Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Pathophysiological role of...
3. Pathophysiological role of...
4. The interface of oxidative...
5. Nrf2 as a multifaceted...
6. Sestrin as a multifunctional...
7. Role of HO-1 in metabolic...
8. Crosstalk between Nrf2,...
9. Conclusion and future...
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(12):4888-4907. doi:10.7150/ijbs.98846 This issue Cite
Review
Antioxidant genes in cancer and metabolic diseases: Focusing on Nrf2, Sestrin, and heme oxygenase 1
Jitendra Shrestha1,2#, Khem Raj Limbu1#, Rashmi Bhandari Chhetri1, Keshav Raj Paudel3, Philip M. Hansbro3, Yoon Sin Oh4, Dong Jae Baek1 ![]() , Sung-Hwan Ki5
, Sung-Hwan Ki5 ![]() , Eun-Young Park1
, Eun-Young Park1 ![]()
1. College of Pharmacy, Mokpo National University, Jeonnam 58554, Republic of Korea.
2. Massachusetts General Hospital Cancer Center, Department of Medicine, Harvard Medical School, Boston, MA 02114, USA.
3. Centre for Inflammation, Centenary Institute and University of Technology Sydney, Faculty of Science, School of Life Sci., Sydney, NSW 2007, Australia.
4. Department of Food and Nutrition, Eulji University, Seongnam 13135, Republic of Korea.
5. College of Pharmacy, Chosun University, Gwangju 61451, Republic of Korea.
# These authors contributed equally.
Received 2024-5-24; Accepted 2024-9-3; Published 2024-9-9
Abstract

Reactive oxygen species are involved in the pathogenesis of cancers and metabolic diseases, including diabetes, obesity, and fatty liver disease. Thus, inhibiting the generation of free radicals is a promising strategy to control the onset of metabolic diseases and cancer progression. Various synthetic drugs and natural product-derived compounds that exhibit antioxidant activity have been reported to have a protective effect against a range of metabolic diseases and cancer. This review highlights the development and aggravation of cancer and metabolic diseases due to the imbalance between pro-oxidants and endogenous antioxidant molecules. In addition, we discuss the function of proteins that regulate the production of reactive oxygen species as a strategy to treat metabolic diseases. In particular, we summarize the role of proteins such as nuclear factor-like 2, Sestrin, and heme oxygenase-1, which regulate the expression of various antioxidant genes in metabolic diseases and cancer. We have included recent literature to discuss the latest research on identifying novel signals of antioxidant genes that can control metabolic diseases and cancer.
Keywords: nuclear factor (erythroid-derived 2)-like 2, Sestrin, heme oxygenase 1, metabolic diseases, cancer
1. Introduction
Oxidative stress arises from an imbalance between the production of reactive oxygen species (ROS) and the cellular antioxidant defense mechanisms [1, 2]. ROS, encompassing superoxide anion (O2•-), hydrogen peroxide (H2O2), and hydroxyl radical (•OH), are indispensable byproducts of cellular metabolism but they can exert deleterious effects when their levels exceed physiological thresholds. Moreover, reactive nitrogen species, such as nitric oxide (•NO) and peroxynitrite (ONOO-), and reactive sulfur species, like hydrogen sulfide (H2S) and persulfides, play crucial roles in modulating redox homeostasis and contributing to oxidative stress [3].
ROS are generated through endogenous processes, such as mitochondrial respiration and immune responses, as well as exogenous factors including environmental pollutants and radiation [4]. Cells possess sophisticated antioxidant defense mechanisms, including both enzymatic (e.g., superoxide dismutase, catalase, and glutathione peroxidase) and non-enzymatic components (e.g., vitamins C and E), to mitigate oxidative stress. Additionally, naturally occurring antioxidant compounds (caffeic acid phenethyl ester, thymoquinone, rutin, catechin, and berberine) are widely used as antioxidant supplements [5-10]. Enzymatic antioxidants, including superoxide dismutase, catalase, glutathione peroxidase, and thioredoxin, are pivotal in conferring protection against oxidative stress due to their efficient ROS-scavenging capabilities [11]. The expression of antioxidant enzymes is stringently regulated by transcription factors, including nuclear factor (erythroid-derived 2)-like 2 (Nrf2), Sestrins, and heme oxygenase-1 (HO-1) [12-14]. Oxidative stress elicits a complex transcriptional response. A plethora of genes, including those involved in antioxidant defense, deoxyribonucleic acid (DNA) repair, and cellular signaling (e.g., NQO1, TXNRD1, PRDX4, TERT, SMUG1, H2AFX, DUSP1, NOS3, TCIRG1, and PFKR), are upregulated in response to ROS. Conversely, other genes (e.g., PFKP, CYR2, and CAT) exhibit attenuated expression under pathological conditions, including cancer [15-17]. Beyond transcriptional changes, oxidative stress can also induce epigenetic modifications. These epigenetic modifications, such as DNA methylation and histone post-translational modifications, represent heritable changes in gene expression without altering the underlying DNA sequence. Targeting these epigenetic alterations presents a promising therapeutic strategy for cancer treatment [18].
Oxidative stress plays a pivotal role in the pathogenesis of various diseases, including metabolic, cardiovascular, cancer, and neurodegenerative disorders [19-21]. In the cardiovascular system, elevated ROS contribute to the etiology of hypertension, vasoconstriction, arrhythmias, and cardiac remodeling through mechanisms involving nitric oxide depletion, calcium homeostasis perturbation, and hypertrophic signaling cascades [22]. Nitric oxide exhibits dichotomous effects, exerting both neuroprotective and neurodegenerative influences, contingent upon its source and downstream signaling pathways. Neuroprotective actions involve the activation of the CREB and protein kinase B (AKT) pathways, while neurodegenerative effects are often mediated by iNOS induction [23]. Metabolic diseases, including type 2 diabetes, obesity, hyperlipidemia and non-alcoholic fatty liver disease, are characterized by metabolic dysregulation and elevated oxidative stress. These conditions share common pathological features, such as insulin resistance, chronic low-grade inflammation, and mitochondrial dysfunction, enhanced protein and lipid oxidation, and compromised antioxidant systems, which collectively promote the production of inflammatory cytokines [24]. While other metabolic disorders exhibit similar characteristics, this review will focus on these diseases to provide a comprehensive understanding of the role of oxidative stress and antioxidant genes in their pathophysiology.
ROS have a dual role in cancer. ROS can promote tumorigenesis by stimulating cancer cell proliferation, invasion, and progression through interactions with cancer-associated fibroblasts. Additionally, ROS can induce genomic instability and epigenetic alterations, contributing to cancer progression [25]. ROS also activate various antioxidant genes within cancer cells as a self-defense mechanism. Conversely, several anticancer therapies exploit this vulnerability by inducing ROS generation, leading to cancer cell death via apoptosis or necrosis [21].
Nrf2, a pivotal transcription factor, is ubiquitously expressed and orchestrates the expression of numerous cytoprotective genes involved in antioxidant defense, detoxification, and redox homeostasis [26, 27]. HO-1, a downstream target of Nrf2, catalyzes the rate-limiting step in heme breakdown, yielding biliverdin and carbon monoxide (CO), along with equimolar iron ions [13]. Sestrin family proteins are crucial stress-induced proteins regulated by the Nrf2/ antioxidant responsive element (ARE) pathway [28]. Sestrin2, conversely, activates Nrf2 by promoting the P62-dependent autophagic pathway through the degradation of the Kelch-like-ECH-associated protein 1 (KEAP1) [29]. The intricate interplay between Nrf2, Sestrins, and HO-1 underscores their critical roles in maintaining cellular redox homeostasis and conferring protection against oxidative stress-related pathologies, including metabolic and inflammatory diseases [30-32].
This review aims to elucidate the fundamental regulatory mechanisms of oxidative stress and antioxidant genes, with particular emphasis on Nrf2, Sestrin, and HO-1, in the pathogenesis of metabolic and neoplastic disorders. Through a comprehensive analysis of the regulatory pathways governing these genes, we endeavor to identify potential therapeutic targets and innovative strategies for the prevention and treatment of these complex pathologies. Understanding the multifaceted roles of Nrf2, Sestrin, and HO-1 in metabolic disturbances and cancer progression is imperative for developing novel therapeutic interventions to improve patient outcomes.
2. Pathophysiological role of oxidative stress in metabolic disease
Under oxidative stress, free radicals that are generated in the cell are superoxide anion radical, hydrogen peroxide (H2O2), nitric oxide, and peroxynitrite [33]. The excessive generation of free radicals induces DNA damage, mitochondrial dysfunction, and protein misfolding [33]. This cellular damage caused by free radicals ultimately impairs the function of the major metabolic organs [34, 35]. Therefore, chronic exposure to ROS inhibits the proper utilization of nutrients, accumulates fat in several organs, and increases blood glucose levels (Figure 1). Oxidative stress is recognized as a cause of metabolic diseases such as obesity, insulin resistance, and diabetes [36] (Figure 1).
The role of oxidative stress in metabolic syndrome. Obesity promotes the deposition of white fat mass, increasing ROS production, and a reduction in antioxidant enzymes and adiposity. Immune cells exposed to oxidative stress generate a variety of inflammatory cytokines that lead to arterial hypertension and atherosclerosis. Energy expenditure in the brain becomes imbalanced, resulting in brain hypertension. Insulin resistance causes an increase in blood glucose levels, which leads to pancreatic beta-cell malfunctioning and decreases beta-cell survival. ROS: Reactive oxygen species, PAI-1: Plasminogen activator inhibitor 1, TNFα: Tumor necrosis factor alpha, IL-6: Interleukin-6, MCP-1: Monocyte chemoattractant protein-1.
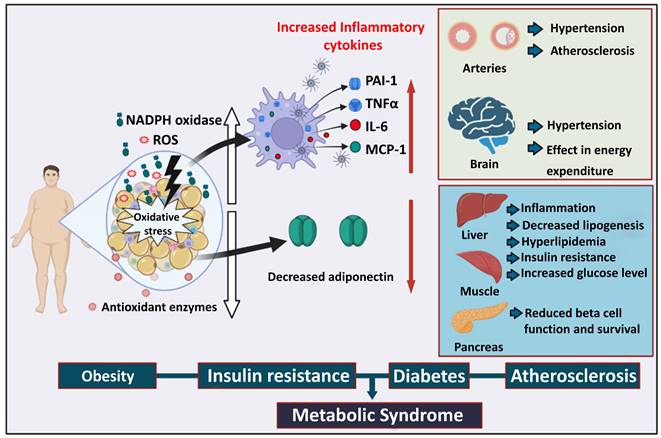
Several reports have suggested a close relationship between adipogenesis and ROS levels [37]. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase is important in initiating adipocyte differentiation from pre-adipocytes and mesenchymal stem cells. The knockdown of NADPH oxidase 4 (NOX4), an isoform of NADPH oxidase, inhibited adipocyte differentiation [38], whereas H2O2-induced oxidative stress and NOX4 overexpression increased lipid storage. The accumulation of ROS in fat due to increased NADPH oxidase causes the inadequate production of adipokines [37]. By increasing the expression of NADPH oxidase and decreasing the expression of antioxidant enzymes, the accumulation of fat (for instance, in the adipose tissue of obese mice) caused oxidative stress, which negatively impacted the other organs like the skeletal muscle, liver, and pancreatic beta cells [34]. Therefore, excessive oxidative stress appears to be associated with the pathogenesis of obesity-associated metabolic syndrome (Figure 1).
Oxidative stress plays an essential role in the dysregulation of glucose uptake and insulin sensitivity under diabetic conditions [39]. H2O2-induced oxidative stress significantly reduces the mRNA and protein expression of the glucose transporter 4 (GLUT4) in adipocytes and muscles [34]. GLUT4 could be involved in peripheral insulin sensitivity and glucose uptake; thus, its expression is important in improving diabetes [40]. Oxidative stress impairs insulin signaling by increasing the serine phosphorylation of insulin receptor substrate-1 (IRS-1) or blocking the pathway between IRS-1 and phosphatidylinositol 3-kinase [39]. Furthermore, excessive oxidative stress decreases the level of adiponectin, an adipokine that regulates insulin sensitivity and inflammatory response [41]. Pancreatic beta cells may be sensitive to ROS because of the low expression of antioxidant enzymes such as CAT and glutathione peroxidase [42]. The insulin gene promoter activity and mRNA expression are suppressed in pancreatic beta cells or isolated rat islets when exposed to an oxidative stress inducer [43]. Therefore, pancreatic beta cells are important targets for oxidative stress-induced diabetes progression [43]. Taken together, excessive oxidative stress plays a critical role in insulin resistance and diabetes progression. Conversely, antioxidant genes appear to exert a protective effect by neutralizing deleterious ROS, thereby protecting pancreatic beta cells from oxidative damage.
Excessive ROS generation leads to oxidative stress, resulting in the development of the chronic inflammatory disease atherosclerosis. Vascular cells under increased oxidative stress experience endothelial dysfunction and more oxidative stress, and they convert low-density lipoprotein (LDL) to oxidized LDL (oxLDL). Macrophages then phagocytose oxLDL, after which they transform into lipid-laden foam cells that release chemokines and cytokines. Lesional macrophages release the inflammatory cytokine interleukin-1β (IL-1β), further exacerbating endothelial dysfunction. This creates a feedback loop that accelerates plaque formation and recruits more monocytes and T lymphocytes into the subendothelial area. These deposits begin as fatty streaks and eventually progress to more intricate forms of atherosclerotic plaque formation [44, 45]. Similarly, autophagy protein 5 (ATG5), thiol oxidative stress, 7-hydroperoxide (7-OOH), and macrophage cellular oxidation have been demonstrated to promote macrophage oxidative stress and increase atherosclerotic plaque formation [46]. According to recent research, tetrapod PdH nanoenzymes interact with macrophages and strongly inhibit ROS, prevented the oxidative modification of trapped LDL and downregulated TNF-α, IL-1β, and IL-6, thereby exerting a strong antiatherosclerosis effect [47]. These findings indicate that increased ROS generation and oxidative stress contribute to the progression of atherosclerosis.
3. Pathophysiological role of oxidative stress in cancer
Cancer is one of the most lethal diseases and is characterized by the transformation of aberrant cells harboring diverse mutations in a set of oncogenes that promote their uncontrolled proliferation and dissemination either in local or distant organs, as well as promoting angiogenesis, immortalization, metastasis, and epithelial-mesenchymal transition (EMT) [48]. The cellular metabolic rate is considerably upregulated to fulfill the massive energy demands of cancer cells that rapidly multiply. The mitochondrial activity of cancer cells remains intact in most cancer types, although glucose metabolism is enhanced in some cancer types and occasionally converted to anaerobic metabolism, demonstrating Warburg's effect [49]. These events in cancer cells ultimately increase the production of various free radicals and ROS within the cancer microenvironment [50]. H2O2 and oxygen free radicals, primarily generated by the mitochondria, are significant ROS in the cancer microenvironment [36, 49].
Considerable literature has described the multifaceted role of oxidative stress in the occurrence of cancer, genetic instability, cancer progression, metastasis, angiogenesis, and cancer treatment by various chemotherapeutic drugs (Figure 2) [21, 51]. Perillo et al., 2020 presented ROS as a double-edged sword in cancer in their review, highlighting the role of ROS in cancer progression through the conversion of fibroblasts into cancer-associated fibroblasts (CAF), which then begin to generate ROS and stimulate cancer cell death pathways by the produced ROS in response to various cancer treatment approaches (Figure 2) [21]. The ROS generated by CAF stimulates the expression of several antioxidant genes that promote cancer cell growth and invasion [52]. Suraweera et al. reviewed the function of antioxidant flavonoids in both the development and treatment of cancer. The function of numerous antioxidant substances has been explored for their specific involvement in cancer, given that the antioxidant system has a dual role in advancing cancer and its treatment [53]. Initially, antioxidant activity is regarded as a crucial factor in promoting tumorigenesis. Mutations in Nrf2 and its regulator KEAP1 have been identified in many cancer types [54]. The breast cancer susceptibility 1 (BRCA1) gene interacts with and increases Nrf2 expression, thereby improving cancer cell survival [55]. In addition, highly efficient peroxiporin activity in pancreatic cancer suggests the presence of an aquaporin AQP5-mediated H2O2 influx, which activates important cell survival and cancer progression signaling networks. At the same time, AQP3/5 silencing significantly reduced cell migration but was recovered by external treatment with H2O2 [56]. These studies support the idea that antioxidant genes and cancer have a complex functional interaction. Although antioxidant gene expression in normal cells serves to mitigate damage induced by ROS, cancer cells can exploit this system for their benefit [57]. First, cancer cells can neutralize ROS produced through their increased metabolic activity or dysfunctional mitochondria. Second, these cells can mitigate ROS generated by anticancer therapies, thereby promoting resistance [58]. The precise mechanisms underlying the functional correlation between antioxidant genes and cancer warrant further exploration.
Dual role of oxidative stress and ROS in cancer progression and prognosis. In the complex landscape of cancer, reactive oxygen species (ROS) play a paradoxical role. Originating from a myriad of environmental sources and inherent metabolic processes, ROS, at the early stages of cancer, act as facilitators for tumorigenesis. Beyond this, they further exacerbate the malignancy by promoting cancer progression, particularly enhancing metastasis and advancing angiogenesis. However, this very nature of ROS is harnessed therapeutically with several anticancer chemotherapeutic agents and irradiation techniques. These treatments exploit ROS production to inflict damage on cancer cells, leading to DNA disruption, protein degradation, and lipid peroxidation. This cascade of cellular damage steers the malignant cells towards a variety of fates, including apoptosis, destructive autophagy, and even outright necrosis. ETC: Electron transport chain, CAF: Cancer-associated fibroblast, FAK: Focal adhesion kinase, MMP2: Matrix metalloproteinase-2, TIT: Tumor induced T-cell, TAM: Tumor associated macrophage, HIF-1α: Hypoxia inducible factor 1α, VEGF: Vascular endothelial growth factor, TIE2: The endothelial-specific receptor, tyrosine kinase with immunoglobulin-like loops and epidermal growth factor homology domain-2, CXCR4:C-X-C chemokine receptor type-4.

Polychlorinated biphenyls are well-known pollutants that accelerate the advancement of hepatocellular cancer by increasing the pyruvate kinase M2 mediated increment of glycolysis via a ROS-mediated mechanism [59]. ROS activates the Distal-less homeobox-2 (Dlx-2)/Snail axis in breast cancer, causing EMT, glycolytic switch, and mitochondrial suppression, which are essential for metastasis. Increased intracellular ROS in colorectal cancer cells caused by fatty acid oxidation stimulates the mitogen-activated protein kinase (MAPK) cascade, causing EMT in ROS high tumor sphere (RH-TS) cells and enhancing metastasis [60]. In breast cancer, ROS increased angiogenesis through the extracellular signal regulated kinase 1/2 (ERK1/2)-HIF-1α-VEGF pathway induced by RRM2 and decreased apoptosis by activating the NF-κB pathway [61]. The vitagenes gene family encodes antioxidant protein families, including (Hsp) Hsp32, Hsp70, thioredoxin, and sirtuin protein systems. Recent studies have demonstrated the neuroprotective effects of dietary antioxidants, including polyphenols and L-carnitine/acetyl-L-carnitine, through activating hermetic pathways [62]. By activating vitagenes, acetylcarnitine can differentially modulate signal transduction cascades, causing apoptosis/cell death in abnormal cancer cells while enhancing defensive enzymes to stop carcinogenesis and neurodegeneration in normal cells [63]. This suggests that ROS plays a pivotal role in cancer cell survival, proliferation, EMT, metastasis, and preventing cancer progression at the initial stage (Figure 2).
4. The interface of oxidative stress in the relationship between metabolic diseases and cancer
Accumulating evidence suggests that both oxidative and antioxidative genes have functional correlations with metabolic diseases and cancer. Metabolic diseases such as obesity linked to diabetes accelerate the progression of cancer and neoangiogenesis because of increased levels of peripheral estrogens, promitogen cytokines, and growth factors. The long-term inflammation associated with obesity promotes ROS generation, thereby causing cellular damage, stimulating the release of proinflammatory cytokines and transcription factors, and facilitating the spread of cancer [64]. According to a recent report, long-term treatment with hypoglycemic drugs such as dipeptidyl peptidase-4 inhibitors in an animal model of breast cancer triggered ROS overproduction and activated the ROS-dependent Nrf2 and HO-1 genes. Nrf2 and HO-1 activation accelerates the metastasis of breast cancer through an oncogenic ROS/Nrf2/HO-1 axis. Moreover, inhibition of the antioxidant gene HO-1 leads to the inhibition of Nrf2 and suppression of breast cancer metastasis-associated genes [65]. Elevated ROS levels lead to mutations in mitochondrial DNA and activate the Protein kinase c (PKC) and MAPK pathways, promoting the deregulation of NF-κB and survival of cancer cells. ROS-dependent PKC amplification both affects the insulin receptor substrate and disturbs insulin signaling, contributing to the development of type 2 diabetes mellitus [66]. Similarly, recent research linked the etiology of numerous malignancies to obesity. Obesity is associated with both the diagnosis of cancer and an increased risk of cancer-related death because of the advanced disease at the time of diagnosis. Transcriptional regulation downstream of the lipid chaperone fatty acid binding protein (FABP4) is connected to cellular redox, with Nrf2 acting as an upstream regulator. In response to FABP4, Nrf2 promotes cellular proliferation by reducing ROS activity [67]. These findings collectively indicate that antioxidant genes are associated with metabolic disease, and they play functional roles in cancer progression.
5. Nrf2 as a multifaceted regulator in metabolic diseases and cancer
5.1 Nrf2 as a master transcription factor for redox homeostasis
Nrf2 is a basic leucine zipper (bZip) transcription factor with a Cap “n” Collar (CNC) structure. Without stress, Nrf2 is retained in the cytoplasm at a low level due to proteasomal degradation [68]. The degradation of Nrf2 is regulated by KEAP1, which is a negative regulator of Nrf2. KEAP1 acts as an adapter protein for the Cullin 3 (CUL3)/Rbx1 ubiquitin-ligase and a sensor to activate Nrf2 [26, 27]. Various stressors or Nrf2 activators alter the cysteine residues in KEAP1. These changes interfere with the binding of KEAP1 and Nrf2, thus allowing Nrf2 to surpass the proteasomal degradation and accumulation. The accumulated Nrf2 translocates to the nucleus and binds to the small Maf proteins to form a heterodimer, which regulates the transcription of various target genes with an ARE in the promoter region [69] (Figure 3).
Classical model of Nrf2 activation and response. In basal conditions, Nrf2 remains bound to Keap1, leading to its ubiquitylation and degradation. AMPK phosphorylation regulates the fate of Nrf2 in normal conditions. In response to oxidative stress, Keap1 is inactivated, resulting in Nrf2 stabilization. Nrf2 then translocates to the nucleus, binds to small Maf proteins, and activates the transcription of downstream target genes such as Sestrin, HO-1, NQO1, γ-GCL, GST etc., through antioxidant response elements (AREs). Nrf2: Nuclear factor erythroid 2-related factor 2, NQO1: NAD(P)H quinone oxidoreductase, γ-GCL: gamma-glutamyl cysteine ligase, HO-1: Heme oxygenase-1, GST: Glutathione S-transferase.
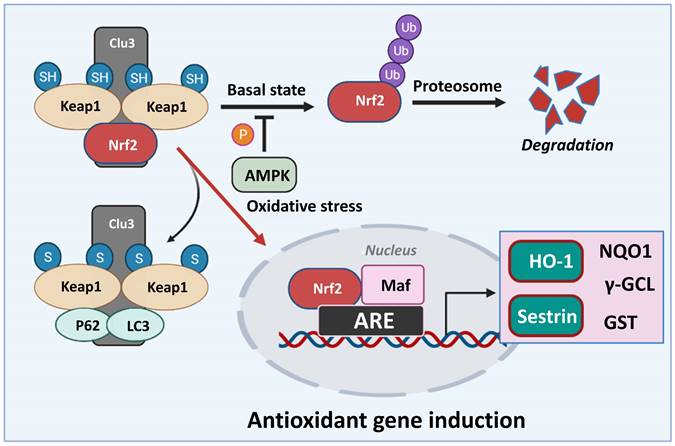
Nrf2 regulates intracellular antioxidant homeostasis by regulating the expression of various proteins. First, Nrf2 modulates the synthesis of glutathione (GSH) by directly regulating the expression of γ-glutamylcysteine synthetase, which is involved in the rate-limiting step of GSH synthesis [27, 70]. Nrf2 also regulates NADPH production and enzymes that mediate the reduction of oxidized GSH [71]. Furthermore, Nrf2 induces a protein responsible for detoxifying various substances that are removed from the body [27]. In addition, Nrf2 contributes to reducing the level of ROS in the cell by regulating the activity of NADPH oxidase [27]. Therefore, the amount of ROS is increased in Nrf2-disrupted cells, which are observed to be more sensitive to oxidative stress.
5.2 Role of Nrf2 in metabolic diseases
5.2.1 Nrf2 and mitochondrial function
Mitochondria are one of the primary sites in cells that produce ROS. They are also essential organelles that initiate cell death signaling by regulating membrane potential. Thus, under oxidative stress conditions, mitochondria can be regarded as a source of oxidative stress and an organelle that induces cell death [72]. In addition, mitochondria produce adenosine triphosphate (ATP) and the importance of ATP in metabolic diseases is widely recognized [73].
Several studies have reported the role of Nrf2 in mitochondrial function. In Nrf2-KO cells, the amount of produced ROS in the mitochondria is increased [74], which may be due to the destruction of the function of complex 1 in the mitochondrial electron transport system. Nrf2 also affects the mitochondrial membrane potential [75]. Decreased mitochondrial membrane potential is important for determining cell death because it is the initiation signal that releases cytochrome c into the cytoplasm [76]. Nrf2 deficiency lowers mitochondrial membrane potential, and the overexpression of Nrf2 by KEAP1-KO upregulates mitochondrial membrane potential [27]. In addition, Nrf2 is deeply involved in mitochondrial biogenesis. Compared to the wild-type, Nrf2-KO mice show reduced mitochondrial DNA in the liver [77]. An important factor for mitochondrial biogenesis is nuclear respiratory factor 1 (NRF1), which is regulated by Nrf2 [78]. The Nrf2 deficiency inhibits the increase in NRF1 mRNA caused by incremental exercise in the mouse muscle [78]. Moreover, Piantadosi et al., demonstrated that Nrf2 can bind with the ARE region in the NRF1 promoter [79]. Since mitochondrial dysfunction is observed in metabolic diseases, the crosstalk between Nrf2 and mitochondria is considered necessary.
5.2.2 Crosstalk between Nrf2 and AMP-activated protein kinase (AMPK)
The goal of cellular metabolism in cells is the production of ATP. AMPK is an enzyme that regulates energy homeostasis by recognizing the amount of ATP. AMPK plays a positive role in various metabolic diseases by inhibiting the ATP consumption reaction while increasing the ATP-producing response [80]. Therefore, crosstalk with the AMPK signal is important for the role of Nrf2 in metabolic diseases. Like Nrf2, AMPK is activated by various intracellular stress conditions. In addition, AMPK activates signals that inhibit inflammation and oxidative stress, similar to those mediated by Nrf2 [80]. Recently, literature on Nrf2 and AMPK signals has accumulated. It has been shown that AMPK directly phosphorylates the Ser550 residues present in the nuclear export signal of Nrf2, thus increasing the nuclear translocation of Nrf2 [81]. Similar results demonstrated that the AMPK activator increases the nuclear level of Nrf2 [82]. Zimmermann et al., 2015 also found that AMPK positively regulates the Nrf2-mediated HO-1 signaling in mouse embryonic fibroblasts [83]. In addition, the upregulation of Nrf2 in KEAP1-KO mice increased the phosphorylation of AMPK in the liver and primary hepatocytes [84]. Taken together, the AMPK signal, which acts as an intracellular energy regulator, is closely related to Nrf2, and this signal crosstalk is expected to contribute to inhibiting metabolic diseases.
5.2.3 Targeting Nrf2 in metabolic diseases
Various experimental results using animal models and Nrf2 activators suggest the role of Nrf2 in metabolic diseases, including obesity and diabetes [85, 86]. Genetic activation of Nrf2 in hypomorphic KEAP1 allele mice fed a high-fat diet ameliorates hepatic gluconeogenesis and lipogenesis, which may be mediated by the activation of AMPK [86]. In contrast, Nrf2-null mice fed a high-fat diet exhibited severe steatohepatitis with cirrhosis due to the upregulation of lipogenic genes and downregulation of β-oxidation genes and AMPK levels [85]. CDDO-Me (2-cyano-3,12-dioxo-oleana-1,9-dien-28-oic acid methyl ester), a potent inducer of Nrf2, reduces body fat, plasma triglycerides, and fatty acids and lowers blood glucose by improving insulin sensitivity in mice fed a high-fat diet and also had an antidiabetic effect in leptin receptor-deficient mice [87]. Although CDDO-Me has been discontinued in Phase III clinical trials, it has significant implications for patients with diabetes [88]. In addition, several compounds that positively affect metabolic disease are accompanied by Nrf2 activation [89, 90]. Nrf2 inducers contribute to the inhibition of diabetes by protecting the pancreatic beta cells from oxidative stress [91]. Apigenin, a naturally occurring compound, was found to bind to Nrf2 and inhibit high-fat diet-induced fatty liver through Nrf2 activation [90]. Hidrox, a polyphenol compound derived from organic olives, has been shown to have antioxidant and anti-inflammatory properties in rotenone-induced oxidative stress [92]. In addition, it has been reported that Ezetimibe, a drug approved for hypercholesterolemia, inhibits diet-induced non-alcoholic steatohepatitis through Nrf2 activation [89]. More disease models are required to be validated and applied to humans, but numerous studies support that the activation of Nrf2 contributes to improving metabolic diseases.
5.3 Role of Nrf2 in cancer
5.3.1 Dichotomous functions of Nrf2 in cancer
Nrf2 is recognized for its cytoprotective properties, safeguarding cells against diverse cellular stresses. However, recent research has suggested a more nuanced role for Nrf2 in cancer. Although Nrf2 activation offers a protective advantage in normal cells, its constitutive upregulation in cancer cells appears to promote tumorigenesis and resistance to chemotherapeutic agents. This upregulation is correlated with chemoresistance to drugs such as 5-fluorouracil (5-FU) in colorectal cancer [93]. Mutations within the Nrf2 pathway, exemplified by those affecting KEAP1, and alternative activation mechanisms contribute to this observed chemoresistance [94]. Somatic mutations in the Nrf2 gene, KEAP1, and the E3 ubiquitin-ligase complex CUL3 have been recognized in many types of cancer, and these mutations are responsible for Nrf2 activation. In addition, Nrf2 activation occurs through alternative splicing of Nrf2 protein lacking a KEAP1-binding site or the overexpression of other proteins that bind with KEAP1. Abnormal Nrf2 activation was found to be responsible for chemoresistance in various cancers, including breast, colorectal, pancreatic, lung, and gastric cancers [95, 96].
Nrf2 activation through diverse mechanisms, such as the interaction between ataxia-telangiectasia group D-associated gene (ATDC) and KEAP1 in pancreatic cancer and the overexpression of progestin and adipo-q receptor family member 4 (PAQR4) in non-small cell lung cancer (NSCLC) cells, promotes cancer progression and treatment resistance [95, 97]. Conversely, Nrf2 knockout enhances chemosensitivity in cervical cancer [91]. The oncogenic potential of Nrf2 is further highlighted by its association with poor prognosis in glioma, while its deletion in glioma stem cells suppresses tumorigenesis.
Ionizing radiation in prostate cancer can activate Nrf2, leading to its nuclear translocation and the upregulation of antioxidant genes (e.g., GCLC, GCLM, HO1), thereby enhancing radiotherapy resistance by increasing GSH synthesis and suppressing apoptosis [98]. Patients with glioma and high Nrf2 expression had poorer prognoses, characterized by shorter disease-free survival and higher tumor grades [99]. Conversely, Nrf2 knockout (Nrf2-KO) in glioma stem cells suppressed their proliferation and downregulated the key tumor-promoting factors Sox2, BMI-1, and cyclin E, leading to inhibited tumorigenesis in nude mice [100]. Similarly, Nrf2-KO in cervical cancer enhanced chemosensitivity and tumor suppression, highlighting its context-dependent role in cancer progression [101]. These findings collectively suggest a role of Nrf2 in cancer; hence, further investigation is required to develop therapeutic strategies.
5.3.2 Targeting Nrf2 in cancer
Despite the context- and cancer-specific roles of Nrf2, various therapeutic approaches target Nrf2 to overcome drug resistance and enhance cancer treatment efficacy. Halofuginone, a febrifugine derivative, has emerged as a potential treatment through inhibiting Nrf2 translation and suppressing its cytoplasmic accumulation, thereby overcoming drug resistance and sensitizing lung cancer cells to chemo- and radiotherapy [102]. Similarly, chalcone derivatives such as 4-methoxy-chalcone (4-MC), hesperidin methylchalcone, and neohesperidin dihydrochalcone target Nrf2, reducing the expression of the downstream antioxidant gene NQO1 and promoting ROS generation, thereby sensitizing lung cancer cells to chemotherapy [103]. In another study, convallatoxin sensitized lung cancer cells to 5-FU by inhibiting Nrf2 via glycogen synthase kinase-3β (GSK-3β) activation [104].
The PI3K/Akt pathway is another target for Nrf2 modulation. CP-673451, which suppresses Nrf2 transcription through PI3K/Akt inhibition, downregulated platelet-derived growth factor receptorβ (PDGFRβ) and induced apoptosis in NSCLC cells, ultimately sensitizing them to cisplatin [105]. Similarly, the combination of xanthohumol and phenethyl isothiocyanate modulated Nrf2 in pancreatic cancer, decreasing cytosolic COX-2 and nuclear STAT-3 to induce apoptosis [106]. Ailanthone, a plant extract, also inhibited Nrf2 in pancreatic cancer cell lines, leading to decreased proliferation and viability [107].
Nrf2 knockdown combined with chemotherapy has displayed promise in cervical cancer. Studies reported that Nrf2 knockdown in combination with cisplatin, paclitaxel, doxorubicin, or 5-FU reduces GSH levels and promotes cancer cell apoptosis by modulating mitochondrial BAX [108]. Cyanidin-3-O-glucoside, when combined with cisplatin in cervical cancer cells, downregulated Nrf2 and its downstream antioxidant proteins, increasing sensitivity to chemotherapy [109]. Artesunate treatment in head and neck cancer effectively inhibited cell growth by enhancing Nrf2 inhibition [110]. Interestingly, some flavonoids, such as quercetin and procyanidin B2, were reported to activate Nrf2, potentially preventing procarcinogen-induced ROS generation and DNA damage, suggesting a cancer-preventive role in the early stages [101]. Together, these studies highlight the potential of targeting Nrf2 for improved cancer therapy outcomes. Inhibiting Nrf2 can increase cancer cell susceptibility to chemotherapy and radiotherapy. Further research is needed to fully understand the multifaceted role of Nrf2 in different cancer types and develop targeted therapeutic strategies.
6. Sestrin as a multifunctional regulator in metabolic disease and cancer
6.1 Sestrin family proteins
Sestrin belongs to a highly conserved gene family consisting of three genes, namely Sestrin1, Sestrin2, and Sestrin3 in vertebrates [89, 111]. Although there is a difference in the expression levels, Sestrins are ubiquitously expressed in several tissues. Sestrin1, named PA26 at the time of identification, was found to be a p53 response gene according to a study on tetracyclin-regulated p53 expression systems [112]. The expression level of Sestrin1 is sensitive to genotoxic stress and depends on p53 [111]. In addition, it is reported that Sestrin1 is induced during serum deficiency, and Forkhead box O (FoxO) proteins are involved in regulating its expression [113]. Sestrin2 was first identified as a stress-response gene by microarray analysis under hypoxic conditions and was named Hi95 [114]. Sestrin2 shows strong homology with Sestrin1 and is the “growth arrest and DNA damage” family PA26. It is regulated as a genotoxic stress response similar to Sestrin1 [111]. It is reported that the expression of Sestrin2 is regulated by hypoxia-inducible factor-1, p53, or Nrf2, depending on the type of stress stimulus and cells [115, 116]. Importantly, overexpression of Sestrin2 inhibited cell death by hypoxia/glucose deprivation or H2O2 [28, 114, 117]. Sestrin3 was found by the in silico analysis of Sestrin2 and was shown to share homology with Sestrin1 and Sestrin2 [118]. Sestrin3 is known to be directly regulated by serum and growth factors, and FoxO3A and FoxO1 were identified as direct regulatory transcription factors [119].
Following the identification of Sestrin, subsequent research demonstrated that its expression increases under stress, offering protection to a wide range of cell types [115, 116]. Notably, Sestrin2 has been implicated in maintaining redox homeostasis while concurrently engaging in crosstalk with diverse intracellular signaling pathways [115, 116]. Considering the well-established role of Sestrin2 in redox homeostasis and its interactions with intracellular signaling pathways, we reviewed its relevance to metabolic disorders.
6.2 Sestrin and redox homeostasis
Sestrin showed low sequence similarity with other proteins at the time of their discovery; therefore, the protein sequence did not contribute to the understanding of the function of Sestrin [115, 116]. However, it has been reported that Sestrin contains a conserved region with the prokaryotic protein AhpD, which plays a role in the regeneration of oxidized peroxiredoxin in bacteria [120]. In mammals, Sestrin also exerts enzymatic activity by inducing the regeneration of peroxiredoxins (Figure 4). Given the oxidative stress, cysteine residues within peroxiredoxins become excessively oxidized, and thus peroxiredoxins lose their antioxidant activity [121]. Reduction of peroxiredoxins by Sestrin re-induces the antioxidant activity of peroxiredoxins [120] (Figure 4). However, research has shown that the regeneration of peroxiredoxins is mainly controlled by sulfiredoxin, which is independent of the Sestrin2 protein [122]. Nevertheless, the antioxidant activity of Sestrin has been reported to be beneficial under various stress conditions [28, 117, 123]. Sestrin1 and Sestrin2 are known to activate antioxidant signals via p53, and the downregulation of p53-mediated antioxidant genes like Sestrin contributes significantly to a mutation in p53-suppressive conditions [124]. Nrf2-dependent induction of Sestrin2 contributes to suppressing cell death by H2O2 [28]. Conversely, Sestrin2 also activates Nrf2 through the degradation of KEAP1, which plays a vital role in liver protection [29]. Moreover, the overexpression of Sestrin2 inhibits methylglyoxal-induced hepatocellular damage at the cellular level and in animal models [125]. FoxO induces sestrin3 and contributes to antioxidant activity [119]. In addition, Sestrin inhibits ROS in macrophages, thus suppressing the inflammatory response [126]. Taken together, it can be concluded that the positive effects of Sestrin on various cells are based on their antioxidant effect.
Multifaced role of Sestrin in the regulation of metabolic and cellular redox homeostasis. Sestrin regulates cellular homeostasis over cellular growth, lipid synthesis (lipogenesis), and the vital process of autophagy, mainly by modulating the activity of the mechanistic target of rapamycin complex 1 (mTORC1) and its downstream targets. mTORC1 is a central regulator of cell growth and metabolism, and its dysregulation is implicated in various diseases. This Sestrin-mediated autophagy pathway regulates ROS levels and triggers redox adaptations under stress conditions. To maintain redox homeostasis, Sestrin stabilizes Nrf2 by inhibiting Keap1 and recycling peroxiredoxin (Prx), enhancing antioxidant defense. Activated (Prx) shows antioxidant activity by neutralizing H2O2 produced in mitochondria. ACC: Acetyl-CoA carboxylase, SREBP: Sterol regulatory element binding protein, SOD: Superoxide dismutase, TSC: Tuberous sclerosis complex.
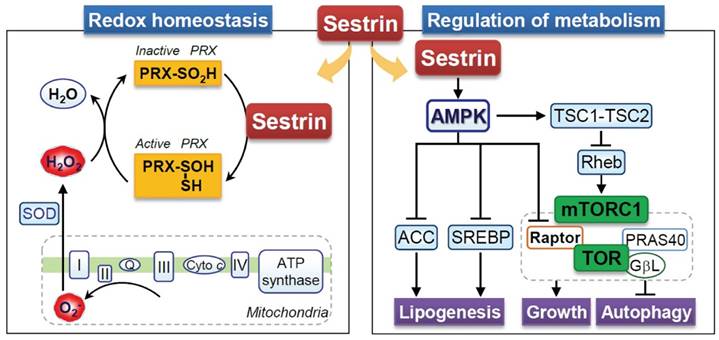
6.3 Sestrin and metabolic homeostasis
The AMPK-mammalian target of the rapamycin (mTOR) pathway is an intracellular energy regulatory signaling pathway that sensitively induces a response in accordance with the redox status [127]. This pathway has been recognized to play an essential role in metabolic diseases [80]. Sestrin is known to be an upstream regulator of the AMPK-mTOR pathway activity; thus, they are expected to play a positive role in metabolic diseases [80] (Figure 4). Ectopic expression of Sestrin1/2 inhibits the TOR complex 1 (TORC1), one of the two mTOR complexes [111]. Sestrin1/2 increases the phosphorylation of AMPK and appears to be due to direct interaction and not its redox activity [111]. The activity of TORC1 is regulated by Rheb, a small GTPase, which is negatively regulated by the TSC1:TSC2 complex [128], and AMPK positively regulates the TSC1:TSC2 complex [127]. As a result, both AMPK and tuberous sclerosis complex phosphorylation by Sestrin1/2 contributes to the inhibition of mTOR activity [111]. Interestingly, Sestrin3 directly activates the mTORC2-Akt signal, which contributes to the enhanced insulin sensitivity of the liver [129]. Based on this, Sestrin has positive effects on metabolic disorders. Studies on Drosophila have shown that defects in Sestrin lead to fat accumulation, mitochondrial damage, and muscle degeneration [130]. In Sestrin-lacking mice, insulin responsiveness was defective, which promoted high-fat diet-induced insulin resistance and diabetes progression [131]. In the muscle cells, Sestrin2 increases autophagy to restore insulin responsiveness through AMPK activation [132]. In addition, the overexpression of Sestrin inhibited the Liver X receptor α activity, indicating the potential inhibition of fat accumulation in the liver [133]. In nutrient-deficient conditions, Sestrin deficiency makes cells more susceptible to cell death, whereas Sestrin overexpression protects cells from metabolic stress caused by nutrient deficiency [117, 123]. Taken together, Sestrin can potentially be a therapeutic target for controlling metabolic disorders.
6.4 Role of Sestrin2 in cancer
6.4.1 Multifaceted roles of Sestrin2 in cancer
Sestrin2, a highly conserved protein, plays an important role in response to various types of cellular and environmental stress like DNA damage, hypoxia, oxidative stress, obesity, and endoplasmic reticulum (ER) stress in various tumors and cancers [134]. Sestrin2 overexpression is found in various cancers, such as endometrial, colorectal, lung, breast, hepatocellular, bone, and pancreatic cancer, and is involved in the defense mechanisms against cancer. Sestrin2 suppresses cancer cell progression through various signaling pathways. Among them, mTORC1 is one of the key signaling molecules highly expressed in cancer cells and enables the proliferation of cancer [135]. The mTORC1 signaling pathway activates the phosphorylation of S6K1 and p40 ribosomal protein kinase s6, inhibiting the translation initiation factor 4E, thereby promoting cancer progression [135]. The tumor suppressor gene p53 contributes to the suppression of cancer progression by suppressing mTORC-signaling through the increased expression of Sestrin2 [136]. In hepatocellular carcinoma, p53 translocation from the cytosol to the nucleus was found to be responsible for Sestrin2 expression, which increased autophagy by activating the Sestrin2-AMPK pathway [137]. Sestrin2 reduced colorectal cancer stem cells by inhibiting the Wnt/β-catenin pathway, suggesting that this mechanism may be a potential target for anticancer therapy [138]. The upregulation of cellular transformation, tumor progression, and migration occurs through the hyperactivation of the p-Akt/mTORC1/p70S60K pathway, suggesting that Sestrin2, as their regulator, is a potential therapeutic target in cancer [139]. In the breast cancer cell line MDA-MB-453ATF4, upregulation of ATF4 upon treatment with Nelfinavir is associated with Sestrin2 expression, which downregulates mTORC1, leading to apoptosis and autophagy [140]. Overexpression of Sestrin2 activates AMPK, which inhibits the phosphorylation of mTORC and the proliferating cell nuclear antigen, resulting in the induction of apoptosis (Caspase-3, -7, and -9) in colorectal cancer cell lines SW620 and LoVo, as well as in an in vivo mouse model [141]. Similarly, in the colorectal cancer cell line HCT116, Sestrin2 overexpression induced apoptosis by inhibiting the mitochondrial membrane potential and generated ROS via the AMPK/p38 signaling pathway [142]. In pancreatic cancer, the upregulation of Sestrin2 inhibited the cancer cells' migration, proliferation, and invasion by activating the p-Nrf2/KEAP1/HO-1/NQO-1 signaling axis [143]. In human bladder and cervical cancer, Sestrin2 expression enhanced the autophagy of cancer cells by modulating the Sestrin2-dependent MAPK8/JNK1/JUN mechanism [144]. In addition, Sestrin2 overexpression inhibits cancer growth by the activation of various signaling pathways such as MAPK4/SAPK/JNK1/Jun kinase, MAPK8/Jun, and the inhibition of x-linked inhibitor protein (XIAP) [145].
According to numerous studies, [146] it has been shown that, in contrast to its role in inhibiting cancer cell growth, Sestrin2 has a progressive role in cancer proliferation, migration, invasion, and chemoresistance. Knockdown of Sestrin2 in the lung cancer cell line A549 suppressed the cancer stemness markers (OCT, SOX2, and Nanog), drug resistance markers, migration markers (Snail, ZEB1, and vimentin), and transcription factors (Nrf2 and HO-1) which indicates that the expression of Sestrin2 upregulated the progression of lung cancer [146]. In glutamine-depleted lung cancer cells, Sestrin2 was upregulated, which reprogrammed the lipid metabolism through differential regulation of mTORC1 and mTORC2 and allowed the glutamine-depleted lung cancer cells to survive by maintaining their energy and redox balance [147]. Furthermore, Sestrin2 expression in hepatocellular carcinoma was found to be responsible for sorafenib drug resistance via the upregulation of phosphorylation of AKT/AMPKα, leading to carcinoma progression [148]. In human squamous cell carcinoma and melanoma cells, UVB radiation increases Sestrin2 levels, thereby enhancing the survival of cancer cells and chemotherapy resistance through the phosphorylation of p-PTEN and phosphorylation of the AKT pathway. Knockdown of Sestrin2 induced chemosensitivity and apoptosis of cancer cells in vitro/in vivo [149]. In osteosarcoma, Sestrin2 activates autophagy by inhibiting mTOR through the PERK-eIF2α-CHOP pathway, inhibits apoptosis through Bcl-2, and subsequently suppresses the response to anticancer drugs and induces resistance [150]. Taken together, these studies suggest that Sestrin2 has both cancer progression and inhibition functions, and further studies of the molecular mechanisms involved can be beneficial for identifying potential therapeutic targets and understanding the physiological properties of cancer.
6.4.2 Targeting Sestrin (Sestrin2 specific) in cancer
Various studies have shown that targeting Sestrin2 represses cancer progression. Among them, one of the anticancer compounds, Fisetin, induces apoptosis through the induction of Sestrin2 via the SESN2/mTORC/Mcl-1 pathway in human head and neck cancer cell lines (MC3, Ca9.22, and HN22) [151]. Treatment with Tanshinone IIA in osteosarcoma increased autophagy to prevent cancer cell proliferation and progression by upregulating Sestrin2 via the HGK/SAPK/JNK1/Jun dependent pathway [152]. These studies illustrated that Sestrin2 has anti-tumor activity in colorectal, bladder, lung, and endometrial cancer and osteosarcoma by suppressing the mTORC1 signaling pathway. Sestrin2 may be a potential biomarker for tumor repression in these cancer types. Usually, in human colorectal cancer cells, the level of Sestrin2 is found to be lower than in adjacent para-cancerous tissue. However, the cancer therapy treatments oxaliplatin and DHA increase the amount of Sestrin2, obtained when Sestrin2 is bound with CHOP to cause upregulation in colorectal cancer and is strongly associated with ER stress [153]. On the other hand, the lentivirus mediated Sestrin2 overexpression significantly enhanced apoptosis in the colorectal cancer cell lines SW620 and LoVo by suppressing the AMPK/mTORC1 pathway [141]. Quercetin treatment in HCT116 and HT-29 colon cancer cells increased Sestrin2 expression, thereby activating the AMPKα1 and mTOR pathways and enhancing ROS levels and apoptosis [154]. In colon cancer cells treated with 5-FU, Sestrin2 was found to inhibit cell proliferation and migration through a p53-dependent mechanism without increasing ROS levels [155]. Moreover, carnosol, a phytochemical compound from rosemary, upregulated Sestrin2 and modulated pERK in HCT116 and SW480 colon cancer cells, thereby promoting the Nrf2/KEAP1 mediated pathway, leading to increased apoptosis and reduced colorectal cancer cell viability [156].
Through the upregulation and stabilization of Sestrin2 mRNA, ChlA-F, a derivative of Cheliensisin A, induces anticancer activity via an autophagy-dependent mechanism to prevent bladder cancer progression [157]. Isorhapontigenin treatment effectively increased Sestrin2 by allowing JUN to bind to the AP-1-binding region of the Sestrin2 promoter via MAPK8/JNK1, which subsequently enhanced autophagy by converting LC-3I to LC-3II and increased apoptosis in human bladder cancer cells. In pancreatic cancer, curcumin treatment remarkably repressed pancreatic cancer cell proliferation, migration, and progression through synergistic activity with Sestrin2 via Nrf2/KEAP1/HO-1/NQO-1 [143]. Various chemical compounds such as 5-FU, excisanin A, curcumin, fisetin, 2-imino-6-methoxy-2H-chromene-3-carbothioamide (IMCA), and quercetin were reported to inhibit cancer cell proliferation through the AMPK/mTORC signaling pathway by upregulating Sestrin2 [145, 158]. Similarly, IMCA increased the Sestrin2 level in Medullary thyroid cancer, increased the phosphorylation of AMPK, and reduced the phosphorylation of p70S6K, an essential enzyme in the mTOR pathway. This study demonstrated that IMCA drugs function via the p53/Sestrin2/AMPK/mTOR signaling pathway [159]. Overall, targeting Sestrin2 in cancer research may be a potential therapeutic strategy to halt cancer growth, invasion, and migration via various molecular signaling pathways.
7. Role of HO-1 in metabolic diseases and cancer
7.1 HO-1 and redox homeostasis
HO-1, a target gene of Nrf2, is a stress-inducible enzyme that catalyzes the degradation of heme into ferrous iron, CO, and biliverdin [160]. Two upstream enhancers (E1 and E2) induce the HO-1 gene to maintain redox homeostasis [160]. Several AREs are localized in both E1 and E2 regions that play crucial roles in the cellular defense mechanisms against oxidative damage. Oxidative stress promotes Nrf2 translocation and ARE binding activity of HO-1 genes. Besides the redox-dependent transcription factors, BTB and CNC homolog 1 (Bach1), NF-κB, and AP-1 also regulate the HO-1 gene expression under ROS, cytokines, and other pathophysiological stimuli [161]. In HO-1-deficient animal and cell models, ROS levels and sensitivity to oxidative stress were increased [162]. Evidence has revealed that regulating HO-1 levels plays a role in improving metabolic diseases, such as obesity, insulin resistance, and diabetes [41].
7.2 Role and targeting of HO-1 in metabolic diseases
Obesity is one of the causes of metabolic diseases, such as insulin resistance, diabetes, and cardiovascular disease; therefore, its control plays an important role in suppressing various metabolic disorders [35]. Several reports have suggested that increased ROS induces pre-adipocyte differentiation and adipogenesis [35, 163]. Because oxidative stress plays a critical role in obesity, regulating HO-1 signaling may be the therapeutic target for treating obesity [35, 163]. Adipocyte-specific HO-1 overexpression attenuates adiposity and adipogenesis in a diet-induced obesity mouse model. The overexpression of HO-1 in adipocytes increased the reprogramming of pre-adipocytes through the regulation of signaling mechanisms of the paternally expressed gene 1 (Peg1)/mesoderm specific transcript (Mest) and Wnt10b/β-catenin /Sonic hedgehog proteins [164].
The induction of HO-1 by chemical treatment benefits body weight loss and insulin resistance improvement in obese animals [165, 166]. Obese mice treated with cobalt protoporphyrin (CoPP), an HO-1 inducer, had a lower body weight and less fat content (visceral and subcutaneous) compared to those in the vehicle-treated obese mice. The increase in HO-1 level reduced adipogenesis in the bone marrow and resulted in a decrease in body weight through the reduction of visceral and subcutaneous fat content [167] (Figure 4). Administration of CoPP in Zucker diabetic fatty (ZDF) rats has been shown to decrease weight gain and fat content [165]. In this report, they suggested that the decreased cannabinoid-1 receptor by HO-1 induction may contribute to reducing fat accumulation in ZDF rats [165]. Although the exact mechanism has not been elucidated, the administration of hemin, another HO-1 inducer, also reduced body weight and retroperitoneal adiposity in ZDF rats. One possibility is that hemin treatment increased the expression of lipoprotein lipase, an enzyme related to lipolysis, and therefore, it may have enhanced lipolysis in ZDF rats [166]. Treatment with butein (flavonoid chalcone) increases HO-1 levels through the p38 MAPK/Nrf2 pathway and reduces adipocyte hypertrophy in mice fed a high-fat diet [168].
Obesity causes adipose tissue hypertrophy, leading to the secretion of various inflammatory cytokines and adipokines, such as TNF-α, IL-1β, resistin, leptin, and adiponectin. This increased protein release regulates insulin resistance and sensitivity [169, 170]. In obese diabetic mice, administration of CoPP induced HO-1 protein levels, resulting in increased insulin sensitivity and improved glucose tolerance. The mechanism of action of CoPP could be multifaceted and attributable to the activation of the HO-1-adiponectin axis [167]. Insulin sensitivity and obesity are accompanied by chronic inflammation and pro-inflammatory cytokines, produced by the infiltrating macrophages in the adipose tissue [171]. It has been shown that adiponectin decreases the generation of pro-inflammatory risk factors.
Type 2 diabetes mellitus is a chronic metabolic disorder characterized by insulin resistance and hyperglycemia. Various pathologies, including defective insulin secretion from pancreatic beta cells, inadequate hepatic glucose production, and peripheral insulin resistance, are involved in the development of the disease, and several therapeutic approaches have been attempted to treat this metabolic syndrome [172]. The expression of HO-1 was reduced in patients with type 2 diabetes, which is associated with insulin sensitivity and oxidative capacity [173, 174] (Figure 5). The upregulation of HO-1 exerts an antidiabetic effect, and the underlying mechanisms include the induction of insulin sensitization and glucose metabolism-related genes, such as GLUT4, adiponectin, AMPK, cAMP, and cGMP. Although the mechanism is not fully understood, treatment with hemin, an HO-1 inducer, increased the adiponectin levels through AMPK signaling activation, and this pathway stimulated the translocation of GLUT4 in Goto-Kakizaki rats, which are non-obese, type 2 diabetic model rats [166].
Role of heme oxygenase-1 (HO-1) as antioxidant protein in obesity and diabetes. HO-1 catalyzes the enzymatic breakdown of stress-inducing heme, producing biliverdin/bilirubin, carbon monoxide (CO), and free iron. Biliverdin/bilirubin, through its effects on hypertrophic adipocyte remodeling and inhibition of reactive oxygen species (ROS)-mediated adipogenesis, demonstrates an attenuating effect on adiposity. CO exhibits anti-inflammatory and anti-apoptotic properties, contributing to cardioprotection by inhibiting vasoconstrictive processes. HO-1 also plays a pivotal role in safeguarding vascular function against the deleterious impact of free iron. In the context of prediabetic obesity, HO-1 displays significant enhancements in the functionality of glucose transporter-4 (GLUT-4), adiponectin, and AMP-activated protein kinase (AMPK), promoting insulin sensitivity and beta-cell protection.

Excess oxidative stress induces damage to cellular DNA and proteins and causes pancreatic beta-cell dysfunction and death during the progression of type 2 diabetes [175]. Studies reported that the HO-1 signaling pathway partially protects beta-cells from palmitic acid-induced lipotoxicity that causes oxidative or ER stress [176]. Nrf2 activation or Bach1 deficiency is involved in regulating intracellular oxidative defense capacity through HO-1 induction [176]. Taken together, HO-1 can potentially be a suitable target for treating obesity, insulin resistance, and diabetes (Figure 5).
7.3 Role of HO-1 in cancer
7.3.1 Functions of HO-1 in cancer
The importance of HO-1 in cancer progression has been highlighted by numerous studies, and its expression is linked to tumor growth, invasiveness, metastasis, angiogenesis, resistance to therapy, tumor immune escape, and poor prognosis through various mechanisms [177]. HO-1 promotes aggressiveness and therapy resistance, resulting in a higher risk of cancer development and therapeutic failure because HO-1 accelerates the production of tumor neovasculature and confers a selective advantage in overcoming increased oxidative stress during carcinogenesis and therapy to tumor cells [178]. Several studies have suggested that HO-1 is involved in tumor induction and can promote tumor growth and metastasis more effectively through catabolites. HO-1 activity provides antioxidant, anti-apoptotic, and cytoprotective properties and removes harmful intracellular heme from the cell intrinsically and promotes anti-inflammation and immune suppression by modulating the tumor microenvironment when the degradation of heme produces catabolites that diffuse out of the cell [179]. In a carcinogenic mouse model and cell line of head and neck cancer, cytoplasmic HO-1 was expressed in the pre-neoplastic lesions and nuclear HO-1 was expressed in tumor tissues. This suggests that the increased expression of HO-1 in the nucleus is associated with tumor growth and may have pro-tumor functions [180]. HO-1 upregulation in melanoma reduced the survival of tumor-bearing mice because melanoma cells develop more condensed tumors with increased viability, proliferation, and metastasis [181]. HL-60R, a drug-resistant acute myeloid leukemia cell line that overexpresses HO-1, showed less sensitivity to cytarabine and daunorubicin than HL-60 cells. In addition, the downregulation of HO-1 increased sensitivity to chemotherapy in the HL-60R cell line, which indicates that the increased expression of HO-1 correlates with chemoresistance in hematological malignancies [182]. Similarly, high HO-1 expression induced IL-6, one of the most important survival factors of cancer cells in multiple myeloma, and increased resistance to lenalidomide treatment [183]. Furthermore, HO-1 promoted lung metastasis by inducing the activation of vascular endothelial growth factor (VEGF) and IL-10 [184]. In a study utilizing colon cancer cells, it was reported that lowering the level of glucose-regulated protein 78 (GPR78) increased the metastatic capacity through the activation of Nrf2/HO-1 signaling and the upregulation of vimentin and downregulation of E-cadherin levels [185]. HO-1 also has an important role in modulating immune responses [186]. In a study with colorectal cancer cells, HO-1 expression decreased the cell-mediated cytotoxicity by lowering the expression of ICAM-1 and CXCL10, which suppressed the adhesion of peripheral blood mononuclear lymphocytes to the colorectal carcinoma cells and the recruitment of T effector cells which regulate anti-tumor immunity [187]. The upregulation of HO-1 activated the Sonic Hedgehog (SHH) signaling pathway. It was found to induce cell proliferation, while the downregulation of HO-1 resulted in decreased cell proliferation in pancreatic cancer (PC) [188]
However, in cancer studies, the role of HO-1 is controversial, as several studies have reported that the activation of HO-1 prevented the proliferation of breast cancer [189] and angiogenesis in prostate cancer [190]. The overexpression of HO-1 altered the levels of matrix metalloproteinase and oncogenic miR-378 within the tumor microenvironment. It contributed to the downregulation of metastasis, cell proliferation, and migration in mucoepidermoid carcinoma of the lung [191]. According to reports by Gandini et al. 2019, HO-1 was found to be related to anti-tumor activity in cell lines and animal and human tumor samples of breast cancer, and the dual effect of HO-1 anti- or pro-tumor activity may be dependent on the subcellular localization of HO-1 [192]. When colon cancer progressed, upregulation of HO-1 was associated with an increase in the survival time of patients, and this effect is due to HO-1 suppressing the survival and migration of colon cancer cells in a p53 tumor suppressor protein-dependent manner [193]. In colorectal cancer, silencing the HO-1 gene resulted in the upregulation of ROS and DNA damage to enhance cancer progression [194]. Overexpression of HO-1 resulted in the upregulation of proangiogenic enzyme thymidine phosphorylase, which significantly elevates angiogenesis in cancer cells, but in in vitro, it suppressed the proliferation and migration of cancer cells [195]. Therefore, more research is required to determine the precise role of HO-1 in diverse stress conditions in cancer.
7.3.2 Targeting HO-1 in cancer
In cancer progression, HO-1 is remarkably overexpressed in different types of cancer and can negatively affect the treatment outcome, particularly with its upregulation in chemotherapy and photodynamic therapy [196]. HO-1 may act as an anti-apoptotic defense against tumors, protecting tumor cells from the oxidative stress produced by excessive nitric oxide produced during the growth of solid tumors. Thus, the HO-1 inhibitor zinc protoporphyrin (ZnPP) significantly reduced tumor growth in a mouse model [197]. Poly (ethylene glycol)-conjugated ZnPP (PEG-ZnPP) has been proposed for use in solid tumor cancers because it has tumor-selective HO-1 inhibitory activity and increases the apoptosis of cancer cells [198]. The increased expression of HO-1 in PC cell lines treated with gemcitabine or radiation has been associated with resistance to chemotherapy and radiation therapy. Thus, inhibition of HO-1 expression increases tumor cells' sensitivity to radiation therapy and chemotherapy, thereby increasing their efficacy against PC cells [199]. Similarly, the inhibition of HO-1 or direct interference with HO-1 metabolites may be a new strategy for PC treatment, as HO-1 inhibition or iron chelation in human PC cell lines has increased the sensitivity and susceptibility to chemotherapy in an in vivo model [200]. The expression of HO-1 protein was higher in colon cancer cell line FHC compared to normal tissues by 1.5-fold as well as the HO-1 mRNA and enzymatic activity. Therefore, the viability of colon cancer cells declined when treated with zinc protoporphyrin, an inhibitor of HO-1, indicating that HO-1 is a valuable biomarker for anti-colon cancer therapy [201]. A series of novel indolyl-chalcone derivatives were synthesized and evaluated for anticancer activity by demonstrating the effect of in vitro and in vivo cell apoptosis of a human lung cancer cell line A549 by activating the Nrf2/HO-1 pathway without any change in normal angiogenesis [202]. The sensitivity to chemotherapy was increased when SLV-1199, a pharmacological inhibitor of HO-1 activity, was used in PANC-1 and DU-145 cell lines [203]. Demonstration of the Nrf2/HO-1 axis may be an important target for breast cancer treatment, as overexpression of miR-140-5p in hypoxic conditions attenuates the progression of hypoxia-induced breast cancer by reducing the expression of Nrf2 [204]. Collectively, HO-1 can be used as an important therapeutic target in cancer therapy, which may provide a novel strategy to increase the therapeutic efficacy of chemotherapy and radiation therapy.
8. Crosstalk between Nrf2, Sestrin2 and HO-1 in oxidative stress
During oxidative stress, various endogenous antioxidant proteins, and vitagene-originated proteins, interact in a hermetic manner to produce the best possible outcome for cells [63]. At the initial stage of oxidative stress, Nrf2 is activated and translocated into the nucleus to bind with Antioxidant Response Elements (AREs). The activation of AREs triggers the upregulation of various antioxidant genes, including HO-1. HO-1 can break down heme, a pro-oxidant molecule, into biliverdin, carbon monoxide, and iron, which have antioxidant and anti-inflammatory properties, thereby reducing oxidative stress and protecting cells from damage [32]. Additionally, the expression of Sestrin family proteins can be regulated by HO-1 by promoting its degradation.
Sestrin2 is a stress-responsive protein that regulates various cellular functions, such as antioxidant defense, autophagy, and mTOR signaling [205]. Recent studies suggest Sestrin2 may regulate HO-1 expression and activity in stressed conditions. It was found that Sestrin2 deficiency led to a reduction in HO-1 expression and increased susceptibility to oxidative stress-induced cell death in cardiomyocytes. A research study has elucidated the prominent involvement of Sestrin2 as a fundamental constituent within the context of major depressive disorder, thereby illuminating its expansive implications across a spectrum of diverse pathological conditions [206]. The precise mechanism underlying the crosstalk between Sestrin 2, and HO-1 is undetermined, although it may involve signaling pathways such as the Nrf2 pathway. Sestrin2 has been shown to stimulate Nrf2 signaling and enhance the expression of Nrf2 target genes such as HO-1, which may contribute to its antioxidant effects [207].
Overall, in stressed settings, the interaction between Nrf2, Sestrin2, and HO-1 may regulate cellular antioxidant defense mechanisms and protect cells from oxidative damage. However, the crosstalk and interaction between these proteins are complex and context-specific, and more studies are needed to fully understand the mechanism underlying this crosstalk and its consequences for cellular stress response.
9. Conclusion and future perspectives
Generally, it is widely known that oxidative stress is highly correlated with the incidence of metabolic diseases and cancer. This review examined the results from the latest research on antioxidant genes that can overcome metabolic diseases and cancers (Table 1). These recent studies may advance our biological understanding of the role of antioxidant genes in human disease. Nrf2 and Nrf2-regulated genes, HO-1, and Sestrin, positively affect metabolic diseases through their antioxidant activities or by controlling other metabolic signals. However, there are many 'grey areas' in understanding the dual roles of Nrf2, Sestrin, and HO-1 in cancer, with some studies revealing positive effects and others suggesting negative effects. This ambiguity indicates that further studies are needed to unravel these genes' complex relationship with various disease states. Despite remaining uncertainties, a detailed compilation of the results of various pathological models, along with discovering compounds that trigger antioxidant-mediated signaling pathways, will lead to the discovery of new targets for treating metabolic diseases and cancer.
Role of antioxidant genes Nrf2, Sestrin2 and HO-1 in metabolic disease and cancer
| Gene | Involved Metabolic Diseases and Regulatory Mechanisms | Involved Cancer and Regulatory Mechanisms | Reference |
|---|---|---|---|
| Nrf2 | Obesity: AMPK activation, reduction of lipogenesis, enhancement of β-oxidation, insulin sensitivity improvement Diabetes: Insulin sensitivity improvement, protection of pancreatic beta cells, reduction of oxidative stress, AMPK activation Non-alcoholic Steatohepatitis (NASH): Reduction of lipogenesis, enhancement of β-oxidation, AMPK activation | Colorectal Cancer: Chemoresistance through Nrf2 activation via KEAP1 and CUL3 mutations Breast Cancer: Chemoresistance via constitutive Nrf2 upregulation, leading to increased antioxidant gene expression Pancreatic Cancer: Chemoresistance enhancement, tumor progression through Nrf2 interaction with ATDC Gastric Cancer: Chemoresistance via Nrf2 activation and antioxidant gene upregulation Cervical Cancer: Increased chemosensitivity and suppression of tumor growth through Nrf2 inhibition or knockout Glioma: Tumor progression and poor prognosis through Nrf2 activation, suppression of tumorigenesis through Nrf2 knockout Non-Small Cell Lung Cancer (NSCLC): Tumor progression and treatment resistance through Nrf2 activation and interaction with PAQR4 | [26], [27], [70], [71], [78], [85], [89], [93], [95], [96], [97], [98] |
| Sestrin2 | Obesity: AMPK activation, inhibition of mTORC1 activity, enhancement of autophagy Diabetes: AMPK activation, improvement of insulin sensitivity, reduction of oxidative stress, inhibition of mTORC1 activity Insulin Resistance: AMPK activation, enhancement of autophagy, inhibition of mTORC1 activity Liver Fat Accumulation: Inhibition of Liver X receptor α, prevention of liver fat accumulation | Endometrial Cancer: mTORC1 inhibition, suppression of cancer cell proliferation, induction of autophagy Colorectal Cancer: mTORC1 inhibition, AMPK suppression, induction of apoptosis, reduction of cancer cell viability Lung Cancer: AKT/mTOR/P70S6K signaling suppression, inhibition of proliferation and migration of cancer cell Breast Cancer: mTORC1 inhibition, suppression of cancer progression, induction of apoptosis Hepatocellular Carcinoma: p53-dependent mechanisms, suppression of cancer progression, induction of autophagy Bone Cancer: mTORC1 inhibition, suppression of cancer cell proliferation, induction of autophagy Pancreatic Cancer: Inhibition of cancer cell migration, proliferation, and invasion through Nrf2/KEAP1/HO-1/NQO-1 signaling Bladder Cancer: Autophagy modulation, induction of apoptosis, suppression of cancer progression Cervical Cancer: Autophagy modulation, suppression of cancer progression, induction of apoptosis Osteosarcoma: mTOR inhibition through p-ERK-eIF2α-CHOP pathway, suppression of apoptosis, induction of chemoresistance Melanoma: Enhancement of cancer cell survival, promotion of chemoresistance, activation of AKT pathway | [28], [29], [80], [131], [132], [133], [134], [137], [138], [139], [140], [141], [143], [144], [149], [150], [157] |
| HO-1 | Obesity: Maintenance of redox homeostasis, inhibition of adipogenesis, regulation of Peg1/Mest and Wnt10b/β-catenin/Sonic hedgehog signaling pathways Diabetes: Protection of pancreatic beta cells, reduction of oxidative stress, induction of glucose metabolism-related genes (e.g., GLUT4, adiponectin), activation of AMPK signaling Insulin Resistance: Improvement of insulin sensitivity, reduction of pro-inflammatory cytokines, activation of the HO-1-adiponectin axis, and AMPK signaling | Melanoma: Promotion of tumor aggressiveness, increased viability, proliferation, and metastasis Acute Myeloid Leukemia: Chemoresistance through HO-1 overexpression, reduced sensitivity to cytarabine and daunorubicin Multiple Myeloma: Induction of IL-6, increased resistance to lenalidomide treatment, survival factor promotion Lung Cancer: Promotion of lung metastasis, activation of VEGF and IL-10 Colon Cancer: Modulation of Nrf2/HO-1 signaling, upregulation of vimentin, downregulation of E-cadherin, suppression of immune responses Pancreatic Cancer: Activation of the Sonic Hedgehog (SHH) signaling pathway, promotion of cell proliferation Prostate Cancer: Controversial role; prevention of angiogenesis, progression of the tumor Breast Cancer: Dual role; promotion or suppression of tumor proliferation depending on angiogenesis and subcellular localization Head and Neck Cancer: Enhancement of cancer cell growth, pro-tumor activity enhancement | [161], [164], [165], [166], [167], [168], [170], [179], [181], [182], [183], [184], [185], [186], [187], [188], [190], [199] |
Acknowledgements
The figures were generated utilizing BioRender.
Funding
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science (RS-2023-00209117). This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. RS-2023-00222390). K.R.P is supported by a fellowship from the Prevent Cancer Foundation (PCF) and the International Association for the Study of Lung Cancer (IASLC). P.M.H. would like to acknowledge Cancer Council NSW, Australia for their funding.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Betteridge DJ. What is oxidative stress? Metabolism. 2000;49:3-8
2. Panth N, Paudel KR, Parajuli K. Reactive Oxygen Species: A Key Hallmark of Cardiovascular Disease. Adv Med. 2016;2016:9152732
3. Taysi S, Tascan AS, Ugur MG, Demir M. Radicals, Oxidative/Nitrosative Stress and Preeclampsia. Mini Rev Med Chem. 2019;19:178-93
4. Serafini M. The role of antioxidants in disease prevention. Medicine. 2006;34:533-5
5. Taysi S, Algburi FS, Taysi ME, Caglayan C. Caffeic acid phenethyl ester: A review on its pharmacological importance, and its association with free radicals, COVID-19, and radiotherapy. Phytother Res. 2023;37:1115-35
6. Taysi S, Algburi FS, Mohammed ZR, Ali OA, Taysi ME. Thymoquinone: A Review on its Pharmacological Importance, and its Association with Oxidative Stress, COVID-19, and Radiotherapy. Mini Rev Med Chem. 2022;22:1847-75
7. Mehta M, Paudel KR, Shukla SD, Shastri MD, Satija S, Singh SK. et al. Rutin-loaded liquid crystalline nanoparticles attenuate oxidative stress in bronchial epithelial cells: a PCR validation. Future Med Chem. 2021;13:543-9
8. Devkota HP, Adhikari-Devkota A, Paudel KR, Panth N, Chellappan DK, Hansbro PM. et al. Tea (Catechins Including (-)-Epigallocatechin-3-Gallate) and Cancer. Nutraceuticals and Cancer Signaling. 2021;2:451-66
9. Paudel KR, Panth N, Manandhar B, Singh SK, Gupta G, Wich PR. et al. Attenuation of Cigarette-Smoke-Induced Oxidative Stress, Senescence, and Inflammation by Berberine-Loaded Liquid Crystalline Nanoparticles: In Vitro Study in 16HBE and RAW264.7 Cells. Antioxidants (Basel). 2022;11:873
10. Sharifi-Rad M, Anil Kumar NV, Zucca P, Varoni EM, Dini L, Panzarini E. et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front Physiol. 2020;11:694
11. He L, He T, Farrar S, Ji L, Liu T, Ma X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell Physiol Biochem. 2017;44:532-53
12. Tonelli C, Chio IIC, Tuveson DA. Transcriptional regulation by Nrf2. Antioxid Redox Signal. 2018;29:1727-45
13. Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci. 2016;73:3221-47
14. Chen Y, Huang T, Yu Z, Yu Q, Wang Y, Hu Ja. et al. The functions and roles of sestrins in regulating human diseases. Cell Mol Biol Lett. 2022;27:1-24
15. Zhao J, Sui X, Shi Q, Su D, Lin Z. Effects of antioxidant intervention in patients with polycystic ovarian syndrome: A systematic review and meta-analysis. Medicine. 2022;101:e30006
16. Zhao Y, Feng HM, Yan WJ, Qin Y. Identification of the Signature Genes and Network of Reactive Oxygen Species Related Genes and DNA Repair Genes in Lung Adenocarcinoma. Front Med. 2022;9:833829
17. Cen K, Wu Z, Mai Y, Dai Y, Hong K, Guo Y. Identification of a novel reactive oxygen species (ROS)-related genes model combined with RT-qPCR experiments for prognosis and immunotherapy in gastric cancer. Front Genet. 2023;14:1074900
18. Wadhwa R, Paudel KR, Shukla S, Shastri M, Gupta G, Devkota HP. et al. Epigenetic Therapy as a Potential Approach for Targeting Oxidative Stress-Induced Non-Small-Cell Lung Cancer. Handbook of Oxidative Stress in Cancer: Mechanistic Aspects. Springer. 2022:1545-1560
19. Rani V, Deep G, Singh RK, Palle K, Yadav UC. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016;148:183-93
20. Shohag S, Akhter S, Islam S, Sarker T, Sifat MK, Rahman MM. et al. Perspectives on the Molecular Mediators of Oxidative Stress and Antioxidant Strategies in the Context of Neuroprotection and Neurolongevity: An Extensive Review. Oxid Med Cell Longev. 2022;2022:7743705
21. Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G. et al. ROS in cancer therapy: The bright side of the moon. Exp Mol Med. 2020;52:192-203
22. Senoner T, Dichtl W. Oxidative stress in cardiovascular diseases: still a therapeutic target? Nutrients. 2019;11:2090
23. Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 2007;8:766-75
24. Jha BK, Sherpa ML, Imran M, Mohammed Y, Jha LA, Paudel KR. et al. Progress in Understanding Metabolic Syndrome and Knowledge of Its Complex Pathophysiology. Diabetology. 2023;4:134-59
25. Chan JSK, Tan MJ, Sng MK, Teo Z, Phua T, Choo CC. et al. Cancer-associated fibroblasts enact field cancerization by promoting extratumoral oxidative stress. Cell Death Dis. 2018;8:e2562-e
26. Suzuki T, Yamamoto M. Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med. 2015;88:93-100
27. Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med. 2015;88:179-88
28. Shin BY, Jin SH, Cho IJ, Ki SH. Nrf2-ARE pathway regulates induction of Sestrin-2 expression. Free Radic Biol Med. 2012;53:834-41
29. Bae SH, Sung SH, Oh SY, Lim JM, Lee SK, Park YN. et al. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab. 2013;17:73-84
30. Shrestha J, Baek D-J, Oh Y-S, Cho S-S, Ki S-H, Park E-Y. Protective Effect of Cudrania tricuspidata Extract against High-Fat Diet Induced Nonalcoholic Fatty Liver Disease through Nrf-2/HO-1 Pathway. Molecules. 2021;26:2434
31. Ma H, Wang X, Zhang W, Li H, Zhao W, Sun J. et al. Melatonin Suppresses Ferroptosis Induced by High Glucose via Activation of the Nrf2/HO-1 Signaling Pathway in Type 2 Diabetic Osteoporosis. Oxid Med Cell Longev. 2020;2020:9067610
32. Zhai CM, Jia BY, Wang ZB, Huai X, Ma ZC, Tian ZK. et al. [Nrf2-NF-κB pathway axes, epigenetic regulation and traditional Chinese medicine (natural medicine) to treat type 2 diabetes]. Zhongguo Zhong Yao Za Zhi. 2016;41:4314-9
33. Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. 2011;15:1583-606
34. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y. et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2017;114:1752-61
35. Abraham NG, Junge JM, Drummond GS. Translational significance of heme oxygenase in obesity and metabolic syndrome. Trends Pharmacol Sci. 2016;37:17-36
36. Shah D, Gandhi M, Kumar A, Cruz-Martins N, Sharma R, Nair S. Current insights into epigenetics, noncoding RNA interactome and clinical pharmacokinetics of dietary polyphenols in cancer chemoprevention. Crit Rev Food Sci Nutr. 2023;63:1755-91
37. Higuchi M, Dusting GJ, Peshavariya H, Jiang F, Hsiao ST-F, Chan EC. et al. Differentiation of human adipose-derived stem cells into fat involves reactive oxygen species and Forkhead box O1 mediated upregulation of antioxidant enzymes. Stem cells Dev. 2013;22:878-88
38. Schröder K, Wandzioch K, Helmcke I, Brandes RP. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler Thromb Vasc Biol. 2009;29:239-45
39. Bloch-Damti A, Bashan N. Proposed mechanisms for the induction of insulin resistance by oxidative stress. Antioxid Redox Signal. 2005;7:1553-67
40. Lin HV, Ren H, Samuel VT, Lee H-Y, Lu TY, Shulman GI. et al. Diabetes in mice with selective impairment of insulin action in Glut4-expressing tissues. Diabetes. 2011;60:700-9
41. Ndisang JF, Jadhav A. Heme oxygenase system enhances insulin sensitivity and glucose metabolism in streptozotocin-induced diabetes. Am J Physiol Endocrinol Metab. 2009;296:E829-E41
42. Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes. 1997;46:1733-42
43. Robertson M, Currie J, Morgan L, Jewell D, Frayn K. Prior short-term consumption of resistant starch enhances postprandial insulin sensitivity in healthy subjects. Diabetologia. 2003;46:659-65
44. Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685-95
45. Poston RN. Atherosclerosis: integration of its pathogenesis as a self-perpetuating propagating inflammation: a review. Cardiovasc Endocrinol Metab. 2019;8:51-61
46. Yang X, Li Y, Li Y, Ren X, Zhang X, Hu D. et al. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front Physiol. 2017;8:600
47. Hu R, Dai C, Dong C, Ding L, Huang H, Chen Y. Living Macrophage-Delivered Tetrapod PdH Nanoenzyme for Targeted Atherosclerosis Management by ROS Scavenging, Hydrogen Anti-inflammation, and Autophagy Activation. ACS Nano. 2022;16:15959-76
48. Roy P, Saikia B. Cancer and cure: A critical analysis. Indian J Cancer. 2016;53:441-442
49. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029-33
50. Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794-8
51. Nishikawa M. Reactive oxygen species in tumor metastasis. Cancer Lett. 2008;266:53-9
52. Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15:411-21
53. T LS, Rupasinghe HPV, Dellaire G, Xu Z. Regulation of Nrf2/ARE Pathway by Dietary Flavonoids: A Friend or Foe for Cancer Management? Antioxidants (Basel). 2020;9:973
54. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN. et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780-3
55. Bae I, Fan S, Meng Q, Rih JK, Kim HJ, Kang HJ. et al. BRCA1 induces antioxidant gene expression and resistance to oxidative stress. Cancer Res. 2004;64:7893-909
56. Rodrigues C, Pimpão C, Mósca AF, Coxixo AS, Lopes D, da Silva IV. et al. Human aquaporin-5 facilitates hydrogen peroxide permeation affecting adaption to oxidative stress and cancer cell migration. Cancers. 2019;11:932
57. Muchtaridi M, Az-Zahra F, Wongso H. Molecular Mechanism of Natural Food Antioxidants to Regulate ROS in Treating Cancer: A Review. Antioxidants. 2024;13:207
58. Liu Y, Li Q, Zhou L, Xie N, Nice EC, Zhang H. et al. Cancer drug resistance: redox resetting renders a way. Oncotarget. 2016;7:42740-61
59. Liang W, Zhang Y, Song L, Li Z. 2, 3′ 4, 4′, 5-Pentachlorobiphenyl induces hepatocellular carcinoma cell proliferation through pyruvate kinase M2-dependent glycolysis. Toxicol Lett. 2019;313:108-19
60. Wang C, Shao L, Pan C, Ye J, Ding Z, Wu J. et al. Elevated level of mitochondrial reactive oxygen species via fatty acid β-oxidation in cancer stem cells promotes cancer metastasis by inducing epithelial-mesenchymal transition. Stem Cell Res Ther. 2019;10:1-16
61. Kansestani AN, Mansouri K, Hemmati S, Zare ME, Moatafaei A. High glucose-reduced apoptosis in human breast cancer cells is mediated by activation of NF-κB. Iran J Allergy Asthma Immunol. 2019;18:153-62
62. Devkota HP, Paudel KR, Lall N, Tomczyk M, Atanasov AG. Editorial: Pharmacology of Plant Polyphenols in Human Health and Diseases. Front Pharmacol. 2022;13:945033
63. Calabrese V, Cornelius C, Dinkova-Kostova AT, Calabrese EJ. Vitagenes, cellular stress response, and acetylcarnitine: relevance to hormesis. Biofactors. 2009;35:146-60
64. Sciacca L, Vigneri R, Tumminia A, Frasca F, Squatrito S, Frittitta L. et al. Clinical and molecular mechanisms favoring cancer initiation and progression in diabetic patients. Nutr Metab Cardiovasc Dis. 2013;23:808-15
65. Li R, Zeng X, Yang M, Xu X, Feng J, Bao L. et al. Antidiabetic Agent DPP-4i Facilitates Murine Breast Cancer Metastasis by Oncogenic ROS-NRF2-HO-1 Axis via a Positive NRF2-HO-1 Feedback Loop. Front Oncol. 2021;11:679816
66. Black HS. Oxidative Stress and ROS Link Diabetes and Cancer. J Mol Pathol. 2024;5:96-119
67. Wirth K, Shinoda S, Sato-Dahlman M, Dickey DM, Bernlohr DA, Ikramuddin S. et al. Fatty acid binding protein 4 regulates pancreatic cancer cell proliferation via activation of nuclear factor E2-related factor 2. Surg Obes Relat Dis. 2022;18:485-93
68. Suzuki T, Takahashi J, Yamamoto M. Molecular Basis of the KEAP1-NRF2 signaling pathway. Mol Cells. 2023;46:133-141
69. Shaw P, Chattopadhyay A. Nrf2-ARE signaling in cellular protection: Mechanism of action and the regulatory mechanisms. J Cell Physiol. 2020;235:3119-30
70. Wild AC, Moinova HR, Mulcahy RT. Regulation of γ-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem. 1999;274:33627-36
71. Wu KC, Cui JY, Klaassen CD. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol Sci. 2011;123:590-600
72. Kausar S, Wang F, Cui H. The role of mitochondria in reactive oxygen species generation and its implications for neurodegenerative diseases. Cells. 2018;7:274
73. Wende AR, Young ME, Chatham J, Zhang J, Rajasekaran NS, Darley-Usmar VM. Redox Biol and the interface between bioenergetics, autophagy and circadian control of metabolism. Free Radic Biol Med. 2016;100:94-107
74. Kovac S, Angelova PR, Holmström KM, Zhang Y, Dinkova-Kostova AT, Abramov AY. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim Biophys Acta. 2015;1850:794-801
75. Bento-Pereira C, Dinkova-Kostova AT. Activation of transcription factor Nrf2 to counteract mitochondrial dysfunction in Parkinson's disease. Med Res Rev. 2021;41:785-802
76. Holmström KM, Baird L, Zhang Y, Hargreaves I, Chalasani A, Land JM. et al. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol Open. 2013;2:761-70
77. Zhang Y-KJ, Wu KC, Klaassen CD. Genetic activation of Nrf2 protects against fasting-induced oxidative stress in livers of mice. PLoS One. 2013;8:e59122
78. Merry TL, Ristow M. Nuclear factor erythroid-derived 2-like 2 (NFE2L2, Nrf2) mediates exercise-induced mitochondrial biogenesis and the anti-oxidant response in mice. J physiol. 2016;594:5195-207
79. Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res. 2008;103:1232-40
80. Gr S, Be K. AMPK in health and disease. Physiol Rev. 2009;89:1025-78
81. Joo MS, Kim WD, Lee KY, Kim JH, Koo JH, Kim SG. AMPK facilitates nuclear accumulation of Nrf2 by phosphorylating at serine 550. Mol Cell Biol. 2016;36:1931-42
82. Liu X-m, Peyton KJ, Shebib AR, Wang H, Korthuis RJ, Durante W. Activation of AMPK stimulates heme oxygenase-1 gene expression and human endothelial cell survival. Am J Physiol Heart Circ Physiol. 2011;300:H84-H93
83. Zimmermann K, Baldinger J, Mayerhofer B, Atanasov AG, Dirsch VM, Heiss EH. Activated AMPK boosts the Nrf2/HO-1 signaling axis—a role for the unfolded protein response. Free Radic Biol Med. 2015;88:417-26
84. Xu J, Donepudi AC, Moscovitz JE, Slitt AL. Keap1-knockdown decreases fasting-induced fatty liver via altered lipid metabolism and decreased fatty acid mobilization from adipose tissue. PLoS One. 2013;8:e79841
85. Meakin PJ, Chowdhry S, Sharma RS, Ashford FB, Walsh SV, McCrimmon RJ. et al. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol Cell Biol. 2014;34:3305-20
86. Slocum SL, Skoko JJ, Wakabayashi N, Aja S, Yamamoto M, Kensler TW. et al. Keap1/Nrf2 pathway activation leads to a repressed hepatic gluconeogenic and lipogenic program in mice on a high-fat diet. Arch Biochem Biophys. 2016;591:57-65
87. Saha PK, Reddy VT, Konopleva M, Andreeff M, Chan L. The triterpenoid 2-cyano-3, 12-dioxooleana-1, 9-dien-28-oic-acid methyl ester has potent anti-diabetic effects in diet-induced diabetic mice and Leprdb/db mice. J Biol Chem. 2010;285:40581-92
88. De Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H. et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med. 2013;369:2492-503
89. Nam KT, Park JS, Kim SH, Lee M, Kim G, Min BS. et al. Ezetimibe, an NPC1L1 inhibitor, is a potent Nrf2 activator that protects mice from diet-induced nonalcoholic steatohepatitis. Free Radic Biol Med. 2016;99:520-32
90. Feng X, Yu W, Li X, Zhou F, Zhang W, Shen Q. et al. Apigenin, a modulator of PPARγ, attenuates HFD-induced NAFLD by regulating hepatocyte lipid metabolism and oxidative stress via Nrf2 activation. Biochem Pharmacol. 2017;136:136-49
91. Uruno A, Furusawa Y, Yagishita Y, Fukutomi T, Muramatsu H, Negishi T. et al. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol Cell Biol. 2013;33:2996-3010
92. Siracusa R, Scuto M, Fusco R, Trovato A, Ontario ML, Crea R. et al. Anti-inflammatory and Anti-oxidant Activity of Hidrox((R)) in Rotenone-Induced Parkinson's Disease in Mice. Antioxidants (Basel). 2020;9:824
93. Sánchez-Ortega M, Carrera AC, Garrido A. Role of NRF2 in Lung Cancer. Cells. 2021;10:1879
94. Robertson H, Dinkova-Kostova AT, Hayes JD. NRF2 and the Ambiguous Consequences of Its Activation during Initiation and the Subsequent Stages of Tumourigenesis. Cancers. 2020;12:3609
95. Purohit V, Wang L, Yang H, Li J, Ney GM, Gumkowski ER. et al. ATDC binds to KEAP1 to drive NRF2-mediated tumorigenesis and chemoresistance in pancreatic cancer. Genes Dev. 2021;35:218-33
96. Fu D, Wang C, Yu L, Yu R. Induction of ferroptosis by ATF3 elevation alleviates cisplatin resistance in gastric cancer by restraining Nrf2/Keap1/xCT signaling. Cell Mol Biol Lett. 2021;26:26
97. Xu P, Jiang L, Yang Y, Wu M, Liu B, Shi Y. et al. PAQR4 promotes chemoresistance in non-small cell lung cancer through inhibiting Nrf2 protein degradation. Theranotics. 2020;10:3767-3778
98. Jayakumar S, Kunwar A, Sandur SK, Pandey BN, Chaubey RC. Differential response of DU145 and PC3 prostate cancer cells to ionizing radiation: Role of reactive oxygen species, GSH and Nrf2 in radiosensitivity. Biochim Biophys Acta. 2014;1840:485-94
99. Zhao M, Xu H, Zhang B, Hong B, Yan W, Zhang J. Impact of nuclear factor erythroid-derived 2-like 2 and p62/sequestosome expression on prognosis of patients with gliomas. Hum Pathol. 2015;46:843-9
100. Zhu J, Wang H, Sun Q, Ji X, Zhu L, Cong Z. et al. Nrf2 is required to maintain the self-renewal of glioma stem cells. BMC Cancer. 2013;13:380
101. Pópulo H, Lopes JM, Soares P. The mTOR Signalling Pathway in Human Cancer. Int J Mol Sci. 2012;13:1886-918
102. Tsuchida K, Tsujita T, Hayashi M, Ojima A, Keleku-Lukwete N, Katsuoka F. et al. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radic Biol Med. 2017;103:236-47
103. Lim J, Lee SH, Cho S, Lee IS, Kang BY, Choi HJ. 4-methoxychalcone enhances cisplatin-induced oxidative stress and cytotoxicity by inhibiting the Nrf2/ARE-mediated defense mechanism in A549 lung cancer cells. Mol Cells. 2013;36:340-6
104. Lee J, Kang JS, Nam LB, Yoo OK, Keum YS. Suppression of NRF2/ARE by convallatoxin sensitises A549 cells to 5-FU-mediated apoptosis. Free Redic Res. 2018;52:1416-23
105. Yang Y, Deng Y, Chen X, Zhang J, Chen Y, Li H. et al. Inhibition of PDGFR by CP-673451 induces apoptosis and increases cisplatin cytotoxicity in NSCLC cells via inhibiting the Nrf2-mediated defense mechanism. Toxicol Lett. 2018;295:88-98
106. Cykowiak M, Kleszcz R. Attenuation of Pancreatic Cancer In Vitro and In Vivo via Modulation of Nrf2 and NF-κB Signaling Pathways by Natural Compounds. Cells. 2021;10:3556
107. Grattarola M, Cucci MA, Roetto A, Dianzani C, Barrera G, Pizzimenti S. Post-translational down-regulation of Nrf2 and YAP proteins, by targeting deubiquitinases, reduces growth and chemoresistance in pancreatic cancer cells. Free Radic Biol Med. 2021;174:202-10
108. Ma X, Zhang J, Liu S, Huang Y, Chen B, Wang D. Nrf2 knockdown by shRNA inhibits tumor growth and increases efficacy of chemotherapy in cervical cancer. Cancer Chemother Pharmacol. 2012;69:485-94
109. Li X, Mu J, Lin Y, Zhao J, Meng X. Combination of cyanidin-3-O-glucoside and cisplatin induces oxidative stress and apoptosis in HeLa cells by reducing activity of endogenous antioxidants, increasing bax/bcl-2 mRNA expression ratio, and downregulating Nrf2 expression. J Food Biochem. 2021;45:e13806
110. Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017;11:254-62
111. Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451-60
112. Buckbinder L, Talbott R, Seizinger BR, Kley N. Gene regulation by temperature-sensitive p53 mutants: identification of p53 response genes. Proc Natl Acad Sci U S A. 1994;91:10640-4
113. Chen C-C, Jeon S-M, Bhaskar PT, Nogueira V, Sundararajan D, Tonic I. et al. FoxOs inhibit mTORC1 and activate Akt by inducing the expression of Sestrin3 and Rictor. Dev Cell. 2010;18:592-604
114. Budanov AV, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H. et al. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene. 2002;21:6017-31
115. Budanov AV, Lee JH, Karin M. Stressin'Sestrins take an aging fight. EMBO Mol Med. 2010;2:388-400
116. Lee JH, Budanov AV, Karin M. Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab. 2013;18:792-801
117. Seo K, Ki SH, Shin SM. Sestrin2-AMPK activation protects mitochondrial function against glucose deprivation-induced cytotoxicity. Cell Signal. 2015;27:1533-43
118. Peeters H, Debeer P, Bairoch A, Wilquet V, Huysmans C, Parthoens E. et al. PA26 is a candidate gene for heterotaxia in humans: identification of a novel PA26-related gene family in human and mouse. Hum Genet. 2003;112:573-80
119. Nogueira V, Park Y, Chen C-C, Xu P-Z, Chen M-L, Tonic I. et al. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer cell. 2008;14:458-70
120. Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science. 2004;304:596-600
121. Yang K-S, Kang SW, Woo HA, Hwang SC, Chae HZ, Kim K. et al. Inactivation of human peroxiredoxin I during catalysis as the result of the oxidation of the catalytic site cysteine to cysteine-sulfinic acid. J Biol Chem. 2002;277:38029-36
122. Woo HA, Bae SH, Park S, Rhee SG. Sestrin 2 is not a reductase for cysteine sulfinic acid of peroxiredoxins. Antioxid Redox Signal. 2009;11:739-45
123. Ben-Sahra I, Dirat B, Laurent K, Puissant A, Auberger P, Budanov A. et al. Sestrin2 integrates Akt and mTOR signaling to protect cells against energetic stress-induced death. Cell Death Differ. 2013;20:611-9
124. Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306-13
125. Seo K, Seo S, Han JY, Ki SH, Shin SM. Resveratrol attenuates methylglyoxal-induced mitochondrial dysfunction and apoptosis by Sestrin2 induction. Toxicol Appl Pharmacol. 2014;280:314-22
126. Yang JH, Kim KM, Kim MG, Seo KH, Han JY, Ka S-O. et al. Role of sestrin2 in the regulation of proinflammatory signaling in macrophages. Free Radic Biol Med. 2015;78:156-67
127. Zhao Y, Hu X, Liu Y, Dong S, Wen Z, He W. et al. ROS signaling under metabolic stress: cross-talk between AMPK and AKT pathway. Mol Cancer. 2017;16:1-12
128. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471-84
129. Tao R, Xiong X, Liangpunsakul S, Dong XC. Sestrin 3 protein enhances hepatic insulin sensitivity by direct activation of the mTORC2-Akt signaling. Diabetes. 2015;64:1211-23
130. Lee JH, Budanov AV, Park EJ, Birse R, Kim TE, Perkins GA. et al. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science. 2010;327:1223-8
131. Lee JH, Budanov AV, Talukdar S, Park EJ, Park HL, Park H-W. et al. Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metab. 2012;16:311-21
132. Li H, Liu S, Yuan H, Niu Y, Fu L. Sestrin 2 induces autophagy and attenuates insulin resistance by regulating AMPK signaling in C2C12 myotubes. Exp Cell Res. 2017;354:18-24
133. Jin SH, Yang JH, Shin BY, Seo K, Shin SM, Cho IJ. et al. Resveratrol inhibits LXRα-dependent hepatic lipogenesis through novel antioxidant Sestrin2 gene induction. Toxicol Appl Pharmacol. 2013;271:95-105
134. Haidurov A, Budanov AV. Sestrin family - the stem controlling healthy ageing. Mech Ageing Dev. 2020;192:111379
135. Murugan AK. mTOR: Role in cancer, metastasis and drug resistance. Semin Cancer Biol. 2019;59:92-111
136. Ro SH, Xue X, Ramakrishnan SK, Cho CS, Namkoong S, Jang I. et al. Tumor suppressive role of sestrin2 during colitis and colon carcinogenesis. Elife. 2016;5:e12204
137. Wang N, Pan W, Zhu M, Zhang M, Hao X, Liang G. et al. Fangchinoline induces autophagic cell death via p53/sestrin2/AMPK signalling in human hepatocellular carcinoma cells. Br J Pharmacol. 2011;164:731-42
138. Wei J, Zheng X, Li W, Li X, Fu Z. Sestrin2 reduces cancer stemness via Wnt/β-catenin signaling in colorectal cancer. Cancer Cell Int. 2022;22:75
139. Xu H, Sun H, Zhang H, Liu J, Fan F, Li Y. et al. An ShRNA Based Genetic Screen Identified Sesn2 as a Potential Tumor Suppressor in Lung Cancer via Suppression of Akt-mTOR-p70S6K Signaling. PLoS one. 2015;10:e0124033
140. Brüning A, Rahmeh M, Friese K. Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated sestrin-2 regulation. Mol Oncol. 2013;7:1012-8
141. Wei JL, Fang M, Fu ZX, Zhang SR, Guo JB, Wang R. et al. Sestrin 2 suppresses cells proliferation through AMPK/mTORC1 pathway activation in colorectal cancer. Oncotarget. 2017;8:49318-28
142. Kim GT, Lee SH, Kim JI, Kim YM. Quercetin regulates the sestrin 2-AMPK-p38 MAPK signaling pathway and induces apoptosis by increasing the generation of intracellular ROS in a p53-independent manner. Int J Mol Med. 2014;33:863-9
143. Fu H, Ni X. Study of the Mechanism by Which Curcumin Cooperates with Sestrin2 to Inhibit the Growth of Pancreatic Cancer. gastroenterol Res Pract. 2021;2021:7362233
144. Liang Y, Zhu J, Huang H, Xiang D, Li Y, Zhang D. et al. SESN2/sestrin 2 induction-mediated autophagy and inhibitory effect of isorhapontigenin (ISO) on human bladder cancers. Autophagy. 2016;12:1229-39
145. Reyes-Farias M, Carrasco-Pozo C. The Anti-Cancer Effect of Quercetin: Molecular Implications in Cancer Metabolism. Int J Mol Sci. 2019;20:3177
146. Chae HS, Gil M, Saha SK. Sestrin2 Expression Has Regulatory Properties and Prognostic Value in Lung Cancer. j Pers Med. 2020;10:109
147. Byun JK, Choi YK, Kim JH, Jeong JY, Jeon HJ, Kim MK. et al. A Positive Feedback Loop between Sestrin2 and mTORC2 Is Required for the Survival of Glutamine-Depleted Lung Cancer Cells. Cell Rep. 2017;20:586-99
148. Dai J, Huang Q. Sestrin 2 confers primary resistance to sorafenib by simultaneously activating AKT and AMPK in hepatocellular carcinoma. Cancer Med. 2018;7:5691-703
149. Zhao B, Shah P, Budanov AV, Qiang L, Ming M, Aplin A. et al. Sestrin2 protein positively regulates AKT enzyme signaling and survival in human squamous cell carcinoma and melanoma cells. J Biol Chem. 2014;289:35806-14
150. Tang Z, Wei X, Li T, Wang W, Wu H, Dong H. et al. Sestrin2-Mediated Autophagy Contributes to Drug Resistance via Endoplasmic Reticulum Stress in Human Osteosarcoma. Front Cell Dev Biol. 2021;9:722960
151. Won DH, Chung SH, Shin JA, Hong KO, Yang IH, Yun JW. et al. Induction of sestrin 2 is associated with fisetin-mediated apoptosis in human head and neck cancer cell lines. J Clin Biochem Nutr. 2019;64:97-105
152. Yen JH, Huang ST, Huang HS, Fong YC, Wu YY, Chiang JH. et al. HGK-sestrin 2 signaling-mediated autophagy contributes to antitumor efficacy of Tanshinone IIA in human osteosarcoma cells. Cell Death Dis. 2018;9:1003
153. Jeong S, Kim DY, Kang SH. Docosahexaenoic Acid Enhances Oxaliplatin-Induced Autophagic Cell Death via the ER Stress/Sesn2 Pathway in Colorectal Cancer. Cancers. 2019;11:982
154. Kim GT, Lee SH, Kim YM. Quercetin Regulates Sestrin 2-AMPK-mTOR Signaling Pathway and Induces Apoptosis via Increased Intracellular ROS in HCT116 Colon Cancer Cells. J Cancer Prev. 2013;18:264-70
155. Seo K, Ki SH, Park EY, Shin SM. 5-Fluorouracil inhibits cell migration by induction of Sestrin2 in colon cancer cells. Arch Pharm Res. 2017;40:231-9
156. Yan M, Vemu B, Veenstra J, Petiwala SM, Johnson JJ. Carnosol, a dietary diterpene from rosemary (Rosmarinus officinalis) activates Nrf2 leading to sestrin 2 induction in colon cells. Integr Mol Med. 2018;5:10
157. Hua X, Xu J, Deng X, Xu J, Li J, Zhu DQ. et al. New compound ChlA-F induces autophagy-dependent anti-cancer effect via upregulating Sestrin-2 in human bladder cancer. Cancer Lett. 2018;436:38-51
158. Ding B, Parmigiani A, Yang C, Budanov AV. Sestrin2 facilitates death receptor-induced apoptosis in lung adenocarcinoma cells through regulation of XIAP degradation. Cell cycle. 2015;14:3231-41
159. Zhang L, Liu W, Wang Q, Li Q, Wang H. New Drug Candidate Targeting the 4A1 Orphan Nuclear Receptor for Medullary Thyroid Cancer Therapy. Molecules. 2018;23:565
160. Abraham NG, Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev. 2008;60:79-127
161. Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem Pharmacol. 2010;80:1895-903
162. Wiesel P, Patel AP, DiFonzo N, Marria PB, Sim CnU, Pellacani A. et al. Endotoxin-induced mortality is related to increased oxidative stress and end-organ dysfunction, not refractory hypotension, in heme oxygenase-1-deficient mice. Circulation. 2000;102:3015-22
163. Lin Y, Berg AH, Iyengar P, Lam TK, Giacca A, Combs TP, Rajala MW, Du X, Rollman B, Li W, Hawkins M, Barzilai N, Rhodes CJ, Fantus IG, Brownlee M, Scherer PE. The hyperglycemia-induced inflammatory response in adipocytes: the role of reactive oxygen species. J Biol Chem. 2005;280:4617-26
164. Cao J, Peterson SJ, Sodhi K, Vanella L, Barbagallo I, Rodella LF. et al. Heme oxygenase gene targeting to adipocytes attenuates adiposity and vascular dysfunction in mice fed a high-fat diet. Hypertension. 2012;60:467-75
165. Nicolai A, Li M, Kim DH, Peterson SJ, Vanella L, Positano V. et al. Heme oxygenase-1 induction remodels adipose tissue and improves insulin sensitivity in obesity-induced diabetic rats. Hypertension. 2009;53:508-15
166. Ndisang J, Jadhav A. Hemin therapy suppresses inflammation and retroperitoneal adipocyte hypertrophy to improve glucose metabolism in obese rats co-morbid with insulin-resistant type-2 diabetes. Diabetes Obes Metab. 2013;15:1029-39
167. Li M, Kim DH, Tsenovoy PL, Peterson SJ, Rezzani R, Rodella LF. et al. Treatment of obese diabetic mice with a heme oxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes. 2008;57:1526-35
168. Wang Z, Ka S-O, Lee Y, Park B-H, Bae EJ. Butein induction of HO-1 by p38 MAPK/Nrf2 pathway in adipocytes attenuates high-fat diet induced adipose hypertrophy in mice. Eur J Pharmacol. 2017;799:201-10
169. Padmalayam I, Suto M. Role of adiponectin in the metabolic syndrome: current perspectives on its modulation as a treatment strategy. Curr Pharm Des. 2013;19:5755-63
170. Xu L, Kitade H, Ni Y, Ota T. Roles of chemokines and chemokine receptors in obesity-associated insulin resistance and nonalcoholic fatty liver disease. Biomolecules. 2015;5:1563-79
171. Lacasa D, Taleb S, Keophiphath M, Miranville A, Clement K. Macrophage-secreted factors impair human adipogenesis: involvement of proinflammatory state in preadipocytes. Endocrinology. 2007;148:868-77
172. Association AD. Standards of medical care in diabetes—2009. Diabetes care. 2009;32:S13
173. Bruce CR, Carey AL, Hawley JA, Febbraio MA. Intramuscular heat shock protein 72 and heme oxygenase-1 mRNA are reduced in patients with type 2 diabetes: evidence that insulin resistance is associated with a disturbed antioxidant defense mechanism. Diabetes. 2003;52:2338-45
174. Chen K, Ou B, Huang Q, Deng D, Xiang Y, Hu F. LncRNA NEAT1 aggravates human microvascular endothelial cell injury by inhibiting the Apelin/Nrf2/HO-1 signalling pathway in type 2 diabetes mellitus with obstructive sleep apnoea. Epigenetics. 2024;19:2293409
175. Kajimoto Y, Kaneto H. Role of oxidative stress in pancreatic β-cell dysfunction. Ann N Y Acad Sci. 2004 p. 168-76
176. Kim M-H, Kim E-H, Jung HS, Yang D, Park E-Y, Jun H-S. EX4 stabilizes and activates Nrf2 via PKCδ, contributing to the prevention of oxidative stress-induced pancreatic beta cell damage. Toxicol Appl Pharmacol. 2017;315:60-9
177. Nitti M, Piras S, Marinari UM, Moretta L, Pronzato MA, Furfaro AL. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants (Basel, Switzerland). 2017;6:29
178. Nitti M, Ivaldo C, Traverso N, Furfaro AL. Clinical Significance of Heme Oxygenase 1 in Tumor Progression. Antioxidants. 2021;10:789
179. Luu Hoang KN, Anstee JE, Arnold JN. The Diverse Roles of Heme Oxygenase-1 in Tumor Progression. Front Immunol. 2021;12:658315
180. Gandini NA, Fermento ME, Salomón DG, Blasco J, Patel V, Gutkind JS. et al. Nuclear localization of heme oxygenase-1 is associated with tumor progression of head and neck squamous cell carcinomas. Exp Mol Pathol. 2012;93:237-45
181. Was H, Cichon T, Smolarczyk R, Rudnicka D, Stopa M, Chevalier C. et al. Overexpression of heme oxygenase-1 in murine melanoma: increased proliferation and viability of tumor cells, decreased survival of mice. Am J Pathol. 2006;169:2181-98
182. Zhe N, Wang J, Chen S, Lin X, Chai Q, Zhang Y. et al. Heme oxygenase-1 plays a crucial role in chemoresistance in acute myeloid leukemia. Hematology. 2015;20:384-91
183. Wu W, Ma D, Wang P, Cao L, Lu T, Fang Q. et al. Potential crosstalk of the interleukin-6-heme oxygenase-1-dependent mechanism involved in resistance to lenalidomide in multiple myeloma cells. FEBS J. 2016;283:834-49
184. Lin HH, Chiang MT, Chang PC, Chau LY. Myeloid heme oxygenase-1 promotes metastatic tumor colonization in mice. Cancer Sci. 2015;106:299-306
185. Chang YJ, Chen WY, Huang CY, Liu HH, Wei PL. Glucose-regulated protein 78 (GRP78) regulates colon cancer metastasis through EMT biomarkers and the NRF-2/HO-1 pathway. Tumour Biol. 2015;36:1859-69
186. Pae HO, Oh GS, Choi BM, Chae SC, Chung HT. Differential expressions of heme oxygenase-1 gene in CD25- and CD25+ subsets of human CD4+ T cells. Biochem Biophys Res Commun. 2003;306:701-5
187. Seo GS, Jiang WY, Chi JH, Jin H, Park WC, Sohn DH. et al. Heme oxygenase-1 promotes tumor progression and metastasis of colorectal carcinoma cells by inhibiting antitumor immunity. Oncotarget. 2015;6:19792-806
188. Han L, Jiang J, Ma Q, Wu Z, Wang Z. The inhibition of heme oxygenase-1 enhances the chemosensitivity and suppresses the proliferation of pancreatic cancer cells through the SHH signaling pathway. Int J Oncol. 2018;52:2101-9
189. Hill M, Pereira V, Chauveau C, Zagani R, Remy S, Tesson L. et al. Heme oxygenase-1 inhibits rat and human breast cancer cell proliferation: mutual cross inhibition with indoleamine 2,3-dioxygenase. FASEB J. 2005;19:1957-68
190. Ferrando M, Gueron G, Elguero B, Giudice J, Salles A, Leskow FC. et al. Heme oxygenase 1 (HO-1) challenges the angiogenic switch in prostate cancer. Angiogenesis. 2011;14:467-79
191. Tertil M, Golda S, Skrzypek K, Florczyk U, Weglarczyk K, Kotlinowski J. et al. Nrf2-heme oxygenase-1 axis in mucoepidermoid carcinoma of the lung: Antitumoral effects associated with down-regulation of matrix metalloproteinases. Free Radic Biol Med. 2015;89:147-57
192. Gandini NA, Alonso EN, Fermento ME, Mascaró M, Abba MC, Coló GP. et al. Heme Oxygenase-1 Has an Antitumor Role in Breast Cancer. Antioxid Redox Signal. 2019;30:2030-49
193. Andrés NC, Fermento ME, Gandini NA, Romero AL, Ferro A, Donna LG. et al. Heme oxygenase-1 has antitumoral effects in colorectal cancer: involvement of p53. Exp Mol Pathol. 2014;97:321-31
194. Seiwert N, Wecklein S, Demuth P, Hasselwander S, Kemper TA, Schwerdtle T. et al. Heme oxygenase 1 protects human colonocytes against ROS formation, oxidative DNA damage and cytotoxicity induced by heme iron, but not inorganic iron. Cell Death Dis. 2020;11:787
195. Tertil M, Skrzypek K, Florczyk U, Weglarczyk K, Was H, Collet G. et al. Regulation and novel action of thymidine phosphorylase in non-small cell lung cancer: crosstalk with Nrf2 and HO-1. PLoS one. 2014;9:e97070
196. Kongpetch S, Kukongviriyapan V, Prawan A, Senggunprai L, Kukongviriyapan U, Buranrat B. Crucial role of heme oxygenase-1 on the sensitivity of cholangiocarcinoma cells to chemotherapeutic agents. PLoS One. 2012;7:e34994
197. Tanaka S, Akaike T, Fang J, Beppu T, Ogawa M, Tamura F. et al. Antiapoptotic effect of haem oxygenase-1 induced by nitric oxide in experimental solid tumour. Br J Cancer. 2003;88:902-9
198. Fang J, Sawa T, Akaike T, Akuta T, Sahoo SK, Khaled G. et al. In vivo antitumor activity of pegylated zinc protoporphyrin: targeted inhibition of heme oxygenase in solid tumor. Cancer Res. 2003;63:3567-74
199. Berberat PO, Dambrauskas Z, Gulbinas A, Giese T, Giese N, Künzli B. et al. Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clin Cancer Res. 2005;11:3790-8
200. Nuhn P, Künzli BM, Hennig R, Mitkus T, Ramanauskas T, Nobiling R. et al. Heme oxygenase-1 and its metabolites affect pancreatic tumor growth in vivo. Mol Cancer. 2009;8:37
201. Kang KA, Maeng YH, Zhang R, Yang YR, Piao MJ, Kim KC. et al. Involvement of heme oxygenase-1 in Korean colon cancer. Tumour Biol. 2012;33:1031-8
202. Zhao X, Dong W, Gao Y, Shin DS, Ye Q, Su L. et al. Novel indolyl-chalcone derivatives inhibit A549 lung cancer cell growth through activating Nrf-2/HO-1 and inducing apoptosis in vitro and in vivo. Sci Rep. 2017;7:3919
203. Mucha O, Podkalicka P, Mikulski M, Barwacz S, Andrysiak K, Biela A. et al. Development and characterization of a new inhibitor of heme oxygenase activity for cancer treatment. Arch Biochem Biophys. 2019;671:130-42
204. Mahajan M, Sitasawad S. miR-140-5p Attenuates Hypoxia-Induced Breast Cancer Progression by Targeting Nrf2/HO-1 Axis in a Keap1-Independent Mechanism. Cells. 2021;11:12
205. Chen Y, Huang T, Yu Z, Yu Q, Wang Y, Hu J. et al. The functions and roles of sestrins in regulating human diseases. Cell Mol Biol Lett. 2022;27:2
206. Aslan E, Demir B, Ulusal H, Sahin S, Taysi S, Elboga G. et al. Sestrin-2 and hypoxia-inducible factor-1 alpha levels in major depressive disorder and its subtypes. Psychopharmacology. 2023;240:1691-704
207. Li R, Huang Y, Semple I, Kim M, Zhang Z, Lee JH. Cardioprotective roles of sestrin 1 and sestrin 2 against doxorubicin cardiotoxicity. Am J Physiol Heart Circ Physiol. 2019;317:H39-H48
Author contact
![]() Corresponding authors: Prof. Dong Jae Baek, dbaekac.kr, College of Pharmacy, Mokpo National University, Jeonnam 58554, Republic of Korea. Prof. Sung-Hwan Ki, shkiac.kr, College of Pharmacy, Chosun University, Gwangju 61451, Republic of Korea. Prof. Eun-Young Park, parkeyac.kr, College of Pharmacy, Mokpo National University, Jeonnam 58554, Republic of Korea.
Corresponding authors: Prof. Dong Jae Baek, dbaekac.kr, College of Pharmacy, Mokpo National University, Jeonnam 58554, Republic of Korea. Prof. Sung-Hwan Ki, shkiac.kr, College of Pharmacy, Chosun University, Gwangju 61451, Republic of Korea. Prof. Eun-Young Park, parkeyac.kr, College of Pharmacy, Mokpo National University, Jeonnam 58554, Republic of Korea.