Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Methods and materials
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(10):3892-3910. doi:10.7150/ijbs.92046 This issue Cite
Research Paper
The Interaction of Calcium-Sensing Receptor with KIF11 Enhances Cisplatin Resistance in Lung Adenocarcinoma via BRCA1/cyclin B1 pathway
Fuhao Wang1,2, Xing Fu1, Ming Chang1, Tianzi Wei1, Risheng Lin1, Haibo Tong3, Xiao Zhang1, Runzhu Yuan1,2, Zhiqing Zhou1, Xin Huang1, Wei Zhang5, Wenmei Su6, Yi Lu1,4 ![]() , Zhen Liang2
, Zhen Liang2 ![]() , Jian Zhang1,4
, Jian Zhang1,4 ![]()
1. School of Medicine, Southern University of Science and Technology, Shenzhen, Guangdong 518055, China.
2. The First Affiliated Hospital, School of Medicine, Southern University of Science and Technology, Shenzhen 518055, China.
3. Faculty of Health Sciences, University of Macau, Taipa, Macao SAR, 999078, China.
4. Joint Laboratory of Guangdong-Hong Kong Universities for Vascular Homeostasis and Diseases, School of Medicine, Southern University of Science and Technology, Shenzhen, Guangdong 518055, China.
5. School of Public Health and Emergency Management, Southern University of Science and Technology, Shenzhen 518055, Guangdong, China.
6. Department of Pulmonary Oncology, Affiliated Hospital of Guangdong Medical University, Zhanjiang, China.
Received 2023-11-9; Accepted 2024-7-5; Published 2024-7-15
Abstract

Cisplatin (DDP) is commonly used in the treatment of non-small cell lung cancer (NSCLC), including lung adenocarcinoma (LUAD), and the primary cause for its clinical inefficacy is chemoresistance. Here, we aimed to investigate a novel mechanism of chemoresistance in LUAD cells, focusing on the calcium-sensing receptor (CaSR). In this study, high CaSR expression was detected in DDP-resistant LUAD cells, and elevated CaSR expression is strongly correlated with poor prognosis in LUAD patients receiving chemotherapy. LUAD cells with high CaSR expression exhibited decreased sensitivity to cisplatin, and the growth of DDP-resistant LUAD cells was inhibited by cisplatin treatment in combination with CaSR suppression, accompanied by changes in BRCA1 and cyclin B1 protein expression both in vitro and in vivo. Additionally, an interaction between CaSR and KIF11 was identified. Importantly, suppressing KIF11 resulted in decreased protein levels of BRCA1 and cyclin B1, enhancing the sensitivity of DDP-resistant LUAD cells to cisplatin with no obvious decrease in CaSR. Here, our findings established the critical role of CaSR in promoting cisplatin resistance in LUAD cells by modulating cyclin B1 and BRCA1 and identified KIF11 as a mediator, highlighting the potential therapeutic value of targeting CaSR to overcome chemoresistance in LUAD.
Keywords: lung adenocarcinoma, chemoresistance, CaSR, BRCA1, cyclin B1, KIF11.
Introduction
Lung cancer remains the deadliest form of cancer globally, with the highest number of fatalities in recent years regardless of gender [1]. It has also been the second leading cause of cancer incidence globally in 2023 [2, 3]. Non-small cell lung cancer (NSCLC) represents the most widespread type of lung cancer, accounting for approximately 85% of cases [4]. Research in the field of lung cancer treatment is of considerable importance and significance [5, 6]. Chemotherapy has been crucial strategy [7], both in early [8] and in advanced [9, 10] NSCLC patients. Platinum chemotherapy is commonly used in the management of lung cancer and is presently acknowledged as the primary treatment option for lung cancer by the American Society of Clinical Oncology (ASCO) and the National Comprehensive Cancer Network (NCCN) [11, 12]; it can be utilized as part of a combination drug strategy to lengthen the overall survival of patients and still prevails as the primary approach for the treatment of lung cancer. Cisplatin, which is widely acknowledged as the most extensively used platinum chemotherapy drug, is commonly employed for treating NSCLC [13], it hinders the DNA replication and repair processes of tumor cells, consequently restraining tumor growth and metastasis [14]. However, the emergence of drug resistance is the primary cause of failure in current clinical treatments for NSCLC. Reversing drug resistance and improving the efficacy of cisplatin are ongoing challenges in clinical applications. Although previous studies have explored the mechanisms of cisplatin resistance in NSCLC, few definitive molecular markers have been identified to predict or reverse resistance [15]. In the clinical management of lung cancer, foreknowledge of how the disease responds to cisplatin is crucial in selecting more efficacious treatments, allowing for significant strides in treating the condition.
Calcium-sensing receptor (CaSR), as a calcium sensor, regulates cell division and many fundamental biological processes [16]. The effect of CaSR on tumors has been investigated in various human malignancies [17]. Recent research indicates that higher CaSR expression in breast cancer may be associated with a worse prognosis and treatment outcome, independent of subtype [18, 19]. In lung cancer, CaSR is also highly expressed in LUAD tissues, and the expression level thereof is associated with the degrees of cancer differentiation and metastasis [20]. CaSR has also been reported as a potential biomarker for predicting bone metastasis and prognosis in lung cancer patients [21]. Therapeutic employment of CaSR targeting can be considered in patients with increased CaSR expression in cancer cells, which exhibit increased tumor malignancy [22]. Moreover, some reviews have reported that CaSR is associated with drug resistance [23, 24]. Nevertheless, little is known about the role of CaSR in lung cancer and its contribution to chemoresistance in lung cancer. CaSR controls cell proliferation by modifying calcium channel expression [25], a key point in cell cycle [26]. Cancer cells rely heavily on the G2/M phase for DNA repair [27] and the capacity for DNA damage repair is intimately connected with the cisplatin resistance observed in NSCLC [28]. However, it remains unclear how CaSR affects the cell cycle and DNA damage repair in tumor cells. Therefore, further investigation is required to determine the significance of CaSR in cancer treatment.
Adenocarcinoma is the most prevalent histological subtype of NSCLC, accounting for a high proportion of NSCLC patients in China [29]. Considering that the mechanism of cisplatin resistance varies among different types of tumors, we established two DDP-resistant LUAD cell sublines by progressively escalating the concentration of cisplatin. We observed high expression of CaSR in both DDP-resistant cell lines, indicating a potential association between CaSR and cisplatin resistance. Moreover, our data revealed a significant correlation between CaSR expression and prognosis in LUAD patients undergoing chemotherapy. Subsequently, we conducted a systematic investigation to elucidate the role of CaSR in the development of cisplatin resistance. Our findings offer new strategies for accurately predicting tumor chemosensitivity, reversing multidrug resistance, and gaining insights into the molecular mechanisms underlying chemoresistance in LUAD cells.
Methods and materials
Cell culture
A549 and H1299 cell lines were obtained from ATCC (Manassas, VA, USA). A549-DDP and H1299-DDP cell lines resistant to cisplatin were established by exposing the cells to progressive concentrations of cisplatin for approximately 2 months. Cells were cultured in RPMI 1640 (Gibco; Carlsbad, CA, USA) with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin (Gibco) in an incubator with 5% CO2 at 37°C. The culture medium of DDP-resistant cells was supplemented with an additional 2 µM cisplatin. All cell lines were tested against contamination by mycoplasma.
Drugs and inhibitors
Cisplatin (HY-17394) and NPS-2143 (HY-10007) were purchased from MedChem Express (Monmouth Junction, NJ, USA). A kinesin family member 11 (KIF11) inhibitor (EG5 Inhibitor V, trans-24; T11155) was purchased from TargetMol (Boston, MA, USA).
Clinical samples
Tissue sections from patient clinical samples were purchased from Zhongke Guanghua intelligent biological technology co. (Xi'an, China).
RNA-seq and data analysis
Total RNA was extracted using 1 mL TRIzol per 10 cm2 plate volume for each RNA sequencing (RNA-seq) sample. All RNA-seq samples were quality controlled and customized for analysis by Gene Denovo Biotechnology Co. (Guangzhou, China). DEseq2 software was utilized to analyze the distinctively expressed genes. Genes with false discovery rates (FDR) < 0.05 and |Log2(fold change)| > 1 were considered differentially expressed genes (DEGs), which were utilized for the subsequent enrichment analysis.
Survival analysis
Survival and prognosis analysis was evaluated by the Kaplan-Meier plotter server [30], which contained independent datasets from the caBIG, GEO, and TCGA repositories.
CCK8 assay
A total of 2000 cells in 100 µl of medium were seeded into 96-well plates. Then, 10 µl of Cell Counting Kit-8 (CCK8, TargetMol) reagent was added to the cells and incubated at 37°C for 2 h. The absorbance was measured at 450 nm using a microplate reader (BioTek; Vermont, USA). For cisplatin resistance detection, cisplatin concentrations were set to 0, 1, 2, 4, 8, 16, 32, or 64 µM.
Clonogenicity assay
Cells were seeded into 6-well dishes at a concentration of 300 cells per dish. After two weeks of growth, the cells were treated with 4% cell fixative (Solarbio; Beijing, China) and stained with crystal violet (Solarbio) for colony counting.
Western blotting
The total protein was lysed by sonication and the protocol used for western blotting was based on a previous report [31]. The primary antibodies were listed in Table S1.
Establishment of CaSR-overexpressing cell lines
To overexpress CaSR, the coding sequence of CaSR was subcloned. The protocol used for establishing the CaSR-overexpressing cell lines was based on a previous report [32].
Cell cycle analysis
Cells from each sample were cultured for cell cycle analysis in 6-cm dishes. After rinsing with PBS, the cells were fixed by placing them in 75% pre-cooled ethanol overnight. After washing with PBS, staining was then performed at 37°C for 30 min (Beyotime; Shanghai, China) and was detected using an AFC2 acoustic focusing cytometer (Invitrogen; Carlsbad, CA, USA).
Apoptosis analysis
A total of 2-5*106 cells were collected for the purpose of apoptosis detection. The cells were digested with trypsin without EDTA and were prepared with the apoptosis detection kit (Yeasen; Shanghai, China). The samples were then detected using an AFC2 acoustic focusing cytometer (Invitrogen; Carlsbad, CA, USA).
Glycolysis stress test
The Agilent Seahorse XF Glycolysis Stress Test Kit (Agilent; CA, USA) was employed to assess the principal parameters of glycolytic flux. The cells were examined on the Agilent Seahorse XF Pro Analyzer (Agilent; CA, USA).
RNAi transfection
Lipofectamine RNAiMax reagent (Invitrogen, 13778150), Opti-MEM medium (Gibco, 31985-062), targeted siRNAs and non-targeted controls from General Biosystems Co. (Anhui, China) were transfected into cells. The working concentrations for all siRNAs were 10-20 nM, and the targeting sequences for each siRNA are listed in Table S2.
Animal model
Female BALB/c nude mice at approximately 4 weeks of age were used for the tumorigenesis assay. Each mouse was subcutaneously injected with a total of 2 × 106 A549-DDP cells on the right side of the back. Injections were performed when tumor diameters reached 4 mm, with 6 mice per group. The weights and tumor volumes of the mice were monitored, and injections were given twice a week for 1 month. Mice were euthanized at study termination, and tumors were removed and weighed. Sections of subcutaneous tumor were used for immunohistochemistry (IHC), and the remaining sections were stored in formalin. The animal protocol received approval from the Institutional Animal Ethics Committee at the Southern University of Science and Technology. All mice were maintained by facility technicians at the Laboratory Animal Center (SUSTech-JY2020140).
Immunohistochemistry
For IHC detection, slides were incubated with the indicated primary antibodies (Table S1). Following incubation with the primary antibodies, the slides were subjected to a secondary antibody incubation using an IHC kit (Biotechnologies) for 20 min. The results were observed utilizing a digital pathology scanner (Leica Microsystems; Wetzlar, Germany). CaSR, KI67, BRCA1, and cyclin B1 positive cells were quantified by measuring staining intensity and percentage using ImageJ software.
Molecular docking
The X-ray crystal structures of CaSR (7SIL) and KIF11 (1IIL) were retrieved from the Protein Data Bank. To ensure the accuracy of the docking results, the proteins were prepared using AutoDockTools-1.5.7[33, 34].
Immunoprecipitation and co-immunoprecipitation
A total of 1 × 107 cells were lysed with NP-40 buffer (Beyotime) and centrifuged at 12000 rpm for 10 min. Antibodies (Table S1) were added, and the samples were shaken overnight at 4°C. Protein G Dynabeads (Invitrogen) were introduced and incubated for 4 h at 4°C, and the isolated samples were then collected for mass spectrometry and western blotting.
Statistical analysis
The results of this study are presented as the means ± SEM. Statistically significant variations between two groups were evaluated utilizing an independent t-test, and comparisons across more than two groups were evaluated using one-way analysis of variance (ANOVA). Survival curves were developed from Kaplan-Meier estimates, and the log-rank test was used to perform survival analysis. P < 0.05 was considered statistically significant (* P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001 and ns, not significant). R software and GraphPad Prism 9 were used for all statistical analyses and visualizations.
Results
CaSR was upregulated in DDP-resistant LUAD cells and associated with poor prognosis of chemotherapy in LUAD
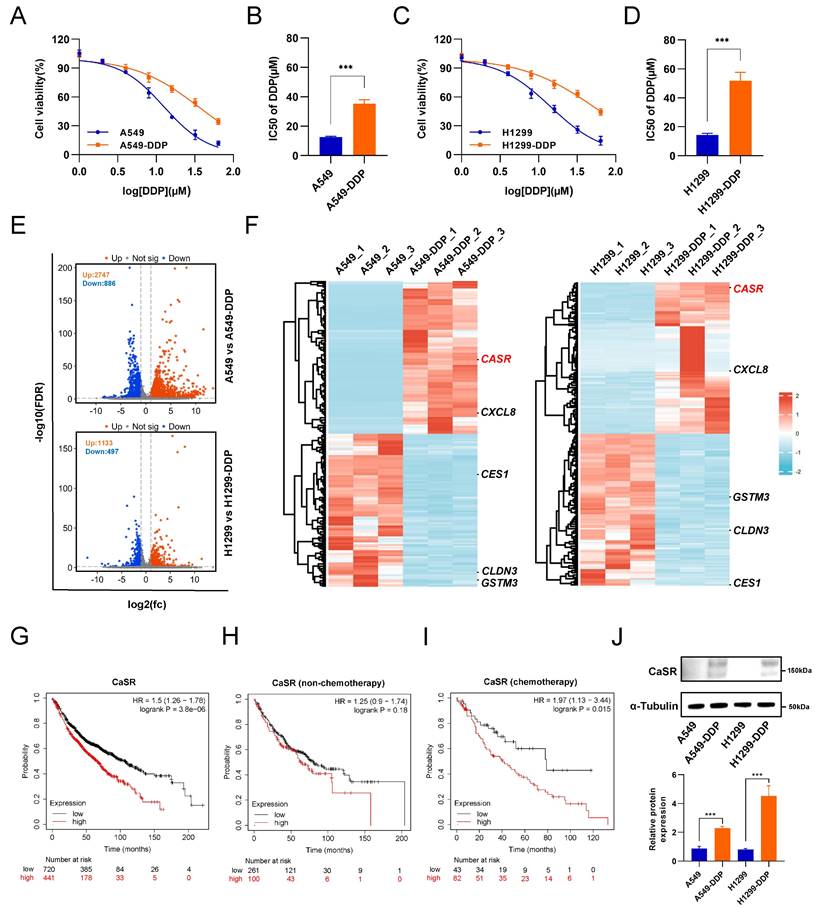
Cisplatin (DDP) remains the predominant therapeutic regimen in LUAD, overcoming cisplatin resistance will liberate its curative potential. Here, we established two LUAD cell lines, based on A549 and H1299 cell lines, with cisplatin resistance using progressively increasing concentrations of cisplatin (Fig. S1A). A comparison of the half-maximal inhibitory concentration (IC50) values revealed that the cisplatin resistance levels of the LUAD cell lines were significantly higher than those of the parental A549 and H1299 cells (Fig. 1A-D). To comprehensively investigate the molecular mechanism of LUAD resistance and identify potential targets to reverse resistance, we sequenced the transcriptomes of the parental and resistant A549 and H1299 cell lines. The results revealed 2747 upregulated genes and 886 downregulated genes in A549-DDP cells compared to parental cells and 1133 upregulated genes and 497 downregulated genes in H1299-DDP cells compared to parental cells (Fig. 1E). We conducted intersection analysis on the top 100 up-regulated and down-regulated differentially expressed genes (DEGs) from the two cisplatin-resistant cells and their parental cells, aiming to identify genes which were commonly up-regulated and down-regulated in A549-DDP and H1299-DDP cells. The results showed there were 3 co-upregulated genes (Calcium-sensing receptor, CaSR; CXC motif chemokine ligand 8, CXCL8; and ARL14EPL) and 6 co-downregulated genes (glutathione S-transferase mu 3, GSTM3; CES4A; carboxylesterase 1, CES1; PPP1R3G; claudin 3, CLDN3; RGL3) (Fig. S1B). Subsequently, we utilized the Kaplan-Meier plotter server to investigate the association between these genes and the prognosis of LUAD patients who received chemotherapy. Survival information was identified only for the CXCL8, CaSR, CLDN3, GSTM3, and CES1 genes in LUAD patients who underwent chemotherapy intervention (Fig.1F, S1C). We observed that no statistically significant correlations were found between CXCL8, CLDN3, GSTM3, and LUAD chemotherapy prognosis (Fig. S1E-G). Patients with high expression of CES1 had a poor prognosis after chemotherapy (Fig. S1H), though high CES1 expression was not detected in our established DDP-resistant LUAD cells (Fig.1F). Therefore, we suppose that CES1 may have a role in other types of chemotherapy. We further focused on CaSR. Surprisingly, our analysis revealed a strong correlation between elevated CaSR expression and survival in LUAD patients (Fig. 1G). Moreover, we also found that high expression of CaSR was associated with a worse prognosis following chemotherapy in breast cancer patients who did not receive endocrine therapy (Fig. S2A) and in ovarian cancer patients who underwent suboptimal debulking surgery (Fig. S2B). Then we hypothesized that CaSR might be associated with chemotherapy prognosis. Upon further examination, we divided the data into two groups based on whether the patients underwent chemotherapy or not. Intriguingly, we found no significant variations in CaSR expression and prognosis among LUAD patients without a history of chemotherapy (Fig. 1H). However, among patients who received chemotherapy, those with high CaSR expression had a worse prognosis (Fig. 1I). According to the GEPIA database, there were some LUAD tumor tissues with high CaSR expression, although there was no statistical difference between tumor and normal tissues in LUAD at the overall level (Fig. S1D). Additionally, our study detected high levels of CaSR protein in our established DDP-resistant LUAD cells (Fig. 1J), and high CaSR expression was detected in clinical samples from LUAD patients with chemotherapy (Fig. S3), suggesting that CaSR may play a role in the progression of chemotherapy in LUAD patients.
CaSR was upregulated in DDP-resistant LUAD cells and associated with poor prognosis in LUAD. Cell viability and the IC50 values of cisplatin in A549-DDP and parental A549 cell lines (A and B) were determined by CCK8 assay. Same with H1299-DDP and parental H1299 cell lines (C and D). Volcano plot showed differentially expressed genes between parental and DDP-resistant LUAD cells (E). Heat map showed the top 100 up-regulated and down-regulated differentially expressed genes (DEGs) from the two cisplatin-resistant cells and their parental cells (F). Kaplan-Meier plots showed the correlation of CaSR with overall survival in LUAD (G), including the prognosis receiving non-chemotherapy (H) and chemotherapy (I). Western blotting results showed high CaSR expression in DDP-resistant LUAD cells (J). Data are depicted as the mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001 and ns, not significant.
High expression of CaSR enhanced cisplatin resistance in LUAD cell lines
As indicated by the data presented above, CaSR could potentially be linked to chemotherapy resistance in LUAD. To investigate the involvement of CaSR in the chemotherapeutic resistance of LUAD, we established CaSR-overexpressing A549 and H1299 cell lines using lentiviruses, with cells stably expressing empty vector used as a negative control (Fig. S4A). Indeed, we found that upregulation of CaSR expression increased cisplatin resistance in LUAD cells to a certain extent (Fig. 2A-D). We performed transcriptome sequencing on stable CaSR-overexpressing and control cell lines to further investigate the involvement of CaSR in the mechanism of chemoresistance. We found that A549 cells overexpressing CaSR had 272 upregulated genes compared to negative control cells. In H1299 cells overexpressing CaSR, 1644 genes were upregulated, and 47 genes were downregulated compared to negative control cells (Fig. 2E).
Overexpression of CaSR affected the cell cycle and cisplatin resistance in LUAD cell lines. Cell viability and the IC50 values of cisplatin in A549-NC and A549-CaSR cell lines (A and B), H1299-NC and H1299-CaSR cell lines (C and D). Volcano plot showed differentially expressed genes between CaSR-overexpressing cell lines and negative control cell lines (E). KEGG enrichment analysis (F), GO enrichment analysis (G), and cell cycle analysis were presented (H and I). CaSR overexpression did not affect the proliferation of LUAD cells, and the panels on the right showed the relative cell viability after 48 h (J). BRCA1 and cell cycle related proteins were determined by western blotting between CASR-overexpressing cell lines and negative control cell lines (K). Data are presented as the mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001, and ns, not significant.
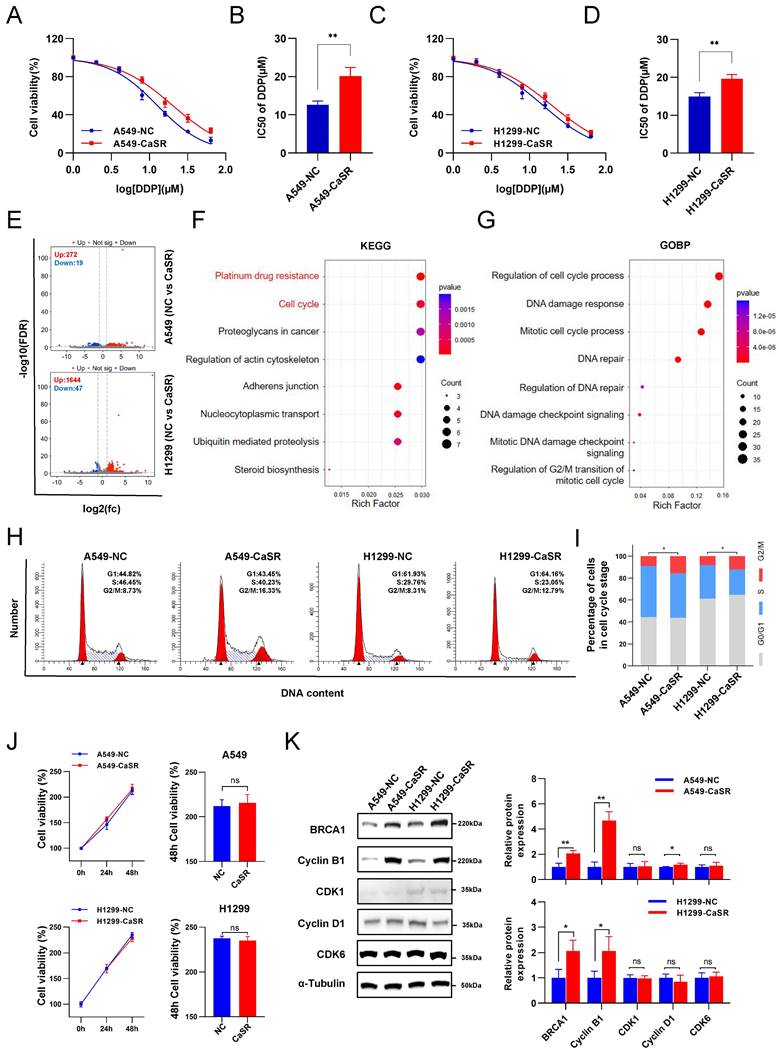
Moreover, we identified 235 co-upregulated genes and 1 co-downregulated gene (Fig. S4B). Enrichment analysis of the Kyoto Encyclopedia of Genes and Genomes (KEGG) signaling pathways revealed that CaSR-overexpressing LUAD showed significant changes in cisplatin drug resistance-related pathways compared to the negative control group (Fig. 2F), including BRCA1(Fig. S4C), which plays an important role in homology-directed repair and therapy resistance [35], and BRCA1 is also an important indicator for predicting the efficacy of chemotherapy in NSCLC [36]. Meanwhile, the enrichment outcomes of Gene Ontology (GO), Reactome, and WikiPathways analyses highlighted the activation of numerous pathways linked to the cell cycle and DNA damage repair (Fig. 2G, S4D, E). These pathways are strongly associated with the development of cisplatin resistance. Based on the transcriptome sequencing results, significant changes in the cell cycle, especially in the G2/M phase, were observed in the CaSR-overexpressing cell lines compared to the NC group (Fig. 2H, I). CaSR overexpression did not have a significant impact on the proliferation of LUAD in either A549 or H1299 cell lines (Fig. 2J). Additionally, there was no significant change observed in the G1 phase (Fig. 2H). Therefore, we hypothesize that the increased percentage of cells in G2/M phase is not due to cell arrest, but rather a result of the cell's self-adaptation and self-regulation. To further investigate this, we examined the expression levels of CDK6 and cyclin D1, two important regulators of the G1 phases [37]. However, we observed either inconsistent changes or no substantial alteration in their expression levels. Moreover, the G2/M phase-related proteins, cyclin-dependent kinase 1 (CDK1), cyclin B1 and breast cancer gene 1 (BRCA1) were also evaluated in the CaSR-overexpressing and control cell lines. We found a pronounced upregulation of BRCA1 in CaSR-overexpressing LUAD cells. Cyclin B1 expression was significantly increased in these cells, though there was no substantial change in CDK1 expression (Fig. 2K). As we know, cyclin B1/CDK1 plays a crucial role in regulating cell cycle progression. When the protein level of cyclin B1 reaches a certain level, it enters the nucleus and forms a complex with CDK1 to affect the cell cycle [38]. Typically, cyclin B1 is the main regulated protein [39], and its increased presence may be closely related to the increase in the proportion of G2/M phase cells. Moreover, cyclin B1 stability is increased by interaction with BRCA1 [40], which supports our results. As the majority of tumor cells depend on the G2/M checkpoint for DNA repair, the increase of BRCA1 and the elevated percentages of CaSR-overexpressing cells in the LUAD cell lines in G2/M phase imply that these cells possess a more adequate DNA repair process.
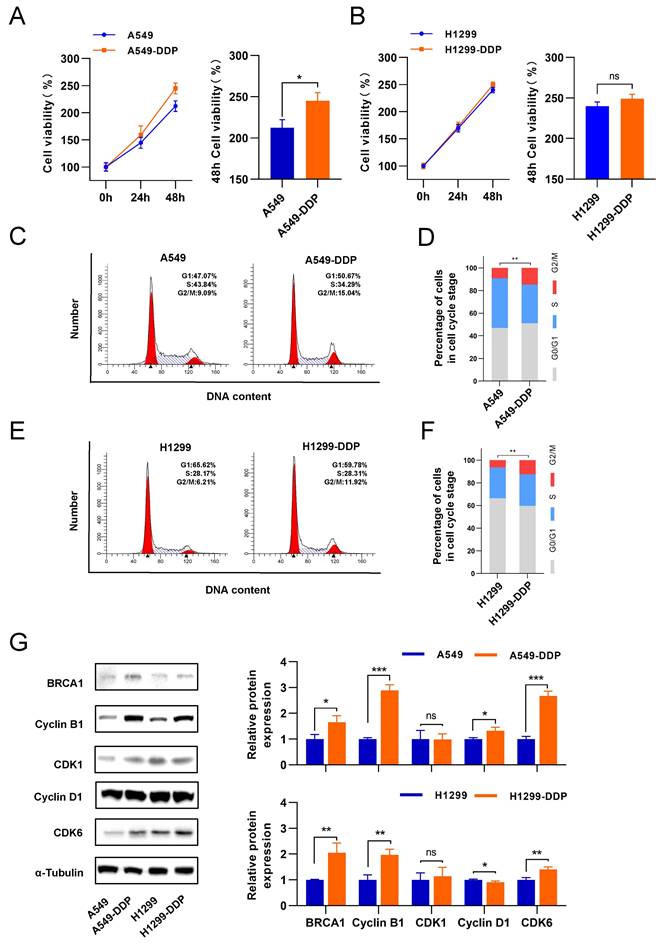
Following the biological phenomenon we observed in CaSR-overexpressing cell lines, we investigated the changes in proliferation in LUAD cells before and after they acquired cisplatin resistance. We found that A549-DDP cells displayed slightly higher proliferation than A549 cells (Fig. 3A). Conversely, the proliferation levels of H1299 and H1299-DDP cells were similar (Fig. 3B). We thought that this difference occurred because of the inherent genomic alterations and molecular signature present in A549 (p53 wild-type) and H1299 (p53-deficient) cells, which are both lung adenocarcinoma cells [41], and the differential changes in the transcriptome after cisplatin treatment may also account for the different proliferation response. Next, the proportion of cells in the G2/M phase among DDP-resistant LUAD cells was significantly increased (Fig. 3C-F). Western blotting results showed that cyclin B1 and BRCA1 were upregulated in the DDP-resistant LUAD cells. In contrast, CDK1 did not show significant changes (Fig. 3G). Taken together with the previous findings in CaSR-overexpressing LUAD cells, we hypothesized that CaSR might affect the cisplatin resistance of LUAD cells through cyclin B1 and BRCA1.
G2/M phase of DDP-resistant LUAD was increased, accompanied by upregulation of cyclin B1 and BRCA1. The proliferation of DDP-resistant LUAD cell lines and their parental cell lines, and the panels on the right showed the relative cell viability after 48 h (A and B). Cell cycle analysis between DDP-resistant LUAD cell lines and their parental cell lines (C-F). BRCA1 and cell cycle related protein were determined by western blotting (G). Data are presented as the mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001, and ns, not significant.
The cisplatin sensitivity in DDP-resistant LUAD cells was reversed by suppressing CaSR expression
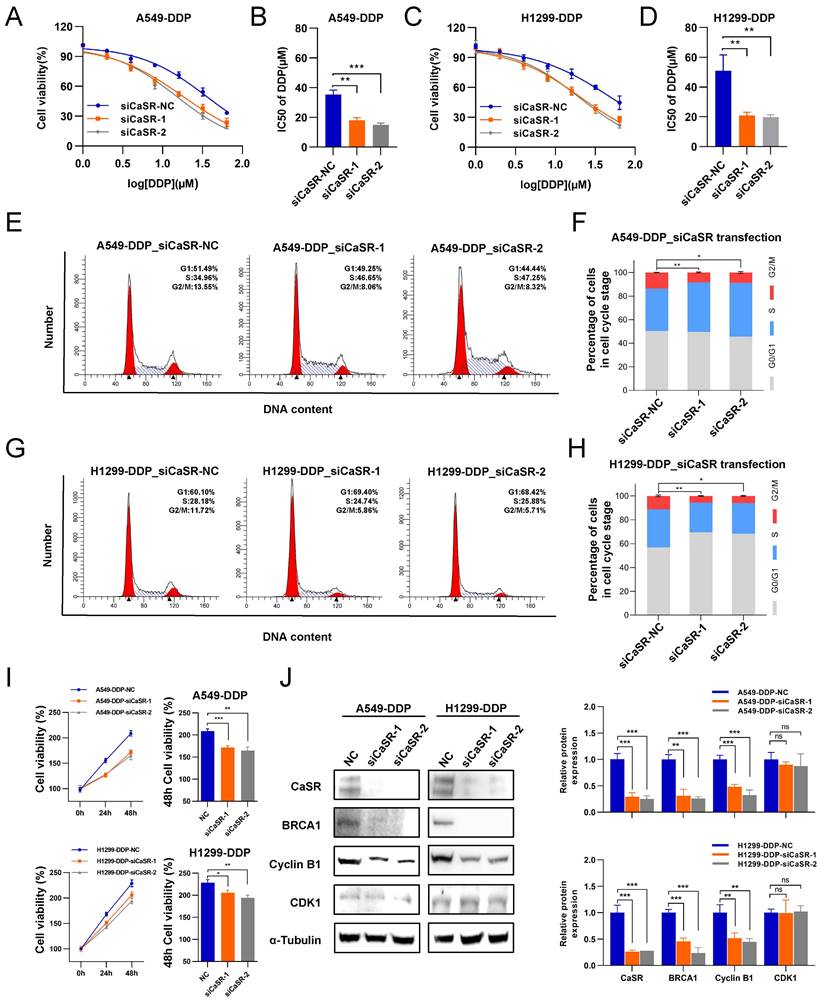
To examine the necessity of CaSR for the acquisition of cisplatin resistance by DDP-resistant LUAD cell lines, we exploited small interfering RNAs (siRNAs) to disrupt CaSR expression in DDP-resistant cells. We found that DDP-resistant LUAD cells became more sensitive to cisplatin after CaSR knockdown (Fig. 4A-D). Furthermore, cell cycle analysis revealed that the proportion of cells in the G2/M phase was decreased after CaSR knockdown (Fig. 4E-H), which also suggested that the DNA damage repair response process was reduced. Cell proliferation assay revealed that the cell growth was affected after CaSR knockdown (Fig. 4I). Moreover, we detected decreased protein expression levels of BRCA1 and cyclin B1 (Fig. 4J). Combining multiple drugs to treat tumors or drug-resistant tumors is a common strategy in cancer treatment, and the current research into G protein-coupled receptor (GPCR)-targeted drugs is well advanced. Therefore, we examined the impact of CASR inhibitors on cisplatin resistance in lung cancer. The CaSR inhibitor NPS-2143 (Fig. S5A) was utilized in the following experiments. We noted that a higher concentration of NPS-2143 (1 µM) had a strong toxic effect on cell viability (Fig. S5B-D). Cisplatin combined with NPS-2143 (10 or 100 nM) was then administered. The clonogenic proliferation assay results revealed that the combination of 100 nM NPS-2143 with cisplatin (10 µM) suppressed the growth of DDP-resistant LUAD cells, and this effect was more pronounced in A549-DDP cells (Fig. 5A).
CaSR was emerged as a potential target for reversing platinum-based chemotherapy resistance in LUAD. A549-DDP and H1299-DDP cells were transfected with siRNA targeting CaSR (siCaSR-1, siCaSR-2) or non-targeting siRNA (siCaSR-NC), CaSR knockdown affected the sensitivity of A549-DDP cells (A and B) and H1299-DDP cells (C and D) to cisplatin. Cell cycle analysis showed that the G2/M phase was decreased after CaSR knockdown both in A549-DDP (E and F) and H1299-DDP cells (G and H).The proliferation ability of A549-DDP and H1299-DDP cells was impacted to a certain extent after CaSR knockdown and the right panels show the relative cell viability at the 48 h (I). The protein levels of CaSR, cyclin B1, CDK1 and BRCA1 were determined by western blotting (J). Data are presented as the mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001 and ns, not significant.
NPS-2143 alleviated the cisplatin resistance of DDP-resistant LUAD cells. The clonogenic proliferation of DDP-resistant cells was disrupted by the CaSR inhibitor NPS-2143 (A). A549-DDP and H1299-DDP cells were treated with 10 µM cisplatin and 100 nM NPS-2143, respectively, or in combination. The sensitivity of A549-DDP (B and C) and H1299-DDP (D and E) cells to cisplatin was affected by the CaSR inhibitor NPS-2143. Cell cycle analysis showed that the changes after A549-DDP and H1299-DDP cells treated with 100 nM CaSR inhibitor NPS-2143 (F and G). The protein levels of CaSR, BRCA1, cyclin B1, and CDK1 were detected after A549-DDP and H1299-DDP cells treated with 100 nM CaSR inhibitor NPS-2143 (H). All datas are showed as the mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001 and ns, not significant.
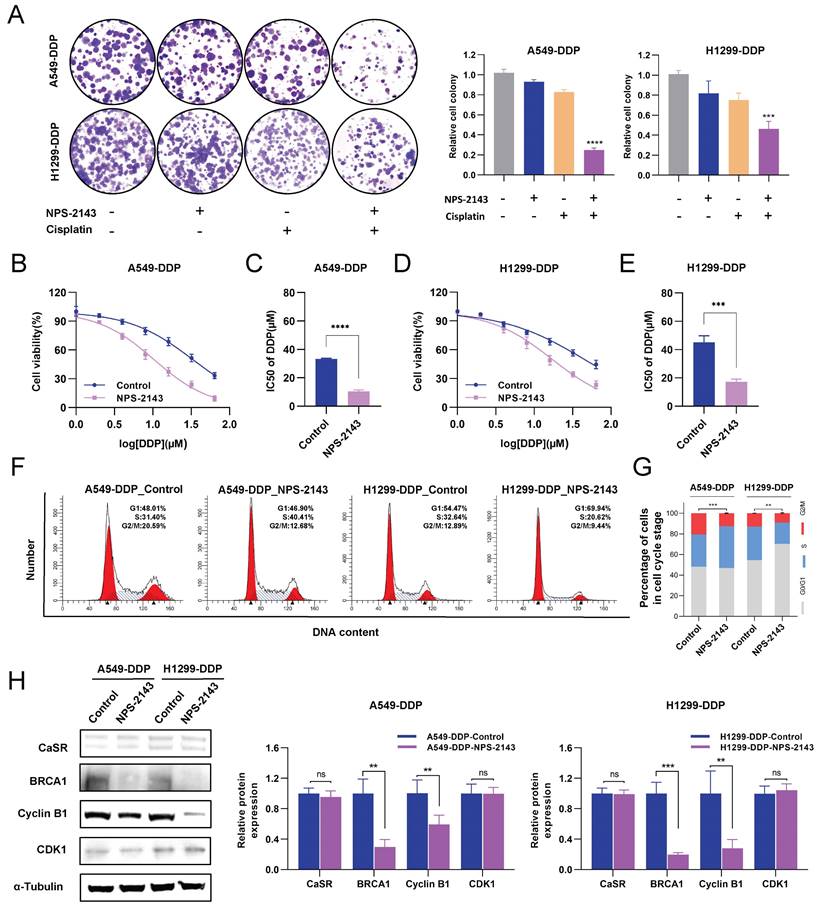
In contrast, the concomitant treatment consisting of 10 nM NPS-2143 did not considerably curtail the proliferation of DDP-resistant LUAD cells (Fig. S5B-D). We then investigated alterations in the cisplatin resistance of DDP-resistant LUAD cells following treatment with 100 nM NPS-2143 and observed that the cells exhibited increased cisplatin sensitivity (Fig. 5B-E), suggesting that a combination of NPS-2143 and cisplatin can mitigate the cisplatin resistance of DDP-resistant LUAD cells. The G2/M phase was similarly reduced following treatment with NPS-2143, as was observed after CaSR knockdown (Fig. 5F, G). Western blotting results showed that there was no notable distinction in CaSR expression after NPS-2143 treatment. Meanwhile, the protein expression levels of BRCA1 and cyclin B1 decreased (Fig. 5H), confirming our hypothesis and indicating that CaSR has effects on the closely related BRCA1 and the cell cycle and is involved in the development of cisplatin resistance in LUAD. Hence, CaSR was determined to be a potential target for reversing platinum-based chemotherapy resistance in LUAD.
NPS-2143 combined with cisplatin significantly repressed the growth of DDP-resistant LUAD cells in vivo
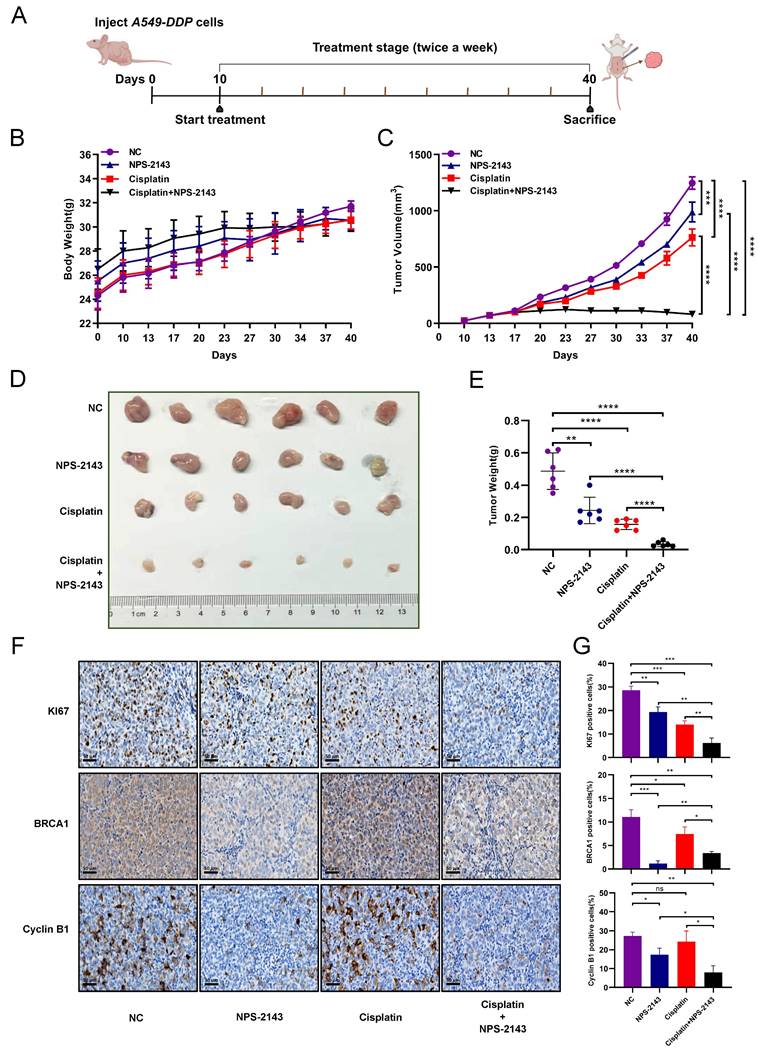
NPS-2143, as a CaSR inhibitor, is commonly used in cancer research [42, 43]. To evaluate the effect of the combination of a CaSR inhibitor and cisplatin on the growth of LUAD in vivo, the established A549-DDP cells were injected subcutaneously into nude mice as tumor xenografts. For approximately 1 month, each group received two weekly injections of PBS, cisplatin (6 mg/kg), NPS-2143 (80 µg/kg), and NPS-2143 (80 µg/kg) in combination with cisplatin (6 mg/kg). Mouse body weights and tumor sizes were recorded as shown in Fig. 6A. There were no significant differences in body weight among the groups before and after they were treated (Fig. 6B). In nude mice, tumor volume growth was significantly inhibited by the combined use of NPS-2143 and cisplatin (Fig. 6C). Correspondingly, the weights of the resected tumors in the drug combination groups were substantially lower than the other tumor weights (Fig. 6D, E). We subsequently conducted IHC assays on excised tumors from each group (Fig. 6F). As a marker for evaluating tumor proliferation, the reduction of KI67 confirmed that the tumor growth was repressed in the combination group. Moreover, in the NPS-2143 treatment group, the expression levels of BRCA1 and cyclin B1 were decreased, reflecting the effect of NPS-2143 to some extent. In the group that received only cisplatin treatment, the expression of BRCA1 was significantly higher compared to the NPS-2143 treatment group and the combination group, but slightly lower compared to the NC group. In comparison to the NC group, the cisplatin treatment group exhibited smaller tumor size (Fig. 6D) and slightly lower expression of BRCA1, indicating that cisplatin (6 mg/kg) had drug effect to a limited extent, but the inhibitory effect on the growth of DDP-resistant cells in vivo was not as effective as in the combination group. In the combination group, as expected, BRCA1 and cyclin B1 expression levels were significantly reduced (Fig. 6F, G). These results demonstrated a noteworthy decrease in the growth of DDP-resistant LUAD cells in vivo when treated with a combination of cisplatin and NPS-2143.
NPS-2143 combined with cisplatin repressed the growth of DDP-resistant LUAD in vivo. Schematic diagram of the experimental procedure in nude mice (A). The A549-DDP cells were subcutaneously injected into nude mice as tumor xenografts. Four groups were treated with cisplatin (6 mg/kg), NPS-2143 (80 µg/kg), PBS and NPS-2143 in combination with cisplatin twice a week for one month, while body weight (B) and tumor (C) size were recorded. At the termination of the experiment, photographs of all tumors were taken (D), and tumor weight was measured (E). The expression of KI67, BRCA1 and cyclin B1 has been determined by IHC (F), followed by statistics and corresponding images (G). Data are presented as the mean ± SEM. Statistical significance was determined by one-way ANOVA with * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001 and ns, not significant.
KIF11 interacted with CaSR and was upregulated in DDP-resistant LUAD cells
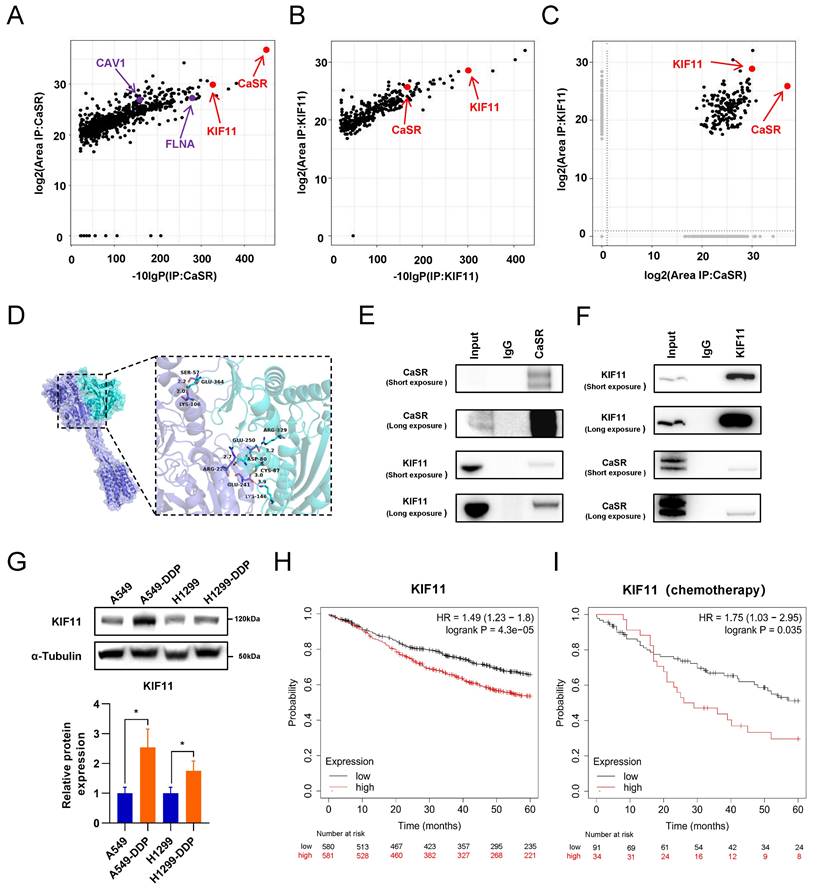
The results of the study described above indicated that the impact of CaSR on cisplatin sensitivity in LUAD may be related to BRCA1 and cyclin B1. However, a direct relationship between CaSR, BRCA1, and cyclin B1 has not yet been well documented. Moreover, how CaSR impacts the repair of DNA damage and the cell cycle is a topic of significant interest. To explore this question, we performed co-immunoprecipitation (coIP) assessments of protein lysates from the A549-DDP cell line using CaSR antibodies to identify potential CaSR-interacting proteins by mass spectrometry (MS). The top 20 scoring proteins in the identification results are listed in Table S3. Among them, filamin A (FLNA) and caveolin 1 (CAV1), which are known to interact with CaSR [44, 45], were shown as reference proteins for successful coIP and MS analyses (Fig. 7A). Considering that BRCA1 and cyclin B1 were found to be affected by CaSR and were associated with the cell cycle and DNA damage repair in the above studies, we selected proteins associated with DNA damage repair or mitosis from the CaSR-interacting proteins. Surprisingly, KIF11, a protein that plays key roles in mitosis and the cell cycle [46], was identified. KIF11-interacting proteins were also identified by MS, and CaSR was among the identified proteins (Fig. 7B, C). The protein identification results that scored in the top 20 are listed in Table S4. Next, molecular docking prediction of CaSR and KIF11 was performed. We found that CaSR and KIF11 have multiple predictable binding sites, most involving ionic bonds (Fig. 7D). We then investigated the interaction between CaSR and KIF11 through coIP assessments to identify interactions with either CaSR or KIF11. KIF11 was clearly detected among the CaSR-interacting proteins (Fig. 7E); likewise, CaSR was detected among the KIF11-interacting proteins (Fig. 7F). KIF11 was found to be highly expressed in various types of malignant tumors, including LUAD (Fig. S6A, B). Upon further investigation, we found that KIF11 expression was higher in DDP-resistant LUAD than in parental cells (Fig. 7G), which appeared to be related to the high expression of CaSR in DDP-resistant LUAD cells. Moreover, KIF11 exhibited a poor correlation with CaSR but strong and positive correlations with BRCA1 and cyclin B1 (Fig. S6C). Kaplan-Meier plotter server analysis also revealed an association between KIF11 and the survival and prognosis of patients with LUAD (Fig. 7H) Importantly, high expression of KIF11 was found to be significantly associated with poor prognosis in LUAD with chemotherapy (Fig. 7I).
KIF11 interacted with CaSR and was upregulated in DDP-resistant LUAD cells. Mass spectrometry (MS) was used to identify proteins (A-C), CaSR-interacting proteins (A), KIF11-interacting proteins (B), and proteins interacting with CaSR or KIF11 are selected based on two screening criteria: The protein was not pulled down by IgG or the protein had less than 2 folds increase in detection in IgG compared to CaSR or KIF11 (C). Protein-protein interaction maps between CaSR and KIF11 (D), CaSR is depicted in dark blue, and KIF11 is depicted in cyan. The corresponding binding points are indicated as clubbed structures of the corresponding colors. And western blotting results showed clear detection of KIF11 in the CaSR-interacting proteins (E) and CaSR in the KIF11-interacting proteins (F). KIF11 was highly expressed in LUAD (G). Kaplan-Meier plots showed the correlation of KIF11 with overall survival in LUAD (H), including the patient underwent chemotherapy (I).
KIF11 acted as a crucial mediator of cisplatin resistance in LUAD induced by CaSR
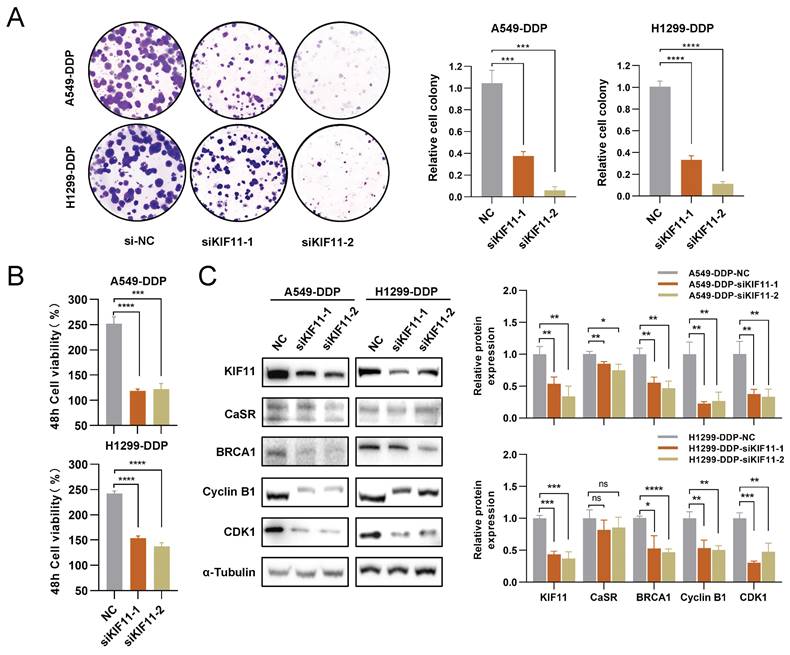
The above investigations revealed that KIF11 exhibited a close association with the prognosis of LUAD under chemotherapy and interacted with CaSR through protein-protein binding. We then investigated the role of KIF11 in CaSR-mediated cisplatin resistance in LUAD cells by disrupting KIF11 expression in DDP-resistant LUAD cells using siRNA transfection. Clonogenicity assay results further demonstrated that KIF11 knockdown significantly inhibited cell growth (Fig. 8A) and cell proliferation assay revealed that the cell growth was almost arrested following KIF11 knockdown (Fig. 8B). The inhibition of cell growth made it challenging to observe the alterations in cisplatin sensitivity of DDP-resistant LUAD cells after KIF11 knockdown, as determined by CCK8 assay. Considering the previous speculation that KIF11 may be the mediator by which CaSR affects BRCA1 and cyclin B1, we further examined the expression levels of BRCA1 and cyclin B1 in DDP-resistant LUAD cells after KIF11 knockdown and observed significant decreases in the expression levels of BRCA1 and cyclin B1 after KIF11 knockdown (Fig. 8C). Furthermore, we investigated the impact of KIF11 knockdown on CaSR and observed a slight reduction in A549-DDP cells (Fig. 8C). However, we did not observe a significant effect of KIF11 knockdown on CaSR expression in H1299-DDP cells. Additionally, CDK1 expression in DDP-resistant LUAD cells was significantly inhibited after KIF11 knockdown, suggesting that KIF11 knockdown may affect more aspects of the cell cycle.
Downregulation of KIF11 inhibited the growth of DDP-resistant LUAD cells. A549-DDP and H1299-DDP cells were transfected with siRNA targeting KIF11 (siKIF11-1, siKIF11-2) or non-targeting siRNA (si-NC). After KIF11 knockdown, the clonogenic proliferation of cisplatin-resistant cells was disrupted (A). The proliferation ability of A549-DDP and H1299-DDP were inhibited and the panels show the relative cell viability at the 48 h (B). Western blotting results showed the changes on protein levels of KIF11, CaSR, cyclin B1, CDK1 and BRCA1 (C). Data are presented as the mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001 and ns, not significant.
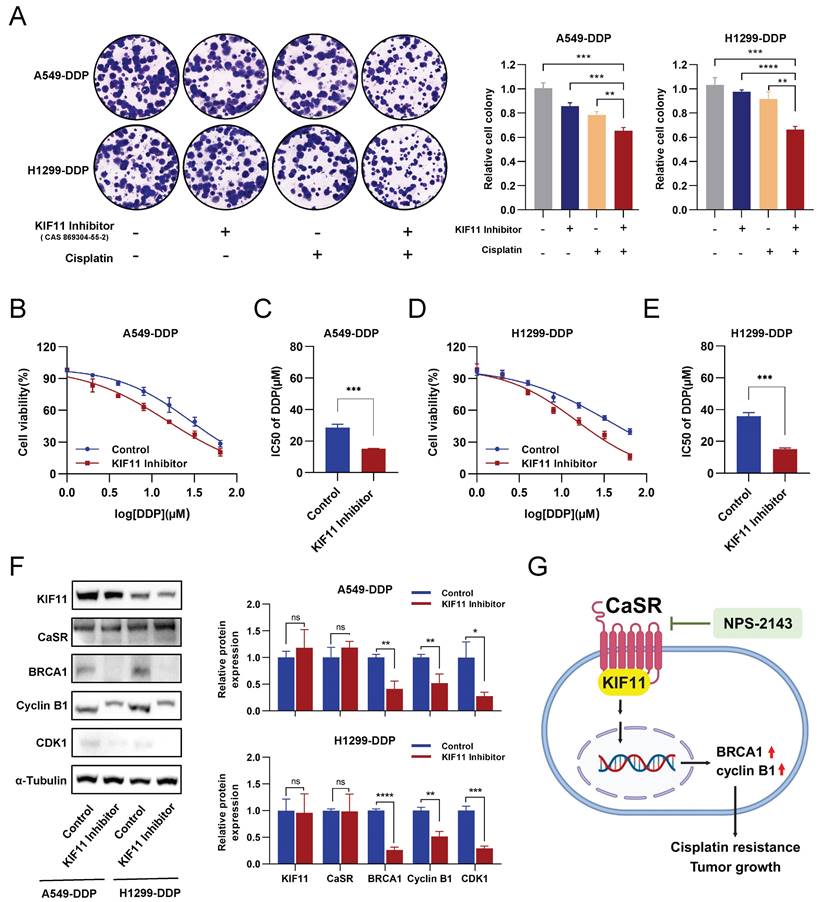
Considering that KIF11 knockdown has a strong effect on cell growth, we then employed KIF11 inhibitors commonly used in cancer research (Fig. S7A). Similarly to KIF11 knockdown, the growth levels of A549-DDP and H1299-DDP cells treated with 10 µM of KIF11 inhibitor were significantly inhibited or even negative (Fig. S7B-D), though the growth inhibition of DDP-resistant LUAD cells receiving KIF11 inhibitor (1 µM) was not obvious (Fig. S7C-D). The clonogenic proliferation assay demonstrated that either the KIF11 inhibitor (1 µM) or cisplatin (10 µM) alone was not significantly effective in inhibiting or eliminating DDP-resistant LUAD cells. In contrast, the combination of the KIF11 inhibitor and cisplatin significantly inhibited the growth of LUAD cells (Fig. 9A). Importantly, similar to treatment with the CaSR inhibitor NPS-2143, DDP-resistant LUAD cells treated with the KIF11 inhibitor (1 µM) showed increased sensitivity to cisplatin (Fig. 9B-E). Western blotting results revealed that treatment with the KIF11 inhibitor (1 µM) did not have an impact on the expression of KIF11 or CaSR in DDP-resistant LUAD cells. Notably, treatment with the KIF11 inhibitor significantly decreased the expression levels of BRCA1 and cyclin B1 (Fig. 9F). To verify the causal relationship, we performed knockdown of BRCA1 in A549-DDP and H1299-DDP cells, observed a significant reduction in cyclin B1 expression upon BRCA1 knockdown. Previous studies have reported the involvement of BRCA1 in the expression and stability of cyclin B1 in breast cancer [40, 47]. Meanwhile, the knockdown of BRCA1 did not result in significant changes in CaSR and KIF11 expression in A549-DDP and H1299-DDP cells (Fig. S8). Based on the observed changes in the sensitivity of DDP-resistant LUAD cell lines to cisplatin after treatment with the KIF11 inhibitor, as well as the detected alterations in the protein levels of CaSR, BRCA1, and cyclin B1, we suggest that the interaction between CaSR and the KIF11 protein could potentially play a role in the pathway through which CaSR influences cisplatin resistance in LUAD cells.
Inhibition of KIF11 reduced cisplatin resistance in DDP-resistant LUAD cells. The clonogenic proliferation of DDP-resistant cells was disrupted by the KIF11 inhibitor (A). A549-DDP cells and H1299-DDP cells were treated with 10 µM cisplatin and 1µM KIF11 inhibitor, respectively, or in combination. Colony formation assay was performed to analyze the cells, and the right panel quantifies the relative number of colonies formed after 14 days. The sensitivity of A549-DDP (B and C) and H1299-DDP (D and E) cells to cisplatin was affected by the KIF11 inhibitor. The protein levels of KIF11, CaSR, cyclin B1, CDK1 and BRCA1 were detected by western blotting (F). The schematics illustrate the proposed mechanism of CaSR activates the cisplatin resistance in LUAD (G). Data are showed as the mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001 and ns, not significant.
Discussion
Lung cancer is the leading cause of cancer-related deaths worldwide and is proving to be a challenging disease to treat. Cisplatin, one of the principal first-choice treatments for NSCLC, efficiently inhibits tumor growth, contributes to the management of the condition, and yields remarkable therapeutic outcomes [48, 49]. However, the development of chemoresistance often leads to treatment failure. Overcoming resistance to cisplatin and enhancing its efficacy continue to be pressing issues in clinical practice. In this study, we analyzed the transcriptomic changes between lung adenocarcinoma cell lines before and after acquiring cisplatin resistance and determined that CaSR was significantly upregulated in DDP-resistant LUAD. The outcome of public database comparisons provided initial evidence that patients with LUAD and clinically elevated levels of CaSR expression were prone to a distressing prognosis following chemotherapy, suggesting that CaSR may play an influential role in the development of tumor resistance to chemotherapy.
CaSR has emerged as an important receptor to target in the treatment of cancer due to the important functions it plays in tumors [50]. In this study, we found that LUAD cell lines overexpressing CaSR had decreased sensitivity to cisplatin compared to the parental cell lines and that platinum drug resistance and cell cycle-related pathways were upregulated at the transcriptional level in the two LUAD cell lines overexpressing CaSR. Simultaneously, pathways related to DNA damage repair were also activated. It is known that cisplatin hinders tumor growth by affecting DNA replication and repair mechanisms within tumor cells [51]. The activation of these pathways suggests that CaSR may function in mediating cisplatin resistance. Further investigation revealed that both DDP-resistant and CaSR-overexpressing LUAD cells exhibited significant increases in cells in the G2/M phase by cell cycle analysis. We know that normal cells typically prolong the G1 phase of cell division during the DNA repair process. However, most tumor cells have a dysfunctional G1 checkpoint and often require a functional G2/M checkpoint for DNA repair [52, 53]. Combined with our results, it is reasonable to speculate that the prolongation of the G2/M phase of these cells gives them more time to self-repair and acquire resistance to cisplatin. We also observed a significant upregulation of cyclin B1, a protein closely associated with the G2/M phase of the cell cycle and chemoresistance. Several studies have demonstrated the crucial involvement of cyclin B1 in promoting cell cycle progression and enhancing the development of drug-resistant tumors. Additionally, researchers have found that cyclin B1 could be linked to the metabolic reprogramming involved in tumor adaptive resistance [54, 55]. Recent studies have indicated that disruption of cyclin B1 can affect the sensitivity of nasopharyngeal carcinoma cells to cisplatin [56]. Meanwhile, high levels of BRCA1 have been detected in LUAD cell lines that are resistant to cisplatin. BRCA1 was the first gene linked to hereditary breast cancer [57]and acts as a tumor suppressor with several functional domains involved in significant biological processes, such as DNA damage repair and regulation of the G2/M checkpoint [58]. BRCA1 has also been implicated in the response to cisplatin resistance. Platinum-induced DNA cross-linking, a significant and lethal form of DNA damage that can lead to double-stranded breaks (DSBs), is well established, and BRCA1 is important in activating DSB repair [59]. According to an analysis of prognostic data from 57 patients with locally advanced bladder cancer, patients with elevated BRCA1 expression were less likely to benefit from cisplatin-based therapy [60]. In addition, under conditions of DNA damage, BRCA1 can increase the stability of cyclin B1 through protein interactions [40]. In NSCLC cells that are resistant to cisplatin, the homologous recombination repair function of BRCA1 has been reported to be enhanced [61], supporting our findings to some extent. Therefore, we hypothesize that CaSR may affect the cisplatin resistance of tumor cells through its effects on cyclin B1 and BRCA1.
To comprehensively assess the role of CaSR, siRNA interference was employed to suppress CaSR expression in DDP-resistant LUAD cells. The sensitivity to cisplatin in DDP-resistant LUAD cells was increased after CaSR knockdown, and the protein expression levels of BRCA1 and cyclin B1 were decreased. We observed that overexpression and knockdown of CaSR all influenced the proportion of cells in the G2/M phase, but leading to varied effects on cell proliferation. As we know, cell proliferation is a multifaceted process influenced by factors such as cell cycle, metabolism and apoptosis [62-64]. Relying solely on cell cycle distribution may not accurately determine the cell's proliferation. Furthermore, we detected the effects of CaSR overexpression on the metabolism and apoptosis of LUAD cells. The overexpression of CaSR in LUAD cells resulted in a reduction in glycolysis and glycolytic capacity (Fig. S4F). Meanwhile, the percentage of apoptosis cells was significantly decreased in LUAD cells overexpressing CaSR (Fig. S4G). Given these findings, it is plausible that the combined effects of these conditions influence the growth status of LUAD cells.
CaSR modulators have various potential applications in treating certain diseases [65]. To further confirm the role of CaSR, we also performed in vitro and in vivo experiments with CaSR inhibitors. The results showed that NPS-2143 could reverse the resistance of DDP-resistant LUAD cells in vitro. In our subsequent experiment, cisplatin combined with NPS-2143 was found to significantly suppress the growth of DDP-resistant LUAD cells in immunodeficient mice. These findings provide recommendations for overcoming cisplatin resistance in LUAD. In addition, both in vitro and in vivo, NPS-2143 showed an effect on the protein levels of BRCA1 and cyclin B1 in DDP-resistant LUAD cells.
Currently, little is known about the relationship between CaSR, BRCA1, and cyclin B1. It has been reported that the administration of the CaSR agonist NPS-R-568 in oocytes can increase the levels of cyclin B1 [66], while CaSR can protect BRCA1-deficient cells from the negative consequences of the loss of BRCA1 function [67]. However, the effects of CaSR on BRCA1 and cyclin B1 have not been previously evaluated. To or knowledge, this was the first study to propose a protein interaction between CaSR and KIF11. Specifically, KIF11 is known to play a critical role in mitosis and is closely related to the G2/M phase [68]. We also found that KIF11 was positively correlated with BRCA1 and cyclin B1 expression in the LUAD gene co-expression database. In addition, KIF11 knockdown or inhibition was observed to decrease the protein expression levels of BRCA1 and cyclin B1 but did not significantly affect the protein expression of CaSR. Meanwhile, depletion of BRCA1 resulted in a significant decrease in cyclin B1 levels, but did not have a significant effect on the expression of CaSR and KIF11 in A549-DDP and H1299-DDP cells. These findings indicate that KIF11 serves as a crucial mediator of CaSR, which can influence cisplatin resistance as well as the expression of BRCA1 and cyclin B1 in LUAD cells. Furthermore, suppression of KIF11 led to a reduction in CDK1 expression and induced substantial cell growth arrest. These findings suggest that the role of KIF11 may be multifaceted and should be taken into consideration. Further studies are needed to determine how CaSR and KIF11 interact to influence their respective functions, how KIF11 influences the expression of BRCA1 and cyclin B1, and whether there are alternative pathways by which CaSR contributes to DNA damage repair and mitosis.
Conclusion
In this study, we identified a novel mechanism of cisplatin resistance in LUAD cells (Fig. 9G). CaSR enhanced cisplatin resistance in LUAD cells by affecting BRCA1 and cyclin B1, through KIF11 interaction. Moreover, CaSR altered the ability of LUAD to combat DNA damage and comprehensively changed the sensitivity of LUAD to cisplatin. Hence, we presented a novel molecular marker for diagnosing chemosensitivity in LUAD and a potential method for reversing multidrug resistance. Our study also suggested that the combination of a CaSR inhibitor and cisplatin may represent a new therapeutic strategy for the treatment of chemoresistant LUAD.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript. We sincerely thank Dr. Xinyu Ye, Dr. Xiaoyi Li, Dr. Shu Yang and Dr. Xinyu Wang for their useful suggestions and technical help. We also acknowledge the technical support provided by the SUSTech Core Research Facilities.
Funding
This work was supported by NSFC Project (81972766, 82173336, 82073388 and 81972420); The Science and Technology Project of Shenzhen (JCYJ20190809161811237, JCYJ20210324104214040); the Natural Outstanding Youth Fund of Guangdong Province (2022B1515020090).
Ethics committee approval
All animal experiments and procedures were approved and performed in accordance with the Animal Experimentation Ethics Committee of Southern University of Science and Technology.
Author contributions
FW, YL, and JZ designed the whole project; FW, MC, TW, RL, HT, XH and RY performed the experiments; FW, WZ, XZ and ZZ analyzed the data; WS provided clinical samples; FW wrote the manuscript; XF, YL, JZ, and ZL revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Wagle NS. et al. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17-48
2. Sung H, Ferlay J, Siegel RL. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209-49
3. Wu F, Wang L, Zhou C. Lung cancer in China: current and prospect. Curr Opin Oncol. 2021;33:40-6
4. Chen Z, Fillmore CM, Hammerman PS. et al. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14:535-46
5. Alduais Y, Zhang H, Fan F. et al. Non-small cell lung cancer (NSCLC): A review of risk factors, diagnosis, and treatment. Medicine (Baltimore). 2023;102:e32899
6. Chen J, Zhang K, Zhi Y. et al. Tumor-derived exosomal miR-19b-3p facilitates M2 macrophage polarization and exosomal LINC00273 secretion to promote lung adenocarcinoma metastasis via Hippo pathway. Clin Transl Med. 2021;11:e478
7. Pirker R. Chemotherapy remains a cornerstone in the treatment of nonsmall cell lung cancer. Curr Opin Oncol. 2020;32:63-7
8. Forde PM, Spicer J, Lu S. et al. Neoadjuvant Nivolumab plus Chemotherapy in Resectable Lung Cancer. N Engl J Med. 2022;386:1973-85
9. Chemotherapy in non-small cell lung cancer. a meta-analysis using updated data on individual patients from 52 randomised clinical trials. Non-small Cell Lung Cancer Collaborative Group. BMJ. 1995;311:899-909
10. Group NM-AC. Chemotherapy in addition to supportive care improves survival in advanced non-small-cell lung cancer: a systematic review and meta-analysis of individual patient data from 16 randomized controlled trials. J Clin Oncol. 2008;26:4617-25
11. Ettinger DS, Wood DE, Aisner DL. et al. Non-Small Cell Lung Cancer, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2022;20:497-530
12. Griesinger F, Korol EE, Kayaniyil S. et al. Efficacy and safety of first-line carboplatin-versus cisplatin-based chemotherapy for non-small cell lung cancer: A meta-analysis. Lung Cancer. 2019;135:196-204
13. Rossi A, Di Maio M. Platinum-based chemotherapy in advanced non-small-cell lung cancer: optimal number of treatment cycles. Expert Rev Anticancer Ther. 2016;16:653-60
14. Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364-78
15. Kryczka J, Kryczka J, Czarnecka-Chrebelska KH. et al. Molecular Mechanisms of Chemoresistance Induced by Cisplatin in NSCLC Cancer Therapy. Int J Mol Sci. 2021;22:8885
16. Brown EM, Gamba G, Riccardi D. et al. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature. 1993;366:575-80
17. Saidak Z, Mentaverri R, Brown EM. The role of the calcium-sensing receptor in the development and progression of cancer. Endocr Rev. 2009;30:178-95
18. Busic-Pavlek I, Dumic-Cule I, Kovacevic L. et al. Calcium-Sensing Receptor Expression in Breast Cancer. Int J Mol Sci. 2023;24:11678
19. Liao J, Schneider A, Datta NS. et al. Extracellular calcium as a candidate mediator of prostate cancer skeletal metastasis. Cancer Res. 2006;66:9065-73
20. Liu L, Fan Y, Chen Z. et al. CaSR Induces Osteoclast Differentiation and Promotes Bone Metastasis in Lung Adenocarcinoma. Front Oncol. 2020;10:305
21. Chai X, Yinwang E, Wang Z. et al. Predictive and Prognostic Biomarkers for Lung Cancer Bone Metastasis and Their Therapeutic Value. Front Oncol. 2021;11:692788
22. Tuffour A, Kosiba AA, Zhang Y. et al. Role of the calcium-sensing receptor (CaSR) in cancer metastasis to bone: Identifying a potential therapeutic target. Biochim Biophys Acta Rev Cancer. 2021;1875:188528
23. Singh N, Liu G, Chakrabarty S. Isolation and characterization of calcium sensing receptor null cells: a highly malignant and drug resistant phenotype of colon cancer. Int J Cancer. 2013;132:1996-2005
24. Zhong T, Zhang W, Guo H. et al. The regulatory and modulatory roles of TRP family channels in malignant tumors and relevant therapeutic strategies. Acta Pharm Sin B. 2022;12:1761-80
25. El Hiani Y, Lehen'kyi V, Ouadid-Ahidouch H. et al. Activation of the calcium-sensing receptor by high calcium induced breast cancer cell proliferation and TRPC1 cation channel over-expression potentially through EGFR pathways. Arch Biochem Biophys. 2009;486:58-63
26. Borowiec AS, Bidaux G, Pigat N. et al. Calcium channels, external calcium concentration and cell proliferation. Eur J Pharmacol. 2014;739:19-25
27. Wang Z, Fan M, Candas D. et al. Cyclin B1/Cdk1 coordinates mitochondrial respiration for cell-cycle G2/M progression. Dev Cell. 2014;29:217-32
28. Zhu C, Xie Y, Li Q. et al. CPSF6-mediated XBP1 3'UTR shortening attenuates cisplatin-induced ER stress and elevates chemo-resistance in lung adenocarcinoma. Drug Resist Updat. 2023;68:100933
29. Chen P, Liu Y, Wen Y. et al. Non-small cell lung cancer in China. Cancer Commun (Lond). 2022;42:937-70
30. Gyorffy B, Surowiak P, Budczies J. et al. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One. 2013;8:e82241
31. Chang M, He Y, Liu C. et al. Downregulation of SEPTIN5 inhibits prostate cancer progression by increasing CD8(+) T cell infiltration. Int J Biol Sci. 2022;18:6035-51
32. Wei T, Lin R, Fu X. et al. Epigenetic regulation of the DNMT1/MT1G/KLF4/CA9 axis synergises the anticancer effects of sorafenib in hepatocellular carcinoma. Pharmacol Res. 2022;180:106244
33. Katchalski-Katzir E, Shariv I, Eisenstein M. et al. Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc Natl Acad Sci U S A. 1992;89:2195-9
34. Morris GM, Huey R, Olson AJ. Using AutoDock for ligand-receptor docking. Curr Protoc Bioinformatics. 2008 Chapter 8: Unit 8 14
35. Anantha RW, Simhadri S, Foo TK. et al. Functional and mutational landscapes of BRCA1 for homology-directed repair and therapy resistance. Elife. 2017;6:e21350
36. Taron M, Rosell R, Felip E. et al. BRCA1 mRNA expression levels as an indicator of chemoresistance in lung cancer. Hum Mol Genet. 2004;13:2443-9
37. Fassl A, Geng Y, Sicinski P. CDK4 and CDK6 kinases: From basic science to cancer therapy. Science. 2022;375:eabc1495
38. Hochegger H, Takeda S, Hunt T. Cyclin-dependent kinases and cell-cycle transitions: does one fit all? Nat Rev Mol Cell Biol. 2008;9:910-6
39. Qu J, Ke F, Liu Z. et al. Uncovering the mechanisms of dandelion against triple-negative breast cancer using a combined network pharmacology, molecular pharmacology and metabolomics approach. Phytomedicine. 2022;99:153986
40. Choi EK, Lim JA, Kim JK. et al. Cyclin B1 stability is increased by interaction with BRCA1, and its overexpression suppresses the progression of BRCA1-associated mammary tumors. Exp Mol Med. 2018;50:1-16
41. Pustovalova M, Alhaddad L, Smetanina N. et al. The p53-53BP1-Related Survival of A549 and H1299 Human Lung Cancer Cells after Multifractionated Radiotherapy Demonstrated Different Response to Additional Acute X-ray Exposure. Int J Mol Sci. 2020;21:3342
42. Schepelmann M, Kupper N, Sladczyk M. et al. Stereo-Specific Modulation of the Extracellular Calcium-Sensing Receptor in Colon Cancer Cells. Int J Mol Sci. 2021;22:10124
43. Zhang ZL, Li ZR, Li JS. et al. Calcium-sensing receptor antagonist NPS-2143 suppresses proliferation and invasion of gastric cancer cells. Cancer Gene Ther. 2020;27:548-57
44. Awata H, Huang C, Handlogten ME. et al. Interaction of the calcium-sensing receptor and filamin, a potential scaffolding protein. J Biol Chem. 2001;276:34871-9
45. Kifor O, Diaz R, Butters R. et al. The calcium-sensing receptor is localized in caveolin-rich plasma membrane domains of bovine parathyroid cells. J Biol Chem. 1998;273:21708-13
46. Rath O, Kozielski F. Kinesins and cancer. Nat Rev Cancer. 2012;12:527-39
47. Somasundaram K. Breast cancer gene 1 (BRCA1): role in cell cycle regulation and DNA repair-perhaps through transcription. J Cell Biochem. 2003;88:1084-91
48. Planchard D, Popat S, Kerr K. et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29:iv192-iv237
49. Hanna N, Johnson D, Temin S. et al. Systemic Therapy for Stage IV Non-Small-Cell Lung Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J Clin Oncol. 2017;35:3484-515
50. Pereira RS, Kumar R, Cais A. et al. Distinct and targetable role of calcium-sensing receptor in leukaemia. Nat Commun. 2023;14:6242
51. Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573-84
52. Chen T, Stephens PA, Middleton FK. et al. Targeting the S and G2 checkpoint to treat cancer. Drug Discov Today. 2012;17:194-202
53. Matheson CJ, Backos DS, Reigan P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol Sci. 2016;37:872-81
54. Xie B, Wang S, Jiang N. et al. Cyclin B1/CDK1-regulated mitochondrial bioenergetics in cell cycle progression and tumor resistance. Cancer Lett. 2019;443:56-66
55. Liu HY, Liu YY, Yang F. et al. Acetylation of MORC2 by NAT10 regulates cell-cycle checkpoint control and resistance to DNA-damaging chemotherapy and radiotherapy in breast cancer. Nucleic Acids Res. 2020;48:3638-56
56. Xie L, Shi F, Li Y. et al. Drp1-dependent remodeling of mitochondrial morphology triggered by EBV-LMP1 increases cisplatin resistance. Signal Transduct Target Ther. 2020;5:56
57. Miki Y, Swensen J, Shattuck-Eidens D. et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66-71
58. Yoshida K, Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004;95:866-71
59. Tarsounas M, Sung P. The antitumorigenic roles of BRCA1-BARD1 in DNA repair and replication. Nat Rev Mol Cell Biol. 2020;21:284-99
60. Font A, Taron M, Gago JL. et al. BRCA1 mRNA expression and outcome to neoadjuvant cisplatin-based chemotherapy in bladder cancer. Ann Oncol. 2011;22:139-44
61. Ray Chaudhuri A, Callen E, Ding X. et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535:382-7
62. Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342-8
63. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029-33
64. Chang L, Fang S, Gu W. The Molecular Mechanism of Metabolic Remodeling in Lung Cancer. J Cancer. 2020;11:1403-11
65. Nemeth EF, Shoback D. Calcimimetic and calcilytic drugs for treating bone and mineral-related disorders. Best Pract Res Clin Endocrinol Metab. 2013;27:373-84
66. Liu C, Wu GQ, Fu XW. et al. The Extracellular Calcium-Sensing Receptor (CASR) Regulates Gonadotropins-Induced Meiotic Maturation of Porcine Oocytes. Biol Reprod. 2015;93:131
67. Singh N, Promkan M, Liu G. et al. Role of calcium sensing receptor (CaSR) in tumorigenesis. Best Pract Res Clin Endocrinol Metab. 2013;27:455-63
68. Qi X, Liu Y, Peng Y. et al. UHRF1 promotes spindle assembly and chromosome congression by catalyzing EG5 polyubiquitination. J Cell Biol. 2023;222:e202210093
Author contact
![]() Corresponding authors: Yi Lu (luy3edu.cn), Zhen Liang (liang.zhencom) and Jian Zhang (zhangjianedu.cn).
Corresponding authors: Yi Lu (luy3edu.cn), Zhen Liang (liang.zhencom) and Jian Zhang (zhangjianedu.cn).