Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Renal lipid homeostasis
Lipid homeostasis imbalance and...
Autophagy of lipid homeostasis...
Conclusion and prospect
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(10):3710-3724. doi:10.7150/ijbs.95216 This issue Cite
Review
Lipid homeostasis in diabetic kidney disease
Ying Wang1,2, Tongtong Liu3, Yun Wu2, Lin Wang2, Shaowei Ding2, Baoluo Hou2, Hailing Zhao1 ![]() , Weijing Liu2
, Weijing Liu2 ![]() , Ping Li1
, Ping Li1 ![]()
1. China-Japan Friendship Hospital, Institute of Medical Science, Beijing, China.
2. Key Laboratory of Chinese Internal Medicine of Ministry of Education and Beijing, Dongzhimen Hospital Affiliated to Beijing University of Chinese Medicine, Beijing, China.
3. Guang'anmen Hospital, China Academy of Chinese Medical Sciences, Beijing, China.
Received 2024-2-9; Accepted 2024-6-21; Published 2024-7-2
Abstract

Lipid homeostasis is crucial for proper cellular and systemic functions. A growing number of studies confirm the importance of lipid homeostasis in diabetic kidney disease (DKD). Lipotoxicity caused by imbalance in renal lipid homeostasis can further exasperate renal injury. Large lipid deposits and lipid droplet accumulation are present in the kidneys of DKD patients. Autophagy plays a critical role in DKD lipid homeostasis and is involved in the regulation of lipid content. Inhibition or reduction of autophagy can lead to lipid accumulation, which in turn further affects autophagy. Lipophagy selectively recognizes and degrades lipids and helps to regulate cellular lipid metabolism and maintain intracellular lipid homeostasis. Therefore, we provide a systematic review of fatty acid, cholesterol, and sphingolipid metabolism, and discuss the responses of different renal intrinsic cells to imbalances in lipid homeostasis. Finally, we discuss the mechanism by which autophagy, especially lipophagy, maintains lipid homeostasis to support the development of new DKD drugs targeting lipid homeostasis.
Keywords: Diabetic kidney disease, Lipid homeostasis, Lipotoxicity, Autophagy.
Introduction
Diabetic kidney disease (DKD) is the most common complication of diabetes mellitus (DM) and the leading cause of end-stage renal disease (ESRD) [1], and it is also one of the fastest growing causes of chronic kidney disease (CKD) morbidity and mortality [2]. DKD is characterized by the accumulation of collagen, fibronectin, and other extracellular matrix (ECM) proteins. Such accumulation leads to inflammation induced by tubulointerstitial fibrosis, glomerular plasma membrane hypertrophy and dilatation, thickening of the glomerular basement membrane, loss of podocyte foot processes, and infiltration of monocytes and macrophages [3]. Disorders of lipid metabolism are associated with renal insufficiency and the pathological features of DKD [4]. Increasing evidence has shown that imbalances in lipid homeostasis and lipotoxicity can lead to kidney injury in DKD [5]. Lipid accumulation in the kidney is related to glomerulosclerosis and tubulointerstitial damage [6, 7]. Significant lipid deposition and lipid droplet (LD) accumulation are found in the kidneys of patients with DKD [8]. Lipid deposition has been reported in diabetic glomerulosclerosis [8, 9]. Lipotoxicity due to fatty acid (FA) deposition and renal tubulointerstitial fibrosis characterized by epithelial-mesenchymal transition (EMT) are hallmarks of DKD [10]. There are differences in the lipid metabolic profile in various stages of DKD [11]. Macroalbuminuria is associated with elevated total cholesterol levels in DKD [12]. Dyslipidemia, which is characterized by high levels of triglyceride-rich lipoproteins, low levels of high-density lipoprotein-cholesterol (HDL-C), and high levels of oxidized low-density lipoprotein (oxLDL), accelerates DKD progression [13, 14]. Therefore, close crosstalk occurs between lipid metabolism and DKD.
Lipids are the primary source of energy for the kidney, which is a highly energy-intensive organ [15]. The total lipid content of the kidney in healthy individuals is about 3% of its wet weight [16], with more than half of the lipid content being phospholipids, about one-fifth being triglycerides, and about one-tenth being nonesterified FAs (NEFAs). In humans, the level of kidney-extracted FAs is linearly related to plasma FA concentrations [16]. The levels of cholesterol, phospholipids, triglycerides, FAs, and sphingolipids are altered in DKD, and their accumulation in the kidney is associated with DKD pathogenesis [17]. FA oxidation (FAO) can be reduced by disrupting the balance between FA synthesis, intake, and consumption, thereby affecting renal lipid metabolism and leading to intracellular lipid accumulation [18]. Additionally, dyslipidemia alters lipid homeostasis by causing apoptosis of podocytes and endothelial cells, macrophage activation, and mesangial matrix hyperplasia, as well as increasing lipoprotein receptor-mediated cholesterol uptake, inhibiting ATP-binding cassette protein 1-mediated cholesterol efflux, and impairing cholesterol synthesis in peripheral cells [19]. However, the renoprotective effect of lipid-lowering therapy remains controversial [20, 21].
Here, we describe the major regulatory molecules of renal lipid metabolism homeostasis, discuss lipid metabolism in various types of renal intrinsic cells, and summarize the current state of research on the role of autophagy in lipid metabolism. As mentioned earlier, the imbalance of lipid homeostasis and lipotoxicity can aggravate DKD progression. This article aims to support the development of new DKD drugs targeting lipid homeostasis.
Renal lipid homeostasis
The mechanisms of renal lipid accumulation may differ among different causes of chronic kidney disease. In glomerulonephritis, inflammation can disrupt normal renal lipid metabolism. However, renal lipid accumulation in DKD is mainly driven by increased glucose and fatty acids levels owing to insulin resistance [22]. Under normal conditions, the uptake, synthesis, and oxidation or efflux of FAs in renal cells are regulated by a series of transcription factors to achieve a balanced and coordinated system to avoid lipid accumulation in the cells [8, 23]. FA synthase (FAS) and acetyl-CoA carboxylase (ACC) catalyze FA synthesis, whereas stearoyl-CoA desaturase 1 is a rate-limiting enzyme that converts saturated FAs to monounsaturated FAs. FAs are translocated to the mitochondria and degraded (through β-oxidation) by carnitine palmitoyltransferase 1 (CPT1) and acyl-CoA oxidase [8].
Lipotoxicity is a metabolic condition caused by the intracellular accumulation of toxic lipid intermediates in nonadipose tissues, thus resulting in cellular dysfunction and potential cell death (lipoapoptosis) [24]. FAs are essential for cell structure, function, and signaling. In the blood, free FAs (FFAs) are transported by serum albumin as complex lipoproteins [25]. In kidney disease patients, oxidative modification of HDL and LDL particles occurs, leading to the formation of small lipoproteins and enhanced synthesis of oxLDL [19]. FFAs produced by adipocytes or released by extracellular lipases are transported into cells by membrane-associated proteins that include scavenger receptor B2 (SR-B2; also known as differentiation antigen 36 [CD36]), FA transporter proteins (FATPs), and FA-binding proteins (FABPs) or by passive diffusion [26]. The discussion on FA uptake, transport, oxidation, and synthesis below also includes cholesterol metabolism and sphingolipid metabolism (Fig. 1).
Fatty acid, cholesterol and sphingolipid metabolism. (1) FAs are taken up by cells through CD36, CXCL16, and FATP, and transported to mitochondria via FABP. They also synthesize TGs in the endoplasmic reticulum, which are stored as LDs. These LDs can be decomposed by lysosomes or undergo lipophagy (not shown in the figure). CPT1 on the outer membrane of the mitochondria serves as a carrier to control FA β-oxidation of medium- and long-chain FAs. The oxidation-related genes are regulated by PPAR and PGC-1α. SREBPs and ChREBPs can affect lipid synthesis. (2) Cholesterol influx mainly depends on SR-A1, CD36, LOX-1, LDLR. Cholesterol production is primarily regulated by SREBP-2 and can be activated in the Golgi apparatus. Free cholesterol can be esterified in the endoplasmic reticulum and stored as LDs that may undergo decomposition by lysosomes or lipophagy (not shown in the figure). Cholesterol efflux is mainly mediated by ABCA1, ABCG1, and SR-BI under regulation from LXR. (3) Sphingolipids include CER, C1P and S1P, etc.,while SMPDL3b regulates cell membrane fluidity associated with podocyte function.
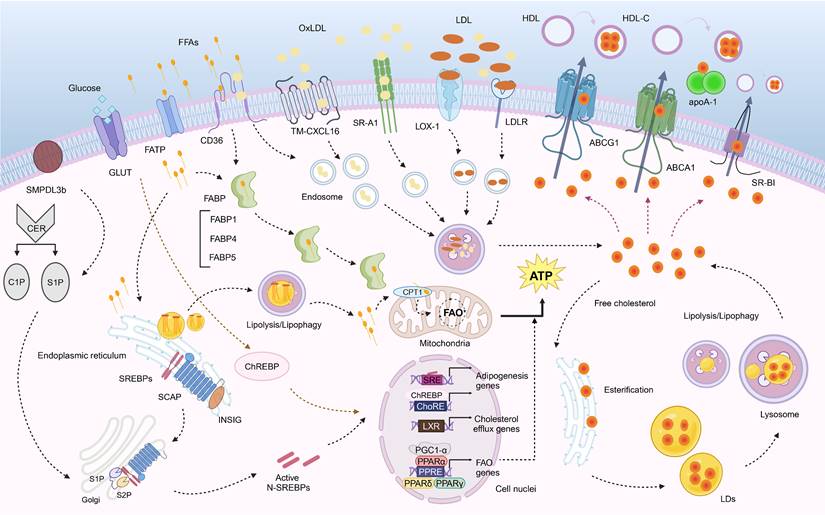
Fatty acid metabolism
Fatty acid uptake and transport
FAs serve as an important source of energy for the body. FAs are taken up and transported to mitochondria for oxidation. This process involves molecules that include CD36, CXCL16, FATP, and FABP. FAs are taken up by proximal tubular cells mainly via CD36 [10]. CD36 is a transmembrane glycoprotein that transports FAs into cells and is expressed primarily in renal tubular epithelial cells (TECs), podocytes, and mesangial cells (MCs) in the kidney, where it can act as a receptor for long-chain FAs, and oxidized lipids and play a role in lipid accumulation, inflammatory signaling, and renal fibrosis [27]. However, there is evidence that the chemokine CXCL16, but not CD36, is the major scavenger receptor mediating oxLDL uptake in human podocytes [28, 29]. OxLDL downregulates integrin α3, increases fibronectin production, and induces reactive oxygen species (ROS) production in human podocytes [29]. FATPs are transmembrane proteins involved in FA uptake and activation. The FATP family consists of six tissue-specific isoforms, of which the kidney predominantly expresses FATP1, FATP2, and FATP4 [30]. FATPs can catalyze the formation of CoA derivatives with long-chain and ultra-long-chain FAs, bile acids, and bile acid precursors as substrates [31]. FATP2, one of the primary FA transporters expressed in renal TECs, induces reprogramming of lipid metabolism, including abnormal FA uptake and FAO defects, triggering renal interstitial fibrosis, which is closely associated with decreased renal function [32]. FABPs are intracellular lipid chaperones involved in the regulation of intracellular lipid transport and responses, and they are related to metabolic and inflammatory pathways [33]. FABPs are small, water-soluble proteins that bind to long-chain FAs and other bioactive ligands to promote intracellular localization [34]. FABP1 is expressed in renal proximal tubular cells and is released into the urine in response to hypoxia caused by reduced peritubular capillary blood flow. FABP2 is responsible for the transport of FFAs in intestinal endothelial cells. Both FABP1 and FABP2 are biomarkers of DKD [35]. Urinary liver-type FA-binding protein (L-FABP) indicates the extent of tubulointerstitial damage [36], and it is an invaluable marker of DKD progression [37].
Fatty acid oxidation
FAO, the catabolic process by which FAs are broken down into acetyl-CoA, is the preferred energy source for higher metabolizing cells and takes place primarily in mitochondria [18]. CPT1A is a carrier of medium- and long-chain FAs into mitochondria, binds FAs to carnitine, and is a key rate-limiting enzyme for FAO [38]. In three models of renal fibrosis (unilateral ureteral obstruction, folic acid nephropathy, and adenine-induced nephrotoxicity), the extent of renal fibrosis was reduced in CPT1A knockin (CPT1A-KI) mice, and a protective effect against fibrosis was seen following inducing CPT1A overexpression [39]. Peroxisome proliferator-activated receptor (PPAR) and PPAR-γ coactivator-1α (PGC-1α) are key transcription factors that regulate the expression of FA uptake- and oxidation-related proteins [40, 41]. PPAR belongs to the type II nuclear hormone receptor superfamily, which is organized into three isoforms: PPAR-α, PPAR-β/δ, and PPAR-γ. It can act as a transcription factor that binds to response elements within the promoters of genes related to glycolipid metabolism [42]. PPAR-α and PPAR-β/δ regulate FAO, whereas PPAR-γ is more closely linked to adipogenesis and lipid storage [43]. PPAR-α promotes FAO and oxidative phosphorylation [44]. In the DKD model, reduced PPAR-α and PPAR-δ expression can lead to decreased FAO [41] and is associated with a decreased estimated glomerular filtration rate (eGFR) [8]. PPAR-γ participates in the maintenance of renal metabolic homeostasis, and PPAR-γ inhibition leads to renal tubular hypertrophy, tubulointerstitial fibrosis, and impaired renal function [45]. PGC-1α participates in cellular lipid metabolism and energy regulation by interacting with PPAR to influence the expression of genes related to lipid synthesis and transport [46]. FABP4, one of the isoforms of FABP, is able to translocate FAs and downregulate its target gene, PPARG, encoding PPAR-γ [47].
Lipogenesis
Enhanced activation of adipogenic genes will promote glomerular and tubular lipid deposition. In the DKD model, increased renal lipid levels were associated with increased expression of renal sterol regulatory element-binding proteins (SREBPs) and carbohydrate response element-binding proteins (ChREBPs) [41, 48-50]. SREBPs (SREBP1a, SREBP1c, and SREBP2) belong to the family of membrane-bound transcription factors involved in the regulation of lipid synthesis, with SREBP1a being responsible for overall lipid synthesis, SREBP1c being responsible for FA and triglyceride synthesis, and SREBP2 specifically controlling steroidogenesis [51]. In DKD patients, increased expression of SREBP1 and SREBP2 causes renal lipid deposition, lipotoxicity, and fibrosis [23]. In unilateral ureteral obstruction rats, blockade of SREBP1/2 signaling using SREBP inhibitors significantly attenuates renal inflammation, necrosis, and fibrosis by affecting lipogenesis and transforming growth factor-β1 (TGF-β1) expression [52]. Junctional adhesion molecule-like protein (JAML) regulates podocyte lipid metabolism through SIRT1-mediated SREBP1 signaling [53]. Farnesoid X receptor (FXR) agonists modulate SREBP1 expression, lipid metabolism, and the expression of renal profibrotic growth factor, proinflammatory cytokines, and oxidative stress enzymes and reduce glomerulosclerosis, tubulointerstitial fibrosis, and proteinuria. ChREBP is a primary mediator of the action of glucose on the expression of adipogenic genes and a key determinant of lipid synthesis in vivo [54]. ChREBP regulates the expression of ACC and FAS and regulates adipogenesis by inducing expression of the glycolytic enzyme pyruvate kinase 1 [55].
Cholesterol metabolism
In addition to FA metabolism, dysregulation of renal cellular cholesterol metabolism has also been linked to lipotoxicity and lipid accumulation in DM, caused by alterations in cholesterol uptake, intracellular synthesis, esterification, and efflux [56]. Cholesterol influx into cells is mediated by several independent receptors, including the class A scavenger receptor CD36, lectin-like oxLDL receptor-1 [57], and the LDL receptor (LDLR) [28]. A highly significant correlation was found between increased expression of LDLR, oxLDL, acetylated LDL (acLDL), and CD36 and progression of DKD and worsening of eGFR [8]. SREBP cleavage-activating protein (SCAP) is thought to be a chaperone for SREBP2, which is transported from the endoplasmic reticulum (ER) to the Golgi apparatus for activation via proteolytic cleavage [58]. 3-Hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase is involved in the maintenance of cholesterol synthesis, which involves SREBP2 [59]. In the kidneys of diabetic rats, LD accumulation and increases in HMG-CoA reductase, LDLR, SREBP2, and SCAP levels were observed [60]. The preprotein convertase chymotrypsin 9 (PCSK9) is a crucial protein in the regulation of lipid metabolism in which SREBP2 is involved [61]. PCSK9 binds to surface LDLR, leading to its degradation and subsequent elevation of plasma LDL-C levels [62, 63]. In a clinical study, mean serum PCSK9 concentrations were higher in CKD patients than in controls [64].
Cholesterol efflux is primarily mediated by ATP-binding cassette transporter proteins, including ATP-binding cassette transporter A1 (ABCA1) and ATP-binding cassette transporter G1 (ABCG1), and SR-BI [65, 66]. ABCA1 promotes cholesterol efflux from cells and inhibits inflammatory responses and is highly expressed in the kidney [67]. Liver X receptor alpha (LXRα) and LXRβ are members of the nuclear receptor family that crucially regulate cholesterol homeostasis. In DKD, the expression of the cholesterol efflux genes ABCA1, ABCG1, and apoE was reduced, and the expression of nuclear receptor LXRα, which regulates cholesterol efflux genes, was also downregulated [68]. ABCA1 deficiency in glomerular endothelial cells exacerbated renal lipid deposition and renal injury in type 2 DM mice, as evidenced by elevated creatinine levels, more severe proteinuria, and more pronounced dilatation of the thylakoid matrix, pedunculopapillary fusion, renal inflammatory injury, and cell death [65]. ABCA1 deficiency is associated with cardiolipin-driven mitochondrial dysfunction, ultimately leading to DKD [69]. PPAR-γ also plays an essential role in regulating the uptake of oxLDL by upregulating CD36 expression and LXR-ABCA1-mediated cholesterol efflux [70, 71]. Apolipoprotein L1 (APOL1) is a secreted HDL that co-localizes with APOA1 in HDL particles, which promotes cholesterol efflux from cells [72].
Sphingolipid metabolism
Sphingolipids are defined as compounds containing long-chain bases or sphingamines. Sphingolipids have also been shown to play an essential role in the onset and development of DKD. Sphingolipids and their metabolites, including ceramide (CER), sphingosine, CER-1-phosphate (C1P), and sphingosine-1-phosphate (S1P), as biologically active signaling molecules, perform vital actions in intracellular signaling and may be involved in the regulation of apoptosis, autophagy, inflammation, immunity, and membrane fluidity [73]. Following 4 days of streptozotocin (STZ) treatment, the levels of neutral ceramidase, SK activity, and S1P were significantly elevated in isolated glomeruli of rats, thus suggesting that S1P may be involved in the early glomerular proliferative response in DKD [74]. Elevated albuminuria and increased connective tissue growth factor expression were found in SK-1 knockout mice as compared to wild-type C57BL/6 mice [75]. CER is a biologically active sphingolipid that is a substrate for the production of C1P and S1P [76]. Increased CER production due to upregulation of serine palmitoyltransferase, a key enzyme involved in de novo CER synthesis, is associated with increased apoptosis in STZ-induced DKD in renal TECs and microvascular endothelial cells [77].
Sphingolipids, especially sphingolipids and sphingolipid sugars, form aggregates in various regions with cholesterol, frequently referred to as lipid rafts [78]. Sphingomyelin phosphodiesterase acid-like 3b (SMPDL3b) is a lipid raft sphingomyelinase that modifies plasma lipid composition, regulates intracellular inflammatory pathways, and controls the ability of circulating factors to affect podocyte function and survival [20]. SMPDL3b overdose resulted in reduced C1P levels and impaired insulin-mediated prosurvival signaling pathways in cultured human podocytes in vitro and in the renal cortex of DKD mice in vivo [79]. In db/db mice, podocyte-specific SMPDL3b deficiency restores renal cortical C1P content, which in turn prevents DKD [79]. The potential role of glycosphingolipid accumulation in DKD suggests that hyperglycemia is associated with enhanced synthesis of glucosylceramide and ganglioside GM3, thus ultimately leading to renal hypertrophy in STZ-induced diabetic rats [80].
Lipid homeostasis imbalance and DKD progression
Lipid molecule signaling imbalances and lipid accumulation in nonadipose tissues can negatively affect homeostasis [81]. There is a growing consensus that ectopic lipids (accumulation of lipids in nonadipose tissue) are associated with structural and functional changes in MCs, podocytes, and proximal renal tubule cells [82]. Crosstalk between cells in kidney homeostasis plays a vital role [83]. Dysregulation of lipid metabolism in renal intrinsic cells is able to contribute more directly to renal lipotoxicity as opposed to dyslipidemia (Fig. 2).
Lipid metabolism in renal cells in DKD. (1) Podocytes primarily rely on anaerobic glycolysis as their main energy source, with lipid metabolism being mainly regulated by CD36, LDLR, CXCL16, and ABCA1. An imbalance in lipid homeostasis can result in endoplasmic reticulum stress, apoptosis, and disruption of the actin cytoskeleton. (2) Renal tubular epithelial cells predominantly utilize FAO as their primary energy source, with lipid metabolism being mainly regulated by ABCA1, ABCG1, SRBI, CB1R, FATP, and KIM-1. An imbalance in lipid homeostasis can lead to metabolic reprogramming, apoptosis, and increased dedifferentiation. (3) Lipid accumulation in mesangial cells is primarily associated with LDL and oxLDL uptake while MAPK, NF-κB, and AP-1 are involved in regulation. An imbalance of lipid homeostasis will lead to endoplasmic reticulum stress, apoptosis, inflammation, foam cells, mesangial cell proliferation and hypertrophy. (4) Caveolae play a crucial role for endothelial cells' uptake of lipids. Lipid metabolism is mainly influenced by FABP, ABCA1, and sphingolipid metabolites. An imbalance of lipid homeostasis will cause endoplasmic reticulum stress, apoptosis, and affect angiogenic responses. (5) Macrophage depolarization is closely related to lipid homeostasis.
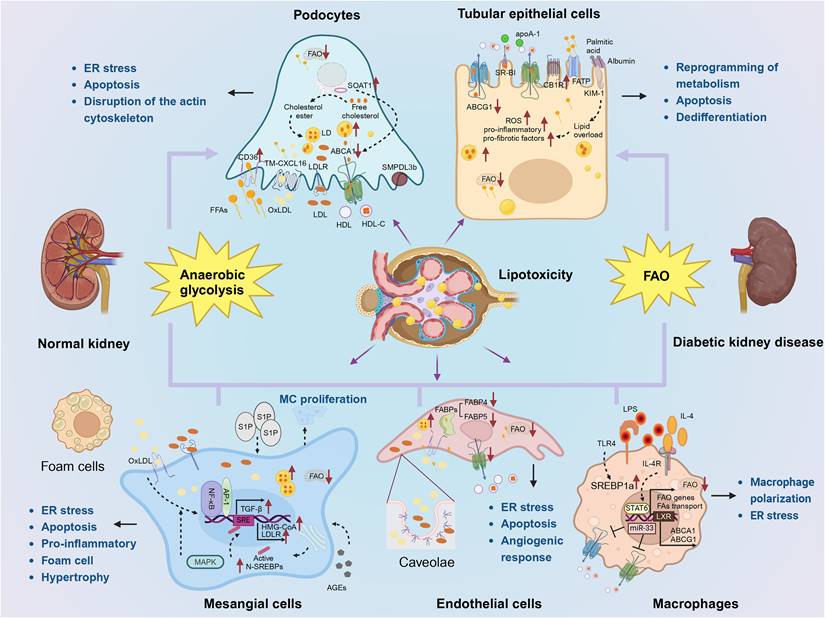
Lipid metabolism in podocytes
Podocytes are essential for maintaining a normally functioning glomerular filtration barrier. Loss of podocytes leads to a reduction in glomerular basement membrane coverage, which initially manifests as albuminuria and eventually as glomerulosclerosis [84]. Podocytes are sensitive to lipid accumulation [69, 85]. Lipid accumulation in podocytes is a major determinant of proteinuric nephropathies, including obesity-associated nephropathy, DKD, and focal segmental glomerulosclerosis [53, 86]. Lipotoxicity and lipid accumulation leading to podocyte damage and apoptosis in DKD patients are characteristic features of DKD, and podocytopenia is an independent predictor of DKD progression [7, 50]. Anaerobic glycolysis and the fermentation of glucose to lactate are key processes providing energy for podocytes [87]. Saturated FFAs lead to ER stress and apoptosis in podocytes [88, 89], whereas stearoyl-CoA desaturase-1 upregulation attenuates ER stress and podocyte apoptosis [90]. Enhanced FFA uptake by podocytes is mediated by increased expression of the C36 scavenger receptor, as well as decreased β-oxidation and intracellular lipid accumulation [28]. Dysregulated transport and oxidation of FFAs, coupled with impaired antioxidant responses, cause structural damage to the podocyte, leading to early DKD glomerulopathy [28]. LDLR is the primary receptor mediating lipid uptake in podocytes, and high glucose dysregulates feedback regulation of the LDLR pathway [20]. Treatment of human podocytes with serum from patients with DKD resulted in more cholesterol accumulation compared with human podocytes exposed to serum from diabetic patients (but not DKD) at the same total cholesterol, HDL-C, and LDL concentrations [91]. Podocyte survival and the integrity of the actin cytoskeleton are compromised following exposure to oxLDL [92, 93]. Elevated tumor necrosis factor (TNF) levels promote free cholesterol-dependent podocyte depletion via ABCA1-mediated reduction of cholesterol efflux and cholesterol esterification by cholesterol-O-acyltransferase 1 (SOAT1) [94, 95]. Inhibition of SOAT1 in human podocytes reduced lipotoxic injury while increasing ABCA1 expression and ABCA1-mediated cholesterol efflux [96]. However, in vitro knockdown of ABCA1 in podocytes and in vivo podocyte-specific deletion of ABCA1 were insufficient to cause podocyte apoptosis and glomerular injury, respectively [94]. SMPDL3b expression is increased in glomeruli from DKD patients and human podocytes treated with DKD serum, which makes podocytes more susceptible to apoptosis through suPAR [97]. Podocin is a slit membrane protein in podocytes. Notably, MEC-2 and podocin bind and collect cholesterol to organize the lipid microenvironment of related ion channel complexes. Cholesterol interaction also regulates the glomerular filtration barrier ion channel activity [98, 99]. Cholesterol can also enhance inflammatory effects by directly activating inflammasomes, inducing lysosomal damage, and causing toll-like receptor (TLR) localization in lipid rafts [100]. SMPDL3b is also involved in TLR33-mediated inflammatory activation [101]. In turn, inflammation can upregulate the expression of LDLR, SCAP, and SREBP-2[102]. Inflammation reduction can also be achieved through endogenous specialized solubilizing lipid mediators (SPMs) and branched-chain fatty acid esters of hydroxy fatty acids [103, 104]. Therefore, lipids and inflammation can interact and regulate each other.
Lipid metabolism in renal tubular epithelial cells
The presence of lipid deposits in diabetic TECs was initially described in 1936[105]. FAs are the primary energy source for TECs [15, 18]. Therefore, FAO in lipid metabolism, in particular, is the primary pathway by which renal tubulointerstitial fibrosis affects TECs [106]. Inhibition of FAO in TECs causes ATP depletion, cell death, dedifferentiation, and intracellular lipid deposition [18].
Decreased FAO in TECs leads to reprogramming of their metabolism, which in turn results in increased apoptosis and dedifferentiation [18]. Lipotoxic manifestations, including ROS, release of proinflammatory and profibrotic factors, and apoptosis, can occur as a result of TEC lipid overload due to excessive dietary intake or dysfunctional lipid depletion or degradation [107, 108]. In a DKD model, tubule-specific deletion of Pacs2, which encodes a protein associated with lipid metabolism, resulted in severe tubular injury accompanied by increased lipid synthesis and uptake and decreased cholesterol efflux [109]. ABCA1, ABCG1, and SR-B1 are expressed in human renal MCs and proximal TECs, all of which mediate cholesterol efflux to ApoA1 and HDL [110]. Albumin itself is not cytotoxic to proximal tubules, but NEFAs bound to albumin trigger apoptosis in proximal tubules [111, 112]. In contrast with adipocytes, which have large stores of intracellular LDs under physiological conditions, TECs contain limited LDs to maintain their energy homeostasis [113]. LDs and LD-associated proteins protect against FA-bound albumin-induced apoptosis by sequestering FFAs [57]. FATP2 is a major apical proximal renal tubule NEFA transporter protein that regulates apoptosis in proximal TECs [114]. Further, FATP2 deficiency does not completely eliminate FA uptake in the proximal renal tubule [114]. Among the FA transport proteins that are not members of the FATP family, kidney injury molecule-1 (KIM-1) is expressed within the proximal tubule membrane and acts as a scavenging receptor for oxidized lipoproteins and apoptotic cells by recognizing exogenous phosphatidylserine [115]. DKD mouse models and in vitro studies have shown that KIM-1 mediates proximal tubule uptake of albumin-bound palmitate [116]. In proximal tubules, palmitate induced proximal tubular cannabinoid expression and enhanced apoptosis, thus suggesting that cannabinoids may mediate proximal tubular lipotoxicity [117]. Specific deletion of cannabinoid receptor 1 in renal proximal tubule cells, although not protecting mice from obesity, significantly attenuates obesity-induced renal lipid accumulation and renal dysfunction, injury, inflammation, and fibrosis [118].
Lipid metabolism in mesangial cells
Prior studies have suggested that MCs exhibit specific binding and uptake of LDL [119] and that lipid accumulation in MCs may be caused by receptor-mediated endocytosis of LDL particles [120]. The mesangial matrix has a high capacity to bind LDL in a nonsaturated manner and to modify LDL (by glycation or oxidation) [121]. oxLDL induces the expression of activator of transcription factor 1 (AP-1) in rat MCs [122], which regulates TGF-β gene expression through the AP-1-binding site [123], and nuclear factor-κB (NF-κB) is also involved in oxLDL-induced enhancement of ACC [124]. Excess oxLDL, especially lipid peroxides and lysophospholipids of oxLDL, may exert cytotoxic effects on MCs, epithelial, and endothelial cells, thereby contributing to a vicious cycle of cellular damage and sclerosis [121]. Some oxLDL can be absorbed by scavenger receptors on MCs and mononuclear macrophages, leading to foam cell formation [121]. oxHDL enhances the proinflammatory properties of MCs in part through CD36 and LDLR-1, and the mitogen-activated protein kinase (MAPK) and NF-κB pathways are also involved in this process [125]. Lipotoxicity can induce the protein expression of arginine methyltransferase 1 to promote ER stress-mediated apoptosis of MCs [126]. In MCs, exposure to advanced glycation end products (AGEs) leads to increased SCAP translocation and transport of SREBP2 to the Golgi apparatus, which in turn leads to mesangial foam cell formation through activation of proteolytic cleavage through enhanced transcription of HMG-CoA and LDLR [59]. In experimental models of DKD, inhibition of glucosylceramide synthase reversed MC hypertrophy by reducing high glucose-induced phosphorylation of SMAD3 and Akt, alleviating fibrosis, and reducing the expression of ECM proteins [127]. S1P stimulates MC proliferation [128], and exposure to the MC matrix induces monocyte differentiation toward a macrophage-like phenotype and promotes LDL oxidation, thereby transforming this lipoprotein into scavenger receptor ligands associated with foam cell formation in the mesangium [129]. Insulin-like growth factor-1 induces lipid accumulation in MCs, reducing their ability to respond to specific migratory and contractile stimuli [130].
Lipid metabolism in endothelial cells
The endothelium is a thin monolayer of cells covering the luminal surface of the vessel wall; it forms a barrier between blood and surrounding tissues and plays an active role in vascular function and homeostasis [131]. Caveolae consist of 50- to 100-nm apical plasma membrane invaginations and are important mediators of endothelial endocytosis, endocytosis, lipid homeostasis, and signaling in endothelial cells [132]; however, high levels of circulating oxLDL affect caveolae lipid composition and/or function [131]. Circulating LDL is absorbed by endothelial cells through receptor-mediated endocytosis or endocytosis [131]. In contrast, the only way for LDL to cross the endothelium is through caveolae-mediated endocytosis [131]. FABP is closely related to endothelial cells. Cellular aging and oxidative stress induce FABP4 expression in microvascular endothelial cells [133], which can enhance the angiogenic response of endothelial cells, including proliferation, migration, and survival [134]. FABP4 generally has higher affinity and selectivity for long-chain FAs than albumin [135]. FABP5 deficiency leads to endothelial cell proliferation and chemotactic migration [134]. ABCA1 deficiency in glomerular endothelial cells exacerbates renal lipid deposition and renal injury in type 2 diabetic mice [65]. ABCA1 overexpression can enhance cholesterol efflux or inhibit ER stress in vitro and significantly protect glomerular endothelial injury stimulated by high glucose and high cholesterol [65]. Sphingolipid metabolites and related enzymes are closely related to the apoptosis, senescence, oxidative stress, and other activities of endothelial cells and affect the regulatory function of endothelial cells in the stress response, angiogenesis, and the inflammatory response [136].
Lipid metabolism in macrophages
Macrophages are cells of the innate immune system, which can be polarized into M1 macrophages (proinflammatory role) and M2 macrophages (anti-inflammatory role) depending on the type of stimulus [137]. Macrophages can maintain tissue homeostasis, induce immune responses, and participate in tissue repair [138, 139]. Studies have shown that lipid synthesis is related to macrophage function. Following lipopolysaccharide stimulation of TLR4, macrophages can increase de novo lipogenesis and also activate SREBP1a expression [140-142]. Macrophages can also increase the uptake of FFAs and lipoproteins following exposure to inflammatory stimuli [143, 144]. IL-4 activates macrophages, and FAMIN proteins link de novo lipogenesis to FAO through an apparent “substrate cycle” [141]. IL-4 signaling activates signal transducer and activator of transcription 6 (STAT6), which promotes lipid transport, FAO, and mitochondrial biogenesis [145]. Macrophage polarization is affected by lipids, which also play an influential role in the control of macrophage function. The accumulation of lipids and inflammatory cytokines can jointly induce ER stress in macrophages [146]. Lipotoxic TEC-derived extracellular vesicles (EVs) induce the expression and release of proinflammatory cytokines such as IL-1β and TNF-α in macrophages and the release of macrophage-derived EVs [147]. LDs are involved in cellular FA homeostasis and the regulation of macrophage function [145]. Deficiency of enzymes involved in FA esterification, including diacylglycerol acyltransferase-1 (DGAT1), increases the proinflammatory response of macrophages [148]. Adipose triglyceride lipase (ATGL) is one of the enzymes involved in the breakdown of triglycerides in macrophage LDs, and deficiency of lipases such as ATGL attenuates the expression of proinflammatory genes (such as IL6) and favors the activation of anti-inflammatory macrophages [149]. Monoacylglycerol lipase is a lipase that decomposes monoacylglycerol into FFAs and glycerol and is involved in macrophage autophagy and inflammation [150]. Inhibition of lysosomal acid lipase (LAL) resulted in M2-type macrophage polarization and reduced mitochondrial oxidative respiration, suggesting that lipolysis is also required for macrophage polarization [151]. PPAR-γ is also involved in the suppression of macrophage proinflammatory responses [152]. miR-33 is a microRNA involved in SREBP signaling. In macrophages, miR-33 can regulate cholesterol homeostasis by inhibiting the expression of genes encoding ABCA1 and ABCG1[153]. LXR regulates reverse cholesterol transport in macrophages by controlling the expression of cholesterol transporters and apolipoproteins, including ABCA1, ABCG1, apoE, and apoC [154, 155].
Autophagy of lipid homeostasis in DKD
Lipid and bioenergy are popular topics in DM research. Lipid metabolism plays an important role in the physiology and pathology of DKD. Autophagy is not only a cellular waste degradation pathway, but it is also a core way to maintain cell and organism homeostasis. Autophagy plays an important role in the pathogenesis of DKD [156]. In the state of continuous hyperglycemia, multiple pathways and mechanisms will lead to the decrease of DKD autophagy activity, including lysosomal autophagy, mitochondrial autophagy, etc., also including podocyte and renal tubular cells of different kidney-resident parenchymal cells of autophagy [157, 158]. Moreover, targeted improvement of autophagy is expected to be a new strategy for the treatment of DKD [159]. Autophagy is involved in the regulation of lipid content. Decreased autophagy promotes lipid accumulation, inhibition of autophagy further increases lipid retention, and autophagy is further impaired as lipid content increases [160]. Here, we focus on autophagy.
The autophagy pathway has an imperative role in physiology, with its primary function being to protect cells or organisms from starvation during nutrient deprivation by enabling them to recycle nutrients from digested organelles and macromolecules, as well as to ensure intracellular homeostasis through the removal of damaged organelles and abnormally folded proteins. Studies have shown that decreased autophagy promotes lipid accumulation, which in turn further inhibits autophagic function, thereby increasing lipid accumulation [160]. Resveratrol can improve lipid metabolism in diabetic nephropathy rats through AMPKα/mTOR-mediated autophagy [161].
Lipids are increasingly involved in the control of biochemical processes and membrane remodeling underlying autophagosome biogenesis and autophagy more generally [162]. Since autophagosome membranes largely lack transmembrane proteins, autophagosome biogenesis is thought to be largely regulated by lipid transfer and lipid modification as well as membrane-associated proteins [163]. Lipids and lipid-metabolizing enzymes mediate the process of autophagy by controlling at least four fundamental aspects (Table 1). First, lipids mediate signaling. They regulate a signaling cascade that converges on the mTOR pathway, which in turn negatively regulates the initiation of autophagy. Central to this signaling cascade is class I PI3K and its product phosphatidylinositol-3,4,5-trisphosphate (PI(3,4,5)P3) [164]. Second, lipids mediate the local recruitment of effectors to the membrane. Lipids act as local signals for membrane binding and control membrane dynamics by specifically recruiting cytoplasmic protein effectors that mediate membrane deformation, swelling, and vesicle trafficking. A typical example of this regulation is PI3P, which controls autophagosome biogenesis and maturation by this mechanism [165, 166]. Third, lipids mediate covalent modification of proteins. Amine-containing phospholipids that include phosphatidylethanolamine covalently bind to members of the Atg8/LC3 family, providing a unique mode of regulation by anchoring these key factors stably to the membrane of phagocytes, mediating their extension and eventual closure [167]. Fourth, lipids can control membrane dynamics independently of protein effectors by directly affecting the physicochemical properties of the lipid bilayer. Examples of such regulation include lipid rafts, cone-shaped lipids such as phosphatidic acid, which predisposes to or induces negative curvature, and cholesterol, which promotes or stabilizes the liquid-ordered phase within the bilayer [168]. Cholesterol is associated with the organization of microdomains within the lysosomal membrane that control the effects of chaperone-mediated autophagy (CMA) and autophagosome-lysosome fusion [169, 170]. Short-term cholesterol depletion leads to a rapid induction of autophagy, and the ER-localized cholesterol transporter GRAMD1C has been proposed as a negative regulator of starvation-induced macroautophagy/autophagy [163]. Sphingolipids are ubiquitous membrane lipids in eukaryotes and are involved in the generation of a variety of membrane structures, including rafts, vesicles, and cytoplasmic vesicles. There are two major sphingolipids involved in autophagy, namely, ceramide and S1P [171]. Exogenous application of short-chain ceramides, including C2-ceramides, stimulates autophagy, probably by promoting de novo synthesis of long-chain ceramides [172, 173]. Long-chain ceramide partially activates autophagy by inhibiting the phosphorylation of Akt/protein kinase B (PKB) in the class I PI3K pathway, reducing mTOR activation and upregulating beclin1 function through JNK1-mediated dissociation of the beclin1-Bcl2 complex [172, 174]; however, the specific mechanism underlying the role of sphingolipids in autophagy needs to be further elucidated. In sum, five major lipid classes are directly associated with autophagy (FAs, phospholipids, glycerides, sphingolipids, and sterols).
Lipids and lipid-metabolizing enzymes mediate the process of autophagy by controlling four fundamental aspects.
| Mediation of autophagy by lipids | Molecules/Targets involved | Biological effects | References |
|---|---|---|---|
| Signaling | mTOR; PI3K; PI (3,4,5) P3 | Negative regulation of autophagy | [164] |
| Local recruitment of effectors to the membrane | PI3P | Control of autophagosome biogenesis and maturation | [165, 166] |
| Covalent modification of proteins | phosphatidylethanolamine; Atg8/LC3 | Extension and closure of the membrane | [167] |
| Membrane dynamics | Lipid rafts | / | [168] |
| Phosphatidic acid | Induction of negative curvature | [168] | |
| Cholesterol | Promotion or stabilization of a liquid-ordered phase within a bilayer | [168] | |
| Control of chaperone-mediated autophagy | [169, 170] | ||
| Control of autophagosome-lysosome fusion | [169, 170] | ||
| GRAMD1C | Negative regulation of autophagy | [163] | |
| Short-chain ceramides | Stimulation of autophagy | [172, 173] | |
| Long-chain ceramide; JNK1; Beclin 1-Bcl2 complex; PI3K; mTOR | Activation of autophagy | [172, 174] |
Lipids themselves can also serve as substrates of autophagy, a pathway in which autophagic lysosomes directly consume cellular lipids in the form of LDs, a phenomenon known as lipophagy (Fig. 3), to achieve tissue energy homeostasis [175, 176]. Synthesis of LDs occurs in the ER [177]. Most cells produce LDs between 0.1 and 10 μm in size. LDs serve as cellular stores of neutral lipids, including triglycerides and cholesteryl esters, which contribute to the initiation of autophagy. The stored neutral lipids are mobilized during autophagy to support autophagic membrane formation [178]. LDs are surrounded by phospholipid monolayers and LD shell proteins of the periplasmic lipoprotein family (PLIN) [179]. The PLIN family consists of five members, PLIN1-5. PLIN1 and PLIN2 are located only on the surface of LDs and are degraded when not bound to LDs. PLIN3 and PLIN4 freely bind to or dissociate from LDs and remain stable even when released into the cytoplasm. PLIN5 is predominantly expressed in a number of highly oxidized tissues in vivo, including the heart, skeletal muscle, and liver. PLINs can participate in lipophagy by regulating the binding of lipases to LDs. Once the lipolysis signal is detected, PLIN1 rapidly phosphorylates and releases CGI-58, which ultimately activates ATGL and initiates lipolysis. PLIN2 and PLIN3 contain a CMA recognition sequence (KFERQ) that binds to a 70-kDa heat-shock protein (HSP70), thereby directing the LD toward the lysosome for CMA degradation [180]. Cytosolic lipases mobilize LDs by disintegrating one triglyceride molecule into three FA molecules and one glycerol molecule through tandem reactions catalyzed by ATGL [181], HSL, and monoacyltriglyceride lipase (MAGL) [182]. ATGL-deficient mice exhibit lipid accumulation in adipose tissue, heart, and liver [183]. ATGL can co-localize with the autophagy marker protein LC3 on the surface of LDs and enhance the binding of LC3 to lysosomes and LDs, thereby enhancing lipophagy activity [184]. Overexpression of HSL has been shown to reverse hepatic steatosis [185]. The Rab GTPase protein family comprises key regulators of intracellular vesicle trafficking. Nearly 30 Rab family members have been identified on the surface of LDs, including Rab7, Rab10, Rab32, and Rab25. Among them, Rab7 is the most important protein [186]. It can mediate the fusion of autophagosome membranes and late endocytic membranes [187]. Mutations in Huntingtin lead to significant accumulation of LDs, suggesting that it plays an essential role in the regulation of lipophagy as an LD-recognizing receptor protein [188].
Pathway of lipid droplet decomposition. a) The KFERQ sequence in PLIN2/3 on the surface of LDs can bind to HSP70 and mediate chaperone-mediated autophagy. b) LDs can be decomposed by lipases ATGL, HSL, and MAGL. c) ATGL co-localizes with the autophagy marker protein LC3 to promote the formation of phagocytosed LDs and autophagosomes, while Rab7 on the surface of LDs promotes fusion between autophagosomes and lysosomes, leading to lipophagy occurrence. All three pathways generate FFAs and facilitate FAO for ATP production as an energy source.
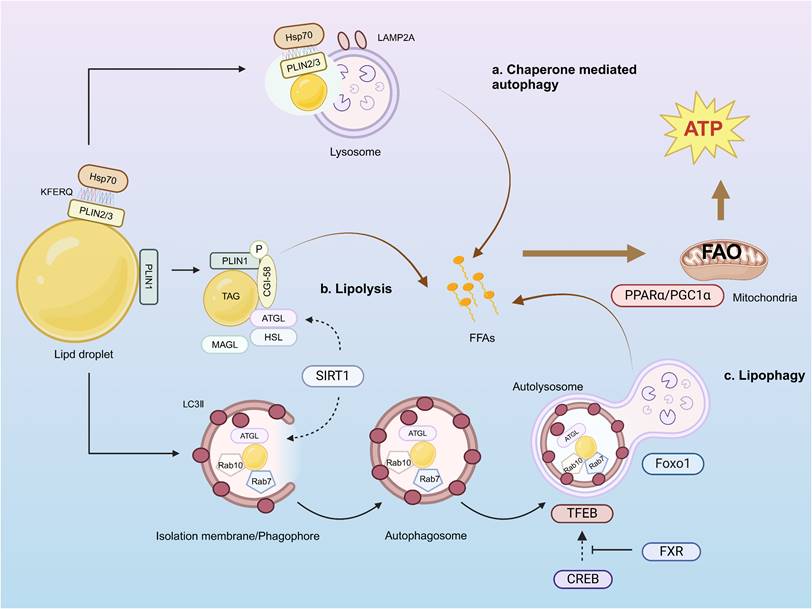
In addition, the occurrence of lipophagy is regulated by different mechanisms. ATGL-mediated signaling can promote autophagy/lipophagy through SIRT1 [184]. Starvation-induced activation of the transcription factor forkhead box protein O1 (FoxO1) regulates lipid content through transcriptional upregulation of LAL-mediated autophagy [189]. Transcription factor EB (TFEB) plays a central role in lipid metabolism by regulating starvation-induced transcription through the PGC1-α-PPAR-α-lipophagy axis, which mediates lipid catabolism [190]. cAMP response element-binding protein (CREB) upregulates autophagy genes, including Atg7, Ulk1, and TFEB, by recruiting the co-activator CRTC2 in the fasting state. In contrast, nutrients inhibit this effect by activating the nuclear receptor FXR [191]. Starvation-induced PPAR-α activation reverses diet-induced inhibition of FXR-driven autophagy [192].
Blocking lipophagy promotes intracellular lipid accumulation, whereas activation of lipophagy leads to clearance of LDs. PNPLA5 is localized to LDs; the PNPLA5 pathway is the optimal pathway to initiate autophagy and is required for autophagy of multiple substrates, including degradation of autophagic junctions, bulk proteolysis, control of the number of mitochondria, and microbial clearance [178]. The unique hydrophobic structural domains of ATG14 and the E2-like enzyme ATG3 were found to be key determinants in permitting surface recruitment of LDs and extending the autophagy machinery [193]. Significant accumulation of LDs was accompanied by a significant reduction in FAO in hepatocytes treated with 3-methyladenine or by knockdown of Atg7 and Atg5[186]. Consistent with this, the accumulation of triglycerides in Atg5-/- mouse embryonic fibroblasts also suggests that an autophagic defect hinders LD degradation [186]. Decreased lipid autophagy and ectopic lipid deposition were observed in renal tubular cells of DKD patients, db/db mice, and HK-2 cells induced by high glucose [194]. Shear stress can stimulate lipid autophagy, promote FA production, and promote mitochondrial ATP production through FAO to maintain renal metabolism [195].
Conclusion and prospect
Currently, renal replacement therapy is the only available treatment modality for ESRD. Patients in whom DKD progresses to ESRD require substantial healthcare resources. Therefore, it is essential to provide early treatment to DKD patients. A growing body of evidence supports the essential role of lipid metabolism in the onset and progression of DKD. Here, we present the regulatory factors involved in the metabolism of different lipid types, including FAs, cholesterol, and sphingolipids, discuss the relationship between lipid metabolism and renal intrinsic cells, and emphasize the roles of targeted autophagy in the regulation of lipid homeostasis in DKD, in order to support the development of novel treatment strategies for DKD targeting lipid metabolism. It is of great value to identify new therapeutic targets and develop therapeutic drugs to improve lipid homeostasis and inhibit the progression of DKD. Numerous studies have shown that improving renal lipid deposition can reduce renal injury; however, the benefits of lipid-lowering therapy for DKD remain controversial. Clinical trials of commonly used lipid-lowering drugs, such as fenofibrate, statins, PCSK9 inhibitors, sodium-glucose transporter-2 inhibitors, glucagon-like peptide-1 receptor agonists, and mineralocorticoid receptor antagonists, have been far less aggressive than animal studies. The benefit may be limited to a reduction in proteinuria, and data on effects on renal lipotoxicity are lacking [84]. Empagliflozin can improve FFA-induced renal tubular injury through the PPAR-γ/CD36 pathway [196]. In a study of potential compounds to treat lipotoxicity in DKD, berberine has been shown to promote mitochondrial FAO in podocytes by activating PGC-1α [197]. Tetrapeptide SS-31, which targets cardiolipin and protects the structure of mitochondrial cristae, protects kidney cells of C57 BL/6 mice after 28 weeks of high-fat diet, and prevents intracellular lipid accumulation [198]. Synthetic S1p analogue FTY720 can alleviate S1P-induced podocyte injury by reducing inflammatory cytokines [199]. Therefore, more systematic and comprehensive studies that focus on the role of lipid-lowering therapy in the treatment of DKD are necessary.
Acknowledgements
Funding
This work was supported by the National Natural Science Foundation of China [Grant Number 82174144, 82174296], Elite Medical Professionals project of China-Japan Friendship Hospital [NO. ZRJY2021-QM15], National High Level Hospital Clinical Research Funding [NO. 2023-NHLHCRFDJMS-05].
Author contributions
Wang Ying and Zhao Hailing conceived and designed the study. Wang Ying and Liu Tongtong wrote the original draft. Zhao Hailing, Liu Weijing, and Li Ping reviewed and edited the manuscript. Wang Ying, Liu Tongtong, Wu Yun, and Wang Lin designed and drew the figures. Ding Shaowei and Hou Baoluo designed the tables. All authors contributed to the article and approved the submitted version.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Umanath K, Lewis JB. Update on Diabetic Nephropathy: Core Curriculum 2018. Am J Kidney Dis. 2018;71:884-95
2. Rayego-Mateos S, Rodrigues-Diez RR, Fernandez-Fernandez B, Mora-Fernandez C, Marchant V, Donate-Correa J. et al. Targeting inflammation to treat diabetic kidney disease: the road to 2030. Kidney Int. 2023;103:282-96
3. Kato M, Natarajan R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat Rev Nephrol. 2019;15:327-45
4. Kim Y, Lim JH, Kim MY, Kim EN, Yoon HE, Shin SJ. et al. The Adiponectin Receptor Agonist AdipoRon Ameliorates Diabetic Nephropathy in a Model of Type 2 Diabetes. J Am Soc Nephrol. 2018;29:1108-27
5. Chen DQ, Chen H, Chen L, Vaziri ND, Wang M, Li XR. et al. The link between phenotype and fatty acid metabolism in advanced chronic kidney disease. Nephrol Dial Transplant. 2017;32:1154-66
6. Bobulescu IA. Renal lipid metabolism and lipotoxicity. Curr Opin Nephrol Hypertens. 2010;19:393-402
7. Jiang T, Wang XX, Scherzer P, Wilson P, Tallman J, Takahashi H. et al. Farnesoid X receptor modulates renal lipid metabolism, fibrosis, and diabetic nephropathy. Diabetes. 2007;56:2485-93
8. Herman-Edelstein M, Scherzer P, Tobar A, Levi M, Gafter U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J Lipid Res. 2014;55:561-72
9. Saito T, Ootaka T, Sato H, Furuta T, Sato T, Soma J. et al. Participation of macrophages in segmental endocapillary proliferation preceding focal glomerular sclerosis. J Pathol. 1993;170:179-85
10. Console L, Scalise M, Giangregorio N, Tonazzi A, Barile M, Indiveri C. The Link Between the Mitochondrial Fatty Acid Oxidation Derangement and Kidney Injury. Front Physiol. 2020;11:794
11. Hirano T. Abnormal lipoprotein metabolism in diabetic nephropathy. Clin Exp Nephrol. 2014;18:206-9
12. Moriwaki Y, Yamamoto T, Shibutani Y, Aoki E, Tsutsumi Z, Takahashi S. et al. Elevated levels of interleukin-18 and tumor necrosis factor-alpha in serum of patients with type 2 diabetes mellitus: relationship with diabetic nephropathy. Metabolism. 2003;52:605-8
13. Hirano T. Lipoprotein abnormalities in diabetic nephropathy. Kidney Int Suppl. 1999;71:S22-4
14. Muntner P, Coresh J, Smith JC, Eckfeldt J, Klag MJ. Plasma lipids and risk of developing renal dysfunction: the atherosclerosis risk in communities study. Kidney Int. 2000;58:293-301
15. Yan Q, Song Y, Zhang L, Chen Z, Yang C, Liu S. et al. Autophagy activation contributes to lipid accumulation in tubular epithelial cells during kidney fibrosis. Cell Death Discov. 2018;4:2
16. Nieth H, Schollmeyer P. Substrate-utilization of the human kidney. Nature. 1966;209:1244-5
17. Njeim R, Alkhansa S, Fornoni A. Unraveling the Crosstalk between Lipids and NADPH Oxidases in Diabetic Kidney Disease. Pharmaceutics. 2023;15:1360
18. Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F. et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21:37-46
19. Ruan XZ, Varghese Z, Moorhead JF. An update on the lipid nephrotoxicity hypothesis. Nat Rev Nephrol. 2009;5:713-21
20. Wahl P, Ducasa GM, Fornoni A. Systemic and renal lipids in kidney disease development and progression. Am J Physiol Renal Physiol. 2016;310:F433-45
21. Agarwal R. Effects of statins on renal function. Am J Cardiol. 2006;97:748-55
22. Mitrofanova A, Merscher S, Fornoni A. Kidney lipid dysmetabolism and lipid droplet accumulation in chronic kidney disease. Nat Rev Nephrol. 2023;19:629-45
23. Chen YY, Chen XG, Zhang S. Druggability of lipid metabolism modulation against renal fibrosis. Acta Pharmacol Sin. 2022;43:505-19
24. Li LO, Klett EL, Coleman RA. Acyl-CoA synthesis, lipid metabolism and lipotoxicity. Biochim Biophys Acta. 2010;1801:246-51
25. Black PN, Sandoval A, Arias-Barrau E, DiRusso CC. Targeting the fatty acid transport proteins (FATP) to understand the mechanisms linking fatty acid transport to metabolism. Immunol Endocr Metab Agents Med Chem. 2009;9:11-7
26. Glatz JF, Luiken JJ. From fat to FAT (CD36/SR-B2): Understanding the regulation of cellular fatty acid uptake. Biochimie. 2017;136:21-6
27. Yang X, Okamura DM, Lu X, Chen Y, Moorhead J, Varghese Z. et al. CD36 in chronic kidney disease: novel insights and therapeutic opportunities. Nat Rev Nephrol. 2017;13:769-81
28. Nosadini R, Tonolo G. Role of oxidized low density lipoproteins and free fatty acids in the pathogenesis of glomerulopathy and tubulointerstitial lesions in type 2 diabetes. Nutr Metab Cardiovasc Dis. 2011;21:79-85
29. Gutwein P, Abdel-Bakky MS, Doberstein K, Schramme A, Beckmann J, Schaefer L. et al. CXCL16 and oxLDL are induced in the onset of diabetic nephropathy. J Cell Mol Med. 2009;13:3809-25
30. Acharya R, Shetty SS, Kumari NS. Fatty acid transport proteins (FATPs) in cancer. Chem Phys Lipids. 2023;250:105269
31. Gimeno RE. Fatty acid transport proteins. Curr Opin Lipidol. 2007;18:271-6
32. Chen Y, Yan Q, Lv M, Song K, Dai Y, Huang Y. et al. Involvement of FATP2-mediated tubular lipid metabolic reprogramming in renal fibrogenesis. Cell Death Dis. 2020;11:994
33. Furuhashi M, Hotamisligil GS. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat Rev Drug Discov. 2008;7:489-503
34. McKillop IH, Girardi CA, Thompson KJ. Role of fatty acid binding proteins (FABPs) in cancer development and progression. Cell Signal. 2019;62:109336
35. Tsai IT, Wu CC, Hung WC, Lee TL, Hsuan CF, Wei CT. et al. FABP1 and FABP2 as markers of diabetic nephropathy. Int J Med Sci. 2020;17:2338-45
36. Kamijo-Ikemori A, Sugaya T, Kimura K. [L-type fatty acid binding protein (L-FABP) and kidney disease]. Rinsho Byori. 2014;62:163-70
37. Panduru NM, Forsblom C, Saraheimo M, Thorn L, Bierhaus A, Humpert PM. et al. Urinary liver-type fatty acid-binding protein and progression of diabetic nephropathy in type 1 diabetes. Diabetes Care. 2013;36:2077-83
38. Song X, Du Z, Yao Z, Tang X, Zhang M. Rhein Improves Renal Fibrosis by Restoring Cpt1a-Mediated Fatty Acid Oxidation through SirT1/STAT3/twist1 Pathway. Molecules. 2022;27:2344
39. Miguel V, Tituana J, Herrero JI, Herrero L, Serra D, Cuevas P. et al. Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J Clin Invest. 2021;131:e140695
40. Tran M, Tam D, Bardia A, Bhasin M, Rowe GC, Kher A. et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest. 2011;121:4003-14
41. Proctor G, Jiang T, Iwahashi M, Wang Z, Li J, Levi M. Regulation of renal fatty acid and cholesterol metabolism, inflammation, and fibrosis in Akita and OVE26 mice with type 1 diabetes. Diabetes. 2006;55:2502-9
42. Tyagi S, Gupta P, Saini AS, Kaushal C, Sharma S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J Adv Pharm Technol Res. 2011;2:236-40
43. Chawla A. Control of macrophage activation and function by PPARs. Circ Res. 2010;106:1559-69
44. Lakhia R, Yheskel M, Flaten A, Quittner-Strom EB, Holland WL, Patel V. PPARalpha agonist fenofibrate enhances fatty acid beta-oxidation and attenuates polycystic kidney and liver disease in mice. Am J Physiol Renal Physiol. 2018;314:F122-F31
45. Lyu Z, Mao Z, Li Q, Xia Y, Liu Y, He Q. et al. PPARgamma maintains the metabolic heterogeneity and homeostasis of renal tubules. EBioMedicine. 2018;38:178-90
46. Liang H, Ward WF. PGC-1alpha: a key regulator of energy metabolism. Adv Physiol Educ. 2006;30:145-51
47. Hotamisligil GS, Bernlohr DA. Metabolic functions of FABPs-mechanisms and therapeutic implications. Nat Rev Endocrinol. 2015;11:592-605
48. Sun L, Halaihel N, Zhang W, Rogers T, Levi M. Role of sterol regulatory element-binding protein 1 in regulation of renal lipid metabolism and glomerulosclerosis in diabetes mellitus. J Biol Chem. 2002;277:18919-27
49. Jun H, Song Z, Chen W, Zanhua R, Yonghong S, Shuxia L. et al. In vivo and in vitro effects of SREBP-1 on diabetic renal tubular lipid accumulation and RNAi-mediated gene silencing study. Histochem Cell Biol. 2009;131:327-45
50. Wang Z, Jiang T, Li J, Proctor G, McManaman JL, Lucia S. et al. Regulation of renal lipid metabolism, lipid accumulation, and glomerulosclerosis in FVBdb/db mice with type 2 diabetes. Diabetes. 2005;54:2328-35
51. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125-31
52. Mustafa M, Wang TN, Chen X, Gao B, Krepinsky JC. SREBP inhibition ameliorates renal injury after unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2016;311:F614-25
53. Fu Y, Sun Y, Wang M, Hou Y, Huang W, Zhou D. et al. Elevation of JAML Promotes Diabetic Kidney Disease by Modulating Podocyte Lipid Metabolism. Cell Metab. 2020;32:1052-62 e8
54. Postic C, Dentin R, Denechaud PD, Girard J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu Rev Nutr. 2007;27:179-92
55. Benhamed F, Denechaud PD, Lemoine M, Robichon C, Moldes M, Bertrand-Michel J. et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J Clin Invest. 2012;122:2176-94
56. Decleves AE, Zolkipli Z, Satriano J, Wang L, Nakayama T, Rogac M. et al. Regulation of lipid accumulation by AMP-activated kinase [corrected] in high fat diet-induced kidney injury. Kidney Int. 2014;85:611-23
57. Urahama Y, Ohsaki Y, Fujita Y, Maruyama S, Yuzawa Y, Matsuo S. et al. Lipid droplet-associated proteins protect renal tubular cells from fatty acid-induced apoptosis. Am J Pathol. 2008;173:1286-94
58. Nohturfft A, DeBose-Boyd RA, Scheek S, Goldstein JL, Brown MS. Sterols regulate cycling of SREBP cleavage-activating protein (SCAP) between endoplasmic reticulum and Golgi. Proc Natl Acad Sci U S A. 1999;96:11235-40
59. Yuan Y, Zhao L, Chen Y, Moorhead JF, Varghese Z, Powis SH. et al. Advanced glycation end products (AGEs) increase human mesangial foam cell formation by increasing Golgi SCAP glycosylation in vitro. Am J Physiol Renal Physiol. 2011;301:F236-43
60. Sun H, Yuan Y, Sun ZL. Cholesterol Contributes to Diabetic Nephropathy through SCAP-SREBP-2 Pathway. Int J Endocrinol. 2013;2013:592576
61. Wicinski M, Zak J, Malinowski B, Popek G, Grzesk G. PCSK9 signaling pathways and their potential importance in clinical practice. EPMA J. 2017;8:391-402
62. Lo Surdo P, Bottomley MJ, Calzetta A, Settembre EC, Cirillo A, Pandit S. et al. Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep. 2011;12:1300-5
63. Zhang DW, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD. et al. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. 2007;282:18602-12
64. Konarzewski M, Szolkiewicz M, Sucajtys-Szulc E, Blaszak J, Lizakowski S, Swierczynski J. et al. Elevated circulating PCSK-9 concentration in renal failure patients is corrected by renal replacement therapy. Am J Nephrol. 2014;40:157-63
65. Zhang J, Wu Y, Zhang J, Zhang R, Wang Y, Liu F. ABCA1 deficiency-mediated glomerular cholesterol accumulation exacerbates glomerular endothelial injury and dysfunction in diabetic kidney disease. Metabolism. 2023;139:155377
66. Jessup W, Gelissen IC, Gaus K, Kritharides L. Roles of ATP binding cassette transporters A1 and G1, scavenger receptor BI and membrane lipid domains in cholesterol export from macrophages. Curr Opin Lipidol. 2006;17:247-57
67. Yin QH, Zhang R, Li L, Wang YT, Liu JP, Zhang J. et al. Exendin-4 Ameliorates Lipotoxicity-induced Glomerular Endothelial Cell Injury by Improving ABC Transporter A1-mediated Cholesterol Efflux in Diabetic apoE Knockout Mice. J Biol Chem. 2016;291:26487-501
68. Tachibana H, Ogawa D, Matsushita Y, Bruemmer D, Wada J, Teshigawara S. et al. Activation of liver X receptor inhibits osteopontin and ameliorates diabetic nephropathy. J Am Soc Nephrol. 2012;23:1835-46
69. Ducasa GM, Mitrofanova A, Mallela SK, Liu X, Molina J, Sloan A. et al. ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. J Clin Invest. 2019;129:3387-400
70. Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93:241-52
71. Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB. et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7:161-71
72. Duchateau PN, Pullinger CR, Orellana RE, Kunitake ST, Naya-Vigne J, O'Connor PM. et al. Apolipoprotein L, a new human high density lipoprotein apolipoprotein expressed by the pancreas. Identification, cloning, characterization, and plasma distribution of apolipoprotein L. J Biol Chem. 1997;272:25576-82
73. Bhat OM, Yuan X, Li G, Lee R, Li PL. Sphingolipids and Redox Signaling in Renal Regulation and Chronic Kidney Diseases. Antioxid Redox Signal. 2018;28:1008-26
74. Geoffroy K, Troncy L, Wiernsperger N, Lagarde M, El Bawab S. Glomerular proliferation during early stages of diabetic nephropathy is associated with local increase of sphingosine-1-phosphate levels. FEBS Lett. 2005;579:1249-54
75. Ren S, Babelova A, Moreth K, Xin C, Eberhardt W, Doller A. et al. Transforming growth factor-beta2 upregulates sphingosine kinase-1 activity, which in turn attenuates the fibrotic response to TGF-beta2 by impeding CTGF expression. Kidney Int. 2009;76:857-67
76. Chalfant CE, Spiegel S. Sphingosine 1-phosphate and ceramide 1-phosphate: expanding roles in cell signaling. J Cell Sci. 2005;118:4605-12
77. Liu G, Han F, Yang Y, Xie Y, Jiang H, Mao Y. et al. Evaluation of sphingolipid metabolism in renal cortex of rats with streptozotocin-induced diabetes and the effects of rapamycin. Nephrol Dial Transplant. 2011;26:1493-502
78. van Meer G. Invisible rafts at work. Traffic. 2004;5:211-2
79. Mitrofanova A, Mallela SK, Ducasa GM, Yoo TH, Rosenfeld-Gur E, Zelnik ID. et al. SMPDL3b modulates insulin receptor signaling in diabetic kidney disease. Nat Commun. 2019;10:2692
80. Zador IZ, Deshmukh GD, Kunkel R, Johnson K, Radin NS, Shayman JA. A role for glycosphingolipid accumulation in the renal hypertrophy of streptozotocin-induced diabetes mellitus. J Clin Invest. 1993;91:797-803
81. Jao TM, Nangaku M, Wu CH, Sugahara M, Saito H, Maekawa H. et al. ATF6alpha downregulation of PPARalpha promotes lipotoxicity-induced tubulointerstitial fibrosis. Kidney Int. 2019;95:577-89
82. de Vries AP, Ruggenenti P, Ruan XZ, Praga M, Cruzado JM, Bajema IM. et al. Fatty kidney: emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. 2014;2:417-26
83. Hu S, Hang X, Wei Y, Wang H, Zhang L, Zhao L. Crosstalk among podocytes, glomerular endothelial cells and mesangial cells in diabetic kidney disease: an updated review. Cell Commun Signal. 2024;22:136
84. Schelling JR. The Contribution of Lipotoxicity to Diabetic Kidney Disease. Cells. 2022;11:3236
85. Falkevall A, Mehlem A, Palombo I, Heller Sahlgren B, Ebarasi L, He L. et al. Reducing VEGF-B Signaling Ameliorates Renal Lipotoxicity and Protects against Diabetic Kidney Disease. Cell Metab. 2017;25:713-26
86. Fornoni A, Merscher S, Kopp JB. Lipid biology of the podocyte-new perspectives offer new opportunities. Nat Rev Nephrol. 2014;10:379-88
87. Brinkkoetter PT, Bork T, Salou S, Liang W, Mizi A, Ozel C. et al. Anaerobic Glycolysis Maintains the Glomerular Filtration Barrier Independent of Mitochondrial Metabolism and Dynamics. Cell Rep. 2019;27:1551-66 e5
88. Lennon R, Pons D, Sabin MA, Wei C, Shield JP, Coward RJ. et al. Saturated fatty acids induce insulin resistance in human podocytes: implications for diabetic nephropathy. Nephrol Dial Transplant. 2009;24:3288-96
89. Sieber J, Lindenmeyer MT, Kampe K, Campbell KN, Cohen CD, Hopfer H. et al. Regulation of podocyte survival and endoplasmic reticulum stress by fatty acids. Am J Physiol Renal Physiol. 2010;299:F821-9
90. Sieber J, Weins A, Kampe K, Gruber S, Lindenmeyer MT, Cohen CD. et al. Susceptibility of podocytes to palmitic acid is regulated by stearoyl-CoA desaturases 1 and 2. Am J Pathol. 2013;183:735-44
91. Merscher-Gomez S, Guzman J, Pedigo CE, Lehto M, Aguillon-Prada R, Mendez A. et al. Cyclodextrin protects podocytes in diabetic kidney disease. Diabetes. 2013;62:3817-27
92. Bussolati B, Deregibus MC, Fonsato V, Doublier S, Spatola T, Procida S. et al. Statins prevent oxidized LDL-induced injury of glomerular podocytes by activating the phosphatidylinositol 3-kinase/AKT-signaling pathway. J Am Soc Nephrol. 2005;16:1936-47
93. Gutwein P, Abdel-Bakky MS, Schramme A, Doberstein K, Kampfer-Kolb N, Amann K. et al. CXCL16 is expressed in podocytes and acts as a scavenger receptor for oxidized low-density lipoprotein. Am J Pathol. 2009;174:2061-72
94. Pedigo CE, Ducasa GM, Leclercq F, Sloan A, Mitrofanova A, Hashmi T. et al. Local TNF causes NFATc1-dependent cholesterol-mediated podocyte injury. J Clin Invest. 2016;126:3336-50
95. Meyer-Schwesinger C. The ins-and-outs of podocyte lipid metabolism. Kidney Int. 2020;98:1087-90
96. Liu X, Ducasa GM, Mallela SK, Kim JJ, Molina J, Mitrofanova A. et al. Sterol-O-acyltransferase-1 has a role in kidney disease associated with diabetes and Alport syndrome. Kidney Int. 2020;98:1275-85
97. Yoo TH, Pedigo CE, Guzman J, Correa-Medina M, Wei C, Villarreal R. et al. Sphingomyelinase-like phosphodiesterase 3b expression levels determine podocyte injury phenotypes in glomerular disease. J Am Soc Nephrol. 2015;26:133-47
98. Schermer B, Benzing T. Lipid-protein interactions along the slit diaphragm of podocytes. J Am Soc Nephrol. 2009;20:473-8
99. Huber TB, Schermer B, Muller RU, Hohne M, Bartram M, Calixto A. et al. Podocin and MEC-2 bind cholesterol to regulate the activity of associated ion channels. Proc Natl Acad Sci U S A. 2006;103:17079-86
100. Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 2015;15:104-16
101. Watanabe S, Hidenori U, Hashimoto S, Riko S, Aizawa T, Tsugawa K. et al. Sphingomyelin Phosphodiesterase Acid-Like 3b is Essential for Toll-Like Receptor 3 Signaling in Human Podocytes. J Membr Biol. 2022;255:117-22
102. Zhang Y, Ma KL, Liu J, Wu Y, Hu ZB, Liu L. et al. Inflammatory stress exacerbates lipid accumulation and podocyte injuries in diabetic nephropathy. Acta diabetologica. 2015;52:1045-56
103. Vartak T, Godson C, Brennan E. Therapeutic potential of pro-resolving mediators in diabetic kidney disease. Adv Drug Deliv Rev. 2021;178:113965
104. Brennan E, Kantharidis P, Cooper ME, Godson C. Pro-resolving lipid mediators: regulators of inflammation, metabolism and kidney function. Nat Rev Nephrol. 2021;17:725-39
105. Kimmelstiel P, Wilson C. Intercapillary Lesions in the Glomeruli of the Kidney. Am J Pathol. 1936;12:83-98 7
106. Goncalves LED, Andrade-Silva M, Basso PJ, Camara NOS. Vitamin D and chronic kidney disease: Insights on lipid metabolism of tubular epithelial cell and macrophages in tubulointerstitial fibrosis. Front Physiol. 2023;14:1145233
107. Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr, Ory DS. et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A. 2003;100:3077-82
108. Izquierdo-Lahuerta A, Martinez-Garcia C, Medina-Gomez G. Lipotoxicity as a trigger factor of renal disease. J Nephrol. 2016;29:603-10
109. Zhao C, Li L, Li C, Tang C, Cai J, Liu Y. et al. PACS-2 deficiency in tubular cells aggravates lipid-related kidney injury in diabetic kidney disease. Mol Med. 2022;28:117
110. Tsun JG, Yung S, Chau MK, Shiu SW, Chan TM, Tan KC. Cellular cholesterol transport proteins in diabetic nephropathy. PLoS One. 2014;9:e105787
111. Kamijo A, Kimura K, Sugaya T, Yamanouchi M, Hase H, Kaneko T. et al. Urinary free fatty acids bound to albumin aggravate tubulointerstitial damage. Kidney Int. 2002;62:1628-37
112. Arici M, Chana R, Lewington A, Brown J, Brunskill NJ. Stimulation of proximal tubular cell apoptosis by albumin-bound fatty acids mediated by peroxisome proliferator activated receptor-gamma. J Am Soc Nephrol. 2003;14:17-27
113. Welte MA. Expanding roles for lipid droplets. Curr Biol. 2015;25:R470-81
114. Khan S, Cabral PD, Schilling WP, Schmidt ZW, Uddin AN, Gingras A. et al. Kidney Proximal Tubule Lipoapoptosis Is Regulated by Fatty Acid Transporter-2 (FATP2). J Am Soc Nephrol. 2018;29:81-91
115. Ichimura T, Asseldonk EJ, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest. 2008;118:1657-68
116. Mori Y, Ajay AK, Chang JH, Mou S, Zhao H, Kishi S. et al. KIM-1 mediates fatty acid uptake by renal tubular cells to promote progressive diabetic kidney disease. Cell Metab. 2021;33:1042-61 e7
117. Lim JC, Lim SK, Han HJ, Park SH. Cannabinoid receptor 1 mediates palmitic acid-induced apoptosis via endoplasmic reticulum stress in human renal proximal tubular cells. J Cell Physiol. 2010;225:654-63
118. Udi S, Hinden L, Earley B, Drori A, Reuveni N, Hadar R. et al. Proximal Tubular Cannabinoid-1 Receptor Regulates Obesity-Induced CKD. J Am Soc Nephrol. 2017;28:3518-32
119. Wasserman J, Santiago A, Rifici V, Holthofer H, Scharschmidt L, Epstein M. et al. Interactions of low density lipoprotein with rat mesangial cells. Kidney Int. 1989;35:1168-74
120. Wheeler DC, Fernando RL, Gillett MP, Zaruba J, Persaud J, Kingstone D. et al. Characterisation of the binding of low-density lipoproteins to cultured rat mesangial cells. Nephrol Dial Transplant. 1991;6:701-8
121. Schlondorff D. Cellular mechanisms of lipid injury in the glomerulus. Am J Kidney Dis. 1993;22:72-82
122. Wu ZL, Wang YC, Zhou Q, Ge YQ, Lan Y. Oxidized LDL induces transcription factor activator protein-1 in rat mesangial cells. Cell Biochem Funct. 2003;21:249-56
123. Wu Z, Zhou Q, Lan Y, Wang Y, Xu X, Jin H. AP-1 complexes mediate oxidized LDL-induced overproduction of TGF-beta(1) in rat mesangial cells. Cell Biochem Funct. 2004;22:237-47
124. Lan Y, Zhou Q, Wu ZL. NF-kappa B involved in transcription enhancement of TGF-beta 1 induced by Ox-LDL in rat mesangial cells. Chin Med J (Engl). 2004;117:225-30
125. Zhang M, Gao X, Wu J, Liu D, Cai H, Fu L. et al. Oxidized high-density lipoprotein enhances inflammatory activity in rat mesangial cells. Diabetes Metab Res Rev. 2010;26:455-63
126. Park MJ, Han HJ, Kim DI. Lipotoxicity-Induced PRMT1 Exacerbates Mesangial Cell Apoptosis via Endoplasmic Reticulum Stress. Int J Mol Sci. 2017;18:1421
127. Subathra M, Korrapati M, Howell LA, Arthur JM, Shayman JA, Schnellmann RG. et al. Kidney glycosphingolipids are elevated early in diabetic nephropathy and mediate hypertrophy of mesangial cells. Am J Physiol Renal Physiol. 2015;309:F204-15
128. Hanafusa N, Yatomi Y, Yamada K, Hori Y, Nangaku M, Okuda T. et al. Sphingosine 1-phosphate stimulates rat mesangial cell proliferation from outside the cells. Nephrol Dial Transplant. 2002;17:580-6
129. Rahman EU, Ruan XZ, Chana RS, Brunskill NJ, Gaya J, Powis SH. et al. Mesangial matrix-activated monocytes express functional scavenger receptors and accumulate intracellular lipid. Nephrol Dial Transplant. 2008;23:1876-85
130. Berfield AK, Andress DL, Abrass CK. IGF-1-induced lipid accumulation impairs mesangial cell migration and contractile function. Kidney Int. 2002;62:1229-37
131. Luchetti F, Crinelli R, Nasoni MG, Benedetti S, Palma F, Fraternale A. et al. LDL receptors, caveolae and cholesterol in endothelial dysfunction: oxLDLs accomplices or victims? Br J Pharmacol. 2021;178:3104-14
132. Shvets E, Ludwig A, Nichols BJ. News from the caves: update on the structure and function of caveolae. Curr Opin Cell Biol. 2014;29:99-106
133. Ha MK, Soo Cho J, Baik OR, Lee KH, Koo HS, Chung KY. Caenorhabditis elegans as a screening tool for the endothelial cell-derived putative aging-related proteins detected by proteomic analysis. Proteomics. 2006;6:3339-51
134. Yu CW, Liang X, Lipsky S, Karaaslan C, Kozakewich H, Hotamisligil GS. et al. Dual role of fatty acid-binding protein 5 on endothelial cell fate: a potential link between lipid metabolism and angiogenic responses. Angiogenesis. 2016;19:95-106
135. Furuhashi M, Fuseya T, Murata M, Hoshina K, Ishimura S, Mita T. et al. Local Production of Fatty Acid-Binding Protein 4 in Epicardial/Perivascular Fat and Macrophages Is Linked to Coronary Atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36:825-34
136. Lai Y, Tian Y, You X, Du J, Huang J. Effects of sphingolipid metabolism disorders on endothelial cells. Lipids Health Dis. 2022;21:101
137. Huang X, Li Y, Fu M, Xin HB. Polarizing Macrophages In Vitro. Methods Mol Biol. 2018;1784:119-26
138. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445-55
139. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723-37
140. Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY. et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol. 2014;15:323-32
141. Cader MZ, Boroviak K, Zhang Q, Assadi G, Kempster SL, Sewell GW. et al. C13orf31 (FAMIN) is a central regulator of immunometabolic function. Nat Immunol. 2016;17:1046-56
142. Olona A, Hateley C, Muralidharan S, Wenk MR, Torta F, Behmoaras J. Sphingolipid metabolism during Toll-like receptor 4 (TLR4)-mediated macrophage activation. Br J Pharmacol. 2021;178:4575-87
143. Posokhova EN, Khoshchenko OM, Chasovskikh MI, Pivovarova EN, Dushkin MI. Lipid synthesis in macrophages during inflammation in vivo: effect of agonists of peroxisome proliferator activated receptors alpha and gamma and of retinoid X receptors. Biochemistry (Mosc). 2008;73:296-304
144. Feingold KR, Shigenaga JK, Kazemi MR, McDonald CM, Patzek SM, Cross AS. et al. Mechanisms of triglyceride accumulation in activated macrophages. J Leukoc Biol. 2012;92:829-39
145. Yan J, Horng T. Lipid Metabolism in Regulation of Macrophage Functions. Trends Cell Biol. 2020;30:979-89
146. Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT. et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010;12:467-82
147. Jiang WJ, Xu CT, Du CL, Dong JH, Xu SB, Hu BF. et al. Tubular epithelial cell-to-macrophage communication forms a negative feedback loop via extracellular vesicle transfer to promote renal inflammation and apoptosis in diabetic nephropathy. Theranostics. 2022;12:324-39
148. Koliwad SK, Streeper RS, Monetti M, Cornelissen I, Chan L, Terayama K. et al. DGAT1-dependent triacylglycerol storage by macrophages protects mice from diet-induced insulin resistance and inflammation. J Clin Invest. 2010;120:756-67
149. Aflaki E, Balenga NA, Luschnig-Schratl P, Wolinski H, Povoden S, Chandak PG. et al. Impaired Rho GTPase activation abrogates cell polarization and migration in macrophages with defective lipolysis. Cell Mol Life Sci. 2011;68:3933-47
150. Habib A, Chokr D, Wan J, Hegde P, Mabire M, Siebert M. et al. Inhibition of monoacylglycerol lipase, an anti-inflammatory and antifibrogenic strategy in the liver. Gut. 2019;68:522-32
151. Huang SC, Everts B, Ivanova Y, O'Sullivan D, Nascimento M, Smith AM. et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15:846-55
152. Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79-82
153. Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N. et al. MiR-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570-3
154. Zelcer N, Tontonoz P. Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest. 2006;116:607-14
155. Repa JJ, Mangelsdorf DJ. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu Rev Cell Dev Biol. 2000;16:459-81
156. Klionsky DJ, Petroni G, Amaravadi RK, Baehrecke EH, Ballabio A, Boya P. et al. Autophagy in major human diseases. EMBO J. 2021;40:e108863
157. Tseng CH, Shah KM, Chiu IJ, Hsiao LL. The Role of Autophagy in Type 2 Diabetic Kidney Disease Management. Cells. 2023;12:2691
158. Ma Z, Li L, Livingston MJ, Zhang D, Mi Q, Zhang M. et al. p53/microRNA-214/ULK1 axis impairs renal tubular autophagy in diabetic kidney disease. J Clin Invest. 2020;130:5011-26
159. Liu WJ, Gan Y, Huang WF, Wu HL, Zhang XQ, Zheng HJ. et al. Lysosome restoration to activate podocyte autophagy: a new therapeutic strategy for diabetic kidney disease. Cell Death Dis. 2019;10:806
160. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M. et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131-5
161. Zhao YH, Fan YJ. Resveratrol improves lipid metabolism in diabetic nephropathy rats. Front Biosci (Landmark Ed). 2020;25:1913-24
162. Dall'Armi C, Devereaux KA, Di Paolo G. The role of lipids in the control of autophagy. Curr Biol. 2013;23:R33-45
163. Charsou C, Ng MYW, Simonsen A. Regulation of autophagosome biogenesis and mitochondrial bioenergetics by the cholesterol transport protein GRAMD1C. Autophagy. 2023;19:2159-61
164. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21-35
165. Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol. 2009;186:773-82
166. Burman C, Ktistakis NT. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010;584:1302-12
167. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107-32
168. Haucke V, Di Paolo G. Lipids and lipid modifications in the regulation of membrane traffic. Curr Opin Cell Biol. 2007;19:426-35
169. Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010;24:3052-65
170. Rodriguez-Navarro JA, Kaushik S, Koga H, Dall'Armi C, Shui G, Wenk MR. et al. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc Natl Acad Sci U S A. 2012;109:E705-14
171. Li Y, Li S, Qin X, Hou W, Dong H, Yao L. et al. The pleiotropic roles of sphingolipid signaling in autophagy. Cell Death Dis. 2014;5:e1245
172. Bedia C, Levade T, Codogno P. Regulation of autophagy by sphingolipids. Anticancer Agents Med Chem. 2011;11:844-53
173. Scarlatti F, Bauvy C, Ventruti A, Sala G, Cluzeaud F, Vandewalle A. et al. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem. 2004;279:18384-91
174. Pattingre S, Bauvy C, Carpentier S, Levade T, Levine B, Codogno P. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J Biol Chem. 2009;284:2719-28
175. Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab. 2011;13:495-504
176. Zhang X, Evans TD, Jeong SJ, Razani B. Classical and alternative roles for autophagy in lipid metabolism. Curr Opin Lipidol. 2018;29:203-11
177. Greenberg AS, Coleman RA, Kraemer FB, McManaman JL, Obin MS, Puri V. et al. The role of lipid droplets in metabolic disease in rodents and humans. J Clin Invest. 2011;121:2102-10
178. Dupont N, Chauhan S, Arko-Mensah J, Castillo EF, Masedunskas A, Weigert R. et al. Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr Biol. 2014;24:609-20
179. Kimmel AR, Brasaemle DL, McAndrews-Hill M, Sztalryd C, Londos C. Adoption of PERILIPIN as a unifying nomenclature for the mammalian PAT-family of intracellular lipid storage droplet proteins. J Lipid Res. 2010;51:468-71
180. Kaushik S, Cuervo AM. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy. 2016;12:432-8
181. Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M. et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383-6
182. Vaughan M, Berger JE, Steinberg D. Hormone-Sensitive Lipase and Monoglyceride Lipase Activities in Adipose Tissue. J Biol Chem. 1964;239:401-9
183. Martinez-Lopez N, Singh R. Autophagy and Lipid Droplets in the Liver. Annu Rev Nutr. 2015;35:215-37
184. Sathyanarayan A, Mashek MT, Mashek DG. ATGL Promotes Autophagy/Lipophagy via SIRT1 to Control Hepatic Lipid Droplet Catabolism. Cell Rep. 2017;19:1-9
185. Reid BN, Ables GP, Otlivanchik OA, Schoiswohl G, Zechner R, Blaner WS. et al. Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J Biol Chem. 2008;283:13087-99
186. Khawar MB, Gao H, Li W. Autophagy and Lipid Metabolism. Adv Exp Med Biol. 2019;1206:359-74
187. Balderhaar HJ, Ungermann C. CORVET and HOPS tethering complexes - coordinators of endosome and lysosome fusion. J Cell Sci. 2013;126:1307-16
188. Tshilenge KT, Aguirre CG, Bons J, Gerencser AA, Basisty N, Song S. et al. Proteomic Analysis of Huntington's Disease Medium Spiny Neurons Identifies Alterations in Lipid Droplets. Mol Cell Proteomics. 2023;22:100534
189. Lettieri Barbato D, Tatulli G, Aquilano K, Ciriolo MR. FoxO1 controls lysosomal acid lipase in adipocytes: implication of lipophagy during nutrient restriction and metformin treatment. Cell Death Dis. 2013;4:e861
190. Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O. et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013;15:647-58
191. Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar S. et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature. 2014;516:108-11
192. Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA. et al. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014;516:112-5
193. Nath S, Dancourt J, Shteyn V, Puente G, Fong WM, Nag S. et al. Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane-curvature-sensing domain in Atg3. Nat Cell Biol. 2014;16:415-24
194. Han Y, Xiong S, Zhao H, Yang S, Yang M, Zhu X. et al. Lipophagy deficiency exacerbates ectopic lipid accumulation and tubular cells injury in diabetic nephropathy. Cell Death Dis. 2021;12:1031
195. Miceli C, Roccio F, Penalva-Mousset L, Morel E, Codogno P, Dupont N. Fluid flow-induced shear stress controls the metabolism of proximal tubule kidney epithelial cells through primary cilium-dependent lipophagy and mitochondria biogenesis. Autophagy. 2020;16:2287-8
196. Huang CC, Chou CA, Chen WY, Yang JL, Lee WC, Chen JB. et al. Empagliflozin Ameliorates Free Fatty Acid Induced-Lipotoxicity in Renal Proximal Tubular Cells via the PPARgamma/CD36 Pathway in Obese Mice. Int J Mol Sci. 2021;22:12408
197. Qin X, Jiang M, Zhao Y, Gong J, Su H, Yuan F. et al. Berberine protects against diabetic kidney disease via promoting PGC-1alpha-regulated mitochondrial energy homeostasis. Br J Pharmacol. 2020;177:3646-61
198. Szeto HH, Liu S, Soong Y, Alam N, Prusky GT, Seshan SV. Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int. 2016;90:997-1011
199. Su K, Zeng P, Liang W, Luo Z, Wang Y, Lv X. et al. FTY720 Attenuates Angiotensin II-Induced Podocyte Damage via Inhibiting Inflammatory Cytokines. Mediators of inflammation. 2017;2017:3701385
Author contact
![]() Corresponding authors: Hailing Zhao (zhly0167com), Weijing Liu (liuweijing-1977com), and Ping Li (lp8675com).
Corresponding authors: Hailing Zhao (zhly0167com), Weijing Liu (liuweijing-1977com), and Ping Li (lp8675com).