy in vivo and in patients | [19,20] |
| BAY-876, STF-31 and WZB117 | Inhibition of GLUT-1 function | In vitro and in vivo studies | High activity in vitro, no toxicity but modest efficacy in vivo | [19,20] |
| 3-bromopyruvate and lonidamine | Catalytic inhibition of HK2 | Phase I, II and III trials | High clinical efficacy but severe toxicity | [54] |
| 2-deoxyglucose (2-DG) | Allosteric and competitive inhibition of HK2 | Phase I, II and III trials | High clinical efficacy but severe toxicity | [54] |
| Wogonin, epigallocatechin-3-gallate, β-escin and tocotrienols | HK2 downregulation | In vitro and in vivo studies, Phase I and II trials | High activity in vitro, no toxicity but modest efficacy in vivo and in patients | [20] |
| Specific anti-HK2 peptides | Selective removal of HK2 from outer mitochondrial membrane | In vitro studies | High activity | [32,36,55,56] |
| 3PO, compound 26, PQP, KAN0438757, PFK15 and PFK158 | Inhibition of PFKB3 function | In vitro and in vivo studies, Phase I trials | High activity in vitro, high efficacy and no toxicity in vivo and in patients | [59] |
| Shikonin and its analogs | Allosteric inhibition of PKM2 | In vitro studies | High activity | [20,86-91] |
| Oxamate | Pyruvate‐competitive inhibition of LDHA | In vitro studies | High activity | [92] |
| Gossypol, FX11 and quinoline 3‐sulfonamides | NADH‐competitive inhibition of LDHA | In vitro and in vivo studies | High in vitro activity and in vivo efficacy | [92] |
| N‐hydroxyindoles | Pyruvate‐ and NADH-competitive inhibition of LDHA | In vitro studies | High activity | [92] |
| Galloflavin | Inhibition of LDHA function | In vitro studies | High activity | [92] |
6. Conclusions and future perspectives
A growing body of evidence suggests that aberrant activation of the PI3K/Akt cascade drives tumor initiation and progression via a profound rewiring of glucose metabolism. In this context, future studies should be focused on a deeper characterization of the critical regulatory nodes connecting this signaling to the glycolytic flux in different malignancies, to identify new metabolic dependencies and vulnerabilities. On the other side, the interactions between the PI3K/Akt pathway and other glycolysis-related metabolic networks should be investigated, to develop a broader view of the tumorigenic implications of Akt constitutive phosphorylation. Indeed, metabolic enzymes are inherently druggable and could represent fundamental therapeutic hotspots, due to their role as essential modulators of cancer cell proliferation and survival.
Finally, it should be taken in consideration that the specific features of the stroma surrounding both the primary and metastatic tumor as well as the nutritional status of the host differentially influence the impact of PI3K/Akt signaling on cancer metabolism, further supporting the usage of combinatorial anti-metabolic strategies in tumor treatment.
Acknowledgements
The authors acknowledge support from the Università degli Studi di Milano through the APC initiative.
Funding
This research was funded by PRIN 2015 (grant number 2015B7M39T_004) and MIUR Progetto di Eccellenza (Department of Pharmacological and Biomolecular Sciences "Rodolfo Paoletti", University of Milan). F.F. was supported by Fondazione Umberto Veronesi.
Author contributions
Writing - original draft preparation, F.F.; writing - review and editing, F.F., G.G. and S.M.; funding acquisition, F.F. and P.L.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Khezri MR, Jafari R, Yousefi K, Zolbanin NM. The PI3K/AKT signaling pathway in cancer: Molecular mechanisms and possible therapeutic interventions. Exp Mol Pathol [Internet]. 2022;127:104787 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0014480022000478
2. Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer [Internet]. 2020;20:74-88 Available at: http://www.nature.com/articles/s41568-019-0216-7
3. Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer [Internet]. 2019;18:26 Available at: https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-019-0954-x
4. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene [Internet]. 2008;27:5497-510 Available at: https://www.nature.com/articles/onc2008245
5. Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT Pathway for Cancer Drug Discovery. Nat Rev Drug Discov [Internet]. 2005;4:988-1004 Available at: http://www.nature.com/articles/nrd1902
6. Hemmings BA, Restuccia DF. PI3K-PKB/Akt Pathway. Cold Spring Harb Perspect Biol [Internet]. 2012;4:a011189-a011189 Available at: http://cshperspectives.cshlp.org/lookup/doi/10.1101/cshperspect.a011189
7. Zhang Y, Kwok-Shing Ng P, Kucherlapati M. et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell [Internet]. 2017;31:820-832.e3 Available at: https://linkinghub.elsevier.com/retrieve/pii/S153561081730168X
8. Tsai P-J, Lai Y-H, Manne RK, Tsai Y-S, Sarbassov D, Lin H-K. Akt: a key transducer in cancer. J Biomed Sci [Internet]. 2022;29:76 Available at: https://jbiomedsci.biomedcentral.com/articles/10.1186/s12929-022-00860-9
9. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science (80- ) [Internet]. 2009;324:1029-33 Available at: https://www.science.org/doi/10.1126/science.1160809
10. Liberti M V, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci [Internet]. 2016;41:211-8 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0968000415002418
11. Chen X, Qian Y, Wu S. The Warburg effect: Evolving interpretations of an established concept. Free Radic Biol Med [Internet]. 2015;79:253-63 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0891584914004183
12. Vazquez A, Oltvai ZN. Molecular Crowding Defines a Common Origin for the Warburg Effect in Proliferating Cells and the Lactate Threshold in Muscle Physiology. Moreno Y, Ed. PLoS One [Internet]. 2011;6:e19538 Available at: https://dx.plos.org/10.1371/journal.pone.0019538
13. Ancey P, Contat C, Meylan E. Glucose transporters in cancer - from tumor cells to the tumor microenvironment. FEBS J [Internet]. 2018;285:2926-43 Available at: https://onlinelibrary.wiley.com/doi/10.1111/febs.14577
14. Wahl H, Daudi S, Kshirsagar M. et al. Expression of metabolically targeted biomarkers in endometrial carcinoma. Gynecol Oncol [Internet]. 2010;116:21-7 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0090825809007835
15. Fang J, Bao Y-Y, Zhou S-H. et al. Recurrent prognostic factors and expression of GLUT-1, PI3K and p-Akt in adenoid cystic carcinomas of the head and neck: Clinicopathological features and biomarkers of adenoid cystic carcinoma. Oncol Lett [Internet]. 2012;4:1234-40 Available at: https://www.spandidos-publications.com/10.3892/ol.2012.895
16. Shen W-Q, Cheng K-J, Bao Y-Y, Zhou S-H, Yao H-T. Expression of Glut-1, HIF-1α, PI3K and p-Akt in a case of ceruminous adenoma. Head Neck Oncol [Internet]. 2012;4:18 Available at: https://headandneckoncology.biomedcentral.com/articles/10.1186/1758-3284-4-18
17. Hong SY, Yu F-X, Luo Y, Hagen T. Oncogenic activation of the PI3K/Akt pathway promotes cellular glucose uptake by downregulating the expression of thioredoxin-interacting protein. Cell Signal [Internet]. 2016;28:377-83 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0898656816300110
18. Fang J, Zhou S-H, Fan J, Yan S-X. Roles of glucose transporter-1 and the phosphatidylinositol 3-kinase/protein kinase B pathway in cancer radioresistance (Review). Mol Med Rep [Internet]. 2015;11:1573-81 Available at: https://www.spandidos-publications.com/10.3892/mmr.2014.2888
19. Chen X, Zhao Y, Lyu S. et al. Identification of novel inhibitors of GLUT1 by virtual screening and cell-based assays. Invest New Drugs [Internet]. 2021;39:1242-55 Available at: https://link.springer.com/10.1007/s10637-021-01109-2
20. Cui Y, Li C, Sang F, Cao W, Qin Z, Zhang P. Natural products targeting glycolytic signaling pathways-an updated review on anti-cancer therapy. Front Pharmacol [Internet]. 2022; 13. Available at: https://www.frontiersin.org/articles/10.3389/fphar. 2022 1035882/full
21. Guo D, Meng Y, Jiang X, Lu Z. Hexokinases in cancer and other pathologies. Cell Insight [Internet]. 2023;2:100077 Available at: https://linkinghub.elsevier.com/retrieve/pii/S2772892723000019
22. Ciscato F, Ferrone L, Masgras I, Laquatra C, Rasola A. Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. Int J Mol Sci [Internet]. 2021;22:4716 Available at: https://www.mdpi.com/1422-0067/22/9/4716
23. Zou Y, Du Y, Cheng C. et al. FAT10 promotes the progression of bladder cancer by upregulating HK2 through the EGFR/AKT pathway. Exp Cell Res [Internet]. 2021;398:112401 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0014482720306546
24. Wang Y, Pan S, He X. et al. CPNE1 Enhances Colorectal Cancer Cell Growth, Glycolysis, and Drug Resistance Through Regulating the AKT-GLUT1/HK2 Pathway. Onco Targets Ther [Internet]. 2021 Volume 14: 699-710. Available at: https://www.dovepress.com/cpne1-enhances-colorectal-cancer-cell-growth-glycolysis-and-drug-resis-peer-reviewed-article-OTT
25. Cui Y, Qin L, Wu J. et al. SIRT3 Enhances Glycolysis and Proliferation in SIRT3-Expressing Gastric Cancer Cells. Shi X, Ed. PLoS One [Internet]. 2015;10:e0129834 Available at: https://dx.plos.org/10.1371/journal.pone.0129834
26. Tian X, Liu D, Zuo X. et al. Hexokinase 2 promoted cell motility and proliferation by activating Akt1/p-Akt1 in human ovarian cancer cells. J Ovarian Res [Internet]. 2022;15:92 Available at: https://ovarianresearch.biomedcentral.com/articles/10.1186/s13048-022-01027-8
27. Chen Q, Li L, Liu X. et al. Hexokinases 2 promoted cell motility and distant metastasis by elevating fibronectin through Akt1/p-Akt1 in cervical cancer cells. Cancer Cell Int [Internet]. 2021;21:600 Available at: https://cancerci.biomedcentral.com/articles/10.1186/s12935-021-02312-0
28. Deng B, Deng J, Yi X, Zou Y, Li C. ROCK2 Promotes Osteosarcoma Growth and Glycolysis by Up-Regulating HKII via Phospho-PI3K/AKT Signalling. Cancer Manag Res [Internet]. 2021 Volume 13: 449-62. Available at: https://www.dovepress.com/rock2-promotes-osteosarcoma-growth-and-glycolysis-by-up-regulating-hki-peer-reviewed-article-CMAR
29. He R, Liu H. TRIM59 knockdown blocks cisplatin resistance in A549/DDP cells through regulating PTEN/AKT/HK2. Gene [Internet]. 2020;747:144553 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0378111920302225
30. Lin C, Chen H, Han R. et al. Hexokinases <scp>II</scp> -mediated glycolysis governs susceptibility to crizotinib in <scp>ALK</scp> -positive non-small cell lung cancer. Thorac Cancer [Internet]. 2021;12:3184-93 Available at: https://onlinelibrary.wiley.com/doi/10.1111/1759-7714.14184
31. Pastorino JG, Shulga N, Hoek JB. Mitochondrial Binding of Hexokinase II Inhibits Bax-induced Cytochrome c Release and Apoptosis. J Biol Chem [Internet]. 2002;277:7610-8 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0021925819823232
32. Majewski N, Nogueira V, Bhaskar P. et al. Hexokinase-Mitochondria Interaction Mediated by Akt Is Required to Inhibit Apoptosis in the Presence or Absence of Bax and Bak. Mol Cell [Internet]. 2004;16:819-30 Available at: https://linkinghub.elsevier.com/retrieve/pii/S1097276504006781
33. Majewski N, Nogueira V, Robey RB, Hay N. Akt Inhibits Apoptosis Downstream of BID Cleavage via a Glucose-Dependent Mechanism Involving Mitochondrial Hexokinases. Mol Cell Biol [Internet]. 2004;24:730-40 Available at: https://journals.asm.org/doi/10.1128/MCB.24.2.730-740.2004
34. Xiong X, Wen Y-A, Mitov MI, C Oaks M, Miyamoto S, Gao T. PHLPP regulates hexokinase 2-dependent glucose metabolism in colon cancer cells. Cell Death Discov [Internet]. 2017;3:16103 Available at: https://www.nature.com/articles/cddiscovery2016103
35. Baffi TR, Cohen-Katsenelson K, Newton AC. PHLPPing the Script: Emerging Roles of PHLPP Phosphatases in Cell Signaling. Annu Rev Pharmacol Toxicol [Internet]. 2021;61:723-43 Available at: https://www.annualreviews.org/doi/10.1146/annurev-pharmtox-031820-122108
36. Ciscato F, Filadi R, Masgras I. et al. Hexokinase 2 displacement from mitochondria-associated membranes prompts Ca 2+ -dependent death of cancer cells. EMBO Rep [Internet]. 2020 21. Available at: https://onlinelibrary.wiley.com/doi/10.15252/embr.201949117
37. Cheung EC, Ludwig RL, Vousden KH. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc Natl Acad Sci [Internet]. 2012;109:20491-6 Available at: https://pnas.org/doi/full/10.1073/pnas.1206530109
38. Wolf A, Agnihotri S, Micallef J. et al. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med [Internet]. 2011;208:313-26 Available at: https://rupress.org/jem/article/208/2/313/40856/Hexokinase-2-is-a-key-mediator-of-aerobic
39. Patra KC, Wang Q, Bhaskar PT. et al. Hexokinase 2 Is Required for Tumor Initiation and Maintenance and Its Systemic Deletion Is Therapeutic in Mouse Models of Cancer. Cancer Cell [Internet]. 2013;24:213-28 Available at: https://linkinghub.elsevier.com/retrieve/pii/S1535610813002882
40. Liu Y, Wu K, Shi L, Xiang F, Tao K, Wang G. Prognostic Significance of the Metabolic Marker Hexokinase-2 in Various Solid Tumors: A Meta-Analysis. Coleman WB, Ed. PLoS One [Internet]. 2016;11:e0166230 Available at: https://dx.plos.org/10.1371/journal.pone.0166230
41. Anderson M, Marayati R, Moffitt R, Yeh JJ. Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer. Oncotarget [Internet]. 2017;8:56081-94 Available at: https://www.oncotarget.com/lookup/doi/10.18632/oncotarget.9760
42. Yang T, Ren C, Qiao P. et al. PIM2-mediated phosphorylation of hexokinase 2 is critical for tumor growth and paclitaxel resistance in breast cancer. Oncogene [Internet]. 2018;37:5997-6009 Available at: http://www.nature.com/articles/s41388-018-0386-x
43. Liu C, Wang X, Zhang Y. The Roles of HK2 on Tumorigenesis of Cervical Cancer. Technol Cancer Res Treat [Internet]. 2019;18:153303381987130 Available at: http://journals.sagepub.com/doi/10.1177/1533033819871306
44. Zhang X-Y, Zhang M, Cong Q. et al. Hexokinase 2 confers resistance to cisplatin in ovarian cancer cells by enhancing cisplatin-induced autophagy. Int J Biochem Cell Biol [Internet]. 2018;95:9-16 Available at: https://linkinghub.elsevier.com/retrieve/pii/S1357272517303163
45. Fan K, Fan Z, Cheng H. et al. Hexokinase 2 dimerization and interaction with voltage-dependent anion channel promoted resistance to cell apoptosis induced by gemcitabine in pancreatic cancer. Cancer Med [Internet]. 2019;8:5903-15 Available at: https://onlinelibrary.wiley.com/doi/10.1002/cam4.2463
46. Huang X, Liu M, Sun H. et al. HK2 is a radiation resistant and independent negative prognostic factor for patients with locally advanced cervical squamous cell carcinoma. Int J Clin Exp Pathol [Internet]. 2015;8:4054-63 Available at: http://www.ncbi.nlm.nih.gov/pubmed/26097593
47. Vartanian A, Agnihotri S, Wilson MR. et al. Targeting hexokinase 2 enhances response to radio-chemotherapy in glioblastoma. Oncotarget [Internet]. 2016;7:69518-35 Available at: https://www.oncotarget.com/lookup/doi/10.18632/oncotarget.11680
48. Yoo J-J, Yu S, Na J. et al. Hexokinase-II Inhibition Synergistically Augments the Anti-tumor Efficacy of Sorafenib in Hepatocellular Carcinoma. Int J Mol Sci [Internet]. 2019;20:1292 Available at: https://www.mdpi.com/1422-0067/20/6/1292
49. Gu JJ, Singh A, Xue K. et al. Up-regulation of hexokinase II contributes to rituximab-chemotherapy resistance and is a clinically relevant target for therapeutic development. Oncotarget [Internet]. 2018;9:4020-33 Available at: https://www.oncotarget.com/lookup/doi/10.18632/oncotarget.23425
50. DeWaal D, Nogueira V, Terry AR. et al. Author Correction: Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat Commun [Internet]. 2018;9:2539 Available at: https://www.nature.com/articles/s41467-018-04182-z
51. Wang L, Xiong H, Wu F. et al. Hexokinase 2-Mediated Warburg Effect Is Required for PTEN- and p53-Deficiency-Driven Prostate Cancer Growth. Cell Rep [Internet]. 2014;8:1461-74 Available at: https://linkinghub.elsevier.com/retrieve/pii/S2211124714006378
52. Nogueira V, Patra KC, Hay N. Selective eradication of cancer displaying hyperactive Akt by exploiting the metabolic consequences of Akt activation. Elife. 2018.
53. Fontana F, Anselmi M, Limonta P. Exploiting the Metabolic Consequences of PTEN Loss and Akt/Hexokinase 2 Hyperactivation in Prostate Cancer: A New Role for δ-Tocotrienol. Int J Mol Sci [Internet]. 2022;23:5269 Available at: https://www.mdpi.com/1422-0067/23/9/5269
54. Garcia SN, Guedes RC, Marques MM. Unlocking the Potential of HK2 in Cancer Metabolism and Therapeutics. Curr Med Chem [Internet]. 2020;26:7285-322 Available at: http://www.eurekaselect.com/168321/article
55. Chiara F, Castellaro D, Marin O. et al. Hexokinase II Detachment from Mitochondria Triggers Apoptosis through the Permeability Transition Pore Independent of Voltage-Dependent Anion Channels. Koch K-W, Ed. PLoS One [Internet]. 2008;3:e1852 Available at: https://dx.plos.org/10.1371/journal.pone.0001852
56. Pantic B, Trevisan E, Citta A. et al. Myotonic dystrophy protein kinase (DMPK) prevents ROS-induced cell death by assembling a hexokinase II-Src complex on the mitochondrial surface. Cell Death Dis [Internet]. 2013;4:e858-e858 Available at: https://www.nature.com/articles/cddis2013385
57. Novellasdemunt L, Tato I, Navarro-Sabate A. et al. Akt-dependent Activation of the Heart 6-Phosphofructo-2-kinase/Fructose-2,6-bisphosphatase (PFKFB2) Isoenzyme by Amino Acids. J Biol Chem [Internet]. 2013;288:10640-51 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0021925820672866
58. Feng Y, Wu L. mTOR up-regulation of PFKFB3 is essential for acute myeloid leukemia cell survival. Biochem Biophys Res Commun [Internet]. 2017;483:897-903 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0006291X17300402
59. Kotowski K, Rosik J, Machaj F. et al. Role of PFKFB3 and PFKFB4 in Cancer: Genetic Basis, Impact on Disease Development/Progression, and Potential as Therapeutic Targets. Cancers (Basel) [Internet]. 2021;13:909 Available at: https://www.mdpi.com/2072-6694/13/4/909
60. Lu H, Chen S, You Z, Xie C, Huang S, Hu X. PFKFB4 negatively regulated the expression of histone acetyltransferase GCN5 to mediate the tumorigenesis of thyroid cancer. Dev Growth Differ [Internet]. 2020;62:129-38 Available at: https://onlinelibrary.wiley.com/doi/10.1111/dgd.12645
61. Sittewelle M, Kappès V, Zhou C, Lécuyer D, Monsoro-Burq AH. PFKFB4 interacts with ICMT and activates RAS/AKT signaling-dependent cell migration in melanoma. Life Sci Alliance [Internet]. 2022;5:e202201377 Available at: https://www.life-science-alliance.org/lookup/doi/10.26508/lsa.202201377
62. Griguer CE, Oliva CR, Gillespie GY. Glucose Metabolism Heterogeneity in Human and Mouse Malignant Glioma Cell Lines. J Neurooncol [Internet]. 2005;74:123-33 Available at: http://link.springer.com/10.1007/s11060-004-6404-6
63. Miralpeix M, Azcon-Bieto J, Bartrons R, Argiles JM. The impairment of respiration by glycolysis in the Lewis lung carcinoma. Cancer Lett [Internet]. 1990;50:173-8 Available at: https://linkinghub.elsevier.com/retrieve/pii/030438359090261U
64. Nissler K, Petermann H, Wenz I, Brox D. Fructose 2,6-bisphosphate metabolism in Ehrlich ascites tumour cells. J Cancer Res Clin Oncol [Internet]. 1995;121:739-45 Available at: http://link.springer.com/10.1007/BF01213320
65. Minchenko OH, Ochiai A, Opentanova IL. et al. Overexpression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-4 in the human breast and colon malignant tumors. Biochimie [Internet]. 2005;87:1005-10 Available at: https://linkinghub.elsevier.com/retrieve/pii/S030090840500101X
66. Minchenko OH. Mechanisms of regulation of PFKFB expression in pancreatic and gastric cancer cells. World J Gastroenterol [Internet]. 2014;20:13705 Available at: http://www.wjgnet.com/1007-9327/full/v20/i38/13705.htm
67. Peng F, Li Q, Sun J-Y, Luo Y, Chen M, Bao Y. PFKFB3 is involved in breast cancer proliferation, migration, invasion and angiogenesis. Int J Oncol [Internet]. 2018; Available at: http://www.spandidos-publications.com/10.3892/ijo. 2018 4257
68. Atsumi T, Chesney J, Metz C. et al. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res [Internet]. 2002;62:5881-7 Available at: http://www.ncbi.nlm.nih.gov/pubmed/12384552
69. Han J, Meng Q, Xi Q, Wang H, Wu G. PFKFB3 was overexpressed in gastric cancer patients and promoted the proliferation and migration of gastric cancer cells. Cancer Biomarkers [Internet]. 2017;18:249-56 Available at: https://www.medra.org/servlet/aliasResolver?alias=iospress&doi=10.3233/CBM-160143
70. Shi W-K, Zhu X-D, Wang C-H. et al. PFKFB3 blockade inhibits hepatocellular carcinoma growth by impairing DNA repair through AKT. Cell Death Dis [Internet]. 2018;9:428 Available at: https://www.nature.com/articles/s41419-018-0435-y
71. Zahra K, Dey T, Ashish, Mishra SP, Pandey U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front Oncol [Internet]. 2020; 10. Available at: https://www.frontiersin.org/article/10.3389/fonc. 2020 00159/full
72. Ye G, Qin Y, Wang S. et al. Lamc1 promotes the Warburg effect in hepatocellular carcinoma cells by regulating PKM2 expression through AKT pathway. Cancer Biol Ther [Internet]. 2019;20:711-9 Available at: https://www.tandfonline.com/doi/full/10.1080/15384047.2018.1564558
73. Chen C, Liu W-R, Zhang B. et al. LncRNA H19 downregulation confers erlotinib resistance through upregulation of PKM2 and phosphorylation of AKT in EGFR-mutant lung cancers. Cancer Lett [Internet]. 2020;486:58-70 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0304383520302445
74. Zheng B, Geng L, Zeng L, Liu F, Huang Q. AKT2 contributes to increase ovarian cancer cell migration and invasion through the AKT2-PKM2-STAT3/NF-κB axis. Cell Signal [Internet]. 2018;45:122-31 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0898656818300275
75. Liu X, Liu H, Zeng L, Lv Y. BRCA1 overexpression attenuates breast cancer cell growth and migration by regulating the pyruvate kinase M2-mediated Warburg effect via the PI3K/AKT signaling pathway. PeerJ [Internet]. 2022;10:e14052 Available at: https://peerj.com/articles/14052
76. Park YS, Kim DJ, Koo H. et al. AKT-induced PKM2 phosphorylation signals for IGF-1-stimulated cancer cell growth. Oncotarget [Internet]. 2016;7:48155-67 Available at: https://www.oncotarget.com/lookup/doi/10.18632/oncotarget.10179
77. Zhang K, Zhang M, Jiang H, Liu F, Liu H, Li Y. Down-regulation of miR-214 inhibits proliferation and glycolysis in non-small-cell lung cancer cells via down-regulating the expression of hexokinase 2 and pyruvate kinase isozyme M2. Biomed Pharmacother [Internet]. 2018;105:545-52 Available at: https://linkinghub.elsevier.com/retrieve/pii/S075333221833453X
78. Yang P, Li Z, Wang Y, Zhang L, Wu H, Li Z. Secreted pyruvate kinase M2 facilitates cell migration via PI3K/Akt and Wnt/β-catenin pathway in colon cancer cells. Biochem Biophys Res Commun [Internet]. 2015;459:327-32 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0006291X15003526
79. Wang C, Jiang J, Ji J. et al. PKM2 promotes cell migration and inhibits autophagy by mediating PI3K/AKT activation and contributes to the malignant development of gastric cancer. Sci Rep [Internet]. 2017;7:2886 Available at: https://www.nature.com/articles/s41598-017-03031-1
80. Zhang S, Wang C, Ju J, Wang C. Extracellular Hsp90α Supports the ePKM2-GRP78-AKT Axis to Promote Tumor Metastasis. Front Oncol [Internet]. 2022; 12. Available at: https://www.frontiersin.org/articles/10.3389/fonc. 2022 906080/full
81. Zhang Q, Zhang J, Yao A. et al. OTUB2 promotes the progression of endometrial cancer by regulating the PKM2-mediated PI3K/AKT signaling pathway. Cell Biol Int [Internet]. 2023;47:428-38 Available at: https://onlinelibrary.wiley.com/doi/10.1002/cbin.11950
82. He C-L, Bian Y-Y, Xue Y. et al. Pyruvate Kinase M2 Activates mTORC1 by Phosphorylating AKT1S1. Sci Rep [Internet]. 2016;6:21524 Available at: https://www.nature.com/articles/srep21524
83. Wu J, Hu L, Chen M, Cao W, Chen H, He T. Pyruvate kinase M2 overexpression and poor prognosis in solid tumors of digestive system: evidence from 16 cohort studies. Onco Targets Ther [Internet]. 2016 Volume 9: 4277-88. Available at: https://www.dovepress.com/pyruvate-kinase-m2-overexpression-and-poor-prognosis-in-solid-tumors-o-peer-reviewed-article-OTT
84. Feng J, Li J, Wu L. et al. Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. J Exp Clin Cancer Res [Internet]. 2020;39:126 Available at: https://jeccr.biomedcentral.com/articles/10.1186/s13046-020-01629-4
85. Du D, Liu C, Qin M. et al. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm Sin B [Internet]. 2022;12:558-80 Available at: https://linkinghub.elsevier.com/retrieve/pii/S2211383521003610
86. Vander Heiden MG, Christofk HR, Schuman E. et al. Identification of small molecule inhibitors of pyruvate kinase M2. Biochem Pharmacol [Internet]. 2010;79:1118-24 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0006295209010600
87. Varghese B, Swaminathan G, Plotnikov A. et al. Prolactin Inhibits Activity of Pyruvate Kinase M2 to Stimulate Cell Proliferation. Mol Endocrinol [Internet]. 2010;24:2356-65 Available at: https://academic.oup.com/mend/article-lookup/doi/10.1210/me.2010-0219
88. Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene [Internet]. 2011;30:4297-306 Available at: https://www.nature.com/articles/onc2011137
89. Ning X, Qi H, Li R. et al. Discovery of novel naphthoquinone derivatives as inhibitors of the tumor cell specific M2 isoform of pyruvate kinase. Eur J Med Chem [Internet]. 2017;138:343-52 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0223523417305184
90. Zhao X, Zhu Y, Hu J. et al. Shikonin Inhibits Tumor Growth in Mice by Suppressing Pyruvate Kinase M2-mediated Aerobic Glycolysis. Sci Rep [Internet]. 2018;8:14517 Available at: https://www.nature.com/articles/s41598-018-31615-y
91. Ning X, Qi H, Li R, Jin Y, McNutt MA, Yin Y. Synthesis and antitumor activity of novel 2, 3-didithiocarbamate substituted naphthoquinones as inhibitors of pyruvate kinase M2 isoform. J Enzyme Inhib Med Chem [Internet]. 2018;33:126-9 Available at: https://www.tandfonline.com/doi/full/10.1080/14756366.2017.1404591
92. Feng Y, Xiong Y, Qiao T, Li X, Jia L, Han Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med [Internet]. 2018;7:6124-36 Available at: https://onlinelibrary.wiley.com/doi/10.1002/cam4.1820
93. Yao F, Zhao T, Zhong C, Zhu J, Zhao H. LDHA is necessary for the tumorigenicity of esophageal squamous cell carcinoma. Tumor Biol [Internet]. 2013;34:25-31 Available at: http://link.springer.com/10.1007/s13277-012-0506-0
94. Matsubara M, Bissell MJ. Inhibitors of Rho kinase (ROCK) signaling revert the malignant phenotype of breast cancer cells in 3D context. Oncotarget [Internet]. 2016;7:31602-22 Available at: https://www.oncotarget.com/lookup/doi/10.18632/oncotarget.9395
95. Zhang X, Ai Z, Chen J. et al. Glycometabolic adaptation mediates the insensitivity of drug-resistant K562/ADM leukaemia cells to adriamycin via the AKT-mTOR/c-Myc signalling pathway. Mol Med Rep [Internet]. 2017;15:1869-76 Available at: https://www.spandidos-publications.com/10.3892/mmr.2017.6189
96. An J, Zhang Y, He J. et al. Lactate dehydrogenase A promotes the invasion and proliferation of pituitary adenoma. Sci Rep [Internet]. 2017;7:4734 Available at: https://www.nature.com/articles/s41598-017-04366-5
97. Li Y, He Z-C, Liu Q. et al. Large Intergenic Non-coding RNA-RoR Inhibits Aerobic Glycolysis of Glioblastoma Cells via Akt Pathway. J Cancer [Internet]. 2018;9:880-9 Available at: http://www.jcancer.org/v09p0880.htm
98. Long W, Gong X, Yang Y, Yang K. Downregulation of PER2 Promotes Tumor Progression by Enhancing Glycolysis via the Phosphatidylinositol 3-Kinase/Protein Kinase B Pathway in Oral Squamous Cell Carcinoma. J Oral Maxillofac Surg [Internet]. 2020;78:1780.e1-1780.e14 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0278239120305607
99. Xu L, Chen J, Jia L, Chen X, Awaleh Moumin F, Cai J. SLC1A3 promotes gastric cancer progression via the PI3K/AKT signalling pathway. J Cell Mol Med [Internet]. 2020;24:14392-404 Available at: https://onlinelibrary.wiley.com/doi/10.1111/jcmm.16060
100. Luan Y, Zhang W, Xie J, Mao J. CDKN2A inhibits cell proliferation and invasion in cervical cancer through LDHA-mediated AKT/mTOR pathway. Clin Transl Oncol [Internet]. 2021;23:222-8 Available at: https://link.springer.com/10.1007/s12094-020-02409-4
101. Qi C-L, Huang M-L, Zou Y. et al. The IRF2/CENP-N/AKT signaling axis promotes proliferation, cell cycling and apoptosis resistance in nasopharyngeal carcinoma cells by increasing aerobic glycolysis. J Exp Clin Cancer Res [Internet]. 2021;40:390 Available at: https://jeccr.biomedcentral.com/articles/10.1186/s13046-021-02191-3
102. Dai M, Wang L, Yang J. et al. LDHA as a regulator of T cell fate and its mechanisms in disease. Biomed Pharmacother [Internet]. 2023;158:114164 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0753332222015530
103. Cao Z, Xu D, Harding J. et al. Lactate oxidase nanocapsules boost T cell immunity and efficacy of cancer immunotherapy. Sci Transl Med [Internet]. 2023 15. Available at: https://www.science.org/doi/10.1126/scitranslmed.add2712
104. Choi H, Yeo M, Kang Y. et al. Lactate oxidase/catalase-displaying nanoparticles efficiently consume lactate in the tumor microenvironment to effectively suppress tumor growth. J Nanobiotechnology [Internet]. 2023;21:5 Available at: https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-022-01762-6
105. Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang C V. MYC, Metabolism, and Cancer. Cancer Discov [Internet]. 2015;5:1024-39 Available at: https://aacrjournals.org/cancerdiscovery/article/5/10/1024/4488/MYC-Metabolism-and-CancerMYC-Metabolism-and-Cancer
106. West MJ, Stoneley M, Willis AE. Translational induction of the c-myc oncogene via activation of the FRAP/TOR signalling pathway. Oncogene [Internet]. 1998;17:769-80 Available at: https://www.nature.com/articles/1201990
107. Csibi A, Lee G, Yoon S-O. et al. The mTORC1/S6K1 Pathway Regulates Glutamine Metabolism through the eIF4B-Dependent Control of c-Myc Translation. Curr Biol [Internet]. 2014;24:2274-80 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0960982214009853
108. Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev [Internet]. 2000;14:2501-14 Available at: http://genesdev.cshlp.org/lookup/doi/10.1101/gad.836800
109. Gregory MA, Qi Y, Hann SR. Phosphorylation by Glycogen Synthase Kinase-3 Controls c-Myc Proteolysis and Subnuclear Localization. J Biol Chem [Internet]. 2003;278:51606-12 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0021925820754268
110. Welcker M, Orian A, Jin J. et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci [Internet]. 2004;101:9085-90 Available at: https://pnas.org/doi/full/10.1073/pnas.0402770101
111. Bouchard C, Marquardt J, Brás A, Medema RH, Eilers M. Myc-induced proliferation and transformation require Akt-mediated phosphorylation of FoxO proteins. EMBO J [Internet]. 2004;23:2830-40 Available at: http://emboj.embopress.org/cgi/doi/10.1038/sj.emboj.7600279
112. Wiegering A, Uthe FW, Jamieson T. et al. Targeting Translation Initiation Bypasses Signaling Crosstalk Mechanisms That Maintain High MYC Levels in Colorectal Cancer. Cancer Discov [Internet]. 2015;5:768-81 Available at: https://aacrjournals.org/cancerdiscovery/article/5/7/768/5243/Targeting-Translation-Initiation-Bypasses
113. Brondfield S, Umesh S, Corella A. et al. Direct and indirect targeting of MYC to treat acute myeloid leukemia. Cancer Chemother Pharmacol [Internet]. 2015;76:35-46 Available at: http://link.springer.com/10.1007/s00280-015-2766-z
114. Safaroghli-Azar A, Bashash D, Kazemi A, Pourbagheri-Sigaroodi A, Momeny M. Anticancer effect of pan-PI3K inhibitor on multiple myeloma cells: Shedding new light on the mechanisms involved in BKM120 resistance. Eur J Pharmacol [Internet]. 2019;842:89-98 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0014299918306216
115. Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckland, NZ) [Internet]. 2015;3:83-92 Available at: http://www.ncbi.nlm.nih.gov/pubmed/27774485
116. Infantino V, Santarsiero A, Convertini P, Todisco S, Iacobazzi V. Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. Int J Mol Sci [Internet]. 2021;22:5703 Available at: https://www.mdpi.com/1422-0067/22/11/5703
117. Zhong H, Chiles K, Feldser D. et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res [Internet]. 2000;60:1541-5 Available at: http://www.ncbi.nlm.nih.gov/pubmed/10749120
118. Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) Signaling Increases the Rate of Hypoxia-Inducible Factor 1α (HIF-1α) Synthesis: Novel Mechanism for HIF-1-Mediated Vascular Endothelial Growth Factor Expression. Mol Cell Biol [Internet]. 2001;21:3995-4004 Available at: https://journals.asm.org/doi/10.1128/MCB.21.12.3995-4004.2001
119. Hudson CC, Liu M, Chiang GG. et al. Regulation of Hypoxia-Inducible Factor 1α Expression and Function by the Mammalian Target of Rapamycin. Mol Cell Biol [Internet]. 2002;22:7004-14 Available at: https://journals.asm.org/doi/10.1128/MCB.22.20.7004-7014.2002
120. Majumder PK, Febbo PG, Bikoff R. et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med [Internet]. 2004;10:594-601 Available at: http://www.nature.com/articles/nm1052
121. Thomas G V, Tran C, Mellinghoff IK. et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med [Internet]. 2006;12:122-7 Available at: http://www.nature.com/articles/nm1337
122. Düvel K, Yecies JL, Menon S. et al. Activation of a Metabolic Gene Regulatory Network Downstream of mTOR Complex 1. Mol Cell [Internet]. 2010;39:171-83 Available at: https://linkinghub.elsevier.com/retrieve/pii/S1097276510004636
123. Li Y, Sun X-X, Qian DZ, Dai M-S. Molecular Crosstalk Between MYC and HIF in Cancer. Front Cell Dev Biol [Internet]. 2020; 8. Available at: https://www.frontiersin.org/articles/10.3389/fcell. 2020 590576/full
124. Sun Q, Chen X, Ma J. et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci [Internet]. 2011;108:4129-34 Available at: https://pnas.org/doi/full/10.1073/pnas.1014769108
125. Jin M-Z, Jin W-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther [Internet]. 2020;5:166 Available at: https://www.nature.com/articles/s41392-020-00280-x
126. Dumas J-F, Brisson L. Interaction between adipose tissue and cancer cells: role for cancer progression. Cancer Metastasis Rev [Internet]. 2021;40:31-46 Available at: https://link.springer.com/10.1007/s10555-020-09934-2
127. Fontana F, Anselmi M, Carollo E. et al. Adipocyte-Derived Extracellular Vesicles Promote Prostate Cancer Cell Aggressiveness by Enabling Multiple Phenotypic and Metabolic Changes. Cells [Internet]. 2022;11:2388 Available at: https://www.mdpi.com/2073-4409/11/15/2388
128. Diedrich JD, Rajagurubandara E, Herroon MK, Mahapatra G, Hüttemann M, Podgorski I. Bone marrow adipocytes promote the Warburg phenotype in metastatic prostate tumors via HIF-1α activation. Oncotarget [Internet]. 2016;7:64854-77 Available at: https://www.oncotarget.com/lookup/doi/10.18632/oncotarget.11712
129. Lunt SY, Vander Heiden MG. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu Rev Cell Dev Biol [Internet]. 2011;27:441-64 Available at: https://www.annualreviews.org/doi/10.1146/annurev-cellbio-092910-154237
130. Lane AN, Fan TW-M. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res [Internet]. 2015;43:2466-85 Available at: https://academic.oup.com/nar/article/43/4/2466/2411261
131. Villa E, Ali E, Sahu U, Ben-Sahra I. Cancer Cells Tune the Signaling Pathways to Empower de Novo Synthesis of Nucleotides. Cancers (Basel) [Internet]. 2019;11:688 Available at: https://www.mdpi.com/2072-6694/11/5/688
132. TeSlaa T, Ralser M, Fan J, Rabinowitz JD. The pentose phosphate pathway in health and disease. Nat Metab [Internet]. 2023;5:1275-89 Available at: https://www.nature.com/articles/s42255-023-00863-2
133. Buj R, Aird KM. Deoxyribonucleotide Triphosphate Metabolism in Cancer and Metabolic Disease. Front Endocrinol (Lausanne) [Internet]. 2018; 9. Available at: http://journal.frontiersin.org/article/10.3389/fendo. 2018 00177/full
134. Saha A, Connelly S, Jiang J. et al. Akt Phosphorylation and Regulation of Transketolase Is a Nodal Point for Amino Acid Control of Purine Synthesis. Mol Cell [Internet]. 2014;55:264-76 Available at: https://linkinghub.elsevier.com/retrieve/pii/S1097276514004523
135. Juvekar A, Hu H, Yadegarynia S. et al. Phosphoinositide 3-kinase inhibitors induce DNA damage through nucleoside depletion. Proc Natl Acad Sci [Internet]. 2016 113. Available at: https://pnas.org/doi/full/10.1073/pnas.1522223113
136. Mannava S, Grachtchouk V, Wheeler LJ. et al. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle [Internet]. 2008;7:2392-400 Available at: http://www.tandfonline.com/doi/abs/10.4161/cc.6390
137. Cunningham JT, Moreno M V, Lodi A, Ronen SM, Ruggero D. Protein and Nucleotide Biosynthesis Are Coupled by a Single Rate-Limiting Enzyme, PRPS2, to Drive Cancer. Cell [Internet]. 2014;157:1088-103 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0092867414004826
138. Yi M, Ban Y, Tan Y, Xiong W, Li G, Xiang B. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 and 4: A pair of valves for fine-tuning of glucose metabolism in human cancer. Mol Metab [Internet]. 2019;20:1-13 Available at: https://linkinghub.elsevier.com/retrieve/pii/S2212877818310342
139. Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell [Internet]. 2006;9:425-34 Available at: https://linkinghub.elsevier.com/retrieve/pii/S1535610806001450
140. Lim HY, Ho QS, Low J, Choolani M, Wong KP. Respiratory competent mitochondria in human ovarian and peritoneal cancer. Mitochondrion [Internet]. 2011;11:437-43 Available at: https://linkinghub.elsevier.com/retrieve/pii/S1567724910002357
141. Scott DA, Richardson AD, Filipp F V. et al. Comparative Metabolic Flux Profiling of Melanoma Cell Lines. J Biol Chem [Internet]. 2011;286:42626-34 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0021925820871506
142. Koppenol WH, Bounds PL, Dang C V. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer [Internet]. 2011;11:325-37 Available at: https://www.nature.com/articles/nrc3038
143. Cerniglia GJ, Dey S, Gallagher-Colombo SM. et al. The PI3K/Akt Pathway Regulates Oxygen Metabolism via Pyruvate Dehydrogenase (PDH)-E1α Phosphorylation. Mol Cancer Ther [Internet]. 2015;14:1928-38 Available at: https://aacrjournals.org/mct/article/14/8/1928/130592/The-PI3K-Akt-Pathway-Regulates-Oxygen-Metabolism
144. Röhrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer [Internet]. 2016;16:732-49 Available at: http://www.nature.com/articles/nrc.2016.89
145. Goo CK, Lim HY, Ho QS, Too H-P, Clement M-V, Wong KP. PTEN/Akt Signaling Controls Mitochondrial Respiratory Capacity through 4E-BP1. Koritzinsky M, Ed. PLoS One [Internet]. 2012;7:e45806 Available at: https://dx.plos.org/10.1371/journal.pone.0045806
146. Fontana F, Limonta P. The multifaceted roles of mitochondria at the crossroads of cell life and death in cancer. Free Radic Biol Med [Internet]. 2021;176:203-21 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0891584921007449
147. Fontana F, Anselmi M, Limonta P. Unraveling the Peculiar Features of Mitochondrial Metabolism and Dynamics in Prostate Cancer. Cancers (Basel) [Internet]. 2023;15:1192 Available at: https://www.mdpi.com/2072-6694/15/4/1192
148. Zheng J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol Lett [Internet]. 2012;4:1151-7 Available at: https://www.spandidos-publications.com/10.3892/ol.2012.928
149. Liang L, Li W, Li X. et al. 'Reverse Warburg effect' of cancer-associated fibroblasts (Review). Int J Oncol [Internet]. 2022;60:67 Available at: http://www.spandidos-publications.com/10.3892/ijo.2022.5357
150. Li Z, Sun C, Qin Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics [Internet]. 2021;11:8322-36 Available at: https://www.thno.org/v11p8322.htm
151. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer [Internet]. 2013;13:227-32 Available at: http://www.nature.com/articles/nrc3483
152. Lee J, Ridgway ND. Substrate channeling in the glycerol-3-phosphate pathway regulates the synthesis, storage and secretion of glycerolipids. Biochim Biophys Acta - Mol Cell Biol Lipids [Internet]. 2020;1865:158438 Available at: https://linkinghub.elsevier.com/retrieve/pii/S1388198119300460
153. Lee J V, Carrer A, Shah S. et al. Akt-Dependent Metabolic Reprogramming Regulates Tumor Cell Histone Acetylation. Cell Metab [Internet]. 2014;20:306-19 Available at: https://linkinghub.elsevier.com/retrieve/pii/S1550413114002691
154. Carrer A, Trefely S, Zhao S. et al. Acetyl-CoA Metabolism Supports Multistep Pancreatic Tumorigenesis. Cancer Discov [Internet]. 2019;9:416-35 Available at: https://aacrjournals.org/cancerdiscovery/article/9/3/416/10656/Acetyl-CoA-Metabolism-Supports-Multistep
155. Khwairakpam A, Shyamananda M, Sailo B. et al. ATP Citrate Lyase (ACLY): A Promising Target for Cancer Prevention and Treatment. Curr Drug Targets [Internet]. 2015;16:156-63 Available at: http://www.eurekaselect.com/openurl/content.php?genre=article&issn=1389-4501&volume=16&issue=2&spage=156
156. Granchi C. ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur J Med Chem [Internet]. 2018;157:1276-91 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0223523418307736
157. Porstmann T, Griffiths B, Chung Y-L. et al. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene [Internet]. 2005;24:6465-81 Available at: https://www.nature.com/articles/1208802
158. Jiang T, Zhang G, Lou Z. Role of the Sterol Regulatory Element Binding Protein Pathway in Tumorigenesis. Front Oncol [Internet]. 2020; 10. Available at: https://www.frontiersin.org/article/10.3389/fonc. 2020 01788/full
159. Ricoult SJH, Yecies JL, Ben-Sahra I, Manning BD. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene [Internet]. 2016;35:1250-60 Available at: https://www.nature.com/articles/onc2015179
160. Kim KH, Song MJ, Yoo EJ, Choe SS, Park SD, Kim JB. Regulatory Role of Glycogen Synthase Kinase 3 for Transcriptional Activity of ADD1/SREBP1c. J Biol Chem [Internet]. 2004;279:51999-2006 Available at: https://linkinghub.elsevier.com/retrieve/pii/S0021925820676372
161. Gouw AM, Margulis K, Liu NS. et al. The MYC Oncogene Cooperates with Sterol-Regulated Element-Binding Protein to Regulate Lipogenesis Essential for Neoplastic Growth. Cell Metab [Internet]. 2019;30:556-572.e5 Available at: https://linkinghub.elsevier.com/retrieve/pii/S1550413119303857
162. Guo D, Bell E, Mischel P, Chakravarti A. Targeting SREBP-1-driven Lipid Metabolism to Treat Cancer. Curr Pharm Des [Internet]. 2014;20:2619-26 Available at: http://www.eurekaselect.com/openurl/content.php?genre=article&issn=1381-6128&volume=20&issue=15&spage=2619
163. Nakamura H, Takada K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci [Internet]. 2021;112:3945-52 Available at: https://onlinelibrary.wiley.com/doi/10.1111/cas.15068
164. Koundouros N, Poulogiannis G. Phosphoinositide 3-Kinase/Akt Signaling and Redox Metabolism in Cancer. Front Oncol [Internet]. 2018; 8. Available at: http://journal.frontiersin.org/article/10.3389/fonc. 2018 00160/full
165. Ilic N, Birsoy K, Aguirre AJ. et al. PIK3CA mutant tumors depend on oxoglutarate dehydrogenase. Proc Natl Acad Sci [Internet]. 2017 114. Available at: https://pnas.org/doi/full/10.1073/pnas.1617922114
166. Hoxhaj G, Ben-Sahra I, Lockwood SE. et al. Direct stimulation of NADP + synthesis through Akt-mediated phosphorylation of NAD kinase. Science (80- ) [Internet]. 2019;363:1088-92 Available at: https://www.science.org/doi/10.1126/science.aau3903
167. Cirone M, D'Orazi G. NRF2 in Cancer: Cross-Talk with Oncogenic Pathways and Involvement in Gammaherpesvirus-Driven Carcinogenesis. Int J Mol Sci [Internet]. 2022;24:595 Available at: https://www.mdpi.com/1422-0067/24/1/595
168. Fontana F, Raimondi M, Marzagalli M. et al. Mitochondrial functional and structural impairment is involved in the antitumor activity of δ-tocotrienol in prostate cancer cells. Free Radic Biol Med [Internet]. 2020 Available at: https://linkinghub.elsevier.com/retrieve/pii/S089158492031145X
169. Dermit M, Casado P, Rajeeve V. et al. Oxidative stress downstream of mTORC1 but not AKT causes a proliferative defect in cancer cells resistant to PI3K inhibition. Oncogene [Internet]. 2017;36:2762-74 Available at: http://www.nature.com/articles/onc2016435
170. Perillo B, Di Donato M, Pezone A. et al. ROS in cancer therapy: the bright side of the moon. Exp Mol Med [Internet]. 2020;52:192-203 Available at: http://www.ncbi.nlm.nih.gov/pubmed/32060354
171. Mishra R, Patel H, Alanazi S, Kilroy MK, Garrett JT. PI3K Inhibitors in Cancer: Clinical Implications and Adverse Effects. Int J Mol Sci [Internet]. 2021;22:3464 Available at: https://www.mdpi.com/1422-0067/22/7/3464
172. Coleman N, Moyers JT, Harbery A, Vivanco I, Yap TA. Clinical Development of AKT Inhibitors and Associated Predictive Biomarkers to Guide Patient Treatment in Cancer Medicine. Pharmgenomics Pers Med [Internet]. 2021 Volume 14: 1517-35. Available at: https://www.dovepress.com/clinical-development-of-akt-inhibitors-and-associated-predictive-bioma-peer-reviewed-fulltext-article-PGPM
Author contact
![]() Corresponding authors: Department of Pharmacological and Biomolecular Sciences "Rodolfo Paoletti", Università degli Studi di Milano, Via Balzaretti 9, 20133 Milan, Italy. E-mail address: fabrizio.fontanait; Phone number: +39 0250318427; Fabrizio Fontana ORCID ID: 0000-0002-9148-0884.
Corresponding authors: Department of Pharmacological and Biomolecular Sciences "Rodolfo Paoletti", Università degli Studi di Milano, Via Balzaretti 9, 20133 Milan, Italy. E-mail address: fabrizio.fontanait; Phone number: +39 0250318427; Fabrizio Fontana ORCID ID: 0000-0002-9148-0884.