Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Ferroptosis: A bird's-eye view
Mechanism of Ferroptosis
Ferroptosis in the...
Ferroptosis in Hepatocellular...
Future Perspectives and...
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(9):3621-3637. doi:10.7150/ijbs.96014 This issue Cite
Review
Current Progress of Ferroptosis Study in Hepatocellular Carcinoma
Xinyue Zhu1, Xudong Sha2, Yan Zang1, Qiaohui Ren1, Shubing Zhang1, Dongyue Ma1, Lianzi Wang1, Junxiao Yao1, Xinyi Zhou1, Li Yu3 ![]() , Tao Li1
, Tao Li1 ![]()
1. Department of Clinical Laboratory, the First Affiliated Hospital of Anhui Medical University, Shushan District, No. 218 Jixi Road, Hefei, 230032, Anhui, China.
2. Department of Pharmacology and Chemical Biology, Shanghai Jiao Tong University School of Medicine, Shanghai, 200025, China.
3. Anhui Province Key Laboratory of Zoonoses, Anhui Medical University, Hefei, 230032, Anhui, China.
Received 2024-3-6; Accepted 2024-6-8; Published 2024-7-1
Abstract

Ferroptosis, an emerging type of programmed cell death, is initiated by iron-dependent and excessive ROS-mediated lipid peroxidation, which eventually leads to plasma membrane rupture and cell death. Many canonical signalling pathways and biological processes are involved in ferroptosis. Furthermore, cancer cells are more susceptible to ferroptosis due to the high load of ROS and unique metabolic characteristics, including iron requirements. Recent investigations have revealed that ferroptosis plays a crucial role in the progression of tumours, especially HCC. Specifically, the induction of ferroptosis can not only inhibit the growth of hepatoma cells, thereby reversing tumorigenesis, but also improves the efficacy of immunotherapy and enhances the antitumour immune response. Therefore, triggering ferroptosis has become a new therapeutic strategy for cancer therapy. In this review, we summarize the characteristics of ferroptosis based on its underlying mechanism and role in HCC and provide possible therapeutic applications.
Keywords: ferroptosis, hepatocellular carcinoma, molecular mechanism, combined treatment
Introduction
Hepatocellular carcinoma (HCC), one of the most prevalent malignancies and a leading cause of cancer-related mortality worldwide [1,2], is characterized by rapid disease onset and progression. HCC accounts for 80%~90% of primary liver cancers [3]. HCC is a highly lethal disease due to its poor prognosis, high recurrence rate, rapid progression and therapeutic resistance, although significant advances have been achieved in diagnosis and treatment. Compared with that of noncancer cells, the metabolism of iron in hepatoma cells usually changes due to iron requirements. Accordingly, ferroptosis, a term coined by Dixon et al. in 2012, is an iron-dependent form of cell death [4].
Ferroptosis, a novel type of regulated cell death (RCD), is caused by iron dependence and the lethal accumulation of lipid peroxidation products, which culminates in membrane destruction and cell death [5]. Ferroptosis differs markedly from apoptosis and other types of regulated cell death and has distinct morphological and mechanistical features [6]. Considerable effort has been devoted to examining the physiological and pathophysiological effects of ferroptosis, which has dramatically expanded our knowledge.
Strikingly, the crux of cancer therapy is how to effectively eliminate cancer cells without impairing normal cells. Inducing cancer cell death, including ferroptosis, is currently the primary treatment in certain contexts [7]. Ferroptosis, a unique cell death mechanism, has sparked great attention in cancer research because targeting ferroptosis might provide a novel approach for treating cancers refractory to conventional therapies, such as those with chemotherapy resistance. Therapy resistance occurs due to the presence of some antagonistic mechanisms in cancer cells. Therefore, targeting ferroptosis and attenuating defence systems could provide an intriguing approach for killing therapy-resistant cancers. Iron overload has been observed in HCC, and research based on an increase in susceptibility to ferroptosis in HCC has been well underway.
In this review, we focus on studies of ferroptosis in the setting of HCC. We first summarize the current understanding of the mechanisms of ferroptosis, including its prerequisites and defensive systems. Next, we highlight several mechanisms involved in and underlying ferroptosis in HCC, including those involving noncoding RNA, NRF2, HIF-1α, P53 and metabolic dysregulation. Thirdly, we gather research findings based on an increase in susceptibility to ferroptosis combined with chemotherapy or immunotherapy to treat HCC. Finally, we highlight the challenges and future directions regarding investigations of ferroptosis.
Ferroptosis: A bird's-eye view
Ferroptosis, which was first proposed in 2012, refers to a form of regulated cell death triggered by excess iron-dependent peroxidation of PUFA-containing phospholipids (PUFA-PLs) and imbalanced oxidoreduction on the cellular membrane [4]. This process has distinct morphological, mechanistical and genetic features that differ from those of apoptosis, necroptosis, pyroptosis and autophagy. Morphologically, ferroptosis is characterized by shrunken mitochondria, increased cell membrane density and a decreased number of mitochondrial cristae, which is consistent with changes in nuclear morphology and membrane integrity [8,9]. In addition, whether cells undergo ferroptosis primarily depends on antagonism between the executive and defensive systems. Ferroptosis occurs when the accumulation of reactive oxygen species (ROS) overrides the antioxidant capacity provided by the antioxidant defensive system [10]. Usually, multiple processes instigate ferroptosis, including iron metabolism, PUFA-PL synthesis and hydroxyl radical generation, which are collectively known as prerequisites for ferroptosis. In contrast, defensive systems, including the GPX4-dependent and the GPX4-independent defensive systems, play pivotal roles in suppressing ferroptosis by trapping lipid peroxyl radicals and neutralizing peroxidative reactions.
Mechanism of Ferroptosis
Ferroptosis is characterized by precisely regulated molecular and signalling machineries. Mechanistically, lipid peroxides execute the ferroptosis process, which involves the interaction between the generation of oxidized phospholipids and the suppression of ferroptosis mainly through glutathione (GSH)-dependent and GSH-independent mechanisms. In this section, we summarize the current understanding of the regulatory networks involved in ferroptosis.
Prerequisites of Ferroptosis
Labile iron pool (LIP)
Over the past few years, the field of iron metabolism has gained momentum due to increased recognition of cellular processes, including DNA synthesis, mitochondrial metabolism and cell proliferation. Iron is also a double-edged sword [11]. The main source of iron is the breakdown of senescent erythrocytes after phagocytosis by macrophages, while a small proportion of iron is absorbed through the intestinal tract from food. Generally, plasma iron is mainly absorbed in the form of divalent iron (Fe2+) in duodenal enterocytes [12]. Divalent iron is oxidized into trivalent iron by ceruloplasmin and then transported to various tissues and cells bound to transferrin [13]. Inorganic trivalent iron (Fe3+) accompanied by transferrin binds to transferrin receptor 1 (TFR1) on the cellular membrane [14] and is subsequently released into the cytoplasm[15]. Subsequently, Fe3+ is reduced back to Fe2+ by the six-segment transmembrane epithelial antigen of prostate 3 (STEAP3) [16], is released into the cytosol by divalent metal transporter 1 (DMT1) and is ultimately stored in the labile iron pool (LIP) [17].
The linchpin involved in ferroptosis is iron deposition [18], and an imbalance in iron homeostasis may trigger ferroptotic cell death. Normally, intracellular iron is balanced in the form of LIP through well-established regulation of iron metabolism, including iron uptake, utilization, storage and release, due to the lack of effective iron excretion mechanisms in the organism [18]. Imbalanced LIPs can induce or inhibit ferroptosis depending on whether the level of iron increases or decreases, respectively. Accumulating evidence has revealed that iron metabolism governs ferroptosis in a variety of ways. On the one hand, excessive iron is toxic and initiates the nonenzymatic Fenton reaction, generating hydroxide and hydroxyl radicals, which can subsequently induce ferroptosis [19,20]; on the other hand, iron can act as a prominent catalyst for enzymes such as cytochrome P450 oxidoreductase (POR) and arachidonate lipoxygenase (ALOX), which participate in lipid peroxidation [21-25]. LIP dysfunction can be rescued by iron chelators. Conversely, the suppression of iron chelators to increase susceptibility to ferroptosis could be a promising therapeutic strategy for cancer treatment. H-Ferritin, a nontoxic form of stored iron, suppresses hepatoma cell sensitivity to RSL3-induced ferroptosis [26].
PUFA-PL synthesis and peroxidation
Ferroptosis is characterized by excessive peroxidation of polyunsaturated fatty acid-containing phospholipids (PUFA-PLs), which are toxic to cellular membranes. The accumulation of lipid peroxides is recognized as a determinant of ferroptosis. PUFA-PL synthesis and peroxidation involve the participation of several rate-limiting enzymes, including acyl-coenzyme A (CoA) synthetase long chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3). PUFAs, such as arachidonic acid and adrenic acid, are ligated to CoA under the catalysis of ACSL4 to produce PUFA-CoA, which is subsequently re-esterified and incorporated into PLs by LPCAT3 to form PUFA-PLs [27,28] (Figure 1). PUFA-PLs are particularly vulnerable to peroxide not only in the presence of bis-allylic moieties in PUFAs [19] but also in the presence of oxygen radicals and ROS stemming from hydrogen peroxide through the Fenton reaction. Eventually, PUFA-PLs disrupt the lipid bilayer membranes, which leads to dysfunction and ferroptosis. Recent findings also revealed that peroxisome-mediated plasmalogen biosynthesis provides another source of PUFAs for lipid peroxidation [29]. In addition, exogenous monounsaturated fatty acids (MUFAs) can block ROS accumulation and suppress ferroptosis, which is characterized by reduced total levels of PUFA-PLs at the plasma membrane, in an ACSL3-dependent manner [30].
Canonical mechanism of ferroptosis. Ferroptosis is a type of regulated cell death characterized by excess iron in the LIP and the lethal accumulation of lipid peroxidation products. Fe3+ is transferred into the cell by TFR1, is converted to Fe2+ in the endosome and is then released from the endosome by DMT1. Iron-containing ferritin is degraded to produce a large amount of free iron via NCOA4-mediated ferritinophagy, resulting in ferroptosis. The Fenton reaction promotes lipid peroxidation by activating lipoxygenases. Cystine is taken up by System Xc- for the synthesis of GSH, which further enhances the anti-lipid peroxidation activity of GPX4. In addition, FSP1-CoQ10 and GCH1-BH4 are two parallel GSH-independent pathways involved in the suppression of ferroptosis.
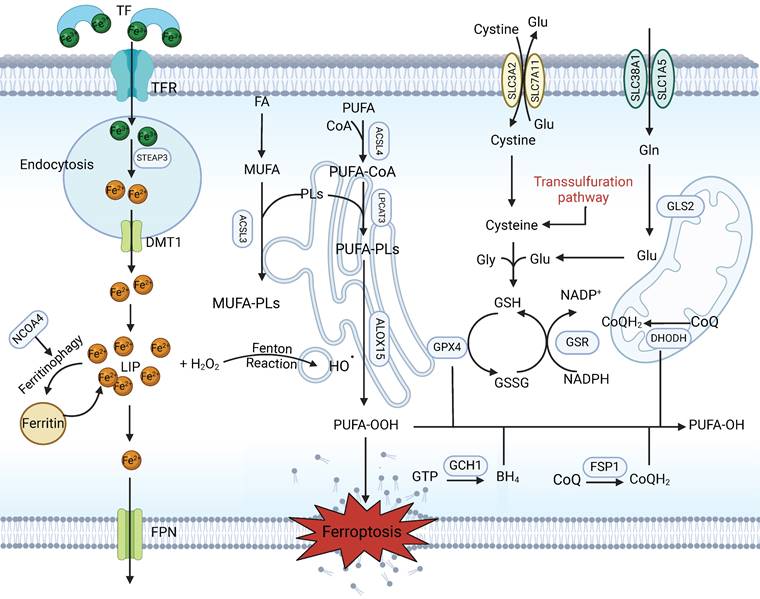
Within this context, ACSL4 is central to this process. ACSL4 was identified by a number of independent studies to be a key metabolic determinant in sensitizing cells to ferroptosis through the selective enrichment of PUFAs, more specifically arachidonic acid (AA)-containing species [28,31]. Ectopic ACSL4 promotes or attenuates ferroptosis. Studies have revealed that PKCβII, a member of the protein kinase C (PKC) family that acts as a sensor of lipid peroxidation, amplifies lipid peroxidation by phosphorylating and activating ACSL4 and is involved in the lipid peroxidation-PKCβII-ACSL4 positive feedback axis, which promotes ferroptosis onset [32]. Wang et al. demonstrated that IFNγ secreted by cytotoxic T lymphocytes (CTLs) and AA coordinately induces ferroptosis via ACSL4 in cancer cells [33]. Interestingly, in a novel study, researchers reported that ACSL4 deficiency prevents ferroptotic cell death in hepatocytes but does not aggravate tumour progression and instead results in less fibrosis and proliferation. Hence, ACSL4-dependent processes have an unanticipated cancer-promoting effect on HCC tumorigenesis [34]. Therefore, further therapies aimed at increasing ferroptosis sensitivity could consider the cancer-promoting function of ACSL4. In addition, the levels of lipid peroxides can be enhanced enzymatically by the lipoxygenase family (including ALOXE3, ALOX5, ALOX12, and ALOX15) in mammalian cells [35,36].
Ferroptosis Defensive Systems
GSH-dependent defensive system
Glutathione (GSH), the most abundant intracellular antioxidant, is a thiol-containing tripeptide derived from three amino acids (glycine, glutamate and cysteine), with cysteine being the rate-limiting cofactor and precursor [37-40]. GSH plays an essential role in maintaining redox homeostasis and neutralizing ROS to limit ferroptosis and serves as the substrate of choice for glutathione peroxidase 4 (GPX4), which is capable of detoxifying lipid peroxides into lipid alcohols [41,42]. GSH deficiency or depletion directly triggers ferroptotic cell death. De novo synthesis of GSH depends on cysteine obtained through cystine (an oxidized dimer of cysteine) uptake through a cystine/glutamate exchange transporter, known as System Xc-; this transporter consists of two subunits, solute carrier family 7 member 11 (SLC7A11) and solute carrier family 3 member 2 (SLC3A2) [43,44] (Figure 1). Therefore, ferroptosis can be potently induced by cysteine deprivation and GPX4 inhibition. Small pharmacological inhibitors, including the GPX4 inhibitor RSL3, erastin and sorafenib, which are direct inhibitors of the System Xc--mediated import function, are widely used for the induction of ferroptosis [4,45].
It is believed that the SLC7A11-GSH-GPX4 axis constitutes the major cellular system that defends against ferroptosis. Dysfunction of this axis can affect the occurrence of ferroptosis. Mounting research has been devoted to examining and clarifying the roles and mechanisms of the ferroptosis defence system in tumour suppression. Some tumour suppressors, including p53 and IFNγ, can hamper SLC7A11 expression and transport activity [46,47]. In contrast, tumour cells take advantage of stress-related transcription factors, including nuclear factor erythroid 2-related factor 2 (NRF2) and activating transcription factor 4 (ATF4), to increase SLC7A11 expression to combat ferroptosis. Some factors, such as 8 open reading frame 76 (C8orf76) and the RNA-binding protein DAZAP1, have been shown to inhibit ferroptosis through increasing the transcription of SLC7A11 in HCC [48,49]. Moreover, enhancing the ubiquitination-mediated degradation of SLC7A11 to induce ferroptosis by increasing the expression of suppressor of cytokine signalling 2 (SOCS2) may enhance the efficacy of HCC radiotherapy and improve patient prognosis [50]. Furthermore, many studies have demonstrated that GSH depletion can reduce lipid peroxide levels and that targeting GSH to decrease oxidative stress can suppress ferroptosis. Further research revealed that TGF-β1/Smad3 signalling plays a role in the repression of SLC7A11 activity and that promoting lipid peroxidation increases vulnerability to GPX4 inhibition in HCC cells [51].
However, some cancer cells remain resistant to ferroptosis in the absence of GPX4 activation [52], which suggests the existence of alternative mechanisms to counter cellular oxidative stress and suppress ferroptosis.
GSH-independent defensive system
Promisingly, ferroptosis suppressor protein 1 (FSP1) has been identified as a potential endogenous ferroptosis suppressor that functions in parallel with GPX4 to confer ferroptosis resistance [53,54]. FSP1, which is a second mainstay of ferroptosis control after GPX4[55], is also known as flavoprotein apoptosis-inducing factor mitochondria-associated 2 (AIFM2). The protein encoded by this gene is located on the plasma membrane, where it suppresses lipid hydroperoxides by catalysing the reduction of ubiquinone (CoQ)[56,57] back to ubiquinol (CoQH2)[54], which can trap lipid peroxyl radicals, thereby blocking PUFA-PL synthesis and ferroptosis. The expression of FSP1 is positively associated with the resistance of cells to GPX4 inhibitors and is essential for maintaining tumour cell growth in the absence of GPX4[54]. Therefore, FSP1 is a potent antioxidant that contributes to ferroptosis resistance [58]. A recent study identified FSP1 as a novel, vulnerable therapeutic target in HCC [59]. Furthermore, disrupting FSP1 is a promising therapeutic approach for HCC patients with high-density lipoprotein-binding protein (HDLBP) or lncFAL expression [60].
In addition, another antiferroptosis system is composed of dihydroorotate dehydrogenase (DHODH), which mechanistically operates in parallel with mitochondrial GPX4 to detoxify lipid peroxidation products and block ferroptosis by reducing CoQ to CoQH2[61]. Unlike FSP1, DHODH is localized to the inner mitochondrial membrane. DHODH has been intensively explored in recent years as a promising target for cancer therapy [62,63]. It has been proposed that overexpression of the oncogenic protein ubiquitin-conjugating enzyme E2T (UBE2T) in HCC leads to the upregulation of DHODH, thereby promoting HCC development [64].
Cancer cells rewire their metabolism and rely on endogenous antioxidants to mitigate lethal oxidative damage to lipids. Recently, a novel pathway named GTP cyclohydrolase 1 (GCH1)-tetrahydrobiopterin (BH4) was reported to be an essential metabolic signalling pathway upon GPX4 inhibition [65,66]. Mechanistically, BH4 is a potent radical-trapping antioxidant that protects lipid membranes from autoxidation, both alone and in combination with vitamin E [67,68]. Early reports have verified that GCH1, which originates from GTP, is a governing rate-limiting enzyme in the synthesis of BH4 and has the outstanding capacity to eliminate lipid peroxidation and prevent RSL3-induced ferroptosis [67,69]. GCH1 overexpression exhibits robust protection against RSL3- and IKE-induced ferroptosis and genetic ablation of GPX4-induced ferroptosis but does not protect cells against inducers of apoptosis and is only marginally effective against necroptosis, which indicates that GCH1 selectively counters ferroptotic cell death [65]. Thus, these results indicate that the GCH1-BH4 pathway acts as an endogenous antioxidant pathway to inhibit ferroptosis through a mechanism independent of the glutathione system. Although studies on the GCH1-BH4 pathway have not been applied to the inhibition of ferroptosis in HCC, the GCH1-BH4 pathway can undoubtedly promote HCC progression [70].
Ferroptosis in the Hepatocellular Carcinoma-associated Signalling Pathway
Noncoding RNA
As a result of the exponential growth in ferroptosis research, the participation of ncRNAs in regulating ferroptosis in HCC has also been reported. Mechanistically, miRNAs function by binding to complementary sequences in key genes involved in ferroptosis and suppressing their expression [71,72] (Figure 2). In one study, miR-23a-3p acted as a direct suppressor of ferroptosis by targeting the 3′‑untranslated region (UTR) of ACSL4 and was found to be responsible for the acquisition of sorafenib resistance in sorafenib‑treated HCC cells [73]. The mechanisms by which lncRNAs and circRNAs govern gene expression are relatively complex. On the one hand, they can act as ceRNAs to sponge miRNAs to block mRNA degradation in ferroptotic cell death and regulate the binding of transcription factors to promoters [74]. For example, as ceRNAs, HCG18 and CircIL4R can sponge miR-450b-5p and miR-541-3p, respectively, to modulate the expression of GPX4 and inhibit ferroptosis in HCC [75,76]. Similarly, circ0097009 acts as a competing endogenous RNA that regulates the expression of SLC7A11 by sponging miR-1261 in HCC [77]. Furthermore, the long noncoding RNA NEAT1 is upregulated in HCC cells after erastin and RSL3 treatment and cooperates with miR-362-3p and its downstream Myo-inositol oxygenase (MIOX), a nonheme ferritin, to form a ceRNA network that can eventually promote ferroptosis [78]. On the other hand, lncRNAs and circRNAs can also function as scaffolds to regulate protein-protein interactions and related downstream signalling pathways. In one study, LINC01134, a promising lncRNA, promoted NRF2 recruitment to the GPX4 promoter region to transcriptionally regulate GPX4. Facilitating ferroptosis by knocking down LINC01134 could be a potential therapeutic strategy for the treatment of HCC [79]. Recently, the ferroptosis-related lncRNA URB1-AS1, which is highly expressed in sorafenib-resistant HCC samples and mitigates sorafenib-induced ferroptosis by inducing ferritin phase separation and reducing the cellular free iron content, was identified through lncRNA sequencing. Silencing URB1-AS1 successfully enhanced the sensitivity of HCC cells to sorafenib in an in vivo tumour model [80].
Noncoding RNAs participate in regulating ferroptosis in HCC. NEAT1 indirectly upregulates MIOX expression by competitively binding to miR-362-3p, thereby promoting erastin- and RSL3-induced ferroptosis in HCC cells. p53 can increase NEAT1 expression. URB1-AS1 mitigates sorafenib-induced ferroptosis by inducing ferritin phase separation and reducing the free iron content. HIF-1α promotes URB1-AS1 expression. The ZNF8/miR-552-5p/ACSL4 and ETS1/miR-23a-3p/ACSL4 axes regulate sensitivity to ferroptosis. LncRNA HEPFAL promotes ferroptosis by modifying SLC7A11 ubiquitination. LINC01134 positively regulates GPX4 through NRF2. Downregulation of HULC induces ferroptosis in liver cancer cells by targeting the miR-3200-5p/ATF4 axis to modulate HCC development. Circ0097009 acts as a ceRNA to regulate the expression of SLC7A11 by sponging miR-1261. The lncRNAs HCG18 and CircIL4R sponge miR-450b-5p and miR-541-3p, respectively, to regulate GPX4. lncFAL reduces vulnerability to ferroptosis by binding to FSP1.
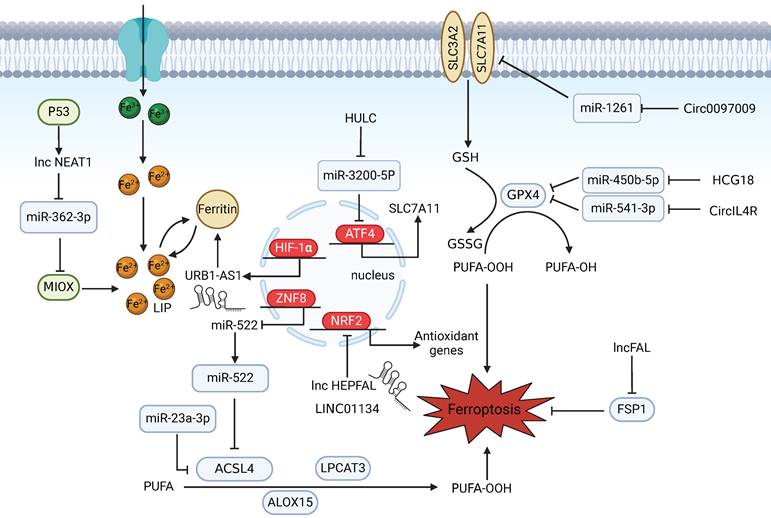
Additionally, some studies have shown that ncRNAs participate in the epigenetic modulation of chromatin to regulate gene expression. Overall, ncRNAs may serve as diagnostic biomarkers for HCC and as potential targets for HCC therapy.
NRF2
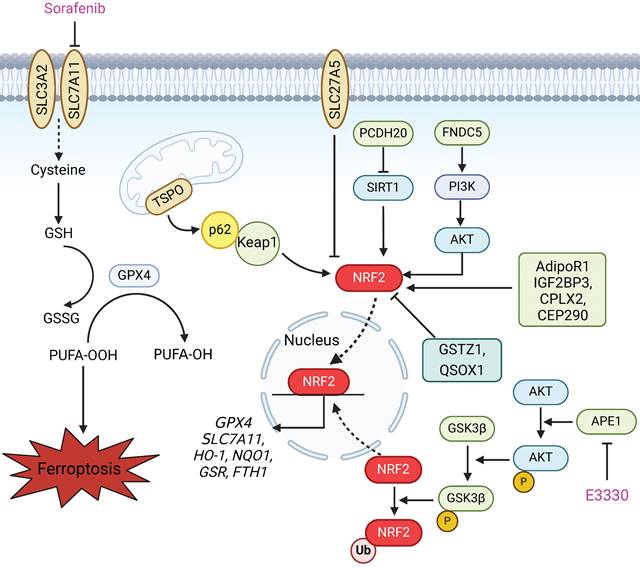
Nuclear factor erythroid 2-related factor 2 (NRF2) is considered a master regulator of the antioxidant response. NRF2 can translocate to the nucleus to initiate the transcription of antioxidant response element (ARE)-containing genes, which are involved in preventing or correcting redox imbalances in the cell in different contexts (Figure 3). Notably, two inducers of ferroptosis, RSL3 and erastin, initiate the ferroptotic cascade by inhibiting glutathione GPX4 and SLC7A11, both of which are downstream targets of NRF2. Furthermore, many other proteins and enzymes associated with glutathione biosynthesis, the antioxidant response, and lipid and iron metabolism, such as HO-1, NQO1, GSR, FTH1, FTL, and ferroportin, which are responsible for preventing lipid peroxidation and thus causing ferroptosis, are target genes of NRF2[81]. Therefore, ferroptotic cell death is largely associated with NRF2 dysfunction. NRF2 status is a key factor that determines the therapeutic response to ferroptosis-targeted therapies and improves resistance to chemotherapeutic drugs in HCC cells [82]. Several studies have shown that the NRF2 status is modulated by a variety of proteins and drugs, which in turn affects the occurrence of ferroptosis in HCC (Figure 3). According to recent research, the mitochondrial translocator protein (TSPO), which is involved in a broad range of mitochondrial functions, inhibits ferroptosis in HCC cells through the P62/KEAP1/NRF2 antioxidant pathway [83]. Protocadherin 20 (PCDH20) can promote ferroptosis by preventing Sirtuin 1 (SIRT1) from deacetylating NRF2, which leads to the downregulation of SLC7A11, GPX4, and GSH in HCC [84]. In addition, ABCC5, a member of the ATP-binding cassette (ABC) transporter family, and fibronectin type III domain containing 5 (FNDC5) can activate NRF2 through the PI3K/AKT pathway, which confers resistance to ferroptosis and reduces the anticancer activity of sorafenib in HCC cells [85,86]. Tiliroside, a potent TANK-binding kinase 1 (TBK1) inhibitor, induces ferroptosis via the P62/KEAP1/NRF2 pathway and eventually increases the sensitivity of HCC cells to sorafenib [87]. A recent study reported that apurinic/apyrimidinic endonuclease 1 (APE1), a key enzyme with dual functions in DNA repair and redox regulation, enhances ferroptosis through regulation of the NRF2/SLC7A11/GPX4 axis in HCC. Research on tumour xenografts in nude mice further demonstrated that the inhibition of APE1 blocks HCC progression and contributes to ferroptosis-based HCC therapy [88]. Thus, the induction of lipid peroxidation and ferroptosis by regulating the NRF2 pathway is a promising strategy for enhancing the anticancer effects of chemotherapy and radiotherapy, especially in therapy-resistant HCC cells.
NRF2 is involved in preventing lipid peroxidation and ferroptosis in HCC. Upon exposure of HCC cells to specific ferroptosis inducers, such as sorafenib, erastin and RSL3, the expression of P62 increases, which prevents degradation of the NRF2 protein by competitively binding to the Keap1 protein. This process also promotes the entry of NRF2 into the nucleus to initiate the transcription of downstream ferroptosis-related proteins, including GPX4, SLC7A11, HO-1, NQO1, GSR and FTH1. Many studies have shown that various proteins affect the sensitivity of HCC cells to ferroptosis via the NRF2 pathway. TSPO inhibits ferroptosis in HCC cells through the P62/KEAP1/NRF2 antioxidant pathway. SLC27A5 inhibits the NRF2/GSR pathway to reduce GSH in sorafenib-sensitive HCC cells. PCDH20 promotes ferroptosis by suppressing the expression of SIRT1 and thus promoting NRF2 acetylation in HCC. FNDC5 activates NRF2 through the PI3K/AKT pathway, conferring resistance to ferroptosis. AdipoR1, IGF2BP3, CPLX2 and CEP290 inhibit ferroptosis in liver cancer cells by activating the NRF2 pathway. GSTZ1 and QSOX1 enhance the sensitivity of HCC cells to sorafenib-induced ferroptosis via inhibition of the NRF2 pathway. Furthermore, APE1 inhibition promotes the degradation of NRF2 through the AKT/GSK3β-mediated ubiquitin‒proteasome pathway and subsequently suppresses the NRF2/SLC7A11/GPX4 axis, thereby facilitating ferroptosis.
HIF-1α
Given that an imbalance between insufficient blood supply and the high oxygen consumption needed to support the rapid growth of tumour cells leads to widespread hypoxia in the tumour microenvironment (TME), hypoxia tightly regulates tumour cell apoptosis, chemoresistance, invasion, immune escape, and many other biological functions [89-91]. In addition, hypoxia is accompanied by the production of ROS, which trigger oxidative stress [92]. Hypoxia-inducible factor 1 alpha (HIF-1α) is a crucial regulator of the response to hypoxic stress in cells. Several pioneering studies have shown that HIF-1α is an important driver of susceptibility to ferroptosis in cancer cells, including HCC cells, by modulating the transcription of numerous genes involved in iron metabolism, lipid metabolism, glycolysis and glutamate metabolism. Recently, evidence has suggested that HIF-1α serves as the main driver of ferroptosis resistance in tumour cells under hypoxic conditions through the generation of a lactate-induced acidic environment and enhancement of the transcription of the glutamate transporter SLC1A1[93]. In HCC, copper metabolism MURR1 domain 10 (COMMD10) inhibits the HIF-1α/CP loop, which enhances ferroptosis and radiosensitivity by disrupting Cu-Fe homeostasis [94]. Moreover, lipid metabolism reprogramming steered by HIF-1α results in a reduction in the synthesis of polyunsaturated fatty acids (PUFAs), which are substrates of lipid peroxidation, and thus reduces sensitivity to ferroptosis. Recently, FASN, an enzyme that regulates the de novo synthesis of fatty acids, was shown to bind to and upregulate HIF-1α and its downstream target SLC7A11 to inhibit HCC ferroptosis and promote sorafenib resistance [95]. Nevertheless, the potential of HIF-1α as a therapeutic target requires further research. The complex relationship between HIF-1α expression and various factors, including the mutational landscape of HCC, the tumour microenvironment, and the administration of other therapeutic agents, requires comprehensive consideration. In conclusion, understanding the links between HIF-1α and ferroptosis provides a novel perspective for understanding HCC pathology and may offer potential novel treatment strategies.
p53
The tumour suppressor p53 is considered the guardian of the genome and participates in multifaceted cellular activities, including cell cycle arrest, senescence, and apoptosis [96]. Remarkably, numerous studies have provided evidence that p53, a hub involved in cellular redox regulation and a therapeutic target in cancer, has various effects on ferroptotic cell death [97]. On the one hand, p53 has been shown to increase sensitivity to ferroptosis through the regulation of multiple downstream targets through either transcriptional or posttranslational mechanisms. First, p53 can decrease the expression of SLC7A11, which is necessary for the uptake of cystine and may contribute to tumour suppression in vitro and in vivo [46]. Second, the induction of glutaminase 2 (GLS2) and spermidine/spermine N1-acetyltransferase 1 (SAT1) at the transcriptional level promotes ferroptosis, favouring glutaminolysis and the formation of lipid peroxides, respectively [98,99]. Dysfunctional p53 signalling is one of the major causes of HCC tumorigenesis and development. Research has shown that other components of the p53 network can also regulate ferroptosis in HCC. For example, p53 can activate ALOX12, a lipoxygenase, indirectly by transcriptional repression of SLC7A11, which results in ALOX12-dependent ferroptosis upon ROS stress and alleviation of tumorigenesis [35]. The protocadherin gene PCDHB14, a novel p53 target gene, promotes ferroptosis to inhibit SLC7A11 expression via the NF-κB signalling pathway in HCC [100]. Another study revealed that Krüppel-associated box (KRAB)-type zinc-finger protein (ZNF498) functions as an oncogene in promoting HCC carcinogenesis by suppressing ferroptosis via interactions with p53, which decreases p53 Ser46 phosphorylation [101]. In addition, a study revealed that Gls2 KO mice have a marked propensity to develop late HCC, which suggests that GLS2 plays a role in ferroptosis and consequent tumour suppression [102]. On the other hand, p53 suppresses ferroptosis through direct inhibition of dipeptidyl peptidase 4 (DPP4) activity [103]. By identifying a new p53 acetylation site at lysine K136, mutations at all five acetylation sites (p53-5KR) completely/greatly reduced the remaining tumour suppressive function of p53[104,105], which indicates that p53 acetylation plays an important role in regulating ferroptosis and inhibiting tumour development. Given that p53 can modulate multiple target genes involved in various biological processes, the exact role of p53 in ferroptosis is likely to depend on the specific context.
Metabolic Dysregulation
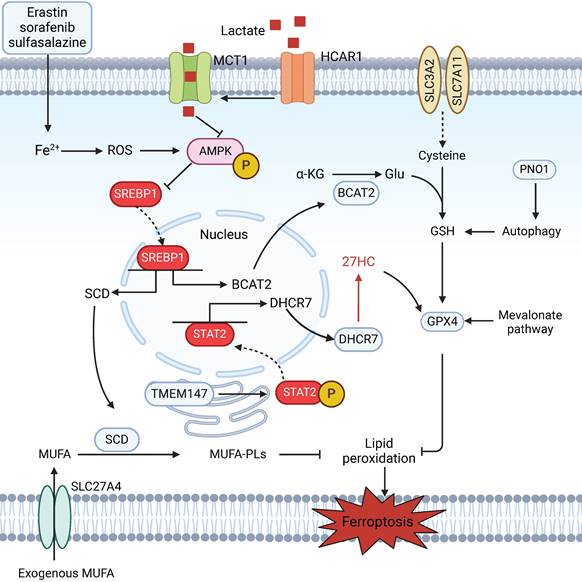
Accumulating evidence indicates that cellular metabolism plays a crucial role in ferroptosis [106,107]. Intracellular iron metabolism is essential for ferroptosis through either iron-dependent oxidases or the Fenton reaction. A recent report suggested that the autophagic degradation of ferritin regulates ferroptosis through the autophagy cargo receptor nuclear receptor coactivator 4 (NCOA4) [108]. Amino acid metabolism also influences the susceptibility of HCC cells to ferroptosis. Branched-chain amino acid aminotransferase 2 (BCAT2) suppresses ferroptosis by mediating the metabolism of sulfur-containing amino acids and regulating intracellular glutamate levels [109] (Figure 4). Additionally, the mevalonate (MVA) pathway not only affects the synthesis of CoQ and GPX4, two key regulators of ferroptosis, by regulating the maturation of selenocysteine tRNA, but also provides several antioxidant intermediates, such as squalene and 7-dehydrocholesterol, which act as radical-trapping agents to suppress lipid peroxidation [110,111]. According to recent reports, the RNA-binding protein partner of NOB1 (PNO1) protects HCC cells from ferroptotic cell death by increasing the synthesis and accumulation of intracellular glutamate [112]. The liver, the central organ of fatty acid metabolism, is enriched with a diverse range of lipids [113]. Interestingly, the occurrence and development of ferroptosis are closely related to lipid metabolism. Aberrant lipid metabolism, such as enhanced the cellular uptake of exogenous PUFAs via the overexpression of the fatty acid transporter enzyme CD36, could increase the cellular PUFA-PL content, thereby promoting lipid peroxidation and ferroptosis. In contrast, treatment with exogenous MUFAs results in a reduction in the amount of PUFA-PLs in cell membranes and inhibition of lipid peroxidation and subsequent ferroptosis [30]. In one study, solute carrier family 27 member 4 (SLC27A4), a fatty acid transporter protein, promoted the selective uptake of MUFAs, which resulted in a reduction in lipid peroxidation and resistance to ferroptosis [114]. In addition, elevated cholesterol levels in the tumour microenvironment (TME) are a common feature of various cancers. A metabolic derivative of cholesterol, 27-hydroxycholesterol, has been reported to trigger ferroptosis resistance by chronic selection of cancer cells with increased uptake and/or lipid biosynthesis, including cholesterol [115]; other studies have shown that the protective role of cholesterol is ascribed to its ability to decrease membrane fluidity and promote lipid raft formation, which affects the diffusion of LPO substrates and subsequent resistance to ferroptosis [116]. Therefore, future research should aim to determine additional connections between metabolic dysregulation and ferroptosis in HCC, which will pave the way for more effective therapeutic strategies.
Metabolic dysregulation mediates ferroptosis in HCC. The AMPK/SREBP1 pathway plays a pivotal role in mediating metabolic homeostasis during ferroptosis. HCAR1/MCT1-mediated lactate uptake promotes ATP production in HCC cells and deactivates AMPK, which leads to the upregulation of SREBP1 and downstream SCD1 to enhance the production of MUFAs; this results in resistance to lipid peroxidation and ferroptosis in HCC cells. In addition, SLC27A4 overexpression promotes the selective uptake of MUFAs in HCC cells. BCAT2 is the key enzyme that regulates intracellular glutamate levels and is a specific inhibitor of ferroptosis. Ferroptosis inducers (erastin, sorafenib, and sulfasalazine) can activate the AMPK/SREBP1 pathway and subsequently inhibit BCAT2 transcription. Moreover, the level of intracellular glutamate could be promoted by PNO1-induced autophagy, which results in the accumulation of GSH and ferroptosis in HCC. The mevalonate pathway affects the synthesis of CoQ and GPX4 by regulating the maturation of selenocysteine tRNA. Increased levels of 27HC also upregulate GPX4 in HCC, leading to ferroptosis resistance via the TMEM147/STAT2/DHCR7 axis.
Ferroptosis in Hepatocellular Carcinoma Therapy
Chemotherapy
Patients with advanced HCC generally have fewer opportunities for surgery, and interventions such as interventional therapies are often limited due to liver function and other reasons. Therefore, targeted therapy is usually the main treatment option, and it is popular because of its minimal side effects, good efficacy, and convenience. Currently, three main targeted drugs are used to treat advanced HCC in China. Among them, first-line treatments include sorafenib and lenvatinib, while second-line treatment includes regorafenib.
Sorafenib is a multitarget, multikinase inhibitor and first-line molecular-target drug for the treatment of advanced HCC that prolongs the median overall survival to 6.5 months [117]. Ferroptosis is essential for sorafenib-induced cell death and has been implicated in the pathology of HCC; thus, ferroptosis induction is a promising novel cancer treatment [118,119]. Notably, sorafenib has been reported to block SLC7A11 function, subsequent ROS accumulation and GSH depletion, in addition to its well-known inhibition of many angiogenesis-associated kinases (e.g., VEGFR, FGFR, PDGFR) [120]. Thus, it is reasonable to speculate that genetic alterations involved in ferroptosis may regulate sorafenib sensitivity and mediate drug resistance during the development or treatment of HCC, which adds to the complexity of the antitumour mechanism of sorafenib (Figure 5). A few studies have explored this hypothesis. YAP/TAZ [121] and C8orf76[48] act as negative regulators of ferroptosis by upregulating SLC7A11 transcription, thus driving resistance to sorafenib in hepatocellular carcinoma. Louandre C et al. reported that, upon exposure to sorafenib, the reduced levels of Rb achieved through stable RNA interference in HCC cells promote the onset of ferroptosis [122]. In one study, the positive-acting enzyme ACSL4, whose expression is independent of sorafenib treatment in HCC cell lines, can serve as a promising predictive and validated biomarker that contributes to the precise treatment of HCC that is superior to that involving Rb [123]. In addition, the ability of sorafenib to regulate a series of downstream effectors, such as MT1 and HBXIP, increases sensitivity to ferroptosis [124,125]. Apart from some research already devoted to deciphering the mechanisms that enhance sorafenib-induced ferroptosis, it is imperative to identify resistance targets and combination drugs, which are essential for overcoming the dilemma of sorafenib resistance due to their limited survival. Here, we summarize some compounds that can target other essential aspects of ferroptosis that may synergize with sorafenib to inhibit HCC (Table 1) and compounds or drugs that trigger ferroptotic cell death that exhibit antitumour effects independent of sorafenib-induced ferroptosis (Table 2).
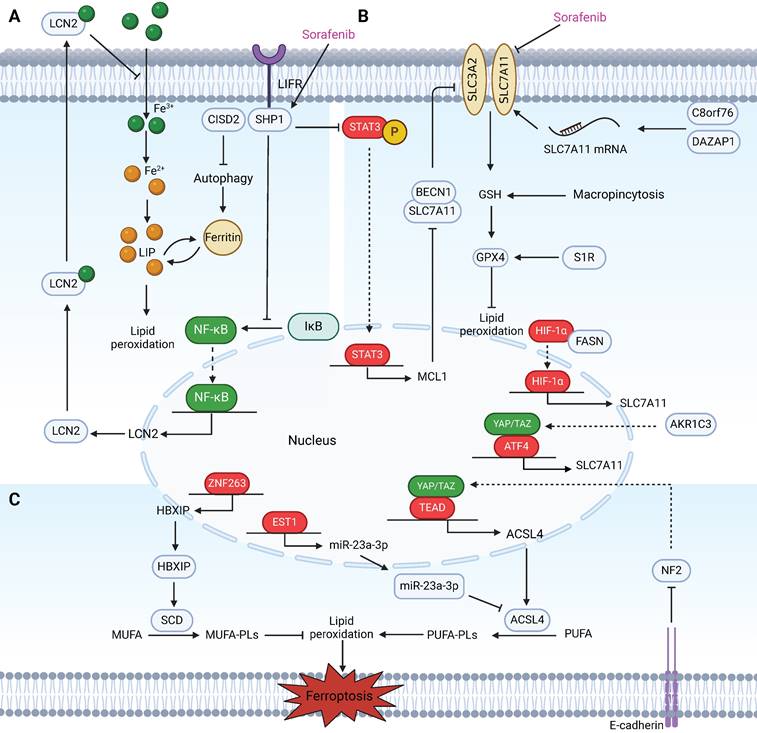
Regulatory factors involved in sensitivity and resistance to ferroptosis in HCC patients treated with sorafenib. These regulatory factors mainly participate in sensitivity and resistance to ferroptosis in HCC cells treated with sorafenib through three pathways. (A) Labile iron pool: Inhibition of CISD2 promotes excessive iron accumulation through autophagy, resulting in sorafenib-induced ferroptosis in resistant cells. LIFR sensitizes HCC cells to sorafenib-induced ferroptosis through NF-κB inhibition and the subsequent downregulation of iron-sequestering LCN2. (B) The biosynthesis of PUFAs: Activation of the HBXIP/SCD axis via coactivation of ZNF263 reduces the anticancer activity of sorafenib and suppresses ferroptosis. MiR-23a-3p acts as a direct suppressor of ferroptosis by targeting the 3'UTR of ACSL4. The ETS1/miR‑23a‑3p/ACSL4 axis contributes to sorafenib resistance in HCC by regulating ferroptosis. Inhibition of the Hippo signalling pathway can activate YAP to promote the transcription of ACSL4, thereby promoting ferroptosis. The interaction mediated by E-cadherin in HepG2 cells suppresses ferroptosis by activating the intracellular NF2 and Hippo signalling pathways. (C) The defensive system against ferroptosis: In sorafenib-resistant HCC cells, YAP/TAZ and ATF4 are activated in the nucleus where they induce SLC7A11 expression. AKR1C3 suppresses ferroptosis through the regulation of YAP/SLC7A11. Binding between BECN1 and SLC7A11 increases, which inhibits the activity of System Xc- and the triggering of ferroptosis in sorafenib-treated HCC cells via the SHP-1/STAT3/MCL1 axis. C8orf76 and DAZAP1 reduce cellular sensitivity to sorafenib by acting on SLC7A11 through different mechanisms. Additionally, targeting the FASN/HIF1α/SLC7A11 pathway could resensitize HCC cells to sorafenib. S1R and macropinocytosis negatively regulate sorafenib-induced ferroptosis.
Agents that synergize with sorafenib.
| Agents | Mechanism | Target | References |
|---|---|---|---|
| Carmustine (BCNU) | Inhibits glutathione reductase (GSR) | GSR | [81] |
| RSL3 | Inhibits GPX4 in GSTZ1-deficient hepatoma cells | GPX4 | [142] |
| Tiliroside | Inhibits TBK1-p62-Keap1-NRF2 pathway | GPX4, G6PD, FTH1 | [87] |
| Metformin | Inhibits p62-Keap1-NRF2 pathway | HO-1 | [143] |
| Camptothecin | Inhibits p62-Keap1-NRF2 pathway | GPX4, GSR, SLC7A11 | [144] |
| Orlistat | Inhibits FASN/HIF1α/SLC7A11 pathway | SLC7A11 | [95] |
| Withaferin A | Inhibits Keap1-NRF2 pathway | SLC7A11 | [145] |
| Artesunate | Induces lysosome-mediated ferritinophagy | FTL | [146] |
| Caryophyllene oxide | Induces ferritinophagy by regulating the NCOA4/FTH1/LC3 pathway | NCOA4 | [147] |
| Haloperidol | Enhances the expression of sigma receptor 1 (S1R) | S1R | [148] |
| Disulfiram (DSF) | Inhibits the compensatory NRF2 elevation | NRF2 | [149] |
| Aspirin | Silences ACSL4 and induces GADD45B expression | ACSL4 | [150] |
| Deferasirox (DFX) | Inhibits NF-κB activity | NF-κB | [151] |
Potential compounds or drugs for HCC therapy via triggering ferroptosis.
| Compounds/ Drugs | Mechanism | Target | References |
|---|---|---|---|
| Gentian violet | Increases p53 levels | p53 | [152] |
| Dihydroartemisinin (DHA) | Promotes PEBP1 protein expression | PEBP1 | [153] |
| ZZW-115 | Induces mitochondrial dysfunction with a ROS overproduction and downregulates key enzymes involved in the GPX-dependent antioxidant systems | TFAM | [154] |
| Anisomycin | Activates p38 MAPK through H3S10 phosphorylation and upregulates NCOA4 | NCOA4 | [155] |
| Electrophilic sesquiterpenes isolated from E. chinense L. (EChLESs) | Controls the expression of NCOA4 at the transcriptional and post-transcriptional levels | NCOA4 | [156] |
| Schizocapsa plantaginea Hance I (SSPH I) | Elevates the expression of SLC7A5 and induces iron accumulation | SLC7A5, TFR and FPN | [157] |
| All-trans retinoic acid (ATRA) | Blocks GSH synthesis | GPX4, FTH1, GCLC, GCLM, SOD-1 | [158] |
| PK11195 | Inhibits TSPO-p62-Keap1-NRF2 pathway | GPX4, HO-1, NQO1, GCLC, GCLM | [83] |
| Polyphyllin I (PPI) | Inhibits NRF2/HO-1/GPX4 antioxidant axis | GPX4, HO-1 | [159] |
| Scutellaria barbata | Promotes iron peroxidation and lipid ROS metabolism by regulating ferroptosis-related genes | GPX4, SLC7A11, IREB2, ACSL4 | [160] |
| Solasonine | Destroys the glutathione redox system by suppression of GPX4 and glutathione synthetase (GSS) | GPX4, GSS | [161] |
| Parthenolide (PTL) | Increases GSH depletion, rapid oxidation of intracellular and mitochondrial thiols, mitochondrial dysfunction, and suppression of GPX4 | GPX4 | [162] |
| Heteronemin | Reduces the expression of GPX4 | GPX4 | [163] |
| Polyphyllin VI | Inhibits the STAT3/GPX4 axis | STAT3, GPX4 | [164] |
| Atractylodin | Inhibits the expression of GPX4 and FTL and upregulates the expression of ACSL4 and TFR1 | GPX4, FTL, ACSL4, TFR1 | [165] |
| Aspirin | Triggers ferroptosis by restricting NF-κB-activated SLC7A11 transcription | SLC7A11 | [166] |
| Solamargine | Inducing ferroptosis by downregulating MTCH1 expression | MTCH1 | [167] |
In recent years, the new targeted therapy drug lenvatinib has shown good efficacy, especially in the treatment of HBV-related HCC, and has become the second first-line drug for the treatment of advanced liver cancer after sorafenib. Compared with sorafenib, lenvatinib significantly improved the objective response rate (ORR), progression-free survival (PFS) and time to progression (TTP) in one study [126]. This finding demonstrated the superior antitumour effect of lenvatinib over sorafenib.
Lenvatinib has been approved as an alternative option for patients who do not respond to sorafenib treatment, and its targets include VEGFR, PDGFR, FGFR1/2/3/4, KIT, and RET [127-130]. An association between ferroptosis and lenvatinib has also been reported. One study revealed that lenvatinib suppressed SLC7A11 and GPX4 expression, which resulted in the accumulation of lipid ROS in Hep3B and HuH7 cells. These findings further confirmed that lenvatinib induces ferroptosis by inhibiting FGFR4. FGFR4 expression in cancer cells is related to the therapeutic efficacy of lenvatinib in patients with HCC. The PFS of FGFR4-positive HCC patients is longer than that of FGFR4-negative patients. Additionally, NRF2 is involved in the regulation of this process [131]. As with sorafenib, resistance to lenvatinib is common. Therefore, the exploration of the potential mechanisms of lenvatinib resistance in HCC is urgently needed. A recent study revealed that HAND2-AS1 promotes the expression of ferroptosis-related genes (TLR4, NOX2, and DUOX2) and promotes ferroptosis to reverse lenvatinib resistance in HepG2 lenvatinib-resistant cells and xenograft models [132]. Lenvatinib can induce ferroptosis, which is also involved in lenvatinib resistance, but related research on this topic is lacking. Therefore, further research is needed to determine whether induction of ferroptosis to reverse lenvatinib resistance is a promising treatment strategy.
Regorafenib, a second-line targeted therapy for HCC, is typically used for liver cancer patients to prolong patient survival after failure of first-line treatment [133]. Regorafenib is a multitargeted tyrosine kinase inhibitor that can inhibit tumour cell proliferation, suppress tumour angiogenesis, and modulate the tumour microenvironment. This drug targets VEGFR, PDGFR, B/C-Raf, KIT, RET, and FGFR1/2 [134]. Currently, no published studies have revealed the connection between regorafenib and ferroptosis in HCC.
Antitumour Immunotherapy
In recent years, breakthroughs have been achieved in the application of immunotherapy in HCC patients. Atezolizumab (an anti-PD-L1 antibody) in combination with bevacizumab (a VEGF antagonist) was approved for first-line treatment of advanced-stage HCC [135]. However, more sufficient medical evidence is still needed for immunotherapy as a first-line treatment. Recent research has revealed that ferroptosis is a form of immunogenic cell death (ICD) and has a certain degree of crosstalk with immunotherapy compared with other types of cell death, including apoptosis, autophagy and necrosis [136]. On the one hand, the ferroptotic pathway can reshape the tumour immune microenvironment and improve anti-PD1 immunotherapy in HCC. Using single-cell RNA sequencing, one study identified a differentially expressed gene, APOC1, which is overexpressed in tumour-associated macrophages (TAMs) in HCC tissues. APOC1 inhibition can promote the transition of M2 macrophages to M1 macrophages through the ferroptotic pathway [137]. Furthermore, for the first time, a recent study revealed ferroptosis upstream of immune cell activation and demonstrated that hepatocyte ferroptosis due to genetic GPX4 ablation causes a complex immune response that includes triggering CD8+ T-cell recruitment and inducing IFN-γ secretion by CD8+ T cells, which enhances PD-L1 expression on tumour cells. Thus, a novel therapeutic option for the treatment of HCC involving the combination of withaferin A (WFA) (a ferroptosis inhibitor), α-PD-1 and SB225002 (a CXCR2 inhibitor) has been proposed [138]. On the other hand, the immune response can trigger tumour cell ferroptosis. For example, in anti-PD-L1 immunotherapy, IFN-γ secreted by activated CD8+ T cells downregulates the expression of SLC3A2 and SLC7A11, impairs the uptake of cystine by tumour cells, and consequently promotes ferroptosis-specific lipid peroxidation in tumour cells [47]. In addition, IFN-γ stimulates ACSL4 and alters the lipid profile of tumour cells, which promotes ACSL4-dependent tumour ferroptosis [139]. Thus, cancer immune checkpoint blockade paired with selective fatty acids is a potential anticancer approach. However, studies have shown that CD8+ T cells lose their antitumour effector function in the TME when they take up fatty acids through CD36, which induces ferroptosis and results in decreased cytotoxic cytokine production by cells. Inhibition of CD36-mediated ferroptosis in combination with ICB greatly enhances the antitumour effects of CD8+ T cells [140]. Therefore, to some extent, ferroptosis also suppresses antitumour immunity and promotes tumour growth.
Many studies have identified key molecules and drugs that can change the efficacy of immunotherapy in patients with HCC. FSP1 inhibitor (iFSP1) exerts a better effect than anti-PD-L1 in prolonging survival time, and the combination of iFSP1 and anti-PD-L1 can further inhibit the progression of HCC in mice [59]. Phosphoglycerate mutase 1 (PGAM1) inhibition and the mitochondrial translocator protein (TSPO) inhibitor PK11195 exert antitumour effects by promoting ferroptosis, and these compounds can synergize with anti-PD-1 immunotherapy in HCC [83,141]. Therefore, the addition of ferroptosis-based therapy to immunotherapy provides a new option for the personalized treatment of HCC patients. Overall, the intersection of ferroptosis and the immune system in HCC provides fertile ground for novel antitumour immunotherapeutic strategies, but the detailed mechanism underlying the relationship between ferroptosis and the TME is not fully understood, and extensive clinical research on how to induce ferroptosis safely and effectively in clinical practice is lacking.
Future Perspectives and Conclusion
Ferroptosis is a novel form of programmed and nonapoptotic cell death driven by cellular metabolism and iron-dependent lipid peroxidation. Recent clarification of molecular pathways and regulatory proteins, such as GPX4, ACSL4, and NRF2, which control ferroptosis, has provided insight into the underlying mechanism of ferroptosis. However, we currently have only a patchy portrait of the importance of several genes involved in preventing or inducing ferroptosis, and several questions about the fine-tuning of ferroptotic cell death remain largely unanswered.
Hepatocellular carcinoma, one of the most prevalent malignancies and a leading cause of cancer-related mortality worldwide, is characterized by rapid disease onset and progression. With increasing research on ferroptosis, tremendous evidence indicates that ferroptosis plays a positive role in anti-HCC therapy and could become a promising therapeutic target. Therefore, this article focused on summarizing the hotspots and major findings in ferroptosis research in HCC, including the current understanding of ferroptosis mechanisms and research based on an increase in susceptibility to ferroptosis combined with chemotherapy or immunotherapy to treat HCC, which could improve anticancer effects.
Different viewpoints suggest that ferroptosis demonstrates unique advantages in its underlying mechanisms and implications for HCC therapy compared with other forms of cell death. Furthermore, ferroptosis has emerged as a potential therapeutic target for the treatment of HCC due to its distinct molecular pathways and vulnerability to pharmacological manipulation. Targeting key regulators of ferroptosis, such as GPX4 or the lipid peroxidation pathway, holds promise for selectively inducing cancer cell death. In contrast, conventional therapies for HCC, such as chemotherapy and radiotherapy, primarily induce apoptosis or necrosis and may not effectively target ferroptosis pathways. Immunotherapy, which harnesses the immune system to target cancer cells, has shown promising results in HCC treatment. Understanding the differences between ferroptosis and other forms of cell death is crucial for optimizing immunotherapeutic strategies. Ferroptosis may contribute to immunogenic cell death and enhance antitumour immune responses, but its interplay with immunotherapy requires further investigation. Finally, the combination of ferroptosis-inducing agents with immunotherapeutic approaches may offer synergistic benefits by promoting tumour cell death while stimulating antitumour immune responses.
In the application of ferroptosis therapy in HCC, treatment tolerance and responsiveness are critical factors that influence the effectiveness and safety of the therapy. Treatment tolerance mainly includes patient health status, the toxicity of ferroptosis inducers or inhibitors used to treat HCC, dosing and the risk of adverse effects of combining ferroptosis-based therapy with other treatment modalities, such as chemotherapy or immunotherapy. Furthermore, HCC is characterized by significant heterogeneity, both within individual tumours and among different patients. Some tumours may be more susceptible to ferroptosis induction due to specific molecular profiles, while others may be resistant. The tumour microenvironment, which includes factors such as hypoxia, inflammation, and stromal components, can modulate ferroptosis sensitivity. Strategies that modulate the tumour microenvironment to enhance treatment responsiveness need to be explored. The mechanisms underlying resistance to ferroptosis therapy, such as the upregulation of antioxidant pathways or alterations in iron metabolism, need to be understood and targeted to improve treatment responsiveness. Overall, achieving an optimal balance between treatment tolerance and treatment responsiveness is essential for the successful application of ferroptosis therapy in HCC. This requires a comprehensive understanding of the underlying mechanisms, patient-specific factors, and careful clinical management. Ongoing research aimed at elucidating these factors and optimizing treatment strategies will be critical for improving outcomes in HCC patients receiving ferroptosis-based therapy.
Abbreviations
LIP: labile iron pool; TF: transferrin; TFR: transferrin receptor 1; STEAP3: STEAP family member 3; DMT1: divalent metal transporter 1; NCOA4: nuclear receptor coactivator 4; FPN: ferroportin; PUFA: polyunsaturated fatty acid; CoA: coenzyme A; PL: lysophosphatide; ACSL4: acyl-CoA synthetase long-chain family member 4; LPCAT3: lysophosphatidylcholine acyltransferase 3; PUFA-OOH: polyunsaturated fatty acid hydroperoxides; MUFA: monounsaturated fatty acid; ACSL3: acyl-CoA synthetase long-chain family member 3; GSH: glutathione; SLC7A11: solute carrier family 7 member 11; SLC3A2: solute carrier family 3 member 2; GPX4: glutathione peroxidase 4; Cys: cysteine; Gly: glycine; Glu: glutamic acid; Gln: glutamine; GSSG: glutathione disulfide; GSR: glutathione reductase; FSP1: ferroptosis suppressor protein 1; GCH-1: guanosine triphosphate cyclohydrolase 1; BH4: tetrahydrobiopterin; CoQ: coenzyme Q (also known as ubiquinone); CoQH2: ubiquinol; DHODH: dihydroorotate dehydrogenase; GLS2: glutamine synthase 2; SLC38A1: solute carrier family 38 member 1; SLC1A5: solute carrier family 1 member 5; NEAT1: nuclear paraspeckle assembly transcript 1; MIOX: Myo-inositol oxygenase; HULC: highly upregulated in liver cancer; ATF4: activating transcription factor 4; ZNF8: Krüppel-associated box (KRAB)-type zinc-finger protein 8; lncRNA HEPFAL: hepatocellular carcinoma ferroptosis associative lncRNA; circIL4R: circ-interleukin-4 receptor; MELK: maternal-fetal leucine zipper kinase; YTHDF2: YTH N6-methyladenosine RNA-binding protein 2; lncFAL: ferroptosis-associated lncRNA; HDLBP: high-density lipoprotein-binding protein; SLC27A5: solute carrier family 27 member 5; TSPO: translocator protein; PCDH20: protocadherin 20; SIRT1: sirtuin 1; FNDC5: fibronectin type III domain containing 5; AdipoR1: adiponectin receptor 1; IGF2BP3: insulin-like growth factor 2 mRNA-binding protein 3; CPLX2: complexin II; CEP290: centrosomal protein 290; GSTZ1: glutathione S-transferase zeta 1; QSOX1: quiescin sulfhydryl oxidase 1; LCN2: lipocalin 2; CISD2: CDGSH iron sulfur domain 2; LIFR: leukemia inhibitory factor receptor; SHP-1: Src homology region 2 domain-containing phosphatase-1; STAT3: signal transducer and activator of transcription 3; MCL1: myeloid cell leukemia-1; BECN1: beclin 1; C8orf76: Chromosome 8 open reading frame 76; DAZAP1: deleted in azoospermia-associated protein 1; PNO1: RNA-binding protein partner of NOB1; S1R: Sigma-1 receptor; HIF-1α: hypoxia-inducible factor 1-alpha; FASN: fatty acid synthase; AKR1C3: Aldo-keto reductase 1C3; YAP: Yes-associated protein 1; TEAD1: TEA domain transcription factor 1; HBXIP: hepatitis B X-interacting protein; ETS1: ETS Proto‑Oncogene 1; SCD: stearoyl-CoA desaturase; SLC27A4: solute carrier family 27 member 4; MTCH1: mitochondrial carrier 1.
Acknowledgements
This review was supported by the Provincial Natural Science Research Project of Anhui Educational Committee (Grant: KJ2020ZD24), the open fund of Key Laboratory of Anti-inflammatory and Immune Medicine Ministry of Education, P.R. China (Anhui Medical University; Grant: KFJJ-2021-04), Anhui Provincial Academic and Technical Leaders and Reserve Candidates Project (Grant: 2022H291), Horizontal Project Funding of Anhui Medical University (Grant: 2023009) and Translational Medicine Research fund of Anhui Province (Grant: 2023zhyx-C24).
Author Contributions
Xinyue Zhu and Yan Zang supervised the general concept of this review. Li Yu and Tao Li revised the manuscript. Qiaohui Ren, Shubing Zhang and Dongyue Ma dedicated in drafting the manuscript. Lianzi Wang and Xudong Sha participated in sorting and organizing the sentences and figures. Xinyi Zhou and Junxiao Yao participated in the literature search. All authors have read and agreed to the published version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Vogel A, Meyer T, Sapisochin G, Salem R, Saborowski A. Hepatocellular carcinoma. Lancet. 2022;400:1345-62
2. Sung H, Ferlay J, Siegel RL. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. Ca Cancer J Clin. 2021;71:209-49
3. Li X, Ramadori P, Pfister D, Seehawer M, Zender L, Heikenwalder M. The immunological and metabolic landscape in primary and metastatic liver cancer. Nat Rev Cancer. 2021;21:541-57
4. Dixon SJ, Lemberg KM, Lamprecht MR. et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell. 2012;149:1060-72
5. Yang WS, SriRamaratnam R, Welsch ME. et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell. 2014;156:317-31
6. Mo Y, Zou Z, Chen E. Targeting ferroptosis in hepatocellular carcinoma. Hepatol Int. 2024;18:32-49
7. Ajoolabady A, Tang D, Kroemer G, Ren J. Ferroptosis in hepatocellular carcinoma: mechanisms and targeted therapy. Br J Cancer. 2023;128:190-205
8. Xie Y, Hou W, Song X. et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369-79
9. Yang WS, Stockwell BR. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem Biol. 2008;15:234-45
10. Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22:381-96
11. Galaris D, Barbouti A, Pantopoulos K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim Biophys Acta Mol Cell Res. 2019;1866:118535
12. Andrews NC, Schmidt PJ. Iron Homeostasis. Annu Rev Physiol. 2007;69:69-85
13. Kouroumalis E, Tsomidis I, Voumvouraki A. Iron as a therapeutic target in chronic liver disease. World J Gastroenterol. 2023;29:616-55
14. Yu Y, Jiang L, Wang H. et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood. 2020;136:726-39
15. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell. 2015;59:298-308
16. Ward DM, Kaplan J. Ferroportin-mediated iron transport: Expression and regulation. Biochim Biophys Acta. 2012;1823:1426-33
17. Dev S, Babitt JL. Overview of iron metabolism in health and disease: Iron metabolism in health and disease. Hemodial Int. 2017;21:S6-20
18. Chen X, Yu C, Kang R, Tang D. Iron Metabolism in Ferroptosis. Front Cell Dev Biol. 2020;8:590226
19. Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. 2019;15:1137-47
20. Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. 2017;482:419-25
21. Koppula P, Zhuang L, Gan B. Cytochrome P450 reductase (POR) as a ferroptosis fuel. Protein & Cell. 2021;12:675-9
22. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci. 2016 113
23. Yan B, Ai Y, Sun Q. et al. Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol Cell. 2021;81:355-369.e10
24. Zou Y, Li H, Graham ET. et al. Cytochrome P450 oxidoreductase contributes tophospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16:302-9
25. Wenzel SE, Tyurina YY, Zhao J. et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell. 2017;171:628-641.e26
26. Asperti M, Bellini S, Grillo E. et al. H-ferritin suppression and pronounced mitochondrial respiration make Hepatocellular Carcinoma cells sensitive to RSL3-induced ferroptosis. Free Radic Biol Med. 2021;169:294-303
27. Dixon SJ, Winter GE, Musavi LS. et al. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem Biol. 2015;10:1604-9
28. Doll S, Proneth B, Tyurina YY. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91-8
29. Zou Y, Henry WS, Ricq EL. et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585:603-8
30. Magtanong L, Ko P-J, To M. et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem Biol. 2019;26:420-432.e9
31. Friedmann Angeli JP, Xavier da Silva TN, Schilling B. CD8+ T cells PUF(A)ing the flames of cancer ferroptotic cell death. Cancer Cell. 2022;40:346-8
32. Zhang H-L, Hu B-X, Li Z-L. et al. PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol. 2022;24:88-98
33. Wang Z. IFNγ regulates ferroptosis with fatty acids. Nat Cell Biol. 2022;24:601-601
34. Grube J, Woitok MM, Mohs A. et al. ACSL4-dependent ferroptosis does not represent a tumor-suppressive mechanism but ACSL4 rather promotes liver cancer progression. Cell Death Dis. 2022;13:704
35. Chu B, Kon N, Chen D. et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21:579-91
36. Qin Y, Pei Z, Feng Z. et al. Oncogenic Activation of YAP Signaling Sensitizes Ferroptosis of Hepatocellular Carcinoma via ALOXE3-Mediated Lipid Peroxidation Accumulation. Front Cell Dev Biol. 2021;9:751593
37. Aquilano K, Baldelli S, Ciriolo MR. Glutathione: new roles in redox signaling for an old antioxidant. Front Pharmacol. 2014;5:196
38. Forman HJ, Zhang H, Rinna A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med. 2009;30:1-12
39. Lee J, Roh J-L. SLC7A11 as a Gateway of Metabolic Perturbation and Ferroptosis Vulnerability in Cancer. Antioxidants. 2022;11:2444
40. Pompella A, Visvikis A, Paolicchi A, Tata VD, Casini AF. The changing faces of glutathione, a cellular protagonist. Biochem Pharmacol. 2003;66:1499-503
41. Bayır H, Anthonymuthu TS, Tyurina YY. et al. Achieving Life through Death: Redox Biology of Lipid Peroxidation in Ferroptosis. Cell Chem Biol. 2020;27:387-408
42. Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. 2019;133:144-52
43. Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12:599-620
44. Sato H, Tamba M, Ishii T, Bannai S. Cloning and Expression of a Plasma Membrane Cystine/Glutamate Exchange Transporter Composed of Two Distinct Proteins. J Biol Chem. 1999;274:11455-8
45. Lachaier E, Louandre C, Godin C. et al. Sorafenib Induces Ferroptosis in Human Cancer Cell Lines Originating from Different Solid Tumors. Anticancer Res. 2014;34:6417-22
46. Lei G, Zhang Y, Hong T. et al. Ferroptosis as a mechanism to mediate p53 function in tumor radiosensitivity. Oncogene. 2021;40:3533-47
47. Wang W, Green M, Choi JE. et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270-4
48. Li D, Pan J, Zhang Y. et al. C8orf76 Modulates Ferroptosis in Liver Cancer via Transcriptionally Up-Regulating SLC7A11. Cancers. 2022;14:3410
49. Wang Q, Guo Y, Wang W. et al. RNA binding protein DAZAP1 promotes HCC progression and regulates ferroptosis by interacting with SLC7A11 mRNA. Exp Cell Res. 2021;399:112453
50. Chen Q, Zheng W, Guan J. et al. SOCS2-enhanced ubiquitination of SLC7A11 promotes ferroptosis and radiosensitization in hepatocellular carcinoma. Cell Death Differ. 2023;30:137-51
51. Kim DH, Kim WD, Kim SK, Moon DH, Lee SJ. TGF-β1-mediated repression of SLC7A11 drives vulnerability to GPX4 inhibition in hepatocellular carcinoma cells. Cell Death Dis. 2020;11:406
52. Viswanathan VS, Ryan MJ, Dhruv HD. et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453-7
53. Doll S, Freitas FP, Shah R. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693-8
54. Bersuker K, Hendricks JM, Li Z. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688-92
55. Wu J, Wang Y, Jiang R. et al. Ferroptosis in liver disease: new insights into disease mechanisms. Cell Death Discov. 2021;7:276
56. Stefely JA, Pagliarini DJ. Biochemistry of Mitochondrial Coenzyme Q Biosynthesis. Trends Biochem Sci. 2017;42:824-43
57. Crane FL. Discovery of ubiquinone (coenzyme Q) and an overview of function. Mitochondrion. 2007;7:S2-7
58. Zeng F, Chen X, Deng G. The anti-ferroptotic role of FSP1: current molecular mechanism and therapeutic approach. Mol Biomed. 2022;3:37
59. Cheu JW-S, Lee D, Li Q. et al. Ferroptosis Suppressor Protein 1 Inhibition Promotes Tumor Ferroptosis and Anti-tumor Immune Responses in Liver Cancer. Cell Mol Gastroenterol Hepatol. 2023;16:133-59
60. Yuan J, Lv T, Yang J. et al. HDLBP-stabilized lncFAL inhibits ferroptosis vulnerability by diminishing Trim69-dependent FSP1 degradation in hepatocellular carcinoma. Redox Biol. 2022;58:102546
61. Mao C, Liu X, Zhang Y. et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586-90
62. Madak JT, Bankhead A, Cuthbertson CR, Showalter HD, Neamati N. Revisiting the role of dihydroorotate dehydrogenase as a therapeutic target for cancer. Pharmacol Ther. 2019;195:111-31
63. Li L, Ng SR, Colón CI. et al. Identification of DHODH as a therapeutic target in small cell lung cancer. Sci Transl Med. 2019;11:eaaw7852
64. Zhu Z, Cao C, Zhang D. et al. UBE2T-mediated Akt ubiquitination and Akt/β-catenin activation promotes hepatocellular carcinoma development by increasing pyrimidine metabolism. Cell Death Dis. 2022;13:154
65. Kraft VAN, Bezjian CT, Pfeiffer S. et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci. 2020;6:41-53
66. Soula M, Weber RA, Zilka O. et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol. 2020;16:1351-60
67. Vasquez-Vivar J, Shi Z, Tan S. Tetrahydrobiopterin in Cell Function and Death Mechanisms. Antioxid Redox Signal. 2022;37:171-83
68. Wei C-C, Crane BR, Stuehr DJ. Tetrahydrobiopterin Radical Enzymology. Chem Rev. 2003;103:2365-84
69. Kim HK, Han J. Tetrahydrobiopterin in energy metabolism and metabolic diseases. Pharmacol Res. 2020;157:104827
70. Zhong G-C, Zhao Z-B, Cheng Y. et al. Epigenetic silencing of GCH1promotes hepatocellular carcinoma growth by activating superoxide anion-mediated ASK1/p38 signaling via inhibiting tetrahydrobiopterin de novo biosynthesis. Free Radic Biol Med. 2021;168:81-94
71. Panni S, Lovering RC, Porras P, Orchard S. Non-coding RNA regulatory networks. Biochim Biophys Acta Gene Regul Mech. 2020;1863:194417
72. Zhang X, Wang L, Li H, Zhang L, Zheng X, Cheng W. Crosstalk between noncoding RNAs and ferroptosis: new dawn for overcoming cancer progression. Cell Death Dis. 2020;11:580
73. Lu Y, Chan Y-T, Tan H-Y. et al. Epigenetic regulation of ferroptosis via ETS1/miR-23a-3p/ACSL4 axis mediates sorafenib resistance in human hepatocellular carcinoma. J Exp Clin Canc Res. 2022;41:3
74. Huang J, Wang J, He H. et al. Close interactions between lncRNAs, lipid metabolism and ferroptosis in cancer. Int J Biol Sci. 2021;17:4493-513
75. Li X, Li Y, Lian P, lv Q, Liu F. Silencing lncRNA HCG18 regulates GPX4-inhibited ferroptosis by adsorbing miR-450b-5p to avert sorafenib resistance in hepatocellular carcinoma. Hum Exp Toxicol. 2023;42:9603271221142818
76. Xu Q, Zhou L, Yang G. et al. CircIL4R facilitates the tumorigenesis and inhibits ferroptosis in hepatocellular carcinoma by regulating the miR-541-3p/GPX4 axis. Cell Biol Int. 2020;44:2344-56
77. Lyu N, Zeng Y, Kong Y. et al. Ferroptosis is involved in the progression of hepatocellular carcinoma through the circ0097009/miR-1261/SLC7A11 axis. Ann Transl Med. 2021;9:675-675
78. Zhang Y, Luo M, Cui X, O'Connell D, Yang Y. Long noncoding RNA NEAT1 promotes ferroptosis by modulating the miR-362-3p/MIOX axis as a ceRNA. Cell Death Differ. 2022;29:1850-63
79. Kang X, Huo Y, Jia S. et al. Silenced LINC01134 Enhances Oxaliplatin Sensitivity by Facilitating Ferroptosis Through GPX4 in Hepatocarcinoma. Front Oncol. 2022;12:939605
80. Gao Y, Tong M, Wong T-L. et al. Long Noncoding RNA URB1-Antisense RNA 1 (AS1) Suppresses Sorafenib-Induced Ferroptosis in Hepatocellular Carcinoma by Driving Ferritin Phase Separation. ACS Nano. 2023;17:22240-58
81. Xu F, Wu X, Chen C. et al. SLC27A5 promotes sorafenib-induced ferroptosis in hepatocellular carcinoma by downregulating glutathione reductase. Cell Death Dis. 2023;14:22
82. Sun X, Ou Z, Chen R. et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells: Hepatobiliary Malignancies. Hepatology. 2016;63:173-84
83. Zhang D, Man D, Lu J. et al. Mitochondrial TSPO Promotes Hepatocellular Carcinoma Progression through Ferroptosis Inhibition and Immune Evasion. Adv Sci. 2023;10:2206669
84. Jun L, Chen W, Han L, Yanmin L, Qinglei Z, Pengfei Z. Protocadherin 20 promotes ferroptosis by suppressing the expression of Sirtuin 1 and promoting the acetylation of nuclear factor erythroid 2-related factor 2 in hepatocellular carcinoma. Int J Biochem Cell Biol. 2023;156:106363
85. Huang W, Chen K, Lu Y. et al. ABCC5 facilitates the acquired resistance of sorafenib through the inhibition of SLC7A11-induced ferroptosis in hepatocellular carcinoma. Neoplasia. 2021;23:1227-39
86. Liu H, Zhao L, Wang M. et al. FNDC5 Causes Resistance to Sorafenib by Activating the PI3K/Akt/Nrf2 Pathway in Hepatocellular Carcinoma Cells. Front Oncol. 2022;12:852095
87. Yang C, Lu T, Liu M. et al. Tiliroside targets TBK1 to induce ferroptosis and sensitize hepatocellular carcinoma to sorafenib. Phytomedicine. 2023;111:154668
88. Du Y, Zhou Y, Yan X. et al. APE1 inhibition enhances ferroptotic cell death and contributes to hepatocellular carcinoma therapy. Cell Death Differ. 2024;31:431-46
89. Chen Z, Shao Y-L, Wang L-L. et al. YTHDF2 is a potential target of AML1/ETO-HIF1α loop-mediated cell proliferation in t(8;21) AML. Oncogene. 2021;40:3786-98
90. Li Q, Ni Y, Zhang L. et al. HIF-1α-induced expression of m6A reader YTHDF1 drives hypoxia-induced autophagy and malignancy of hepatocellular carcinoma by promoting ATG2A and ATG14 translation. Signal Transduct Target Ther. 2021;6:76
91. Yang Z, Yu W, Huang R, Ye M, Min Z. SIRT6/HIF-1α axis promotes papillary thyroid cancer progression by inducing epithelial-mesenchymal transition. Cancer Cell Int. 2019;19:17
92. Smith KA, Waypa GB, Schumacker PT. Redox signaling during hypoxia in mammalian cells. Redox Biol. 2017;13:228-34
93. Yang Z, Su W, Wei X. et al. HIF-1α drives resistance to ferroptosis in solid tumors by promoting lactate production and activating SLC1A1. Cell Rep. 2023;42:112945
94. Yang M, Wu X, Hu J. et al. COMMD10 inhibits HIF1α/CP loop to enhance ferroptosis and radiosensitivity by disrupting Cu-Fe balance in hepatocellular carcinoma. J Hepatol. 2022;76:1138-50
95. Li Y, Yang W, Zheng Y. et al. Targeting fatty acid synthase modulates sensitivity of hepatocellular carcinoma to sorafenib via ferroptosis. J Exp Clin Canc Res. 2023;42:6
96. Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med. 2019;133:162-8
97. Eriksson SE, Ceder S, Bykov VJN, Wiman KG. p53 as a hub in cellular redox regulation and therapeutic target in cancer. Verma CS, Ed. J Mol Cell Biol. 2019;11:330-41
98. Ou Y, Wang S-J, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci. 2016;113:E6806-12
99. Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci. 2010;107:7455-60
100. Liu Y, Ouyang L, Mao C. et al. PCDHB14 promotes ferroptosis and is a novel tumor suppressor in hepatocellular carcinoma. Oncogene. 2022;41:3570-83
101. Zhang X, Zheng Q, Yue X. et al. ZNF498 promotes hepatocellular carcinogenesis by suppressing p53-mediated apoptosis and ferroptosis via the attenuation of p53 Ser46 phosphorylation. J Exp Clin Canc Res. 2022;41:79
102. Suzuki S, Venkatesh D, Kanda H. et al. GLS2 Is a Tumor Suppressor and a Regulator of Ferroptosis in Hepatocellular Carcinoma. Cancer Res. 2022;82:3209-22
103. Xie Y, Zhu S, Song X. et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017;20:1692-704
104. Kon N, Ou Y, Wang S-J, Li H, Rustgi AK, Gu W. mTOR inhibition acts as an unexpected checkpoint in p53-mediated tumor suppression. Genes Dev. 2021;35:59-64
105. Wang S-J, Li D, Ou Y. et al. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016;17:366-73
106. Stockwell BR, Friedmann Angeli JP, Bayir H. et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171:273-85
107. Angeli JPF, Shah R, Pratt DA, Conrad M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol Sci. 2017;38:489-98
108. Hou W, Xie Y, Song X. et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425-8
109. Wang K, Zhang Z, Tsai H. et al. Branched-chain amino acid aminotransferase 2 regulates ferroptotic cell death in cancer cells. Cell Death Differ. 2021;28:1222-36
110. Garcia-Bermudez J, Baudrier L, Bayraktar EC. et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature. 2019;567:118-22
111. Freitas FP, Alborzinia H, Dos Santos AF. et al. 7-Dehydrocholesterol is an endogenous suppressor of ferroptosis. Nature. 2024;626:401-10
112. Hu X, He Y, Han Z. et al. PNO1 inhibits autophagy-mediated ferroptosis by GSH metabolic reprogramming in hepatocellular carcinoma. Cell Death Dis. 2022;13:1010
113. Alves-Bezerra M, Cohen DE. Triglyceride Metabolism in the Liver. In: Terjung R, Ed. Compr Physiol. 2017;8:1-8
114. Li Z. SLC27A4-mediated selective uptake of mono-unsaturated fatty acids promotes ferroptosis defense in hepatocellular carcinoma. Free Radic Biol Med. 2023;201:41-54
115. Liu W, Chakraborty B, Safi R, Kazmin D, Chang C, McDonnell DP. Dysregulated cholesterol homeostasis results in resistance to ferroptosis increasing tumorigenicity and metastasis in cancer. Nat Commun. 2021;12:5103
116. Zhao X, Lian X, Xie J, Liu G. Accumulated cholesterol protects tumours from elevated lipid peroxidation in the microenvironment. Redox Biol. 2023;62:102678
117. Bruix J, Takayama T, Mazzaferro V. et al. Adjuvant sorafenib for hepatocellular carcinoma after resection or ablation (STORM): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2015;16:1344-54
118. Nie J, Lin B, Zhou M, Wu L, Zheng T. Role of ferroptosis in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2018;144:2329-37
119. Tang W, Chen Z, Zhang W. et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: theoretical basis and therapeutic aspects. Signal Transduct Target Ther. 2020;5:87
120. Dixon SJ, Patel DN, Welsch M. et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. 2014;3:e02523
121. Gao R, Kalathur RKR, Coto-Llerena M. et al. YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis. Embo Mol Med. 2021;13:e14351
122. Louandre C, Marcq I, Bouhlal H. et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015;356:971-7
123. Feng J, Lu P, Zhu G. et al. ACSL4 is a predictive biomarker of sorafenib sensitivity in hepatocellular carcinoma. Acta Pharmacol Sin. 2021;42:160-70
124. Houessinon A, François C, Sauzay C. et al. Metallothionein-1 as a biomarker of altered redox metabolism in hepatocellular carcinoma cells exposed to sorafenib. Mol Cancer. 2016;15:38
125. Zhang L, Li X, Shi X. et al. Sorafenib triggers ferroptosis via inhibition of HBXIP/SCD axis in hepatocellular carcinoma. Acta Pharmacol Sin. 2023;44:622-34
126. Nair A, Reece K, Donoghue MB. et al. FDA Supplemental Approval Summary: Lenvatinib for the Treatment of Unresectable Hepatocellular Carcinoma. Oncologist. 2021;26:e484-91
127. Tohyama O, Matsui J, Kodama K. et al. Antitumor Activity of Lenvatinib (E7080): An Angiogenesis Inhibitor That Targets Multiple Receptor Tyrosine Kinases in Preclinical Human Thyroid Cancer Models. J Thyroid Res. 2014;2014:1-13
128. Yamamoto Y, Matsui J, Matsushima T. et al. Lenvatinib, an angiogenesis inhibitor targeting VEGFR/FGFR, shows broad antitumor activity in human tumor xenograft models associated with microvessel density and pericyte coverage. Vasc Cell. 2014;6:18
129. Matsuki M, Hoshi T, Yamamoto Y. et al. Lenvatinib inhibits angiogenesis and tumor fibroblast growth factor signaling pathways in human hepatocellular carcinoma models. Cancer Med. 2018;7:2641-53
130. Kudo M. Lenvatinib May Drastically Change the Treatment Landscape of Hepatocellular Carcinoma. Liver Cancer. 2018;7:1-19
131. Iseda N, Itoh S, Toshida K. et al. Ferroptosis is induced by lenvatinib through fibroblast growth factor receptor-4 inhibition in hepatocellular carcinoma. Cancer Sci. 2022;113:2272-87
132. Song Z, Zhang Y, Luo W. et al. HAND2-AS1 Promotes Ferroptosis to Reverse Lenvatinib Resistance in Hepatocellular Carcinoma by TLR4/NOX2/DUOX2 Axis. Curr Cancer Drug Targets. 2024 24
133. Bruix J, Qin S, Merle P. et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389:56-66
134. Wilhelm SM, Dumas J, Adnane L. et al. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Intl Journal of Cancer. 2011;129:245-55
135. Finn RS, Qin S, Ikeda M. et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med. 2020;382:1894-905
136. Efimova I, Catanzaro E, Van Der Meeren L. et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J Immunother Cancer. 2020;8:e001369
137. Hao X, Zheng Z, Liu H. et al. Inhibition of APOC1 promotes the transformation of M2 into M1 macrophages via the ferroptosis pathway and enhances anti-PD1 immunotherapy in hepatocellular carcinoma based on single-cell RNA sequencing. Redox Biology. 2022;56:102463
138. Conche C, Finkelmeier F, Pešić M. et al. Combining ferroptosis induction with MDSC blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut. 2023;72:1774-82
139. Liao P, Wang W, Wang W. et al. CD8+ T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell. 2022;40:365-378.e6
140. Ma X, Xiao L, Liu L. et al. CD36-mediated ferroptosis dampens intratumoral CD8+ T cell effector function and impairs their antitumor ability. Cell Metab. 2021;33:1001-1012.e5
141. Zheng Y, Wang Y, Lu Z. et al. PGAM1 Inhibition Promotes HCC Ferroptosis and Synergizes with Anti-PD-1 Immunotherapy. Adv Sci. 2023;10:2301928
142. Wang Q, Bin C, Xue Q. et al. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021;12:426
143. Tang K, Chen Q, Liu Y, Wang L, Lu W. Combination of Metformin and Sorafenib Induces Ferroptosis of Hepatocellular Carcinoma Through p62-Keap1-Nrf2 Pathway. J Cancer. 2022;13:3234-43
144. Elkateb AS, Nofal S, Ali SA, Atya HB. Camptothecin Sensitizes Hepatocellular Carcinoma Cells to Sorafenib- Induced Ferroptosis Via Suppression of Nrf2. Inflammation. 2023;46:1493-511
145. Zhang Y, Tan Y, Liu S. et al. Implications of Withaferin A for the metastatic potential and drug resistance in hepatocellular carcinoma cells via Nrf2-mediated EMT and ferroptosis. Toxicol Mech Methods. 2023;33:47-55
146. Li Z, Dai H, Huang X. et al. Artesunate synergizes with sorafenib to induce ferroptosis in hepatocellular carcinoma. Acta Pharmacol Sin. 2021;42:301-10
147. Xiu Z, Zhu Y, Han J. et al. Caryophyllene Oxide Induces Ferritinophagy by Regulating the NCOA4/FTH1/LC3 Pathway in Hepatocellular Carcinoma. Front Pharmacol. 2022;13:930958
148. Bai T, Wang S, Zhao Y, Zhu R, Wang W, Sun Y. Haloperidol, a sigma receptor 1 antagonist, promotes ferroptosis in hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2017;491:919-25
149. Ren X, Li Y, Zhou Y. et al. Overcoming the compensatory elevation of NRF2 renders hepatocellular carcinoma cells more vulnerable to disulfiram/copper-induced ferroptosis. Redox Biol. 2021;46:102122
150. Xia H, Lee KW, Chen J. et al. Simultaneous silencing of ACSL4 and induction of GADD45B in hepatocellular carcinoma cells amplifies the synergistic therapeutic effect of aspirin and sorafenib. Cell Death Discov. 2017;3:17058
151. Jomen W, Ohtake T, Akita T. et al. Iron chelator deferasirox inhibits NF-κB activity in hepatoma cells and changes sorafenib-induced programmed cell deaths. Biomed Pharmacother. 2022;153:113363
152. Chen J, Zhao F, Yang H. et al. Gentian violet induces apoptosis and ferroptosis via modulating p53 and MDM2 in hepatocellular carcinoma. Am J Cancer Res. 2022;12:3357-72
153. Su Y, Zhao D, Jin C. et al. Dihydroartemisinin Induces Ferroptosis in HCC by Promoting the Formation of PEBP1/15-LO. Xu Q, Ed. Oxid Med Cell Longev. 2021;2021:1-22
154. Huang C, Santofimia-Castaño P, Liu X. et al. NUPR1 inhibitor ZZW-115 induces ferroptosis in a mitochondria-dependent manner. Cell Death Discov. 2021;7:269
155. Chen W, Yang W, Zhang C. et al. Modulation of the p38 MAPK Pathway by Anisomycin Promotes Ferroptosis of Hepatocellular Carcinoma through Phosphorylation of H3S10. Luo L, Ed. Oxid Med Cell Longev. 2022;2022:1-20
156. Zhu Z, Xu X, Shen C. et al. A novel sesquiterpene lactone fraction from Eupatorium chinense L. suppresses hepatocellular carcinoma growth by triggering ferritinophagy and mitochondrial damage. Phytomedicine. 2023;112:154671
157. Huang D, Dong X, Li J. et al. Steroidal saponin SSPH I induces ferroptosis in HepG2 cells via regulating iron metabolism. Med Oncol. 2023;40:132
158. Sun Y, He Y, Tong J. et al. All-trans retinoic acid inhibits the malignant behaviors of hepatocarcinoma cells by regulating ferroptosis. Genes Dis. 2022;9:1742-56
159. Yang R, Gao W, Wang Z. et al. Polyphyllin I induced ferroptosis to suppress the progression of hepatocellular carcinoma through activation of the mitochondrial dysfunction via Nrf2/HO-1/GPX4 axis. Phytomedicine. 2024;122:155135
160. Li Y, Zhang J, Zhang K. et al. Scutellaria barbata Inhibits Hepatocellular Carcinoma Tumorigenicity by Inducing Ferroptosis of Hepatocellular Carcinoma Cells. Front Oncol. 2022;12:693395
161. Jin M, Shi C, Li T, Wu Y, Hu C, Huang G. Solasonine promotes ferroptosis of hepatoma carcinoma cells via glutathione peroxidase 4-induced destruction of the glutathione redox system. Biomed Pharmacother. 2020;129:110282
162. LoBianco FV, Krager KJ, Johnson E. et al. Parthenolide induces rapid thiol oxidation that leads to ferroptosis in hepatocellular carcinoma cells. Front Toxicol. 2022;4:936149
163. Chang W-T, Bow Y-D, Fu P-J. et al. A Marine Terpenoid, Heteronemin, Induces Both the Apoptosis and Ferroptosis of Hepatocellular Carcinoma Cells and Involves the ROS and MAPK Pathways. Angeloni C, Ed. Oxid Med Cell Longev. 2021;2021:1-12
164. Huang Q, Li J, Ma M. et al. High-throughput screening identification of a small-molecule compound that induces ferroptosis and attenuates the invasion and migration of hepatocellular carcinoma cells by targeting the STAT3/GPX4 axis. Int J Oncol. 2023;62:42
165. He Y, Fang D, Liang T. et al. Atractylodin may induce ferroptosis of human hepatocellular carcinoma cells. Ann Transl Med. 2021;9:1535-1535
166. Wang Y, Feng J, Zhao L. et al. Aspirin triggers ferroptosis in hepatocellular carcinoma cells through restricting NF-κB p65-activated SLC7A11 transcription. Acta Pharmacol Sin. 2023;44:1712-24
167. Zhang L, Wang J, Deng W, Gui F, Peng F, Zhu Q. Solamargine Induces Hepatocellular Carcinoma Cell Apoptosis and Ferroptosis via Regulating STAT1/MTCH1 Axis. Biochem Genet. 2024
Author contact
![]() Corresponding authors: Tao Li, MD, Department of Clinical Laboratory, the First Affiliated Hospital of Anhui Medical University, Hefei, 230032, Anhui Province, China. Email: taoliedu.cn. Or Li Yu, PhD, Anhui Province Key Laboratory of Zoonoses, Anhui Medical University, Hefei, 230032, Anhui Province, China. Email: lilyyu33com.
Corresponding authors: Tao Li, MD, Department of Clinical Laboratory, the First Affiliated Hospital of Anhui Medical University, Hefei, 230032, Anhui Province, China. Email: taoliedu.cn. Or Li Yu, PhD, Anhui Province Key Laboratory of Zoonoses, Anhui Medical University, Hefei, 230032, Anhui Province, China. Email: lilyyu33com.