Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Fluoropyrimidine-Related Adverse...
Methods
DPYD Genetic Complexity and the...
Case Reports and Case Series
Discussion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(10):3742-3759. doi:10.7150/ijbs.97686 This issue Cite
Review
Clinical Implementation of Rare and Novel DPYD Variants for Personalizing Fluoropyrimidine Treatment: Challenges and Opportunities
Elena De Mattia1, Noemi Milan1, Yehuda G. Assaraf2, Giuseppe Toffoli1 ![]() , Erika Cecchin1
, Erika Cecchin1
1. Experimental and Clinical Pharmacology, Centro di Riferimento Oncologico di Aviano (CRO) IRCCS, via Franco Gallini n. 2, 33081 Aviano (PN), Italy.
2. The Fred Wyszkowski Cancer Research Laboratory, Faculty of Biology, Technion-Israel Institute of Technology, Haifa 3200003, Israel.
Received 2024-4-24; Accepted 2024-6-11; Published 2024-7-2
Abstract

Fluoropyrimidines (FLs) [5-Fluorouracil, Capecitabine] are used in the treatment of several solid tumors. Dihydropyrimidine dehydrogenase (DPD) is the rate-limiting enzyme for FL detoxification, and its deficiency could lead to severe, life-threatening or fatal toxicity after FL administration. Testing with a pharmacogenetic panel of four deleterious variants in the dihydropyrimidine dehydrogenase gene (DPYD) (DPYD*2A, DPYD*13, c.2846A > T, c.1129-5923C > G) prior to FL treatment, is recommended by scientific consortia (e.g., CPIC, DPWG) and drug regulatory agencies (e.g., EMA). However, this panel identifies < 20% of patients at risk of severe FL-related toxicity. Cumulative recent evidence highlights the potential clinical value of rare (minor allele frequency < 1%) and novel DPYD genetic variants for identifying an additional fraction of DPD-deficient patients at increased risk of severe FL-related toxicity. In this review, we aimed to comprehensively describe the available evidence regarding the potential clinical predictive role of novel and rare DPYD variants as toxicity markers in FL-treated patients, and to discuss the challenges and opportunities in tailoring FL treatment based upon clinical application of such markers. Although we must overcome existing barriers to the clinical implementation, the available data support that comprehensive assessment of the DPYD sequence, including rare and novel genetic variants, may significantly enhance the pre-emptive identification of at-risk patients, compared to the current targeted approach.
Keywords: DPYD, rare variant, fluoropyrimidine, toxicity, next-generation sequencing, clinical implementation
Fluoropyrimidine-Related Adverse Drug Reactions and DPYD Testing
Since the 1950s, 5-fluorouracil (5-FU) has been one of the most commonly prescribed anticancer drugs for the treatment of various solid tumors [1, 2]. Belonging to the antimetabolite fluoropyrimidine (FL) class, 5-FU and its oral prodrug capecitabine act as false high-affinity substrates for thymidylate synthase, thereby inhibiting pyrimidine biosynthesis in cells displaying high proliferation rates. Metabolites of 5-FU exhibit potent cytotoxic activity due to the biosynthetic depletion of endogenous thymidine, along with direct damage to DNA and RNA, leading to cell death and tumor growth suppression. While the pharmacological efficacy of FLs is well-established, the narrow therapeutic index is a major issue in the management of chemotherapy. Although the vast majority of patients can be safely treated with FLs, 20-30% will develop severe or even life-threatening untoward toxicity during the course of chemotherapy, resulting in treatment delays and patient discomfort [3].
Inter-individual differences in the pharmacokinetics and pharmacodynamics pathways of FLs could play a role in the observed variability in therapeutic outcome. The metabolic pathway of FLs includes a number of proteins such as ATP-binding cassette (ABC), solute carrier transporters (SLC), nuclear receptors, and enzymes (e.g., thymidine phosphorylase, uridine monophosphate synthase, cytidine deaminase), that have been previously reported to have genetic polymorphisms which could affect drug bioavailability and exposure to some extent. Moreover, thymidylate synthase, which is the drug target, and 5,10-methylenetetrahydrofolate reductase, which is involved in the pharmacodynamic effect of the drug, have been reported to play a relevant pharmacogenetic role in the toxicity and efficacy of FLs [4-6].
One more specific cause of FL-related toxicity is inefficient catabolism of the drug, which is mainly mediated by the detoxification enzyme dihydropyrimidine dehydrogenase (DPD). Many efforts have been made to better characterize the genetic basis of this metabolic defect [7-9]. Currently, only four single-nucleotide polymorphisms in the dihydropyrimidine dehydrogenase gene (DPYD) are classified as clinically relevant, and listed in international pharmacogenetics guidelines for drug dose recommendations—such as those provided by the Clinical Pharmacogenetics Implementation Consortium (CPIC) [10] and the Royal Dutch Association for the Advancement of Pharmacy (DPWG) [11]. The following variants are known for their role in impairing DPD activity: DPYD*2A (rs3918290) and DPYD*13 (rs55886062), which are associated with nearly complete protein deficiency in homozygotes [12], and c.2846A>T (rs67376798) and c.1129-5923C>G (rs75017182, tagging HapB3), which are associated with moderate loss of protein function [7]. On May 2020, this compelling evidence prompted the European regulatory agency to publish its own pharmacogenetic recommendations to improve appropriate FL use. EMA now recommends using a reduced initial dose of FLs in patients with DPD deficiency, as determined either by phenotyping (i.e., measuring plasma uracil concentration) or by genotyping patients with the four-variant DPYD panel [13].
Notably, the four-variant DPYD panel presents a high specificity (between 99% and 100%) but a low sensitivity (1-12%) for detecting patients at risk of toxicity [8], suggesting that other factors—genetic or otherwise—are potentially involved in the development of FL toxicity, and indicating a need for further investigation of the DPYD genotype. It also should be noted that the 4-variant test presents a high specificity, with carriers of these four alleles presenting a high risk for severe toxicity, and a low sensitivity with non-carriers still presenting a high risk of unpredictable toxicity occurrence [14]. Considering the reported complexity of DPYD genetics, it is likely that many additional rare genetic variants may contribute to the observed interindividual variability in the risk of toxicity development [15]. Moreover, about 7% of Europeans are carriers of at least one of the four variants included in the international guidelines, while some of these variants are very rare in populations of African or Asian origin, where additional variants might play a more relevant role [10].
In the present article, we systematically review the available literature regarding the role of genetic DPYD variants in identifying patients at high risk of toxicity following FL administration. In addition to the four currently recommended variants, we also focused on variants that are defined as “rare” in the studied population [minor allele frequency (MAF) < 1%] or that have never been previously reported. We also discuss the potential hurdles for the clinical implementation of using these rare and novel variants as markers untoward toxicity.
Methods
We performed a systematic literature search for all published studies addressing the role of genetic germline DPYD variants that were classified by the authors as “rare” or “novel”, and that were potentially related to severe (grade 3-5) or lethal toxic reactions to FL-based treatment in cancer patients. In general, “rare” was defined as a variant not classifiable as a common polymorphism, and “novel” was defined as a variant absent from the available public databases (i.e., NCBI SNP database, Ensembl) [16, 17] at the time of study publication. For each identified study, we recorded information related to patients' status regarding the routinely tested DPYD markers (DPYD*2A, *13, c.2846A > T, c.1129-5923C >G-HapB3). This systematic review was carried out according to Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines, and the protocol was registered in the International Prospective Register of Systematic Reviews (PROSPERO 2024 CRD42024501461 Available from: https://www.crd.york.ac.uk/prospero/display_record.php?ID=CRD42024501461).
Three databases—MEDLINE (PubMed), Cochrane Library, and Web of Science Core Collection (Clarivate)—were searched for relevant articles, with the last search update on March 1, 2024. Since MEDLINE included all of the articles returned by the other two databases, we referred only to MEDLINE. Search algorithms included the keywords “DPYD”, “rare variant”, “fluoropyrimidine (5-fluorouracil or capecitabine)”, and “toxicity”, combined with Boolean operators (OR/AND). Additional studies were identified by manually searching the references of relevant articles. To evaluate the studies retrieved using this search strategy, two independent authors (EDM and EC) screened the titles and/or abstracts, and selected suitable studies according to the inclusion and exclusion criteria. Then, full-text versions of these potentially eligible articles were retrieved and independently assessed by these two authors. Disagreements were settled by a third researcher (GT). When studies overlapped, the publication with the largest number of patients was included in this systematic review.
Inclusion criteria were that the studies were published in English in a peer-reviewed journal, and contained data regarding the topic of the present review. Given the qualitative nature of this work, we included case reports, case series, and descriptive analyses. Systematic reviews, reviews, and conference abstracts were excluded. Similarly, we excluded papers that did not address the topic of the search—e.g., in vitro studies, epidemiologic analyses of the distribution of DPYD variants by ethnicity, experiences with implementation of DPYD variants in clinical practice, analysis of somatic DPYD mutations, and studies without FL-based therapy or toxicity as a clinical endpoint. Figure 1 shows the flowchart of our literature search. Ultimately, a total of 27 studies were included and discussed in the present systematic review.
PRISMA flow diagram. *One study was both a case report and a clinical study.
For each eligible study, the following items were recorded in a pre-piloted form: the country where the study was based; the analytical method used to detect DPYD variants; the clinical description of cases (for case reports and case series) or the patient population (for clinical studies), including data about the cancer type and patient sex/gender; therapy characteristics (e.g., 5-FU or capecitabine administration, monotherapy or combined therapy, and clinical setting); strategies used for the functional prediction of detected variants; method of DPD phenotyping; and a summary of the main findings of the article. Data related to the patients' status for the routinely tested DPYD markers was recorded when available (Table 1 and Table 2).
Case series and case report: DPYD gene sequencing in patients with severe fluoropyrimidine-related toxicity.
| Analytical method | Cases description | Therapy | SNP Functional prediction | DPD phenotyping | Country (Ethnicity) | Main findings | Outcome | Ref |
|---|---|---|---|---|---|---|---|---|
| CASE SERIES | ||||||||
| Full gene sequencing (NGS) | Case 1: 65-year-old female with LARC. A few days after starting adjuvant treatment she experienced grade2-3 toxicities. CAPE was stopped and switched to mFOLFOX but the patient again developed early grade 3 toxicity that requiring hospitalization. | Neoadjuvant CAPE + RT. Adjuvant XELOX switched after toxicity to mFOLFOX-6 | Literature search | Saudi Arabia (NS) | The patient was heterozygous for the rare DPYD variant rs371313778, c.2434G>A (p.Val812lle), which has normal activity in vitro with uncertain phenotypic significance. This variant has never before been identified in a case of toxicity. | FOLFOX was restarted with 5-FU at 50% dose reduction with further titration in subsequent cycles. The planned adjuvant treatment was completed. | [29] [30] | |
| Whole gene sequencing (NGS) | Case 2: 64-year-old female, a cousin of case 1, with stage III colon adenocarcinoma, sigmoid primary. Intermediate DPYD metabolizer. | Adjuvant XELOX, with CAPE at 40% dose-reduction based on family history of severe toxicity FL-related. | Literature search | Saudi Arabia (NS) | The patient was homozygous for the rare DPYD variant c.1601G>A (p.Ser534Asn, DPYD*4) previously linked to clinical DPD deficiency. | The reduced initial dose prevented the development of severe toxicity. CAPE dose was then increased by 5-10% but the patient developed toxicity requiring hospitalization. Therapy was continued with CAPE at 60% of standard dose. | ||
| Whole gene sequencing (NGS) | Case 3: 66-year-old males with LARC. CAPE was stopped at week 4 of treatment due to severe toxicities requiring hospitalization. | Neoadjuvant CAPE + RT | Saudi Arabia (NS) | The patient was heterozygous for the very rare DPYD variant c.257C>T (p.Pro86Leu) previously linked to clinical DPD deficiency. | The patient concluded the planned therapy with only RT. | |||
| MLPA and array-based comparative genomic hybridization analysis; All exons + flanking intronic regions + intron 10 sequencing (Sanger method) | 8 pts (F, n=3; M, n=5) with GI cancers (62.5%, CRC; 25.5%, rectal cancer; 12.5%, esophageal cancer) and reduced DPD activity: 6 pts experienced early-onset grade 3-4 toxicity and 2 pts lethal grade 5 toxicity. One additional female patient with pre-treatment detected impaired DPD activity was treated with reduced FL dose without developing severe toxicity. | 8 pts received standard dose of FL-containing regimen (UFT, n=1; 5-FU, n=1; CAPE, n=6) ± RT. One patient received a CAPE dose reduced by 50%. | RNA (cDNA) sequencing; Functional analysis of recombinantly-expressed DPD mutants; Crystal DPD structure analysis | DPD enzyme activity assay on PBMCs | Dutch (n=8); Danish (n=1) pts (Caucasian) | All 9 pts possessed a strongly reduced DPD activity (9-53%). A total of 21 DPYD aberrations, including 3 novel (c.1740+2T>C [p.Ser509_Lys580del], c.2407_2427del [p.Leu803_Gly809del], c.2843T>C [p.Ile948Thr]) and 4 very rare (c.321+1G>A [p.Cys79Thrfs*8], c.851G>T [p.Gly284Val], c.1280T>C [p.Val427Ala], c.2766+87G>A) were detected. All but one of these variants (c.2766+87G>A) were reported to have a functional impact on DPD activity. A novel aberration, the c.851_1524dup aberrant protein, was also detected. | -- | [46] |
| Exon sequencing (Sanger method) in case of aberrant dHPLC pattern | 3 female pts with CRC experiencing early-onset grade≥3 toxicity. Negative for DPYD*2A and c.2846A>T. | Pts1: 5-FU + LV + IRI + BV; Pts2: 5-FU + FA + OXA Pts3: adjuvant CAPE + OXA | Literature search; DPD structure analysis | Italy (NS) | 3 novel non-synonymous DPYD variants (c.2509-2510insC [Leu -> Pro], c.1801G>C [Gly-> Ser], c.680G>A [Ser -> Gly]) were detected and possibly associated with a poor-metabolizer phenotype. These novel mutations could represent a detrimental variant and increase the effect of other SNPs identified in the same patient. | Treatment was discontinued / started with FL dose reduction. | [31] | |
| Exon 14 RFLP and DNA sequencing (Sanger method) | Case 1: 76-year-old woman with colon cancer experiencing grade 3-4 toxicity and hospitalization after the third cycle. | Adjuvant 5-FU + Calcium folinate | Literature search; DPD structure analysis | Sweden (NS) | The patient was heterozygous for the functionally-relevant rare DPYD variant c.1796T>C (p.Met599Thr). | 5-FU was reintroduced with a 50% dose reduction, without severe toxicity. | [41] | |
| Case 2: 75-year-old woman with colon cancer experiencing grade 2-3 toxicity after two thirds of the treatment | Adjuvant 5-FU + FA | RNA (cDNA) sequencing | Sweden (NS) | The patient was heterozygous for the DPYD splice site c.IVS14+ 17A>G variant which, however, does not appear to influence the splicing process and thus explains the observed toxicity. | Treatment was interrupted. | |||
| Full coding region screening by dHPLC. Data validation by sequencing | 4 patients with breast (n=3) or colorectal (n=1) cancer experiencing early-onset grade 3-4 toxicity. | Adjuvant CMF (n=2) Neoadjuvant FEC (n=1) Adjuvant 5-FU +FA (n=1) | Crystal DPD structure analysis | DPD enzyme activity assay on PBMCs | Germany (Caucasian) | In addition to the known and more frequent variants (c.85T>C [p.Cys29Arg], c.496A>G [p.Met166Val], c.1601G>A [p.Ser534Asn], c.1627A>G [p.Ile543Val, DPYD*5A], c.1896T>C [p.Phe632Phe]) a novel mutation (e.g., c.775A>G [p.Lys259Glu]) was detected. Rare variants DPYD combinations (e.g., c.85T>C + c.496A>G + c.1601G>A + c.1627A>G or c.496A>G + c.1601G>A) were detected and potentially associated with reduced DPD activity. | -- | [35] |
| CASE REPORT | ||||||||
| Whole-Genome sequencing (NGS). Data validation by Sanger sequencing | 59-year-old female with metastatic colon cancer experiencing grade 4 life-threatening toxicity following the first cycle of treatment. Negative for DPYD*2A, *13, c.2846A>T, c.1129-5923T>G- HapB3. | Neoadjuvant CAPE + OXA + BV | In silico tools: DPYD-Varifier; | DPD Enzyme Activity Assay | India (Indian, South Asian) | The patient was heterozygous for the very rare DPYD missense variant (rs755416212, c.704G>A [p.Arg235Gln]), which is predicted to be deleterious and significantly reduce DPD activity by 88%. | The patient was started on mFOLFOX regimen with a 75% reduced dose of 5-FU and no toxicity was observed. | [39] |
| Exon 14 sequencing; cDNA coding sequence sequencing (Sanger method) | 81-year-old female patient with metastatic breast cancer experiencing grade 3-4 toxicity requiring hospitalization and suspension of CAPE following the first cycle of treatment. Negative for DPYD*2A, *13, c.2846A>T, c.1236G>A-HapB3. | CAPE for metastatic progression | In silico tools: SIFT, MutationTaster, PolyPhen-2, PROVEAN, HSF system; SwissModel web tools; RNA (cDNA) sequencing and real-time quantitative PCR assays; | Determination of U, UH2 and UH2/U ratio; | France (NS) | A partial DPD deficiency was determined. A reduced DPYD mRNA levels was detected. A novel DPYD variant c.1903A>G [p.Asn635Asp] was identified in a heterozygous state. However, the deleteriousness of this variant is doubt. Two other already known variants (i.e., rs1801265, c.85T>C, [p.Cys29Arg]; rs2297595, c.496A>G, [p.Met166Val] with potential clinical significance were identified. | The re-introduction of CAPE at lower doses was associated with a recurrence of severe toxicity, so treatment was stopped and new therapies were adopted. | [42] |
| All exons sequencing (Sanger method) | 79-year-old female with breast cancer experiencing life-threatening toxicity after 1 week of treatment. The CAPE was suspended. Negative for DPYD*2A, *13, c.2846A>T, | Second-line CT based on CAPE | In silico tools: HSF system; RNA (cDNA) sequencing; | Determination of U, UH2 and UH2/U ratio. | Spain (Caucasian) | A novel rare functionally-relevant c.2242+1G>T splicing variant was detected. This variant produces a shorter mRNA and protein, which leads to a non-functional DPD protein and could explain the severe toxicity. | The patient was no longer treated with FL. | [34] |
| All exons + flanking intronic regions sequencing (Sanger method) Copy number variation by MLPA analysis | 59-year-old female patient with a sigmoid adenocarcinoma. A heterozygosity for the DPYD*2A variant and a complete DPD deficiency (i.e., DPD activity in PBMCs) were determined pre-treatment. | Adjuvant CAPE + OXA (CAPE started with dose at 0.8% of originally planned dose) | Literature search; | DPD activity in PBMCs | The Netherlands (NS) | The DPYD*1/*2A genotype did not explain the complete DPD deficiency. A novel amplification of DPYD exons 17 and 18 was detected and associated with the DPD deficiency. | The pre-emptive dose reduction has prevented the development of potentially life-threatening toxicity. | [36] |
| Exome sequencing (NGS) | 49-year-old female patient with resected stage III carcinoma of sigmoid colon developing grade≥3 toxicity after the first cycle requiring hospitalization. | Adjuvant CAPE + OXA | In silico tools: HSF system | Hong Kong (NS) | The patient was heterozygous for the novel intronic c.321+2T>C variant, a pathogenic splicing variant resulting in a non-functional allele. | CT was restarted with FOLFOX and with a dose of 5-FU reduced by 30%. The dose was then titrated, and the patient tolerated the subsequent cycles. | [44] | |
| All exons sequencing (Sanger method) | 37-year-old female with metastatic breast cancer who experienced early onset severe toxicities requiring hospitalization. | Adjuvant CAPE + trastuzumab | Literature search; DPD structure analysis | Italy (Caucasian) | The patient was a heterozygous carrier of four variants: c.257C>T [p.Pro86Leu], c.496A>G [p.Met166Val], c.1850C>T [p.Thr617Met] and c.2194G>A [p.Val732Ile]. The novel missense variant c.1850C>T (p.Thr617Met) in combination with the rare c.257C>T (p.Pro86Leu) was probably responsible for the severe life-threatening toxicity. The two variants could be deleterious. | The patient died due to multiorgan failure. | [32] | |
| Promoter region + coding exons sequencing (Sanger method) | 63-year-old female patient with Lieberkühn adenocarcinoma developing lethal toxicity 8 days after the first infusion of 5-FU. Patient was routinely tested for the DPYD*2A, *13, c.2846A>T, c.464T>A. | Adjuvant FOLFOX | In silico tools: UMD-Predictor; Literature search | Determination of U, UH2 and UH2/U ratio | France (Caucasian) | The patient, with complete DPD deficiency, was heterozygous for two DPYD variants: a novel 8-bp duplication (c.168_175dupGAATAATT, [p.Phe59Ter]) and DPYD*13. The novel variation resulted in a stop codon (p.Phe59Ter) and was likely responsible for the lethal toxicity. | She died 17 days after 5-FU administration. | [43] |
| All exons + adjacent intronic regions sequencing (Sanger method). Data validation by Pyrosequencing. | 73-year-old female with colon cancer developing lethal toxicity 7 days after the first cycle. | Adjuvant 5-FU + LV | Protein truncation test in bacterial expression vector. | Determination of U, UH2 and UH2/U ratio and 5-FU level | Spanish (NS) | The patients, with decreased DPD enzyme activity, was wild-type for 22 potentially relevant variants analyzed by targeted genotyping. The rare novel functionally-relevant variant c.464T>A (p.Leu155Ter) was identified by sequencing and was the potential cause of life- threatening toxicity. | She died 19 days after 5-FU administration. | [40] |
| All exons sequencing (Sanger method) | 35-year-old male with advanced caecum cancer developing a multiple organ dysfunction 2 days after onset of CT requiring hospitalization. | Adjuvant 5-FU + FA | Literature search | Germany (NS) | The patient was carrier of the rare variant c.1601G>A (p.Ser534Asn, *4) in addition to the more frequent non-pathogenetic c.85T>C (p.Cys29Arg) and c.1627A>C (p.Ile543Val) variants. The c.1601G>A was detected together with an intronic mutation (c.IVS13+40G>A); reduced enzyme activity was consistently observed for both variants. | The toxic symptoms, which required intensive treatment, eventually led to a full recovery. | [38] | |
| Coding exons sequencing (Sanger method) | 42-year-old female with advanced ovarian carcinoma developing grade 4 toxicities 4 days after the first single injection of 5-FU. | Palliative 5-FU + LV treatment for symptomatic liver metastases | DPD structure analysis | The Netherlands (NS) | The patient was heterozygous for the novel variant c.61C>T (p.Arg21Ter) that determines non-functional protein without any residual activity. The patient was also heterozygous for the DPYD*2A variant. The two variants were probably located on different alleles, thus causing a complete DPD deficiency DPD and the rapid onset of the lethal toxicity. | She died 21 days after the first push of 5-FU. | [45] | |
| All exons + + 5' and 3' UTRs + promoter region by dHPLC. Data validation by sequencing (Sanger method) | 53-year-old female with LARC experiencing severe toxicity after day 1 of CT; on the third day the 5-FU was discontinued (case 3 in the study). | Neoadjuvant 5-FU + RT | Literature search | DPD enzyme activity measured in human PBM cells. | USA (Caucasian) | The patient was heterozygous for the novel c.545T>A (p.Met182Lys) variation in addition to the rare c.2329 G>T (p.Ala777Ser) and the more frequent DPYD*5 (c.1627A>G, [p.Ile543Val]). Since DPYD*5 has not been shown to lead to any change in DPD enzyme activity, the partial DPD deficiency observed in the patient is due to c.545T>A, and/or c.2329G>T. | The patient died 1 week after the start of the 5-FU treatment. | [33] |
| cDNA sequencing; RFLP | 57-year-old female with breast cancer experiencing severe toxicity during 5-FU treatment | -- | Expression analysis in Escherichia coli; | DPD enzyme activity measured in human PBM cells; Determination of pyrimidines and their derivates. | Japan (Japanese, East Asian) | DPD activity is significantly decreased. The patient was heterozygous for 3 novel mutations (c.62G>A [p.Arg21Gln, DPYD*12], c.1003G>T [p.Val335Leu, DPYD*11], c.1156G>T [p.Glu386Ter]). Analysis of the family genome revealed that p.Arg21Gln and p.Glu386Ter are located on the same allele and p.Val335Leu on the other allele. The p.Val335Leu and p.Glu386Ter resulted in a significant loss of enzymatic activity and no activity, respectively. p.Arg21Gln showed no effect on the functionality of the enzyme. | -- | [37] |
Abbreviations: 5-FU, 5-fluorouracil; BV, bevacizumab; CAPE, capecitabine; CMF, cyclophosphamide + methotrexate + 5-FU; CRC, colorectal cancer; CT, chemotherapy; dHPLC, denaturing high-performance liquid chromatography; DPD (DPYD), dihydropyrimidine dehydrogenase; FA, folinic acid; FEC, 5-FU + epirubicin + cyclophosphamide; FOLFOX, 5-FU + OXA + leucovorin; GI, gastrointestinal; HSF Human Splicing Finder, IRI, irinotecan; LARC, locally advanced rectal cancer; LV, leucovorin; mFOLFOX, modified FOLFOX; MLPA, multiplex ligation-dependent probe amplification; MAF, minor allele frequency; NGS, next generation sequencing; NS, not specified; OXA, oxaliplatin; PBMCs, a peripheral blood mononuclear cell; PROVEAN, Protein Variation Effect Analyzer; Pts, patients; RFLP, Restriction fragment length polymorphism; RT, radiotherapy; SIFT, Scale-Invariant Feature Transform; SNP, single nucleotide polymorphism; U, uracil; UFT, Tegafur/uracil; UH2, dihydrouracil; XELOX, capecitabine + oxaliplatin.
Characteristics of studies investigating a strategy based on DPYD gene sequencing to improve the prediction of fluoropyrimidine-related toxicity risk.
| Analytical method | Patients population | Gender | Therapy | SNP Functional prediction | DPD phenotyping | Country (Ethnicity) | Main findings | Ref |
|---|---|---|---|---|---|---|---|---|
| All exons + splice junctions + 5' and 3' UTRs + proximal promoter region sequencing (NGS). Data validation by Sanger sequencing. | 213 cancer pts (63.8%, colon; 19.2% rectum; 17%, other cancers) Negative for the DPYD*2A, *13, c.2846A>T, c.1236G>A-HapB3. Cases (n=109): grade≥3 toxicity Controls (n=104): no toxicity | F (51.2%); M (48.8%) | 5-FU: 85.5% CAPE: 14.5% Monotherapy (7.0%), Combined therapy (93.0%); Concomitant drug: OXA (38.5%); IRI (35.7%); others (18.8%). | In silico tools: APF, PredictSNP algorithm, LOFTEE, SpliceAI, MicroSNiPer, MirSNP database; Structural modeling | Determination of U, UH2 and UH2/U ratio | Italy (Caucasian) | Carriers of at least one rare missense DPYD variant (MAF<0.01) had an increased risk in the first cycle (OR:16.20; P=0.013) and during the entire course of CT (OR:11.06; P=0.025) of developing grade≥3 toxicity. | [21] |
| Coding exons + flanking intron regions sequencing (NGS) | 301 cancer pts (68.1%, CRC; 23.3% stomach; 17%, other cancers). Cases (n=55): grade3-4 toxicity Controls (n=246): grade0-2 toxicity | F (40.5%); M (59.5%) | 5-FU-based therapy Monotherapy (28.9%), Combined therapy (71.1%); | In silico tools: SIFT, Polyphen-2 | Japan (Japanese, East Asian) | Carriers of at least one rare DPYD variant (MAF<0.01) causing loss of function in silico had an increased risk of developing grade≥3 toxicity in the first two cycles (P=0.003). | [54] | |
| Exome sequencing (Sanger method) | 33 cancer pts (27.3%, breast; 72.7%, digestive tract cancer) Negative for DPYD*2A, *13, c.2846A>T, c.1236G>A-HapB3. Cases (n=11): grade≥3 toxicity within the first two cycles Controls (n=22): no grade≥3 | F (81.8%); M (18.2%) | CAPE (63.6%) 5-FU (36.4%) Monotherapy (42.4%), Combined therapy (57.6%) | Literature search; In silico tools: SpliceAI, RegSNPs-intron | Determination of U, UH2 and UH2/U ratio | Spain (NS) | The functionally-relevant rare rs367619008 (c.187A>G, [p.Lys63Glu]) and rs200643089 (c.2324T>G, [p.Leu775Trp]) variants, together with the more frequent rs76387818 (c.1084G>A, [p.Val362Ile]) variant, increased the percentage of explained toxicities from 20-30% (with the 4 recommended markers) to 38-48%. An intronic rare variant potentially pathogenic (rs944174134, c.322-63G>A) was also identified. | [52] |
| All exons + intron/exon boundaries sequencing (NGS) | 94 cancer pts (81.9%, colon; 18.1%, other cancers) | F (42.5%); M (57.5%) | na | In silico tools: HSF system, redictSNP algorithm | 5-FU degradation rate assay | Italy (NS) | Exon sequencing, with information also on rare variants (MAF<0.05), allowed to recognize an additional 22.5% of pts carrying variants possible cause of DPD deficiency (5-FU degradation rate assay) compared to screening only the recommended variants (DPYD*2A, *13, HapB3 and c.2846A>T [p.Asp949Val]), which identify only 20% of DPD deficiencies. | [47] |
| All exome + flanking intronic regions + 3'/ 5'UTR sequencing (NGS) | 243 advanced breast cancer pts | F (100.0%) | CAPE Monotherapy (88.5%), Combined therapy (11.5%) | In silico tools: UMD-Predictor system, HSF | Determination of U, UH2 and UH2/U ratio | France (NS) | The inclusion of seven rare (MAF<1%) in vitro deleterious DPYD alleles, all mutually exclusive (*2A, *13, c.2846A>T [p.Asp949Val], c.1475C>T [p.Ser492Leu], c.1774C>T [p.Arg592Trp], c.1025A>G [p.Asp342Gly], c.300C>A [p.Phe100Leu]) improved the performance of the genotyping test compared to the inclusion of only the 3 consensual variants (*2A, *13, c.2846A>T) for both grade 3-4 (sensitivity 26.7%, PPV 72.7%, RR 7.6, P<0.001) and grade 4 toxicities (sensitivity 60%, PPV 27.3%, RR 31.4, P=0.001). | [48] |
| Whole sequencing of the coding exon and flanking intron regions (Sanger method) | 41 cancer pts (17.1%, stomach; 17.1% rectum; 51.2% colon; 4.9%, breast; 9.7%, pancreas) Cases (n=27): grade≥ 3 toxicity within the first three cycles. Controls (n=14): grade ≤ 1 toxicity during at least eight treatment cycles. Negative for DPYD*2A, *13, c.2846A>T, c.1236G>A-HapB3. | F (51.2%); M (48.8%) | CAPE (61.0%) 5-FU (39.0%) Concomitant drug: OXA (n=31); IRI (n=4); targeted agents (n=10) | In silico tools: SIFT, Polyphen2 and ClinVar; mRNA expression levels analysis; RNA (cDNA) sequencing; Structural modeling; Literature Search | Determination of U concentration | Spain (NS) | A novel rare nonsense functionally relevant variant (c.2197insA [p.Thr733AsnfsTer14]) was identified and most likely associated with DPD deficiency and early severe toxicity. In the case group, a 3'UTR novel variant (c*159A>G) with possible functional impact was also detected and was a good candidate to explain the observed toxicity. Two other very rare variants (c.1218G>A [p.Met406Ile], c.2071G>T [p.Val691Leu]) were found in case group but both with controversial functional results. | [53] |
| Coding regions sequencing (NGS) KASPar technology for genotyping all study population. | 968 cancer pts (89.0%, colon; 11.0% rectum) 'HiTox' (n=100): early-onset grade 3-4 'LoTox'(n=100): no toxicity | F (43.0%); M (57.0%) | CAPE ± BV | In silico tools: SIFT, Polyphen, PhyloP, MutationTaster; Literature search | United Kingdom (Caucasian) | HiTox and LoTox groups were sequenced and a rare DPYD missense variant (c.1651G>A; [p.Ala551Thr]) was identified in the Hitox group. This variant was found in only one of the 968 pts who experienced grade 4 hematological toxicity. The variant was predicted to be strongly damaging and was reported to be causal for toxicity. | [50] | |
| All exons + intronic neighborhood regions sequencing (Sanger method) | 28 cancer pts (57.1%, colon; 17.9% rectum; 21.4% breast; 3.6% stomach) developing grade≥3 within the first three cycles. Negative for the DPYD*2A, *13, c.2846A>T, c.1236G>A-HapB3 | F (57.1%); M (42.9%) | 5-FU: 35.7% CAPE: 64.3% Monotherapy (42.9%), Combined therapy (57.1%); Concomitant drug: OXA (39.3%); IRI (10.7%); others (14.3%). | In silico tools: SIFT, PolyPhen-2, DPYD-verifier, HSF system; Structural modeling | Determination of U, UH2 and UH2/U ratio | Spain (NS) | Description and functionally characterization of the common and rare variants, including two very rare mutations (c.2087G>A, [p.Arg696His] and the functionally-relevant c.2324T>G, [p.Leu775Trp]). 25 (25/28, 90%) pts had a least 1 variant in DPYD coding sequence, and about half of them were potentially deleterious. | [49] |
| Coding regions sequencing (Sanger method) | 15 cancer pts with DPD deficiency and developing severe toxicity within the first or second cycle. Pts were tested for the DPYD*2A, *13, c.2846A>T, c.464T>A. | 5-FU or CAPE | In silico tools: UMD-Predictor; Literature search | Determination of U, UH2 and UH2/U ratio | France (Caucasian) | In addition to the three tested variants, some rare/ novel deleterious DPYD missense variants (c.257C>T [p.Pro86Leu], c.623G>A [p.Arg208Gln], c.1027A>C [p.Thr343Pro]), not included in the genotyping screening method, were also detected. | [43] | |
| All exons + exon/intron boundaries sequencing (Sanger method) dHPLC and MALDI-TOF-based assays for genotyping all study population. | 683 pts with GI or breast cancer. 573 pts developed grade 0-2 toxicity; 110 pts developed grade 3-4 toxicity)**. Pts were tested for the DPYD*2A **All 28 pts with grade 4 and 28 pts with grade 3 toxicity as well as 28 control patients (grade 0-2) were sequenced. Genetic variants apparently associated with grade 3-4 toxicity were then genotyped in a larger population. | F (43.9%); M (56.1%) | 5-FU ± FA or levamisole | In silico tools: PolyPhen | Germany (NS) | In addition to DPYD*2A, a total of 12 further exonic mutations were identified including four novel variants (c.623G>A [Arg208Gln], c.775A>G[p.Lys259Glu], c.1391T>C [p.Val464Ala], c.2858G>C [p.Cys953Ser]). The novel functionally-relevant variant c.2858G>C was identified in one patient with grade 4 mucositis and was not detected in any other patient. | [51] |
Abbreviations: 5-FU, 5-fluorouracil; APF, ADME-optimized Prediction Framework; BV, bevacizumab; CAPE, capecitabine; CRC, colorectal cancer; CT, chemotherapy; dHPLC, denaturing high-performance liquid chromatography; DPD (DPYD), dihydropyrimidine dehydrogenase; HSF, Human Splicing Finder, IRI, irinotecan; LOFTEE, Loss-Of-Function Transcript Effect Estimator; MAF, minor allele frequency; NGS, next generation sequencing; NS, not specified; OXA, oxaliplatin; PPV, positive predictive value; Pts, patients; RR, relative risk; SIFT, Scale-Invariant Feature Transform; SNP, single nucleotide polymorphism; U, uracil; UH2, dihydrouracil; UTR, untranslated region.
Since the present work is a review of qualitative studies, the risk of bias quality assessment is not applicable.
DPYD Genetic Complexity and the Role of Rare Genetic Variants
Compared to conventional targeted genotyping strategies, recently developed high-throughput next-generation sequencing (NGS) technologies have provided a more complete picture of the variability of candidate genes by elucidating the full spectrum of variants, including rare ones (MAF < 1%). The Exome Aggregation Consortium has reviewed data from international population-scale sequencing programs (e.g., 1000 Genomes Program), revealing that most human germline variants are rare or novel [18] and that the probability of a variant being deleterious is inversely related to its frequency [19]. Therefore, focusing on rare variants could increase the likelihood of finding functionally deleterious and impactful ones.
A growing body of recently published data shows that rare and novel variants may also have significant clinical value for precision medicine [20-24]. In particular, a series of studies investigated the genetic variability of clinically relevant genes (i.e., transporters, phase I and II enzymes, and nuclear receptors), and demonstrated that approximatively 30-40% of the overall functional variability of these genes is caused by rare variants that are not commonly captured by targeted genotyping approaches [25-27]. These same studies also revealed that rare pharmacogenetic variants are highly enriched in mutations predicted to be functionally relevant, and that these rare variants are responsible for much of the unexplained inter-individual variability in drug metabolism phenotypes, pharmacokinetics, and risk of adverse drug reactions.
A recent study compared sequencing-based and targeted genotyping-based approaches and demonstrated that, among pharmacogenes, DPYD had the largest number of unique potentially clinically relevant variants that were missed by standard targeted genotyping [28]. The sequencing design adopted in this analysis captures the genetic variability of all exons, splice junctions, upstream and downstream regulatory regions for each gene as well as also includes probes corresponding to additional sites present for several commercial pharmacogenetic microarray platforms. The study found that approximately 6% of the patient cohort (n = 631/10,030) carried a DPYD variant that was missed by the in silico targeted genotyping panel, and over 100 additional DPYD variants were identified by sequencing with respect to the targeted-approach, each found in only one or a few patients [28]. The further analysis of Zhou et al., revealed that, unlike other pharmacogenes (e.g., TPMT), a very high number of variants must be interrogated for DPYD gene in order to explain the functional variability based on in vitro and in vivo data [15]. For instance, 421 DPYD variants need to be interrogated to explain 99% of the DPD deficiency [15]. Notably, the four routinely tested DPYD variants identified only a minimal percentage of the genetically determined DPD deficiency, and approximately 17% of FL-related severe toxicity. These findings have important implications for genotype-guided prescribing, demonstrating that comprehensive sequencing of the DPYD gene could improve the identification of genetic variants that cause FL toxicity.
Case Reports and Case Series
Our search identified a number of published case reports and case series [29-46] that examined the potential genetic causes of FL-related extreme adverse reactions, including death. Most of these investigations applied a retrospective direct sequencing approach to identify previously uninvestigated variants in the DPYD gene that could be responsible for the observed severe toxic reactions (Table 1). In most studies, the sequencing included all exons and in some cases the adjacent intronic region as well, while a broader design and sequencing of the whole gene was only performed in a few studies. The papers described single or a few cases from clinical practice, mainly involving patients with gastrointestinal or breast cancer, who received FLs (i.e., 5-FU or capecitabine) alone or in combination with other drugs, and who experienced early-onset grade ≥3 or even lethal toxicity. Five out of twelve case reports described lethal toxicity cases. Comprehensive DPYD sequencing was often performed because the patients were found to have a constitutive deficiency in DPD activity that was not related to, or not completely explained by, the four routinely tested DPYD variants. In most cases, the analysis revealed some genetic variants in DPYD that the authors classified as “rare” or “novel” variants, and which were hypothesized to explain the observed adverse effects.
Defining the functional impact of the detected DPYD variants is a critical step towards determining whether they caused an observed toxic side effect. In most of the cases reviewed herein, the identified variants were classified as “functionally relevant” based on different approaches, including in silico tools, in vitro and molecular analyses, crystal DPD structure analysis, and literature search. Interestingly, in one case, the identification of a rare DPYD variation in one patient helped to personalize the FL-based therapy for another family member diagnosed with cancer, thus preventing potential severe toxic effects [29, 30]. In another case, the extreme toxic event described did not appear to be related to a single variant, but rather to a rare haplotype combination of more frequent DPYD variants: c.85T>C + c.496A>G + c.1601G>A + c.1627A>G or c.496A>G + c.1601G>A [35]. Table 1 presents the outcomes of the toxic events, either resulting in treatment interruption or dose adjustment.
Case-control Clinical Studies
Several single case reports or small case series identified previously uninvestigated DPYD variants as potential causative markers of extreme FL-related toxicity, thereby encouraging further investigation on a larger scale. In this context, a number of case control studies were conducted to explore the possibility of performing comprehensive genetic analysis of DPYD to highlight “rare” or “novel” variants as risk factors for severe toxicity after FL-based treatment [21, 43, 47-54] (Table 2). Four studies [43, 49, 51, 53] reported the functional characterization of several DPYD variants—including a number of rare and novel variants—that were identified by sequencing all exons and flanking intronic regions of DPYD gene using the Sanger method in a group of patients with DPD deficiency, who developed early-onset severe toxicity. One of these studies [49] also reported that 90% of the screened patients (25/28) had at least one variant in the DPYD coding sequence, about half of which were potentially deleterious based on functional prediction performed using multiple in silico tools and protein structure modeling. In another investigation [50], the coding DPYD region was sequenced in 968 cancer patients using the NGS method. It was found that one of the patients had a very rare DPYD missense variant (c.G1651G>A; [p.Ala551Thr]), which was predicted to be strongly deleterious by in silico tools and literature review, and was considered to have caused the observed grade 4 hematological toxicity episode.
Three more recent studies [47, 48, 52] have attempted to quantify the increased percentage of toxicity cases that are explained by full DPYD exon sequencing, compared to the genotyping of the recommended four-variant panel. In a study of 94 cancer patients, De Luca et al., [47] showed that the detection of rare DPYD variants (MAF < 0.05) by exon sequencing resulted in a 2.5% improvement in the identification of patients with DPD deficiency, compared to screening only the four recommended DPYD variants, which identified 20% of DPD-deficient patients. In a smaller investigation of 33 cancer patients, Soria-Chacartegui et al., [52] applied DPYD coding region sequencing, and discovered the functionally relevant rare variants rs367619008 (c.187A>G, [p.Lys63Glu]) and rs200643089 (c.2324T>G, [p.Leu775Trp]), together with the more frequent variant rs76387818 (c.1084G>A, [p.Val362Ile]). The functional prediction was performed using in silico tools and literature review. These authors highlighted a larger increase in the identification of patients with toxicity—from 20-30% when using only the four recommended markers, to 38-48% with the additional identified variants. In another study of 243 advanced breast cancer patients receiving capecitabine, Etienne-Grimaldi et al., [48] investigated the value of integrating the three routinely tested variants according to the French pharmacogenetic guidelines (*2A, *13, and c.2846A>T) with an additional four rare variants having an in vitro verified deleterious effect (c.1475C>T, c.1774C>T, c.1025A>G, and c.300C>A) detected by direct sequencing of all exons plus flanking intronic part and the 3'/5' untranslated region (UTR) of DPYD gene. This approach improves the performance of genotyping for identifying patients developing grade 3-4 toxicities (sensitivity: 26.7%; PPV: 72.7%; RR: 7.6; P < 0.001) or only grade 4 toxicities (sensitivity: 60%; PPV: 27.3%; RR: 31.4; P = 0.001).
In two more recent case-control studies [21, 54], researchers have tried to statistically estimate the power of rare and novel variants to predict FL-related toxicity risk. In a study of about 200 Caucasian cancer patients treated with FL-based therapy, De Mattia et al., [21] demonstrated that the burden of rare and novel DPYD variants is significantly higher among patients with toxicity, compared to patients without toxicity by sequencing all exons, splice junctions, 3'/5' UTR and proximal promoter region of the DPYD gene. Specifically, among carriers of at least one rare missense DPYD variant, the risk of developing grade 3 toxicity or greater was 16-fold higher during the first treatment cycle (P = 0.013), and 11-fold higher throughout the course of chemotherapy (P = 0.025). Yokoi et al., [54] obtained similar results in a study including 301 Japanese cancer patients who received 5-FU alone or in combination with other drugs and sequencing the coding exons and flanking intron regions of the DPYD gene. Their analysis revealed that patients carrying at least one rare DPYD variant (MAF < 0.01) related to an in silico loss of function exhibited an increased risk of developing grade 3 toxicity or greater during the first two treatment cycles (P = 0.003).
Analysis of the Identified DPYD Rare and Novel Variants
Table 3 presents detailed descriptions of each rare and novel DPYD variant that was highlighted by the herein reviewed case reports, case series, and clinical studies, and that was potentially associated with severe or lethal FL-related toxicity. Notably, the listed classification of these variants as “rare”, “very rare”, or “novel” (i.e., absent from available public database) are as provided by the authors of papers, and based on the available information at the time of paper publication. These classifications have been revised based on the data currently available in public databases, such as dbsnp [17] or Ensembl [16]. A total of 46 germline aberrations were identified in the DPYD gene, of which, 24/46 (52.2%) were classified by the authors as novel, and 21/46 (45.7%) as rare or very rare; one variant was not classified. Of the 24 novel variants, 10 were assigned an rs ID after study publication. Most of the identified variants were missense (32/46, 69.6%). The remaining variants included stop-gain variants (5/46, 10.9%), a deletion (1/46, 2.2%), an intronic variant (1/46, 2.2%), splice site variants (5/46, 10.9%), a variant within the 3' region (1/46, 2.2%) and a exons amplification (1/46, 2.2%).
List of the rare and novel DPYD variants potentially associated with fluoropyrimidine-related toxicity that have been reported in published case reports, case series and clinical studies selected for this review.
| DPYD variant§ | Typology | Rs ID | Classification by authors# | Variant Allele Frequency& | Ref | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| All | African (AFR) | American (AMR) | East Asian (EAS) | European (EUR/NFE) | South Asian (SAS) | ||||||
| NM_000110.4:c.61C>T | NP_000101.2:p.Arg21Ter | Stop-gained | rs72549310* | Novel | <0.001 | <0.001 | 0 | 0 | <0.001 | 0 | [45] |
| NM_000110.4:c.62G>A | NP_000101.2:p.Arg21Gln, DPYD*12 | Missense | rs80081766* | Novel | <0.001 | 0 | 0 | 0 | <0.001 | <0.001 | [37] |
| NM_000110.3:c.168_175dupGAATAATT | NP_000101.2:p.Phe59Ter | Stop-gained | NA | Novel | [43] | ||||||
| NM_000110.4:c.187A>G | NP_000101.2:p.Lys63Glu | Missense | rs367619008 | Rare | <0.001 | 0 | 0 | <0.001 | <0.001 | 0 | [52] |
| c.2197insA (sequence aligned with NC_000001.11 GRCh38.p13) | p.Thr733AsnfsTer14 | Stop-gained | NA | Novel | [53] | ||||||
| NM_000110.4:c.257C>T | NP_000101.2:p.Pro86Leu | Missense | rs568132506 | Rare/Very Rare | <0.001 | 0 | <0.001 | 0 | <0.001 | <0.001 | [43] [29] [32] |
| NM_000110.4 c.300C>A | NP_000101.2:p.Phe100Leu | Missense | NA | Rare | [48] | ||||||
| NM_000110.3 c.321+2T>C | Splice-site | NA | Novel | [44] | |||||||
| NM_000110.4:c.321+1G>A | NP_000101.2:p.Cys79Thrfs*8 | Splice-site | rs746368304 | Rare | <0.001 | 0 | 0 | 0 | <0.001 | 0 | [46] |
| NM_000110.4:c.464T>A | NP_000101.2:p.Leu155Ter | Stop-gained | rs2101026231* | Novel | na | na | na | na | na | na | [40] |
| NM_000110.4:c.545T>A | NP_000101.2:p.Met182Lys | Missense | rs779728902* | Novel | <0.001 | 0 | <0.001 | 0 | <0.001 | 0 | [33] |
| NM_000110.4:c.623G>A | NP_000101.2:c.Arg208Gln | Missense | rs376073289* | Novel/Rare | <0.001 | 0 | 0 | <0.001 | <0.001 | 0 | [43] [51] |
| c.680G>A | Ser -> Gly | Missense | NA | Novel | [31] | ||||||
| NM_000110.4:c.704G>A | NP_000101.2:p.Arg235Gln | Missense | rs755416212 | Very Rare | <0.001 | 0 | 0 | 0 | <0.001 | 0 | [39] |
| NM_000110.4:c.775A>G | NP_000101.2:p.Lys259Glu | Missense | rs45589337* | Novel | 0.006 | 0.001 | 0.003 | 0 | 0.008 | 0.005 | [35] [51] |
| NM_000110.4:c.851G>T | NP_000101.2:p.Gly284Val | Missense | rs777220476 | Rare | <0.001 | 0 | <0.001 | 0 | <0.001 | 0 | [46] |
| NM_000110.4:c.1003G>T | NP_000101.2:p.Val335Leu, DPYD*11 | Missense | rs72549306* | Novel | <0.001 | <0.001 | <0.001 | <0.001 | 0 | 0 | [37] |
| NM_000110.4:c.1025A>G | NP_000101.2:p.Asp342Gly | Missense | rs769709846 | Rare | <0.001 | 0 | 0 | 0 | <0.001 | 0 | [48] |
| NM_000110.3:c.1027A>C | NP_000101.2:p.Thr343Pro | Missense | NA | Novel | [43] | ||||||
| NM_000110.4:c.1156G>T | NP_000101.2:p.Glu386Ter | Stop-gained | rs78060119* | Novel | <0.001 | 0 | 0 | 0 | <0.001 | <0.001 | [37] |
| NM_000110.4:c.1218G>A | NP_000101.2:p.Met406Ile | Missense | rs61622928 | Very rare | 0.005 | 0.065 | 0.004 | 0 | <0.001 | <0.001 | [53] |
| NM_000110.4:c.1280T>C | NP_000101.2:p.Val427Ala | Missense | rs200693895 | Rare | <0.001 | 0 | 0 | 0 | <0.001 | <0.001 | [46] |
| NM_000110.4:c.1391T>C | NP_000101.2:p.Val464Ala | Missense | rs370707404* | Novel | <0.001 | <0.001 | 0 | 0 | <0.001 | 0 | [51] |
| NM_000110.4:c.1475C>T | NP_000101.2:p.Ser492Leu | Missense | rs72549304 | Rare | <0.001 | <0.001 | 0 | 0 | <0.001 | <0.001 | [48] |
| NM_000110.4:c.1601G>A | NP_000101.2:p.Ser534Asn, DPYD*4 | Missense | rs1801158 | Rare | 0.015 | 0.004 | 0.011 | <0.001 | 0.020 | 0.009 | [29] [38] [35] |
| NM_000110.4:c.1651G>A | NP_000101.2:p.Ala551Thr | Missense | rs777425216 | Rare | <0.001 | <0.001 | 0 | 0 | <0.001 | 0 | [50] |
| NM_000110.3:c.1740+2T>C | NP_000101.2:p.Ser509_Lys580del | Splice-site | NA | Novel | [46] | ||||||
| NM_000110.4:c.1774C>T | NP_000101.2:p.Arg592Trp | Missense | rs59086055 | Rare | <0.001 | <0.001 | 0 | 0.002 | <0.001 | <0.001 | [48] |
| NM_000110.4:c.1796T>C | NP_000101.2:p.Met599Thr | Missense | rs147601618 | Rare | <0.001 | <0.001 | 0 | 0 | <0.001 | 0 | [41] |
| c.1801G>C | Gly-> Ser | Missense | NA | Novel | [31] | ||||||
| NM_020442.6:c.1850C>T | NP_065175.4:p.Thr617Met | Missense | rs367837827* | Novel | <0.001 | 0 | 0 | 0.002 | <0.001 | <0.001 | [32] |
| c.1903A>G (RefSeq: NM_000110, Transcript ID: ENST00000370192.7) | p.Asn635Asp | Missense | NA | Novel | [42] | ||||||
| NM_000110.4:c.2071G>T | NP_000101.2:p.Val691Leu | Missense | rs202212118 | Very rare | <0.001 | 0 | <0.001 | 0 | <0.001 | 0 | [53] |
| NM_000289.6:c.2087G>A | NP_000280.1:p.Arg696His | Missense | rs41291971 | Very rare | 0.010 | 0.003 | 0.006 | 0.002 | 0.016 | 0.002 | [49] |
| NM_000110.4:c.2242+1G>T | Splice-site | NA | Novel | [34] | |||||||
| NM_000110.4:c.2324T>G | NP_000101.2:p.Leu775Trp | Missense | rs200643089 | Rare/Very rare | <0.001 | 0 | <0.001 | 0 | <0.001 | 0 | [52] [49] |
| NM_000110.4:c.2329G>T | NP_000101.2:p.Ala777Ser | Missense | rs672601276 | Rare | <0.001 | 0 | <0.001 | 0 | <0.001 | 0 | [33] |
| NM_000110.3c.2407_2427del | NP_000101.2:p.Leu803_Gly809del | Deletion | NA | Novel | [46] | ||||||
| NM_000110.4:c.2434G>A | NP_000101.2:p.Val812lle | Missense | rs371313778 | Rare | <0.001 | 0.001 | <0.001 | <0.001 | <0.001 | <0.001 | [30] |
| c.2509-2510insC | Leu -> Pro | Missense | NA | Novel | [31] | ||||||
| NM_000110.4:c.2766+87G>A | Intronic | rs556768807 | Rare | <0.001 | 0 | 0 | 0 | <0.001 | 0 | [46] | |
| NM_000110.4:c.2843T>C | NP_000101.2:p.Ile948Thr | Missense | NA | Novel | [46] | ||||||
| c.2858G>C (NC_000001.8 was used as reference sequence) | Cys953Ser | Missense | NA | Novel | [51] | ||||||
| IVS14+17A>G | Splice-site | NA | NA | [41] | |||||||
| c*159A>G (sequence aligned with NC_000001.11 GRCh38.p13) | 3'UTR | NA | Novel | [53] | |||||||
| Exons 17 and 18 (Ref Seq NM_000110.3; Ensembl ENST00000370192) | amplification | Novel | [36] | ||||||||
Abbreviations: NA, not available; UTR, untranslated region
§ When the information about a transcript was not available, the variation change was given as reported by the authors in the original paper.
&Frequency data obtained by Ensembl [51] from gnomADe database or, when the data are not available, from 1000 Genomes European population database.
# The classification as “rare”, “very rare”, or “novel” (i.e., absent from available public database) provided according to the authors of the studies, and based on the information available at the time of the original paper publication.
*The variant was classified as novel at the time of publication of the original paper; an rs ID was assigned only afterwards.
Interestingly, five missense variations (c.257C>T [p.Pro86Leu], c.623G>A [p.Arg208Gln], c.775A>G [p.Lys259Glu], c.1601G>A [p.Ser534Asn, DPYD*4], and c.2324T>G [p.Leu775Trp]) were highlighted by more than one study, supporting their potential functional effects and roles in predicting the risk of severe toxicity, which warrants further investigation. One of these variants, c.1601G>A, has already been extensively investigated and is most common in the European population with respect to other ethnic groups [55]. Notably, this variant has been linked to altered DPD activity and FL-associated toxicity, but the available evidence regarding its clinical validity has not yet been confirmed. A meta-analysis published in 2015 did not confirm a significant association between the c.1601G>A variant and severe FL-related toxicity; however, all included studies reported that variant carriers had a relative risk of toxicity over 1.0, suggesting some effect on toxicity [56]. The most recently published papers have presented controversial results regarding the potential utility of implementing c.1601G>A as a marker in clinical practice, thus leaving an open question [55, 57, 58].
Approaches for Functional Prediction of DPYD Genetic Variants
One challenge of using sequence-level data in the context of clinical implementation is the need to reliably and rapidly classify the identified novel and rare DPYD variants based on their functional impact on enzyme activity. Several strategies have been utilized in the above-discussed studies (Table 1), including in silico tool prediction, literature search, analysis of crystal DPD structure, and various kinds of molecular analyses (e.g., cDNA sequencing and in vitro functional assay). Although experimental assays in recombinant cell lines represent the gold standard for functional evaluation of pharmacogenetic variants, these methods are low-throughput, costly, time-consuming, and require trained laboratory staff, which render them unsuitable for point-of-care use [59, 60]. Recently developed high-throughput experimental functional assays—such as deep mutational scanning to analyze protein-coding mutations—are not yet suitable for prompt functional prediction [60]. Moreover, epidemiological analyses require adequately large sample sizes to achieve statistically significant results, which is not easily achievable for rare variants [59, 60].
Computational predictions appear to be the most suitable method for timely assessment of the phenotypic effects of rare and novel variants. A multitude of algorithms are available to evaluate the deleterious impact of a given variant based upon its sequence conservation, structural data, physicochemical properties of amino acid substitutions, and functional genomics information [61, 62]. However, there exists a need for algorithms specifically designed for evaluating pharmacogenetic variants, since commonly used algorithms generally underperform when assessing variants of pharmacogenetic relevance [61, 62].
Machine-learning based approaches have enabled advances in this field. Specifically, a DPYD-specific variant classifier (DPYD-Varifier) has been trained on 156 missense DPYD variants with matched DPD activity in vitro data [63], achieving 85% prediction accuracy on a set of novel missense variants, hence outperforming other widely used in silico prediction tools, such as PROVEAN, SIFT, and PolyPhen-2. Zhou et al., recently developed a quantitative ensemble classifier—the ADME-optimized prediction framework (APF) algorithm—which is intended to assign the deleteriousness of missense variants in ADME-related pharmacogenes, including DPYD. This novel tool was trained exclusively on experimentally characterized pharmacogenetic variants. Compared to in vitro tests, it reportedly achieved a sensitivity and specificity of 93% for predicting loss-of-function and functionally neutral pharmacogenomic variants, outperforming conventional in silico tools, such as SIFT, Polyphen-2, PROVEAN, and CADD [15, 64]. Notably, APF also performs very well on DPYD variants, even though no DPYD data were used for model training [59, 60]. Interestingly, APF and DPYD-Varifier have shown an overall good agreement for the prediction of population-specific frequencies of DPD metabolizer phenotypes [15]. However, additional efforts are required to further improve these novel in silico algorithms, and to overcome their current limits. Significant advances may derive from the use of large-scale training data sets, e.g., large-scale experimental mutagenesis screening, increased quantities of systematically collected large-scale functional evidence, and the possibility of testing on population-scale genomic biobanks with correlated electronic medical records. The functional classification of missense variants could also potentially be improved by the recently developed artificial intelligence-based structural prediction tools (e.g., AlphaMissense) [65], which open promising new opportunities. Despite progress in the functional classification of missense variants, the available in silico prediction tools for pharmacogenes remain to underperform for non-missense variants, e.g., for synonymous, nonsense, frameshift, splice, and non-coding variants [61, 62]. Although efforts are being made to improve these tools, much work needs to be conducted in order to enable their implementation in a clinical context.
Challenges for Clinical Implementation of an NGS Approach for FL Precision Dosing
Considering the potential value of rare and novel DPYD variants for predicting FL-related toxicity risk, one major opportunity for the near future will be the translation of that information into clinical practice, to enable personalized therapy (Figure 2). However, some critical issues persist, and represent a challenge for pharmacogenetic research. Notably, the currently available data, although promising, are mainly derived from case reports or case-control studies with limited sample sizes, and remain to be validated in sufficiently powered prospective clinical studies (Tables 1 and 2). This will certainly require envisioning new models of prospective clinical pharmacogenetic trials, with the aim of pin-pointing the impact of a genetic variant based on only its minor allele frequency and its predicted functional effect. Clinical translation will also require concentrated efforts by scientific pharmacogenomic consortia to translate the evidence into clinical guidelines, with dose adjustment recommendations for carriers of rare and novel DPYD variants. In this context, there exists a need to develop reliable bioinformatic tools that enable quantitative functional prediction of rare and novel uncharacterized DPYD variants, as discussed in the previous section.
Roadmap for the clinical implementation of DPYD sequencing for optimization of fluoropyrimidine dose adjustment.
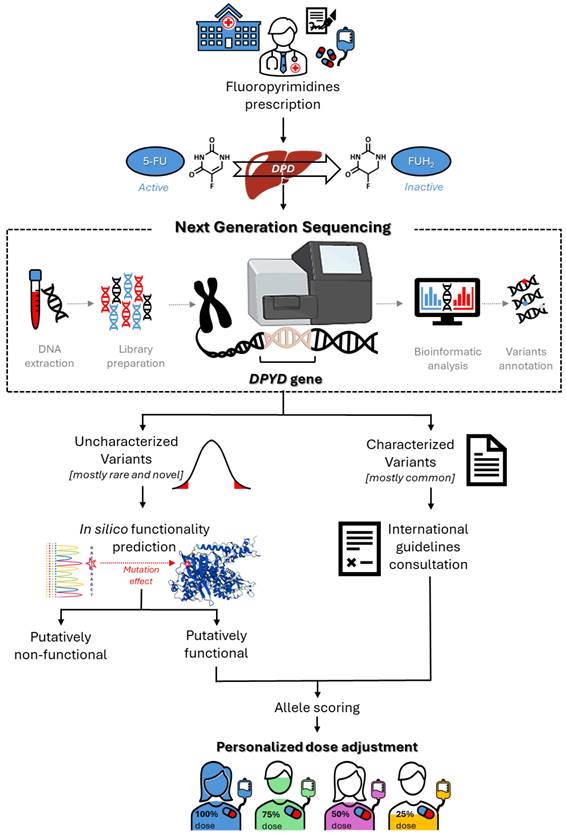
The implementation of rare DPYD variants as predictive biomarkers in clinical practice will also require formal assessment of the cost-effectiveness of sequencing-based analytical approaches, compared to targeted genetic analysis. It would be also worth considering the cost-effectiveness of the required infrastructural investments (e.g., laboratories with state-of-the-art equipment, and expertise) for timely and reliable genotype analyses. However, considering the increasingly widespread use of NGS technology, with associated lowering of costs and increased presence of equipment in laboratories, the adoption of DPYD-sequencing as routine clinical practice seems to be a realistic scenario. Regarding the turnaround time, NGS-based testing workflows (i.e., DNA extraction, library preparation, sequencing, data analysis, and reporting) typically require a few weeks [66], which limits their use for preemptive guidance of personalized prescribing in acute cases. However, a recent study reported that this turnaround time could be reduced to three business days by using a novel automated NGS assay, making the clinical application of NGS methods more realistic in the near future [67]. Notably, smaller laboratories may require a longer time due to the need to obtain a minimum number of samples to perform NGS analysis. An additional shortcoming that must be addressed before NGS-based tests are introduced into clinical practice is the need for a more comprehensive analysis of analytical performance—at least including the performance characteristics recommended by international agencies (e.g., least accuracy, precision, limit of detection, specificity)—and the availability of regulatory guides for standardizing reports on the analytical validation of NGS approaches [68].
Discussion
Overall, the published data reveal that standard screening of routinely tested DPYD variants is sometimes insufficient to prevent severe or fatal toxicity of FLs. Evidence suggests that more comprehensive DPYD sequencing approaches may potentially be useful for detecting rare and novel risk genetic variants, identifying a greater proportion of patients with DPD deficiency, who are likely at risk of developing adverse events related to FL treatment.
The preliminary published reports highlight the advantage that a sequencing-based approach provides a complete picture of the variability of the gene of interest (i.e., DPYD), thereby reducing bias attributable to test design, compared to a targeted genotyping-based test. It should be also noted that a sequencing-based approach allows for future re-analysis of the data using the most recent pharmacogenetic information that was not available at the time of the initial test. The published clinical cases also suggest that extensive DPYD sequencing could help to overcome the ethnic-specific distribution of DPYD variants described by some studies [15, 39, 46]. Notably, testing by targeted genotyping may miss rare but potentially clinically relevant variants, and this is particularly evident among populations that have been historically underrepresented in research and genetic sequencing cohorts. For example, of the four DPYD variants that are validated for pre-treatment dose adjustment in patients of European descent, none has yet been identified in an Eastern Asian population [54, 69]. Moreover, these four DPYD genetic markers present a low frequency among patients of African ancestry, where other variants may play an important role [70, 71]. Extensive DPYD sequencing could be beneficial for detecting potentially relevant variants in different ethnic contexts, leading to both improved and more inclusive patient care [15, 30, 39, 46].
It should be highlighted that in addition to the DPYD variants identified in published studies that may be associated with FL-related toxicity (Table 3), other genetic DPYD alterations and rearrangements detected by gene sequencing have been described and reported as the cause of DPD deficiency and related pathologic syndromes [72-75]. These molecular DPYD variations should be further investigated for their potential clinical role in predicting the risk of developing severe toxicity after FLs administration. The most promising among them is a DPYD exon 4 deletion detected by gene screening in the Finnish population and associated with DPD deficiency and preliminary FL-related toxicity, emphasizing that the potential value of DPYD copy number variation is currently underestimated [73, 76]. It is presently unclear how variants' rearrangement in the haplotype structure might impact the phenotypic effect on DPD activity. Recent data demonstrate how variants with controversial phenotypic effects present significant levels of linkage disequilibrium (LD), and could be combined in haplotypes, allowing the stratification of patients according to both DPD activity [77] and risk of FL-related adverse drug reaction [78]. In addition, advances in the NGS technology could be helpful in the definition of haplotype structures. Although previous research indicated that the two variants linked to the HapB3 haplotype, c.1129-5923C>G and c.1236G>A, are in perfect LD, a recent paper has highlighted rare cases of incomplete linkage within the three variants. This finding could raise a question as to the current DPYD clinical testing strategies that use c.1236G>A as a surrogate for the causative variant c.1129-5923C>G [79]. This adds an additional level of complexity that must be addressed by future studies.
Considering that the variability in DPD enzyme activity can only be partly explained by genetic variants, several DPD-phenotyping methods have been implemented in clinical practice to identify more patients with DPD deficiency [80]. In general, DPD phenotyping has shown higher sensitivity compared to DPYD genotyping, but prospective studies are still required to better define this aspect [80, 81]. The gold standard for DPD phenotyping is the determination of DPD enzyme activity in peripheral blood mononuclear cells. As alternatives, several methods have been developed based upon analyzing the concentrations of dihydrouracil (UH2) and uracil (U), and their ratio in a patient's plasma, as surrogate markers for intracellular DPD activity. However, these tests have limitations, including a lack of consensus regarding the threshold of U concentration or UH2/U ratio to determine DPD deficiency, the requirement for specific equipment that is not readily available at all hospitals, and the fact that the results are strongly influenced by the blood collection time (e.g., U is influenced by circadian rhythm and food) and sample processing (e.g., the sample must be processed immediately due to the limited stability of U and UH2). These issues complicate the implementation of this DPD-phenotyping method in clinical practice, reinforcing the need for further research efforts to improve the guiding of FL dosing according to DPYD genetic testing.
Given the complexity of the metabolic pathway of FLs—with DPD and several other enzymes and metabolic proteins involved in determining drug bioavailability and exposure—a polygenic approach based on rare and novel variants should also be considered. In our recent case-control study [82], we demonstrated that the burden of rare dihydropyrimidinase (DPYS) variants was significantly higher in patients with toxicity compared to controls, and that the presence of at least one rare DPYS variant was associated with an approximately four-fold higher risk of severe toxicity. These data demonstrated that the rare mutational burden of DPYS—a gene that cooperates closely with DPYD in the catabolic pathway of FLs—is another promising pharmacogenetic marker for precision dosing of FL, which may improve treatment personalization, especially in cancer patients with normal DPD activity. Overall, further pharmacogenetic analyses should be performed to investigate a combined algorithm based on the burden of rare variants in multiple FL-related genes.
Another point to consider is that, in addition to genetic factors, non-genetic factors could also influence the risk of developing FL-related toxicity and should be taken into account as covariates. For example, a recent meta-analysis [83] has shown that some clinical variables such as gender, age, body mass index, administration schedule of FL and associated chemotherapeutic agents have significant predictive value. In particular, an increased risk of toxicity was observed in women, which is consistent with the lower lymphocytic DPD enzyme activity and 5-FU clearance described in this gender. Aging, low body surface area, use of 5-FU bolus and concomitant administration of other anticancer drugs were also associated with higher FL-related toxicity. In conclusion, although targeted genotyping detects the most clinically significant DPYD variants, sequencing-based approaches also enable the detection of potentially deleterious rare and novel DPYD variants that collectively affect many patients, and could predispose the patient to severe FL-related toxicity. Moreover, the adoption of sequencing-based strategies could enable more accurate patient metabolizer phenotype classification in historically understudied populations, where targeted genotyping may miss clinically relevant variants. Once the current critical issues are overcome, NGS-based strategies may allow significant improvement of the pre-treatment identification of high-risk patients, thus facilitating adequate dose-adjustment, and potentially improving patients' quality of life and reducing medical costs.
Abbreviations
5-FU: 5-fluorouracil; APF: ADME-optimized prediction framework; DPD, DPYD: dihydropyrimidine dehydrogenase; DPYS: dihydropyrimidinase; FL: fluoropyrimidine; MAF: minor allele frequency; U: uracil; UH2: dihydrouracil.
Acknowledgements
Funding
This work was supported by the Italian Ministry of Health (Ricerca Corrente).
Author contributions
Elena De Mattia: Conceptualization, literature search, formal analysis, data curation, and writing - Original Draft; Noemi Milan: visualization; Yehuda G. Assaraf: writing - Review & Editing; Giuseppe Toffoli: resources and supervision; Erika Cecchin: conceptualization, literature search, formal analysis, and writing - Review & Editing.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Glimelius B, Stintzing S, Marshall J, Yoshino T, de Gramont A. Metastatic colorectal cancer: Advances in the folate-fluoropyrimidine chemotherapy backbone. Cancer Treat Rev. 2021;98:102218
2. Yang L, Yang J, Kleppe A, Danielsen HE, Kerr DJ. Personalizing adjuvant therapy for patients with colorectal cancer. Nat Rev Clin Oncol. 2024;21:67-79
3. Van Cutsem E, Twelves C, Cassidy J, Allman D, Bajetta E, Boyer M. et al. Oral capecitabine compared with intravenous fluorouracil plus leucovorin in patients with metastatic colorectal cancer: results of a large phase III study. J Clin Oncol. 2001;19:4097-106
4. Cecchin E, De Mattia E, Ecca F, Toffoli G. Host genetic profiling to increase drug safety in colorectal cancer from discovery to implementation. Drug Resist Updat. 2018;39:18-40
5. De Mattia E, Dreussi E, Cecchin E, Toffoli G. Pharmacogenetics of the nuclear hormone receptors: the missing link between environment and drug effects? Pharmacogenomics. 2013;14:2035-54
6. Scarabel L, Bignucolo A, Toffoli G, Cecchin E, De Mattia E. Pharmacogenetics Role of Genetic Variants in Immune-Related Factors: A Systematic Review Focusing on mCRC. Pharmaceutics. 2022;14:2468
7. Offer SM, Fossum CC, Wegner NJ, Stuflesser AJ, Butterfield GL, Diasio RB. Comparative functional analysis of DPYD variants of potential clinical relevance to dihydropyrimidine dehydrogenase activity. Cancer Res. 2014;74:2545-54
8. Toffoli G, Giodini L, Buonadonna A, Berretta M, De Paoli A, Scalone S. et al. Clinical validity of a DPYD-based pharmacogenetic test to predict severe toxicity to fluoropyrimidines. Int J Cancer. 2015;137:2971-80
9. Dalle Fratte C, Polesel J, Roncato R, De Mattia E, Ecca F, Bignucolo A. et al. DPYD Gene Activity Score Predicts Dose-Limiting Toxicity in Fluoropyrimidine-Treated Colorectal Cancer Patients. J Mol Clin Med. 2018;1:143-9
10. Amstutz U, Henricks LM, Offer SM, Barbarino J, Schellens JHM, Swen JJ. et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing: 2017 Update. Clin Pharmacol Ther. 2018;103:210-6
11. Lunenburg C, van der Wouden CH, Nijenhuis M, Crommentuijn-van Rhenen MH, de Boer-Veger NJ, Buunk AM. et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction of DPYD and fluoropyrimidines. Eur J Hum Genet. 2020;28:508-17
12. Offer SM, Wegner NJ, Fossum C, Wang K, Diasio RB. Phenotypic profiling of DPYD variations relevant to 5-fluorouracil sensitivity using real-time cellular analysis and in vitro measurement of enzyme activity. Cancer Res. 2013;73:1958-68
13. EMA. EMA recommendations on DPD testing prior to treatment with fluorouracil, capecitabine, tegafur and flucytosine. European Medicines Agency. Published April 30, 2020. Accessed March 25, 2024. https://www.ema.europa.eu/en/news/ema-recommendations-dpd-testing-prior-treatment-fluorouracil-capecitabine-tegafur-flucytosine
14. Innocenti F, Mills SC, Sanoff H, Ciccolini J, Lenz HJ, Milano G. All You Need to Know About DPYD Genetic Testing for Patients Treated with Fluorouracil and Capecitabine: A Practitioner-Friendly Guide. JCO Oncol Pract. 2020;16:793-8
15. Zhou Y, Dagli Hernandez C, Lauschke VM. Population-scale predictions of DPD and TPMT phenotypes using a quantitative pharmacogene-specific ensemble classifier. Br J Cancer. 2020;123:1782-9
16. Ensembl web site. Available from: https://www.ensembl.org/index.html
17. NCBI-dbSNP. Available from: https://www.ncbi.nlm.nih.gov/snp/
18. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285-91
19. Goldstein DB, Allen A, Keebler J, Margulies EH, Petrou S, Petrovski S. et al. Sequencing studies in human genetics: design and interpretation. Nat Rev Genet. 2013;14:460-70
20. Apellaniz-Ruiz M, Lee MY, Sanchez-Barroso L, Gutierrez-Gutierrez G, Calvo I, Garcia-Estevez L. et al. Whole-exome sequencing reveals defective CYP3A4 variants predictive of paclitaxel dose-limiting neuropathy. Clin Cancer Res. 2015;21:322-8
21. De Mattia E, Silvestri M, Polesel J, Ecca F, Mezzalira S, Scarabel L. et al. Rare genetic variant burden in DPYD predicts severe fluoropyrimidine-related toxicity risk. Biomed Pharmacother. 2022;154:113644
22. Gray B, Baruteau AE, Antolin AA, Pittman A, Sarganas G, Molokhia M. et al. Rare Variation in Drug Metabolism and Long QT Genes and the Genetic Susceptibility to Acquired Long QT Syndrome. Circ Genom Precis Med. 2022;15:e003391
23. Ramsey LB, Bruun GH, Yang W, Trevino LR, Vattathil S, Scheet P. et al. Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Res. 2012;22:1-8
24. Xiao Q, Zhou Y, Winter S, Buttner F, Schaeffeler E, Schwab M. et al. Germline variant burden in multidrug resistance transporters is a therapy-specific predictor of survival in breast cancer patients. Int J Cancer. 2020;146:2475-87
25. Ingelman-Sundberg M, Mkrtchian S, Zhou Y, Lauschke VM. Integrating rare genetic variants into pharmacogenetic drug response predictions. Hum Genomics. 2018;12:26
26. Kozyra M, Ingelman-Sundberg M, Lauschke VM. Rare genetic variants in cellular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Genet Med. 2017;19:20-9
27. Scharfe CPI, Tremmel R, Schwab M, Kohlbacher O, Marks DS. Genetic variation in human drug-related genes. Genome Med. 2017;9:117
28. Lopes JL, Harris K, Karow MB, Peterson SE, Kluge ML, Kotzer KE. et al. Targeted Genotyping in Clinical Pharmacogenomics: What Is Missing? J Mol Diagn. 2022;24:253-61
29. Bukhari N, Alshangiti A, Tashkandi E, Algarni M, Al-Shamsi HO, Al-Khallaf H. Fluoropyrimidine-Induced Severe Toxicities Associated with Rare DPYD Polymorphisms: Case Series from Saudi Arabia and a Review of the Literature. Clin Pract. 2021;11:467-71
30. Bukhari N, Azam F, Alfawaz M, Zahrani M. Identifying a Novel DPYD Polymorphism Associated with Severe Toxicity to 5-FU Chemotherapy in a Saudi Patient. Case Rep Genet. 2019;2019:5150725
31. Del Re M, Michelucci A, Di Leo A, Cantore M, Bordonaro R, Simi P. et al. Discovery of novel mutations in the dihydropyrimidine dehydrogenase gene associated with toxicity of fluoropyrimidines and viewpoint on preemptive pharmacogenetic screening in patients. EPMA J. 2015;6:17
32. Del Re M, Quaquarini E, Sottotetti F, Michelucci A, Palumbo R, Simi P. et al. Uncommon dihydropyrimidine dehydrogenase mutations and toxicity by fluoropyrimidines: a lethal case with a new variant. Pharmacogenomics. 2016;17:5-9
33. Ezzeldin H, Johnson MR, Okamoto Y, Diasio R. Denaturing high performance liquid chromatography analysis of the DPYD gene in patients with lethal 5-fluorouracil toxicity. Clin Cancer Res. 2003;9:3021-8
34. Garcia-Gonzalez X, Lopez-Tarruella S, Garcia MI, Gonzalez-Haba E, Blanco C, Salvador-Martin S. et al. Severe toxicity to capecitabine due to a new variant at a donor splicing site in the dihydropyrimidine dehydrogenase (DPYD) gene. Cancer Manag Res. 2018;10:4517-22
35. Gross E, Ullrich T, Seck K, Mueller V, de Wit M, von Schilling C. et al. Detailed analysis of five mutations in dihydropyrimidine dehydrogenase detected in cancer patients with 5-fluorouracil-related side effects. Hum Mutat. 2003;22:498
36. Henricks LM, Siemerink EJM, Rosing H, Meijer J, Goorden SMI, Polstra AM. et al. Capecitabine-based treatment of a patient with a novel DPYD genotype and complete dihydropyrimidine dehydrogenase deficiency. Int J Cancer. 2018;142:424-30
37. Kouwaki M, Hamajima N, Sumi S, Nonaka M, Sasaki M, Dobashi K. et al. Identification of novel mutations in the dihydropyrimidine dehydrogenase gene in a Japanese patient with 5-fluorouracil toxicity. Clin Cancer Res. 1998;4:2999-3004
38. Lazar A, Mau-Holzmann UA, Kolb H, Reichenmiller HE, Riess O, Schomig E. Multiple organ failure due to 5-fluorouracil chemotherapy in a patient with a rare dihydropyrimidine dehydrogenase gene variant. Onkologie. 2004;27:559-62
39. Ly RC, Schmidt RE, Kiel PJ, Pratt VM, Schneider BP, Radovich M. et al. Severe Capecitabine Toxicity Associated with a Rare DPYD Variant Identified Through Whole-Genome Sequencing. JCO Precis Oncol. 2020 4
40. Morel A, Boisdron-Celle M, Fey L, Laine-Cessac P, Gamelin E. Identification of a novel mutation in the dihydropyrimidine dehydrogenase gene in a patient with a lethal outcome following 5-fluorouracil administration and the determination of its frequency in a population of 500 patients with colorectal carcinoma. Clin Biochem. 2007;40:11-7
41. Ofverholm A, Arkblad E, Skrtic S, Albertsson P, Shubbar E, Enerback C. Two cases of 5-fluorouracil toxicity linked with gene variants in the DPYD gene. Clin Biochem. 2010;43:331-4
42. Palmirotta R, Lovero D, Delacour H, Le Roy A, Cremades S, Silvestris F. Rare Dihydropyrimidine Dehydrogenase Variants and Toxicity by Floropyrimidines: A Case Report. Front Oncol. 2019;9:139
43. Thomas F, Hennebelle I, Delmas C, Lochon I, Dhelens C, Garnier Tixidre C. et al. Genotyping of a family with a novel deleterious DPYD mutation supports the pretherapeutic screening of DPD deficiency with dihydrouracil/uracil ratio. Clin Pharmacol Ther. 2016;99:235-42
44. Tong CC, Lam CW, Lam KO, Lee VHF, Luk MY. A Novel DPYD Variant Associated with Severe Toxicity of Fluoropyrimidines: Role of Pre-emptive DPYD Genotype Screening. Front Oncol. 2018;8:279
45. van Kuilenburg AB, Baars JW, Meinsma R, van Gennip AH. Lethal 5-fluorouracil toxicity associated with a novel mutation in the dihydropyrimidine dehydrogenase gene. Ann Oncol. 2003;14:341-2
46. van Kuilenburg AB, Meijer J, Maurer D, Dobritzsch D, Meinsma R, Los M. et al. Severe fluoropyrimidine toxicity due to novel and rare DPYD missense mutations, deletion and genomic amplification affecting DPD activity and mRNA splicing. Biochim Biophys Acta Mol Basis Dis. 2017;1863:721-30
47. De Luca O, Salerno G, De Bernardini D, Torre MS, Simmaco M, Lionetto L. et al. Predicting Dihydropyrimidine Dehydrogenase Deficiency and Related 5-Fluorouracil Toxicity: Opportunities and Challenges of DPYD Exon Sequencing and the Role of Phenotyping Assays. Int J Mol Sci. 2022;23:13923
48. Etienne-Grimaldi MC, Boyer JC, Beroud C, Mbatchi L, van Kuilenburg A, Bobin-Dubigeon C. et al. New advances in DPYD genotype and risk of severe toxicity under capecitabine. PLoS One. 2017;12:e0175998
49. Garcia-Gonzalez X, Kaczmarczyk B, Abarca-Zabalia J, Thomas F, Garcia-Alfonso P, Robles L. et al. New DPYD variants causing DPD deficiency in patients treated with fluoropyrimidine. Cancer Chemother Pharmacol. 2020;86:45-54
50. Rosmarin D, Palles C, Pagnamenta A, Kaur K, Pita G, Martin M. et al. A candidate gene study of capecitabine-related toxicity in colorectal cancer identifies new toxicity variants at DPYD and a putative role for ENOSF1 rather than TYMS. Gut. 2015;64:111-20
51. Schwab M, Zanger UM, Marx C, Schaeffeler E, Klein K, Dippon J. et al. Role of genetic and nongenetic factors for fluorouracil treatment-related severe toxicity: a prospective clinical trial by the German 5-FU Toxicity Study Group. J Clin Oncol. 2008;26:2131-8
52. Soria-Chacartegui P, Villapalos-Garcia G, Lopez-Fernandez LA, Navares-Gomez M, Mejia-Abril G, Abad-Santos F. et al. Clinical Relevance of Novel Polymorphisms in the Dihydropyrimidine Dehydrogenase (DPYD) Gene in Patients with Severe Fluoropyrimidine Toxicity: A Spanish Case-Control Study. Pharmaceutics. 2021;13:2036
53. Villalvazo P, Marzal-Alfaro B, Garcia-Alfonso P, Revuelta-Herrero JL, Thomas F, Lopez-Tarruella S. et al. DPYD Exome, mRNA Expression and Uracil Levels in Early Severe Toxicity to Fluoropyrimidines: An Extreme Phenotype Approach. J Pers Med. 2021;11:792
54. Yokoi K, Nakajima Y, Matsuoka H, Shinkai Y, Ishihara T, Maeda Y. et al. Impact of DPYD, DPYS, and UPB1 gene variations on severe drug-related toxicity in patients with cancer. Cancer Sci. 2020;111:3359-66
55. Lau DK, Fong C, Arouri F, Cortez L, Katifi H, Gonzalez-Exposito R. et al. Impact of pharmacogenomic DPYD variant guided dosing on toxicity in patients receiving fluoropyrimidines for gastrointestinal cancers in a high-volume tertiary centre. BMC Cancer. 2023;23:380
56. Meulendijks D, Henricks LM, Sonke GS, Deenen MJ, Froehlich TK, Amstutz U. et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: a systematic review and meta-analysis of individual patient data. Lancet Oncol. 2015;16:1639-50
57. Del Re M, Cinieri S, Michelucci A, Salvadori S, Loupakis F, Schirripa M. et al. DPYD*6 plays an important role in fluoropyrimidine toxicity in addition to DPYD*2A and c.2846A>T: a comprehensive analysis in 1254 patients. Pharmacogenomics J. 2019;19:556-63
58. Meulendijks D, Henricks LM, Jacobs BAW, Aliev A, Deenen MJ, de Vries N. et al. Pretreatment serum uracil concentration as a predictor of severe and fatal fluoropyrimidine-associated toxicity. Br J Cancer. 2017;116:1415-24
59. Zhou Y, Koutsilieri S, Eliasson E, Lauschke VM. A paradigm shift in pharmacogenomics: From candidate polymorphisms to comprehensive sequencing. Basic Clin Pharmacol Toxicol. 2022;131:452-64
60. Zhou Y, Tremmel R, Schaeffeler E, Schwab M, Lauschke VM. Challenges and opportunities associated with rare-variant pharmacogenomics. Trends Pharmacol Sci. 2022;43:852-65
61. Zhou Y, Fujikura K, Mkrtchian S, Lauschke VM. Computational Methods for the Pharmacogenetic Interpretation of Next Generation Sequencing Data. Front Pharmacol. 2018;9:1437
62. Zhou Y, Lauschke VM. Computational Tools to Assess the Functional Consequences of Rare and Noncoding Pharmacogenetic Variability. Clin Pharmacol Ther. 2021;110:626-36
63. Shrestha S, Zhang C, Jerde CR, Nie Q, Li H, Offer SM. et al. Gene-Specific Variant Classifier (DPYD-Varifier) to Identify Deleterious Alleles of Dihydropyrimidine Dehydrogenase. Clin Pharmacol Ther. 2018;104:709-18
64. Zhou Y, Mkrtchian S, Kumondai M, Hiratsuka M, Lauschke VM. An optimized prediction framework to assess the functional impact of pharmacogenetic variants. Pharmacogenomics J. 2019;19:115-26
65. Cheng J, Novati G, Pan J, Bycroft C, Zemgulyte A, Applebaum T. et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science. 2023;381:eadg7492
66. Lee Y, Clark EW, Milan MSD, Champagne C, Michael KS, Awad MM. et al. Turnaround Time of Plasma Next-Generation Sequencing in Thoracic Oncology Patients: A Quality Improvement Analysis. JCO Precis Oncol. 2020 4
67. Sheffield BS, Beharry A, Diep J, Perdrizet K, Iafolla MAJ, Raskin W. et al. Point of Care Molecular Testing: Community-Based Rapid Next-Generation Sequencing to Support Cancer Care. Curr Oncol. 2022;29:1326-34
68. Huebner T, Steffens M, Scholl C. Current status of the analytical validation of next generation sequencing applications for pharmacogenetic profiling. Mol Biol Rep. 2023;50:9587-99
69. Maekawa K, Saeki M, Saito Y, Ozawa S, Kurose K, Kaniwa N. et al. Genetic variations and haplotype structures of the DPYD gene encoding dihydropyrimidine dehydrogenase in Japanese and their ethnic differences. J Hum Genet. 2007;52:804-19
70. Afolabi BL, Mazhindu T, Zedias C, Borok M, Ndlovu N, Masimirembwa C. et al. Pharmacogenetics and Adverse Events in the Use of Fluoropyrimidine in a Cohort of Cancer Patients on Standard of Care Treatment in Zimbabwe. J Pers Med. 2023;13:588
71. da Rocha JEB, Lombard Z, Ramsay M. Potential Impact of DPYD Variation on Fluoropyrimidine Drug Response in sub-Saharan African Populations. Front Genet. 2021;12:626954
72. Bunyan DJ, Robinson DO, Tyers AG, Huang S, Maloney VK, Grand FH. et al. X-Linked Dominant Congenital Ptosis Cosegregating with an Interstitial Insertion of a Chromosome 1p21.3 Fragment into a Quasipalindromic Sequence in Xq27.1. Open Journal of Genetics. 2014;4:415-25
73. Saarenheimo J, Wahid N, Eigeliene N, Ravi R, Salomons GS, Ojeda MF. et al. Preemptive screening of DPYD as part of clinical practice: high prevalence of a novel exon 4 deletion in the Finnish population. Cancer Chemother Pharmacol. 2021;87:657-63
74. Van Kuilenburg AB, Meinsma R, Beke E, Bobba B, Boffi P, Enns GM. et al. Identification of three novel mutations in the dihydropyrimidine dehydrogenase gene associated with altered pre-mRNA splicing or protein function. Biol Chem. 2005;386:319-24
75. Willemsen MH, Valles A, Kirkels LA, Mastebroek M, Olde Loohuis N, Kos A. et al. Chromosome 1p21.3 microdeletions comprising DPYD and MIR137 are associated with intellectual disability. J Med Genet. 2011;48:810-8
76. Wigle TJ, Medwid S, Ross C, Schwarz UI, Kim RB. DPYD Exon 4 Deletion Associated with Fluoropyrimidine Toxicity and Importance of Copy Number Variation. Curr Oncol. 2023;30:663-72
77. Hamzic S, Scharer D, Offer SM, Meulendijks D, Nakas C, Diasio RB. et al. Haplotype structure defines effects of common DPYD variants c.85T > C (rs1801265) and c.496A > G (rs2297595) on dihydropyrimidine dehydrogenase activity: Implication for 5-fluorouracil toxicity. Br J Clin Pharmacol. 2021;87:3234-43
78. Medwid S, Wigle TJ, Kim RB. Fluoropyrimidine-associated toxicity and DPYD variants c.85T>C, c.496A>G, and c.1236G>A: impact of haplotype. Cancer Chemother Pharmacol. 2023;91:97-102
79. Turner AJ, Haidar CE, Yang W, Boone EC, Offer SM, Empey PE. et al. Updated DPYD HapB3 haplotype structure and implications for pharmacogenomic testing. Clin Transl Sci. 2024;17:e13699
80. Knikman JE, Gelderblom H, Beijnen JH, Cats A, Guchelaar HJ, Henricks LM. Individualized Dosing of Fluoropyrimidine-Based Chemotherapy to Prevent Severe Fluoropyrimidine-Related Toxicity: What Are the Options? Clin Pharmacol Ther. 2021;109:591-604
81. Arrive C, Fonrose X, Thomas F, Roth G, Jacquet E, Brice A. et al. Discrepancies between dihydropyrimidine dehydrogenase phenotyping and genotyping: What are the explanatory factors? Br J Clin Pharmacol. 2023;89:2446-57
82. De Mattia E, Polesel J, Silvestri M, Roncato R, Scarabel L, Calza S. et al. The burden of rare variants in DPYS gene is a novel predictor of the risk of developing severe fluoropyrimidine-related toxicity. Hum Genomics. 2023;17:99
83. Le Teuff G, Cozic N, Boyer JC, Boige V, Diasio RB, Taieb J. et al. Dihydropyrimidine dehydrogenase gene variants for predicting grade 4-5 fluoropyrimidine-induced toxicity: FUSAFE individual patient data meta-analysis. Br J Cancer. 2024;130:808-18
Author contact
![]() Corresponding author: Dr. Giuseppe Toffoli MD, Director, Experimental and Clinical Pharmacology, Centro di Riferimento Oncologico di Aviano (CRO) IRCCS, via Franco Gallini n. 2, 33081 Aviano (PN), Italy. E-mail: gtoffoliit; Telephone +39-0434-659612; Fax +39-0434-659799.
Corresponding author: Dr. Giuseppe Toffoli MD, Director, Experimental and Clinical Pharmacology, Centro di Riferimento Oncologico di Aviano (CRO) IRCCS, via Franco Gallini n. 2, 33081 Aviano (PN), Italy. E-mail: gtoffoliit; Telephone +39-0434-659612; Fax +39-0434-659799.