Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Overview of AIC
2. Mitochondrial quality control...
3. Natural products regulate...
Conclusions
Funding
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(2):507-523. doi:10.7150/ijbs.103810 This issue Cite
Review
New insights into mitochondrial quality control in anthracycline-induced cardiotoxicity: molecular mechanisms, therapeutic targets, and natural products
Weili Li1,#, Yuqin Zhang1,#, Yan Wei1,#, Guanjing Ling1, Yawen Zhang1, Yilin Li1, Shujuan Guo1, Nannan Tan6, Lin Ma6, Wei Li1, Qianbin Sun1, Wei Wang3,4,5, ![]() , Yong Wang2,4,5,
, Yong Wang2,4,5, ![]()
1. School of Traditional Chinese Medicine, Beijing University of Chinese Medicine, Beijing 100029, China.
2. Dongzhimen Hospital, Beijing University of Chinese Medicine, Beijing 100029, China.
3. Guangzhou University of Chinese Medicine, Guangzhou 510006, China.
4. Beijing Key Laboratory of TCM Syndrome and Formula, Beijing University of Chinese Medicine, Beijing 100029, China.
5. Key Laboratory of TCM Syndrome and Formula (Beijing University of Chinese Medicine), Ministry of Education, Beijing 100029, China.
6. Anhui University of Traditional Chinese Medicine, Anhui 230012, China.
#These authors contributed equally to this manuscript.
Received 2024-9-17; Accepted 2024-11-23; Published 2025-1-1
Abstract

Anthracyclines (ANTs) are widely used in cancer therapy, particularly for lymphoma, sarcoma, breast cancer, and childhood leukemia, and have become the cornerstone of chemotherapy for various malignancies. However, it is associated with fatal and dose-dependent cardiovascular complications, especially cardiotoxicity. Mitochondrial quality control mechanisms, encompassing mitophagy, mitochondrial dynamics, and mitochondrial biogenesis, maintain mitochondrial homeostasis in the cardiovascular system. Recent studies have highlighted that mitochondrial quality control mechanisms play considerable roles in ANTs-induced cardiotoxicity (AIC). In addition, natural products targeting mitochondrial quality control mechanisms have emerged as potential therapeutic strategies to alleviate AIC. This review summarizes the types, incidence, prevention, treatment, and pathomechanism of AIC, delves into the molecular mechanisms of mitochondrial quality control in the pathogenesis of AIC, and explores natural products that target these mechanisms, so as to provide potential targets and therapeutic drugs for address the clinical challenges in AIC prevention and treatment, where no effective medicines are available.
Keywords: anthracycline, cardiotoxicity, mitochondrial quality control, mitophagy, mitochondrial dynamics, mitochondrial biogenesis, natural products
1. Overview of AIC
1.1 Types of AIC
The discovery of the first anthracycline compound daunorubicin from Streptomyces peucetius in the 1950s was a major breakthrough in the realm of oncology [1], and thousands of daunorubicin and doxorubicin (DOX) analogues were subsequently produced, fostering a new era in cancer therapeutics [2]. Nowadays, the most clinically pertinent anthracyclines (ANTs), notably DOX, daunorubicin, darubicin, and epirubicin, have emerged as the cornerstone of chemotherapy for an extensive spectrum of malignancies, encompassing lymphoma, sarcoma, breast cancer, and childhood leukemia, among others [3]. While ANTs have undeniably improved long-term survival rates among cancer patients, their efficacy comes at the expense of severe, dose-dependent cardiovascular complications, particularly cardiotoxicity [4]. Anthracycline-induced cardiotoxicity (AIC) includes a broad spectrum of cardiac injury resulting from ANTs exposure, ranging from subclinical cardiomyocyte injury to heart failure (HF). According to the time of onset, AIC was categorized into three types [5-9]: (1) Acute AIC, which occurred within 2 weeks after the end of a single dose or course of ANTs treatment and manifested as supraventricular arrhythmia, transient cardiac dysfunction, and alterations in electrocardiogram. (2) Early-onset Chronic AIC, with onset within 1 year. It is the most prevalent clinically type of cardiotoxicity and usually manifests as dilated and hypokinetic cardiomyopathy, often progressing to HF. (3) Delayed-onset Chronic AIC, occurring years or even decades following chemotherapy, this form of AIC is characterized by the clinical manifestations of HF, cardiomyopathy, and arrhythmia, underscoring the long-term impact of anthracyclines on cardiac health.
1.2 Incidence of AIC
The incidence of AIC varied according to different definitions. Specifically, when cardiotoxicity was defined as clinical congestive HF (CHF), the incidence of DOX at 400, 550, and 700 mg/m2 was 3%, 7%, and 18%, respectively [10]. Subsequent investigations and analyses further corroborated this trend, revealing an escalation in CHF incidence to 4.7%, 26%, and 48% at the same dosage levels [11]. Notably, the incidence of clinically asymptomatic cardiotoxicity far surpassed that of symptomatic CHF, with a decline in absolute value ≥ 10% in left ventricular ejection fraction (LVEF) from baseline and to below 50% occurring in 9%, 18%, 38%, and 65% of patients receiving DOX doses of 250 mg/m2, 350 mg/m2, 450 mg/m2, and 550 mg/m2, respectively [11]. These early retrospective studies resulted in a lifetime cumulative exposure of DOX limit to less than 450 mg/m2 in patients not currently receiving thoracic radiotherapy. In a contemporary prospective study involving 2625 patients receiving ANTs-based chemotherapy, cardiotoxicity (defined as echocardiographic decrease in LVEF ≥ 10% to absolute value less than 50%) occurred in 9% of patients [9]. The use of cardiac biomarkers, such as troponin I, reduces the incidence of cardiotoxicity by detecting early myocardial changes before the reduction in LVEF occurring. Among patients receiving high-dose chemotherapy, predominantly ANTs, a staggering 30% exhibited elevated troponin I levels within 72 hours post-chemotherapy cycle, which is highly predictive of future left ventricular dysfunction [12]. Nevertheless, the susceptibility of different individuals to cardiotoxicity becomes difficult to predict due to genetic variability and the differentiation of cardiovascular risk factors [13].
1.3 Risk stratification of AIC
In contemporary oncology clinical practice, it is highly recommended to conduct a comprehensive cardiovascular toxicity risk assessment prior to initiating potentially cardiotoxic antitumor therapies. This decision-making process must weigh the benefits against the risks associated with treatment, ultimately aiming to not only mitigate the incidence and mortality rates of cardiovascular diseases, but also reduce the interruption of cancer treatment caused by cardiovascular events [14]. Risk factors for cardiovascular toxicity mainly include history of heart disease, abnormal baseline of myocardial biomarkers, underlying diseases, age, and lifestyle [15]. Notably, the Heart Failure Association of the European Society of Cardiology Cardio-Oncology Study Group and International Cardio-Oncology Society jointly released the HFA-ICOS cardiotoxicity risk score, which provides a risk stratification tool for oncologists, hematology oncologists and cardiologists to evaluate the cardiovascular toxicity caused by different chemotherapy drugs [16]. The HFA-ICOS cardiotoxicity risk score encapsulates baseline risk stratification before the receipt of the following cancer treatment: Anthracycline-based chemotherapy, Human epidermal growth factor receptor-2 (HER-2) targeted therapy, Vascular endothelial growth factor inhibitors, Targeted kinase inhibitors, Proteasome inhibitors, Immunomodulatory drugs, Rapidly accelerated fibrosarcoma (RAF) and mitogen-activated extracellular signal-regulated kinase (MEK) inhibitors, and androgen-deprivation therapy. However, the HFA-ICOS cardiovascular risk assessment tool also has limitations. For example, the existing known risk factors cannot fully explain the interindividual differences in susceptibility to cardiovascular toxicity, hinting at genetic susceptibility as an emerging potential risk factor. Wang et al. have found that roundabout guidance receptor 2 (ROBO2) is a novel susceptibility gene for pediatric patients with cardiovascular toxicity, which promotes cardiac fibrosis through the Slit-Robo-Smad pathway, leads to disordered remodeling of extracellular matrix and exacerbates heart failure, suggesting that genetic factors will be an important basis for risk stratification in the future [17]. Therefore, further clinical studies should incorporate the following elements to more accurately detect the risk of cancer treatment exposure: 1) sophisticated cardiovascular imaging algorithms; 2) apply cutting-edge multi-omics techniques to establish genetic markers of potential risk associated with cancer therapy exposure; And 3) utilize an in vitro bioplatform of induced pluripotent stem cells to reimage cardiomyocytes from cancer patients and test their susceptibility to certain cancer therapies.
1.4 Prevention and treatment of AIC
Two primary prevention approaches for AIC are: 1) to reduce cardiotoxicity by continuous infusion, or liposome capsule administration. 2) Concomitant use of cardioprotective agent (e.g., dexrazoxane (DXZ)). Secondary prevention strategies include angiotensin converting enzyme inhibitors (ACEI), angiotensin receptor blocker (ARB), angiotensin receptor neprilysin inhibitors (ARNI), β-blockers, and statin. Primary treatment drugs for AIC includes DXZ, ACEI, ARB, ARNI, β-blockers and sodium-dependent glucose transporters 2 (SGLT-2) inhibitors.
Liposome encapsulation alters pharmacokinetics and tissue distribution without compromising antitumor efficacy [18]. Two types of liposomal DOX are employed clinically: pegylated or unpegylated, with pegylated being more widely used in the United States. Pegylation involves the covalent attachment of surface-bound methoxy polyethylene glycol to the liposomal phospholipid bilayer. This prevents the liposome from being engulfed by the monocyte-macrophage system, reducing its antigenicity and prolonging its half-life [19]. However, the number of clinical studies is small, and data on long-term follow-up are lacking, which may have contributed to the limited widespread clinical application of liposomal DOX.
DXZ is the only cardioprotective agent approved by the Food and Drug Administration (FDA) for AIC [20]. It is a potent cardioprotective agent against ANTs in a variety of cancer types in children and adults [21, 22]. The ability of DXZ to chelate iron is thought to be a major mechanism of myocardial protection [23]. However, other iron chelators such as deferasirox are not cardioprotective [24]. Lyu et al. demonstrated that DXZ exerted cardioprotection by tightly binding to the adenosine triphosphate (ATP)-binding site of topoisomerase II (Top II), thereby preventing ANTs from binding to the Top II complex [25]. Some studies have pointed out that DXZ may interfere with the antitumor efficacy of ANTs [26], but which have not been replicated in other clinical trials, and animal studies suggest that DXZ and ANTs have synergistic anticancer effects [27, 28]. Another controversy about DXZ is the potential risk of increasing the occurrence of secondary malignant tumors [29], but which has not been found in several subsequent studies [30, 31].
ACEI/ARB/ARNI or β-blocker has not been widely used due to lack of clinical evidence. Several clinical studies have investigated ACEI/ARB/ARNI or β-blockers for the prevention and treatment of AIC. A clinical trial in breast cancer patients found that candesartan did not prevent LVEF reduction but was associated with a small reduction in left ventricular end-diastolic volume and preservation of global longitudinal strain [32]. A study involving breast cancer patients receiving ANTs-based chemotherapy found that carvedilol had no effect on the incidence of early LVEF reduction but resulted in a significant reduction in troponin levels and a reduction in the incidence of diastolic dysfunction [33]. In the latest trial, the combination of candesartan and carvedilol had no significant protective effect on LVEF in patients at high risk for cardiotoxicity from ANTs-based chemotherapy [34].
SGLT-2 inhibitors were the cornerstone drugs for the treatment of chronic HF. Recent studies have shown that SGLT-2 inhibitor empagliflozin reduced the inflammation, fibrosis, ferroptosis and apoptosis of myocardial cells related to ANTs chemotherapy, and prevented the decline of cardiac function [35, 36]. A retrospective clinical study has suggested that the incidence of cardiac events was reduced in 3033 cancer patients treated with ANTs combined with SGLT-2 inhibitors [37]. Therefore, SGLT-2 inhibitors have been added as cardioprotective agents for AIC, but it has not been widely applied due to insufficient clinical evidence.
1.5 Molecular mechanisms of AIC
The molecular mechanisms of AIC have been proposed to include reactive oxygen species (ROS) production, topoisomerase inhibition, mitochondrial dysfunction, and multiple forms of cell death (Figure 1). Among them, the most widely accepted hypothesis is the generation of excess ROS through electron exchange between the anthraquinone and cellular electron donors [38, 39]. ANTs also form complexes with iron that undergo redox cycles and generate oxygen free radicals [40]. Although in vivo and in vitro studies have confirmed an increase in intracellular ROS production after ANTs treatment, neither antioxidants nor iron chelators can prevent cardiomyopathy [24, 41, 42].
Molecular mechanisms of AIC. The intricate pathological mechanisms underlying AIC encompass extensive damage to diverse cardiac cell types, notably cardiomyocytes, macrophages, fibroblasts, and endothelial cells, among others. This cellular injury is characterized by the accumulation of reactive oxygen species, the impairment of mitochondria, and the inhibition of topoisomerases, collectively culminating in cell death, including apoptosis, necrosis, and pyroptosis.
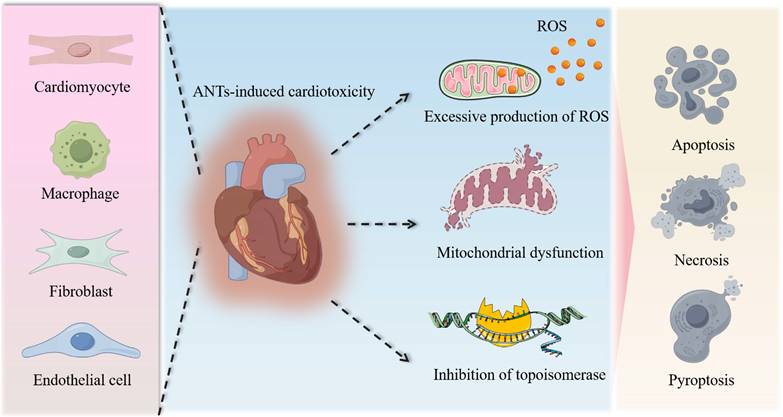
ANTs are mitochondrial toxin that target and accumulate in mitochondria [43]. This accumulation is largely attributed to the cationic nature of ANTs, which have high affinity for cardiolipin, a negatively charged major phospholipid component locating in the inner mitochondrial membrane. The binding of ANTs to cardiolipin results in the formation of an irreversible complex that is retained in the mitochondrial inner membrane [44, 45]. Proteins of the electron transport chain need to bind to cardiolipin to function properly, and more superoxide is produced due to disruption of the cardiolipin protein interface by ANTs, resulting in dysfunction of the mitochondrial respiratory chain. In addition, other membrane proteins, such as those responsible for carnitine transfer, can also be adversely affected by ANTs, leading to decrease in mitochondrial function [46]. ANTs also significantly reduced the oxidation of long-chain fatty acids, increased glucose metabolism, and comprehensively changed the metabolic state from aerobic to anaerobic metabolism [47]. Some studies have also reported that DOX treatment affects myocardial mitochondrial gene expression, leading to defects in the respiratory chain/oxidative phosphorylation system of myocardial mitochondria and reduced ATP production [48]. The above damage to mitochondrial function eventually leads to pathological changes in mitochondrial ultrastructure [49]. The formation of ROS induced by ANTs forms a vicious circle with defects in the respiratory chain/oxidative phosphorylation system, reduced ATP production, and conversion of metabolic substrate utilization, which aggravate mitochondrial damage and bioenergy crisis, and eventually lead to myocardial cell death. Considering that mitochondria are extremely abundant in cardiomyocytes and that the heart has lower levels of antioxidant enzymes such as catalase and superoxide dismutase (SOD) than other organs [50], it is reasonable that the heart is more sensitive to the ROS production induced by ANTs.
Top II unwinds deoxyribonucleic acid (DNA) strands during DNA replication, transcription or recombination [51]. There are two types of Top II: Top IIα and Top IIβ. Top IIα, mainly found in proliferating cells, is required for DNA replication, and is thought to be the molecular basis for the tumor-killing activity of ANTs [52]. In contrast, Top IIβ is present in all resting cells, including cardiomyocytes. Top IIβ has recently been identified as a key mediator of AIC [53]. ANTs disrupt the normal catalytic cycle of Top IIβ and causes DNA double-strand breaks, which further changes the transcriptome and leads to defects in mitochondrial biogenesis and increased ROS, as well as disarrangement and vacuolization of myofibrils in cardiomyocytes [54]. Among them, Top 2β and ANTs significantly reduce the expression of peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC-1α) and peroxisome proliferator-activated receptor-γ coactivator 1-β (PGC-1β), resulting in mitochondrial biogenesis defects [54]; Top IIβ is required for ANTs-induced tumor protein 53 (p53) activation and apoptotic pathways, and ANTs-induced ROS production is also dependent on Top IIβ-mediated reduction of antioxidant enzyme gene transcription [54]. In addition, ANTs decreased the expression of uncoupling proteins 2 and 3 that regulate mitochondrial ROS production [55]. Most importantly, deletion of cardiac Top IIβ protected mice from ANTs-induced cardiomyopathy.
Notably, AIC is a disease resulting from the combined participation and crosstalk among injuries to multiple cell types, including cardiomyocytes, endothelial cells, macrophages, and fibroblasts. For instance, DOX induces DNA double-strand breaks in endothelial cells, fibroblasts, and cardiomyocytes, which may be particularly associated with signaling pathways regulated by RAC1/CDC42 [56]. DOX also induces persistent DNA damage and a fibrotic phenotype in vascular endothelial cells [57]. Autophagy dysregulation is observed in DOX-treated endothelial cells, and restoring autophagic flux ameliorates endothelial-to-mesenchymal transition and eliminates ROS [39]. In chronic AIC models, the cGAS-STING pathway is specifically activated in endothelial cells and macrophages. Endothelial cell-specific knockout of STING regulates cardiomyocyte mitochondrial bioenergetics, thereby significantly preventing the occurrence of cardiotoxicity [58].
2. Mitochondrial quality control in AIC
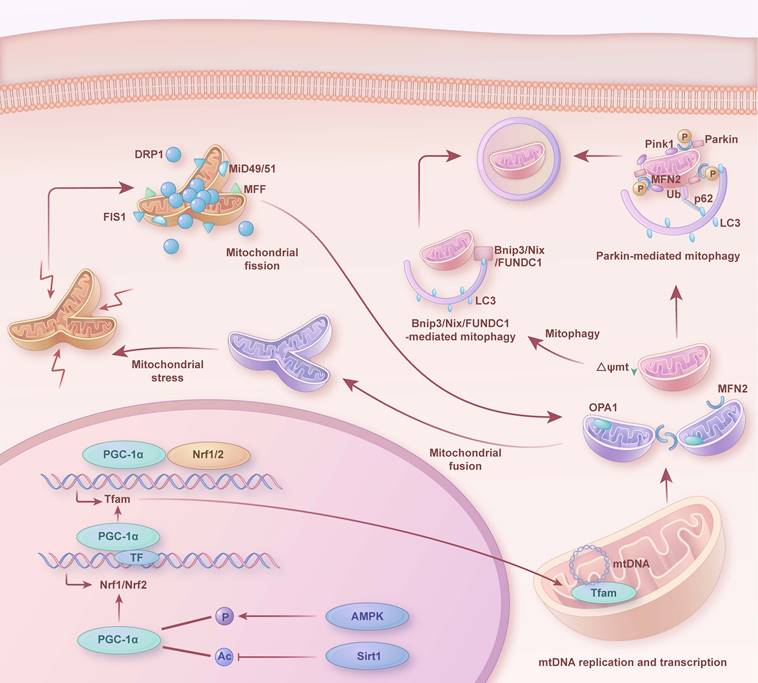
As depicted in Figure 2, in a healthy state, mitochondria preserve their morphology, distribution, and quantity through continuous division and fusion. Abnormal mitochondria are removed by mitophagy (such as PTEN-induced kinase 1 (Pink1)/Parkinson juvenile disease protein 2 (Parkin), FUN14 domain containing 1 (FUNDC1), adenovirus E1B 19kDa interacting protein 3 (Bnip3), and NIP3-like protein X (Nix)-mediated mitophagy) to maintain cell homeostasis. Simultaneously, mitochondrial biogenesis supplies fresh components to replace damaged or senescent mitochondria. A coordinated interplay exists among mitophagy, mitochondrial biogenesis, and mitochondrial dynamics. Specifically, upon mitochondrial damage, mitochondrial fission separates damaged mitochondrial components from healthy mitochondria, producing unequal daughter mitochondria. The damaged daughter mitochondria subsequently undergo mitophagy, where they are engulfed and degraded. Conversely, healthy daughter mitochondria receive signals of high energy demand, mitochondrial fusion generates new, enlarged mitochondria. While, mitochondrial biogenesis continuously furnishes fresh components to the mitochondria, and coordinates with mitochondrial dynamics and mitophagy to uphold the renewal, quantity, morphology, and functionality of mitochondria. This intricate balance ensures the normal metabolism and survival of cells.
Mitochondrial quality control mechanisms in heart. Dynamic changes and interactions of mitochondrial biogenesis, mitophagy, mitochondrial fission and fusion in the healthy heart. Ac: acetylation; P: phosphorylation; mtDNA: mitochondrial DNA; Ub: ubiquitin.
Mitochondria are multifaceted organelles that are essential in every organ of the body and are most abundant in the heart, where they control the metabolism and survival of cardiac cells [59]. Mitochondrial dysfunction has been identified as a major driver of AIC. ANTs-treated cardiomyocytes showed mitochondrial structural and functional abnormalities (such as organelle fragmentation, cristae loss and matrix destruction), and increased DNA damage and generation of ROS [60]. To maintain mitochondrial integrity and function, a variety of cellular mechanisms have evolved, including processes such as mitophagy, mitochondrial dynamics (fission/fusion), mitochondrial biogenesis, and are collectively referred to as mitochondrial quality control mechanisms [61, 62]. Impaired mitochondrial quality control mechanisms aggravate mitochondrial dysfunction, and recent studies have shown that ANTs-induced dysregulation of mitochondrial quality control mechanisms may be a major contributing factor to AIC [63, 64]. Here, we review the research progress on dysregulated mitochondrial quality control in AIC, as shown in Figure 3 and 4.
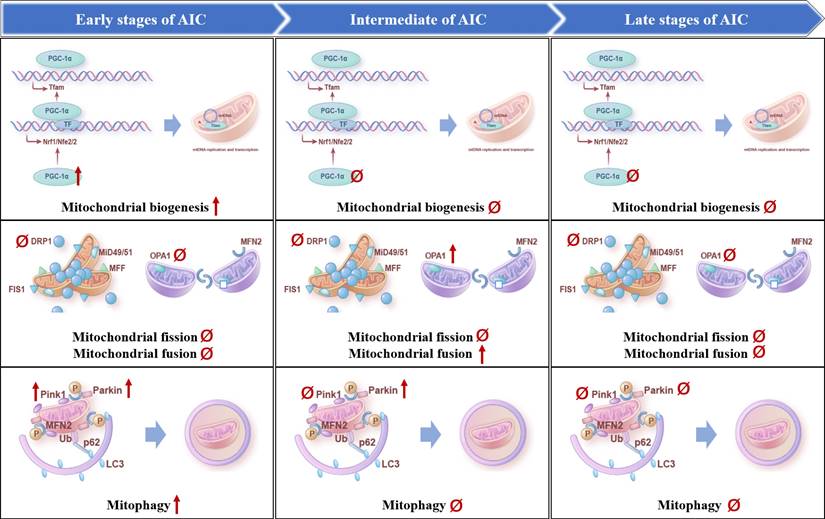
Evolution of mitochondrial quality control mechanisms in AIC. The temporal changes in mitochondrial biogenesis, mitophagy, mitochondrial fission and mitochondrial fusion occurred in the heart during the development of AIC, respectively, including the early stage at 1 week after DOX treatment, the middle stage at 9 weeks after DOX treatment, and the late stage at 15 weeks after DOX treatment. Upward arrows represent up-regulation, downward arrows represent down-regulation, and Ø represents no change.
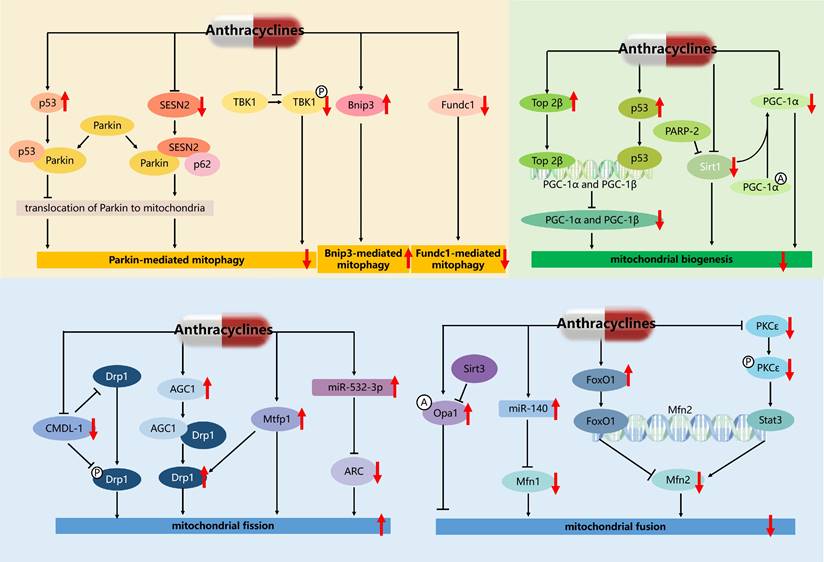
Regulatory network of mitochondrial quality control mechanisms in AIC. The red downward arrow represents the inhibitory effect of anthracyclines, and the red upward arrow represents the activating effect of anthracyclines. A: acetylation; P: phosphorylation.
2.1 Mitophagy in AIC
As shown in Figure 2, mitophagy is an organelle-specific autophagy, which is specifically used to remove damaged mitochondria and unwanted mitochondria to alleviate mitochondrial damage stress. Mitophagy has been classified into classicalPink1Parkin-mediated autophagy and microtubule associated protein 1 light chain 3 (LC3) receptor-mediated autophagy [65]. Thereinto, the latter requires the involvement of several mitophagy receptors, including Bnip3, Nix, and FUNDC1.
Several studies have shown that ANTs induce the dysregulation of mitophagy in cardiomyocytes, leading to abnormal mitochondrial structure and function and cell death [66-70]. Among them, activated Bnip3 by DOX triggers mitochondrial fragmentation, mitophagy, and necrosis [68], downregulated FUNDC1 by DOX blocks autophagic flux by inhibiting autophagosome biogenesis [71], and reduced or activated Parkin and Pink1 by DOX disturbs mitophagy flux [69, 70]. In especial, Parkin-knockout mice showed impaired mitophagy and aggravated DOX-induced mitochondrial damage and cardiotoxicity [66], suggesting the key role of Pink1/Parkin-mediated mitophagy in AIC. However, the role of DOX on the regulation of Pink1 and Parkin remains controversial. For example, in the hearts of mice treated with DOX, Pink1 and Parkin proteins translocating on mitochondria were downregulated, and their mediated mitophagy was inhibited, resulting in dysfunctional mitochondrial accumulation and cardiomyocyte death [66]. On the contrary, DOX promotes mitophagy in AC16 cardiomyocytes, as shown by increased mitochondrial translocation of Pink1 and Parkin proteins [72]. The above results suggest that the changes of mitophagy and involved proteins may be related to the dosage of ANTs, the method of animal model preparation, the detection technology, and the pathological stage of AIC. Latest evidence provides the first longitudinal analysis of the temporal changes in the heart that occur during the development of AIC (1-15 weeks after completion of 5 weekly intraperitoneal injections of DOX at the dose of 5 mg/kg) in mice [73] (Figure 3). The results showed that the Pink1 and Parkin were prominently overexpressed at the early stage of AIC. PINK1 expression subsequently returned to baseline levels, whereas Parkin overexpression was sustained at week 9 and normalized at week 15 [73]. These results indicated that mitophagy was activated compensatively at the defensive stage at the early stage of AIC (1 week after injection), while mitophagy was gradually weakened to lower than the normal level at the middle (9 weeks after injection) and late stage (15 weeks after injection).
In addition, some studies have also reported potential therapeutic targets for the treatment of AIC by targeting mitophagy [66, 74-76] (Figure 4). For example, cytosolic p53 binds to Parkin and disturbs its translocation to damaged mitochondria and their subsequent clearance by mitophagy, and facilitates mitochondrial dysfunction and HF in AIC [66]. These results raise the possibility that therapeutic activation of mitophagy by promoting the translocation of Parkin to mitochondria may ameliorate AIC. Accordingly, Sestrin2 (SESN2) interacting with Parkin and sequestosome (p62), promoted accumulation of Parkin to mitochondria and then alleviated DOX-caused inhibition of Parkin-mediated mitophagy [75]. And both knockout of SESN2 by sgRNA and DOX treatment resulted in the inhibition of Parkin-mediated mitophagy, marked cardiomyocytes apoptosis, and mitochondria dysfunction. Ectopic expression of SESN2 effectively protected against DOX-induced cardiomyocyte apoptosis, mitochondrial injury, and cardiac dysfunction. Additionally, cardiomyocyte-specific TANK-blinding kinase 1 (TBK1) deletion aggravated chronic AIC via inhibition of mitophagy, denoting a pivotal role of TBK1 in DOX-induced mitochondrial injury and cardiotoxicity possibly through its phosphorylation and P62-mediated mitophagy [76]. These results established SESN2, TBK1, and p53 as key players in mitophagy and provided a potential therapeutic approach to AIC.
2.2 Mitochondrial biogenesis in AIC
As shown in Figure 2, mitochondrial biogenesis is a complex nuclei-mitochondrial process that relies on a complex gene regulatory network [77]. This process involves four major steps, including transcriptional activation of nuclear-encoded mitochondrial genes, mitochondrial translocation of the corresponding proteins, amplification of mitochondrial deoxyribonucleic acid (mtDNA), and synthesis of mitochondrial phospholipids [78]. PGC-1α plays a central role in a regulatory network positively governing the transcriptional control of mitochondrial biogenesis and respiratory function [77, 79].
Substantial evidences suggest that mitochondrial biogenesis is impaired in AIC. For example, it has been reported that DOX downregulates PGC-1α and its downstream proteins, such as nuclear respiratory factor 1 (Nrf1) and mitochondrial transcription factor A (TFAM) [80], and activating mitochondrial biogenesis by PGC-1α agonist, CO/heme oxygenase (CO/HO) system or miR-23a inhibitor rescues AIC [48, 81, 82]. Specifically, miR-23a expression was significantly increased by DOX concentration-dependently. Inhibition of miR-23a markedly attenuates cardiomyocyte damage by directly targeting PGC-1α or p-dynamin-related protein (Drp1), thereby inhibiting mitochondria-dependent apoptosis [82]. Latest research reported that PGC-1α was significantly up-regulated at 1 week after DOX treatment (5 weekly intraperitoneal injections of DOX at the dose of 5 mg/kg), regressing to baseline expression levels at subsequent 9 and 15 week after DOX treatment [73] (Figure 3). We hypothesized that there may be a compensatory activation in PGC-1α in early AIC.
Further study found that transcriptional regulation and post-translational modifications of PGC-1α in AIC are essential for regulating mitochondrial biogenesis and function (Figure 4). In particular, DOX promoted Top 2β binding to the promoters of PGC-1α and PGC-1β to block their transcription, and deletion of Top 2β rescued DOX-induced DNA damage and mitochondrial biogenesis defects [54]. DOX also induces DNA damage and telomere dysfunction leading to the activation of p53, which binds to and inhibits the promoters of PGC-1α and PGC-1β, resulting in impaired mitochondrial biogenesis [79, 83]. In addition, post-translational modifications regulate PGC-1α activity as well as mitochondrial biogenesis and function. PGC-1α is directly deacetylated by sirtuin 1 (Sirt1) and activated in response to changes in energy expenditure. A recent study showed that DOX downregulates Sirt1, PGC-1α, and their downstream proteins, leading to impaired mitochondrial biogenesis and cardiac dysfunction. At the same time, the reduction of Sirt1 also leads to the increase of acetylation level and inactivation of PGC-1α, which further reduces the expression of PGC-1α downstream proteins [80]. Another research has reported that depletion of poly (ADP-ribose) polymerase 2 (PARP-2) prevented DOX-induced mitochondrial dysfunction and enhanced mitochondrial biogenesis through Sirt1 activation both in aortas and aortic smooth muscle (MOVAS) cells [84]. These results suggest that Top 2β/p53/Sirt1-PGC-1α signaling plays an important role in DOX-induced mitochondrial biogenesis defects.
2.3 Mitochondrial dynamics in AIC
Mitochondria change their size, number, density, and morphology through fission and fusion in response to the metabolic state of cardiomyocytes, and the dynamic balance between these two processes is referred to as mitochondrial dynamics [85, 86]. As shown in Figure 2, mitochondrial fission not only meets the high energy demands of cardiomyocytes, but also separates dysfunctional mitochondrial parts from the mitochondrial network to maintain mitochondrial health [85]. Mitochondrial fusion generates new mitochondria with a heterogeneous membrane potential and diversity of mtDNA, proteins, and metabolites [87, 88]. Endogenous or exogenous lesions disrupt the balance between mitochondrial fission and fusion, with devastating consequences for cardiomyocyte function and cardiac health. Mitochondrial fission is initiated by the phosphorylation of Drp1, which is recruited to the outer mitochondrial membrane (OMM) by interacting with specific adaptor proteins mitochondrial fission protein 1 (Fis1), mitochondrial fission factor (Mff), mitochondrial dynamics proteins of 49 kDa (MiD49), and mitochondrial dynamics proteins of 51 kDa (MiD51) [89]. Drp1 then undergoes guanosine triphosphate (GTP)-dependent oligomerization to form ring structures that surround and shrink the mitochondria, producing two daughter mitochondria [89]. Mitochondrial fusion between OMM and inner mitochondrial membrane (IMM) is mediated by mitofusion (Mfn) 1/2 and optic atrophy type 1 (Opa1), respectively [90, 91]. Mfn1/2 interacts with homologs on the OMM of the corresponding mitochondria, and Opa1 regulates IMM fusion and mitochondrial cristae remodeling and maintenance [92].
At present, the dysregulation of Drp1-mediated mitochondrial dynamics and mitophagy in cardiovascular diseases has been reported controversially. In the transverse aortic constriction (TAC) mouse heart, transient activation of mitophagy is followed by downregulation of mitophagy and mitochondrial dysfunction. However, Drp1 heterozygous knockout blocks mitophagy and exacerbates the development of mitochondrial dysfunction and HF after TAC [93]. In ischemic/reperfusion heart injury, cardiomyocyte-specific knockout of Drp1 induces mitochondrial elongation, inhibits mitophagy, and leads to mitochondrial dysfunction [94]. On the contrary, several studies found that DOX resulted in the downregulation of mitochondrial fusion proteins Mfn1, Mfn2, and Opa1, and the up-regulation of Drp1 [95], leading to mitochondrial fragmentation [96]. Mfn2 overexpression restored mitochondrial fusion and attenuated DOX-induced apoptosis, oxidative stress, and cardiac dysfunction [97]. Latest research provides the first longitudinal analysis of the temporal changes in the heart that occur during the development of AIC (1-15 weeks after completion of 5 weekly intraperitoneal injections of DOX at the dose of 5 mg/kg) in mice [73] (Figure 3). The results showed that the increased mitochondrial size in the intermediate disease stage (9 week after injection) was accompanied by up-regulated expression of the mitochondrial fusion protein Opa1, followed by down-regulated expression at 15 weeks. While, no significant changes were detected at any disease stage in the expression of Drp1. These different conclusions may be attributed to the dosage of ANTs, the method of animal model preparation, the detection technology, and the pathological stage of AIC, which suggests that we should strictly control the time and dose of drug action when using drug intervention.
Some studies have reported potential regulatory targets for the treatment of AIC by targeting mitochondrial dynamics (Figure 4). For example, cardiomyocyte mitochondrial dynamic-related long non-coding RNAs 1 (CMDL-1) binding to Drp1 and phosphorylated Drp1 to prevent DOX-induced mitochondrial fission, may serve as a potential therapeutic target in AIC [98]. Significantly upregulated aspartate-glutamate carrier1 (AGC1) interacts with Drp1 protein to upregulate Drp1 expression, and contributes to subsequent excessive mitochondrial fission in AIC. And AGC1 knockdown protected mice from DOX-induced cardiomyopathy by preventing mitochondrial fission, while the overexpression of AGC1 in the mouse heart led to impairment of cardiac function [99]. DOX up-regulated mitochondrial fission protein 1 (Mtfp1) expression and induced a significant percentage of cells to undergo mitochondrial fission and apoptosis, and knockdown of Mtfp1 prevented cardiomyocyte from undergoing mitochondrial fission by preventing Drp1 accumulation in mitochondria [100]. DOX treatment of cardiomyocytes resulted in hyperacetylation of endogenous Opa1 at lysine 926 and 931 residues, mitochondrial fragmentation, and subsequent cardiomyocyte death, which were reverted back by overexpressing cells with sirtuin 3 (Sirt3) deacetylating Opa1 and elevating its GTPase activity [101]. Moreover, the suppression of Mfn2-mediated mitochondrial fusion was induced by DOX-elicited upregulation of forkhead box O1 (FoxO1), which inhibited the transcription of Mfn2 by binding to its promoter sites [97]. While, cardiac-specific Mfn2 transgenic mice showed preserved mitochondrial fusion and attenuated myocardial injury upon DOX exposure [97]. A recent study showed that targeting protein kinase C-ε(PKCε) and enabling its phosphorylation and activation by pharmacological intervention, activated the transcription factor signal transducer and activator of transcription 3 (Stat3) which binds to the Mfn2 promoter and up-regulates its expression, and enhanced mitochondrial fusion and cardiac function [102]. Therefore, the development of new therapeutic strategies of targeted regulation of mitochondrial dynamics genes, such as Drp1, Opa1, and Mfn2, is promising for overcoming the cardiotoxicity of ANTs.
In addition, miRNA plays a role in key mitochondrial dynamic proteins (Figure 4). Increased miR-140 in DOX-treated cardiomyocytes decreased Mfn1 expression by targeting the 3'untranslational region of Mfn1, and knockdown of miR-140 inhibited mitochondrial fission [103]. Enforced expression of apoptosis repressor with caspase recruitment domain (ARC) in vivo and in vitro attenuated DOX-induced cardiomyocyte mitochondrial fission, apoptosis, and cardiotoxicity [104]. Furthermore, miR-532-3p was found to directly target ARC and participated in DOX-induced mitochondrial fission and apoptosis [104]. Therefore, the development of new therapeutic strategies based on miR-140 and miR-532-3p is promising for overcoming the cardiotoxicity of ANTs.
Effects of ANTs on cardiac mitochondrial quality control mechanisms.
| Animals | ANTs treatment | Changes of mitochondrial quality control in AIC | Changes of heart or cardiomyocytes in AIC | References |
|---|---|---|---|---|
| Mouse | DOX was administrated in five equal intraperitoneal injections (each containing 2.5 mg▪kg-1) over a period of 2 weeks, and killed them 6 weeks after injection. | Pink1 and Parkin proteins translocating on mitochondria were downregulated, and their mediated mitophagy was inhibited. | Dysfunctional mitochondrial accumulation and cardiomyocyte death. | [66] |
| Postnatal rat cardiac myocytes | Treated with 10 μM DOX for 18 h. | Drp1 was phosphorylated (Drp1 616) and coincided with excessive mitochondrial fragmentation and mitophagy. | Increased necrotic cell death of cardiac myocytes. | [68] |
| Adult zebrafish and embryos | Zebrafish embryos were incubated with DOX (90 μM), and adult zebrafish were intraperitoneally injected with DOX (20 μg▪g-1). | Parkin activation and excessive mitophagy. | Pericardial edema, myocardial vacuolization, and apoptosis. | [69] |
| Rat | DOX was administered intraperitoneally at 5 mg▪kg-1 weekly for consecutive 6 weeks. | DOX disrupted Pink1/Parkin-mediated mitophagy and caused lysosomal impairment. | Aggravated mitochondrial oxidative stress, decreased antioxidant levels, and disrupted mitochondrial membrane potential (MMP). | [70] |
| Mouse | The mice were administrated with DOX at 6 mg▪kg-1 through the tail vein on days 0, 2, and 4. | DOX blocked autophagic flux by inhibiting autophagosome biogenesis, which was attributed to the downregulation of FUNDC1 and disruption of mitochondrial-endoplasmic reticulum contacts structures. | Cardiac remodeling and cardiac dysfunction. | [71] |
| AC16 cardiomyocytes | Treated with 0-250 nM DOX for 24 h. | Pink1/Parkin-mediated mitophagy activation and PGC-1α-mediated mitochondrial biogenesis inhibition. | Mitochondrial ultrastructural alterations, mitochondrial superoxide accumulation, and decreased MMP and mitochondrial DNA copy number. | [72] |
| Mouse | DOX was administered intraperitoneally at 5 mg▪kg-1 weekly for consecutive 5 weeks, followed by serial echocardiography for 15 weeks. | Pink1 and Parkin were overexpressed at the early stage of AIC. Pink1 expression subsequently returned to baseline levels, whereas Parkin overexpression was sustained at week 9 and normalized at week 15. PGC-1α was significantly upregulated at 1 week after DOX treatment, regressing to baseline at subsequent 9 and 15 week. Increased mitochondrial size in the intermediate stage (9 week) was accompanied by upregulated mitochondrial fusion protein Opa1, followed by downregulated expression at 15 weeks. While, no significant changes of Drp1 were detected at any stage. | Cardiac atrophy, reduced cardiomyocyte area, and decreased LVEF. | [73] |
| Mouse | DOX was administered at single dose (15 mg▪kg-1) through the tail vein. | Increased MsrB2 protein level disturbed mitophagy pathway. | Cardiac dysfunction, remodeling, fibrosis, cardiomyocyte hypertrophy, apoptosis, and oxidative stress. | [74] |
| Rat | DOX was administrated intraperitoneally by three times at 4 mg▪kg-1 over a period of 15 days. | DOX induced the inhibition of Parkin-mediated mitophagy by decreasing the interactions of SESN2 with Parkin and p62. | Cardiac dysfunction and cardiomyocytes injury indicated by morphological changes, disorganized myocardium, and increased cell gap, extracellular matrix, cytoplasmic vacuolization, and fibrosis. | [75] |
| Mouse | DOX was administered intraperitoneally once weekly for four weeks at 6 mg▪kg-1. | DOX induced mitophagy disorder, which was significantly exacerbated by Tbk1 knockdown. | DOX decreased TBK1 phosphorylation, resulting in compromised myocardial function, obvious mortality, and overt interstitial fibrosis. | [76] |
| Mouse | DOX was administered once weekly for four weeks at 5 mg▪kg-1 via the tail vein. | DOX inhibited the expression and activity of PGC-1α by promoting the expression of Sirt1, thereby increasing the expression of PGC-1α-regulated mitochondrial biogenesis and fatty acid oxidation genes. | Cardiac dysfunction, myocardial structural damage, and apoptosis. | [80] |
| Mouse | DOX was intraperitoneally administered at single dose (15 mg▪kg-1). | The mouse hearts of AIC fail to upregulate the nuclear program for mitochondrial biogenesis and its associated anti-apoptosis proteins, leading to severe mtDNA depletion. | Sarcomere destruction, apoptosis, necrosis, excessive wall stress, and fibrosis. | [48] |
| Mouse | DOX was intraperitoneally administered at single dose (25 mg▪kg-1), and killed them 16 h later. | Cardiomyocyte-specific deletion of Top2b protected cardiomyocytes from DOX-induced DNA double strand breaks and transcriptome changes that are responsible for defective mitochondrial biogenesis and ROS formation in AIC. | Cardiomyocyte-specific deletion of Top2b protected mice from the development of DOX-induced progressive HF. | [54] |
| Mouse | DOX was intraperitoneally administered at single dose (7.5 mg▪kg-1), and killed them 7 days later. | DOX induced DNA damage and telomere dysfunction leading to the activation of p53 which binds to and inhibits the promoters of PGC-1α and PGC-1β, resulting in impaired mitochondrial biogenesis. | Deteriorated cardiac systolic function. | [79] |
| Mouse | DOX was intraperitoneally administered at single dose (10 mg▪kg-1), and killed them 7 days later. | Decreased the expression of PGC-1α. | DCM-like phenotype with marked morphologic alteration in cardiac tissue and functional derangements. | [81] |
| Neonatal rat ventricular myocytes | Treated with 1μM DOX for 24 h. | DOX reduced PGC-1α protein and increased p-Drp1 protein, which were largely reversed by miR-23a inhibitor. | Reduced cell viability and MMP and increased cell death and ROS production. | [82] |
| Mouse | DOX was intraperitoneally administered at single dose (15 mg▪kg-1), and killed them 5 days later. | Decreased mtDNA copy number and expressions of key regulators for mitochondrial biogenesis. | Deteriorated cardiac systolic function. | [83] |
| Mouse | DOX was intraperitoneally administered at single dose (25 mg▪kg-1), and killed them 2 days later. | DOX induced mitochondrial dysfunction and mitochondrial biogenesis inhibition, which were largely reversed by the depletion of PARP-2 through Sirt1 activation. | Deteriorated vascular smooth muscle function. | [84] |
| Mouse | DOX was administered intraperitoneally at 4 mg▪kg-1 weekly for consecutive 3 weeks, and killed them 6 weeks after the first injection. | DOX induced Drp1 activation and mitochondrial fission through NOX1 and NOX4 activation. | DOX-induced DCM characterization disclosed that NLRP3 inflammasome activation and pyroptosis. | [95] |
| Rat | DOX was administered intraperitoneally at 2 mg▪kg-1 weekly for consecutive 7 weeks. | DOX decreased the expression of mitochondrial fusion proteins (Mfn1, Mfn2, Opa1) and increased the expression of mitochondrial fission proteins Drp1 and the activation of mitophagy. | Augmented mPTP susceptibility and apoptotic signaling. | [96] |
| Mouse | DOX was administered intraperitoneally at 5 mg▪kg-1 weekly for consecutive 3 weeks. | DOX inhibited Mfn2-mediated mitochondrial fusion. | Increased cell apoptosis and oxidative stress, and cardiac dysfunction. | [97] |
| Rat | DOX was administered intraperitoneally at 4 mg▪kg-1 weekly for consecutive 6 weeks. | DOX induced mitochondrial fission in cardiomyocytes. | Cardiac hypertrophy. | [98] |
| Mouse | DOX was administered intraperitoneally at 2.5 mg▪kg-1 three times weekly for 2 weeks, and killed them 4 weeks later. | DOX increased Drp1 Ser616 phosphorylation and mitochondrial fission. | DOX induced mitochondrial dysfunction, cardiac apoptosis and DCM. | [99] |
| HL-1 cardiac cells | Treated with 1μM DOX for 24 h. | DOX upregulated Mtfp1 expression and induced a significant percentage of cells to undergo mitochondrial fission. | Increased apoptosis of cardiac cells. | [100] |
| Neonatal rat cardiomyocytes | Treated with 1μM DOX for 24 h. | DOX treatment resulted in hyperacetylation of endogenous Opa1 and mitochondrial fragmentation, which were reverted back by overexpressing cells with Sirt3 deacetylating Opa1 and elevating its GTPase activity. | Increased death of neonatal rat cardiomyocytes. | [101] |
| Neonatal rat cardiomyocytes | Treated with 1μM DOX for 1-12 h. | Decreased Mfn1 expression and activated mitochondrial fission. | Increased apoptosis of neonatal rat cardiomyocytes. | [103] |
| Mouse | DOX was intraperitoneally administered at 4 mg▪kg-1 weekly for 4 weeks, and killed them 7 days later. | DOX induced cardiomyocyte mitochondrial fission. | Increased cardiomyocyte apoptosis. | [104] |
3. Natural products regulate mitochondrial quality control to prevent AIC
It is evident that, compared to natural products, currently approved clinical drugs typically undergo meticulous research and validation processes, boasting well-defined mechanisms of action and therapeutic effects. They have undergone strict regulatory oversight and quality control, ensuring their effectiveness and safety. However, as highlighted in this article, during the course of clinical application, dexrazoxane, a currently approved medication, has incrementally exhibited adverse effects such as secondary malignancies, prompting limitations on its utilization. Consequently, further clinical evidence is necessary to substantiate its clinical effectiveness and safety. In recent years, ACEI/ARB/ARNI or β-blockers, and SGLT inhibitors have been found to have new indications for preventing and treating anthracycline-induced cardiac dysfunction, but more clinical evidence is required to conclusively establish their efficacy and long-term safety. AIC, an emerging health concern, confronts challenges such as the scarcity of currently approved therapeutic options and uncertainties regarding their effectiveness and safety. Therefore, there is an urgent need to develop more effective and safe medications. Natural products, with their unique advantages of diverse biological activities, low toxicity, and abundant resources, hold immense promise as a novel avenue for the research and development of novel pharmaceuticals aimed at preventing and treating AIC. However, compared to the established cardiovascular medications, the clinical translation of natural products encounters significant hurdles. Specifically, certain products exhibit low bioavailability, and necessitates extensive foundational research and clinical trials to affirm their efficacy and safety. This comprehensive validation process entails considerable time and financial investments. In light of the pivotal significance of mitochondrial quality control in AIC elucidated above, targeted regulation of mitophagy, mitochondrial dynamics, and mitochondrial biogenesis emerges as a promising avenue for both the prevention and therapeutic intervention of AIC. This comprehensive review endeavors to encapsulate the recent advance made in the realm of natural compounds targeting mitophagy, mitochondrial dynamics, and mitochondrial biogenesis to protect against AIC as shown in Table 2.
Natural products regulate mitochondrial quality control to prevent AIC.
| Drugs | Animals | ANTs treatment | Effects on mitochondrial quality control | Effects on heart or cardiomyocytes | References |
|---|---|---|---|---|---|
| Harpagoside | Mouse | DOX was administered once weekly for four weeks at 5 mg▪kg-1 via the tail vein. | Inhibiting the binding of p53 and mitophagy protein Parkin to promote mitophagy | Improving cardiac function and reducing myocardial cell apoptosis and oxidative stress. | [105] |
| Aucubin | Mouse | DOX was administered once weekly for four weeks at 5 mg▪kg-1 via the tail vein. | Activating autophagy by the crosstalk between NRF2 and HIPK2. | Improving cardiac function and reducing myocardial cell apoptosis and oxidative stress. | [106] |
| Luteolin | Adult murine cardiomyocytes | 1 μM DOX for 24 h. | Promoting mitochondrial Drp1 Ser616 phosphorylation and upregulates TFEB expression, thereby to activate mitophagy. | Inhibiting apoptosis, accumulation of ROS, and loss of MMP. | [107] |
| Salidroside | Mouse | DOX was administered intraperitoneally at a single dose of 10 mg▪kg-1. | Promoting mitochondrial biogenesis. | Inhibiting DOX-induced mitochondrial ROS, Fe2+, and lipid peroxidation as well as restored MMP by improving mitochondrial iron-sulfur clusters and restoring mitochondrial OXPHOS complexes thereby to improve mitochondrial function. | [110] |
| Cannabidiol | Mouse | DOX was administered intraperitoneally at a single dose of 20 mg▪kg-1. | Activating myocardial mitochondrial biogenesis manifested as increased mitochondrial copy number, and mRNA expression of PGC-1α, PPARα, and estrogen-related receptor alpha (ERRα). | Inhibiting myocardial injury, myocardial oxidative and nitrative stress, myocardial cell death, and cardiac dysfunction. | [116] |
| Ferruginol | Mouse | DOX was administered once weekly for four weeks at 5 mg▪kg-1 via the tail vein. | Enhancing the transcription factor activity of PGC-1α by promoting the expression of deacetylase Sirt1, thereby to increase the expression of PGC-1α-mediated mitochondrial biogenesis and fatty acid oxidation genes. | Enhancing cardiac function and reducing myocardial structural damage and apoptosis. | [80] |
| Cryptotanshinone | Rat | DOX was administered intraperitoneally once every 2 days for six times at 1.25 mg▪kg-1. | Promoting mitochondrial biogenesis-relative factors PGC-1α, NRF-1, and TFAM. | Significantly promoting the energy production of ATP by increasing the complexes activities, raising MMP, and inhibiting superoxide anion free radical. | [118] |
| Liensinine | Mouse | DOX was administered intraperitoneally at single dose of 15 mg▪kg-1. | Inhibiting Drp1-mediated excess mitochondrial fission, mitochondrial fragmentation, and mitophagy. | Alleviating DOX-induced oxidative stress, cytochrome C leakage, cardiomyocyte apoptosis, as well as improving mitochondrial function and cardiomyocyte contractile function. | [120] |
| Paeonol | Rat | DOX was administered intraperitoneally at the dosage of 5 mg▪kg-1 on days 1, 6, and 11. | Promoting Mfn2-mediated mitochondria fusion by directly targeting PKCε to activate the transcription factor Stat3, which binds to the Mfn2 promoter and up-regulated its transcriptional expression. | Restoring mitochondrial function and cardiac performance. | [102] |
| Irisin | Mouse | DOX was administered intraperitoneally once weekly for four weeks at 4 mg▪kg-1. | Activating AMPK-Nrf2 signaling axis to promote mitochondrial fusion. | Alleviating DOX-induced mortality, body weight loss, myocardial atrophy, damage, and oxidative stress, and cardiac remodeling and dysfunction. | [123] |
| Ellagic acid | Postnatal rat cardiac myocytes | 10 μM DOX for 18 h. | Reducing mitochondria-associated Bnip3, resulting in significantly less mitochondrial fission and cell death. | Suppressing mitochondrial injury and necrotic cell death of cardiac myocytes. | [68] |
| Luteolin | H9c2 and AC16 cells | 1 μM DOX for 24 h. | Decreasing mitochondrial fission protein Drp1 and Drp1 Ser616 phosphorylation. | Ameliorating DOX-induced toxicity and oxidative stress. | [124] |
| Resveratrol | Mouse | DOX was administered intraperitoneally once weekly for four weeks at 8 mg▪kg-1. | Increasing the expression of mitochondrial electron transport chain complexes, Mfn1, and Mfn2 in mice. | Preventing DOX-induced LV remodeling, cardiac dysfunction, and oxidative stress in mice. | [125] |
| Honokiol | Mouse | DOX was administered intraperitoneally once every 15 days for three times at 5 mg▪kg-1. | Promoting mitochondrial fusion. | Preventing DOX-induced ROS production, mitochondrial damage, and cell death | [128] |
3.1 Natural products regulate mitophagy to prevent AIC
Harpagoside is the main component of the Scrophulariae, which exerts its salutary effects by impeding the interaction between the tumor suppressor p53 and mitophagy protein Parkin to promote mitophagy and reduce AIC [105]. Similarly, aucubin, also derived from Chinese herb Scrophulariae, modulates autophagy and apoptosis through intricate crosstalk between nuclear factor erythroid 2-related factor 2 (NRF2) and Homeodomain interacting protein kinase 2 (HIPK2) signaling pathways to alleviate AIC [106]. Luteolin is a natural product extracted from vegetables and fruits with a pleiotropic range of biological effects, including antioxidant, anti-tumor, and anti-inflammatory properties. Luteolin has been reported to attenuate AIC, including apoptosis, accumulation of ROS, and loss of mitochondrial membrane potential (MMP). Specifically, luteolin achieves these benefits by promoting mitochondrial Drp1 Ser 616 phosphorylation and upregulating transcription factor EB (TFEB) expression, thereby stimulating mitophagy [107].
3.2 Natural products regulate mitochondrial biogenesis to prevent AIC
Salidroside, a phenylpropanoid glycoside isolated from the medicinal plant Rhodiola rosea, has garnered attention for its multifaceted biological activities including anti-aging, anti-cancer, anti-inflammatory, and antioxidative functions [108, 109]. A recent investigation has unveiled that salidroside significantly improved DOX-induced cardiac dysfunction, ferroptosis-like cell damage, and fibrosis. Mechanistically, salidroside suppressed DOX-triggered mitochondrial ROS generation, Fe2+ accumulation, and lipid peroxidation as well as restored MMP by promoting mitochondrial biogenesis, improving mitochondrial iron-sulfur clusters, and restoring mitochondrial oxidative phosphorylation (OXPHOS) complexes, thereby improving mitochondrial function [110]. Cannabidiol, a major non-psychoactive phytocannabinoid in cannabis [111], is an effective treatment for some forms of epilepsy and pain [112-115]. Cannabidiol has been shown to safeguard against DOX-induced cardiomyopathy by regulating mitochondrial function and biogenesis [116]. Ferruginol, a diterpenoid compound, enhances cardiac function and reduces myocardial structural damage and apoptosis. Specifically, ferruginol augments the transcription factor activity of PGC-1α by enhancing the expression of deacetylase Sirt1, thereby promoting the expression of PGC-1α-mediated mitochondrial biogenesis and fatty acid oxidation genes [80]. Cryptotanshinone, a key bioactive constituent extracted from Salvia militorrhiza, possesses various pharmacological activities, including cardiovascular protection, neuroprotection, anti-fibrosis, anti-metabolic disorder, anti-tumor, and anti-inflammatory activities [117]. A study has reported that cryptotanshinone protects against DOX-induced mitochondrial dysfunction in cardiomyocytes. Mechanically, cryptotanshinone promotes ATP production, inhibits superoxide anion free radicals, and promotes the expression of mitochondrial biogenesis related factors PGC-1α, NRF-1, and TFAM [118]. Collectively, these natural products, by virtue of their ability to promote mitochondrial biogenesis in the setting of AIC, effectively mitigate oxidative stress and enhance mitochondrial function in cardiomyocytes, offering promising avenues for therapeutic intervention.
3.3 Natural products regulate mitochondrial dynamics to prevent AIC
Liensinine, an isoquinoline alkaloid derived from plants, is a recently recognized mitophagy inhibitor that synergizes in combination with DOX for the treatment of breast cancer. It was also found that liensinine had a protective effect against DOX-induced cardiomyopathy [119]. Liensinine alleviates DOX-induced cardiac dysfunction and apoptosis by reining in Drp1-orchestrated excessive mitochondrial fission [120]. Paeonol is a bioactive compound extracted from the root of Paeonia albiflora and has a long history of clinical use. Currently, the dosage forms of paeonol approved by the Food and Drug Administration of China for human use include tablets, injections, and ointments. Oral and injection administrations of paeonol effectively quell inflammation/pain-associated ailments, ranging from rheumatoid arthritis to headaches and muscular discomfort [121]. Moreover, paeonol harbors significant pharmacological promise for safeguarding cardiac and mitochondrial health [122]. Indeed, paeonol enhanced Mfn2-mediated mitochondrial fusion and restored mitochondrial and cardiac function. Mechanistically, paeonol promoted Mfn2-mediated mitochondria fusion by directly targeting PKCε to activate the transcription factor Stat3, which bound to the Mfn2 promoter in a direct manner and up-regulated its transcriptional expression. In addition, paeonol did not affect the antitumor effect of DOX on a variety of tumor cell [102]. Irisin, a novel exercise-mediated myokine, plays an important role in cardiovascular diseases by regulating cellular energy metabolism. Irisin combats DOX-induced oxidative stress injury in cardiomyocytes by improving mitochondrial dynamics and enhancing the endogenous antioxidant system in an adenosine 5'-monophosphate (AMP)-activated protein kinase (AMPK)-Nrf2-dependent manner, thereby protecting the heart from DOX-induced cardiotoxicity [123]. Ellagic acid, a natural polyphenolic compound, targets mitochondrial fission by impeding Bnip3 to attenuate DOX-induced phosphorylation of Drp1 Ser616 [68].
Similarly, luteolin, another natural polyphenol, ameliorates DOX-induced toxicity in H9c2 and AC16 cells by suppressing DOX-induced Drp1 expression and phosphorylation at Ser 616 [124]. Resveratrol, a natural polyphenol, was found to increase the expression of mitochondrial fusion proteins Mfn1 and Mfn2, restore mitochondrial dysfunction, and mitigate oxidative stress, thereby safeguarding against DOX-related cardiac injury [125]. Honokiol, a component of the genus Magnolia including M. officinalis, obovata, and grandifolia, has been found to be an effective antioxidant and possess the antiangiogenic, anti-inflammatory, and antitumor properties [126, 127]. Here, we found that honokiol promotes mitochondrial fusion by preserving Opa1 and Mfn1 levels, improves mitochondrial function, and reduces mitochondrial DNA damage [128]. Collectively, these natural products accommodating mitochondrial fusion and mitochondrial fission in AIC could effectively improve cardiac and mitochondrial function in cardiomyocytes, offering promising therapeutic avenues.
Conclusions
ANTs, as the cornerstone of chemotherapy for an array of malignancies, undeniably enhance the long-term survival of cancer patients, albeit at the expense of severe and dose-dependent cardiovascular complications, especially cardiotoxicity. The prevalence of AIC has escalated in recent years, ushered in an epidemic of cardio-oncology. This review delves into the current clinical types, incidence, and prevention and treatment strategies of AIC, and meticulously maps out the intricate molecular mechanism networks underlying AIC, in order to illuminate potential novel molecular targets for the development of prevention and treatment agents for AIC. Of particular note, mitochondrial dysfunction has been identified as a major driver of AIC. Impaired mitochondrial quality control mechanisms aggravate mitochondrial dysfunction and become a major contributing factor to AIC. Consequently, we also review the research progress of dysregulated mitochondrial quality control in AIC and natural products that specifically target these mechanisms, so as to provide potentially effective new drugs for the prevention and treatment of AIC in clinical practice.
During the meticulous review process, we discerned that the clinical manifestations corresponding to different AIC subtypes exhibit marked variability. Acute AIC mainly shows arrhythmia on the basis of preserved cardiac function, while chronic AIC shows cardiomyopathy characterized by abnormal cardiac function, often culminating in irreversible myocardial damage. Most clinical trials focus on patients with chronic AIC, and calculate the incidence of AIC and the remission rate of test drugs at different cumulative doses. We found that the incidence of AIC was significantly correlated with the cumulative dose of ANTs and the definition criteria of AIC. Therefore, the dose of ANTs should be strictly regulated during clinical use, and the changes of serum biochemical markers and cardiac function should be monitored throughout the course of chemotherapy, adhering to the principle of early detection and early treatment to reduce the incidence of cardiotoxicity in patients. Nevertheless, amidst the clinical prevention and treatment strategies for AIC, the high-cost liposomal DOX, SGLT-2 inhibitor, ACEI, ARB, ARNI, and β-blocker with insufficient clinical evidence have not been widely used, and only DXZ has been widely employed in the prevention of clinical AIC, but its concomitant side effects and effectiveness have also been questioned.
In conclusion, the effectiveness and safety of existing drugs for AIC still need to be supported by more clinical evidence, and more new drugs for the prevention and treatment of AIC are urgently needed to be developed. Furtherly, a profound understanding of the pathogenesis and drug targets of AIC is essential for advancing the development of therapeutic agents against AIC. As a regulatory mechanism to maintain mitochondrial homeostasis, the importance of mitochondrial quality control in AIC has been continuously confirmed. Among them, there are contradictory views on the activation and inhibition of mitophagy and the balance between mitochondrial fission and fusion, which may stem from disparities in the detection technology, the stage of the disease, and other confounding factors. Recent advancements have witnessed the emergence of natural products that specifically target mitochondrial quality control, demonstrating promising cardioprotective effects. But the translation of these agents into clinical practice poses major challenges, stemming from their potential adverse effects and the elusiveness of mechanisms of action, as well as the variability of mitochondrial biology in response to various stress conditions. Therefore, the focus of further research is to determine the precise treatment regimen and specific targets of action of different drugs targeting and regulating quality control mechanisms, as well as the clinical efficacy and safety of these drugs in AIC patients.
Funding
This work was supported by Noncommunicable Chronic Diseases-National Science and Technology Major Project [grant numbers 2023ZD0502605], Beijing-tianjin-hebei Basic Research Cooperation project [grant numbers J230033], the National Natural Science Foundation of China [grant numbers 82374420, 82174364], the Science and Technology Department of Beijing University of Chinese Medicine [grant numbers 2022-JYB-JBZR-001], and Science and Technology Department of Beijing University of Chinese Medicine [grant numbers BZY-BZX-2022-08].
Competing Interests
The authors have declared that no competing interest exists.
References
1. Di Marco A, Cassinelli G, Arcamone F. The discovery of daunorubicin. Cancer Treat Rep. 1981;65(Suppl 4):3-8
2. Arcamone F, Cassinelli G, Fantini G, Grein A, Orezzi P, Pol C. et al. Adriamycin, 14-hydroxydaunomycin, a new antitumor antibiotic from S. peucetius var. caesius. Biotechnol Bioeng. 1969;11:1101-10
3. Smith LA, Cornelius VR, Plummer CJ, Levitt G, Verrill M, Canney P. et al. Cardiotoxicity of anthracycline agents for the treatment of cancer: systematic review and meta-analysis of randomised controlled trials. BMC Cancer. 2010;10:337
4. Khouri MG, Douglas PS, Mackey JR, Martin M, Scott JM, Scherrer-Crosbie M. et al. Cancer therapy-induced cardiac toxicity in early breast cancer: addressing the unresolved issues. Circulation. 2012;126:2749-63
5. Giantris A, Abdurrahman L, Hinkle A, Asselin B, Lipshultz SE. Anthracycline-induced cardiotoxicity in children and young adults. Crit Rev Oncol Hematol. 1998;27:53-68
6. Grenier MA, Lipshultz SE. Epidemiology of anthracycline cardiotoxicity in children and adults. Semin Oncol. 1998;25:72-85
7. Lipshultz SE, Alvarez JA, Scully RE. Anthracycline associated cardiotoxicity in survivors of childhood cancer. Heart. 2008;94:525-33
8. Yeh ETH, Bickford CL. Cardiovascular complications of cancer therapy: incidence, pathogenesis, diagnosis, and management. J Am Coll Cardiol. 2009;53:2231-47
9. Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, Veglia F. et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation. 2015;131:1981-8
10. Von Hoff DD, Layard MW, Basa P, Davis HL, Von Hoff AL, Rozencweig M. et al. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med. 1979;91:710-7
11. Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003;97:2869-79
12. Cardinale D, Sandri MT, Martinoni A, Borghini E, Civelli M, Lamantia G. et al. Myocardial injury revealed by plasma troponin I in breast cancer treated with high-dose chemotherapy. Ann Oncol. 2002;13:710-5
13. Linschoten M, Teske AJ, Cramer MJ, van der Wall E, Asselbergs FW. Chemotherapy-Related Cardiac Dysfunction: A Systematic Review of Genetic Variants Modulating Individual Risk. Circ Genom Precis Med. 2018;11:e001753
14. Zamorano JL, Lancellotti P, Rodriguez Muñoz D, Aboyans V, Asteggiano R, Galderisi M. et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur J Heart Fail. 2016;37:2768-2801
15. Okwuosa TM, Keramida K, Filippatos G, Yancy CW. Cancer therapy and the heart; the necessity to calibrate risk. Eur J Heart Fail. 2020;22:1961-5
16. Lyon AR, Dent S, Stanway S, Earl H, Brezden-Masley C, Cohen-Solal A. et al. Baseline cardiovascular risk assessment in cancer patients scheduled to receive cardiotoxic cancer therapies: a position statement and new risk assessment tools from the Cardio-Oncology Study Group of the Heart Failure Association of the European Society of Cardiology in collaboration with the International Cardio-Oncology Society. Eur J Heart Fail. 2020;22:1945-60
17. Wang X, Singh P, Zhou L, Sharafeldin N, Landier W, Hageman L. et al. Genome-Wide Association Study Identifies ROBO2 as a Novel Susceptibility Gene for Anthracycline-Related Cardiomyopathy in Childhood Cancer Survivors. J Clin Oncol. 2023;41:1758-69
18. Gabizon AA. Stealth liposomes and tumor targeting: one step further in the quest for the magic bullet. Clin Cancer Res. 2001;7:223-5
19. Waterhouse DN, Tardi PG, Mayer LD, Bally MB. A comparison of liposomal formulations of doxorubicin with drug administered in free form: changing toxicity profiles. Drug Saf. 2001;24:903-20
20. USFaDA. Drug Safety and Availability. p. FDA statement on dexrazoxane. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/020212s017lbl.pdf
21. Lipshultz SE, Scully RE, Lipsitz SR, Sallan SE, Silverman LB, Miller TL. et al. Assessment of dexrazoxane as a cardioprotectant in doxorubicin-treated children with high-risk acute lymphoblastic leukaemia: long-term follow-up of a prospective, randomised, multicentre trial. Lancet Oncol. 2010;11:950-61
22. Speyer JL, Green MD, Zeleniuch-Jacquotte A, Wernz JC, Rey M, Sanger J. et al. ICRF-187 permits longer treatment with doxorubicin in women with breast cancer. J Clin Oncol. 1992;10:117-27
23. Schroeder PE, Hasinoff BB. The doxorubicin-cardioprotective drug dexrazoxane undergoes metabolism in the rat to its metal ion-chelating form ADR-925. Cancer Chemother Pharmacol. 2002;50:509-13
24. Hasinoff BB, Patel D, Wu X. The oral iron chelator ICL670A (deferasirox) does not protect myocytes against doxorubicin. Free Radic Biol Med. 2003;35:1469-79
25. Lyu YL, Kerrigan JE, Lin C-P, Azarova AM, Tsai Y-C, Ban Y. et al. Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007;67:8839-46
26. Swain SM, Whaley FS, Gerber MC, Weisberg S, York M, Spicer D. et al. Cardioprotection with dexrazoxane for doxorubicin-containing therapy in advanced breast cancer. J Clin Oncol. 1997;15:1318-32
27. Wadler S, Green MD, Muggia FM. Synergistic activity of doxorubicin and the bisdioxopiperazine (+)-1,2-bis(3,5-dioxopiperazinyl-1-yl)propane (ICRF 187) against the murine sarcoma S180 cell line. Cancer Res. 1986;46:1176-81
28. Asselin BL, Devidas M, Chen L, Franco VI, Pullen J, Borowitz MJ. et al. Cardioprotection and Safety of Dexrazoxane in Patients Treated for Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia or Advanced-Stage Lymphoblastic Non-Hodgkin Lymphoma: A Report of the Children's Oncology Group Randomized Trial Pediatric Oncology Group 9404. J Clin Oncol. 2016;34:854-62
29. Tebbi CK, London WB, Friedman D, Villaluna D, De Alarcon PA, Constine LS. et al. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin's disease. J Clin Oncol. 2007;25:493-500
30. Vrooman LM, Neuberg DS, Stevenson KE, Asselin BL, Athale UH, Clavell L. et al. The low incidence of secondary acute myelogenous leukaemia in children and adolescents treated with dexrazoxane for acute lymphoblastic leukaemia: a report from the Dana-Farber Cancer Institute ALL Consortium. Eur J Cancer. 2011;47:1373-9
31. Salzer WL, Devidas M, Carroll WL, Winick N, Pullen J, Hunger SP. et al. Long-term results of the pediatric oncology group studies for childhood acute lymphoblastic leukemia 1984-2001: a report from the children's oncology group. Leukemia. 2010;24:355-70
32. Heck SL, Mecinaj A, Ree AH, Hoffmann P, Schulz-Menger J, Fagerland MW. et al. Prevention of Cardiac Dysfunction During Adjuvant Breast Cancer Therapy (PRADA): Extended Follow-Up of a 2×2 Factorial, Randomized, Placebo-Controlled, Double-Blind Clinical Trial of Candesartan and Metoprolol. Circulation. 2021;143:2431-40
33. Avila MS, Ayub-Ferreira SM, de Barros Wanderley MR, das Dores Cruz F, Gonçalves Brandão SM, Rigaud VOC. et al. Carvedilol for Prevention of Chemotherapy-Related Cardiotoxicity: The CECCY Trial. J Am Coll Cardiol. 2018;71:2281-90
34. Henriksen PA, Hall P, MacPherson IR, Joshi SS, Singh T, Maclean M. et al. Multicenter, Prospective, Randomized Controlled Trial of High-Sensitivity Cardiac Troponin I-Guided Combination Angiotensin Receptor Blockade and Beta-Blocker Therapy to Prevent Anthracycline Cardiotoxicity: The Cardiac CARE Trial. Circulation. 2023;148:1680-90
35. Sabatino J, De Rosa S, Tammè L, Iaconetti C, Sorrentino S, Polimeni A. et al. Empagliflozin prevents doxorubicin-induced myocardial dysfunction. Cardiovasc Diabetol. 2020;19:66
36. Quagliariello V, De Laurentiis M, Rea D, Barbieri A, Monti MG, Carbone A. et al. The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc Diabetol. 2021;20:150
37. Gongora CA, Drobni ZD, Quinaglia Araujo Costa Silva T, Zafar A, Gong J, Zlotoff DA. et al. Sodium-Glucose Co-Transporter-2 Inhibitors and Cardiac Outcomes Among Patients Treated With Anthracyclines. JACC Heart Fail. 2022;10:559-67
38. Doroshow JH. Effect of anthracycline antibiotics on oxygen radical formation in rat heart. Cancer Res. 1983;43:460-72
39. Pan J-A, Zhang H, Lin H, Gao L, Zhang H-L, Zhang J-F. et al. Irisin ameliorates doxorubicin-induced cardiac perivascular fibrosis through inhibiting endothelial-to-mesenchymal transition by regulating ROS accumulation and autophagy disorder in endothelial cells. Redox Biol. 2021;46:102120
40. Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV. et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest. 2014;124:617-30
41. Hasinoff BB, Patel D. The iron chelator Dp44mT does not protect myocytes against doxorubicin. J Inorg Biochem. 2009;103:1093-101
42. Van Vleet JF, Ferrans VJ, Weirich WE. Cardiac disease induced by chronic adriamycin administration in dogs and an evaluation of vitamin E and selenium as cardioprotectants. Am J Pathol. 1980;99:13-42
43. Wu BB, Leung KT, Poon EN-Y. Mitochondrial-Targeted Therapy for Doxorubicin-Induced Cardiotoxicity. International Journal of Molecular Sciences. 2022;23:1912
44. Goormaghtigh E, Chatelain P, Caspers J, Ruysschaert JM. Evidence of a complex between adriamycin derivatives and cardiolipin: possible role in cardiotoxicity. Biochem Pharmacol. 1980;29:3003-10
45. Goormaghtigh E, Huart P, Brasseur R, Ruysschaert JM. Mechanism of inhibition of mitochondrial enzymatic complex I-III by adriamycin derivatives. Biochim Biophys Acta. 1986;861:83-94
46. Kashfi K, Israel M, Sweatman TW, Seshadri R, Cook GA. Inhibition of mitochondrial carnitine palmitoyltransferases by adriamycin and adriamycin analogues. Biochem Pharmacol. 1990;40:1441-8
47. Carvalho RA, Sousa RPB, Cadete VJJ, Lopaschuk GD, Palmeira CMM, Bjork JA. et al. Metabolic remodeling associated with subchronic doxorubicin cardiomyopathy. Toxicology. 2010;270:92-8
48. Suliman HB, Carraway MS, Ali AS, Reynolds CM, Welty-Wolf KE, Piantadosi CA. The CO/HO system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J Clin Invest. 2007;117:3730-41
49. Cole MP, Chaiswing L, Oberley TD, Edelmann SE, Piascik MT, Lin S-M. et al. The protective roles of nitric oxide and superoxide dismutase in adriamycin-induced cardiotoxicity. Cardiovasc Res. 2006;69:186-97
50. Li X, Lin Y, Wang S, Zhou S, Ju J, Wang X. et al. Extracellular Superoxide Dismutase Is Associated With Left Ventricular Geometry and Heart Failure in Patients With Cardiovascular Disease. J Am Heart Assoc. 2020;9:e016862
51. Liu LF, Wang JC. Supercoiling of the DNA template during transcription. Proc Natl Acad Sci U S A. 1987;84:7024-7
52. Carpenter AJ, Porter ACG. Construction, characterization, and complementation of a conditional-lethal DNA topoisomerase IIalpha mutant human cell line. Mol Biol Cell. 2004;15:5700-11
53. Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science (New York, NY). 1984;226:466-8
54. Zhang S, Liu X, Bawa-Khalfe T, Lu L-S, Lyu YL, Liu LF. et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18:1639-42
55. Bugger H, Guzman C, Zechner C, Palmeri M, Russell KS, Russell RR. Uncoupling protein downregulation in doxorubicin-induced heart failure improves mitochondrial coupling but increases reactive oxygen species generation. Cancer Chemother Pharmacol. 2011;67:1381-8
56. Kücük P, Abbey L, Schmitt J, Henninger C, Fritz G. Cardiomyocytes, cardiac endothelial cells and fibroblasts contribute to anthracycline-induced cardiac injury through RAS-homologous small GTPases RAC1 and CDC42. Pharmacol Res. 2024;203:107165
57. Nam J-K, Kim AR, Choi S-H, Kim J-H, Choi KJ, Cho S. et al. An antibody against L1 cell adhesion molecule inhibits cardiotoxicity by regulating persistent DNA damage. Nat Commun. 2021;12:3279
58. Luo W, Zou X, Wang Y, Dong Z, Weng X, Pei Z. et al. Critical Role of the cGAS-STING Pathway in Doxorubicin-Induced Cardiotoxicity. Circ Res. 2023;132:e223-e42
59. Kolwicz SC, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. 2013;113:603-16
60. Wallace KB, Sardão VA, Oliveira PJ. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ Res. 2020;126:926-41
61. Eldeeb MA, Thomas RA, Ragheb MA, Fallahi A, Fon EA. Mitochondrial quality control in health and in Parkinson's disease. Physiol Rev. 2022;102:1721-55
62. Tang C, Cai J, Yin X-M, Weinberg JM, Venkatachalam MA, Dong Z. Mitochondrial quality control in kidney injury and repair. Nat Rev Nephrol. 2021;17:299-318
63. Wu L, Wang L, Du Y, Zhang Y, Ren J. Mitochondrial quality control mechanisms as therapeutic targets in doxorubicin-induced cardiotoxicity. Trends Pharmacol Sci. 2023;44:34-49
64. Hull TD, Boddu R, Guo L, Tisher CC, Traylor AM, Patel B. et al. Heme oxygenase-1 regulates mitochondrial quality control in the heart. JCI Insight. 2016;1:e85817
65. Kubli DA, Gustafsson ÅB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. 2012;111:1208-21
66. Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T. et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 2013;4:2308
67. Dhingra R, Margulets V, Chowdhury SR, Thliveris J, Jassal D, Fernyhough P. et al. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc Natl Acad Sci U S A. 2014;111:E5537-E44
68. Dhingra A, Jayas R, Afshar P, Guberman M, Maddaford G, Gerstein J. et al. Ellagic acid antagonizes Bnip3-mediated mitochondrial injury and necrotic cell death of cardiac myocytes. Free Radic Biol Med. 2017;112:411-22
69. Zhou J, Lu Y, Li Z, Wang Z, Kong W, Zhao J. Sphingosylphosphorylcholine ameliorates doxorubicin-induced cardiotoxicity in zebrafish and H9c2 cells by reducing excessive mitophagy and mitochondrial dysfunction. Toxicol Appl Pharmacol. 2022;452:116207
70. Pandey S, Kuo W-W, Shen C-Y, Yeh Y-L, Ho T-J, Chen R-J. et al. Insulin-like growth factor II receptor-α is a novel stress-inducible contributor to cardiac damage underpinning doxorubicin-induced oxidative stress and perturbed mitochondrial autophagy. Am J Physiol Cell Physiol. 2019;317:C235-C43
71. He W, Sun Z, Tong G, Zeng L, He W, Chen X. et al. FUNDC1 alleviates doxorubicin-induced cardiotoxicity by restoring mitochondrial-endoplasmic reticulum contacts and blocked autophagic flux. Theranostics. 2024;14:3719-38
72. Yin J, Guo J, Zhang Q, Cui L, Zhang L, Zhang T. et al. Doxorubicin-induced mitophagy and mitochondrial damage is associated with dysregulation of the PINK1/parkin pathway. Toxicol In Vitro. 2018;51:1-10
73. Díaz-Guerra A, Villena-Gutiérrez R, Clemente-Moragón A, Gómez M, Oliver E, Fernández-Tocino M. et al. Anthracycline Cardiotoxicity Induces Progressive Changes in Myocardial Metabolism and Mitochondrial Quality Control: Novel Therapeutic Target. JACC CardioOncol. 2024;6:217-32
74. Lu L, Shao Y, Wang N, Xiong X, Zhai M, Tang J. et al. Follistatin-like protein 1 attenuates doxorubicin-induced cardiomyopathy by inhibiting MsrB2-mediated mitophagy. Mol Cell Biochem. 2024;479:1817-1831
75. Wang P, Wang L, Lu J, Hu Y, Wang Q, Li Z. et al. SESN2 protects against doxorubicin-induced cardiomyopathy via rescuing mitophagy and improving mitochondrial function. J Mol Cell Cardiol. 2019;133:125-37
76. Yu W, Deng D, Li Y, Ding K, Qian Q, Shi H. et al. Cardiomyocyte-specific Tbk1 deletion aggravated chronic doxorubicin cardiotoxicity via inhibition of mitophagy. Free Radic Biol Med. 2024;222:244-58
77. Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611-38
78. Zhou H, Ren J, Toan S, Mui D. Role of mitochondrial quality surveillance in myocardial infarction: From bench to bedside. Ageing Res Rev. 2021;66:101250
79. Sahin E, Colla S, Liesa M, Moslehi J, Müller FL, Guo M. et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359-65
80. Li W, Cao J, Wang X, Zhang Y, Sun Q, Jiang Y. et al. Ferruginol Restores SIRT1-PGC-1α-Mediated Mitochondrial Biogenesis and Fatty Acid Oxidation for the Treatment of DOX-Induced Cardiotoxicity. Frontiers In Pharmacology. 2021;12:773834
81. Shipra Tembhre MK, Hote MP Bhari N, Lakshmy R Kumaran SS. PGC-1α Agonist Rescues Doxorubicin-Induced Cardiomyopathy by Mitigating the Oxidative Stress and Necroptosis. Antioxidants (Basel). 2023;12:1720
82. Du J, Hang P, Pan Y, Feng B, Zheng Y, Chen T. et al. Inhibition of miR-23a attenuates doxorubicin-induced mitochondria-dependent cardiomyocyte apoptosis by targeting the PGC-1α/Drp1 pathway. Toxicol Appl Pharmacol. 2019;369:73-81
83. Miyagawa K, Emoto N, Widyantoro B, Nakayama K, Yagi K, Rikitake Y. et al. Attenuation of Doxorubicin-induced cardiomyopathy by endothelin-converting enzyme-1 ablation through prevention of mitochondrial biogenesis impairment. Hypertension. 2010;55:738-46
84. Szántó M, Rutkai I, Hegedus C, Czikora Á, Rózsahegyi M, Kiss B. et al. Poly(ADP-ribose) polymerase-2 depletion reduces doxorubicin-induced damage through SIRT1 induction. Cardiovasc Res. 2011;92:430-8
85. Wai T, Langer T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol Metab. 2016;27:105-17
86. Dorn GW, Vega RB, Kelly DP. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015;29:1981-91
87. Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science (New York, NY). 2012;337:1062-5
88. Yang L, Long Q, Liu J, Tang H, Li Y, Bao F. et al. Mitochondrial fusion provides an 'initial metabolic complementation' controlled by mtDNA. Cell Mol Life Sci. 2015;72:2585-98
89. Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlén P. et al. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011;30:2762-78
90. Ban T, Kohno H, Ishihara T, Ishihara N. Relationship between OPA1 and cardiolipin in mitochondrial inner-membrane fusion. Biochim Biophys Acta Bioenerg. 2018;1859:951-7
91. Noone J, O'Gorman DJ, Kenny HC. OPA1 regulation of mitochondrial dynamics in skeletal and cardiac muscle. Trends Endocrinol Metab. 2022;33:710-21
92. Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M. et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160-71
93. Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu C-P. et al. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure. Circulation. 2016;133:1249-63
94. Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J. et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015;116:264-78
95. Zeng C, Duan F, Hu J, Luo B, Huang B, Lou X. et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol. 2020;34:101523
96. Marques-Aleixo I, Santos-Alves E, Torrella JR, Oliveira PJ, Magalhães J, Ascensão A. Exercise and Doxorubicin Treatment Modulate Cardiac Mitochondrial Quality Control Signaling. Cardiovasc Toxicol. 2018;18:43-55
97. Ding M, Shi R, Cheng S, Li M, De D, Liu C. et al. Mfn2-mediated mitochondrial fusion alleviates doxorubicin-induced cardiotoxicity with enhancing its anticancer activity through metabolic switch. Redox Biol. 2022;52:102311
98. Aung LHH, Chen X, Cueva Jumbo JC, Li Z, Wang S-Y, Zhao C. et al. Cardiomyocyte mitochondrial dynamic-related lncRNA 1 (CMDL-1) may serve as a potential therapeutic target in doxorubicin cardiotoxicity. Mol Ther Nucleic Acids. 2021;25:638-51
99. Xia Y, Jin J, Chen A, Lu D, Che X, Ma J. et al. Mitochondrial aspartate/glutamate carrier AGC1 regulates cardiac function via Drp1-mediated mitochondrial fission in doxorubicin-induced cardiomyopathy. Transl Res. 2023;261:28-40
100. Aung LHH, Li R, Prabhakar BS, Li P. Knockdown of Mtfp1 can minimize doxorubicin cardiotoxicity by inhibiting Dnm1l-mediated mitochondrial fission. J Cell Mol Med. 2017;21:3394-404
101. Samant SA, Zhang HJ, Hong Z, Pillai VB, Sundaresan NR, Wolfgeher D. et al. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol Cell Biol. 2014;34:807-19
102. Ding M, Shi R, Fu F, Li M, De D, Du Y. et al. Paeonol protects against doxorubicin-induced cardiotoxicity by promoting Mfn2-mediated mitochondrial fusion through activating the PKCε-Stat3 pathway. J Adv Res. 2023;47:151-62
103. Li J, Li Y, Jiao J, Wang J, Li Y, Qin D. et al. Mitofusin 1 is negatively regulated by microRNA 140 in cardiomyocyte apoptosis. Mol Cell Biol. 2014;34:1788-99
104. Wang JX, Zhang XJ, Feng C, Sun T, Wang K, Wang Y. et al. MicroRNA-532-3p regulates mitochondrial fission through targeting apoptosis repressor with caspase recruitment domain in doxorubicin cardiotoxicity. Cell Death Dis. 2015;6:e1677
105. Li W, Wang X, Liu T, Zhang Q, Cao J, Jiang Y. et al. Harpagoside Protects Against Doxorubicin-Induced Cardiotoxicity via P53-Parkin-Mediated Mitophagy. Front Cell Dev Biol. 2022;10:813370
106. Li W, Cao J, Zhang Y, Ling G, Tan N, Wei Y. et al. Aucubin alleviates doxorubicin-induced cardiotoxicity through crosstalk between NRF2 and HIPK2 mediating autophagy and apoptosis. Phytomedicine. 2024;127:155473
107. Xu H, Yu W, Sun S, Li C, Zhang Y, Ren J. Luteolin Attenuates Doxorubicin-Induced Cardiotoxicity Through Promoting Mitochondrial Autophagy. Front Physiol. 2020;11:113
108. Kanupriya Prasad D, Sai Ram M Kumar R, Sawhney RC Sharma SK. et al. Cytoprotective and antioxidant activity of Rhodiola imbricata against tert-butyl hydroperoxide induced oxidative injury in U-937 human macrophages. Mol Cell Biochem. 2005;275:1-6
109. Wang H, Ding Y, Zhou J, Sun X, Wang S. The in vitro and in vivo antiviral effects of salidroside from Rhodiola rosea L. against coxsackievirus B3. Phytomedicine. 2009;16:146-55
110. Chen H, Zhu J, Le Y, Pan J, Liu Y, Liu Z. et al. Salidroside inhibits doxorubicin-induced cardiomyopathy by modulating a ferroptosis-dependent pathway. Phytomedicine. 2022;99:153964
111. Mechoulam R, Shani A, Edery H, Grunfeld Y. Chemical basis of hashish activity. Science (New York, NY). 1970;169:611-2
112. Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R. et al. Trial of Cannabidiol for Drug-Resistant Seizures in the Dravet Syndrome. N Engl J Med. 2017;376:2011-20
113. Thiele E, Marsh E, Mazurkiewicz-Beldzinska M, Halford JJ, Gunning B, Devinsky O. et al. Cannabidiol in patients with Lennox-Gastaut syndrome: Interim analysis of an open-label extension study. Epilepsia. 2019;60:419-28
114. Alaia MJ, Hurley ET, Vasavada K, Markus DH, Britton B, Gonzalez-Lomas G. et al. Buccally Absorbed Cannabidiol Shows Significantly Superior Pain Control and Improved Satisfaction Immediately After Arthroscopic Rotator Cuff Repair: A Placebo-Controlled, Double-Blinded, Randomized Trial. Am J Sports Med. 2022;50:3056-63
115. Verrico CD, Wesson S, Konduri V, Hofferek CJ, Vazquez-Perez J, Blair E. et al. A randomized, double-blind, placebo-controlled study of daily cannabidiol for the treatment of canine osteoarthritis pain. Pain. 2020;161:2191-202
116. Hao E, Mukhopadhyay P, Cao Z, Erdélyi K, Holovac E, Liaudet L. et al. Cannabidiol Protects against Doxorubicin-Induced Cardiomyopathy by Modulating Mitochondrial Function and Biogenesis. Mol Med. 2015;21:38-45
117. Wu Y-H, Wu Y-R, Li B, Yan Z-Y. Cryptotanshinone: A review of its pharmacology activities and molecular mechanisms. Fitoterapia. 2020;145:104633
118. Zhang Y, Chen L, Li F, Wang H, Yao Y, Shu J. et al. Cryptotanshinone protects against adriamycin-induced mitochondrial dysfunction in cardiomyocytes. Pharm Biol. 2016;54:237-42
119. Zhou J, Li G, Zheng Y, Shen H-M, Hu X, Ming Q-L. et al. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy. 2015;11:1259-79
120. Liang X, Wang S, Wang L, Ceylan AF, Ren J, Zhang Y. Mitophagy inhibitor liensinine suppresses doxorubicin-induced cardiotoxicity through inhibition of Drp1-mediated maladaptive mitochondrial fission. Pharmacol Res. 2020;157:104846
121. Zhang L, Li D-C, Liu L-F. Paeonol: pharmacological effects and mechanisms of action. Int Immunopharmacol. 2019;72:413-21
122. Liu C, Han Y, Gu X, Li M, Du Y, Feng N. et al. Paeonol promotes Opa1-mediated mitochondrial fusion via activating the CK2α-Stat3 pathway in diabetic cardiomyopathy. Redox Biol. 2021;46:102098
123. Zhuo C, Xin J, Huang W, Zhang D, Yan X, Li R. et al. Irisin protects against doxorubicin-induced cardiotoxicity by improving AMPK-Nrf2 dependent mitochondrial fusion and strengthening endogenous anti-oxidant defense mechanisms. Toxicology. 2023;494:153597
124. Shi Y, Li F, Shen M, Sun C, Hao W, Wu C. et al. Luteolin Prevents Cardiac Dysfunction and Improves the Chemotherapeutic Efficacy of Doxorubicin in Breast Cancer. Front Cardiovasc Med. 2021;8:750186
125. Dolinsky VW, Rogan KJ, Sung MM, Zordoky BN, Haykowsky MJ, Young ME. et al. Both aerobic exercise and resveratrol supplementation attenuate doxorubicin-induced cardiac injury in mice. Am J Physiol Endocrinol Metab. 2013;305:E243-E53
126. Fried LE, Arbiser JL. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid Redox Signal. 2009;11:1139-48
127. Weng TI, Wu HY, Kuo CW, Liu SH. Honokiol rescues sepsis-associated acute lung injury and lethality via the inhibition of oxidative stress and inflammation. Intensive Care Med. 2011;37:533-41
128. Pillai VB, Kanwal A, Fang YH, Sharp WW, Samant S, Arbiser J. et al. Honokiol, an activator of Sirtuin-3 (SIRT3) preserves mitochondria and protects the heart from doxorubicin-induced cardiomyopathy in mice. Oncotarget. 2017;8:34082-98
Author contact
![]() Corresponding authors: E-mail addresses: wangwei26960com (Wei Wang), wangyongedu.cn (Yong Wang).
Corresponding authors: E-mail addresses: wangwei26960com (Wei Wang), wangyongedu.cn (Yong Wang).